

Abstract
Medin is a principal component of localized amyloid found in the vasculature of individuals over 50 years old. Its amyloid aggregation has been linked to endothelial dysfunction and vascular inflammation, contributing to the pathogenesis of various vascular diseases. Despite its significance, the structures of medin monomer, oligomer, and fibril remain elusive, and the dynamic processes of medin aggregation are not fully understood. In this study, we comprehensively investigated the medin folding and dimerization dynamics and conformations utilizing the atomistic discrete molecular dynamics simulations. Our simulation results suggested that the folding initiation of the medin involved the formation of β-sheets around medin30–41 and medin42–50, with subsequent capping of other segments to their β-sheet edges. Medin monomer typically consisted of three or four β-strands, along with a dynamic N-terminal helix. Two isolated medin peptides readily aggregated into β-sheet-rich dimer, displaying a strong aggregation propensity. Dimerization of medin not only enhanced the β-sheet conformations but also led to the formation of β-barrel oligomers. The aggregation tendencies of medin1–18 and medin19–29 were relatively weak. However, the segments of medin30–41 and medin42–50 played a crucial role as they primarily formed a β-sheet core and facilitated medin1–18 and medin19–29 to form intra- and inter-peptide β-sheets. The findings highlight the critical role of the medin30–41 and medin42–50 regions in stabilizing the monomer structure and driving medin amyloid aggregation. These regions could potentially serve as promising targets for designing anti-amyloid inhibitors against amyloid aggregation of medin. Additionally, our study provides a full picture of the monomer conformations and dimerization dynamics for medin, which will help better understand the pathology of medin aggregation.
Graphical Abstract
Introduction
Medin is a 50-amino-acid protein derived from the milk fat globule-EGF factor 8 protein1. It serves as a principal component of aortic medial amyloid (AMA)2, which is the most common type of localized amyloid found within the medial layer of the aorta3, 4. AMA frequently occurs in individuals aged 50 years and older and is associated with age-related vascular stiffness5–7. Although the exact role of medin in AMA pathogenesis is not fully understood, mounting evidence suggests the aggregation of medin may contribute to the degeneration of the arterial wall, leading to arterial stiffening and cerebrovascular dysfunction3, 6, 8. Degenhardt et al. have shown that mice with a genetic deficiency of medin exhibit a lack of vascular aggregates and prevent the age-related decline in cerebrovascular function, suggesting that the aggregation of medin may cause cerebrovascular dysfunction4. Cerebral arteriole medin may serve as a novel risk factor or biomarker for Alzheimer’s disease (AD) and vascular dementia9, 10. A recent study demonstrated that medin co-localizes with amyloid-β (Aβ) deposits and promotes vascular β-amyloidosis both in vitro and in vivo1. Therefore, uncovering the molecular mechanisms underlying medin aggregation is crucial for understanding the pathogenesis of age-related cerebrovascular dysfunction and vascular dementia, which also benefits for the development of novel therapeutic strategies for preventing or treating these conditions.
The aggregation kinetics of medin1, 11, similar to other amyloid peptides such as Aβ12, 13 and human amylin14, 15, exhibits a typical sigmoid curve with three phases: a lag phase of nucleation of monomers into oligomers and proto-fibrils, a growth phase of rapid elongation of fibrils, and an equilibrium plateau of mature fibrils. Prior studies using circular dichroism (CD) spectroscopy and nuclear magnetic resonance (NMR) have suggested that the monomeric form of medin primarily adopts random coil and β-sheet structures16–18, although some partial helix structures have also been observed18. The NMR measurements combined with ab initio protein modeling have revealed that the soluble monomeric medin consists of a stable core composed of three β-strands, with shorter and more labile strands located at the C-termini17. The CD measurements showed that the medin aggregates were saturated with β-sheet structures up to 65%19. The aggregation assays of various truncated medin peptides have shown that the amyloid-prone region is located in the last 18–19 C-terminal amino acids20. Specifically, electron microscopy and Congo red staining measurements indicated that medin1–12, medin14–22, medin16–24, and medin1–25 segments did not aggregate into amyloid fibrils, whereas medin32–41, medin42–49, and medin31–50 readily formed amyloid fibrils20. The formation of fibrils by the medin42–49 peptide was observed by Gazit et al. through electron microscopy21, and Middleton et al. subsequently used solid-state NMR spectroscopy and X-ray fiber diffraction to establish that the fibrils consist of parallel, in-register β-sheets that assemble in a face-to-back manner22. Another X-ray crystallography study has demonstrated that both medin19–25 and medin31–37 adopt β-sheet conformations within a cyclic peptide derived from residues 19–37 of medin23. However, the structures of full-length medin monomer, oligomer, and fibril are still unknown, and the dynamics as well as conformations of medin during amyloid aggregation have yet to be established.
To investigate the aggregation dynamics of medin, we propose a comprehensive study of the monomer structures and dimerization dynamics of the full-length medin peptide using atomistic discrete molecular dynamics24, 25 (DMD) simulations. DMD is a rapid and predictive molecular dynamics algorithm that has been extensively employed in the study of amyloid aggregation, encompassing both pathological26–28 and functional29, 30 amyloid peptides. The folding dynamics simulations showed that the folding initiation of the medin monomer involved the formation of β-sheets around medin30–41 and medin42–50, with subsequent capping of other segments to their β-sheet edges. The conformation of the medin monomer typically consisted of three or four β-strands with a dynamic N-terminal helix, in agreement with prior NMR measurements17. Dimerization simulations demonstrated that two isolated medin peptides readily aggregated into β-sheet-rich oligomers, displaying a strong aggregation propensity characteristic of classic amyloid-prone peptides3, 20. Dimerization of medin not only enhanced the β-sheet conformations but also led to the formation of β-barrel oligomers. This observation of β-barrel pore aggregates was consistent with prior experimental measurements11. The analysis of residue-pairwise contact frequency indicated that the aggregation tendencies of medin1–18 and medin19–29 were relatively weak, which was in line with previous aggregation studies of various truncated medin segments20. However, in the medin dimer, the segments of medin30–41 and medin42–50 played a crucial role as they primarily formed a β-sheet core. This core structure further facilitated medin1–18 and medin19–29 to form intra- and inter-peptide β-sheets. In conclusion, our findings highlight the critical role of the medin30–41 and medin42–50 regions in stabilizing the monomer structure and driving medin amyloid aggregation. These regions could potentially serve as promising targets for designing anti-amyloid inhibitors to counteract the pathogenesis of amyloid-related vascular disorders3, 4. Additionally, our simulation results have provided valuable insights into the conformation and self-assembly dynamics of full-length medin peptide, which will help us to better understand the pathological aggregation of medin.
Materials and methods
Molecular systems.
The sequence of human medin used in our simulations is 1RLDKQGNFNA 11WVAGSYGNDQ 21WLQVDLGSSK 31EVTGIITQGA 41RNFGSVQFVA50, which has been widely employed in previous studies1, 4. Since the solution structure of the medin monomer remains elusive, we initiated the simulations with a fully extended conformation. To explore the structural characteristics of the medin monomer, we performed 30 independent simulations, each starting from a different initial state with varied velocities. Each monomeric medin simulation had a duration of up to 600 ns. From these simulations, we identified the top 10 most populated monomeric structures, which accounted for approximately 45.7% of the conformations, and utilized them as initial structures for the dimerization simulations. To ensure thorough conformational sampling, we carried out 60 independent DMD simulations for the dimeric medin, employing diverse initial states in terms of coordinates, orientations, and velocities. For each simulation, two peptides were randomly selected from the top 10 most populated medin monomer structures and placed within an 8.0 nm cubic box, ensuring an inter-molecular distance of more than 1.5 nm. The simulation time for each independent dimerization simulation was also up to 600 ns.
DMD simulations.
Our simulations were conducted using the implicit-solvent united-atom discrete molecular dynamics (DMD) method in the canonical NVT ensemble at a temperature of 300 K. The Medusa force field, which has been extensively validated for its accuracy in predicting protein stability changes and protein-ligand binding affinities31–33, was employed. The force field considers bonded interactions (covalent bonds, bond angles, and dihedrals) as well as non-bonded interactions (van der Waals and electrostatic terms). The DMD algorithm, a unique type of MD algorithm, replaces continuous potential functions with discrete step functions34. The Medusa force field’s van der Waals parameters are based on the CHARMM force field35, and the implicit solvation model used to represent water is the EEF1 model developed by Lazaridis and Karplus36. Hydrogen bond interactions are explicitly modeled using a reaction-like approach33, and screened electrostatic interactions are represented using the Debye-Hückel approximation with a Debye length of 10 Å. Temperature control is achieved using the Anderson thermostat37. The units of mass, time, length, and energy in our simulations are 1 Da, ~50 fs, 1 Å, and 1 kcal/mol, respectively. Academic researchers can access the DMD software through Molecules In Action, LLC (www.moleculesinaction.com).
