

Abstract
Bone tissue is critically lagging behind soft tissues and biofluids in our effort to advance precision medicine. The main challenges have been accessibility and the requirement for deleterious decalcification processes that impact the fidelity of diagnostic histomorphology and hinder downstream analyses such as fluorescence in-situ hybridization (FISH). We have developed an alternative fixation chemistry that simultaneously fixes and decalcifies bone tissue. We compared tissue morphology, immunohistochemistry (IHC), cell signal phosphoprotein analysis, and FISH in 50 patient matched primary bone cancer cases that were either formalin fixed and decalcified, or Theralin fixed with and without decalcification. Use of Theralin improved tissue histomorphology, while overall IHC was comparable to formalin fixed, decalcified samples. Theralin fixed samples showed a significant increase in protein and DNA extractability, supporting technologies such as laser-capture microdissection and reverse phase protein microarrays. Formalin fixed bone samples suffered from a fixation artifact where protein quantification of β-Actin directly correlated with fixation time. Theralin fixed samples were not affected by this artifact. Moreover, Theralin fixation enabled standard FISH staining in bone cancer samples, while no FISH staining was observed in formalin fixed samples. We conclude that the use of Theralin fixation unlocks the molecular archive within bone tissue allowing bone to enter the standard tissue analysis pipeline. This will have significant implications for bone cancer patients, in whom personalized medicine has yet to be implemented.
INTRODUCTION
In recent years, bone has stepped out of its typical functional niche as the skeletal framework and site for hematopoiesis. Multiple hormones originate from bone that regulate glucose homeostasis, energy expenditure, phosphate metabolism, and food intake. This extends its function to that of a complex pleiotropic endocrine organ which communicates with cells in diverse organs such as the brain, kidneys and pancreas 1–4. Bone is also at the core of diseases such as cancer, where more than one third of cancer patients that die each year show bone metastases 5. In fact, bone is a primary metastatic site for breast cancer, the second most common cancer worldwide 6. It is therefore critical to unlock the molecular archive of bone tissue for in-depth molecular and standard clinical histology analysis. Nonetheless, bone remains one of the most difficult tissues to evaluate and treat due to challenging biopsy procedures and its calcified matrix, which requires demineralization prior to regular clinical laboratory processing. Different decalcification strategies have been developed, including solubilizing calcium with acid, chelating calcium with ethylenediaminetetraacetic acid (EDTA), cation ion exchange resins, or ammonium phosphate and ammonium bicarbonate extraction 7–14. Depending on the decalcification strategy chosen, different undesirable artifacts, such as the destruction of nucleic acids and protein epitopes, severely limit downstream analysis 15,16. In fact, standard of care diagnostics, such as fluorescence in-situ hybridization (FISH), are prohibited in bone tissue.
Advancing precision medicine in cancer requires the tissue specific analysis of low abundance and labile biomarkers, such as phosphorylated proteins, in individual patients. Owing to the fact that a large percentage of current lead compounds for targeted cancer therapy are protein kinase inhibitors 17, quantification of cell signaling proteins in soft tissue tumors is becoming increasingly important. But bone tissue is sidelined in the advancement of personalized medicine because of its inaccessibility for standard proteomic and genomic downstream analysis.
It is known that a significant percentage of primary breast tumors and their metastases show discordance in their estrogen, progesterone and HER2 receptor status 18. Unfortunately, patients with a HER2 negative primary breast tumor and HER2 positive bone metastasis are never identified because HER2 FISH assays will not work after standard decalcification and formalin fixation. Since HER2 FISH is the clinical gold standard for determining eligibility for HER2 therapy, these patients can never benefit from standard of care therapy.
We developed Theralin, a single-step, room temperature tissue preservative that is compatible with paraffin embedding and standard clinical and research downstream analysis methods such as immunohistochemistry 19. Theralin is a precipitating fixative that contains: (i) permeation enhancers that decrease the tissue penetration rate, (ii) reversible cross-linkers that stably cross-link biomolecules during tissue preservation while allowing full biomolecule recovery during extraction, (iii) kinase and phosphatase inhibitors that prohibit ex-vivo alterations of protein phosphorylation, and (iv) an osmotically balanced buffer and a carboxylic acid that maintain tissue morphology during the fixation process. This tissue fixative solves the problem of rapid biomarker preservation at room temperature while maintaining tissue histomorphology and antigenicity in soft tissues. Here, we set out to study if this same tissue preservation chemistry could be applied to bone tissue. Due to our serendipitous discovery that Theralin decalcifies bone, we wanted to determine if Theralin could simultaneously retain labile protein biomarkers and tissue morphology while solubilizing the calcified matrix. This would obviate the need for a separate demineralization step and possibly enable rigorous and standardized diagnostic assays such as FISH that were previously hindered by standard formalin fixation coupled with decalcification of bone tissues.
MATERIALS AND METHODS
Sample collection
50 biopsies, performed between February and June 2011 at the Rizzoli Orthopedic Institute, Bologna, Italy, were collected and the bone-containing material divided into six 1 × 0.5 × 0.5 cm blocks. Tissue blocks were processed in parallel according to the following protocols: (1) fixation with Theralin, (2) fixation with Theralin, followed by standard decalcification, and (3) fixation with formalin, followed by standard decalcification. Samples were fixed in 6–7 ml of fixative (either Theralin or 4% neutral buffered formalin) at room temperature for 24–48 hours. Decalcification was performed using a mixture of 5.1% formic acid and 2.5% nitric acid. The time of decalcification varied between samples depending on bone content and was determined by palpation, a commonly used method for decalcification endpoint determination in clinical pathology laboratories20 and established standard method at the Rizzoli Orthopedic Institute, Bologna, Italy, where all clinical samples were collected. Two blocks for each method were processed in parallel with one block being evaluated at the Rizzoli Institute, Italy, (clinical IHC, FISH) and a matching block evaluated at George Mason University, Fairfax, USA, (laser capture microdissection, reverse phase protein microarrays, IHC) or the University of Manitoba, Canada (telomere FISH), resulting in a total of 300 tissue blocks available for this study.
Control samples for FISH analysis: Human tibia, procured following an amputation, and Balb/c mouse (Envigo, Indianapolis, IN, USA) ribs were fixed for 7 to 14 days in either Theralin or 10% neutral buffered formalin. Formalin fixed samples were decalcified in formic acid for 24–48 hours.
Control samples for correlation of β-Actin reactivity with fixation time: U266 cells were grown to 80% confluence and incubated in serum-free media overnight. Following, cells were either lysed directly or fixed with Theralin or 10% neutral buffered formalin for 10 minutes and 2 hours before lysis. Liver samples from three ICR (CD-1) mice (Envigo, Indianapolis, IN, USA) were fixed for 1 day or 7 days in either Theralin or 10% neutral buffered formalin. β-Actin levels were quantified using reverse phase protein microarrays (see below).
Mice were housed according to standard animal care procedures with water and chow provided ad libitum and euthanized with CO2. Animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the protocol was approved by the George Mason University institutional animal care and use committee. All animal tissue samples were taken as a terminal event post euthanasia, and all efforts were made to minimize suffering. Human surgical tissue specimens were collected from patients under written informed consent following the protocols by the Rizzoli Orthopedic Institute (samples 1–50, Supplemental Table 1) or the institutional review boards of Inova Fairfax Hospital and George Mason University (sample 51).
