

Abstract
Trypanosoma brucei, the causative agent of Human African Trypanosomiasis (HAT) and animal trypanosomiases, cycles between a bloodstream form in mammals and a procyclic form in the gut of its insect vector. We previously discovered that the human bromodomain inhibitor I-BET151 causes transcriptome changes that resemble the transition from the bloodstream to the procyclic form. In particular, I-BET151 induces replacement of variant surface glycoprotein (VSG) with procyclin protein. While modest binding of I-BET151 to TbBdf2 and TbBdf3 has been demonstrated, it is unknown whether I-BET151 binds to other identified T. brucei bromodomain proteins and/or other targets. To identify target(s) in T. brucei, we have synthesized I-BET151 derivatives maintaining the key pharmacophoric elements with functionality useful for chemoproteomic approaches. We identified compounds that are potent in inducing expression of procyclin, delineating a strategy towards the design of drugs against HAT and other trypanosomiases. Furthermore, these derivatives represent useful chemical probes to elucidate the molecular mechanism underlying I-BET151-induced differentiation.
Keywords: Human African trypanosomiasis, Bromodomain, Inhibitor, Chemical probes
1. Introduction
Trypanosoma brucei is a protozoan parasite that infects humans and ungulates in sub-Saharan Africa and is therefore referred to as the African trypanosome. Two subspecies (T. brucei gambiense and T. brucei rhodesiense) cause Human African Trypanosomiasis (HAT, also known as African sleeping sickness), which is almost always fatal if untreated. There are currently six compounds in use for the treatment of HAT: pentamidine (Wispelwey and Pearson, 1991) (administered i.v. or aerosolised (Jesuthasan et al., 1987)), suramin (administered by i.v. (see PubMed Health)), melarsoprol (administered i.v.) (Bisser et al., 2007), an arsenic-containing drug known to have numerous side effects, an eflornithine (i.v.)/nifurtimox (oral) combination therapy (NECT), (Priotto et al., 2009) and fexinidazole (De Rycker et al., 2023). While the first five drugs cause a variety of adverse effects and can be challenging to administer, fexinidazole, a 2-substituted 5-nitroimidazole, can be used as a short-term, safe and effective oral treatment (Torreele et al., 2010; Deeks, 2019). However, fexinidazole is not effective against animal trypanosomiases, and these animal diseases impose a severe economic burden on communities that are dependent upon cattle for subsistence and farming (Alsan, 2015). Thus, there is a continuing need to develop novel, safe, and affordable drugs against animal trypanosomiases to alleviate the economic burden they impose on Sub-Saharan Africa.
The life cycle of T. brucei requires adaptation to distinct environments in the tsetse fly (its insect vector) and mammalian hosts (Rico et al., 2013). Within these two host organisms, T. brucei progresses through a series of proliferative and quiescent developmental forms, which significantly vary in their transcriptome and proteome. These differences are reflected in vast developmental changes in metabolism, morphology, cell surface molecules, and virulence among others (Gunasekera et al., 2012) (Dejung et al., 2016) (Queiroz et al., 2009). While the phenotypic changes in the distinct stages have been well characterized, the regulatory mechanisms underlying these changes on the molecular level are not fully understood (Matthews et al., 2004). Because trypanosomes transcribe their genome from polycistronic transcription units that typically contain functionally unrelated genes and because they lack apparent sequence-specific transcription factors, (Nguyen et al., 2012) the majority of research has focused on post-transcriptional gene regulatory mechanisms (Clayton, 2016). Large changes in gene expression are observed at the mRNA level during differentiation (Queiroz et al., 2009) (Vasquez et al., 2014) (Jensen et al., 2014), mediated in part by the interplay between cis-elements such as 5’ and 3’ untranslated regions (Pasion et al., 1996) (Hotz et al., 1997) (Schürch et al., 1997) (Jojic et al., 2018) and trans-factors such as RNA-binding proteins (Clayton, 2016) (Antwi et al., 2016) (Mugo and Clayton, 2017). Together, alternative splicing, RNA editing, mRNA stability, and translational efficiency contribute to the developmental regulation of gene expression (Vasquez et al., 2014) (Jensen et al., 2014) (Nilsson et al., 2010) (McDermott et al., 2019). However, a fundamental understanding of the molecular pathways that link environmental cues to changes in gene expression and ultimately in trypanosome phenotype is still lacking.
We recently characterized the effect of the human bromodomain inhibitor I-BET151 on bloodstream trypanosomes (Schulz et al., 2015). Upon exposure of the bloodstream form of T. brucei to I-BET151, we observed drastic changes in gene expression and phenotype that mimic those seen in parasites differentiating from the long slender bloodstream form to the insect-stage procyclic form (Schulz et al., 2015). Among the most prominent of these changes was remodeling of the surface proteins, wherein the variant surface glycoprotein (VSG) responsible for antigenic variation and immune evasion was replaced with the insect-stage invariant protein procyclin. This phenotype inspired us to explore the idea that drug-induced differentiation may constitute a novel chemotherapeutic approach for sleeping sickness, an idea that has also been described by others (Schulz et al., 2015) (Wenzler et al., 2016). Indeed, we observed that the bloodstream-specific processes of immune evasion were severely compromised upon I-BET151 treatment (Schulz et al., 2015). When we injected I-BET151-treated trypanosomes into mice, 80% of mice survived, which would otherwise completely succumb to infection with untreated T. brucei parasites (Schulz et al., 2015). These proof-of-concept experiments demonstrate that drug-induced differentiation is a powerful, novel concept to combat African trypanosomiasis.
To validate I-BET151 targets in the African trypanosome and to identify pharmacophoric features of I-BET151 that are critical or dispensable for its differentiation-inducing effect, we report here the synthesis of modified I-BET151 derivatives in which we introduced functionalities to enable target identification using chemoproteomic approaches. To confirm that I-BET151 derivatives retained the biological activity observed with I-BET151, we compared their effect on expression of the insect stage-specific procyclin protein in bloodstream form T. brucei parasites. We have identified AP-09-008 and AP-08-188, which show potent functional activity in a procyclin reporter assay, and robust procyclin expression on the surface of the parasite. AP-09-008 and AP-08-188 are I-BET151 derivatives that are modified at its 3,5-dimethylisoxazole moiety by replacement with a benzoic acid or imidazolone moiety by a carboxylic acid, respectively, which are conjugated to hydrophobic and hydrophilic linkers providing optimal tool compounds readily converted into various chemical probes (Fig. 1). Based on their functional activity, we expect that these compounds and resulting chemical probes will be useful tool compounds to help understand the mode of action of I-BET151 in inducing differentiation.
Fig. 1.
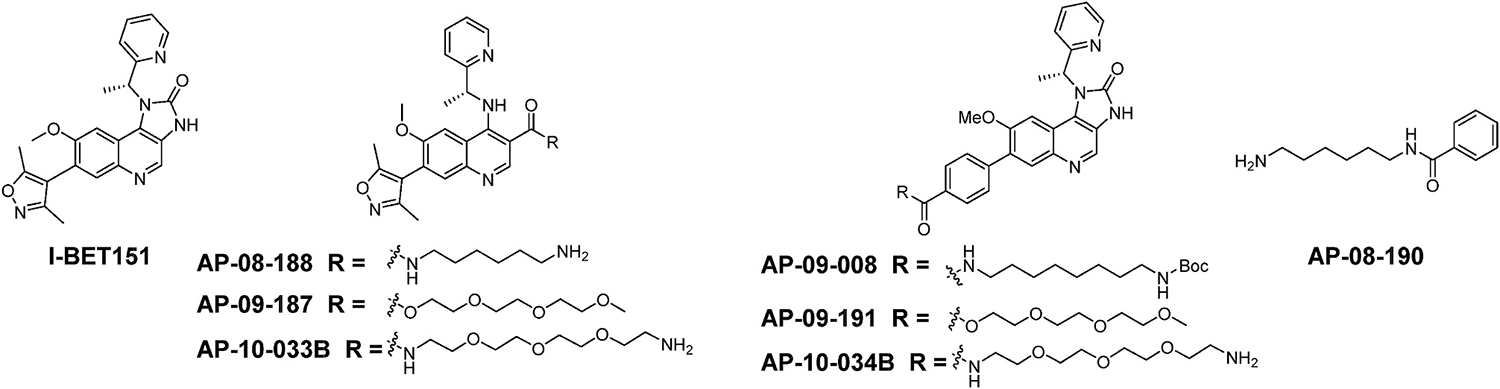
Chemical probes used in this study.