Our previous studies have demonstrated the predictive power of the DMD simulation with the Medusa force field and the EEF1 implicit solvation model. This includes successful folding simulations that capture native states with secondary and tertiary structures25, 33, as well as computationally derived conformational ensembles that align with single-molecule FRET measurements in the dynamics of multi-domain proteins38, 39. Furthermore, our recent amyloid aggregation simulations, encompassing calcitonin peptides (hCT, sCT, phCT, and TL-hCT)26 and amylin peptides (hIAPP, hIAPP(S20G), and rIAPP)27, successfully replicated experimentally observed variations in amyloid tendencies, underscoring the predictive capabilities of DMD simulations with the Medusa force field protocol for studying amyloid peptide aggregation. The accuracy of DMD with the Medusa force field in studying the aggregation of functional suckerin amyloid peptides29 and pathological amylin amyloid peptides27 was extensively benchmarked through comparisons with standard MD simulations using force fields such as GROMOS9640, OPLS-AA41, AMBER99SB-ILDN42, and CHARMM36m43. The DMD algorithm, known for its enhanced sampling efficiency, has been widely utilized by our group12, 44–47 and other researchers48–50 to investigate protein folding and amyloid aggregation phenomena. Therefore, the DMD with the Medusa force field was chosen to investigate the conformational dynamics of the medin monomer and dimer.
Analysis Methods.
The secondary structure of the protein was determined using the dictionary of secondary structure of proteins (DSSP) method51. Residue-pairwise contacts were defined as pairwise interactions between non-sequential sidechain or main chain atoms if their distance was within 0.65 nm. Hydrogen bonds were identified when the distance between the backbone N and O atoms was ⩽3.5 Å and the N-H•••O angle was ⩾120°52. Cluster analysis was conducted using the Daura algorithm53 with a cutoff of 0.50 nm for backbone atom deviations. If the β-strand segments of an oligomer were capable of creating a closed cycle, with each β-strand linked to two adjacent β-strands through a minimum of two hydrogen bonds, the oligomer was classified as a β-barrel oligomer12, 27, 46. To visualize the conformational landscape, a two-dimensional (2D) free energy surface, also known as the potential mean force (PMF) surface, was constructed. This surface was generated by calculating the negative logarithm of the probability, −RT ln P(x, y), where P(x, y) represents the probability of a conformation having specific values for parameters x and y.
Results and discussion
Equilibrium assessment for the folding simulation of medin monomer.
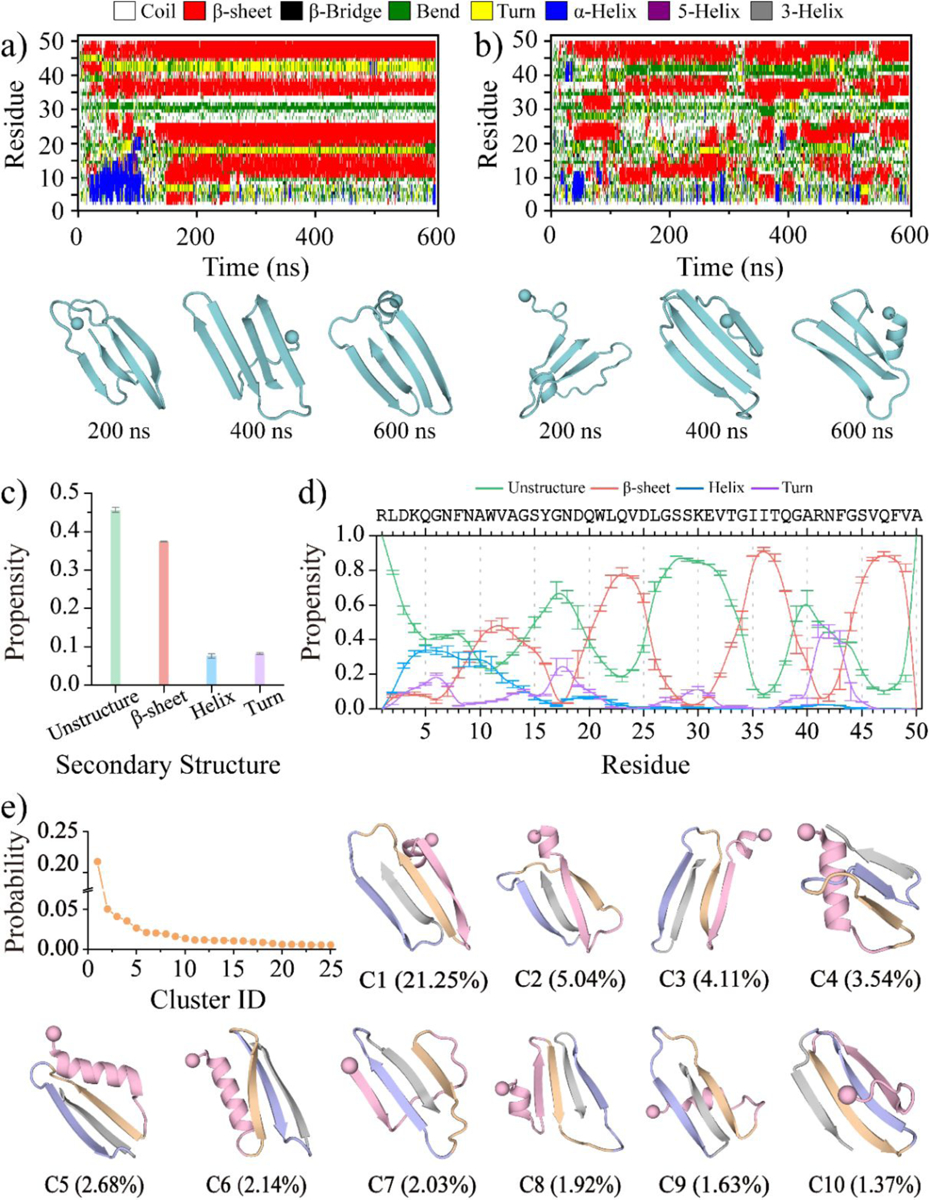
The conformational dynamics of medin monomers were explored through 30 independent 600 ns DMD simulations. The equilibrium of the conformational states was assessed based on structural parameters such as radius of gyration (Rg), number of backbone hydrogen bonds and heavy atom contacts, and secondary structure content over time (Figure. S1). The absence of significant conformational changes in the last 300 ns of the simulations indicated that they reached a reasonable equilibrium. Consequently, only the data from the final 300 ns of each independent simulation trajectory were utilized for the subsequent conformational analysis to minimize potential biases stemming from the initial structures.
The presence of C-terminal β-sheets triggered medin to fold into β-sheet-rich conformations with three or four β-strands and a dynamic N-terminal helix.
The time evolution of the secondary structure for each residue of medin, starting from a fully extended state, revealed the initial adoption of a β-sheet conformation around the C-terminus (Figure 1a&b). This conformational arrangement facilitated the folding of other regions, resulting in a β-sheet-rich structure comprising three or four strands formed by residues 8–50. The β-sheets formed by the C-terminal residues of the medin monomer were observed to exhibit higher stability compared to other regions, which was consistent with previous study on the structure of the medin monomer17. Additionally, transient helical conformations (including α-helix, 5-helix, and 3-helix) involving the N-terminal residues were dynamically observed during the folding process, in agreement with prior experimental measurements18.
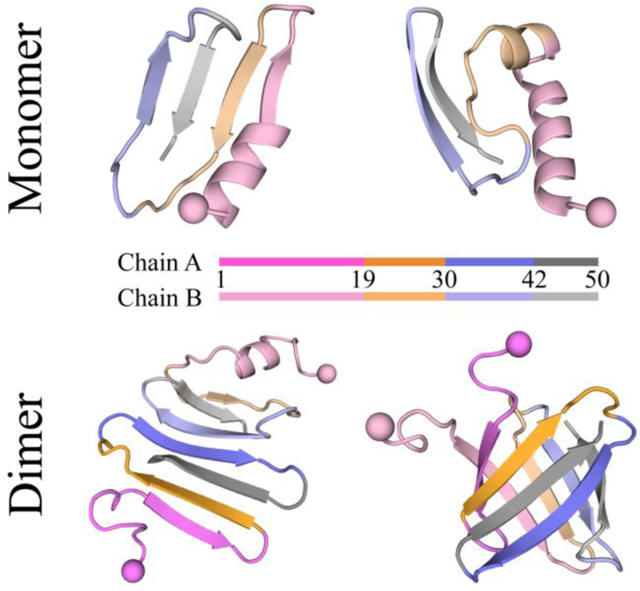
Figure 1. Conformational dynamics analysis of the medin monomer.
Time evolution of the secondary structure for each residue of the medin monomer a&b). Snapshots along the simulation trajectory are shown every 200 ns. Two trajectories are randomly selected from thirty independent DMD simulations to illustrate the folding dynamics. Average secondary structure content of unstructured (coil and bend), β-sheet, helix, and turn conformations in the medin monomer during the last 300 ns of DMD simulations c). Propensity of each residue in the medin monomer to adopt unstructured (coil and bend), β-sheet, and helix conformations d). Probability and corresponding structure of the top ten most populated clusters for the medin monomer e). All three types of helical structures, including α-helix, 5-helix, and 3-helix, are summarized as helix. According to the β-sheet propensity, the entire peptide is divided into four regions: medin1–18, medin19–29, medin30–41, and medin42–50, which are color-coded as pink, orange, purple, and gray, respectively.