Fluorescence In-situ Hybridization (FISH)
FISH was performed at two locations, using different protocols. At the Rizzoli Orthopedic Institute, FISH was performed using the LSI FOXO1 Dual Color Break-apart DNA probe (13q14) (Abbott Molecular, Des Plaines, IL, USA) and the SPEC MDM2/CEN 12 Dual Color probe (12q14.3–12q15) (ZytoVision GmbH, Bremerhaven, Germany) according to the manufacturer’s protocol. Tissue sections of 4 μm were mounted on positively charged slides (Dako, Glostrup, Denmark). Slides were heated overnight (60°C), dewaxed in xylene, and treated with an ethanol-to-water series. This was followed by incubation in TE solution (TRIS 5 mM-EDTA 1 mM) at 96°C for 15 minutes, rinsed in distilled water, and digested with pepsin (0.04%) in 0.01N HCl at 37°C for 5 to 15 minutes, then washed again in distilled water. Slides were finally dehydrated in ethanol (96%) and air dried. Next, the probes were applied to the target area and the slides were coverslipped and sealed with rubber cement. The samples and probes were co-denaturated in Dako Hybridizer (Dako, Glostrup, Denmark) at 85°C for 1 minute and incubated overnight at 37°C. The following day, the coverslips were removed and the slides were washed 2 min at 73°C in 0,4 X SSC/0.3%NP40 and 1 min at room temperature in 2 X SSC/0.1% NP40. The slides were then left to dry in the dark at room temperature; the nuclei were subsequently counterstained in Vectashield Antifade solution with DAPI (Vector Laboratories, Inc. Burlingame CA, USA). Fluorescence signals were counted using an OLYMPUS BX41 fluorescence microscope (Olympus, Hamburg Germany), at 100X under oil immersion using an appropriate filter set. A minimum of 100 tumor cell nuclei with intact morphology, as determined by DAPI counterstaining, were counted in the previously marked neoplastic area. A positive result was defined as the presence of a visible red and green signals in more than 10% of the cells.
At the University of Manitoba, slides were deparaffinized and dehydrated in 100% ethanol, then incubated in 1 M NaSCN for 30 min at 80°C in a water bath. After rinse in double distilled water, slides were incubated in 3.7% buffered formalin solution (Sigma-Aldrich, Oakville, ON, Canada) in 2x SSC buffer (pH 7.6) for 10 min at RT and washed two times in 2xSSC for 5 min each. Totally 50 µg/ml pepsin was added to 0.01 M 37°C prewarmed HCL and slides were incubated for 6 min followed by two washes in 2xSSC for 5 min each. After exposure to 3.7% buffered formalin solution (Sigma-Aldrich, Oakville, ON, Canada) in 2x SSC buffer (pH 7.6) for 10 min at RT and two washes in 2xSSC, tissues were dehydrated in ethanol. 5 µl of Cy3-labeled peptide nucleic acid telomere (PNA) probe (Dako, Glostrup,Denmark) was applied to each slide to detect (T2AG3)n repeats. Slides and probe were incubated at 80°C for 3 min to denature DNA, followed by hybridization at 30°C for 2 hr using the Hybrite chamber (Vysis, Abbott Diagnostics Mississauga, ON, Canada). Next, slides were washed twice in 70% formamide in 10 mM Tris (pH 7.4) followed by successive washings in PBS at RT for 1 min, in 0.13 SSC at 55°C for 5 minutes and in 2x SSC in 0.05% Tween 20 twice at RT. Slides were counterstained with 40,6-diamino-2-phenylindole (DAPI, Invitrogen, Burlington, ON, Canada) (0.1 µl/ml), washed in doubled distilled water and dehydrated in ethanol. Before imaging, slides were mounted to coverslips using Vestashield antifade mounting medium (Vector Laboratories, Burlington, ON, Canada).
Fluorescent images were captured using the AxioImager Z1 microscope, an AxioCamMR3 camera and a Plan-Apochromat 63x/1.40 Oil DIC M27 lens (all from Carl Zeiss, Canada). Acquisition time was 300 msec for Cy3 (telomeres signals) and 100 msec for DAPI. For each nucleus, images were acquired from 60 optical planes (z-stacks) through the nucleus with a sampling distance in the xy axis of 107 nm and z axis of 200 nm using ZEN 2 blue edition software (Carl Zeiss, Canada). Images were deconvolved using a constrained iterative algorithm (Schaefer et al., 2001) available in the ZEN software (Zeiss).
Laser capture microdissection
Tissue was cut into 8 μm sections and laser capture microdissection (LCM) conducted as previously described 21. Care was taken to selectively microdissect tumor cells without inclusion of necrotic tissue, blood vessels, body fluids, bone matrix and normal bone cells. Cells were microdissected from seven representative cases based on comparable cell content across all three fixation types: one chordoma, three giant cell tumors, one bone metastasis from carcinoma, one bone metastasis from melanoma, and one aneurysmal bone cyst. Microdissected tissue was lysed using an extraction buffer made of a 10% (v/v) solution of Tris(2-carboxyethyl)phosphine (TCEP; Pierce, Rockford, IL) in Tissue Protein Extraction Reagent (T-PERTM, Pierce)/2X SDS Tris-glycine buffer (Invitrogen, Carlsbad, CA). Extraction buffer was added to LCM caps and boiled for 10 minutes, followed by manual flushing of LCM caps with extraction buffer for 1 minute and another incubation of the extraction buffer alone at 100°C for ten minutes. Lysed samples were kept at −80°C until printing.
Reverse phase protein microarray construction and staining
Reverse phase protein microarrays (RPPA) were printed and stained as previously described 22. Following printing of the lysates, slides were stored dessicated at −20°C before staining. All antibodies used (Supplemental Table 2) were extensively validated for a specific band at the appropriate molecular weight by Western blotting prior to their use for RPPA. Raw spot analysis was performed using ImageQuant 5.2 (Molecular Dynamics), followed by postprocessing using the Reverse-Phase Protein Microarray Analysis Suite (developed in-house 23).
Immunohistochemistry
At George Mason University, tissue sections were baked at 56°C for 30 minutes and deparaffinized in xylene with rehydration in a series of graded alcohols (100%, 95%, 70%) with a final rinse in water. Following the appropriate heat-induced epitope retrieval per antibody (Supplemental Table 2), slides were stained in a Dako Autostainer using the EnvisionSystem+HRP staining kit (Dako). Nuclear counterstaining was performed using hematoxylin (Sigma Aldrich, St. Louis, MO, USA) and Scott’s Tap Water Substitue (Electron Microscopy Services, Hatfield, PA, USA).
At the Rizzoli Institute, 4μm thick tissue sections were cut, heated at 58 °C for 2 hours, deparaffinised and immunostained on a Ventana BenchMark following the manufacturer’s guidelines (Ventana Medical Systems, Tucson AZ, USA). Antibody detection was performed using UltraView Universal DAB Detection Kit (Ventana Medical Systems, Tucson AZ, USA). Pretreatment for antigen retrieval was performed at 95°C with Tris-EDTA pH 8 for 20 minutes. When necessary, endogenous tissue peroxidase was blocked by treating the sections with 0.3% H2O2. The slides were stained with hematoxylin and then rehydrated and coverslipped. Appropriate positive and negative controls were included in each run. For antibodies used and respective dilutions, see Supplemental Table (Supplemental Table 2).
Tissue scores and statistical analysis
Tissue morphology was scored by clinical pathologists based on nuclear and bone detail (excellent, good, moderate, poor) according to the following criteria. Nuclear details ranged from excellent (very well stained nuclei with visible chromatin detail) to poor (poorly defined chromatin detail or over-decalicified “pink-stained” simil-necrotic nuclei). Bone evaluation ranged from excellent (very well stained bone matrix with clearly visible osteoblasts and osteoclasts) to poor (“blue”-stained poorly decalcified bone matrix or over-decalcified bone with “pink-stained” simil-necrotic nuclei of the osteoblasts and osteoclasts).
Immunohistochemical stains were scored by pathologists as not evaluable (NV), negative (neg), weak (1+), moderate (2+), and strong (3+), according to the number of positive mononuclear cells and their intensity, with respective threshold values of <5%, 5–20%, and >20%.