2. Results
Synthesis of I-BET151 derivatives and controls as tool compounds.
To enable rigorous, unbiased chemical proteomics approaches for the identification and validation of I-BET151 targets that cause the differentiation phenotype in T. brucei, we have synthesized the modified I-BET151 derivatives AP-08-188 and AP-09-008, which maintain the core pharmacophoric features of I-BET151 while incorporating a tethered alkyl linker that substitutes either the imidazolone (AP-08-188) or the 3,5-dimethylisoxazole moiety of I-BET151 (AP-09-008) (Fig. 1). We also synthesized a negative control ligand, AP-08-190, to mimic the linker alone, which was used to rule out non-specific activity due to the linker and additional compounds with PEG-modified linkers to aid in water solubility. The synthesis of AP-09-008 utilized an approach similar to that used for I-BET151 (Fig. 1). (Mirguet et al., 2012)
The key quinoline-3-carboxylate intermediate, AP-09-003B (Scheme 1), was synthesized by Michael addition of 3-bromo-4-methoxyaniline with diethyl 2-(ethoxymethylene) malonate to generate intermediate AP-08-290, followed by thermal ring cyclization to provide the quinoline AP-08-294. Conversion of the 4-hydroxyl to a 4-chloro provided AP-08-296, which was then treated with (R)-1-(pyridin-2-yl) ethan-1-amine under refluxing conditions to install the amine via aromatic nucleophilic substitution to provide AP-08-297. Hydrolysis of the ethyl ester and conversion of the resulting carboxylic acid to the acyl azide followed by Curtius rearrangement provided the core quinoline cyclic urea, AP-09-001.
Scheme 1.
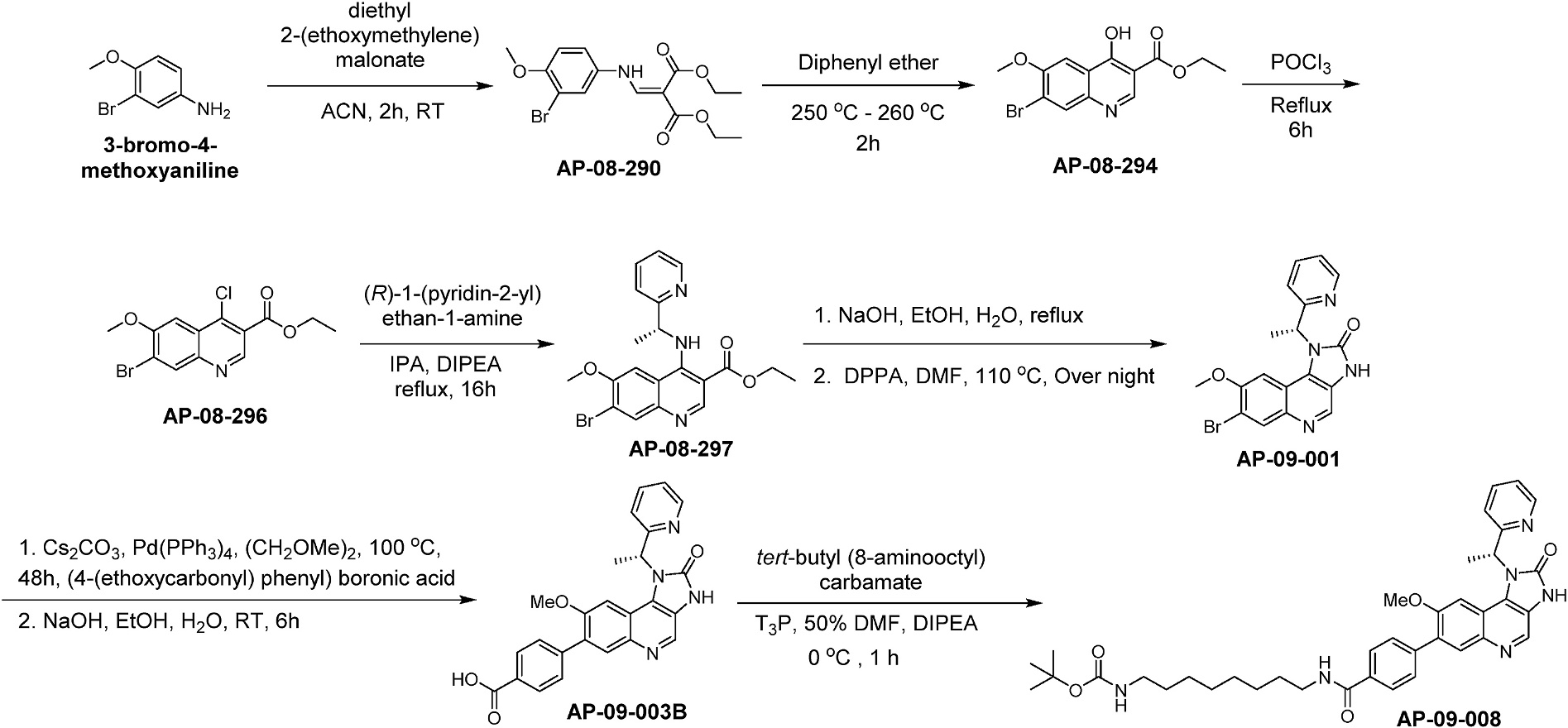
Synthesis of chemical probe AP-09-008.
AP-09-001 was then reacted with (4-(ethoxycarbonyl) phenyl) boronic acid under Suzuki cross coupling conditions followed by basic hydrolysis of the ethyl ester to provide AP-09-003B. AP-09-003B differs from I-BET151 by replacement of the 3,5-dimethylisoxazol-4-yl moiety attached to the 7-position of the quinoline with a benzoic acid. This enables the facile attachment of various linkers through amide coupling. Thus, mono N-Boc protected octyl diamine was coupled to AP-09-003B to provide AP-09-008, an I-BET151 derivative containing an eight-carbon alkyl linker terminating with an N-Boc protected amine. The protected amine of AP-09-008 can be readily deprotected to provide an amine which can be used for attachment to a support (i.e., Sepharose or Streptavidin coated beads) or conjugated to biotin, for use as a chemoproteomics probe. Also, AP-09-003B is a useful intermediate for the synthesis of chemical probes via attachment of various linkers that differ in physical chemical properties and length, which can be used to make bi-functional molecules through attachment of a fluorophore as a chemical probe for fluorescence-based binding assays, or attachment of an E3 ligase-recruiting ligand to synthesize a Proteolysis-targeting chimera (PROTAC).
Synthesis of AP-08-188 (Scheme 2) was accomplished following the reported procedure (Mirguet et al., 2012) to provide quinoline-3-carboxylic acid, AP-08-184. Mono-N-Boc-hexane-1, 6-diamine was then coupled to AP-08-184 using T3P activated ester conditions followed by N-Boc deprotection under acid conditions to provide the quinoline amine, AP-08-188.
Scheme 2.
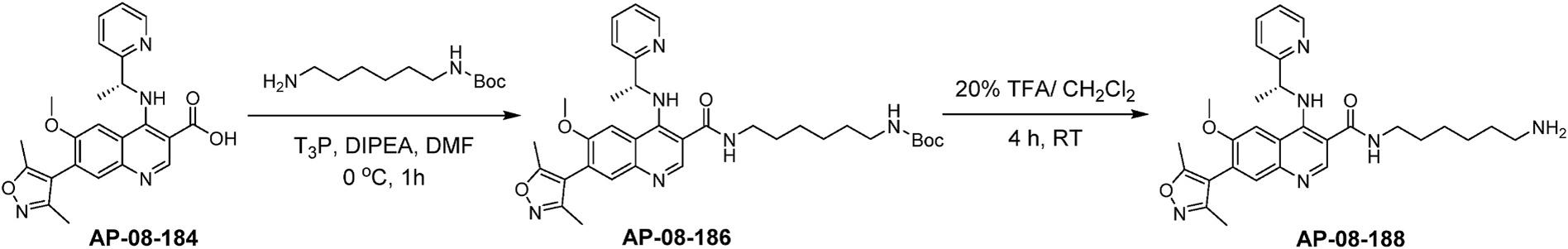
Synthesis of chemical probe AP-08-188.