The ensemble average secondary structure analysis of the medin monomer, based on the last 300 ns of simulation data from 30 independent DMD trajectories, revealed that the predominant conformations were unstructured and β-sheet, with probabilities of approximately 45.7% and 38.4%, respectively (Figure 1c). Prior experimental measurements also suggested that medin monomer primarily assumed random coil and β-sheet structures16–18. The average content of helix and turn structures was relatively low, at approximately 7.6% and 8.2%, respectively (Figure 1c). Consistent with prior studies, medin monomer was also experimentally observed to exhibit partial helical conformations18. The conformation of the medin monomer was further examined using a free energy landscape analysis based on the total number of backbone hydrogen bonds and the average β-sheet ratio (Figure S2). The conformational free energy landscape displayed a broad energy basin, encompassing a range of ~10–30 backbone hydrogen bonds and a β-sheet ratio of ~15%−55%. Within this energy basin, conformations with a high β-sheet ratio exhibited lower free energy compared to conformations with a low β-sheet content (such as helical structures), suggesting a higher stability and preference for β-sheet structures.
To further explore the structural characteristics of each region within the medin monomer, we conducted an analysis of the average secondary structure propensity for each residue (Figure 1d). The distribution of β-sheet propensity revealed the presence of four distinct β-strand regions: residues 1–18 (denote as medin1–18), residues 19–29 (denote as medin19–29), residues 31–41 (denote as medin31–41), and residues 42–50 (denote as medin31–41). Notably, the β-sheet propensity was relatively weaker for the segment of medin1–18 compared to the latter three regions. Specifically, the average β-sheet propensity for medin1–18 was approximately 22.2%, while for fragments of medin19–29, medin30–41, and medin42–50, the β-sheet propensities were approximately 42.7%, 43.6%, and 54.7%, respectively. Additionally, residues 1–16 exhibited a higher propensity for dynamic helical structures, with an average propensity of approximately 20.0%.
Furthermore, we performed a conformational cluster analysis using the Daura algorithm53, employing a cutoff of 0.50 nm for backbone atom deviations. Among the medin monomer conformations, we identified the top ten most populated clusters, which accounted for 45.7% of the total conformations (Figure 1e). Examination of the central structures of these clusters revealed that medin1–18 capable of adopting both helical and β-sheet structures, while the other regions of medin19–29, medin30–41, and medin42–50 predominantly adopted β-sheet conformations. The conformational PMF analysis for mdin1–18 further showed that this region could fold into both partial helix and β-sheet structures (Figure S3).
The segments of medin30–41 and medin42–50 consistently exhibited β-sheet conformations within the medin monomer, whereas the conformations of medin1–18 and medin19–29 were more varied.
The conformational energy landscape was analyzed by examining the relationship between the β-sheet content of the entire peptide (residues 1–50) and the average β-sheet ratio for each segment, including medin1–18, medin19–29, medin30–41, and medin42–50 (Figure 2). The analysis was performed using the last 300 ns of simulation data from 30 independent trajectories. Both low (less than 10%) and high (greater than 30%) β-sheet content conformations with low free energy were observed for medin1–18 and medin19–29 (Figure 2a&b). The low β-sheet content conformations in these regions predominantly occurred within the range of 10%~30% β-sheet ratio for the medin monomer, while the high β-sheet content conformations were primarily observed in structures where the whole medin peptide had a β-sheet content greater than 40%. In contrast, segment of medin30–41 and medin42–50 predominantly adopted conformations with a β-sheet ratio greater than 40%, regardless of whether the β-sheet content of the entire peptide was low or high (Figure 2c&d). Overall, the β-sheet content of the entire peptide was primarily influenced by the conformations of medin1–18 and medin19–29, as the regions of medin30–41 and medin42–50 consistently adopted β-sheet structures.
Figure 2. The conformational free energy landscape analysis for the medin monomer.
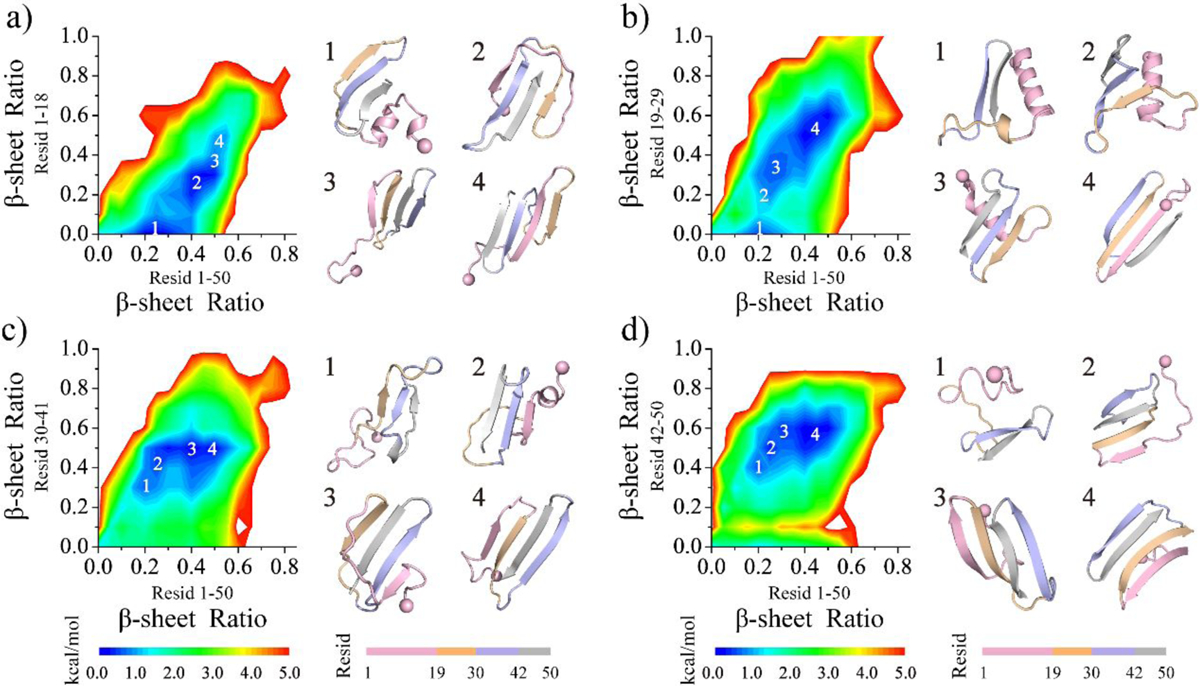
The potential mean force of the saturated medin monomer is plotted as a function of the β-sheet ratio for the entire peptide (residues 1–50) as well as for the segment of medin1–18 a), medin19–29 b), medin30–41 c), and medin42–50 d). The conformational analysis is performed using the last 300 ns of simulation data from 30 independent DMD trajectories. Segments of medin1–18, medin19–29, medin30–41, and medin42–50 are color-coded as pink, orange, purple, and gray, respectively.
Residues 30–50 formed a stable β-hairpin core, facilitating the folding of other regions into β-sheet structures by capping to the β-hairpin edges through hydrophobic interactions.
The conformation of the medin monomer was further investigated through residue-pairwise contact frequency analysis using structures obtained from saturation states (Figure 3). The N-terminal residues 1–19 of medin monomer exhibited a strong inclination towards a helical pattern along the diagonal, particularly in the region encompassing residues 1–13 (snapshots 1 in Figure 3), which was consistent with the findings from the secondary structure analysis (Figure 1d). Three β-hairpin motifs were observed, formed by residues 10–16 vs. 19–25, 22–29 vs. 32–39, and 32–38 vs. 44–50, with contact patterns perpendicular to the diagonal. These motifs involved interactions among hydrophobic residues (snapshots 2, 3, 6 in Figure 3). Notably, the β-hairpin motif formed by residues 30–50, containing the most hydrophobic amino acids within the segments of residues 30–41 and 42–50, exhibited the highest average frequency (>60%) compared to other structured contact patterns, indicating its exceptional stability. Residues 8–15 or 19–25 were found to cap the stable β-hairpin edges, facilitating the formation of β-sheet structures (snapshots 4 and 5 in Figure 3). Strong hydrophobic interactions between residues 30–41 and 42–50 played a crucial role in stabilizing the β-hairpin conformation, acting as a critical core that assisted other regions in folding into β-sheet structures by interacting with the β-hairpin edges.
Figure 3. Residue-pairwise contact frequency analysis of the medin monomer.
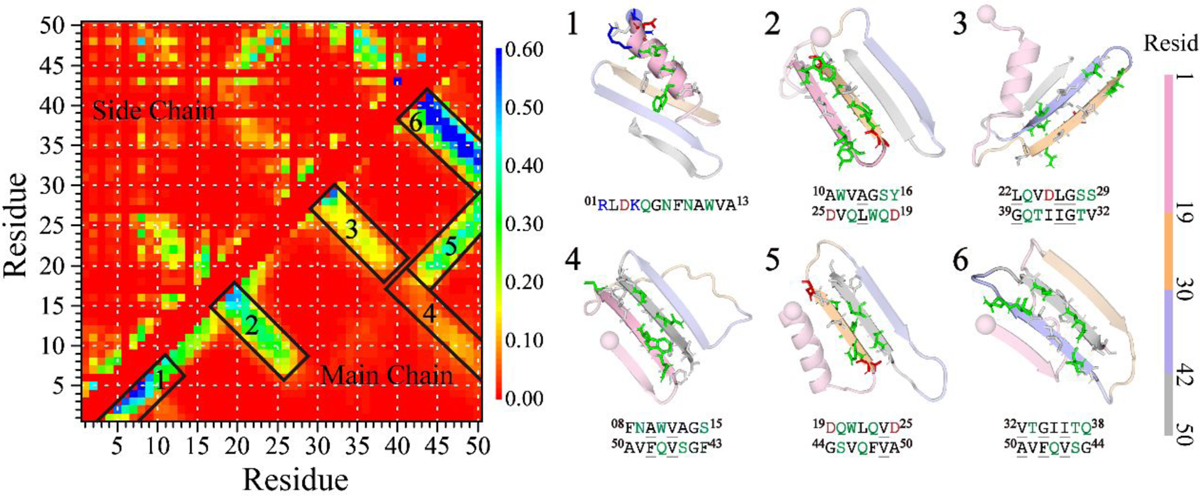
The left panel shows the residue-pairwise contact frequency between main-chain heavy atoms (upper diagonal) and side-chain heavy atoms (lower diagonal) within the medin monomer. Representative structured motifs with high contact frequency patterns, mainly corresponding to helices or β-sheets, are labeled as 1–6 and displayed on the right. The contact frequency map is generated using the last 300 ns trajectories of 30 independent DMD simulations after reaching a steady state.