Two-way unsupervised hierarchical clustering analysis was prepared using R 24. Mean comparisons of proteins levels and protein phosphorylation were conducted using Wilcoxon rank-sum or Student’s t-test, depending on data normality (Shapiro-Wilk test). Data correlation (R2) was determined using GraphPad Prism (version 6.05, GraphPad Software Inc.), which was also used to prepare all of the bar graphs, box plots, and scatter plots. A p < 0.05 was chosen to indicate significance.
RESULTS
Effect of Theralin fixation on sample processing time.
Due to reduced bony matrix content 6 of the 50 samples (12%) did not require decalcification. All remaining formalin fixed samples were decalcified prior to sectioning. Fixation with Theralin obviated the need for a separate decalcification step. Four samples (9%) were found to be under-decalcified following fixation with Theralin. This did not correlate with fixation time or any potential underlying clinical/pathological feature. Moreover, the tissue morphology was scored equal to or better than patient matched samples that were formalin fixed and decalcified for all four samples. Of the samples that required decalcification after formalin fixation, exact fixation/decalcification times were recorded for 19 samples (43% of total of 44), ranging from 6.5 – 45 hours per sample for Theralin fixed and 18.5 – 112 hours per sample for formalin fixed samples. Overall, processing times were decreased by 0 – 96 hours per sample (median 9.0 hours, average 22.6 hours) for Theralin fixed bone tissue compared with formalin fixed tissue (Fig. 1A).
Figure 1. Theralin fixation obviates the need for bone tissue decalcification and provides better clinical histomorphology.
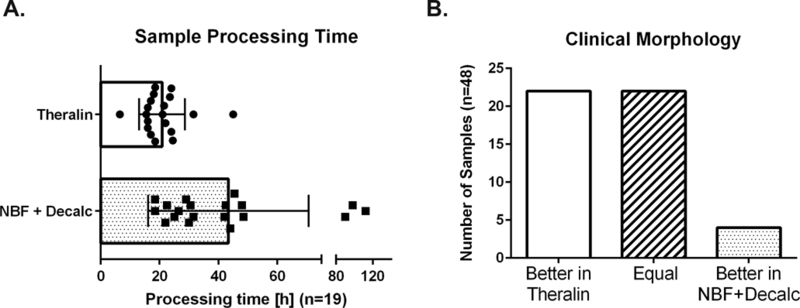
(A) Sample processing time of 19 bone tumor samples fixed in Theralin compared to standard formalin fixation combined with decalcification. Processing time includes fixation and decalcification (if applied). (B) Overall scores of tissue morphology following either Theralin fixation without decalcification or standard formalin fixation with decalcification. (NBF = neutral buffered formalin)
As demonstrated, the carboxylic acid component in Theralin is therefore able to decalcify bone simultaneously with tissue fixation. This obviated the need for a separate tissue processing step for decalcification, significantly decreasing the overall processing time.
Effect of Theralin fixation on clinical histomorphology.
Of the 50 cases, 48 were evaluated by pathologists for adequate preservation of morphology and graded as excellent, good, moderate, and poor based on semi-quantitative evaluation of nuclear detail and bone (nuclear and chromatin detail, bone matrix staining, and clearly identifiable osteoblasts and osteoclasts). Of these, 22 cases (46%) demonstrated better quality morphology after Theralin fixation without decalcification, 4 (8%) showed better morphology after NBF fixation with downstream decalcification, and 22 (46%) cases had equal quality morphology between both preservation treatments (Fig. 1B). The factors that contributed towards the improved morphology after NBF fixation in these four samples are unclear, since the four samples did not overlap with the under-decalcified samples after Theralin fixation, did not belong to a single disease type (osteosarcoma (2), osteochondroma (1), bone metastasis from melanoma (1)), and had different decalcification requirements following formalin fixation (no decalcification (1), 8 hours (1), 24 hours (2)).
Comparison of immunohistochemical stains between Theralin and formalin fixed bone tissue.
As part of the routine clinical diagnostic workflow 17 different immunohistochemical stains were performed depending on the diagnostic requirements for each case, resulting in a total of 192 stained sections. To increase robustness of the analysis we limited the data to proteins that were stained in more than 2 cases, which reduced the total number of stained sections to 125. IHC stains were scored by pathologists according to the number of positive mononuclear cells and their intensity, with respective threshold values of <5%, 5–20%, and >20%. The majority of IHC stains was scored to be of equal quality, with six proteins showing more cases with better IHC stains after Theralin fixation (CD68, Osterix, Brachi, Sox9, P-GP C494, P-GP JSP1) and four proteins with more cases with better IHC stains following fixation with formalin and decalcification (Vimentin, EMA, S100, P-GP MRK16) (Fig. 2). Overall, the relative combined IHC score for both fixation methods was equal in 65%, better for Theralin in 19% and better for formalin with decalcification in 16%.
Figure 2. Overall immunohistochemistry scores are comparable.
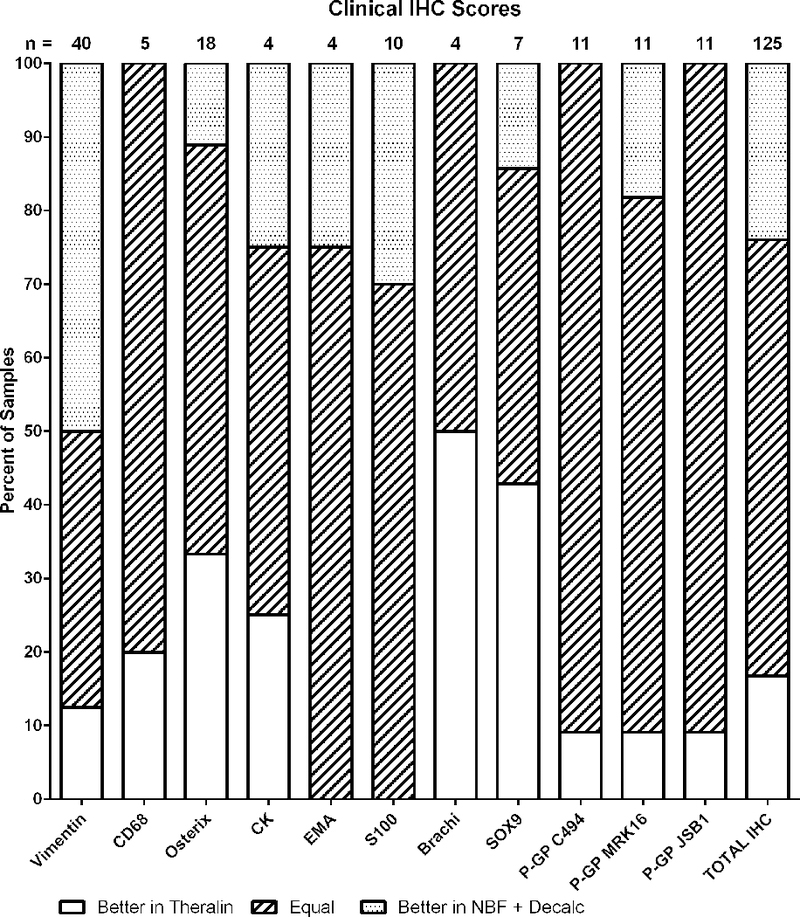
Pathologist scores of IHC stains from patient matched Theralin fixed and formalin fixed, decalcified bone tumor samples.
We also performed additional immunostains for phosphoproteins (ERK Thr202/Tyr204, CREB Ser133, PTEN Ser 380, Acetyl-Co Carboxylase Ser79) and other tumor relevant protein targets (Progesterone receptor, Hif1a, Ki-67, LC3B) on selected cases (Fig. 3). While phosphoprotein stains were comparable between Theralin-fixed and formalin-fixed tissue samples, no immunostain was visible in formalin-fixed, decalcified tissue for Ki-67. This was in agreement with the clinical immunostains performed at the Rizzoli Institute, where two cases were positively scored for Ki-67 after Theralin fixation, but only one of the cases was positive after standard processing in formalin. Anecdotally, we also had one case with positive progesterone receptor staining after Theralin fixation, with no staining after formalin fixation.