The tri-polyethylene glycol (PEG) esters and amides of AP-09-003B and AP-08-184 were synthesized to increase their water solubility. This was accomplished by coupling these acids with 2-(2-(2-methoxyethoxy)ethoxy)ethan-1-ol (tri-PEG-alcohol) using water soluble carbodiimide to provide esters (AP-09-187 and AP-09-191) and coupling with 2,2’-((oxybis(ethane-2,1-diyl))bis(oxy))bis(ethan-1-amine) (tri-PEG-diamine) using the T3P amide coupling conditions to provide amides (AP-10-033B and AP-10-034B) (Scheme 3).
Scheme 3.
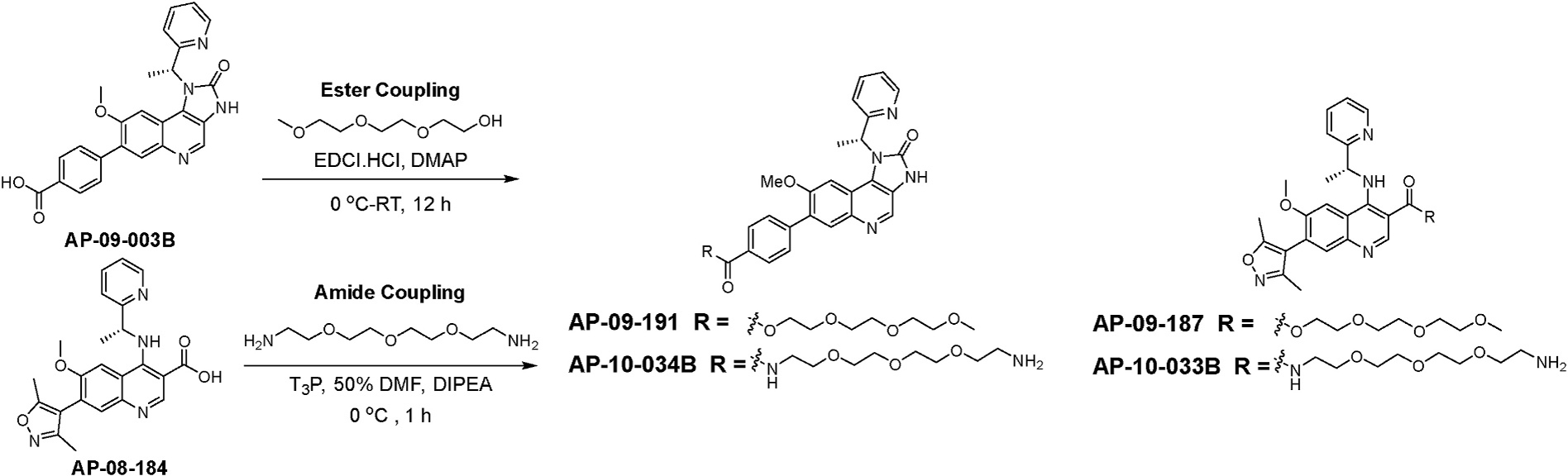
Synthesis of tri-PEG esters; AP-09-191and AP-09-187 and the tri-PEG amides AP-10-033B and AP-10-034B.
2.1. Treatment of bloodstream stage parasites with I-BET151 derivatives induces expression of the insect-stage specific procyclin protein
Bloodstream T. brucei parasites are covered with a dense coat of one specific Variant Surface Glycoprotein (VSG) selectively expressed from a large genomic repertoire of ~2500 VSG genes, (Cross et al., 2014) while insect-stage parasites instead express invariant procyclin proteins on their surface.
Treatment of bloodstream stage T. brucei parasites with the bromodomain inhibitor I-BET151 increases transcript levels of EP1 which, in contrast to the VSG genes, represents one of a small number of genes that code for insect-stage procyclin proteins. I-BET151 treatment of bloodstream parasites strongly increases the levels of procyclin protein on the surface of the parasite (Schulz et al., 2015). To test whether the I-BET151 derivatives affect transcript levels of EP1, we took advantage of a published reporter strain where GFP is knocked into one endogenous EP1 allele. An increase in GFP level for this reporter indicates a concomitant increase in EP1 transcript levels (Walsh et al., 2020). Treatment after 2 or 3 days of bloodstream parasites with I-BET151 resulted in a dose and time sensitive increase in EP1/GFP expression in a flow cytometric assay (Fig. 2, S1 Figure, S1 Table A–B). Treatment of bloodstream parasites with the I-BET151 derivatives likewise showed a dose responsive increase in EP1/GFP expression in the flow cytometric assay, which measures both the mean fluorescence intensity (Fig. 2) and the percent of the population falling within a GFP positive gate (S1 Figure). The percent of GFP positive parasites was highest for a 5 μM treatment with AP-08-188 and AP-09-008; at 2 days this level exceeded treatment with 5 μM I-BET151, while at 3 days the percent of GFP positive cells was slightly higher than for 5 μM treatment with I-BET151 (S1 Figure). The mean fluorescence intensity (Fig. 2) for a 5 μM treatment was highest for AP-08-188, with AP-09-008 and I-BET151 also showing a strong increase at 3 days. AP-09-191 and AP-10-033B also increased mean fluorescence intensity of GFP, but only at a higher 20 μM treatment at 3 days (Fig. 2, S1 Figure). The negative control compound AP-08-190 had a minimal effect even at a 20 μM treatment dose, with only a 1.2-fold change in mean fluorescence intensity compared to DMSO (Fig. 2, S1 Figure, S1 Table A–B). This data suggests that the I-BET151 derivatives phenocopy I-BET151, with AP-09-008 and AP-08-188 being the most potent at activating transcription of EP1/GFP.
Fig. 2. I-BET151 derivatives induce EP1/GFP expression in T. brucei reporter parasites.