Dimerization dynamics displayed a strong propensity of medin to undergo aggregation as a classic amyloid-prone peptide.
To investigate the aggregation dynamics and conformational behavior of medin, we conducted 60 independent 600 ns DMD simulations for two medin peptides. As the experimentally characterized structure of medin remains elusive, the central structures of the top 10 most populated monomeric conformations, which accounted for approximately 45.7% of the total conformations, were chosen as representative medin monomer structures. Two structures were randomly selected from these top 10 conformations and placed in an 8.0 nm cubic simulation box, ensuring a minimum inter-peptide distance of 1.5 nm. The time evolution of inter-peptide hydrogen bonds and contacts, along with the average secondary structure content, as well as snapshots from two randomly selected trajectories, revealed the rapid aggregation of the two isolated medin peptides into a β-sheet-rich dimer (Figures 4a&b). The stability of the dimer was facilitated by the formation of inter-peptide hydrogen bonds and contacts. Furthermore, the dimerization dynamics were examined in all 60 independent simulations, analyzing the number of inter-peptide contacts and hydrogen bonds over time (Figure S4). The results showed that inter-peptide contacts and hydrogen bonds were rapidly formed within the first 100 ns in all trajectories, indicating the strong propensity of medin to undergo aggregation as a classic amyloid-prone peptide20. These findings emphasized the crucial role of intermolecular interactions, specifically the formation of inter-peptide contacts and hydrogen bonds, in driving the aggregation process of medin.
Figure 4. Dimerization dynamics and β-barrel formation analysis of medin.
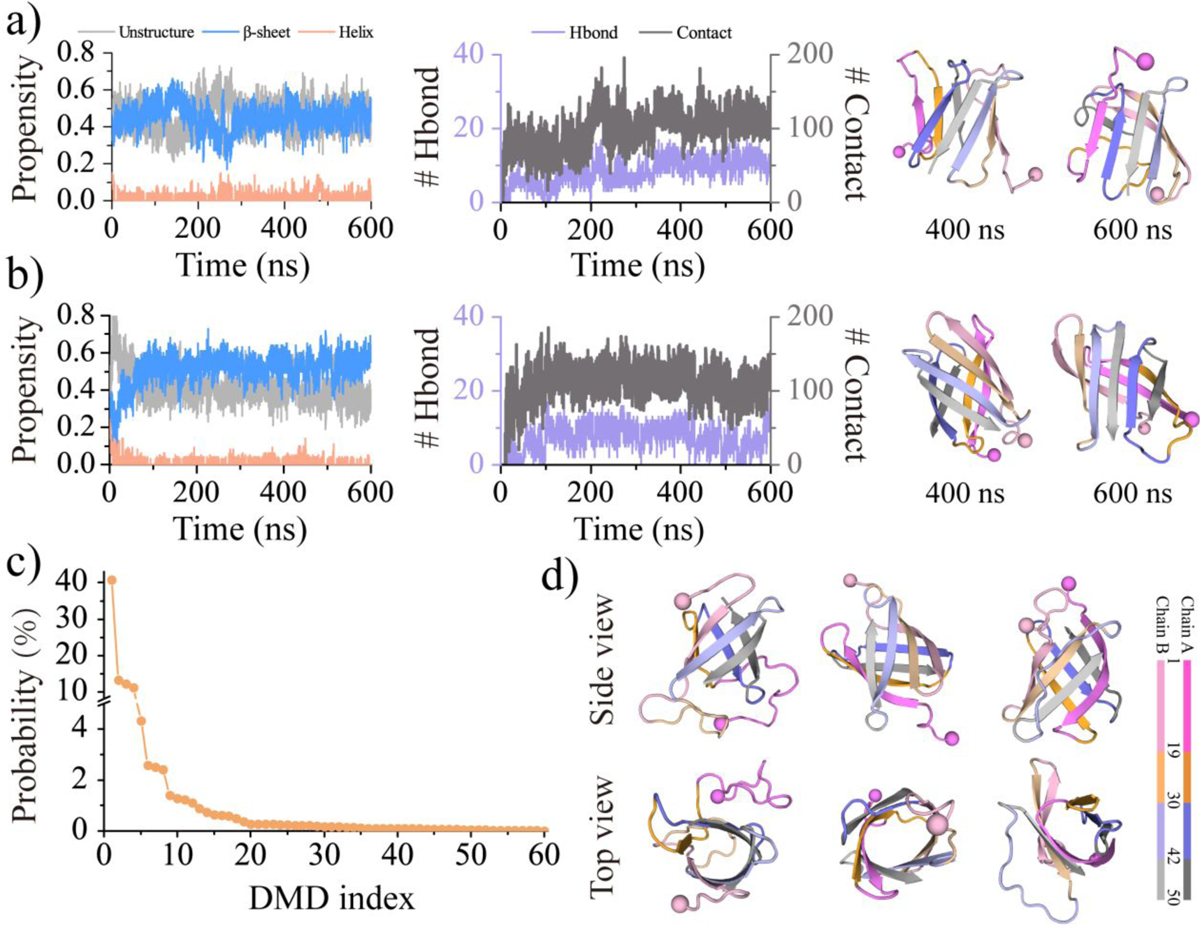
The dimerization dynamics of medin are monitored by the time evolution of the secondary structure content (left panel), the number of inter-molecular contacts and hydrogen bonds (middle panel), along with representative snapshots at the simulation time of 400 and 600 ns (right panel) a&b). Two trajectories are randomly selected from sixty independent simulations to illustrate the dimerization dynamics of medin. The frequency of β-barrel oligomers observed in each simulation trajectory is sorted in descending order based on the probability of β-barrel formation c). Three β-barrel conformations randomly selected from the top 3 most populated β-barrel trajectories are provided in side and top views d).
Dimerization resulted in medin forming β-barrel conformations.
The presence of β-barrel oligomers was initially observed during the aggregation of a fragment from αB-crystalline54, suggesting their potential cytotoxicity in amyloidosis. Both experimental and computational studies provided support for the formation of β-barrel intermediates in toxic fragments45, 46, 54–56 and full-length12, 26, 27, 57–59 for amyloid peptides, such as hIAPP27, 57 and Aβ12,58, 59. For instance, wild-type Aβ showed a higher propensity for β-barrel formation compared to the AD-protective A2T substitution but lower than the AD-causative mutations D7N and E22G12. Moreover, the introduction of the S20G substitution in hIAPP increased its amyloidogenicity and cytotoxicity, resulting in a significant increase in β-barrel formation compared to the wild-type peptide27, 57. These findings shed light on the potential role of β-barrel intermediates in the pathogenesis of amyloid-related disorders. Furthermore, prior experimental measurements along with computational modeling demonstrated that medin oligomers can induce ionic membrane permeability by forming β-barrel (i.e., β-sheet pore) conformations11. Interestingly, the formation of β-barrel oligomers was also observed during dimerization (Figures 4b&S5c). Although the probability of medin forming β-barrel conformations was heterogeneous among different trajectories, the direct observation of β-barrel oligomers indicated their presence as one of the medin aggregation intermediates (Figures 4c&d).
Dimerization promoted the formation of β-sheet conformations in medin by reducing the presence of N-terminal helical structures.
The time evolution of radius of gyration, the number of inter-peptide contacts and hydrogen bonds, as well as the secondary structure contents, indicated that the dimerization simulations reached their corresponding steady states during the last 200 ns (Figure S5). Therefore, the simulation data from the last 200 ns of each independent DMD trajectory were used for the analysis of conformational dynamics. Compared to the medin monomers, the average β-sheet propensity in the medin dimer increased from ~38.4% to 44.3% (Figures 5a). The probability distribution function of the average β-sheet content of medin peptide in monomer and dimer demonstrated that dimerization enhanced the β-sheet conformations (Figure S6a). Oligomerization-enhanced β-sheet conformations of medin were also consistent with prior experimental measurements16–19. Conversely, the average helix content in the medin dimer was significantly reduced to only 1.6%, in contrast to the 7.6% observed in the medin monomer. The secondary structure propensities of each residue within the medin dimer were compared to those of the medin monomer to investigate the effects of dimerization on the conformation of medin (Figures 5b&S7). The analysis revealed a significant suppression of the helical propensity of medin1–18 in the dimer, accompanied by an enhancement of its β-sheet propensity. The average helix and β-sheet ratio probability distribution of each medin1–19 segment in the monomer and dimer further confirmed that dimerization promoted the β-sheet conformation and suppressed the helix conformation for the medin1–18 segment (Figures 5c&S6a). Additionally, the average β-sheet propensities for medin19–29 were also enhanced (Figures 5b&d). Since the residues in medin30–41 and medin42–50 were already predominantly in β-sheet conformations in the monomer, dimerization did not induce significant structural changes in these regions (Figures 5b&e–f). These results suggested that inter-peptide interactions, specifically the formation of inter-peptide contacts and hydrogen bonds, enhance the β-sheet structures in the regions of medin1–18 and medin19–29.