Figure 3. Representative images comparing tissue morphology and IHC stains of Theralin fixed and formalin fixed, decalcified bone tumor samples.
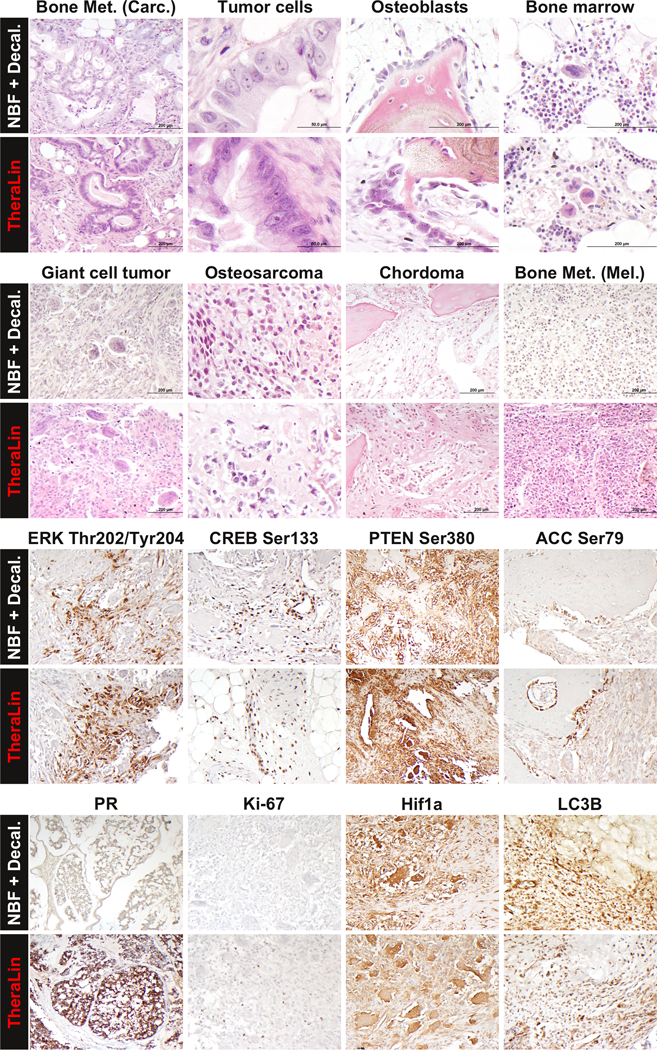
Patient matched tissue sections were stained with either Hematoxylin and eosin (top half) or for selected IHC targets (bottom half).
Protein and DNA extractability.
To evaluate the feasibility of profiling cancer cell specific signal proteins in fixed bone samples, we performed tumor cell enrichment by laser-capture microdissection on seven cases. Cases were chosen to represent different tumor types and ensure equal quality tumor cell material for microdissection in patient matched samples per fixation type. It is well known that formalin fixed tissue requires extended high-temperature protein extraction methods for adequate protein yield 19,25. However, due to the limited lysis volume per laser capture microdissection cap (about 3–7 μl), boiling for one hour or more is not feasible. To increase protein yield we used an adapted protocol in which the microdissected tissue was heated at 100ºC for 20 minutes in SDS/TCEP extraction buffer 19,26. The total amount of protein that was extractable per microdissected area was doubled following Theralin fixation (2.2 fold) or following Theralin fixation with downstream decalcification (1.9 fold) compared to formalin fixation with decalcification (Fig. 4).
Figure 4. Protein and DNA extractability is increased from Theralin fixed tissue.
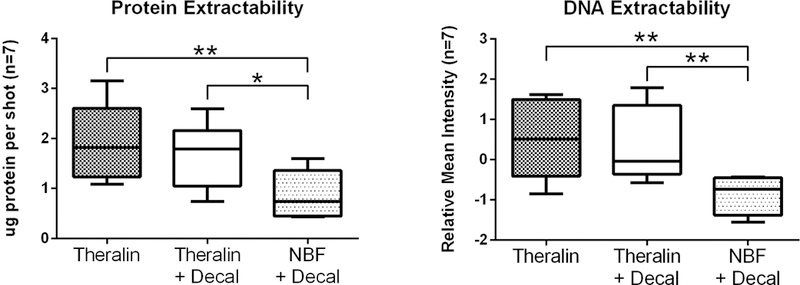
Patient matched bone tumor samples (n=7) were either Theralin fixed, Theralin fixed and decalcified, or formalin fixed and decalcified. An equal number of tumor cells were laser-capture microdissected and cell lysates printed on reverse phase protein microarrays. Formalin fixation followed by decalcification yielded significantly less protein and DNA than Theralin fixation with or without decalcification. (boxes = 25th to 75th percentile with whiskers going to the smallest and largest value, Decal = decalcification, NBF = formalin fixation, * p<0.05, ** p<0.005)
DNA can be used for assay normalization to account for data variances. Proteomic analyses, such as reverse phase protein microarrays (RPPA) or western blotting, require data normalization to an invariant entity. Erythrocytes contribute total protein content to a lysate which can skew total protein levels. However, erythrocytes are devoid of a nucleus and therefore lack nuclear DNA, which allows us to use DNA for normalization when samples such as bone marrow aspirates are contaminated with blood 23. Furthermore, DNA provides molecular information as well as having utility in data analysis. To enable molecular profiling of bony tissues, including FISH, we needed to verify that DNA yield was adequate. Therefore, we also compared the amount of measurable DNA per microdissected area in Theralin and formalin fixed bone samples. We found a 2.8 fold increase in DNA in Theralin fixed samples and 2.7 fold increase in Theralin fixed, decalcified samples, compared to formalin fixed, decalcified samples (Fig. 4). It is important to point out that we did not specifically extract DNA from samples using established nucleic acid isolation protocols but rather printed whole cell lysates onto nitrocellulose slides 22. The DNA per sample was then denatured and permanently cross-linked to the nitrocellulose as previously described 23. This approach is critical to enable normalization of RPPA data with DNA content per sample.
To evaluate whether the reduced protein yield from formalin fixed and decalcified samples was selective for specific classes of proteins, we then quantified 46 individual protein endpoints that represent receptor tyrosine kinases, intracellular kinases (phosphorylated and non-phosphorylated), hormone receptors, cytoskeletal proteins and cleaved caspases. Of these proteins, 21 showed a significantly reduced level in formalin-fixed, decalcified samples compared to matched Theralin-fixed, non-decalcified or decalcified samples (Fig. 5). This reduction in protein yield appeared to be independent of the class of protein evaluated. No significant differences were found between decalcified and non-decalcified Theralin fixed tissue.
Figure 5. The measurable protein level per microdissected tumor area is reduced indiscriminately across protein classes following formalin fixation.
