

SUMMARY
Infiltration of regulatory T (Treg) cells, an immunosuppressive population of CD4+ T cells, into solid cancers represents a barrier to cancer immunotherapy. Chemokine receptors are critical for Treg cell recruitment and cell-cell interactions in inflamed tissues, including cancer, and thus are an ideal therapeutic target. Here, we show in multiple cancer models that CXCR3+ Treg cells were increased in tumors compared to lymphoid tissues, exhibited an activated phenotype, and interacted preferentially with CXCL9-producing BATF3+ dendritic cells (DC1s). Genetic ablation of CXCR3 in Treg cells disrupted DC1-Treg cell interactions and concomittantly increased DC-CD8+ T cell interactions. Mechanistically, CXCR3 ablation in Treg cells increased tumor antigen-specific cross-presentation by DC1s, increasing CD8+ T cell priming and reactivation in tumors. This ultimately impaired tumor progression, especially in combination with anti-PD-1 checkpoint blockade immunotherapy. Overall, CXCR3 is shown to be a critical chemokine receptor for Treg cell accumulation and immune suppression in tumors.
eTOC Blurb:
Regulatory T (Treg) cell accumulation in tumors suppresses antitumor immunity, but the mechanisms are incompletely understood. Here, Moreno Ayala et al. show that CXCR3 expression on Treg cells puts them in close proximity to CXCL9-producting dendritic cells in tumors, where they suppress tumor antigens presentation and impede the anti-tumor CD8+ T cell response.
Graphical Abstract
INTRODUCTION
Cancer immunotherapies aim to stimulate the immune system to reject tumors. A significant obstacle to the success of cancer immunotherapy is immunosuppression within tumors. Treg cells are a subset of CD4+ T cells with suppressive activity and defined by the expression of the forkhead box transcription factor FOXP3. Although Treg cells maintain homeostatic frequencies of 5–10% in blood and lymphoid tissue, they will accumulate within tumors, often exceeding 50% of all T cells1–4. Higher frequencies of Treg cells within tumors are associated with worse outcomes for patients across many types of cancer5–8. Thus, Treg cell function within tumors is likely a barrier to successful immunotherapy.
A key unresolved question in the field is how Treg cells accumulate within tumors and how they suppress antitumor immune responses. Identifying molecules and pathways responsible for Treg cell accumulation and function in tumors may allow for targeting Treg cells in a tumor-specific manner, avoiding autoimmune toxicities associated with systemic Treg cell ablation9. The expression of specific chemokine receptors on Treg cells supports their recruitment to, and cell-cell interactions within, inflamed environments10–14, and likely tumors6,15. I ntratumoral Treg cells can be distinguished from peripheral Treg cells and other effector T cells by high expression of the chemokine receptors CCR4 and CCR86,16–19. This has led to testing anti-CCR4 and anti-CCR8 antibodies for selective targeting of intratumoral Treg cells to prevent their accumulation in tumors, with promising results in mouse cancer models but tempered results in cancer patients20–22. However, the activity of anti-CCR4 and anti-CCR8 antibodies may be independent of the chemokine receptor’s role in promoting Treg accumulation or function in tumors and instead depend on these antibodies’ capacity to deplete intratumoral Treg cells23–27. Indeed, Treg-specific deletion of CCR8 did not impact Treg cell accumulation in tumors or prevent Treg-supported tumor progression28,29. CCR4 and CCR8 expression are linked to a Th2-like Treg phenotype and are generally associated with Treg cells in organ tissues7,10. Thus, expression of these chemokine receptors on intratumoral Treg cells may be related to general tissue infiltration by Treg cells and not critical for antitumor immune control.
The chemokine receptor CXCR3 responds to the chemokines CXCL9, CXCL10, and CXCL11, which are induced by interferons and critical for immune cell control of cancer30,31. However, unlike CCR4 and CCR8, which are increased on Treg cells compared to effector T cells in tumors, CXCR3 is expressed on many effector cells in tumors, including CD4+ and CD8+ T cells and NK cells6. Intratumoral CXCR3+ CD8+ T cells are essential for tumor control, especially in the context of responsiveness to immunotherapy32–35. CXCR3 expression on CD8+ T cells is important for mediating interactions with dendritic cells (DCs) in tumors, particularly BATF3+ type 1 DCs (DC1s), which can produce CXCL9 and CXCL1032,36–38. Thus, CXCR3+ Treg cells may be particularly important for regulating these antitumor CD8+ T cells to impede the effectiveness of immunotherapy.
The transcription factor T-BET drives CXCR3 expression39–41. T-BET-deficiency in Treg cells abrogates CXCR3 expression and results in defective immune control in models of infection and autoimmunity, particularly in the control of IFN-γ-associated Th1 responses11,14,39,40. The expression of T-BET influences many properties of Treg cells, including their proliferation, maintenance, and suppressive activity, which all likely contribute to the defects observed in Treg cells lacking T-BET39,40,42,43. CXCR3+ Treg cells localize within specific niches in lymphoid and non-lymphoid tissues12,36,44,45, and T-BET-deficiency disrupts their localization. These results led to the hypothesis that CXCR3 expression on Treg cells is critical for homing to and interacting with the CD4+ Th1 effector cells they regulate. However, the contribution of CXCR3 expression to Treg cell function has rarely been examined directly46,47, and thus far has not been shown to be critical for Treg suppression of antitumor CD8+ T cell immunity. CXCR3+ Treg cells are enriched in human ovarian and liver carcinomas48,49, and they are expanded in murine models of lung carcinoma and in patients with oral and non-small cell lung cancers50,51, consistent with a role for CXCR3+ Treg cells in regulating antitumor immunity.
Here we investigated the role of CXCR3 expression on Treg cells in promoting cancer progression. Genetic ablation of CXCR3 on Treg cells led to impaired tumor growth, increased antitumor CD8+ T cells, and synergized with anti-PD-1 immunotherapy. Mechanistically, CXCR3 was required for Treg cells to co-localize with BATF3+ DC1s in tumors where they reduced DC1 cross-presentation of tumor antigen to antitumor CD8+ T cells, limiting antitumor immunity. Our results reveal that CXCR3 function has an opposing immunosuppressive activity due to its expression on Treg cells and highlight CXCR3+ Treg cells as a therapeutic target for promoting cancer immunotherapy.
RESULTS
CXCR3+ Treg cells are generated in response to tumors.
CXCR3 is associated with IFN-γ expression and CD8+ T cell responses against cancer32,33,52. Therefore, CXCR3 expression on Treg cells may also be important for controlling the cytotoxic T-cell response to cancer39,40,46. To test whether CXCR3 is expressed on Treg cells in response to tumors originating from different tissues, we inoculated mice with MC38 (colon carcinoma), EL4 (thymic lymphoma), and 9464D (neuroblastoma) and assessed Treg phenotypes. While the frequencies of Treg cells and CD8+ T cells varied across tumor types, we observed a positive correlation between the number of Treg cells and CD8+ T cells infiltrating all tumor models (Figures S1A–S1D), suggesting a positive relationship between Treg cells and CD8+ T cells in tumors. CXCR3 expression on Treg cells increased with proximity to tumors, with negligible CXCR3 expression in peripheral lymph nodes (pLN), but increased percentages of CXCR3+ Treg cells in tumor-draining lymph nodes (dLN) and tumors (Figures 1A–1B and S1E–S1F). We also observed elevated percentages of CXCR3+ cells among effector CD4+ T cells (eCD4: CD4+Foxp3−) and CD8+ T cells, mainly within tumors (Figures 1C–D and S1E–S1F). An evaluation of the dynamics of Treg cell frequency and CXCR3 expression during cancer progression revealed that the percentage Treg cells in tumors reached 40% of CD4+ T cells on day 14 and remained high, whereas in the dLN, Treg frequency increased later, beginning at day 14 and continuing throughout tumor progression (Figure 1E). However, in tumors and dLNs, CXCR3 expression on Treg cells peaked at day 14 and declined thereafter (Figure 1E), which correlated with CXCR3+CD8+ T cell infiltration into tumors (Figure S1G). Therefore, CXCR3 expression on Treg cells may be important for regulating CXCR3+CD8+ T cell antitumor responses.
Figure 1. CXCR3+ Treg cells are generated in response to tumors.

(A) Flow cytometric analysis of FOXP3 versus CXCR3 expression in live CD45+CD4+ T cells in tumor draining lymph node (dLN) and tumor (MC38).
(B-D) Quantification of perecent CXCR3+ T cells in peripheral lymph node (pLN), dLN and tumor in Treg cells (B), eCD4+ (CD4+Foxp3−, C), and CD8+ T cells (D) on day 15 post MC38 tumor inoculation (n=16 pooled from four independent experiments).
(E) Time course of Treg cell (Foxp3+CD25+/Live CD45+CD4+, left panel) and CXCR3+ Treg cell (right panel) frequency over the course of tumor growth in dLN (light purple) and tumor (dark purple) (n=3–4 mice/group, representative of two independent experiments).
(F-G) Left panel: Flow cytometric staining for CD44 versus CXCR3 in FOXP3+CD25+ of Live CD45+ cells in dLN (F) and MC38 tumors (G). Right panels: Quantification of specified proteins in CD44− (black), CD44+CXCR3− (blue), and CD44+CXCR3+ (red) Treg cells (n=4–5 mice pooled from two independent experiments).
Data represents mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 from two-way ANOVA followed by Dunnet’s multiple comparisons test. Also see Figure S1.
Next, we evaluated the differentiation and activation state of CD44+, antigen-experienced, CXCR3+ versus CXCR3− Treg cells in the dLN and tumor at the peak of Treg infiltration into tumors. CD69, a marker of recently activated Treg cells with a better capacity to maintain immune tolerance53, CD103, a marker that defines an effector/memory-like Treg population that can prevent allograft rejection54, and KLRG1, PD-1, and CTLA4, additional markers of effector Treg cells55,56, were all increased on CXCR3+ versus CXCR3− Treg cells (Figures 1F and G). CTLA4 and PD-1 expression are especially relevant because they are immunotherapeutic checkpoint targets and directly modulate Treg suppressive activity56–59. The high prevalence of CXCR3+ Treg cells in the tumors and their associated activation phenotype suggested they may play a role in cancer progression and warranted further investigation.
Treg-specific CXCR3-deficiency impedes cancer.
To test whether there is a functional requirement for CXCR3 expression in Treg cell-mediated control of antitumor T cell responses, we generated Treg-specific CXCR3-deficient mice using an innovative strategy that takes advantage of X chromosome inactivation in female mice. Cxcr3, like Foxp3, is located on the X chromosome. Using a germline disrupted Cxcr3 allele combined with a Foxp3TM2-GFP reporter linked on one X chromosome and a Foxp3DTR-GFP allele on the other X chromosome, we generated Treg-specific Cxcr3KO mice by treatment with diphtheria toxin (DT) (Figures 2A, and S2A–S2B)32,35,60. Treg cells expressing the Foxp3DTR-GFP allele are readily distinguished from those expressing the Foxp3TM2-GFP allele because of the considerably brighter GFP fluorescence from the Foxp3TM2-GFP allele (Figure 2A). We began DT treatment one week before MC38 injection and then continued DT administration during tumor progression by dosing every other day (Figure 2B). Upon analysis of mice at the time of tumor inoculation (day 0, Figure 2B), we did not observe any difference in the frequencies or activation state of Treg, CD4+, or CD8+ T cell populations due to one week of DT treatment, and no systemic changes after three weeks of DT treatment, except for the expected reduction in CXCR3+ Treg cells (Figures S2C–S2I). However, we found that mice lacking CXCR3+ Treg cells with DT treatment exhibited delayed tumor growth in all three cancer models (MC38, EL4, and 9464D) compared to mice treated with PBS, which retained CXCR3+ Treg cells (Figure 2C). To rule out the possibility that DT-mediated Treg ablation impacted tumor growth rather than CXCR3-deficiency in Treg cells, we evaluated tumor growth in mice where CXCR3 was left intact on Treg cells. Depleting approximately half of the Treg cells with DT, while retaining CXCR3 in the remaining Treg cells, did not impact tumor growth (Figure 2D). Finally, to determine whether tumor growth was affected by Treg ablation in the context of CXCR3 heterozygous mice (Cxcr3+/−), we generated female mice wherein the Foxp3DTR-GFP allele was linked in cis with the Cxcr3KO allele such that DT treatment left CXCR3+ Treg cells intact in the context of Cxcr3+/− mice. Even in this scenario, there was no impact on tumor growth (Figure 2E). Altogether, these results reveal a critical role for CXCR3 expression on Treg cells in promoting cancer progression.