Figure 5. Comparing the secondary structure properties between medin monomer and dimer.
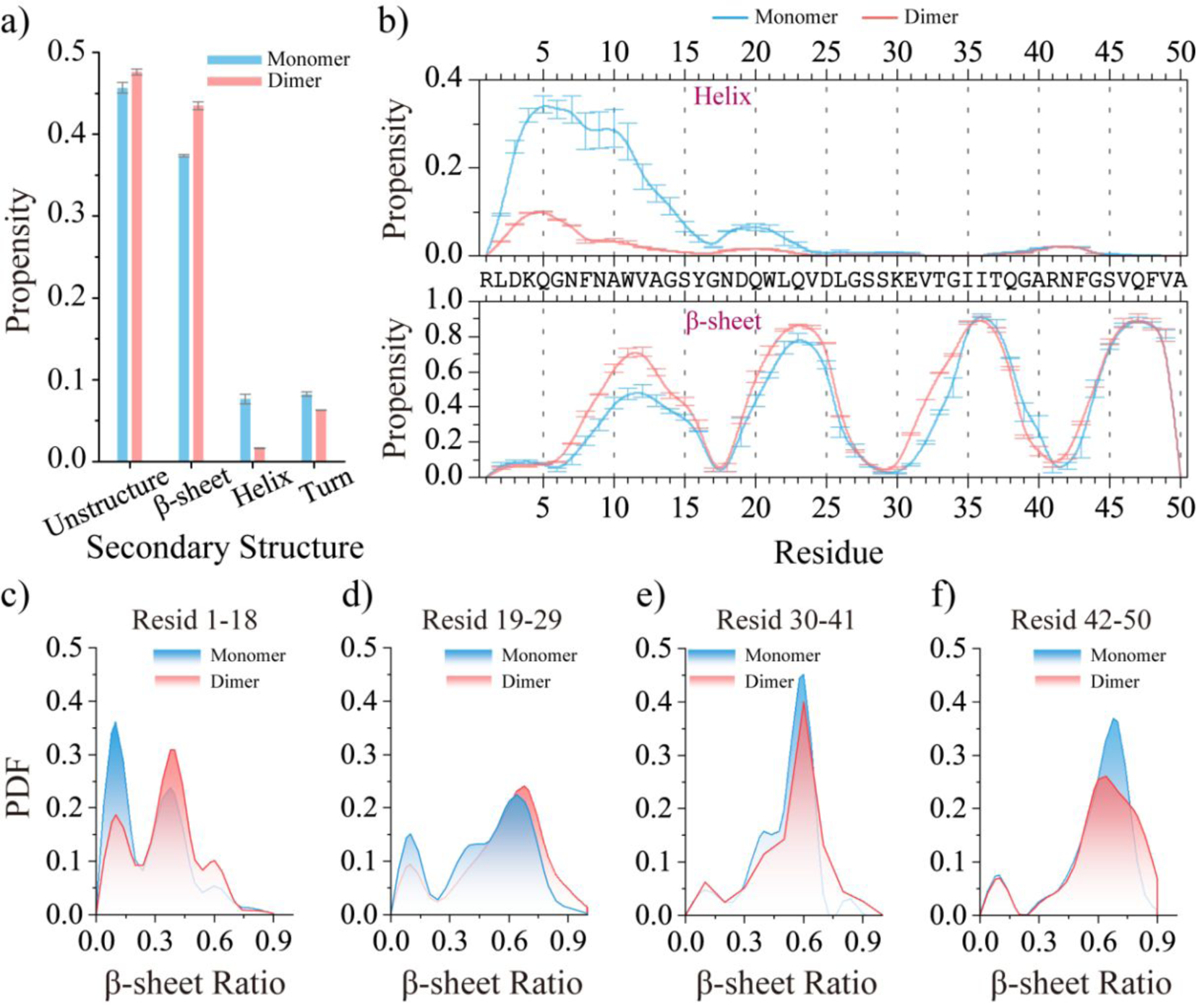
The average secondary structure propensity of monomeric and dimeric medin a). The average propensity of each residue to adopt helix (top) and β-sheet (bottom) conformations in the medin monomer and dimer b). The probability distribution of the β-sheet ratio for the segment of medin1–18 c), medin19–29 d), medin30–41e), and medin42–50 f) in medin monomer and dimer. To minimize potential bias from initial states, only the final 300 ns and 200 ns of the medin monomer and dimer simulations, respectively, were used for the ensemble average secondary structure analysis.
Segments of medin30–41 and medin42–50 played critical roles in the aggregation of medin.
To uncover the key interactions driving medin aggregation, we analyzed both intra- and inter-peptide residue-wise contact frequency maps within the saturated conformations of the medin dimer (Figure 6a). Similar to the medin monomer (Figure 3), three β-hairpin contact patterns, perpendicular to the diagonal, involving regions of residues 7–28, 19–39, and 30–50 were also observed in the medin dimer (snapshots 1, 2, and 5 in Figure 6a). Dimerization enhanced the β-hairpin propensity of residues 7–28, suppressing the helical formations of medin1–19, particularly for residues 10–19. Additionally, the intra-peptide β-sheet conformations formed by capping residues 18–26 and 7–14 to the segment of medin42–50 were also observed in the medin dimer (snapshots 3 and 4 in Figure 6a), similar to those present in the medin monomer.
Figure 6. Contact frequency analysis of the medin dimer.
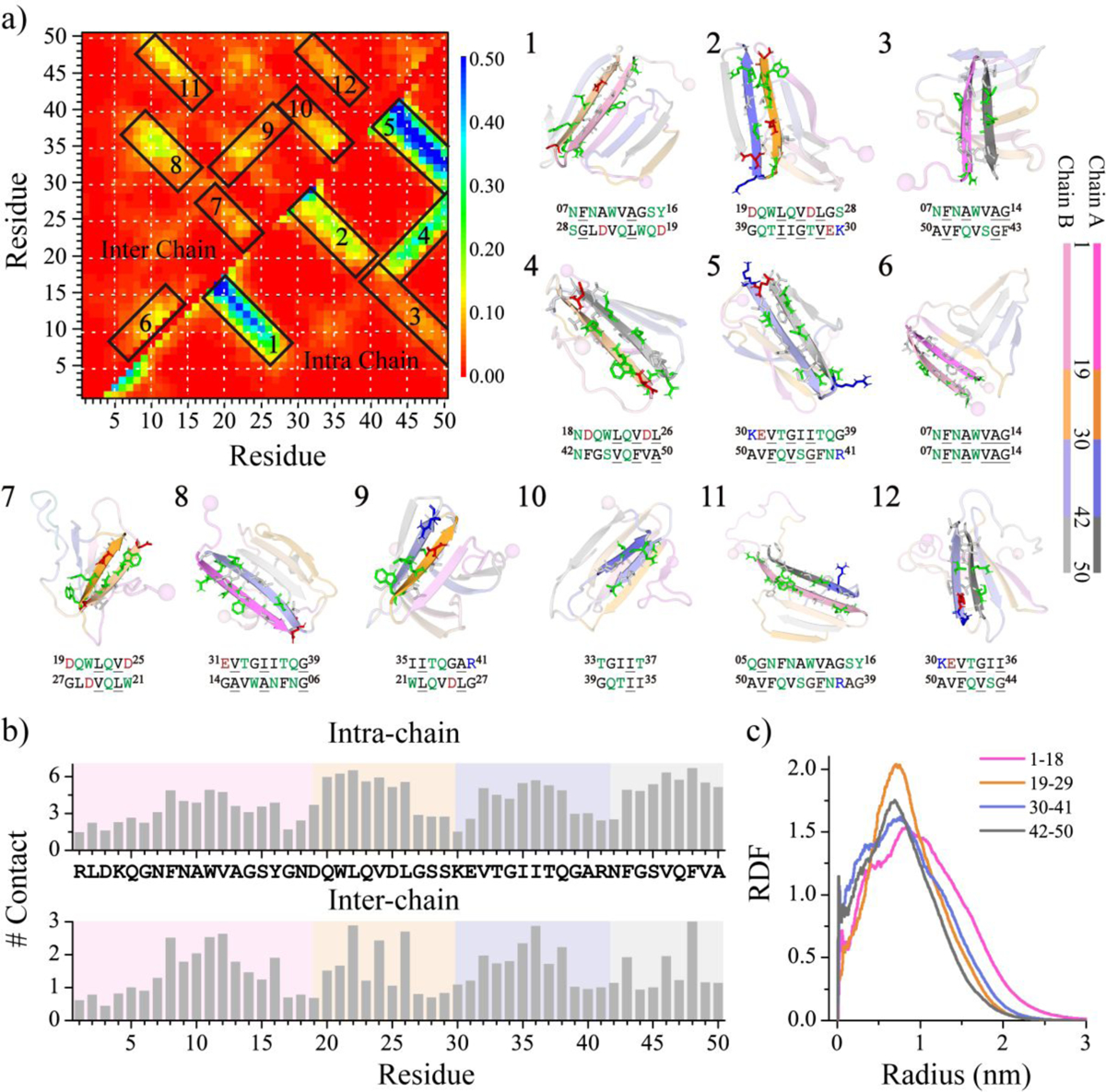
Residue-pairwise contact frequencies between peptides are shown in the upper diagonal, while contact frequencies within each peptide are shown in the lower diagonal a). Representative structural motifs with high-contact-frequency patterns predominantly associated with helices or β-sheets, labeled as 1–12 in the contact frequency map, are also presented. The average number of intra-peptide and inter-peptide contacts per residue in the medin dimer b). The radius distribution function of Cα atoms from each medin region corresponds to the geometric center of the dimer c). All the above analyses were performed using the last 300 ns of trajectories from 50 independent DMD simulations, reaching a saturation state.