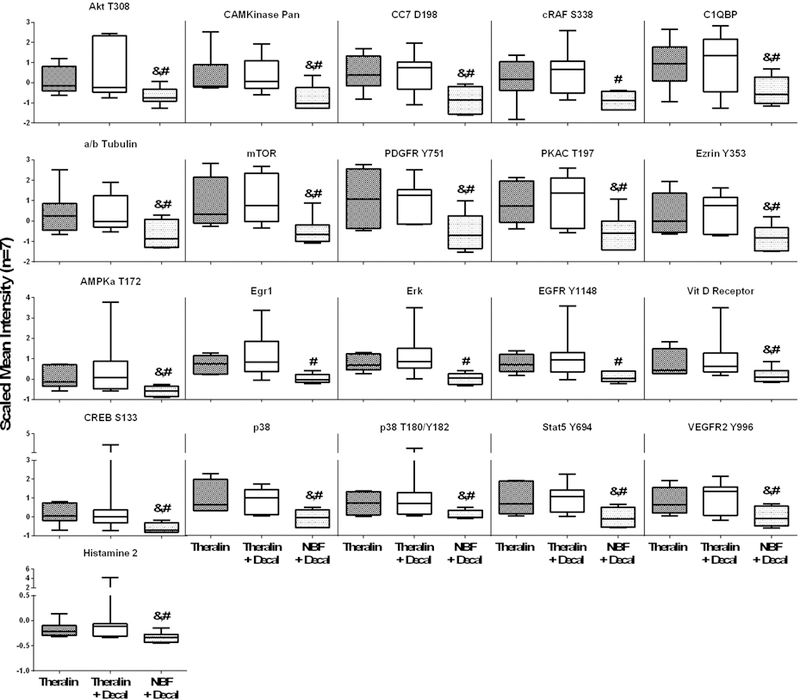
Patient matched bone tumor samples (n=7) were either Theralin fixed (dark gray bar), Theralin fixed and decalcified (white bar), or formalin fixed and decalcified (light gray bar) and tumor cells laser-capture microdissected. Protein levels were measured using reverse phase protein microarrays and normalized to the microdissected area. (boxes = 25th to 75th percentile with whiskers going to the smallest and largest value, # p<0.05 versus Theralin fixation, & p<0.05 versus Theralin fixation and decalcification)
Comparable relative protein abundance.
We then evaluated if the fixation process changes protein phosphorylation. To adjust for the reduced protein extractability from formalin fixed samples, we normalized all protein levels to the total protein amount extracted from each sample. With the exception of β-Actin and Doublecortin, the abundance of all 46 phosphorylated and non-phosphorylated proteins showed no significant difference between Theralin and formalin fixed bone tissue samples. However, following unsupervised two-way hierarchical clustering, formalin fixed bone samples clustered separately from their respective matched Theralin fixed samples in five out of seven cases. In contrast, Theralin fixed samples, either decalcified or not, clustered together in six out of seven cases (Fig. 6).
Figure 6. Formalin fixed and decalcified samples cluster away from patient matched Theralin fixed and Theralin fixed and decalcified samples during 2-way unsupervised hierarchical clustering.
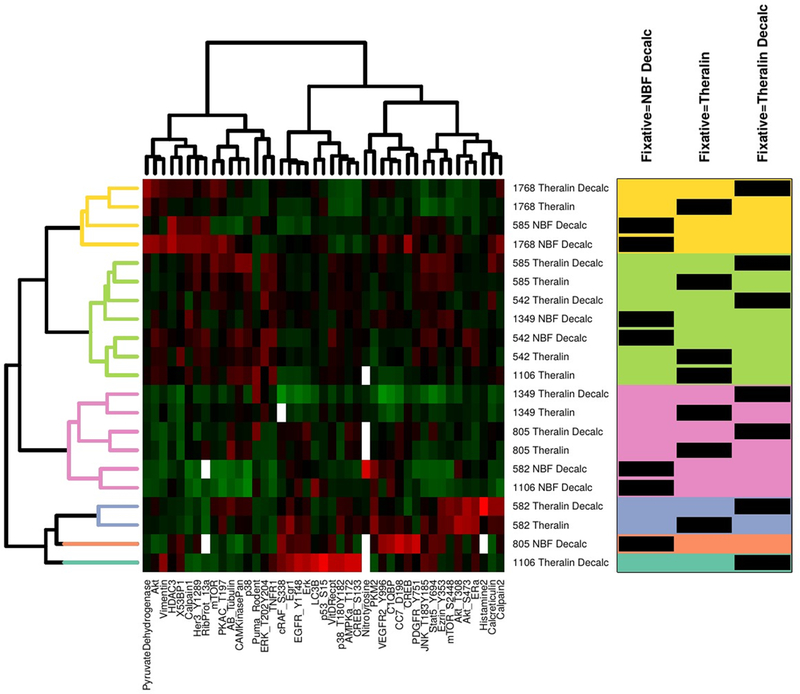
Tumor cells from seven patient matched samples that were either Theralin fixed, Theralin fixed and decalcified, or formalin fixed and decalcified were laser-capture microdissected and lysates printed on reverse phase protein microarrays. Relative protein levels were normalized to the total extracted protein content per sample due to the reduced protein extractability from formalin fixed samples. Numbers in center column represent patient IDs. Box on right shows fixation type per sample (black bars) within each cluster (color shaded areas). (Decalc = decalcified, NBF = formalin fixed)
Formalin fixation artifacts falsely elevate measured β-Actin levels.
Both, β-Actin and Doublecortin, were found to be significantly increased in formalin fixed, decalcified tissue (Fig. 7A, Supplemental Fig. 1). This increase was independent of decalcification itself, as shown by comparing Theralin fixed with Theralin fixed and decalcified tissue (Fig. 7A, Supplemental Fig. 1). Because of its widespread use as loading control in western blot assays, we evaluated whether fixation time had any impact on β-Actin antibody recognition. While time of fixation showed no effect for Theralin fixed, non-decalcified tissue (R2=0.2, p>0.45), β-Actin levels did correlate with time of fixation in formalin (R2=0.8, p<0.05) (Fig. 7B). To determine if this observation extends to other tissues and cells we fixed U266 cells in Theralin or formalin for either ten minutes or two hours. No difference in β-Actin levels between Theralin fixed cells and non-fixed cells was found, while U266 cells fixed in formalin showed a significant increase in β-Actin that was compounded by fixation time (Fig. 7C). Next, we fixed mouse liver for up to seven days in either Theralin or formalin and compared β-Actin levels with frozen tissue. Theralin fixed tissue correctly represented β-Actin levels independent of fixation time, while formalin fixed tissue showed significantly falsely elevated levels of β-Actin that were dependent on fixation time (Fig. 7D). Using a different antibody clone against β-Actin showed the same trend of increased reactivity with increasing fixation time (Supplemental Fig. 1). Doublecortin reactivity was not dependent on fixation time for either of the two antibodies tested (Supplemental Fig. 1).
Figure 7. Formalin fixation introduces a fixation artifact, whereby measurable β-Actin levels correlate with fixation time.
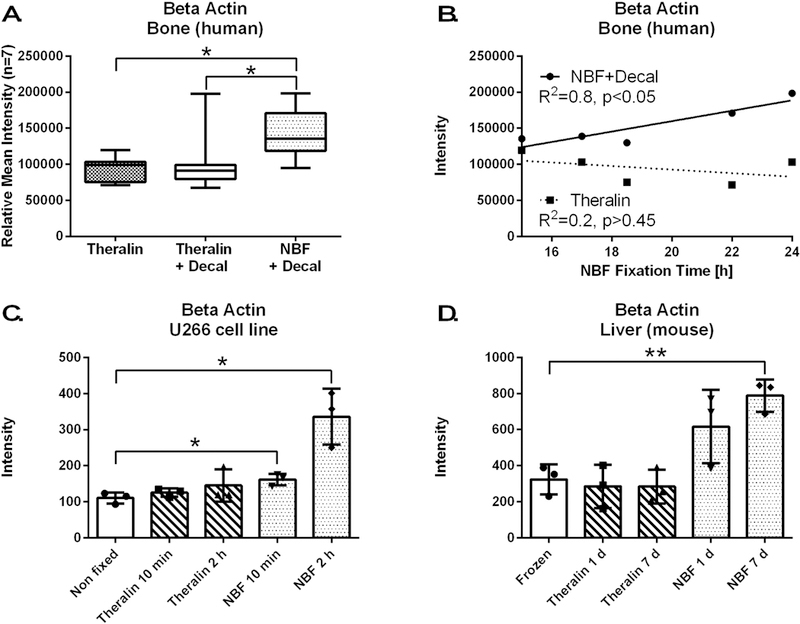
(A) β-Actin is increased following formalin fixation and decalcification of bone tumor samples compared to Theralin fixed or Theralin fixed and decalcified patient matched samples (n=7, laser-capture microdissected tumor cells, boxes = 25th to 75th percentile with whiskers going to the smallest and largest value, * p<0.05). (B) Subset of patient samples from (A) where exact fixation times were available (n=5) show a direct correlation between fixation time and the level of measurable β-Actin when fixed in formalin but not after Theralin fixation. (C) Fixing U266 cells with formalin for either 10 min or 2 h causes an increase in measurable β-Actin versus non-fixed cells. Theralin fixation does not alter β-Actin levels, independent of fixation time (±SD, * p<0.05). (D) Fixing mouse liver with formalin for 1 day or 7 days increases measurable β-Actin versus frozen tissue. Theralin fixation kept β-Actin levels at frozen tissue levels irrespective of fixation time (±SD, * p<0.01).