FIGURE 2. Treg-specific CXCR3-deficiency impedes cancer.

(A) Left: genotype of female mice depicting the allele configuration of Foxp3 and Cxcr3 on the X chromosomes. Right: Schematic depicting the resulting Treg phenotypes from this genotype due to random X inactivation before and after DT treatment. Complementary flow cytometric analysis of CXCR3 versus Foxp3 GFP reporters in Live CD45+CD4+ cells from dLN before and after DT treatment is shown below schematic.
(B) Experimental strategy to deplete Foxp3DTR-GFP-expressing Treg cells in female mice with DT.
(C-E) Left: genotype of female mice and representative flow cytometric analysis of CXCR3 versus Foxp3 GFP reporters in Live CD45+CD4+ cells from tumors with DT treatment. Right: tumor growth curves of MC38, EL4, and 9464D tumors in indicated mice treated with PBS or DT (n=7–8 mice/group, representative of three independent experiments for MC38 and two independent experiments for EL4 and 9464D). Results from Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3KO mice that generate Cxcr3KO Treg cells with DT (C). Results from Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3+ mice that maintain Cxcr3+ Treg cells with DT (D). Results from Foxp3DTR-GFP;Cxcr3KO/Foxp3TM2-GFP;Cxcr3+ mice that maintain Cxcr3+ Treg cells with DT (E).
Data represents mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, and not significant (n.s.) from two-way ANOVA with Sidak’s multiple comparisons. Also see Figure S2.
Enhanced CD8 tumor immunity with CXCR3-deficient Treg cells.
To investigate why CXCR3+ Treg cells are required to promote tumor progression, we first assessed the CD8+ T cell response within tumors. Across all tumor models, we found an increased frequency of intratumoral CD8+ T cells in mice with Cxcr3-deficient Treg cells (Figures 3A and S3A–S3B). Next, we followed the expansion of a tumor-specific CD8+ T cell population that responds to a cancer-associated endogenous retroviral peptide using the p15E/Kb tetramer stain61. We found an increase in these tumor-specific CD8+ T cells in the context of Cxcr3KO Treg cells, compared to CXCR3+ Treg-bearing mice, particularly within the tumor tissues (Figure 3B). In line with our previous results, we did not observe an increase in bulk CD8+ T cells or p15E-specific CD8+ T cells in tumors or lymphoid tissues using our DT strategy alone and CXCR3+ Treg cells were left intact (Figures S3C and S3D). Thus, loss of CXCR3 on Treg cells correlated with increased antitumor CD8+ T cell numbers in tumors. The importance of the enhanced CD8+ T cell response for tumor control in the absence of CXCR3+ Treg cells was supported by the fact that antibody-mediated depletion of CD8+ T cells prevented the tumor growth control observed with Cxcr3KO Treg cells (Figure 3C).
Figure 3. Enhanced CD8 tumor immunity with Cxcr3KO Treg cells.

(A-B) Quantification of total intratumoral CD8+ T cells from Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP; Cxcr3KO female mice harboring Cxcr3+ (+PBS, black) or Cxcr3KO (+DT, red) Treg cells in MC38 tumors (day 28) (n=6–8 per group, data pooled from two independent experiments).
(B) Tumor-specific p15E-reactive CD8+ T cells were measured from indicated organs (day 15, MC38) (n=4–5 mice per group, data representative of two independent experiments). Mice without tumors (naïve) served as a negative control for p15E/Kb tetramer stain (grey). Representative flow cytometric analysis of intratumoral pE15 tetramer stain in PBS and DT treated mice (right panel).
(C) Growth of MC38 tumors in control females (+PBS, black), control females treated with anti-CD8 depleting antibody (+PBS +α-CD8, green), Cxcr3KO Treg cell females (+DT, red), or Cxcr3KO Treg cell females treated with an anti-CD8 depleting antibody (+DT +α-CD8, blue) (n=8 mice per group, representative of two independent experiments). Representative flow cytometric analysis of CD4+ and CD8+ cells in spleens of control or anti-CD8 treated groups (right panel).
(D) IFN-γ+TNFa+CD8+ of Live CD45+ in tumors (day 15, MC38) from indicated mice after no stimulation (no stim) or stimulation with PMA and Ionomycin (P+I) ex vivo (n=4–5 per group).
(E) PD-1 expression on intratumoral CD8+ T cells (day 28, MC38) (data pooled from three independent experiments).
(F) Growth of MC38 tumors in mice with Cxcr3+ (+PBS, black) or Cxcr3KO Treg cells (+DT, red), or treated with anti-PD-1 in mice with Cxcr3+ (+α-PD-1, purple) or Cxcr3KO Treg cells (+DT+α-PD-1, green) (n=6–7 mice per group).
Data represent mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001 from two-way ANOVA followed by Dunnet’s multiple comparisons test, two-way ANOVA with Sidak’s multiple comparisons, or student’s t tests. Tumor growth assessed using multiple regression analyses. Also see Figure S3.
While Cxcr3-deficiency in Treg cells increased CD8+ T cell infiltration into tumors, it did not lead to significantly increased activity of CD8+ T cells as assessed by IFN-γ and TNF-α production upon ex vivo restimulation (Figures 3D and S3E). Furthermore, in the context of Cxcr3KO Treg cells, tumor-infiltrating CD8+ T cells still expressed high levels of PD-1 (Figure 3E), and tumor control was enhanced when Cxcr3KO Treg cell-bearing mice were treated with anti-PD-1 beginning at day 7 of tumor progression (Figure 3F)62. Together, these data suggest that CXCR3+ Treg cells promote cancer by inhibiting the accumulation of antitumor CD8+ T cells in tumors.
CXCR3 is required for Treg accumulation and activation in tumors.
CXCR3+ Treg cells accumulated in tumors and exhibited a heightened activation phenotype (Figure 1). Examination of intratumoral Treg frequency in the absence of CXCR3 revealed a modest reduction in Treg frequency within tumors (Figures 4A–4B and S4A–S4B). This was a consequence of Cxcr3-deficiency in Treg cells and not due to the DT treatment strategy, as intratumoral Treg frequency was unchanged in mice treated with DT that retained CXCR3+ Treg cells (Figure S4C). We also used our Cxcr3KO Treg cell strategy to evaluate CXCR3 function in Treg cells in a genetically engineered model of sarcomagenesis (Figure S4D)63. Here, DT treatment began after sarcomas formed due to their 3–4 month latency period (Figure S4E). In this setting, we again observed increased intratumoral CD8+ T cells and reduced Treg cells with Cxcr3-deficiency (Figures S4F–S4H), highlighting the role of CXCR3 in Treg cells in a non-transplantable cancer model.
Figure 4. CXCR3 is required for Treg accumulation and activation in tumors.

(A-B) Quantification of intratumoral Treg cell frequency or their PD-1 and CTLA-4 expression from Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3KO female mice harboring Cxcr3+ (+PBS, black) or Cxcr3KO (+DT, red) Treg cells at day 15 (A) or day 28 (B) of MC38 tumor progression (n=7 mice per group, pooled from two independent experiments).
(C) Schematic describing the generation of CXCR3+ and Cxcr3ΔTregcells by in vitro differentiation, fluorescent dye-labeling, and co-transfer into MC38 tumor-bearing mice for localization analysis.
(D) Analysis of Treg cells for CXCR3 and T-BET expression prior to adoptive transfer.
(E) Representative flow cytometry plots from dLN and MC38 tumors for transferred Cxcr3+ and Cxcr3KO Treg cells.
(F) Absolute number of Cxcr3+ (black) or Cxcr3KO Treg cells (red) cells recovered. Lines connect Treg cells collected from the same mouse (data pooled from three independent experiments).
Data represents mean ± SEM; *p < 0.05, **p < 0.01 by unpaired (or paired analysis in F) Student’s t tests. Also see Figure S4.
We also consistently observed reductions in PD-1 and CTLA4 co-inhibitory receptor expression on Cxcr3KO Treg cells throughout tumor progression, suggestive of reduced activation of intratumoral Treg cells that lack CXCR3 (Figures 4A and 4B). To determine whether CXCR3 expression supports Treg infiltration and activation within tumors, we compared Cxcr3+ versus Cxcr3KO Treg cells in a competitive setting using Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3KO female mice wherein both populations of Treg cells were present in the absence of DT (Figure S4I). In accordance with our previous results, the overall frequency of Treg cells was not changed between control and Cxcr3KO Treg cell populated mice. However, when we examined the proportion of each population of Treg cells that make up the total Treg pool, Cxcr3KO Treg cells made up a smaller proportion of Treg cells in the dLN and tumor compared to Cxcr3+ Treg cells from control mice (Figures S4J and S4K). Furthermore, PD-1 and CTLA4 expression were reduced in Cxcr3KO Treg cells within tumors in this competitive setting (Figures S4J and S4K). Therefore, Cxcr3KO Treg cells appear to be at a disadvantage in becoming activated within the tumor microenvironment.
To directly test whether Cxcr3-deficient Treg cells were impaired in their capacity to accumulate in tumor tissues, we performed adoptive transfer experiments of labeled Cxcr3+ versus Cxcr3KO Treg cells together into MC38 tumor-bearing mice and assessed their localization 24 hours later. To do this, we sorted naïve Treg cells from Cxcr3+ versus Cxcr3KO mice and differentiated them in vitro to become Th1-like (T-bet+) Treg cells by treatment with IL-12 and IFN-γ after anti-CD3/anti-CD28 activation (Figure 4C). As seen in Figure 4D, the addition of IL-12 and IFN-γ generated CXCR3+T-BET+ Treg cells from wildtype mice and CXCR3–T-BET+ Treg cells from Cxcr3KO mice. This strategy ensured that cells were similar in differentiation and activation state, with the only difference being the expression of CXCR3 before co-transfer. This was important, as T-bet-deficiency in Treg cells disrupts Treg suppression of Th1 responses, but the specific role of CXCR3 in Treg function was never directly tested14,39,40. We harvested the dLN and tumors to compare the number of Treg cells of each type recovered in this competitive setting. We found that Cxcr3KO Treg cells were significantly enriched in the dLN but reduced compared to Cxcr3+ Treg cells within tumors (Figures 4E and 4F). These results suggest that CXCR3 expression on Treg cells is critical for their accumulation within MC38 tumors after migrating from the draining lymph nodes.
CXCR3 facilitates dendritic cell interaction in the tumor microenvironment.