The inter-peptide contact frequency analysis revealed that interactions among residues 1–29 were significantly weak. Only two faint β-sheet contact patterns were observed in this region: one was a parallel β-sheet between residues 7–14 and residues 7–14 (snapshot 6 in Figure 6a), and the other was an anti-parallel β-sheet between residues 19–25 and residues 21–27 (snapshot 7 in Figure 6a). These results suggested that the self-assembly propensity around this region was relatively weak, which was consistent with prior experimental measurements20. Two obvious anti-parallel β-sheet contact patterns were identified between the capping residues of medin1–19 and medin30–41, as well as medin42–50. Specifically, residues 6–14 formed contacts with residues 31–39, and residues 5–16 interacted with residues 39–50 (snapshots 8 and 11 in Figure 6a). Moreover, intermolecular binding between medin19–30 and medin31–41, forming a parallel β-sheet pattern, was also observed with the β-strands around residues 21–27 and residues 35–41 (snapshot 9 in Figure 6a). Prior experiments demonstrated that medin16–24 and medin1–25 didn’t aggregate20, but all the segments of residue 19–37 displayed a strong tendency to form aggregating β-sheet structures23, indicating that medin31–41 may assist residues 19–30 in assembling β-sheet conformations. Additionally, with the strongest β-sheet propensity observed in medin30–41 and medin42–50, inter-peptide β-sheets formed by these two regions were also identified (snapshots 10 and 12 in Figure 6a). The average number of intra- and inter-peptide contacts was also analyzed (Figure 6b). The hydrophobic residues had more intra- and inter-peptide contacts compared to other residues (e.g., polar and charged residues), indicating the interactions among hydrophobic residues played a crucial role in medin aggregation. In addition, residues that demonstrated a strong propensity for forming intra-peptide contacts also exhibited a pronounced tendency to establish inter-peptide contacts. This phenomenon drove the aggregation of medin into β-sheet-rich structures, including β-barrel intermediates, in order to maximize residue-pairwise interactions for these residues.
The radius distribution function of the Cα atoms from each medin segment, corresponding to their geometry center, was analyzed to characterize the conformational features of the medin dimer (Figure 6c). Residues from medin30–41 and medin42–50 were found to preferentially bury inside the dimer, while medin1–19 usually distributed on the outer layer. Additionally, the solvent-accessible surface area (SASA) per residue of the medin monomer and dimer was also studied (Figure S8). The comparison between the medin monomer and dimer showed an increase in SASA around residues 2–9 and a noticeable decrease in SASA around residues 10–15, 20–28, and 30–38 (Figure S8b). However, due to the strong occupation of the surfaces of residues from medin42–50 in assisting the other regions in forming β-sheet conformations, dimerization only resulted in a weak SASA decrease around medin42–50. Taking into account the analyses of secondary structure propensity and residue pairwise contact frequency, it can be deduced that medin30–41 and medin42–50 mainly assumed a β-sheet core buried inside the dimer, while interactions among hydrophobic residues drove the segments of medin1–19 and medin20–29 to cap the elongation edges of the β-sheet core, forming β-sheet conformations.
The dimerization free energy landscape analysis suggested that dimer formation was more favorable than having two isolated monomers, as dimers typically exhibited lower potential energy.
To better understand the energetics of medin aggregation, we computed the potential of mean force as a function of the total number of inter-peptide contacts and potential energy. We utilized all the 600 ns simulation data from 60 independent trajectories for the conformational free energy landscape analysis to capture all the ensemble characteristics during the entire dimerization process (Figure 7a). The dimerization free energy landscape exhibited two distinct free energy basins: one narrow basin corresponding to the isolated monomers and one broad basin corresponding to the dimeric state (Figures 7a&c). For the most favorable conformations (i.e., with the lowest free energy) in each basin, the medin dimer had lower potential energy than the two isolated monomers. Dimeric medin featured a broad free energy basin, indicating that the conformations of the medin dimer were very diverse. Interestingly, most of the β-barrel conformations exhibited lower potential energy than the other dimer conformations, as well as the two isolated monomers (Figures 7b). These results suggested that dimerization resulted in the two medin peptides adopting lower energy conformations. Therefore, the aggregated states were expected to be more favorable compared to the isolated states.
Figure 7. Dimerization free energy landscape analysis of medin.
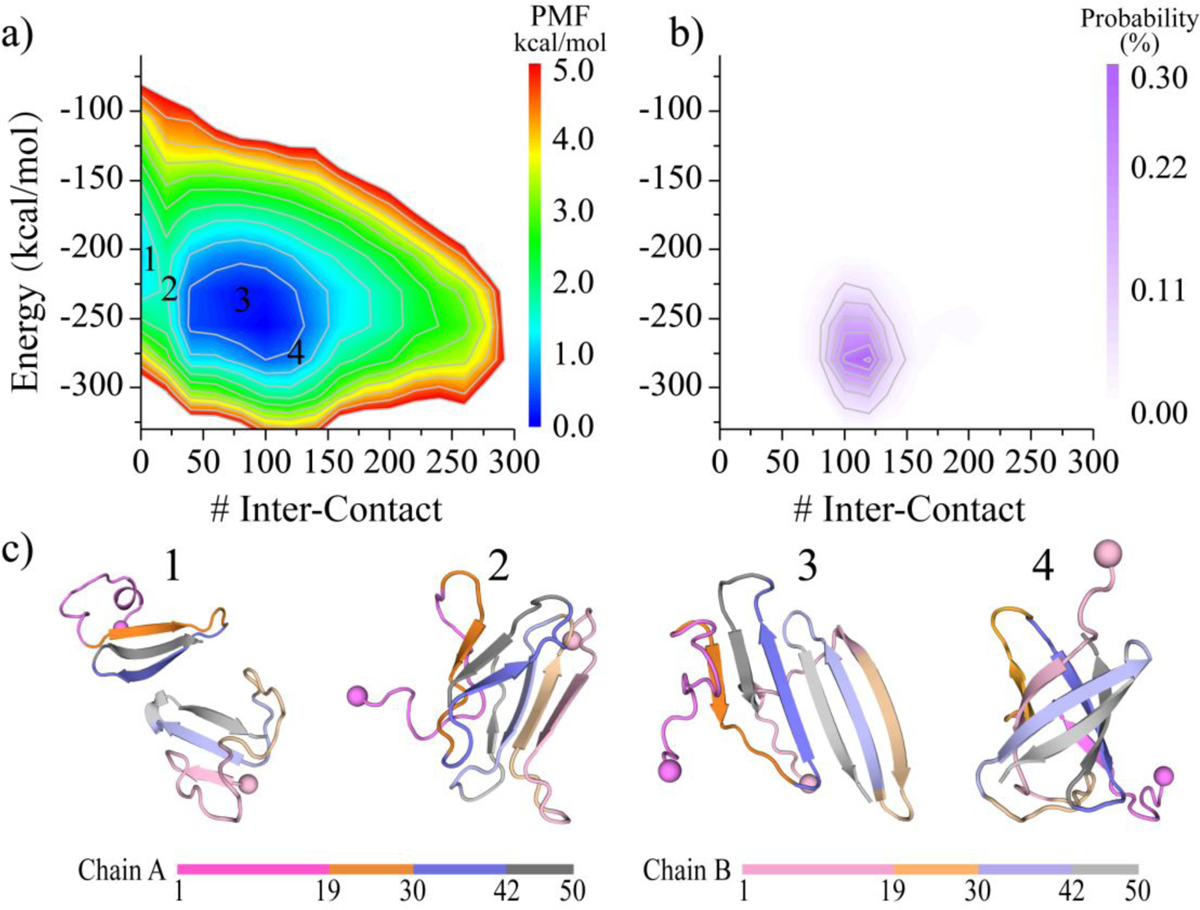
The dimerization free energy landscape is calculated as a function of the number of inter-peptide contacts and potential energy a). The probability distribution of β-barrel conformation is also shown on the same coordinates as the corresponding free energy landscape b). Four snapshots labeled 1–4 on the free energy landscape surface are also presented to illustrate the corresponding conformations c). To capture the entire dimerization process, all the 600 ns data from 60 trajectories are used.
Conclusions.