Theralin fixation enables FISH analysis in bone samples.
We next evaluated the impact of Theralin fixation on downstream fluorescent in-situ hybridization analysis (FISH). Seven osteosarcoma cases were selected, including three high grade central, one low grade central with areas of progression into high grade, one parosteal, and one periosteal osteosarcoma case, as well as a lymph node metastasis from a high grade central osteosarcoma. FISH staining was performed on Theralin fixed, not-decalcified and formalin fixed, decalcified samples, using one of two probes per case. The FOXO1 Dual Color Break-apart DNA probe is typically used to identify rearrangements of the chromosomal locus 13q14 that contains the FOXO1 gene, while the MDM2/CEN 12 Dual Color probe detects MDM2 gene amplifications. Of the five Theralin fixed cases stained with the FOXO1 probe, two showed positive staining and three were negative (Fig. 8). None of the formalin fixed, decalcified samples showed staining. Of the two Theralin fixed cases stained with MDM2/CEN, one was positive (central low grade, with areas of progression into high grade osteosarcoma) and one negative (high grade central osteosarcoma). As before, no FISH staining was observed in patient matched formalin fixed, decalcified samples.
Figure 8. Theralin fixation enables FISH staining in bone tissue.
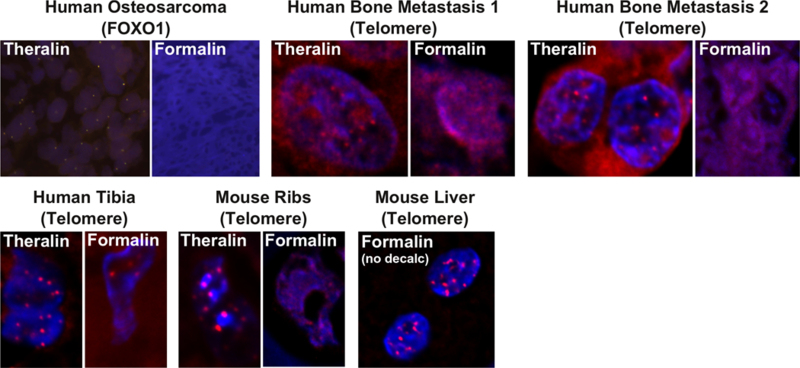
Top panel: Representative images of patient matched, Theralin fixed or formalin fixed, decalcified, human bone tumor samples that were stained with a probe for FOXO1 or telomeres. Theralin fixed samples demonstrate clear FISH staining, while formalin fixed, decalcified samples are non-interpretable. Bottom panel: Representative images of mouse bone samples that were fixed with Theralin or formalin + decalcification and stained for telomeres. In addition, a mouse liver was fixed with formalin without decalcification and stained for telomeres. The positive stain indicates that the formic acid/nitric acid decalcification process is the cause for lack of staining in formalin fixed, decalcified bone samples. Telomere images shown here depict a region of interest zooming onto one or two single nuclei to visualize the telomere signals in red.
To limit any potential impact of tissue inhomogeneities in gene amplification or gene rearrangement between the patient matched sample blocks we next selected two bone metastasis cases and hybridized sections with fluorochrome-coupled (Cy3) Telomere PNA probes and counterstained with DAPI. No telomere staining was observed after formalin fixation and decalcification, while samples fixed with Theralin without decalcification demonstrated clear telomere staining inside nuclei (Fig. 8). The same was observed for mouse rib and human tibia samples, with clear staining in Theralin fixed samples and no or significantly reduced staining in formalin samples. We then stained a mouse liver sample that was fixed with formalin but not decalcified. Telomeres were clearly visible in this sample, indicating that it is the separate formic acid/nitric acid decalcification processing step that hinders FISH analysis.
DISCUSSION
The objective of this study was to critically evaluate a novel fixative, Theralin, for molecular profiling of bone tissue. Precision medicine is advancing asymmetrically, with analytical methods focusing on soft tissue and biofluids, while bone tissue is critically lagging behind27. Overcoming the critical hurdle that prohibits standard clinical assays such as FISH with bone specimens would immediately open an arsenal of already approved treatments and tests available to bone cancer patients. Likewise, unlocking bone for proteomics analysis is essential to identify the driver cell signaling pathways in bone cancer that can be targeted with currently available, or novel, kinase and phosphatase inhibitors.
We developed a tissue fixation chemistry, Theralin, that maintains tissue histomorphology and posttranslationally modified proteins. Our serendipitous discovery that Theralin simultaneously fixes and decalcifies bone tissue, which we attribute to the carboxylic acid component of the fixative, obviates the need for a separate decalcification step and therefore required evaluation of bony tissue. We have shown that Theralin not only eliminates processing complexity, but also decreases sample processing time by an average of 23 hours. This is especially advantageous in a routine clinical setting, where samples from multiple different tissues need to be processed simultaneously. A significant number of bone samples demonstrated better quality morphology after Theralin fixation (46% of samples), contrasting previous observations, where Theralin fixed tissues had equivalent, but not better, tissue morphology compared to standard formalin fixation19. This indicates that the main cause for the inferior quality morphology of formalin fixed bone tissue was the required process of decalcification in formic/nitric acid, not the formalin fixation itself.
Formalin fixation is known to permanently cross-link amino groups28, which requires incubating samples under reducing conditions at high temperature for extended periods of time for adequate protein extraction25,29,30. This becomes a challenge when extracting proteins in small extraction buffer volumes (<10 μl) from very small amounts of tissue, for example after laser-capture microdissection (LCM)31. Using an adapted extraction protocol that was permissive for small buffer volumes following LCM, we found a two-fold increase in protein yield from Theralin-fixed tissue compared to formalin-fixed bone tissue. This was independent of downstream decalcification, as shown by the fact that even Theralin-fixed and formic/nitric acid decalcified tissue had the same increase in protein yield compared to formalin-fixed/decalcified tissue. This indicates that protein extractability is mainly dependent on the nature of the initial tissue fixation, which differs significantly between both fixatives. While formalin fixation is based on permanently cross-linking biomolecules, Theralin is a precipitating fixative that contains reversible cross-linkers19. This is further supported by our observation that proteins belonging to a very diverse set of classes were found to be reduced in formalin-fixed versus Theralin-fixed samples. Of the 21 affected proteins (46% of all proteins quantified), 11 were phosphorylated proteins, 9 were total proteins and one was a cleaved caspase (caspase 7), spanning different biochemical groups, such as cell surface receptors and receptor tyrosine kinases, cytosolic kinases and cleaved proteins, as well as different biological functions, such as apoptosis, cell growth, stress signaling and the cytoskeleton. No difference was found for any protein quantified between Theralin-fixed and formic/nitric acid decalcified samples versus just Theralin-fixed samples.
We also found a three-fold increase in DNA yield from Theralin-fixed tissue compared to formalin-fixed bone tissue. However, it is important to point out that our data does not allow direct conclusions regarding the yield of nucleic acids from formalin-fixed, decalcified bone samples using nucleic acid specific isolation protocols. Rather, our methodological approach of cross-linking proteins and nucleic acids from whole cell lysates directly to nitrocellulose slides is specific to and critical for reverse phase protein microarray analysis and allows data normalization with DNA content per sample23. We have previously described the advantages of using DNA for protein level normalization purposes, especially in the context of bone marrow aspirates that are contaminated with blood23. While the protein contribution by erythrocytes can skew total protein levels within a sample, their lack of a nucleus makes DNA normalization largely blind to the highly variable presence of erythrocytes23. Quantifying low abundance biomolecules therefore requires significantly less material from Theralin fixed bone tissue compared to formalin fixed, decalcified samples. Low-abundance proteins are a significant source of cancer-specific biomarkers32, and the improved sensitivity of protein detection described here will help to unlock the proteomic archive of bone tissue.