As a chemokine receptor, Cxcr3-deficiency may be necessary for Treg accumulation in tumors, particularly due to CXCR3-mediated interactions with the specific cells they regulate within the tumor microenvironment. A major cell type that Treg cells interact with and regulate are DCs64–67. DCs are known to produce the CXCR3 ligands CXCL9 and CXCL10 in C57BL/6 mice32,68. Therefore, we hypothesized that CXCR3 in Treg cells functions by promoting Treg co-localization with DCs to regulate them. To evaluate interactions between Cxcr3+ versus Cxcr3KO Treg cells with DCs in situ, we crossed a CD11c-YFP allele into female Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3KO or Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3+ mice to identify Treg cells and CD11c+ DCs by confocal microscopy. We visualized tumor cells using MC38 cells transduced with Cyan Fluorescent Protein (MC38-CFP) and used fluorescent anti-CD8 antibody staining to track CD8+ T cells. Treatment of these tumor-bearing mice with DT allowed us to quantify interactions between Foxp3TM2-GFP-expressing Cxcr3+ versus Cxcr3KO Treg cells with CD11c+ cells, as well as CD8+ T cells with CD11c+ cells (Figure 5A). As we had seen by flow cytometry, confocal microscopic analysis revealed a reduction in Treg cells within tumors when Treg cells lacked CXCR3, as well as an increase in CD8+ cells, but this occurred without a change in total CD11c cells (Figures 5B, 5C, 5G, and 5H). To quantify our analysis, we assessed cell-cell interactions by counting cells in tight proximity in multiple z-stacks (Figures 5D–F). On a per-cell basis, Cxcr3-deficient Treg cells were less likely to be found interacting with a CD11c+ cell than were CXCR3-competent Treg cells (Figure 5G). With respect to CD8+ T cells, we found a significant increase in CD8+ cells and, on a per-cell basis, an increase in the likelihood of a CD8+ T cell interacting with a CD11c+ cell in the context of Cxcr3+ versus Cxcr3KO Treg cells (Figure 5H), indicative of an inverse relationship between Treg-CD11c and CD8-CD11c interactions. The Treg-CD11c interaction results were also recapitulated in a competitive setting where female mice of these genotypes were not treated with DT and Cxcr3+ Treg cells remained present in both settings (Figures S5A–S5D). However, as expected, the presence of wildtype Cxcr3+ Treg cells in this setting abrogated the increased CD8-CD11c interactions observed when all Treg cells were Cxcr3-deficient (Figure S5E). These results indicate that CXCR3 expression on Treg cells enhances their co-localization with CD11c cells in the tumor microenvironment. The consequence of these interactions may be to suppress CD11c-CD8 T cell interactions within tumors. We performed flow cytometric analysis to determine whether Cxcr3+ Treg cells modulated the frequency of DCs or their expression of co-stimulatory molecules but did not find significant differences in total DC, DC1 (CD103+) or DC2 (CD11b+) in the tumor (Figure S5H–S5J) or dLN (Figure S5K–S5M), suggesting that Cxcr3+ Treg cells do not regulate the frequency or co-stimulatory capacity of DCs to mediate immune suppression.
Figure 5. CXCR3 expression on Tregs facilitates CD11c+ cell interaction in tumors.

(A) Genotype of female mice and schematic depicting the resulting Treg phenotypes after DT treatment. Mice were treated with DT beginning 7 days prior to tumor inoculation and throughout tumor progression.
(B-C) Representative confocal microscopy images from WT (B) and KO (C) mice as described in (A) at 15 days of tumor progression showing MC38-CFP (blue), CD11c-YFP (yellow), Foxp3TM2-GFP (green), and CD8+ (red) cells. Arrowheads point to cell-cell interactions between Tregs and CD11c+ cells (representative of 60–70 images analyzed from 3 mice per group pooled from 3 independent experiment). Scale bar = 50 μm.
(D-F) High magnification examples of counted cell-cell interactions between Treg-CD11c cells (D, E) and CD8-CD11c cells (E, F). Scale bar = 15 μm.
(G-H) Total numbers of Tregs, CD11c cells (G) or CD8+ cells (H) were counted per field at 20X magnification. Cell-cell interactions were examined at higher magnification and confirmed through multiple z-stacks. Cell-cell interactions were normalized based on the number of Tregs (G) or CD8+ cells (H) per field to yield a probability of interaction per Treg or CD8+ cell (representative of 60–70 images analyzed from 3 mice per group and pooled from 3 independent experiments).
Data represents mean ± SEM; *p < 0.05, **p < 0.01 ***p < 0.001 by Student’s t test.
Type 1 DCs are required for the preferential localization of CXCR3+ Treg cells in tumors.
The ligands for CXCR3 in C57BL/6 mice, CXCL9 and CXCL10 (CXCL11 is null), can be produced by conventional type 1 DCs (DC1), a CD11c+ population of cells defined by the expression of the transcription factor BATF337,69. Therefore, we first examined CXCL9 and CXCL10 expression on CD11c+ cells within tumors by immunofluorescence in MC38-CFP tumors from Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3+;CD11c-YFP female mice treated with DT as in Figure 5 (Figures 6A–6D). We found that most CXCL9+ cells were also CD11c-YFP+, and our analysis showed a strong positive correlation between the number of CXCL9+CD11c+ cells and wildtype Treg cells per field that was reduced in mice with Cxcr3KO Treg cells (Figure 6E). We did not observe a positive correlation between the number of CXCL10+CD11c+ (CXCL9-) cells and wildtype or Cxcr3KO Treg cells per field (Figure S6A). This suggests that CXCL9 is particularly important for the localization of CXCR3+ Treg cells.
Figure 6. Type 1 DCs are required for the preferential localization of CXCR3+ Tregs in tumors.

(A-D) Representative confocal microscopy images from Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP; Cxcr3+;CD11c-YFP mice treated with DT and analyzed at 15 days of tumor progression showing CD11c-YFP (yellow) and Foxp3TM2-GFP (green) (A), anti-CXCL10 and MC38-CFP (B), anti-CXCL9 and MC38-CFP (C), and a composite image (D). Arrowheads point to Tregs (green), CXCL9+ (red), and CXCL10+ (white) cells. Scale bar = 20 μm.
(E) Correlation between the number of Foxp3TM2-GFP-expressing CXCR3+ Tregs (blue) from DT treated Foxp3DTR-GFP; Cxcr3+/Foxp3TM2-GFP;Cxcr3+;CD11c-YFP mice or Foxp3TM2-GFP-expressing CXCR3KO Tregs (red) from DT treated Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3KO;CD11c-YFP mice versus CXCL9+CD11c+ cells per field at 20X magnification in MC38 tumors at day 15 of tumor progression (representative of 50–60 images analyzed from 3 mice per group and pooled from 3 independent experiments).
(F) MC38 tumors from DT-treated mice bearing CXCR3+ or CXCR3KO Tregs were harvested at day 15 of tumor progression and CXCL9 and CXCL10 were quantified by ELISA (n= 6–7 mice per group, pooled from two independent experiments).
(G) MC38 tumors from WT or Batf3KO mice were tested for CXCL9 and CXCL10 levels as in (G) (n= 3–4 mice per group, pooled from two independent experiments).
(H) Representative confocal microscopy images from Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3+ mice with CXCR3+ Tregs at 15 days of tumor progression showing MC38-CFP (blue), anti-XCR1 (yellow), Foxp3TM2-GFP (green), and anti-CXCL9+ (red) cells. Scale bar = 5 μm.
(I-J) Fraction of CXCL9+ cells that are XCR1+ (CXCL9+XCR1+/CXCL9+) assessed by immunofluorescence (I). Total Tregs (J) and XCR1-Treg interactions (K) were examined at higher magnification and confirmed through multiple z-stacks. Cell-cell interactions were normalized based on the number of Tregs (K) per field to yield a probability of interaction per Treg. Cells were counted per field at 20X magnification (representative of 22–24 images analyzed from 4 mice per group).
(L) MC38 tumors from WT (black), Batf3KO (grey), or WT mice treated with PBS (black), anti-CD8 (blue), or anti-IFN-γ (green) were tested for CXCL9 by ELISA (L, n= 4 mice per group).
(M-N) Flow cytometric analysis for intracellular CXCL9 protein in DC1 and DC2 from MC38 tumors in mice as described in (L) and quantified in (N, n= 4 mice per group).
(O) Flow cytometric analysis for intracellular CXCL9 protein in DC1 and DC2 from MC38 tumors in mice with CXCR3+ (green) or CXCR3KO (red) Tregs after treatment with DT or in Batf3KO (grey) mice and quantified (n=3–4 mice per group).
(P) Schematic describing the generation of CXCR3+ and CXCR3KO Tregs by in vitro differentiation, fluorescent dye-labeling, and co-transfer into MC38 tumor-bearing C57BL/6 wildtype or Batf3KO mice for localization analysis after 24 hours.
(Q) Absolute number of CXCR3+ (black filled) or CXCR3KO (red filled) Tregs recovered in dLN and tumor 24 hours after transfer into C57BL/6 wildtype mice (WT) or number of CXCR3+ (black hatched) or CXCR3KO (red hatched) Tregs recovered after transfer to Batf3KO mice. Lines connect CXCR3+ and CXCR3KO Tregs collected from the same mouse (n=3 mice per genotype/experiment and all data pooled from three independent experiments).
Data represents mean ± SEM; *p < 0.05, **p < 0.01 by Student’s t test. For correlation in (F), a simple linear regression was employed and slopes between regressions were compared.
We next examined whether Cxcr3-deficiency in Treg cells impacted the number of CD11c+ cells in tumors or their production of CXCR3 ligands. We did not observe any differences in the abundance of CXCL10+CD11c+ or CXCL9+CD11c+ cells in the context of Cxcr3KO compared to Cxcr3+ Treg-bearing mice (Figure S6B). We also did not find significant changes in CXCL9 or CXCL10 protein in explanted tumors from Cxcr3KO compared to Cxcr3+ Treg-bearing mice (Figure 6F). However, we found that tumor-bearing Batf3−/− mice, which lack DC1s, exhibited a near complete loss of CXCL9 production, whereas CXCL10 was unaffected (Figures 6G and S6C–S6D). This may suggest that BATF3+ DC1s produce CXCL9, which is required for interacting with CXCR3+ Treg cells in tumors.
To focus our analysis on DC1s, we used anti-XCR1 immunofluorescence staining to specifically mark DC1s while co-staining for CXCL9 in MC38-CFP tumors from Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3KO compared to Foxp3DTR-GFP;Cxcr3KO/Foxp3TM2-GFP;Cxcr3+ mice treated with DT (Figure 6H). We found that CXCL9+ cells were almost uniformly XCR1+, indicative that DC1s produce CXCL9 (Figure 6I). Furthermore, we failed to detect CXCL9+ cells (or XCR1+ cells) in tumors from Batf3−/− mice (Figure S6E). While we did not observe significant differences between the total number of DCs in mice with Cxcr3KO versus Cxcr3+ Treg cells (Figure S6F), we did observe a reduction in Treg-XCR1 interactions (Figure 6J–6K). Finally, CXCL9+XCR1+ cell frequency was also increased in mice with Cxcr3+ compared to Cxcr3KO Treg cells (Figure S6G), implicating CXCL9 modulation in the setting of Cxcr3KO Treg cells.
T cell secretion of IFN-γ induces the production of CXCL9 in DCs and myeloid cells47,70. To determine whether this is the mechanism of CXCL9 induction in DCs and myeloid cells in MC38 tumors, we inoculated tumors in Batf3−/− or wildtype mice treated with PBS, anti-CD8 depleting, or anti-IFN-γ blocking antibodies and measured CXCL9 secretion from tumors. We found that CXCL9 was reduced in the absence of DC1s or with anti-CD8 or anti-IFN-γ treatment (Figure 6L). Flow cytometry revealed decreased CXCL9 production specifically in DC1s with antibody treatments (Figures 6M, 6N, and S6H–S6J). These results suggest that the IFN-γ production from CD8+ T cells may be responsible for CXCL9 production in DC1s, and this may instigate CXCR3+ Treg interaction with DC1s. Nevertheless, comparisons of mice with Cxcr3+ versus Cxcr3KO Treg cells did not reveal significant differences in CXCL9 production by DC1s, DC2s, or other myeloid cells by flow cytometry (Figures 6O and S6K). Finally, we noted no change in CXCR3 expression from Treg cells or CD8+ T cells in dLN or tumors of Batf3−/− compared to wildtype mice (Figure S6L), indicating that DC1s were not required for CXCR3 expression in T cells47,70, but that CXCR3 on Treg cells is required to properly engage and regulate DC1s.
To directly test whether CXCR3+ Treg cells required BATF3+ DC1s for preferential accumulation in tumors, we again utilized adoptive co-transfer experiments of labeled CXCR3+ versus CXCR3KO Treg cells generated in vitro but transferred the Treg cells into wildtype or Batf3−/− recipient mice and assessed their localization after 24 hours (Figure 6P). As demonstrated previously (Figure 4), CXCR3+ Treg cells accumulated in tumors better than CXCR3KO Treg cells when transferred to wildtype mice (Figure 6Q). However, the preferential localization of CXCR3+ Treg cells in tumors was abolished when transferred into Batf3−/− mice that lacked DC1s, whereas dLN localization was unaffected by the lack of DC1s (Figure 6Q). This suggests that CXCL9 production from BATF3+ DC1s is required for the preferential localization of CXCR3+ Treg cells within MC38 tumors.
Lack of CXCR3+ Treg cells boosts DC1 cross-presentation.