In conclusion, our atomistic DMD simulations have provided valuable insights into both the folding and dimerization dynamics of medin. The folding dynamics simulation of the medin monomer demonstrated that the formation of β-sheets around medin30–41 and medin42–50 triggered the folding of medin into β-sheet-rich conformations, with the medin1–18 and medin19–29 segments capping their β-sheet edges. The conformational analysis indicated that the medin monomer primarily consists of three or four β-strands, along with a dynamic helix around the N-terminus. Additionally, two isolated medin peptides readily aggregated into β-sheet-rich oligomers, showcasing a strong aggregation propensity characteristic of classic amyloid-prone peptides20. Dimerization of medin promoted the formation of β-sheet conformations and reduced the presence of N-terminal helical structures, leading to the emergence of β-barrel conformations. The observation of β-barrel pore oligomers was also supported by prior experimental measurements11. Furthermore, the specific analysis of inter-peptide contacts suggested that the self-assembly tendency among medin1–18 and medin19–29 segments were relatively weak, consistent with prior experimental studies20. In the medin dimer, the segments of medin30–41 and medin42–50 primarily formed a buried β-sheet core, which could further facilitate medin1–18 and medin19–29 in forming intra- and inter-peptide β-sheets. The region of medin1–18 was usually distributed on the outer layer. The dimerization free energy landscape analysis suggested that dimer formation was energetically more favorable than having two isolated monomers, as dimers typically exhibited lower potential energy. Overall, our study sheds light on the molecular mechanisms underlying both medin monomer folding and the formation of stable β-sheet structures in the medin dimer. These findings provide a better understanding of medin’s role in the pathogenesis of amyloid-related vascular disorders4, 18. The knowledge gained from these simulations may have implications for the development of potential therapeutic strategies to target medin aggregation.
Supplementary Material
Acknowledgments.
This work was supported in part by the National Natural Science Foundation of China (Grant No. 11904189 and 82171527), Ningbo Top Medical and Health Research Program (Grant No. 2022020304), PhD Research Initiation Project of Lihuili Hospital (Grant No.: 2023BSKY-HFJ), and US National Institutes of Health R35GM145409. Computer simulations were partially supported by the multi-scale computational modeling core of NIH P20GM121342. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NSFC, NIH, and NSF.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the website. Assessment of equilibrium for the medin monomer folding dynamics smimulation (Figure S1); Conformational free energy landscape analysis of the medin monomer (Figure S2); Conformation analysis of the medin1–18 within medin monomer (Figure S3); Dimerization dynamics analysis of medin (Figure S4); Equilibrium analysis of the dimerization simulation for medin peptides (Figure S5); Comparing the conformation of medin peptide within dimer and monomer (Figure S6); Secondary structure analysis for each residue from medin monomer and dimer (Figure S7); Solvent accessible surface area analysis for each residue from medin monomer and dimer (Figure S8) (PDF).
The authors declare no competing financial interest.
Data and Software Availability.
DMD simulation engine is available at Molecules In Action, LLC. (www.moleculesinaction.com). Initial conformations, input parameter and topology files, as well as the output PDB files of five representative DMD simulations for each system are available (https://doi.org/10.5281/zenodo.8213354).
References
- (1).Wagner J; Degenhardt K; Veit M; Louros N; Konstantoulea K; Skodras A; Wild K; Liu P; Obermuller U; Bansal V; Dalmia A; Hasler LM; Lambert M; De Vleeschouwer M; Davies HA; Madine J; Kronenberg-Versteeg D; Feederle R; Del Turco D; Nilsson KPR; Lashley T; Deller T; Gearing M; Walker LC; Heutink P; Rousseau F; Schymkowitz J; Jucker M; Neher JJ Medin co-aggregates with vascular amyloid-beta in Alzheimer’s disease. Nature 2022, 612 (7938), 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Haggqvist B; Naslund J; Sletten K; Westermark GT; Mucchiano G; Tjernberg LO; Nordstedt C; Engstrom U; Westermark P Medin: an integral fragment of aortic smooth muscle cell-produced lactadherin forms the most common human amyloid. Proc Natl Acad Sci U S A 1999, 96 (15), 8669–8674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Peng S; Larsson A; Wassberg E; Gerwins P; Thelin S; Fu X; Westermark P Role of aggregated medin in the pathogenesis of thoracic aortic aneurysm and dissection. Lab Invest 2007, 87 (12), 1195–1205. [DOI] [PubMed] [Google Scholar]
- (4).Degenhardt K; Wagner J; Skodras A; Candlish M; Koppelmann AJ; Wild K; Maxwell R; Rotermund C; von Zweydorf F; Gloeckner CJ; Davies HA; Madine J; Del Turco D; Feederle R; Lashley T; Deller T; Kahle P; Hefendehl JK; Jucker M; Neher JJ Medin aggregation causes cerebrovascular dysfunction in aging wild-type mice. P Natl Acad Sci USA 2020, 117 (38), 23925–23931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Peng S; Glennert J; Westermark P Medin-amyloid: a recently characterized age-associated arterial amyloid form affects mainly arteries in the upper part of the body. Amyloid 2005, 12 (2), 96–102. [DOI] [PubMed] [Google Scholar]
- (6).Karamanova N; Truran S; Serrano GE; Beach TG; Madine J; Weissig V; Davies HA; Veldhuizen J; Nikkhah M; Hansen M; Zhang W; D’Souza K; Franco DA; Migrino RQ Endothelial Immune Activation by Medin: Potential Role in Cerebrovascular Disease and Reversal by Monosialoganglioside-Containing Nanoliposomes. J Am Heart Assoc 2020, 9 (2), e014810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang M; Wang HH; Lakatta EG Milk fat globule epidermal growth factor VIII signaling in arterial wall remodeling. Curr Vasc Pharmacol 2013, 11 (5), 768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Davies HA; Caamano-Gutierrez E; Chim YH; Field M; Nawaytou O; Ressel L; Akhtar R; Madine J Idiopathic degenerative thoracic aneurysms are associated with increased aortic medial amyloid. Amyloid 2019, 26 (3), 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Migrino RQ; Karamanova N; Truran S; Serrano GE; Davies HA; Madine J; Beach TG Cerebrovascular medin is associated with Alzheimer’s disease and vascular dementia. Alzh Dement-Dadm 2020, 12 (1), e12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ikemoto-Uezumi M; Zhou H; Kurosawa T; Yoshimoto Y; Toyoda M; Kanazawa N; Nakazawa T; Morita M; Tsuchida K; Uezumi A Increased MFG-E8 at neuromuscular junctions is an exacerbating factor for sarcopenia-associated denervation. Aging Cell 2022, 21 (1), e13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Younger S; Jang H; Davies HA; Niemiec MJ; Garcia JGN; Nussinov R; Migrino RQ; Madine J; Arce FT Medin Oligomer Membrane Pore Formation: A Potential Mechanism of Vascular Dysfunction. Biophys J 2020, 118 (11), 2769–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sun Y; Kakinen A; Wan X; Moriarty N; Hunt CPJ; Li Y; Andrikopoulos N; Nandakumar A; Davis TP; Parish CL; Song Y; Ke PC; Ding F Spontaneous Formation of beta-sheet Nano-barrels during the Early Aggregation of Alzheimer’s Amyloid Beta. Nano Today 2021, 38, 101125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cohen SI; Linse S; Luheshi LM; Hellstrand E; White DA; Rajah L; Otzen DE; Vendruscolo M; Dobson CM; Knowles TP Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A 2013, 110 (24), 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang M; Sun Y; Cao X; Peng G; Javed I; Kakinen A; Davis TP; Lin S; Liu J; Ding F; Ke PC Graphene quantum dots against human IAPP aggregation and toxicity in vivo. Nanoscale 2018, 10 (42), 19995–20006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang Y; Hu T; Wei J; Yin X; Gao Z; Li H Inhibitory activities of flavonoids from Scutellaria baicalensis Georgi on amyloid aggregation related to type 2 diabetes and the possible structural requirements for polyphenol in inhibiting the nucleation phase of hIAPP aggregation. Int J Biol Macromol 2022, 215, 531–540. [DOI] [PubMed] [Google Scholar]
- (16).Olofsson A; Borowik T; Grobner G; Sauer-Eriksson AE Negatively charged phospholipid membranes induce amyloid formation of medin via an alpha-helical intermediate. J Mol Biol 2007, 374 (1), 186–194. [DOI] [PubMed] [Google Scholar]
- (17).Davies HA; Rigden DJ; Phelan MM; Madine J Probing Medin Monomer Structure and its Amyloid Nucleation Using (13)C-Direct Detection NMR in Combination with Structural Bioinformatics. Sci Rep 2017, 7, 45224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Davies HA; Phelan MM; Wilkinson MC; Migrino RQ; Truran S; Franco DA; Liu LN; Longmore CJ; Madine J Oxidative Stress Alters the Morphology and Toxicity of Aortic Medial Amyloid. Biophys J 2015, 109 (11), 2363–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Davies HA; Madine J; Middleton DA Comparisons with amyloid-beta reveal an aspartate residue that stabilizes fibrils of the aortic amyloid peptide medin. J Biol Chem 2015, 290 (12), 7791–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Larsson A; Soderberg L; Westermark GT; Sletten K; Engstrom U; Tjernberg LO; Naslund J; Westermark P Unwinding fibril formation of medin, the peptide of the most common form of human amyloid. Biochem Biophys Res Commun 2007, 361 (4), 822–828. [DOI] [PubMed] [Google Scholar]