Improper formalin fixation can lead to artifacts in tissue morphology 16,33,34. For example, fixed tissue can shrink, causing empty spaces between formerly attached tissue areas, biomolecules can diffuse out of the fixed tissue 34, and pigments can form, interfering with the interpretation of IHC stains. In addition, formalin fixation can lead to a falsely elevated identification of DNA mutations due to the introduction of random base damage along templates 35. Here, we have identified an additional formalin fixation artifact that has significant implications for proteomic data quantification. We have shown that fixation time in formalin, but not Theralin, directly correlates with β-Actin levels. Formaldehyde cross-links biomolecules first at reactive sites composed of amines, purines and thiols, followed by the formation of methylene bridges that involve amides, asparagines and guanidine and tyrosine carbon rings36. The number of crosslinks increases with time and may take up to 30 days to finalize36. This has significant implications for proteins with repetitive motifs or proteins, such as β-Actin, that polymerize to form long filaments. Antibodies that are generated to specifically bind cross-linked protein epitopes will “see” more of said protein as fixation time and cross-linking increases. We believe that this effect is causing the correlation of β-Actin levels with formalin fixation time that we observed here. Antibody vendors typically do not share the exact procedure for antibody creation and the pre-processing or the sequence of the antigenic peptides used. It therefore becomes critical to ensure equal formalin fixation times for samples within a study if the downstream analysis includes relative protein quantification. However, in a clinical environment, this is very difficult to achieve. Theralin did not alter detectable protein levels with increased fixation time, which supports its use as an alternative fixative for formalin if the study goal includes protein quantification from clinical samples.
We also measured a significantly increased level of Doublecortin in formalin fixed versus Theralin fixed samples (Supplemental Fig. 1). At this time it is unclear whether both of the antibodies that were tested preferentially bind formalin cross-linked Doublecortin, which does not polymerize into long filaments and should therefore not benefit from longer formalin fixation times, or if Doublecortin is better preserved in formalin fixed samples. Future studies will have to address this question.
A significant percentage of primary breast tumors and their metastases show discordance in their estrogen, progesterone and HER2 receptor status37–40,18,41, with previously reported discordance rates of 7–54%, 20–54%, and 3–33%, respectively. Routinely determining the receptor status of bone metastases could significantly change patient management, as demonstrated in a pilot study, where 20% of breast cancer patients received altered care from their primary physician after including IHC/FISH diagnosis of bone metastases41. While regular IHC can be performed on bone tissue, FISH will not work after standard acid decalcification. Therefore, HER2 FISH, which is the current gold standard in HER2 receptor status determination, has to be done on non-decalcified core biopsies41. But, depending on the site of metastasis, this is generally not practicable. Another alternative is biopsy decalcification with EDTA, which has shown promising preservation of DNA for FISH in selected studies42,43. In fact, one previous study compared HER2 FISH with IHC in breast cancer bone metastasis samples that were decalcified with EDTA, finding high concordance between both methods44. However, a major disadvantage of decalcification with EDTA is its slow action, requiring several days to weeks with regular solution replacement, even when supporting the process by constant oscillation or ultrasonic baths43,44. The fixation technology described here enables rigorous, fast, and standardized clinical IHC and FISH assays to be routinely performed on bone tissue samples.
Interestingly, of the two osteosarcoma cases that were stained with MDM2/CEN, only the central low grade osteosarcoma with areas of progression into high grade showed positive staining. MDM2, which is frequently amplified in low grade, but not high grade osteosarcoma, is a sensitive and specific marker in combination with CDK4 for the subclass of high grade osteosarcomas that have progressed from low grade osteosarcomas45–47.
Enabling FISH analysis for bone tissue is of special concern for breast cancer patients, because bone is a primary metastatic site for breast cancer, regardless of the breast cancer subtype6. There is an unknown proportion of women who harbor HER2 positive bone metastasis but cannot receive efficacious anit-HER2 therapy because HER2 FISH will not work after standard acid decalcification and formalin fixation. We propose that using Theralin for bone tissue preservation meets this critical need because it unlocks decalcified bone tissue for FISH analysis while maintaining the performance of standard IHC. Processing bone tumor samples with Theralin or formalin showed comparable overall results for 125 IHC stains in a regular clinical diagnostics laboratory setting. This is in agreement with previous results, where fixation of soft tissue with Theralin maintains or enhances staining of key cancer diagnostic IHC antigens, including Ki-67, estrogen receptor alpha and progesterone receptor19. HER2 protein epitopes were preserved exceptionally well for IHC in Theralin fixed breast tumor samples19. Although the current data demonstrate that Theralin fixation enables FISH in bone tissue, this study set consisted primarily of primary bone tumors and therefore did not permit us to specifically evaluate HER2 FISH analysis in a breast metastasis setting. A future study will have to evaluate the concordance of HER2 FISH results obtained after using the fixation technology described here and standard buffered formalin, according to published ASCO and CAP guidelines48,49.
In conclusions, we have developed a tissue fixation chemistry, Theralin, that simultaneously fixes and decalcifies bone tissue, obviating the need for a separate decalcification step while improving tissue histomorphology for diagnosis. Theralin fixation significantly shortens the time necessary for tissue processing and eliminates the processing step that hinders FISH in bone. Thus, the use of Theralin enables routine FISH analysis in bone. Furthermore, Theralin greatly enhances the extractability of protein and DNA from bone samples, which significantly reduces the necessary tissue input necessary for downstream applications. In addition, Theralin overcomes a serious formalin fixation artifact, whereby the measured quantity of repetitive proteins such as β-Actin depends directly on fixation time, preventing comparison between tissues with different formalin fixation times. This has significant implications for antibody based proteomic methods that may depend on normalization by a “housekeeping” protein such as β-Actin.
A weakness of the present study is that a relatively low number of cases was distributed among many different types of bone cancer rather than a large sample size of any one individual type of cancer. We used this diverse sample set to test a range of diagnostic biomolecules and analysis technologies that are known to be impacted by bone processing methods. The data presented here provide strong justification that our fixative technology is of general utility by improving tissue histomorphology and phosphoprotein analysis, and enabling FISH. Based on this foundation, we can now apply this fixative in clinical trials for investigating the discordance between primary tumor and bone metastasis in tumor types with high prevalence of bone metastases (i.e. breast cancer), or assessing drug efficacy in bony tissue, as well as discovering biomarkers in bony tissue.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Jennifer Freeland, Sharmini Muralitharan, and Haiping Liu for their helpful guidance and discussions regarding FISH protocols.
Funding: This work was supported in part by George Mason University, Inova Fairfax Hospital, Rizzoli Orthopedic Institute, and two National Institutes of Health grants to LAL (R21CA125698–01A1 and R33CA157403–01) from the National Cancer Institute program “Innovations in cancer cample preparation”.
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
LAL and VE are inventors of the fixation technology and, as university employees, may receive patent royalties per university policies related to US patent 8,460,859 B2: Tissue preservation and fixation method.