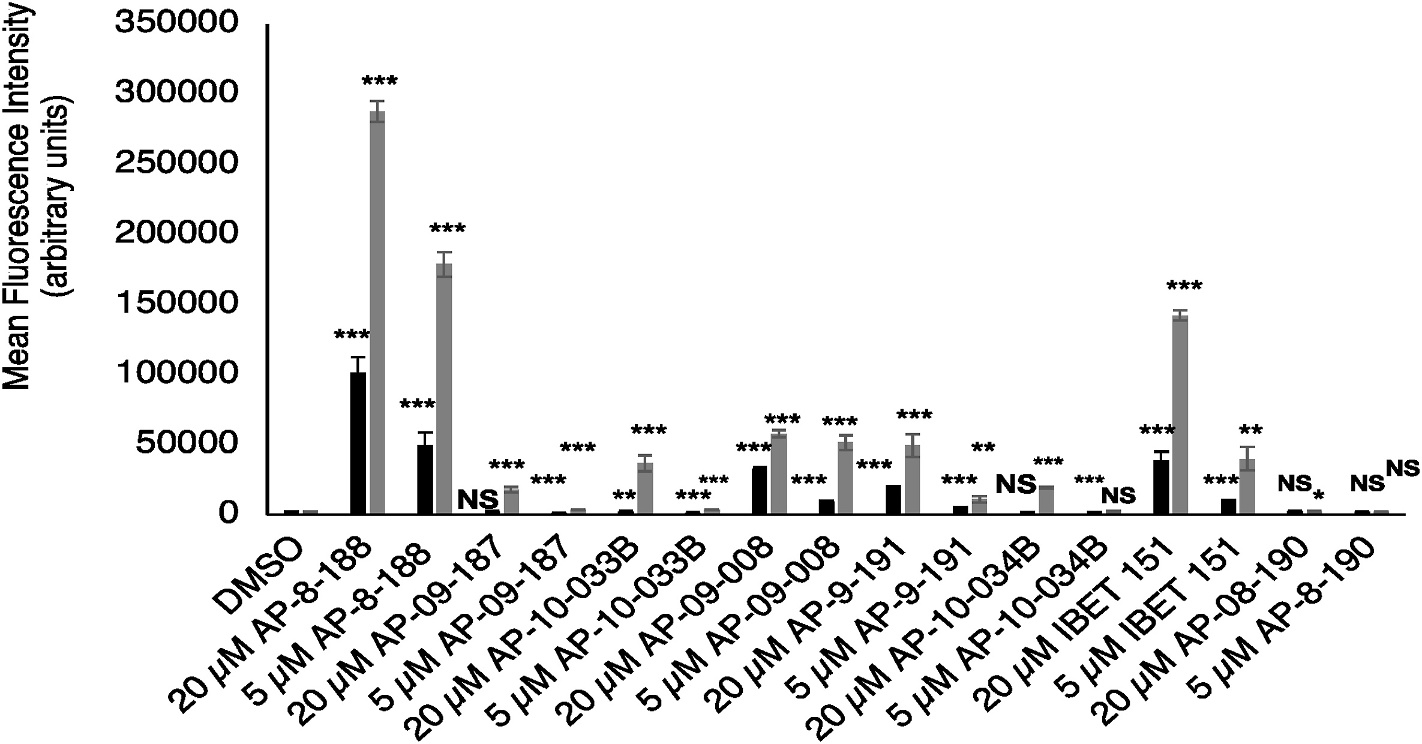
Bar plot of mean fluorescence intensity measurements derived from flow cytometry of EP1/GFP reporter parasites after 2-day (black bars) or 3-day (gray bars) treatment with indicated compounds and concentrations. DMSO was used as a vehicle control. Error bars show standard deviation of the mean from 3 biological replicates. *** indicates a p-value <0.001 derived from a 2 tailed unpaired t-test of values for drug treatment compared to the DMSO control. ** indicates a p-value <0.01 and * indicates a p-value <0.05. NS, not significant.
We then tested whether treatment with I-BET151 derivatives increased procyclin protein on the surface of parasites using an anti-procyclin antibody for those compounds that showed a phenotype for EP1/GFP expression; we evaluated expression levels after a 3-day treatment since this showed the strongest effects in the GFP reporter assay. This is an important test as some compounds have been shown to increase expression of EP1/GFP without inducing procyclin protein replacement on the parasite surface (Walsh et al., 2020). We observed that both AP-08-188 and AP-09-008 increased expression of procyclin protein on the surface of the parasite at 5 μM, similar to I-BET151, as measured by both the percentage of procyclin positive parasites (Fig. 3A) and the mean fluorescence intensity for the procyclin antibody signal (Fig. 3B, S2 Figure, S2 Table).
Fig. 3. AP-08-188, AP-09-008, and AP-09-191 increase expression of insect-stage specific procyclin protein.
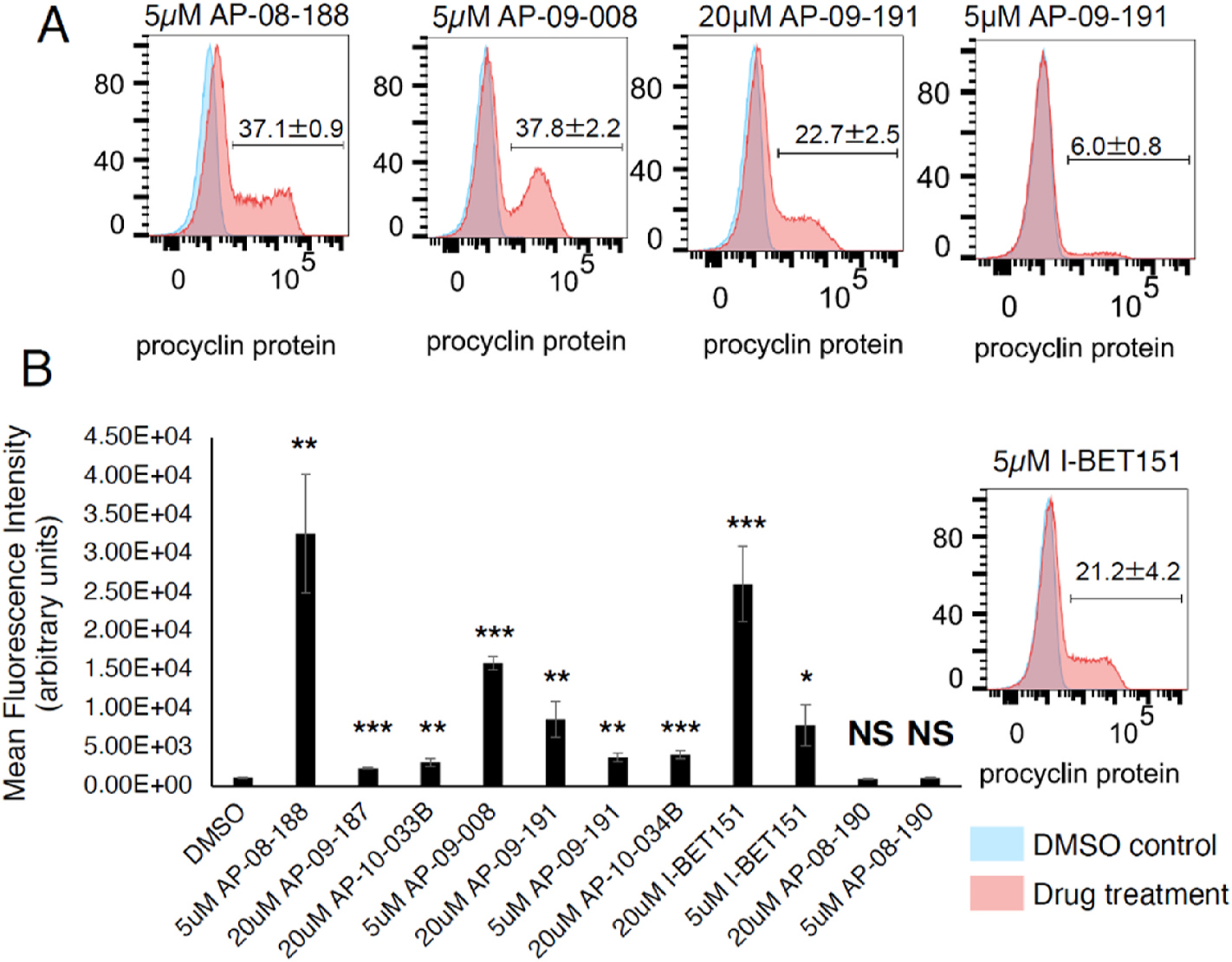
A.) Flow cytometry plot of T. brucei parasites treated with indicated drug for 3 days and stained with an anti-EP1 procyclin protein antibody. Number displayed on flow cytometry plot indicates percent of population falling within the indicated gate plus or minus the standard deviation from 3 biological replicates. DMSO was used as a negative control. B.) Bar plot quantifying the mean fluorescence intensity from the procyclin antibody measurements. Error bars represent standard error of the mean. *** indicates a p-value <0.001 from a two-tailed unpaired T-test compared to DMSO. ** indicates a p-value <0.01. * indicates a p-value <0.05. NS, not significant.
At the 5 μM treatment the percent of procyclin positive parasites was slightly higher for both AP-08-188 and AP-09-008 than for 5 μM treatment with I-BET151, while the mean fluorescence intensity was highest for AP-08-188. 20 μM treatment with AP-09-191 also increased expression of procyclin protein on the parasite surface, while 20 μM treatment with AP-09-187, AP-10-033B, and 10–034B were statistically significant for increased procyclin protein surface expression, but induced surface remodeling to a much lesser degree (Fig. 3, S2 Figure, S2 Table). Neither DMSO nor the control compound AP-08-190 increased procyclin surface expression (S2 Figure, S2 Table). These data suggest that the modified I-BET151 derivatives are behaving in a similar manner as I-BET151 where AP-08-188 and AP-09-008 are slightly more potent for inducing procyclin protein expression on the surface of parasites.