- (21).Reches M; Gazit E Amyloidogenic hexapeptide fragment of medin: homology to functional islet amyloid polypeptide fragments. Amyloid 2004, 11 (2), 81–89. [DOI] [PubMed] [Google Scholar]
- (22).Madine J; Copland A; Serpell LC; Middleton DA Cross-beta spine architecture of fibrils formed by the amyloidogenic segment NFGSVQFV of medin from solid-state NMR and X-ray fiber diffraction measurements. Biochemistry 2009, 48 (14), 3089–3099. [DOI] [PubMed] [Google Scholar]
- (23).Howitz WJ; Wierzbicki M; Cabanela RW; Saliba C; Motavalli A; Tran N; Nowick JS Interpenetrating Cubes in the X-ray Crystallographic Structure of a Peptide Derived from Medin(19–36). J Am Chem Soc 2020, 142 (37), 15870–15875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dokholyan NV; Buldyrev SV; Stanley HE; Shakhnovich EI Discrete molecular dynamics studies of the folding of a protein-like model. Fold Des 1998, 3 (6), 577–587. [DOI] [PubMed] [Google Scholar]
- (25).Shirvanyants D; Ding F; Tsao D; Ramachandran S; Dokholyan NV Discrete molecular dynamics: an efficient and versatile simulation method for fine protein characterization. J Phys Chem B 2012, 116 (29), 8375–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Liu Y; Wang Y; Zhang Y; Zou Y; Wei G; Ding F; Sun Y Structural Perturbation of Monomers Determines the Amyloid Aggregation Propensity of Calcitonin Variants. J Chem Inf Model 2023, 63 (1), 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang Y; Liu Y; Zhang Y; Wei G; Ding F; Sun Y Molecular insights into the oligomerization dynamics and conformations of amyloidogenic and non-amyloidogenic amylin from discrete molecular dynamics simulations. Phys Chem Chem Phys 2022, 24 (36), 21773–21785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Huang F; Wang Y; Zhang Y; Wang C; Lian J; Ding F; Sun Y Dissecting the Self-assembly Dynamics of Imperfect Repeats in alpha-Synuclein. J Chem Inf Model 2023, 63 (11), 3591–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Liu Y; Wang Y; Tong C; Wei G; Ding F; Sun Y Molecular Insights into the Self-Assembly of Block Copolymer Suckerin Polypeptides into Nanoconfined beta-Sheets. Small 2022, 18 (34), e2202642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sun Y; Ding F Thermo- and pH-responsive fibrillization of squid suckerin A1H1 peptide. Nanoscale 2020, 12 (11), 6307–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yin S; Biedermannova L; Vondrasek J; Dokholyan NV MedusaScore: an accurate force field-based scoring function for virtual drug screening. J Chem Inf Model 2008, 48 (8), 1656–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yin S; Ding F; Dokholyan NV Eris: an automated estimator of protein stability. Nat Methods 2007, 4 (6), 466–467. [DOI] [PubMed] [Google Scholar]
- (33).Ding F; Tsao D; Nie H; Dokholyan NV Ab initio folding of proteins with all-atom discrete molecular dynamics. Structure 2008, 16 (7), 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Urbanc B; Borreguero JM; Cruz L; Stanley HE Ab initio discrete molecular dynamics approach to protein folding and aggregation. Methods Enzymol 2006, 412, 314–338. [DOI] [PubMed] [Google Scholar]
- (35).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M Charmm - a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J Comput Chem 1983, 4 (2), 187–217. [Google Scholar]
- (36).Lazaridis T; Karplus M Effective energy functions for protein structure prediction. Curr Opin Struct Biol 2000, 10 (2), 139–145. [DOI] [PubMed] [Google Scholar]
- (37).Andersen HC Molecular-Dynamics Simulations at Constant Pressure and-or Temperature. J Chem Phys 1980, 72 (4), 2384–2393. [Google Scholar]
- (38).Yanez Orozco IS; Mindlin FA; Ma J; Wang B; Levesque B; Spencer M; Rezaei Adariani S; Hamilton G; Ding F; Bowen ME; Sanabria H Identifying weak interdomain interactions that stabilize the supertertiary structure of the N-terminal tandem PDZ domains of PSD-95. Nat Commun 2018, 9 (1), 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hamilton GL; Saikia N; Basak S; Welcome FS; Wu F; Kubiak J; Zhang C; Hao Y; Seidel CAM; Ding F; Sanabria H; Bowen ME Fuzzy supertertiary interactions within PSD-95 enable ligand binding. Elife 2022, 11, e77242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Oostenbrink C; Soares TA; van der Vegt NF; van Gunsteren WF Validation of the 53A6 GROMOS force field. Eur Biophys J 2005, 34 (4), 273–284. [DOI] [PubMed] [Google Scholar]
- (41).Kaminski GA; Friesner RA; Tirado-Rives J; Jorgensen WL Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. Journal of Physical Chemistry B 2001, 105 (28), 6474–6487. [Google Scholar]
- (42).Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78 (8), 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 2017, 14 (1), 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhang Y; Liu Y; Zhao W; Sun Y Hydroxylated single-walled carbon nanotube inhibits beta2m(21)(−)(31) fibrillization and disrupts pre-formed proto-fibrils. Int J Biol Macromol 2021, 193 (Pt A), 1–7. [DOI] [PubMed] [Google Scholar]
- (45).Sun Y; Huang J; Duan X; Ding F Direct Observation of beta-Barrel Intermediates in the Self-Assembly of Toxic SOD1(28–38) and Absence in Nontoxic Glycine Mutants. J Chem Inf Model 2021, 61 (2), 966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sun Y; Kakinen A; Xing Y; Faridi P; Nandakumar A; Purcell AW; Davis TP; Ke PC; Ding F Amyloid Self-Assembly of hIAPP8–20 via the Accumulation of Helical Oligomers, alpha-Helix to beta-Sheet Transition, and Formation of beta-Barrel Intermediates. Small 2019, 15 (18), e1805166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wang Y; Xu J; Huang F; Yan J; Fan X; Zou Y; Wang C; Ding F; Sun Y SEVI Inhibits Abeta Amyloid Aggregation by Capping the beta-Sheet Elongation Edges. J Chem Inf Model 2023, 63 (11), 3567–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Emperador A; Orozco M Discrete Molecular Dynamics Approach to the Study of Disordered and Aggregating Proteins. J Chem Theory Comput 2017, 13 (3), 1454–1461. [DOI] [PubMed] [Google Scholar]
- (49).Xiao X; Wang Y; Seroski DT; Wong KM; Liu R; Paravastu AK; Hudalla GA; Hall CK De novo design of peptides that coassemble into beta sheet-based nanofibrils. Sci Adv 2021, 7 (36), eabf7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH Solving protein structures using short-distance cross-linking constraints as a guide for discrete molecular dynamics simulations. Sci Adv 2017, 3 (7), e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kabsch W; Sander C Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22 (12), 2577–2637. [DOI] [PubMed] [Google Scholar]
- (52).Zhang Y; Wang Y; Liu Y; Wei G; Ding F; Sun Y Molecular Insights into the Misfolding and Dimerization Dynamics of the Full-Length alpha-Synuclein from Atomistic Discrete Molecular Dynamics Simulations. ACS Chem Neurosci 2022, 13 (21), 3126–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Daura X; Gademann K; Jaun B; Seebach D; van Gunsteren WF; Mark AE Peptide folding: When simulation meets experiment. Angew Chem Int Edit 1999, 38 (1–2), 236–240. [Google Scholar]
- (54).Laganowsky A; Liu C; Sawaya MR; Whitelegge JP; Park J; Zhao M; Pensalfini A; Soriaga AB; Landau M; Teng PK; Cascio D; Glabe C; Eisenberg D Atomic view of a toxic amyloid small oligomer. Science 2012, 335 (6073), 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Do TD; LaPointe NE; Nelson R; Krotee P; Hayden EY; Ulrich B; Quan S; Feinstein SC; Teplow DB; Eisenberg D; Shea JE; Bowers MT Amyloid beta-Protein C-Terminal Fragments: Formation of Cylindrins and beta-Barrels. J Am Chem Soc 2016, 138 (2), 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Sun Y; Ge X; Xing Y; Wang B; Ding F beta-barrel Oligomers as Common Intermediates of Peptides Self-Assembling into Cross-beta Aggregates. Sci Rep 2018, 8 (1), 10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sun Y; Kakinen A; Xing Y; Pilkington EH; Davis TP; Ke PC; Ding F Nucleation of beta-rich oligomers and beta-barrels in the early aggregation of human islet amyloid polypeptide. Biochim Biophys Acta Mol Basis Dis 2019, 1865 (2), 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Wu J; Blum TB; Farrell DP; DiMaio F; Abrahams JP; Luo J Cryo-electron Microscopy Imaging of Alzheimer’s Amyloid-beta 42 Oligomer Displayed on a Functionally and Structurally Relevant Scaffold. Angew Chem Int Ed Engl 2021, 60 (34), 18680–18687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Pan J; Han J; Borchers CH; Konermann L Structure and dynamics of small soluble Abeta(1–40) oligomers studied by top-down hydrogen exchange mass spectrometry. Biochemistry 2012, 51 (17), 3694–3703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DMD simulation engine is available at Molecules In Action, LLC. (www.moleculesinaction.com). Initial conformations, input parameter and topology files, as well as the output PDB files of five representative DMD simulations for each system are available (https://doi.org/10.5281/zenodo.8213354).