Our results suggest that DC1s are responsible for the accumulation of CXCR3+ Treg cells in tumors. DC1s are capable of cross-presenting tumor antigens on MHC class I (MHCI). This capability is thought to explain why DC1s are critical for eliciting strong CD8+ T cell responses against cancer71. Therefore, we hypothesized that CXCR3+ Treg cells regulate DC1s capacity to stimulate antitumor CD8+ T cell responses. To test this hypothesis, we inoculated Foxp3DTR-GFP;Cxcr3+/Foxp3TM2-GFP;Cxcr3KO female mice, pre-treated with DT, with MHC I-deficient (B2m−/−) MC38 tumor cells that expressed ovalbumin (MC38-OVA-B2m−/−). This model was used to ensure that the OVA peptide SIINFEKEL could not be directly presented by the cancer cells and only cross-presented by antigen-presenting cells (Figure 7A). We then tracked OVA-specific antitumor CD8+ T cell responses using SIIN/Kb tetramers (Figure 7A). We found a significant increase in SIIN/Kb+CD8+ T cells within tumors and dLN in the context of Cxcr3KO Treg cells (Figure 7B, 7C, and S7A–S7B). However, in Foxp3DTR-GFPCxcr3KO/Foxp3TM2-GFP;Cxcr3+ control female mice treated with DT, where CXCR3+ Treg cells remained, we did not observe an increase in SIIN/Kb+CD8+ T cells within tumors or dLN of these mice (Figures 7D and S7B). Finally, we observed a significant reduction of SIIN/Kb+CD8+ T cells within tumors from Batf3−/− mice (Figures 7E and S7C).
Figure 7. Lack of CXCR3+ Treg cells boosts cross-presentation in tumors.

(A) Experimental setup to assess tumor antigen cross-presentation by analyzing SIIN/H-2Kb presentation on antigen presenting cells (25D1.16 antibody) and CD8+ SIIN/Kb-tetramer+ T cell responses to ovalbumin expressed from β2m-deficient tumors.
(B-E) Representative flow cytometric analysis (B) and quantification (C-E) of the frequencies of intratumoral SIIN/Kb-tetramer-specific CD8+ T cells in indicated mice (day 15–20, MC38) (C-D, n=5–6 mice per group, data pooled from three independent experiments; E, n=3–4 mice per group, data representative of two independent experiments).
(F-G) Representative flow cytometric analysis (F) and quantification (G) of SIIN/H-2Kb presentation on intratumoral DC1 (XCR1+CD103+) (left) and DC2 (right) using 25D1.16 from MC38-β2m-deficient tumors that do no express OVA (OVA-negative, black) or OVA-expressing tumors in Foxp3DTR-GFPCxcr3KO/Foxp3TM2-GFP;Cxcr3+ (CXCR3+, green) and Foxp3DTR-GFPCxcr3+/Foxp3TM2-GFP;Cxcr3KO mice treated with DT (CXCR3KO, red) or PBS (CXCR3+, grey) (n=4–5 mice per DT-treated group pooled from two experiments).
Data represents mean ± SEM; *p < 0.05, **p < 0.01 from one-way ANOVA followed by Dunnet’s multiple comparisons test. Also see Figure S7.
However, the increased number of SIIN/Kb+CD8+ T cells was only an indirect measure of cross-presentation by DCs. Therefore, we used an antibody (25D1.16) that binds to SIIN-loaded H-2Kb molecules to directly monitor tumor antigen cross-presentation on antigen presenting cells by flow cytometry (Figure 7A). We found that only DC1s and no other antigen presenting cell measured (DC2s, macrophages, monocytes, pDCs) in Cxcr3KO Treg-bearing mice exhibited increased SIIN/H-2Kb (Figures 7F, 7G, and S7D–S7E). Finally, by treating Cxcr3KO Treg cell mice with anti-CD8 depleting antibodies, we found that DC1 frequencies and tumor antigen cross-presentation were not altered (Figures S7F–S7H), suggesting that CD8+ T cell tumor infiltration was not directly responsible for increased tumor antigen cross-presentation by DC1s. Altogether, these results demonstrate that co-localization of CXCR3+ Treg cells with DC1s limits DC1 cross-presentation of tumor antigens, impeding antitumor CD8+ T cell control of cancer progression.
DISCUSSION
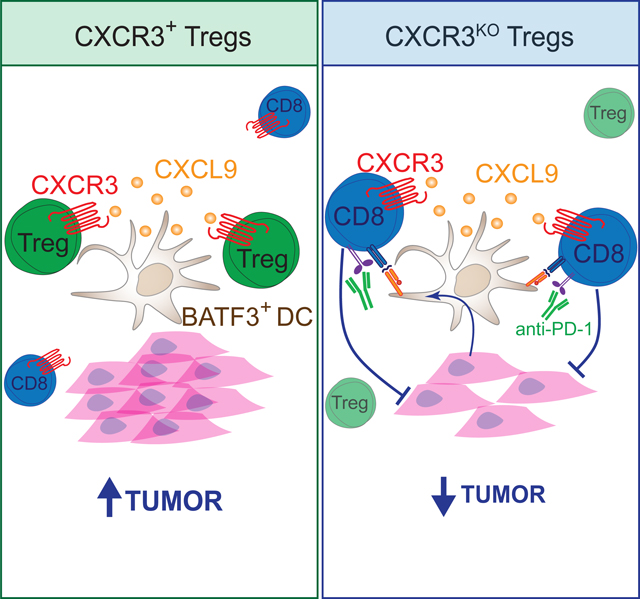
Treg cell accumulation in cancer correlates with worse prognoses. Mechanisms that reduce the number of intratumoral Treg cells are effective in controlling cancer progression in preclinical mouse cancer models without inducing systemic toxicity3,26,72–74. While these methods target distinctive features of intratumoral Treg cells or highly activated Treg cells, targeting the mechanisms that drive the accumulation of Treg cells in tumors or their localization with cell-interacting partners is feasible and may represent a complementary strategy to treat Treg-infiltrated tumors. This study investigated the role of the CXCR3 chemokine receptor in mediating Treg cell support of cancer progression. We found that Cxcr3-deficiency in Treg cells, while not impacting systemic immune homeostasis, did unleash stronger CD8+ T cell responses against multiple types of cancer, leading to slower cancer progression. We revealed that CXCR3 expression on Treg cells guides their interaction with BATF3+ DC1s within tumors to suppress DC1 priming of CD8+ T cells in the tumor dLN or their reactivation directly in tumors by impeding tumor antigen cross-presentation. While CXCR3 is not exclusively expressed on Treg cells within tumors, we showed that it is critical for Treg cell suppression of antitumor CD8+ T cells and thus is functionally relevant for immunosuppression by Treg cells in the context of cancer.
A critical role for CXCR3 in Treg cell function has been proposed previously. However, CXCR3 function has only been inferred by assessing Treg activity in the absence of the transcription factor T-BET, which directly induces CXCR3 expression. While T-BET-driven expression of CXCR3 has been proposed to guide Treg localization to sites of IFN-γ-driven inflammation and to specifically inhibit Th1 effector CD4+ T cell responses75,76, the hypothesis has not been directly tested. A recent study showed that Th1-like Treg cells control CD8+ T cell priming in the tumor dLN, but it appeared to be independent of CXCR3 function47. Only in the setting of autoimmunity of the kidney (crescentic glomerulonephritis), using Foxp3-driven CRE-specific deletion of T-betfl/fl or Cxcr3fl/fl alleles, was it definitively shown that CXCR3 alone was required for Treg cell recruitment to the kidney to reduce the severity of autoimmunity42,46. In our present work, we utilized a novel strategy that takes advantage of X chromosome inactivation to generate mice with Treg-specific Cxcr3-deficiency to show that CXCR3 expression alone is required for Treg interactions with DC1s to control CD8+ T cell responses against cancer. Collectively, our study reveals that an IFN-induced chemokine (CXCL9) has both stimulatory and suppressive actitivity due to interactions with CXCR3 on both CD8+ T cells and Treg cells.
CXCR3+ Treg cells have been found enriched in a multitude of human and mouse cancers48–51, implicating CXCR3+ Treg cells as a target for cancer immunotherapy. Here, we found that CXCR3 was increased on Treg cells in tumor-draining lymph nodes and, most prominently, within tumors, where CXCR3+ Treg cells constituted a highly activated population of antigen-experienced cells. However, many reports suggest that other chemokine receptors are instrumental for Treg function in cancer. Indeed, CCR4 was the first chemokine receptor described to be highly expressed on intratumoral Treg cells6,21,26, and was shown to be critical for Treg accumulation in tumors16,77,78. This led to clinical trials using an anti-CCR4 antibody, Mogamulizumab, which depleted Treg cells and increased tumor-specific CD8+ T cells in adult T-cell leukemia-lymphoma patients26,79. More recently, CCR8 was found increased on Treg cells from breast cancer tissue19, as well as other types of cancer18. Antibody-mediated depletion of CCR8+ Treg cells, as was also shown with CCR4 antibodies, now constitutes a promising strategy to treat cancer23,25,28. However, CCR8 expression on Treg cells was found to be dispensable for Treg cell accumulation in tumors and their suppression of antitumor immune responses28,29. Therefore, the critical chemokine receptors expressed on Treg cells needed to control the immune response to cancer have not been fully defined by prior studies.
In addition, while targeting individual chemokine receptors such as CCR416,77, CCR825,28,29, or CXCR3 (in this study) can reduce intratumoral Treg cell infiltration and slow tumor growth, these approaches have not been as effective as methods that more dramatically deplete intratumoral Treg cells3,72,73,80. This may suggest that dynamic expression of multiple chemokine receptors is involved in Treg accumulation in cancer and thus the examination of specific chemokine receptors in isolation is incomplete. Furthermore, co-expression of CXCR3 and other chemokine receptors, such as CCR4 or CCR8, has been observed13,81,82, and we also observed co-expression of other chemokine receptors in concert with CXCR3 on Treg cells in tumors (not shown). Therefore, while a single chemokine receptor may not be sufficient for Treg cell recruitment and function in tumors, our results do indicate that CXCR3 expression on Treg cells is required for their full suppressive function in many cancers.
Tumors are highly organized pseudo-organs. It is now appreciated that the spatial organization of its heterogeneous cells and components are a crucial feature of tumor biology83–87. Chemokines may act like a “zip code” to guide immune cells to specific niches for cell-cell interactions and immune activation88. In general, Th1 responses in cancer patients, characterized by IFN signaling, are associated with a better prognosis89. CXCR3 ligands, namely CXCL9 and CXCL10, are induced by IFN. Human tumors lacking CXCR3 ligands have reduced CD8+ T cell infiltration90. Several studies have pointed to CXCR3 ligand expression from DCs as critical for orchestrating the immune response to cancer, and particularly important for the efficacy of immunotherapy32,37,91. Moreover, a subset of DCs, the type I conventional DCs (DC1s), distinguished by their expression of the transcription factor BATF3, have been shown to take up tumor antigens within tumors and transport them to the draining lymph nodes to mediate the priming of antitumor CD8+ T cell responses92,93. However, these same DC1s are likely targets of suppressive mechanisms that promote tumor escape, in particular, they may be targeted by Treg cells that can modulate the potency of their antigen presenting activity94. Here, we found extensive interactions between Treg cells and XCR1+ DC1s, which produced the bulk of CXCL9 released in tumors and depended on CXCR3 expression on Treg cells to interact. The dynamics of CXCR3 expression on Treg cells during tumor progression also mirrored the expression pattern on CD8+ T cells, and in the absence of CXCR3+ Treg cells, CD8+ T cell interactions with XCR1+ cells in tumors were increased. Thus, competition between CXCR3+ Treg cells and CD8+ T cells for access to DC1s in tumors and draining lymph nodes may underlie Treg-mediated control of CD8+ T cell responses to cancer. Ultimately, the functional consequence of these DC1-CXCR3+ Treg cell interactions was reduced tumor antigen cross-presentation by DC1s in tumors and dLN.