Growth inhibition of the T. brucei parasites was evaluated after 2-day treatment with the I-BET151 derivatives by analyzing the percent of cells in the live gate using flow cytometry data on forward and side scatter. The percentage of live cells in the population was significantly lower for AP-08-188 and AP-09-008 when compared to DMSO controls (Fig. 4; S3 Table A–B). We observed that AP-09-008 and AP-08-188 resulted in growth inhibition with GEC50s of 1.27 ± 0.046 μM and 1.74 ± 0.044 μM, respectively compared to I-BET151 with a GEC50 = 31.71 ± 7.50 μM, where GEC50 represents the concentration of a drug that reduces total cell growth by 50% (S3 Figure, S4 Table and S5 Table). Growth inhibition was not quantified for the other compounds because they showed minimal growth inhibition at 5 μM.
Fig. 4. AP-09-008 and AP-08-188 inhibit growth in T. brucei parasites.
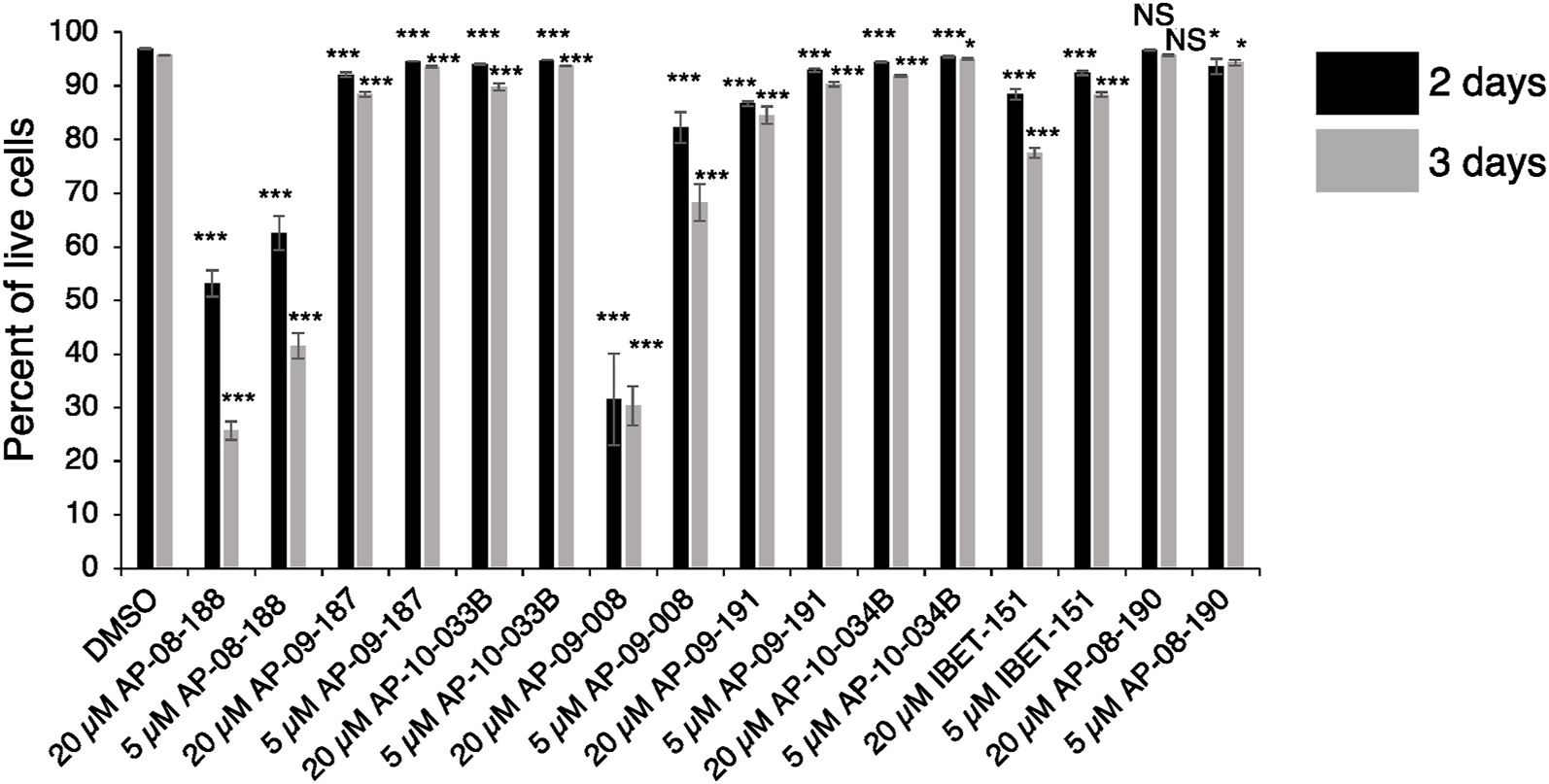
Bar graph showing flow cytometry data for the percent of live cells based on gating for forward and side scatter following treatment for 48h or 72h with the indicated drugs and concentrations. Error bars show standard deviation of the mean from 3 biological replicates. *** indicates a p-value <0.001 derived from a 2 tailed unpaired t-test of values for drug treatment compared to treatment with DMSO. ** indicates a p-value <0.01 and * indicates a p-value <0.05. NS, not significant.
Because I-BET151 had previously been shown to bind weakly to TbBdf2 and TbBdf3, (Schulz et al., 2015) the new derivatives were evaluated for in vitro binding using Surface Plasmon Resonance (SPR) (Table 1) and Isothermal Titration Calorimetry (ITC) methods (Table 2 and S4 Figure). The I-BET151 derivatives were evaluated against TbBdf2, and TbBdf3, and human BRD4 (hBRD4) as a control, against which I-BET151 was originally developed as an inhibitor (Dawson et al., 2011).
Table 1.
Binding of I-BET151 and its derivatives to bromodomain proteins measured by SPR. The standard error is reported.
| hBRD4 | ||
|---|---|---|
|
| ||
| ID | SPR KD, μM | Rmax |
|
| ||
| I-BET151 | 0.036 ± 0.022 | 217 ± 11 |
| AP-08-188 | 0.29 ± 0.17 | 135 ± 27 |
| AP-09-187 | 8.5 ± 4.1 | 173 ± 20 |
| AP-09-008 | 6.1 ± 3.6 | 14.2 ± 1.8 |
| AP-09-191 | 60 ± 26 | 200 (locked) |
|
|
|
|
| TbBdf2 | ||
| ID | SPR KD, μM | Rmax |
|
|
|
|
| I-BET151 | 33 ± 25 | 100 (locked) |
| AP-08-188 | 45 ± 31 | 85 ± 35 |
| AP-09-187 | ||
| AP-09-008 | 1.68 ± 0.34 | 27.1 ± 1.2 |
| AP-09-191 | 44 ± 31 | 200 (locked) |
|
|
|
|
| TbBdf3 | ||
| ID | SPR KD, μM | Rmax |
|
|
|
|
| I-BET151 | 37 ± 22 | 100 (locked) |
| AP-08-188 | 30 ± 21 | 143 ± 51 |
| AP-09-187 | ||
| AP-09-008 | 1.43 ± 0.32 | 36.9 ± 1.9 |
| AP-09-191 | 56 ± 31 | 200 (locked) |
Table 2.