REFERENCES
- 1.Mosialou I et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543, 385–390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee NK et al. Endocrine regulation of energy metabolism by the skeleton. Cell 130, 456–469 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferron M et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142, 296–308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhattacharyya N, Chong WH, Gafni RI & Collins MT Fibroblast growth factor 23: state of the field and future directions. Trends Endocrinol. Metab. TEM 23, 610–618 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mundy GR Metastasis to bone: causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2, 584–593 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Wu Q et al. Breast cancer subtypes predict the preferential site of distant metastases: a SEER based study. Oncotarget 8, 27990–27996 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morris RE & Benton RS Studies on demineralization of bone. I. The basic factors of demineralization. Am. J. Clin. Pathol 26, 579–595 (1956). [DOI] [PubMed] [Google Scholar]
- 8.Morris RE & Benton RS Studies on demineralization of bone. II. The effect of electrolytic technics in demineralization. Am. J. Clin. Pathol 26, 596–603 (1956). [DOI] [PubMed] [Google Scholar]
- 9.Morris RE & Benton RS Studies on demineralization of bone. III. The effect of ion exchange resins and versenate in demineralization. Am. J. Clin. Pathol 26, 771–777 (1956). [DOI] [PubMed] [Google Scholar]
- 10.Walsh L, Freemont AJ & Hoyland JA The effect of tissue decalcification on mRNA retention within bone for in-situ hybridization studies. Int. J. Exp. Pathol 74, 237–241 (1993). [PMC free article] [PubMed] [Google Scholar]
- 11.Alers JC, Krijtenburg PJ, Vissers KJ & van Dekken H Effect of bone decalcification procedures on DNA in situ hybridization and comparative genomic hybridization. EDTA is highly preferable to a routinely used acid decalcifier. J. Histochem. Cytochem. Off. J. Histochem. Soc 47, 703–710 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Bass BP, Engel KB, Greytak SR & Moore HM A review of preanalytical factors affecting molecular, protein, and morphological analysis of formalin-fixed, paraffin-embedded (FFPE) tissue: how well do you know your FFPE specimen? Arch. Pathol. Lab. Med 138, 1520–1530 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Case NM The use of a cation exchange resin in decalcification. Stain Technol 28, 155–158 (1953). [DOI] [PubMed] [Google Scholar]
- 14.Cleland TP & Vashishth D Bone protein extraction without demineralization using principles from hydroxyapatite chromatography. Anal. Biochem 472, 62–66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benton RS & Morris RE Studies on demineralization of bone. IV. Evaluation of morphology and staining characteristics of tissues after demineralization. Am. J. Clin. Pathol 26, 882–898 (1956). [DOI] [PubMed] [Google Scholar]
- 16.Bindhu P, Krishnapillai R, Thomas P & Jayanthi P Facts in artifacts. J. Oral Maxillofac. Pathol. JOMFP 17, 397–401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broxterman HJ & Georgopapadakou NH New cancer therapeutics: target-specific in, cytotoxics out? Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother 7, 79–87 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Pérez-Fidalgo JA et al. An evaluation of the impact of technical bias on the concordance rate between primary and recurrent tumors in breast cancer. Breast Edinb. Scotl 22, 974–979 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Mueller C et al. One-step preservation of phosphoproteins and tissue morphology at room temperature for diagnostic and research specimens. PloS One 6, e23780 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skinner RA, Hickmon SG, Lumpkin CK, Aronson J & Nicholas RW Decalcified Bone: Twenty Years of Successful Specimen Management. J. Histotechnol 20, 267–277 (1997). [Google Scholar]
- 21.Gallagher RI, Blakely SR, Liotta LA & Espina V Laser capture microdissection: Arcturus(XT) infrared capture and UV cutting methods. Methods Mol. Biol. Clifton NJ 823, 157–178 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Mueller C, Liotta LA & Espina V Reverse phase protein microarrays advance to use in clinical trials. Mol. Oncol 4, 461–481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiechi A et al. Improved data normalization methods for reverse phase protein microarray analysis of complex biological samples. BioTechniques 0, 1–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2012). [Google Scholar]
- 25.Becker K-F et al. Quantitative protein analysis from formalin-fixed tissues: implications for translational clinical research and nanoscale molecular diagnosis. J. Pathol 211, 370–378 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Espina V et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PloS One 5, e10240 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhattacharyya S, Byrum S, Siegel ER & Suva LJ Proteomic analysis of bone cancer: a review of current and future developments. Expert Rev. Proteomics 4, 371–378 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Kiernan. Formaldehyde, formalin, paraformaldehyde and gluteraldehyde: What they are and what they do. Microsc. Today 8, 8–12 (2000). [Google Scholar]
- 29.Becker K-F et al. Extraction of Phosphorylated Proteins from Formalin-Fixed Cancer Cells and Tissues. Open Pathol. J 2, 46–52 (2008). [Google Scholar]
- 30.Ostasiewicz P, Zielinska DF, Mann M & Wiśniewski JR Proteome, phosphoproteome, and N-glycoproteome are quantitatively preserved in formalin-fixed paraffin-embedded tissue and analyzable by high-resolution mass spectrometry. J. Proteome Res 9, 3688–3700 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Emmert-Buck MR et al. Laser capture microdissection. Science 274, 998–1001 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Byrum S, Montgomery CO, Nicholas RW & Suva LJ The promise of bone cancer proteomics. Ann. N. Y. Acad. Sci 1192, 222–229 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Chatterjee S Artefacts in histopathology. J. Oral Maxillofac. Pathol. JOMFP 18, S111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khan S, Tijare M, Jain M & Desai A Artifacts in Histopathology: A Potential Cause of Misinterpretation. Res. Rev. J. Dent. Sci 2, 23–31 (2014). [Google Scholar]
- 35.Quach N, Goodman MF & Shibata D In vitro mutation artifacts after formalin fixation and error prone translesion synthesis during PCR. BMC Clin. Pathol 4, 1 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thavarajah R, Mudimbaimannar VK, Elizabeth J, Rao UK & Ranganathan K Chemical and physical basics of routine formaldehyde fixation. J. Oral Maxillofac. Pathol. JOMFP 16, 400 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ibrahim MFK et al. Strategies for obtaining bone biopsy specimens from breast cancer patients – Past experience and future directions. J. Bone Oncol 5, 180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amir E et al. Discordance between receptor status in primary and metastatic breast cancer: an exploratory study of bone and bone marrow biopsies. Clin. Oncol. R. Coll. Radiol. G. B 20, 763–768 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Amir E et al. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 30, 587–592 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aurilio G et al. Discordant hormone receptor and human epidermal growth factor receptor 2 status in bone metastases compared to primary breast cancer. Acta Oncol. Stockh. Swed 52, 1649–1656 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Simmons C et al. Does confirmatory tumor biopsy alter the management of breast cancer patients with distant metastases? Ann. Oncol. Off. J. Eur. Soc. Med. Oncol 20, 1499–1504 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alers JC, Krijtenburg PJ, Vissers KJ & van Dekken H Effect of bone decalcification procedures on DNA in situ hybridization and comparative genomic hybridization. EDTA is highly preferable to a routinely used acid decalcifier. J. Histochem. Cytochem. Off. J. Histochem. Soc 47, 703–710 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Neat MJ, Moonim MT, Dunn RG, Geoghegan H & Foot NJ Fluorescence in situ hybridisation analysis of bone marrow trephine biopsy specimens; an additional tool in the diagnostic armoury. J. Clin. Pathol 66, 54–57 (2013). [DOI] [PubMed] [Google Scholar]
- 44.Zustin J et al. HER-2/neu analysis in breast cancer bone metastases. J. Clin. Pathol 62, 542–546 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Righi A et al. MDM2 and CDK4 expression in periosteal osteosarcoma. Hum. Pathol 46, 549–553 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Dujardin F et al. MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc 24, 624–637 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Yoshida A et al. MDM2 and CDK4 immunohistochemical coexpression in high-grade osteosarcoma: correlation with a dedifferentiated subtype. Am. J. Surg. Pathol 36, 423–431 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Wolff AC et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 25, 118–145 (2007). [DOI] [PubMed] [Google Scholar]
- 49.Hanna W et al. Updated recommendations from the Canadian National Consensus Meeting on HER2/neu testing in breast cancer. Curr. Oncol. Tor. Ont 14, 149–153 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.