Altogether, we demonstrated a novel role for CXCR3 expression on Treg cells in the context of cancer. Cxcr3-deficiency on Treg cells allowed for decoupling of Treg-DC1 interactions. The most relevant consequence was increased accumulation of tumor antigen-specific CD8+ T cells due to increased access to, and activation by, DC1s. The efficacy of checkpoint blockade was also enhanced by Cxcr3-deficiency in Treg cells. Therefore, successful immunotherapies that increase IFN/CXCL9 production must consider disrupting the recruitment of CXCR3+ Treg cells. Total CXCR3 blockade is challenged by its role in other immune cells that express CXCR3, such as T cells and NK cells, that fight cancer. Targeting epigenetic modulators, such as Ezh2, may counteract these challenges by both reprogramming Treg functionality while also increasing CXCL9 and CXCL10 expression from tumor cells3,95,96. Alternatively, specifically targeting CXCR3+ Treg cells could be achieved using bi-specific antibodies that target a Treg-specific receptor, such as CTLA-4 or CD25, and a CXCR3 antibody that is activated specifically in the tumor microenvironment97,98. Here we have clarified a critical mechanism underlying Treg accumulation across multiple tumor types and have shown that blocking CXCR3 function in Treg cells has therapeutic potential by promoting antitumor CD8+ T cell immunity.
Limitations of the study
While our study provides insight into the mechanism of CXCR3 function in mediating Treg promotion of cancer, the therapeutic potential of targeting CXCR3 in established tumors is untested. Here we showed that the removal of wildtype Treg cells in the context of Cxcr3KO Treg cells in established sarcomas from genetically engineered mice led to decreased frequencies of Treg cells and increased frequencies of CD8+ T cells (Figures S4D–H), but we did not measure the impact on tumor growth. This was due to the rapid growth rate of tumors, which only allowed for one week of treatment with DT before a humane endpoint. Optimization of these experiments will be needed for further investigation. Nevertheless, our model to generate Cxcr3KO Treg cells, wherein we must ablate wildtype Treg cells, is problematic and unrealistic for testing the therapeutic efficacy of targeting CXCR3 on Treg cells. Alternative strategies to those employed here will be needed to address the therapeutic potential of targeting CXCR3 on Treg cells in established tumors.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Michel DuPage (dupage@berkeley.edu).
Materials availability
MC38-OVA-B2m−/− were generated in this study. All will be made available from the lead contact upon request.
Data and code availability
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
In vivo animal studies
All the mouse experiments used comparisons between littermates or age-matched control mice. Foxp3DTR-GFP mice express human diphtheria toxin receptor and EGFP genes from the Foxp3 locus without disrupting expression of the endogenous Foxp3 gene65. These mice were kindly provided by Dr. Rudensky (Memorial Sloan Kettering Institute). Foxp3TM2-GFP mice were provided by Dr. Chatila (Boston Children’s)99. CXCR3KO mice were obtained from Jackson laboratories (JAX:005796), this strain was originally created and characterized by Deltagen, Inc. For diphtheria toxin treatments, female mice were treated with a dose 1μg i.p. every other day up to three times per week. For tumor studies, syngeneic C57BL/6 mice were inoculated with 1×105 MC38-GFP cells engineered to express luciferase and GFP3, 2×105 EL4 cells, or 2×106 9464D100 cells in 100 μL of PBS subcutaneously. Tumor measurements were performed blindly across the entire experiment by a single operator measuring three dimensions of the tumor with calipers three times per week. All the experiments were conducted according to the Institutional Animal Care and Use Committee guidelines of the University of California, Berkeley.
Cell lines
MC38 and MC38 expressing ovalbumin (OVA) were kindly provide by Dr. Bluestone101. MC38 cell lines were also engineered to express luciferase and GFP3. EL4 cells were obtained from Cell Culture core at UC Berkeley. 9464D were kindly provided by Dr. Weiss (UCSF)100. MC38-B2m-deficient cell lines were generated in this study using B2m-specific guides cloned into PX458 (see below)102. All cell lines were maintained in DMEM (GIBCO) supplemented with 10% FBS, sodium pyruvate (GIBCO), 10mM HEPES (GIBCO), and penicillin-streptomycin (GIBCO). Tumor cells were grown at 37°C with 5% CO2.
METHOD DETAILS
MC38-B2m−/− Generation
To generate MC38-B2m-deficient cell lines we employed B2m-specific guides cloned into PX458102. CRISPR-Cas9 guide sequence for B2m: AGTCGTCAGCATGGCTCGCT. Briefly, to generate knockout cell lines, plasmids were transiently transfected using LipoD293™ In Vitro DNA Transfection Reagent (SignaGen Laboratories). GFP+ (from PX458 transfection) cells were sorted one day after nucleofection to ensure that all cells in culture received the Cas9 plasmid. One-week later, cells were stained with MHC Class I (H-2Kb) (Clone AF6–88.5.5.3, Biolegend) MHC I negative cells were sorted using a FACS Aria cell sorter. MHC IKO cells were sorted again the following week to >98% purity.
Murine Lymphocyte Isolation
Resected tumors and lung tissues were minced to 1 mm3 fragments and digested in RPMI media supplemented with 4-(2-hydroxyethyl)-1-piperazi-neethanesulfonic acid (HEPES), 20 mg/mL DNase I (Roche), and 125 U/mL collagenase D (Roche) using an orbital shaker for ~45 min at 37°C. Cells from lymphoid organs were prepared by mechanical disruption between frosted slides. All the cell suspensions were passed through 40 μm filters before cell staining or in vitro stimulation. Cytokine staining was performed with 3–5×106 cells after 210 min of in vitro stimulation in Opti-MEM media supplemented with Brefeldin A (eBioscience), 10 ng/ml phorbol 12-myristate 13-acetate (PMA) (Sigma), and 0.25 μM ionomycin (Sigma). Fixation/permeabilization of cells was conducted for intracellular staining using the eBioscience Foxp3 fixation/permeabilization kit (BioLegend) or Tonbo Foxp3 / Transcription Factor Staining Buffer Kit. In some experiments, e.g. representative staining of in vitro differentiated cells prior to adoptive transfer, cells were fixed with 2% PFA in ice for 10 minutes.
Flow Cytometry
Flow cytometry was performed on an BD LSR Fortessa X20 (BD Biosciences), LSRFortessa (BD Biosciences) or Cytek Aurora, and datasets were analyzed using FlowJo software (Tree Star). Single cell suspensions were prepared in ice-cold FACS buffer (PBS with 2mM EDTA and 1% FBS) and subjected to red blood cell lysis using ACK buffer (150mM NH4Cl, 10mM KHCO3, 0.1mM Na2EDTA, pH7.3). Dead cells were stained with Live/Dead Fixable Blue or Aqua Dead Cell Stain kit (Molecular Probes) in PBS for 20 minutes at 4°C. Cell surface antigens were stained for 1 hour at 4°C using a mixture of fluorophore-conjugated antibodies. Surface marker stains for murine samples were carried out with anti-mouse CD3 (17A2, BioLegend), anti-mouse CD4 (RM4–5, BioLegend), anti-mouse CD8a (53–6.7, BioLegend), anti-CD25 (PC61, BioLegend), anti-mouse CD44 (IM7, BioLeged), anti-mouse CD45 (30-F11, BioLegend), anti-mouse CD103 (2E7, BioLegend), anti-mouse CXCR3 (CXCR3–173, BioLegend), anti-mouse KLRG1 (2F1, BioLegend), anti-mouse PD1 (RMP1–30, BioLegend), anti-mouse MHCI ((H-2Kb, AF6–88.5.5.3, BioLegend)), anti-mouse MHCII (M5/114.15.2, BioLegend), anti-H-2Kb MuLV p15E Tetramer-KSPWFTTL (MBL), anti-Kb-A2/SIINFEKEL tetramer (NIH tetramer core), OVA257–264 (SIINFEKL) peptide bound to H-2Kb (25-D1.16, BioLegend), anti-XCR1 (ZET, BioLegend) in PBS, 2% FBS, for 1 hour at 4°C. Cells were fixed using the eBioscience Foxp3/Transcription Factor staining buffer set (eBioscience), prior to intracellular staining. Intracellular staining was performed using anti-mouse CTLA4 (UC10–4B9, BioLegend), anti-mouse Foxp3 (FJK-16S, eBioscience), anti-mouse T-bet (4B10, BioLegend), anti-mouse TNF-a (MP6-XT22, BioLegend), anti-mouse IFNg (XMG1.2, eBioscience), anti-CXCL9 (MIG-2F5.5, BioLegend), 1 hour, at 4°C, according to manufacturer’s instructions. Cells were re-suspended in FACS buffer and filtered through a 70-μm nylon mesh before data acquisition. Datasets were analyzed using FlowJo software (Tree Star).
In vivo antibody-mediated cell depletion or cytokine blockade
For tumor progression studies CD8 depletion was achieved by intraperitoneal injection of 250 μg per mouse of the anti-aCD8 monoclonal antibody clone YST-169.4 (BioXcell, Catalog # BE0117) two days prior to tumor inoculation, followed by additional doses every 6 days thereafter. For PD-1 depletion, intraperitoneal injection of 200 μg/mouse of clone RMP1–14 (BioXcell, Catalog: BE0146) was done twice per week. For studies that involved myeloid compartment characterization, we used anti-mouse CD8b, 250 μg per mouse, clone 53–5.8 for CD8 T cell depletions (Leinco Technologies, Catalog # C2832). IFN-γ blockade was achieved using anti-mouse IFN-γ, clone XMG1.2, 250 μg per mouse (Leinco Technologies, Catalog # I-1119). For CD8b and IFN-γ depletions, mice received the antibodies two days and one day prior to tumor inoculation, followed by additional doses every 6 days until the end of the experiment.
Genetically Engineered Model of Cancer (GEMM).
Foxp3DTR-GFP and Foxp3TM2-GFP;Cxcr3KO alleles were crossed to KrasLSL-G12D/+;p53fl/fl mice. Briefly, in accordance with previous published data63, we generated sarcomas in mice by intramuscular (leg) injection of 5×10^4 lentiviral particles expressing Cre recombinase (Lenti-Cre). Tumors were palpable 3–4 months after Lenti-Cre injection and four doses of DT was administrated every two days until the end of the experiment.
Cytokines measurements
Tumors from mice were dissected 15–20 days post tumor inoculation. One million cells were plated in 100–200 μl of OPTIMEM in a 96 well plate and incubated at 37 C for 4 hours. Supernatants were collected and ELISA was performed using Mouse CXCL9/MIG DuoSet ELISA (Catalog #: DY492, R&D Systems) or Mouse CXCL10/IP-10/CRG-2 DuoSet ELISA (Catalog #: DY466, R&D Systems), following protocol’s instructions. For detection of CXCL9 by flow cytometry (MIG-2F5.5, BioLegend), we performed intracellular staining of single cell suspensions from tissues after a 4 hour incubation with BFA as done for cytokine production flow cytometry assays.
Adoptive transfer experiments
For in vitro T cell culture, spleens and lymph nodes were collected from Foxp3 reporter mice and enriched for CD4 T cells by negative selection using EasySep magnetic bead kit (STEMCELL Technologies. Naïve Tregs (CD4+CD8−CD62L+Foxp3GFP+ cells) were then sorted using an Aria Fusion sorter (BD Biosciences) with a 70μm nozzle. Tregs were activated with anti-CD3 and anti-CD28 coated beads (Dynabeads Mouse T-Activator CD3/CD28, Invitrogen) at a ratio of 1:3 (cell:bead) and kept at a concentration of 106 cells/ml in DMEM medium supplemented with 10% FBS, non-essential amino acids, sodium pyruvate, L-glutamine, HEPES, 55 μM β-ME and 2,000 IU/ml recombinant human IL-2 (TECIM™, Hoffman-La Roche provided by NCI repository, Frederick National Laboratory for Cancer Research). For Th1 Treg differentiation, 48 hours after sorting, media was supplemented with 40 ng of recombinant mouse IFN-γ (Biolegend, Cat# 575304) and recombinant mouse IL-12 Protein (R&D Systems, 419-ML). 4×105-6×105 WT and CXCR3KO Tregs were stained with ViaFluor 405 (Biotum) or CellTrace Far Red (Molecular Probes) dyes according to the manufacturer’s instructions. Dyes were interchanged between experiments to prevent bias in the results due to potential differences in staining. Tregs were co-transferred to WT or Batf3−/− MC38 tumor-bearing mice at 1:1 Tregs WT or CXCR3KO ratio by intravenous injection. 24 hours later, mice were euthanized, and tissues removed for flow cytometry analysis.