Binding of I-BET151 and its derivatives to bromodomain proteins measured by ITC. The error of the fit of the binding isotherm is reported. ND: not determined.
| ID | hBRD4, ITC KD | TbBdf2, ITC KD | TbBdf3, ITC KD |
|---|---|---|---|
|
| |||
| I-BET151 | 0.32 ± 0.10 μM | 94 ± 18 μM | 68 ± 9 μM |
| AP-08-188 | 0.68 ± 0.15 μM | No binding | 87 ± 21 μM |
| AP-09-187 | 1.5 ± 0.3 μM | No binding | 100 ± 70 μM |
| AP-09-008 | ND | ND | ND |
| AP-09-191 | No binding | 66 ± 11 μM | 22 ± 11 μM |
SPR indicated weak binding of the I-BET151 derivatives with KD values in the micromolar range to TbBdf2 and TbBdf3 (Table 1), while I-BET151 displayed high affinity to hBRD4, consistent with previous data (Schulz et al., 2015; Dawson et al., 2011). The two 3,5-dimethyl benzisoxazole containing analogs AP-08-188 and AP-09-187 showed weak binding to TbBdf2 and TbBdf3, but AP-08-188 potently bound to hBRD4 with a dissociation constant, KD = 0.29 ± 0.17 μM. The I-BET151 analog with the alkyl benzoate amide tail, AP-09-008, showed low micromolar binding to all of the bromodomain proteins. The SPR method is measuring the total mass of the material on the sensor chip and therefore the binding is proportional to the mass of the protein attached to the chip and the mass of the analyte, i.e., AP-09-008 in the flow. Theoretically, the estimated Rmax value is expected to be between 80 and 200 RU, and we observed that the Rmax values for AP-09-008 were low. The low Rmax values may be due to poor solubility or high lipophilicity, suggesting this requires further investigation. The I-BET151 analog with the more water-soluble tri-PEG benzoate tail, AP-09-191 showed weak binding to all three bromodomain proteins.
Isothermal Titration Calorimetry (ITC) is a label-free method for measuring binding through the release or absorption of heat upon binding. This technique provides binding affinity (KD), enthalpy changes (ΔH), and binding stoichiometry (n), where the enthalpy change due to binding is roughly equivalent to the amount of energy lost or gained during the binding event. The ITC binding affinity (KD) of hBRD4, TbBdf2, and TbBdf3 for I-BET151 is consistent with previous reports (Schulz et al., 2015; Dawson et al., 2011). The ITC method showed selective binding of TbBdf2 to the I-BET151 derivative AP-09-191, in which the 3,5-dimethyl benzisoxazole was replaced, while no binding was detected to AP-08-188 and AP-09-187, in which the imidazolone moiety was replaced by a linker. These ITC data agree well with the crystal structure of TbBdf2 in complex with I-BET151, where the imidazolone moiety is deeply buried, while the 3,5-dimethyl benzisoxazole is solvent exposed (Schulz et al., 2015). In contrast to the preferred binding of TbBdf2 to one set of I-BET151 derivatives, TbBdf3 can weakly bind to both sets (AP-08-188, AP-09-187, and AP-09-191). Binding to AP-09-008 could not be determined by ITC due to its insolubility at the concentration of 200 μM used for the small-molecule ligands in this assay. Although KD values were estimated using both methods, we hesitate to present them as definitive because of the limitations of these assays for weak interactions.
Altogether, we have synthesized I-BET151 derivatives containing hydrophobic or hydrophilic linkers extending from either end of the molecule useful for the further synthesis of chemical probes. Some of these I-BET151 derivatives induce the expression of insect-stage specific procyclin surface protein and phenocopy I-BET151, thus confirming they may be useful tool compounds to help define the mechanism of I-BET151 and its potential binding partners in the parasite.
3. Discussion
We have synthesized several I-BET151 derivatives that maintain key in vivo phenotypes of I-BET151 and possess added linker functionality useful for synthesizing various chemical probes for chemoproteomic approaches. In the design of these probes, we have focused on two sets of compounds, in which the introduction of the linker functionality has either modified the 3,5-dimethyl benzisoxazole or the imidazolone moiety of I-BET151. Testing these tool compounds in vivo and in vitro towards T. brucei bromodomains, which have been shown to interact with I-BET151 at modest affinity, can provide valuable insight into 1) functionally important elements of I-BET151 and 2) the importance of bromodomains as I-BET151 targets. AP-09-008, an I-BET151 derivative that maintains the imidazolone moiety and provides an alkyl benzoate linker, strongly induces procyclin expression. Similarly, AP-08-188, an I-BET151 derivative that maintains the 3,5-dimethyl benzisoxazole moiety and incorporated an alkyl linker replacing the imidazolone motif, also exhibits high potency in inducing the differentiation phenotype. The in vivo results with AP-08-188 and AP-09-008 suggest that these derivatives possess important pharmacophoric elements of I-BET151 and represent lead molecules for the further development of valuable tool compounds for chemoproteomic studies to help identify the major targets of I-BET151 in vivo. It remains to be determined whether I-BET151 targets trypanosome bromodomain proteins other than TbBdf2 and TbBdf3, and whether I-BET151 targets non-bromodomain proteins that contribute to the differentiation phenotype.
The data show that treatment with the I-BET151 derivatives increases expression of EP1, suggesting these derivatives maintain important features of I-BET151. Some of the derivatives (AP-09-187, AP-10-033B, AP-10-034B) show robust EP1/GFP expression only at a relatively high concentration of 20 μM, while others are more potent and show strong phenotypes at 5 μM (AP-08-188, AP-09-008, and AP-09-191). The lipophilic nature of the derivatives with the alkyl linkers, AP-08-188 and AP-09-008, might facilitate greater or faster uptake in parasites, strengthening the phenotype. AP-09-008 is the most lipophilic as determined by the calculated cLog P = 6.65, compared to AP-08-188 which has a cLog P = 2.96; both compounds have functional activity in increasing expression of insect-stage specific procyclin (Figs. 2 and 3) similar to I-BET151. A 3-day treatment generally showed higher levels of EP1/GFP expression compared to a 2-day treatment (Fig. 2, S1 Figure, S1 Table). Since the expression of EP1 increases during the course of differentiation, the parasites with higher EP1/GFP expression may represent parasites that have committed further to the procyclic fate.
We speculate that the phenotypes for procyclin surface expression for AP-09-008 and AP-08-188 may be slightly stronger than the other compounds because they inhibit parasite growth to a greater extent than I-BET151 or the other derivatives (Fig. 4, S3 Figure). Bloodstream parasites naturally undergo growth arrest as they form stumpy intermediates that start the transition to the insect life cycle stage (Reuner et al., 1997). Thus, the growth arrest that results from treatment with I-BET151, AP-09-008, and AP-08-188 may mimic the natural growth arrest that occurs during differentiation and may make the replacement of the surface coat with procyclin more favorable than in rapidly dividing parasites. It is interesting that AP-08-188, which contains a lipophilic alkyl amine linker, shows a dose and time responsive effect on T. brucei parasite viability which is more pronounced than the highly lipophilic AP-09-008, suggesting that the 3,5-dimethyl benzisoxazole moiety, a key pharmacophoric feature in I-BET151, is critical. In addition, the conversion to the tri-PEG linkers from AP-08-188 to AP-09-187 and AP-10-033B reduced the effect on parasite viability and expression of insect-stage specific procyclin protein, suggesting a hybrid linker may be more favorable in designing our probes.
Based on these data, the I-BET151 derivatives AP-09-008 and AP-08-188 and their more water-soluble derivatives will be useful compounds to elucidate the molecular mechanism underlying I-BET151-induced life-cycle stage reprogramming and the pathways of T. brucei differentiation. Identifying and pharmacologically interfering with I-BET151 targets may also delineate a new strategy towards the design of drugs against HAT and other trypanosomiases. Thus, these chemical biology probes may prove helpful in future studies to further understand the role of TbBdf proteins in trypanosome biology and to develop alternate therapeutic strategies.
4. Materials and methods
4.1. T. brucei culture growth and strains
We used the Lister 427 L224 strain of bloodstream T. brucei parasites with dual Bloodstream Expression Site (BES) markers that expresses VSG3 from BES7 (with a NeoR gene downstream of the promoter) and VSG2 from BES1 (with a PuroR gene downstream of the BES1 promoter) (Hertz-Fowler et al., 2008). EP1/GFP reporter experiments were performed with the above strain also containing the EP1/GFP reporter construct generated in this report (Walsh et al., 2020). Bloodstream parasites were cultured in HMI-9 at 37 °C with 5% CO2.
4.2. Flow cytometry
Flow cytometry experiments were performed using a Novocyte 2000R from Acea biosciences (now Agilent). Parasites were stained with anti-EP1 (Cedarlane CLP001A) for 10 min on ice, then washed twice with HMI-9 prior to analysis.