Tumor tissue cryo sectioning
Tumors were removed and fixed in 4% paraformaldehyde in ice in the dark for four hours, washed twice with PBS, then dehydrated in 10% sucrose, 20% sucrose, and 30% sucrose for at least 6 hours each at 4°C. Tumors were imbedded in OCT (Fisher) cut side-down and frozen in a slurry of dry ice and ethanol, then stored at −80°C until sectioning. Tumors were cut into 12-micron sections on a cryostat microtome (Leica Biosystems) onto SuperFrost Plus slides (Fisher Scientific) and stored at −80°C until used for immunofluorescence.
Immunofluorescence
Sections were permeabilized with tissue staining buffer (PBS with 0.3% Triton X-100, 0.1 M glycine, and 1% BSA) and blocked with tissue staining buffer plus 5% normal rat serum and FcγII/III CD16/32 antibody (2.4G2; BioLegend Catalog 101320), before staining with anti-CD8 alpha antibody (Clone YTS169.4, Abcam Catalog ab22378) ON, at 4°C, washed with PBS followed by incubation with IgG (H+L) Cross-Adsorbed Goat anti-Rat, DyLight™ 594, (Invitrogen, Catalog PISA510020) or a cocktail of CXCL10 goat antibody103 and CXCL9 Recombinant Rabbit Monoclonal Antibody (Clone: 11H1L14, Invitrogen Catalog 701117) followed by Alexa Fluor 647-conjugated bovine anti-goat IgG (Jackson Immunoresearch) and Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 594 (Invitrogen Catalog A32740). Anti-XCR1-biotin staining was performed ON at 4C (1:50, Clone ZET, BioLegend Catalog 148212) followed by two hours incubation with Streptavidin-Alexa Fluor™ 647 Conjugate (1:100, Invitrogen, Catalog S32357). Stained tissue sections were mounted with Vectashield® Mounting Medium, (Vector Laboratories). Images were taken using Zeiss LSM 880 NLO OPO microscope. Imaging analysis was performed using Imaris software.
Statistical Methods
p values were obtained from unpaired two-tailed Student’s t tests for all statistical comparisons between two groups, and data were displayed as mean ± SEMs. Paired t tests were used in all adoptive transfer of Treg tracking experiments. For multiple comparisons, one-way ANOVA was used. For tumor growth curves, two-way ANOVA was used with Sidak’s multiple comparisons test performed at each time point or by multiple regression analysis p values are denoted in figures by *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 BUV737 Clone 17A2 | BD | Cat#: 612803 |
| CD4 BV605 Clone RM4-5 | BioLegend | Cat#:100548 |
| CD4 APC Clone RM4-S | BioLegend | Cat#:100516 |
| CD8 BUV 737 Clone 53-6.7 | BD | Cat#:564297 |
| CD8a PerCP-Cy5.5 Clone 53-6.7 | BioLegend | Cat#:100734 |
| CD25 BV785 Clone PC61 | BioLegend | Cat#:102051 |
| CD25 PerCP-Cy5.5 Clone PC61 | BioLegend | Cat#:102030 |
| CD44 APCe-780 Clone IM7 | eBioscience/ Thermo | Cat#:47-0441-82 |
| CD44 PerCP-Cy5.5 Clone I7 | BioLegend | Cat#:103036 |
| CD45 BUV-395 Clone 30-F11 | BD | Cat#:564279 |
| CD69 PE-Cy7 Clone H1.2F3 | BioLegend | Cat#:104510 |
| CD103 BV605 2E7 | BioLegend | Cat#:121433 |
| CD103 BV510 2E7 | BioLegend | Cat#:121423 |
| CXCL9 PE MIG-2F5.5 | BioLegend | Cat#:515604 |
| CXCR3 (CD183) PE CXCR3-173 | BioLegend | Cat#:126506 |
| CXCR3 (CD183) APC CXCR3-173 | BioLegend | Cat#:126512 |
| KLRG1 BV510 2F1/KLRG1 | BioLegend | Cat#:138421 |
| KLRG1 APC 2F1/KLRG1 | BioLegend | Cat#:138412 |
| PD1 (CD279) BV711 29F.1A12 | BioLegend | Cat#:135231 |
| CTLA-4 (CD152) BV605 UC10-4B9 | BioLegend | Cat#:106323 |
| Foxp3-e450 FJK-16S | eBioscience/ Thermo | Cat#:48-5773-82 |
| Foxp3 FITC FJK-16S | eBioscience/ Thermo | Cat#:11-5773-82 |
| T-bet PE-Cy7 4B10 | BioLegend | Cat#:644824 |
| TNFα PerCP-Cy5.5 MP6-XT22 | BioLegend | Cat#:506322 |
| IFNg e450 XMG1.2 | eBioscience/ Thermo | Cat#:48-7311-82 |
| H-2Kb MuLV p15E Tetramer-KSPWFTTL-PE | MBL | Cat#: TB-M507-1 |
| K(b)-A2/ova.SIINFEKL-APC | NIH | N/D |
| MHC Class I (H-2Kb) AF6-88.5.5.3 | BioLegend | Cat#: 17-5958-80 |
| MHC Class II (I-A/I-E) | BioLegend | Cat#: 10-7628 |
| XCR1 Alexa Fluor® 647 anti-mouse/rat XCR1 Antibody | BioLegend | Cat#: 148214 |
| Biotin anti-mouse/rat XCR1 Antibody | BioLegend | Cat#: 148212 |
| OVA257-264 (SIINFEKL) peptide bound to H-2Kb Monoclonal Antibody | Thermo Scientist | Cat#: 13-5743-81 |
| CD16/32 antibody | BioLegend | Cat#:101320 |
| InVivoMAb anti-mouse CD8α | BioXcell | Cat#: BE0117 |
| InVivoMAb anti-mouse PD-1 (CD279) | BioXcell | Cat#: BE0146 |
| Anti-Mouse CD8b.2 Purified In vivo GOLD™ | Leinco Technologies | Cat# C2832 |
| Anti-Mouse IFN-γ Purified In vivo GOLD™ | Leinco Technologies | Cat# I-1119 |
| aCD8 alpha Clone YTS169.4 | Abcam | Cat#: ab22378 |
| Goat anti-Rat, DyLight™ 594 | Invitrogen | Cat#:PISA510020 |
| CXCL9 Recombinant Rabbit Monoclonal Antibody Clone: 11H1L14 | Invitrogen | Cat#:701117 |
| Alexa Fluor 647-conjugated bovine anti-goat IgG | Jackson Immunoresearch | Cat#: 805-607-008 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ Plus 594 | Invitrogen | Cat#:A32740 |
| Mouse CXCL10/IP-10/CRG-2 Antibody | R&D | Cat#:AF-466-NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| eBioscience Foxp3 fixation/permeabilization kit | eBioscience | Cat#:00-5523-00 |
| Tonbo Foxp3 / Transcription Factor Staining Buffer Kit | Tonbo Bioscience | Cat#:TNB-0607-KIT |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15713S |
| DNase I | Roche | Cat#:10104159001 |
| Collagenese IV | Roche | Cat#:11088882001 |
| Diphtheria Toxin | Sigma-Aldrich | Cat#:322326 |
| DMEM High Glu w/Gl w/ Pyr | Gibco | Cat# 11995-065 |
| 100 X Penicilin-Streptomycin | Gibco | Cat# 15140-122 |
| HEPES | Gibco | Cat#: 15630-080 |
| MEM Non-Essential Amino Acids Solution (100X | Gibco | Cat#: 11140-050 |
| Fetal Bovine Serum, Heat Inactivated | Omega Scientist | Cat#: FB-01 |
| Recombinant human IL-2 Teceleukin (Tecin™) | Hoffmann-LAROCHE | NCI repository (Frederick National Laboratory for Cancer Research) |
| LEAF™ Purified anti-mouse IFN-γ [XMG1.2] | BioLegend | Cat# 505802 |
| Recombinant Mouse IL-12 Protein | R&D | Cat#: 419-ML |
| Protein Transport Inhibitor (Containing Monensin), BD GolgiStop™ | BD Biosciences | Cat#: 554724 |
| eBioscience™ Fixable Viability Dye eFluor™ 780 | eBioscience | Cat#:65-0865-14 |
| LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit | Molecular Probes | Cat#: L34962 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit | Molecular Probes | Cat# L34965 |
| ViaFluor® 405 Cell Proliferation Kit | Biotium | Cat#:30068 |
| CellTrace™ Far Red Cell Proliferation Kit, for flow cytometry | Molecular Probes | Cat#: C34564 |
| Streptavidin, Alexa Fluor™ 647 Conjugate | Invitrogen | Cat#: S32357 |
| Triton X-100 | Fisher Scientific | Cat#: BP151-100 |
| SuperFrost Plus slides | Fisher Scientific | Cat# 12-550-15 |
| Critical Commercial Assays | ||
| CD4 Enrichment EasySep magnetic bead kit | STEMCELL Technologies | Cat#: 19852A |
| Dynabeads™ Mouse T-Activator CD3/CD28 for T-Cell Expansion and Activation | Gibco | Cat#: 11456D |
| Mouse CXCL10/IP-10/CRG-2 DuoSet ELISA | R&D | Cat#:DY466-05 |
| Mouse CXCL9/MIG DuoSet ELISA | R&D | Cat#:DY492-05 |
| DuoSet Ancillary Reagent Kit 2 | R&D | Cat#: DY008 |
| LipoD293™ In Vitro DNA Transfection Reagent | SignaGen Laboratories | Cat#: SL100668 |
| Experimental Models: Cell lines | ||
| MC38 | Bluestone Laboratory | N/D |
| EL4 | Cell Culture facility UC Berkeley | N/D |
| 9464D | Bill Weiss Laboratory | N/D |
| MC38-OVA | Bluestone Laboratory | N/D |
| MC38-OVA-B2mKO | This paper | N/D |
| Experimental Models: Organisms/Strains | ||
| Foxp3DTR-GFP | Rudensky Laboratory | RRID:IMSR_JAX:016958 |
| CXCR3KO mice | The Jackson Laboratory | RRID:IMSR_JAX:005796 |
| Foxp3TM2-GFP | Chatila Laboratory | RRID:IMSR_JAX:006772 |
| C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Software | ||
| FlowJo (v10) | TreeStar | https://www.flowjo.com |
| Imaris Image Analysis Software | BitPlane | http://www.bitplane.com/ |
| GradhPad prism | Dotmatics | https://www.graphpad.com/scientific-software/prism/ |
| Illustrator | Adobe | https://www.adobe.com/products |
Highlights:
CXCR3 is elevated on Treg cells within draining lymph nodes and tumors
CXCR3+ Treg cellss co-localize with CXCL9-producing type 1 DCs in tumors
Disrupting CXCR3 in Tregs increases tumor antigen cross-presentation by DC1s in tumors
Loss of CXCR3 in Treg cells boosts tumor CD8+ T cells and slows cancer progression
ACKNOWLEDGEMENTS
We thank David Raulet, Gregory Barton, Ellen Robey, Lin He and Janneke Peeters for reviewing the manuscript and providing feedback; Hector Nolla, Alma Valleros, Kartoosh Heydari and Anita Wong Lin of the UC Berkeley Cancer Research Laboratory Flow Cytometry Facility; Holly Aaron and Feather Ives of the UC Berkeley Cancer Research Laboratory Molecular Imaging Center. This research was supported by 5T32AI100829-09 to M.A.M.A. and 1DP2CA247830-01. M.D. is a Pew-Stewart Scholar and a St. Baldrick’s Scholar with generous support from Hope with Hazel.