4.3. IC50 growth inhibition
Bloodstream parasites were treated for 2 days at varying concentrations of I-BET151, AP-08-188, AP-09-001, and AP-09-008. 2000 parasites were plated in a 200 μl volume of HMI9 (10,000 cells/ml) at the indicated concentrations of each drug and grown for 48 h at 37 °C with 5% CO2. Following treatment, parasites were stained with 5 μM Sytox Orange (Fisher Scientific S11368) and incubated for 15 min at 37 °C prior to flow cytometric analysis with a fixed volume. Flow cytometry plots were gated using a live/Sytox Orange negative gate as in this reference (Walsh et al., 2020). Data were analyzed as in this reference (Walsh et al., 2020) using GRMetric, an R package for calculation of dose response metrics based on growth rate inhibition (Hafner et al., 2016).
4.4. SPR binding kinetics and affinity analysis
Compounds were evaluated for binding to bromodomain proteins by Surface Plasmon Resonance (SPR). TbBdf2, TbBdf3, and hBRD4 were expressed and purified as His6-tagged proteins as previously described (Schulz et al., 2015) (Nicodeme et al., 2010). Proteins were attached to the SPR sensor chip using the CLICK chemistry coupling method where the proteins are labeled with DBCO-PEG4-NHS ester followed by reaction with the azide-linked carboxylate SPR chip. Thus, 25 μL of each protein at 100 μM was incubated with 300 μM DBCO-PEG4-NHS ester at 3-fold molar excess (300 μM) for 2 h at room temperature. The reaction was quenched with ethanolamine (50 mM final), and then the proteins were desalted through a 7K desalting column into phosphate-buffered saline (PBS) with 0.5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and 0.005% Tween 20. The azide-functionalized SPR chip (AZHC1500M) using ddH20 as the running buffer was mounted and washed with 0.1 M sodium borate, 1 M NaCl for 3 min. Electrostatic pre-concentration was used to achieve a high density of each protein on the chip. Then 2 μM of bromodomain proteins (either hBRD4, TbBdf2, or TbBdf3) in 10 mM sodium acetate, pH 4.0 was injected for 60 s at 30 μL/min flow rate. The chip was washed with buffer for 3 min (PBS with TCEP and Tween). After the wash, the amount of protein covalently bound to the chip was assessed. After 60 s of injection, there was ~6000 RU bound to the chip for each protein.
The SPR experiment was run under the following conditions. The running buffer was 10 mM HEPES, pH 7.4, 150 mM NaCl, 0.5 mM TCEP, 0.005% Tween20, and 5% DMSO. Each compound was serially diluted 1:3 in DMSO at 20-fold final concentration. Final concentrations tested were between 0.1 and 100 μM. The DMSO dilutions were diluted 1:20 in running buffer without DMSO such that the final DMSO concentration in the samples was 5%. The association time was 90 s, the dissociation time was 240 s, and the flow rate was 30 μL/min. After each injection, the needle was washed in 50% DMSO. Solvent correction cycles were included to adjust for DMSO effects. Each SPR experiment was performed in duplicate, and the standard error is reported in Table 1.
4.5. Isothermal Titration Calorimetry
ITC measurements were performed at 15 °C using a Malvern PEAQ ITC calorimeter (Malvern Panalytical) or a NanoITC (TA instruments). TbBdf2 and TbBdf3 protein samples were extensively dialyzed against a buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, 0.5 mM TCEP, and 1% DMSO, while hBRD4 was dialyzed against a buffer containing 20 mM HEPES, pH 7.5, 300 mM NaCl, 0.5 mM TCEP, and 1% DMSO. Typically, 2.0 mM protein was injected in increments of 2–3 μL into 0.2 mL of 200 μM ligand in the cell. Baseline-corrected data were analyzed with PEAQ ITC Data Analysis or NanoAnalyze software. The molar ratio n was set to 1.0 based on the previously established stoichiometries (Schulz et al., 2015) (Dawson et al., 2011), which resulted in better curve fitting. Each protein-ligand titration was performed at least twice. The reported data of Table 2 are from single experiments using the error of the fit of the binding isotherm.
4.6. Compound synthesis
Detailed synthetic procedures and spectral characterization are in the Supporting Information.
Supplementary Material
Acknowledgements
We thank the members of the Debler, Schulz, and Salvino labs for their helpful comments. We are grateful to Stoyan Milev for help with ITC experiments. We thank the X-Ray Crystallography & Molecular Interactions Facility at the Sidney Kimmel Cancer Center, which is supported in part by National Cancer Institute Cancer Center Support Grant P30 CA56036 and S10 OD017987. This work was supported by a National Institute of Allergy and Infectious Diseases (NIAID) grants from the National Institutes of Health (NIH) (R21AI154191 and R01AI165840 to E.W.D.), (R21AI154191 to D.S. and J.M.S.), S10OD030245-01 (J.M.S), and P30 CA010815-53 (J.M.S).
Financial support
This work was supported by NIH grants R21 AI154191-02 (JMS, EWD, and DS), R01 AI165840 (EWD), P30 CA56036 (EWD), S10 OD017987 (EWD), S10OD030245-01 (JMS), and P30 CA010815-53 (JMS).
Footnotes
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Joseph M. Salvino, Erik W. Debler, Danae Schulz reports financial support was provided by National Institutes of Health. Joseph M. Salvino reports a relationship with Alliance Discovery, Inc, Barer Institute, Context Therapeutics, Syndeavor Theappeutics that includes: consulting or advisory and equity or stocks. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.crchbi.2023.100047.
Data availability
Data will be made available on request.
References
- Alsan M, 2015. The effect of the TseTse fly on african development. Am. Econ. Rev. 105 (1), 382–410. 10.1257/aer.20130604. [DOI] [Google Scholar]
- Antwi EB, Haanstra JR, Ramasamy G, Jensen B, Droll D, Rojas F, Minia I, Terrao M, Mercé C, Matthews K, Myler PJ, Parsons M, Clayton C, 2016. Integrative analysis of the Trypanosoma brucei gene expression cascade predicts differential regulation of mRNA processing and unusual control of ribosomal protein expression. Epub 20160426 BMC Genom 17, 306. 10.1186/s12864-016-2624-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisser S, N’Siesi FX, Lejon V, Preux PM, Van Nieuwenhove S, Miaka Mia Bilenge C, Būscher P, 2007. Equivalence trial of melarsoprol and nifurtimox monotherapy and combination therapy for the treatment of second-stage Trypanosoma brucei gambiense sleeping sickness. Epub 20061221 J. Infect. Dis. 195 (3), 322–329. 10.1086/510534. [DOI] [PubMed] [Google Scholar]
- Clayton CE, 2016. Gene expression in kinetoplastids. Epub 20160510 Curr. Opin. Microbiol. 32, 46–51. 10.1016/j.mib.2016.04.018. [DOI] [PubMed] [Google Scholar]
- Cross GA, Kim HS, Wickstead B, 2014. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Epub 20140630 Mol. Biochem. Parasitol. 195 (1), 59–73. 10.1016/j.molbiopara.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ, Kouzarides T, 2011. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Epub 20111002 Nature 478 (7370), 529–533. 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rycker M, Wyllie S, Horn D, Read KD, Gilbert IH, 2023. Anti-trypanosomatid drug discovery: progress and challenges. Epub 20220822 Nat. Rev. Microbiol. 21 (1), 35–50. 10.1038/s41579-022-00777-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeks ED, 2019. Fexinidazole: first global approval. Drugs 79 (2), 215–220. 10.1007/s40265-019-1051-6. [DOI] [PubMed] [Google Scholar]