Footnotes
DECLARATION OF INTERESTS
M.A.M.A. is currently employed by Revolution Medicines (Redwood City, CA).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Liu C, Workman CJ, and Vignali DA (2016). Targeting regulatory T cells in tumors. FEBS J 283, 2731–2748. 10.1111/febs.13656. [DOI] [PubMed] [Google Scholar]
- 2.Nunez NG, Tosello Boari J, Ramos RN, Richer W, Cagnard N, Anderfuhren CD, Niborski LL, Bigot J, Meseure D, De La Rochere P, et al. (2020). Tumor invasion in draining lymph nodes is associated with Treg accumulation in breast cancer patients. Nat Commun 11, 3272. 10.1038/s41467-020-17046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, Silveria S, Harwin T, Abnousian A, Pagani M, et al. (2018). Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep 23, 3262–3274. 10.1016/j.celrep.2018.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DuPage M (2011). Endogenous T cell responses to antigens expressed in lung adenocarcinoma delay malignant tumor progression. Cancer Cell 19 (Supplemental data). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shang B, Liu Y, Jiang SJ, and Liu Y (2015). Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep 5, 15179. 10.1038/srep15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cinier J, Hubert M, Besson L, Di Roio A, Rodriguez C, Lombardi V, Caux C, and Menetrier-Caux C (2021). Recruitment and Expansion of Tregs Cells in the Tumor Environment-How to Target Them? Cancers (Basel) 13. 10.3390/cancers13081850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wing JB, Tanaka A, and Sakaguchi S (2019). Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 50, 302–316. 10.1016/j.immuni.2019.01.020. [DOI] [PubMed] [Google Scholar]
- 8.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. (2004). Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10, 942–949. 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka A, and Sakaguchi S (2017). Regulatory T cells in cancer immunotherapy. Cell Res 27, 109–118. 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sather BD, Treuting P, Perdue N, Miazgowicz M, Fontenot JD, Rudensky AY, and Campbell DJ (2007). Altering the distribution of Foxp3(+) regulatory T cells results in tissue-specific inflammatory disease. J Exp Med 204, 1335–1347. 10.1084/jem.20070081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferreira C, Barros L, Baptista M, Blankenhaus B, Barros A, Figueiredo-Campos P, Konjar S, Laine A, Kamenjarin N, Stojanovic A, et al. (2020). Type 1 Treg cells promote the generation of CD8(+) tissue-resident memory T cells. Nat Immunol 21, 766–776. 10.1038/s41590-020-0674-9. [DOI] [PubMed] [Google Scholar]
- 12.Seung E, Cho JL, Sparwasser T, Medoff BD, and Luster AD (2011). Inhibiting CXCR3-dependent CD8+ T cell trafficking enhances tolerance induction in a mouse model of lung rejection. J Immunol 186, 6830–6838. 10.4049/jimmunol.1001049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oo YH, Weston CJ, Lalor PF, Curbishley SM, Withers DR, Reynolds GM, Shetty S, Harki J, Shaw JC, Eksteen B, et al. (2010). Distinct roles for CCR4 and CXCR3 in the recruitment and positioning of regulatory T cells in the inflamed human liver. J Immunol 184, 2886–2898. 10.4049/jimmunol.0901216. [DOI] [PubMed] [Google Scholar]
- 14.Tan TG, Mathis D, and Benoist C (2016). Singular role for T-BET+CXCR3+ regulatory T cells in protection from autoimmune diabetes. Proc Natl Acad Sci U S A 113, 14103–14108. 10.1073/pnas.1616710113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ozga AJ, Chow MT, and Luster AD (2021). Chemokines and the immune response to cancer. Immunity 54, 859–874. 10.1016/j.immuni.2021.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marshall LA, Marubayashi S, Jorapur A, Jacobson S, Zibinsky M, Robles O, Hu DX, Jackson JJ, Pookot D, Sanchez J, et al. (2020). Tumors establish resistance to immunotherapy by regulating Treg recruitment via CCR4. J Immunother Cancer 8. 10.1136/jitc-2020-000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuo K, Itoh T, Koyama A, Imamura R, Kawai S, Nishiwaki K, Oiso N, Kawada A, Yoshie O, and Nakayama T (2016). CCR4 is critically involved in effective antitumor immunity in mice bearing intradermal B16 melanoma. Cancer Lett 378, 16–22. 10.1016/j.canlet.2016.04.039. [DOI] [PubMed] [Google Scholar]
- 18.Haruna M, Ueyama A, Yamamoto Y, Hirata M, Goto K, Yoshida H, Higuchi N, Yoshida T, Kidani Y, Nakamura Y, et al. (2022). The impact of CCR8+ regulatory T cells on cytotoxic T cell function in human lung cancer. Sci Rep 12, 5377. 10.1038/s41598-02209458-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, Chudakov DM, and Rudensky AY (2016). Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 45, 1122–1134. 10.1016/j.immuni.2016.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong DS, Rixe O, Chiu VK, Forde PM, Dragovich T, Lou Y, Nayak-Kapoor A, Leidner R, Atkins JN, Collaku A, et al. (2022). Mogamulizumab in Combination with Nivolumab in a Phase I/II Study of Patients with Locally Advanced or Metastatic Solid Tumors. Clin Cancer Res 28, 479–488. 10.1158/1078-0432.CCR-21-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshie O (2021). CCR4 as a Therapeutic Target for Cancer Immunotherapy. Cancers (Basel) 13. 10.3390/cancers13215542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saito T, Kurose K, Kojima T, Funakoshi T, Sato E, Nishikawa H, Nakajima J, Seto Y, Kakimi K, Iida S, et al. (2021). Phase Ib study on the humanized anti-CCR4 antibody, KW-0761, in advanced solid tumors. Nagoya J Med Sci 83, 827–840. 10.18999/nagjms.83.4.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Campbell JR, McDonald BR, Mesko PB, Siemers NO, Singh PB, Selby M, Sproul TW, Korman AJ, Vlach LM, Houser J, et al. (2021). Fc-Optimized Anti-CCR8 Antibody Depletes Regulatory T Cells in Human Tumor Models. Cancer Res 81, 2983–2994. 10.1158/0008-5472.CAN-20-3585. [DOI] [PubMed] [Google Scholar]
- 24.Wang T, Zhou Q, Zeng H, Zhang H, Liu Z, Shao J, Wang Z, Xiong Y, Wang J, Bai Q, et al. (2020). CCR8 blockade primes antitumor immunity through intratumoral regulatory T cells destabilization in muscle-invasive bladder cancer. Cancer Immunol Immunother 69, 1855–1867. 10.1007/s00262-020-02583-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kidani Y, Nogami W, Yasumizu Y, Kawashima A, Tanaka A, Sonoda Y, Tona Y, Nashiki K, Matsumoto R, Hagiwara M, et al. (2022). CCR8-targeted specific depletion of clonally expanded Treg cells in tumor tissues evokes potent tumor immunity with long-lasting memory. Proc Natl Acad Sci U S A 119. 10.1073/pnas.2114282119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, Ezoe S, Kanakura Y, Sato E, Fukumori Y, et al. (2013). Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc Natl Acad Sci U S A 110, 17945–17950. 10.1073/pnas.1316796110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, Doi T, Suzuki S, Isobe M, Funakoshi T, et al. (2015). Phase Ia Study of FoxP3+ CD4 Treg Depletion by Infusion of a Humanized Anti-CCR4 Antibody, KW-0761, in Cancer Patients. Clin Cancer Res 21, 4327–4336. 10.1158/1078-0432.CCR-15-0357. [DOI] [PubMed] [Google Scholar]
- 28.Van Damme H, Dombrecht B, Kiss M, Roose H, Allen E, Van Overmeire E, Kancheva D, Martens L, Murgaski A, Bardet PMR, et al. (2021). Therapeutic depletion of CCR8(+) tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy. J Immunother Cancer 9. 10.1136/jitc-2020-001749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Whiteside SK, Grant FM, Gyori DS, Conti AG, Imianowski CJ, Kuo P, Nasrallah R, Sadiyah F, Lira SA, Tacke F, et al. (2021). CCR8 marks highly suppressive Treg cells within tumours but is dispensable for their accumulation and suppressive function. Immunology 163, 512–520. 10.1111/imm.13337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Minn AJ, and Wherry EJ (2016). Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 165, 272–275. 10.1016/j.cell.2016.03.031. [DOI] [PubMed] [Google Scholar]
- 31.Mowat C, Mosley SR, Namdar A, Schiller D, and Baker K (2021). Antitumor immunity in mismatch repair-deficient colorectal cancers requires type I IFN-driven CCL5 and CXCL10. J Exp Med 218. 10.1084/jem.20210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, and Luster AD (2019). Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 50, 1498–1512 e1495. 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, Ku AW, Frelinger JG, Odunsi K, Gajewski TF, et al. (2015). Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun 6, 7458. 10.1038/ncomms8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, Lanitis E, Duraiswamy J, Tanyi JL, Benencia F, et al. (2019). Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 35, 885–900 e810. 10.1016/j.ccell.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vonderhaar EP, Barnekow NS, McAllister D, McOlash L, Eid MA, Riese MJ, Tarakanova VL, Johnson BD, and Dwinell MB (2021). STING Activated Tumor-Intrinsic Type I Interferon Signaling Promotes CXCR3 Dependent Antitumor Immunity in Pancreatic Cancer. Cell Mol Gastroenterol Hepatol 12, 41–58. 10.1016/j.jcmgh.2021.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maurice NJ, McElrath MJ, Andersen-Nissen E, Frahm N, and Prlic M (2019). CXCR3 enables recruitment and site-specific bystander activation of memory CD8(+) T cells. Nat Commun 10, 4987. 10.1038/s41467-019-12980-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spranger S, Dai D, Horton B, and Gajewski TF (2017). Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31, 711–723 e714. 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.House IG, Savas P, Lai J, Chen AXY, Oliver AJ, Teo ZL, Todd KL, Henderson MA, Giuffrida L, Petley EV, et al. (2020). Macrophage-Derived CXCL9 and CXCL10 Are Required for Antitumor Immune Responses Following Immune Checkpoint Blockade. Clin Cancer Res 26, 487–504. 10.1158/1078-0432.CCR-19-1868. [DOI] [PubMed] [Google Scholar]
- 39.Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, Hoyos BE, Putintseva EV, Chaudhry A, Dikiy S, et al. (2017). Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425. 10.1038/nature22360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 10, 595–602. 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kornete M, Mason ES, Girouard J, Lafferty EI, Qureshi S, and Piccirillo CA (2015). Th1-Like ICOS+ Foxp3+ Treg Cells Preferentially Express CXCR3 and Home to beta-Islets during Pre-Diabetes in BDC2.5 NOD Mice. PLoS One 10, e0126311. 10.1371/journal.pone.0126311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nosko A, Kluger MA, Diefenhardt P, Melderis S, Wegscheid C, Tiegs G, Stahl RA, Panzer U, and Steinmetz OM (2017). T-Bet Enhances Regulatory T Cell Fitness and Directs Control of Th1 Responses in Crescentic GN. J Am Soc Nephrol 28, 185–196. 10.1681/ASN.2015070820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Warunek J, Jin RM, Blair SJ, Garis M, Marzullo B, and Wohlfert EA (2021). Tbet Expression by Regulatory T Cells Is Needed to Protect against Th1-Mediated Immunopathology during Toxoplasma Infection in Mice. Immunohorizons 5, 931–943. 10.4049/immunohorizons.2100080. [DOI] [PubMed] [Google Scholar]
- 44.Kohlmeier JE, Cookenham T, Miller SC, Roberts AD, Christensen JP, Thomsen AR, and Woodland DL (2009). CXCR3 directs antigen-specific effector CD4+ T cell migration to the lung during parainfluenza virus infection. J Immunol 183, 4378–4384. 10.4049/jimmunol.0902022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Magnuson AM, Kiner E, Ergun A, Park JS, Asinovski N, Ortiz-Lopez A, Kilcoyne A, Paoluzzi-Tomada E, Weissleder R, Mathis D, and Benoist C (2018). Identification and validation of a tumor-infiltrating Treg transcriptional signature conserved across species and tumor types. Proc Natl Acad Sci U S A 115, E10672–E10681. 10.1073/pnas.1810580115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paust HJ, Riedel JH, Krebs CF, Turner JE, Brix SR, Krohn S, Velden J, Wiech T, Kaffke A, Peters A, et al. (2016). CXCR3+ Regulatory T Cells Control TH1 Responses in Crescentic GN. J Am Soc Nephrol 27, 1933–1942. 10.1681/ASN.2015020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zagorulya M, Yim L, Morgan DM, Edwards A, Torres-Mejia E, Momin N, McCreery CV, Zamora IL, Horton BL, Fox JG, et al. (2023). Tissue-specific abundance of interferon-gamma drives regulatory T cells to restrain DC1-mediated priming of cytotoxic T cells against lung cancer. Immunity 56, 386–405 e310. 10.1016/j.immuni.2023.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li CX, Ling CC, Shao Y, Xu A, Li XC, Ng KT, Liu XB, Ma YY, Qi X, Liu H, et al. (2016). CXCL10/CXCR3 signaling mobilized-regulatory T cells promote liver tumor recurrence after transplantation. J Hepatol 65, 944–952. 10.1016/j.jhep.2016.05.032. [DOI] [PubMed] [Google Scholar]