- Dejung M, Subota I, Bucerius F, Dindar G, Freiwald A, Engstler M, Boshart M, Butter F, Janzen CJ, 2016. Quantitative proteomics uncovers novel factors involved in developmental differentiation of trypanosoma brucei. Epub 20160224 PLoS Pathog. 12 (2), e1005439. 10.1371/journal.ppat.1005439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekera K, Wüthrich D, Braga-Lagache S, Heller M, Ochsenreiter T, 2012. Proteome remodelling during development from blood to insect-form Trypanosoma brucei quantified by SILAC and mass spectrometry. Epub 20121016 BMC Genom 13, 556. 10.1186/1471-2164-13-556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Niepel M, Chung M, Sorger PK, 2016. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Epub 20160502 Nat. Methods 13 (6), 521–527. 10.1038/nmeth.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, Brooks K, Churcher C, Fahkro S, Goodhead I, Heath P, Kartvelishvili M, Mungall K, Harris D, Hauser H, Sanders M, Saunders D, Seeger K, Sharp S, Taylor JE, Walker D, White B, Young R, Cross GA, Rudenko G, Barry JD, Louis EJ, Berriman M, 2008. Telomeric expression sites are highly conserved in Trypanosoma brucei. Epub 20081027 PLoS One 3 (10), e3527. 10.1371/journal.pone.0003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotz HR, Hartmann C, Huober K, Hug M, Clayton C, 1997. Mechanisms of developmental regulation in Trypanosoma brucei: a polypyrimidine tract in the 3’-untranslated region of a surface protein mRNA affects RNA abundance and translation. Nucleic Acids Res. 25 (15), 3017–3026. 10.1093/nar/25.15.3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BC, Ramasamy G, Vasconcelos EJ, Ingolia NT, Myler PJ, Parsons M, 2014. Extensive stage-regulation of translation revealed by ribosome profiling of Trypanosoma brucei. Epub 20141020 BMC Genom. 15 (1), 911. 10.1186/1471-2164-15-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesuthasan AJ, Datta AK, Hamilton R, Elliott M, Guz A, Weinberg J, 1987. Aerosolised pentamidine. Lancet 2 (8565), 971–972. 10.1016/s0140-6736(87)91455-3. [DOI] [PubMed] [Google Scholar]
- Jojic B, Amodeo S, Bregy I, Ochsenreiter T, 2018. Distinct 3’ UTRs regulate the life-cycle-specific expression of two TCTP paralogs in Trypanosoma brucei. Epub 20180510 J. Cell Sci 131 (9). 10.1242/jcs.206417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews KR, Ellis JR, Paterou A, 2004. Molecular regulation of the life cycle of African trypanosomes. Trends Parasitol. 20 (1), 40–47. 10.1016/j.pt.2003.10.016. [DOI] [PubMed] [Google Scholar]
- McDermott SM, Carnes J, Stuart K, 2019. Editosome RNase III domain interactions are essential for editing and differ between life cycle stages in Trypanosoma brucei. Epub 20190606 RNA 25 (9), 1150–1163. 10.1261/rna.071258.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O, Lamotte Y, Donche F, Toum J, Gellibert F, Bouillot A, Gosmini R, Nguyen VL, Delannée D, Seal J, Blandel F, Boullay AB, Boursier E, Martin S, Brusq JM, Krysa G, Riou A, Tellier R, Costaz A, Huet P, Dudit Y, Trottet L, Kirilovsky J, Nicodeme E, 2012. From ApoA1 upregulation to BET family bromodomain inhibition: discovery of I-BET151. Epub 20120208 Bioorg. Med. Chem. Lett 22 (8), 2963–2967. 10.1016/j.bmcl.2012.01.125. [DOI] [PubMed] [Google Scholar]
- Mugo E, Clayton C, 2017. Expression of the RNA-binding protein RBP10 promotes the bloodstream-form differentiation state in Trypanosoma brucei. Epub 20170811 PLoS Pathog. 13 (8), e1006560. 10.1371/journal.ppat.1006560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TN, Nguyen BN, Lee JH, Panigrahi AK, Günzl A, 2012. Characterization of a novel class I transcription factor A (CITFA) subunit that is indispensable for transcription by the multifunctional RNA polymerase I of Trypanosoma brucei. Epub 20121026 Eukaryot. Cell 11 (12), 1573–1581. 10.1128/ec.00250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A, 2010. Suppression of inflammation by a synthetic histone mimic. Epub 20101110 Nature 468 (7327), 1119–1123. 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson D, Gunasekera K, Mani J, Osteras M, Farinelli L, Baerlocher L, Roditi I, Ochsenreiter T, 2010. Spliced leader trapping reveals widespread alternative splicing patterns in the highly dynamic transcriptome of Trypanosoma brucei. Epub 20100805 PLoS Pathog. 6 (8), e1001037. 10.1371/journal.ppat.1001037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasion SG, Hines JC, Ou X, Mahmood R, Ray DS, 1996. Sequences within the 5’ untranslated region regulate the levels of a kinetoplast DNA topoisomerase mRNA during the cell cycle. Mol. Cell Biol. 16 (12), 6724–6735. 10.1128/mcb.16.12.6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, Ghabri S, Baudin E, Buard V, Kazadi-Kyanza S, Ilunga M, Mutangala W, Pohlig G, Schmid C, Karunakara U, Torreele E, Kande V, 2009. Nifurtimox-eflornithine combination therapy for second-stage African Trypanosoma brucei gambiense trypanosomiasis: a multicentre, randomised, phase III, non-inferiority trial. Epub 20090624 Lancet 374 (9683), 56–64. 10.1016/s0140-6736(09)61117-x. [DOI] [PubMed] [Google Scholar]
- Queiroz R, Benz C, Fellenberg K, Hoheisel JD, Clayton C, 2009. Transcriptome analysis of differentiating trypanosomes reveals the existence of multiple post-transcriptional regulons. Epub 20091026 BMC Genom. 10, 495. 10.1186/1471-2164-10-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuner B, Vassella E, Yutzy B, Boshart M, 1997. Cell density triggers slender to stumpy differentiation of Trypanosoma brucei bloodstream forms in culture. Mol. Biochem. Parasitol. 90 (1), 269–280. 10.1016/s0166-6851(97)00160-6. [DOI] [PubMed] [Google Scholar]
- Rico E, Rojas F, Mony BM, Szoor B, Macgregor P, Matthews KR, 2013. Bloodstream form pre-adaptation to the tsetse fly in Trypanosoma brucei. Epub 20131114 Front. Cell. Infect. Microbiol. 3, 78. 10.3389/fcimb.2013.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Mugnier MR, Paulsen EM, Kim HS, Chung CW, Tough DF, Rioja I, Prinjha RK, Papavasiliou FN, Debler EW, 2015. Bromodomain proteins contribute to maintenance of bloodstream form stage identity in the african trypanosome. Epub 20151208 PLoS Biol. 13 (12), e1002316. 10.1371/journal.pbio.1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schürch N, Furger A, Kurath U, Roditi I, 1997. Contributions of the procyclin 3’ untranslated region and coding region to the regulation of expression in bloodstream forms of Trypanosoma brucei. Mol. Biochem. Parasitol. 89 (1), 109–121. 10.1016/s0166-6851(97)00107-2. [DOI] [PubMed] [Google Scholar]
- Torreele E, Bourdin Trunz B, Tweats D, Kaiser M, Brun R, Mazué G, Bray MA, Pécoul B, 2010. Fexinidazole–a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. Epub 20101221 PLoS Neglected Trop. Dis. 4 (12), e923. 10.1371/journal.pntd.0000923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez JJ, Hon CC, Vanselow JT, Schlosser A, Siegel TN, 2014. Comparative ribosome profiling reveals extensive translational complexity in different Trypanosoma brucei life cycle stages. Epub 20140117 Nucleic Acids Res. 42 (6), 3623–3637. 10.1093/nar/gkt1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh ME, Naudzius EM, Diaz SJ, Wismar TW, Martchenko Shilman M, Schulz D, 2020. Identification of clinically approved small molecules that inhibit growth and affect transcript levels of developmentally regulated genes in the African trypanosome. Epub 20200313 PLoS Neglected Trop. Dis 14 (3), e0007790. 10.1371/journal.pntd.0007790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzler T, Schumann Burkard G, Schmidt RS, Mäser P, Bergner A, Roditi I, Brun R, 2016. A new approach to chemotherapy: drug-induced differentiation kills African trypanosomes. Epub 20160302 Sci. Rep. 6, 22451. 10.1038/srep22451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wispelwey B, Pearson RD, 1991. Pentamidine: a review. Infect. Control Hosp. Epidemiol. 12 (6), 375–382. 10.1086/646360. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.