- 49.Redjimi N, Raffin C, Raimbaud I, Pignon P, Matsuzaki J, Odunsi K, Valmori D, and Ayyoub M (2012). CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res 72, 4351–4360. 10.1158/0008-5472.CAN-120579. [DOI] [PubMed] [Google Scholar]
- 50.Kachler K, Holzinger C, Trufa DI, Sirbu H, and Finotto S (2018). The role of Foxp3 and Tbet co-expressing Treg cells in lung carcinoma. Oncoimmunology 7, e1456612. 10.1080/2162402X.2018.1456612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santegoets SJ, Duurland CL, Jordanova ES, van Ham JJ, Ehsan I, van Egmond SL, Welters MJP, and van der Burg SH (2019). Tbet-positive regulatory T cells accumulate in oropharyngeal cancers with ongoing tumor-specific type 1 T cell responses. J Immunother Cancer 7, 14. 10.1186/s40425-019-0497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vollmer T, Schlickeiser S, Amini L, Schulenberg S, Wendering DJ, Banday V, Jurisch A, Noster R, Kunkel D, Brindle NR, et al. (2021). The intratumoral CXCR3 chemokine system is predictive of chemotherapy response in human bladder cancer. Sci Transl Med 13. 10.1126/scitranslmed.abb3735. [DOI] [PubMed] [Google Scholar]
- 53.Yu L, Yang F, Zhang F, Guo D, Li L, Wang X, Liang T, Wang J, Cai Z, and Jin H (2018). CD69 enhances immunosuppressive function of regulatory T-cells and attenuates colitis by prompting IL-10 production. Cell Death Dis 9, 905. 10.1038/s41419-018-0927-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao D, Zhang C, Yi T, Lin CL, Todorov I, Kandeel F, Forman S, and Zeng D (2008). In vivo-activated CD103+CD4+ regulatory T cells ameliorate ongoing chronic graf-tversus-host disease. Blood 112, 2129–2138. 10.1182/blood-2008-02-140277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng G, Yuan X, Tsai MS, Podack ER, Yu A, and Malek TR (2012). IL-2 receptor signaling is essential for the development of Klrg1+ terminally differentiated T regulatory cells. J Immunol 189, 1780–1791. 10.4049/jimmunol.1103768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kamada T, Togashi Y, Tay C, Ha D, Sasaki A, Nakamura Y, Sato E, Fukuoka S, Tada Y, Tanaka A, et al. (2019). PD-1(+) regulatory T cells amplified by PD-1 blockade promote hyperprogression of cancer. Proc Natl Acad Sci U S A 116, 9999–10008. 10.1073/pnas.1822001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tan CL, Kuchroo JR, Sage PT, Liang D, Francisco LM, Buck J, Thaker YR, Zhang Q, McArdel SL, Juneja VR, et al. (2021). PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J Exp Med 218. 10.1084/jem.20182232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ha D, Tanaka A, Kibayashi T, Tanemura A, Sugiyama D, Wing JB, Lim EL, Teng KWW, Adeegbe D, Newell EW, et al. (2019). Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U S A 116, 609–618. 10.1073/pnas.1812186116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wei SC, Anang NAS, Sharma R, Andrews MC, Reuben A, Levine JH, Cogdill AP, Mancuso JJ, Wargo JA, Pe’er D, and Allison JP (2019). Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci U S A 116, 22699–22709. 10.1073/pnas.1821218116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winkler AE, Brotman JJ, Pittman ME, Judd NP, Lewis JS Jr., Schreiber RD, and Uppaluri R (2011). CXCR3 enhances a T-cell-dependent epidermal proliferative response and promotes skin tumorigenesis. Cancer Res 71, 5707–5716. 10.1158/0008-5472.CAN-110907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hos BJ, Camps MGM, van den Bulk J, Tondini E, van den Ende TC, Ruano D, Franken K, Janssen GMC, Ru A, Filippov DV, et al. (2019). Identification of a neoepitope dominating endogenous CD8 T cell responses to MC-38 colorectal cancer. Oncoimmunology 9, 1673125. 10.1080/2162402X.2019.1673125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He X, and Xu C (2020). PD-1: A Driver or Passenger of T Cell Exhaustion? Mol Cell 77, 930–931. 10.1016/j.molcel.2020.02.013. [DOI] [PubMed] [Google Scholar]
- 63.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, and Jacks T (2012). Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409. 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vignali DA, Collison LW, and Workman CJ (2008). How regulatory T cells work. Nat Rev Immunol 8, 523–532. 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim JM, Rasmussen JP, and Rudensky AY (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8, 191–197. 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 66.Onishi Y, Fehervari Z, Yamaguchi T, and Sakaguchi S (2008). Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A 105, 10113–10118. 10.1073/pnas.0711106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, and Bar-Sagi D (2017). Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Antitumor Immunity in Pancreatic Cancer. Cell Rep 20, 558–571. 10.1016/j.celrep.2017.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Groom JR, and Luster AD (2011). CXCR3 in T cell function. Exp Cell Res 317, 620–631. 10.1016/j.yexcr.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Metzemaekers M, Vanheule V, Janssens R, Struyf S, and Proost P (2017). Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front Immunol 8, 1970. 10.3389/fimmu.2017.01970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, and Luster AD (2012). CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37, 1091–1103. 10.1016/j.immuni.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, and Sancho D (2020). Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 20, 7–24. 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 72.Grinberg-Bleyer Y, Oh H, Desrichard A, Bhatt DM, Caron R, Chan TA, Schmid RM, Klein U, Hayden MS, and Ghosh S (2017). NF-kappaB c-Rel Is Crucial for the Regulatory T Cell Immune Checkpoint in Cancer. Cell 170, 1096–1108 e1013. 10.1016/j.cell.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 210, 1695–1710. 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, and Korman AJ (2013). Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res 1, 32–42. 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 75.Duhen T, Duhen R, Lanzavecchia A, Sallusto F, and Campbell DJ (2012). Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood 119, 4430–4440. 10.1182/blood-2011-11-392324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cretney E, Kallies A, and Nutt SL (2013). Differentiation and function of Foxp3(+) effector regulatory T cells. Trends Immunol 34, 74–80. 10.1016/j.it.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 77.Maeda S, Murakami K, Inoue A, Yonezawa T, and Matsuki N (2019). CCR4 Blockade Depletes Regulatory T Cells and Prolongs Survival in a Canine Model of Bladder Cancer. Cancer Immunol Res 7, 1175–1187. 10.1158/2326-6066.CIR-18-0751. [DOI] [PubMed] [Google Scholar]
- 78.Sarkar T, Dhar S, Chakraborty D, Pati S, Bose S, Panda AK, Basak U, Chakraborty S, Mukherjee S, Guin A, et al. (2022). FOXP3/HAT1 Axis Controls Treg Infiltration in the Tumor Microenvironment by Inducing CCR4 Expression in Breast Cancer. Front Immunol 13, 740588. 10.3389/fimmu.2022.740588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saito M, Ishii T, Urakawa I, Matsumoto A, Masaki A, Ito A, Kusumoto S, Suzuki S, Takahashi T, Morita A, et al. (2020). Robust CD8+ T-cell proliferation and diversification after mogamulizumab in patients with adult T-cell leukemia-lymphoma. Blood Adv 4, 2180-2191. 10.1182/bloodadvances.2020001641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bos PD, Plitas G, Rudra D, Lee SY, and Rudensky AY (2013). Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med 210, 2435–2466. 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim CH, Rott L, Kunkel EJ, Genovese MC, Andrew DP, Wu L, and Butcher EC (2001). Rules of chemokine receptor association with T cell polarization in vivo. J Clin Invest 108, 1331–1339. 10.1172/JCI13543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, Smyth MJ, Kuchroo VK, and Anderson AC (2015). TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest 125, 4053–4062. 10.1172/JCI81187. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 83.Fu T, Dai LJ, Wu SY, Xiao Y, Ma D, Jiang YZ, and Shao ZM (2021). Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J Hematol Oncol 14, 98. 10.1186/s13045-021-01103-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sautes-Fridman C, Petitprez F, Calderaro J, and Fridman WH (2019). Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer 19, 307–325. 10.1038/s41568-019-0144-6. [DOI] [PubMed] [Google Scholar]
- 85.Palucka AK, and Coussens LM (2016). The Basis of Oncoimmunology. Cell 164, 1233–1247. 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550. 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, et al. (2015). Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Antitumor T Cell Responses. Immunity 43, 579–590. 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van der Woude LL, Gorris MAJ, Halilovic A, Figdor CG, and de Vries IJM (2017). Migrating into the Tumor: a Roadmap for T Cells. Trends Cancer 3, 797–808. 10.1016/j.trecan.2017.09.006. [DOI] [PubMed] [Google Scholar]
- 89.Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C, Koski G, Disis ML, Czerniecki BJ, and Kodumudi K (2021). Differentiation and Regulation of TH Cells: A Balancing Act for Cancer Immunotherapy. Front Immunol 12, 669474. 10.3389/fimmu.2021.669474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, and Gajewski TF (2009). Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69, 3077–3085. 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kuhn NF, Lopez AV, Li X, Cai W, Daniyan AF, and Brentjens RJ (2020). CD103(+) cDC1 and endogenous CD8(+) T cells are necessary for improved CD40L-overexpressing CAR T cell antitumor function. Nat Commun 11, 6171. 10.1038/s41467-020-19833-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. (2016). Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938. 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336. 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cancel JC, Crozat K, Dalod M, and Mattiuz R (2019). Are Conventional Type 1 Dendritic Cells Critical for Protective Antitumor Immunity and How? Front Immunol 10, 9. 10.3389/fimmu.2019.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, Zhang R, Turka L, Marson A, and Bluestone JA (2015). The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 42, 227–238. 10.1016/j.immuni.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W, et al. (2015). Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 527, 249–253. 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dovedi SJ, Elder MJ, Yang C, Sitnikova SI, Irving L, Hansen A, Hair J, Jones DC, Hasani S, Wang B, et al. (2021). Design and Efficacy of a Monovalent Bispecific PD-1/CTLA4 Antibody That Enhances CTLA4 Blockade on PD-1(+) Activated T Cells. Cancer Discov 11, 1100–1117. 10.1158/2159-8290.CD-20-1445. [DOI] [PubMed] [Google Scholar]
- 98.Autio KA, Boni V, Humphrey RW, and Naing A (2020). Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin Cancer Res 26, 984–989. 10.1158/10780432.CCR-19-1457. Additional STAR methods references: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Haribhai D, Lin W, Relland LM, Truong N, Williams CB, and Chatila TA (2007). Regulatory T cells dynamically control the primary immune response to foreign antigen. J Immunol 178, 2961–2972. 10.4049/jimmunol.178.5.2961. [DOI] [PubMed] [Google Scholar]
- 100.Kroesen M, Nierkens S, Ansems M, Wassink M, Orentas RJ, Boon L, den Brok MH, Hoogerbrugge PM, and Adema GJ (2014). A transplantable TH-MYCN transgenic tumor model in C57Bl/6 mice for preclinical immunological studies in neuroblastoma. Int J Cancer 134, 1335–1345. 10.1002/ijc.28463. [DOI] [PubMed] [Google Scholar]
- 101.Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, Chhabra A, Silveria SL, George BM, King IC, et al. (2018). Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 359, 1037–1042. 10.1126/science.aar3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nicolai CJ, Wolf N, Chang IC, Kirn G, Marcus A, Ndubaku CO, McWhirter SM, and Raulet DH (2020). NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci Immunol 5. 10.1126/sciimmunol.aaz2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goldberg MF, Roeske EK, Ward LN, Pengo T, Dileepan T, Kotov DI, and Jenkins MK (2018). Salmonella Persist in Activated Macrophages in T Cell-Sparse Granulomas but Are Contained by Surrounding CXCR3 Ligand-Positioned Th1 Cells. Immunity 49, 1090–1102 e1097. 10.1016/j.immuni.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.