

Abstract
The development of physical dependence and addiction disorders due to misuse of opioid analgesics is a major concern with pain therapeutics. We developed a mouse model of oxycodone exposure and subsequent withdrawal in the presence or absence of chronic neuropathic pain. Oxycodone withdrawal alone triggered robust gene expression adaptations in the nucleus accumbens, medial prefrontal cortex and ventral tegmental area, with numerous genes and pathways selectively affected by oxycodone withdrawal in mice with peripheral nerve injury. Pathway analysis predicted that histone deacetylase (HDAC) 1 is a top upstream regulator in opioid withdrawal in nucleus accumbens and medial prefrontal cortex. The novel HDAC1/HDAC2 inhibitor, Regenacy Brain Class I HDAC Inhibitor (RBC1HI), attenuated behavioral manifestations of oxycodone withdrawal, especially in mice with neuropathic pain. These findings suggest that inhibition of HDAC1/HDAC2 may provide an avenue for patients with chronic pain who are dependent on opioids to transition to non-opioid analgesics.
Synthetic opioids are used successfully to alleviate acute and postoperative pain1. By contrast, for chronic neuropathic pain conditions, opioid analgesics show limited efficacy and only alleviate a subset of symptoms2–6. Moreover, prolonged use of opioids leads to severe side effects, including hyperalgesia and physical dependence, and often transition to addiction1,7. The dopaminergic mesocorticolimbic brain circuitry is highly dysregulated under states of physical dependence and addiction8,9. The brain’s reward circuitry also plays a prominent role in the perception and processing of chronic pain symptoms in humans as well as in preclinical models2,10–13. While numerous studies have investigated the mechanisms underlying opioid actions in components of the reward circuitry1,14,15, much less is known about the lasting actions of opioids under chronic neuropathic pain states. Several human studies have documented changes in the activity of the nucleus accumbens (NAc) and other reward circuitry components that correlate with chronic pain states11,16. The prevalence of addiction and physical dependence to synthetic opioids is especially high among patients with chronic pain. Clinical studies document exacerbation of pain states among opioid-dependent chronic pain patients and report pain reduction upon detoxification from opioids17–19. Indeed, current treatments for opioid use disorders, such as buprenorphine/naloxone, may promote hyperalgesia in patients with chronic pain20. Furthermore, since many pain patients obtain opioids illicitly, there is risk for acute withdrawal if the buprenorphine/naloxone regimen is not carefully tapered20. Methadone substitution therapy requires frequent clinic visits, which is particularly difficult for patients with chronic pain19,20. Therefore, the identification of non-opioid medications that alleviate pain while attenuating withdrawal would be a major advance in the field.
Our earlier work demonstrated robust gene expression adaptations in the murine NAc and medial prefrontal cortex (mPFC) in response to both peripheral nerve injury21 and antidepressant drug treatment under nerve injury states22. Furthermore, interventions in gene expression or in the activity of brain reward subregions robustly affect both behavioral manifestations of peripheral nerve injury and the efficacy of therapeutic compounds13,22–25. However, to date there has been no investigation of how coincident chronic pain affects transcriptional responses to chronic opioid administration.
To better understand the transcriptomic effects of chronic opioid exposure and physical dependence under chronic pain states in the brain reward circuitry, we administered high doses of oxycodone for 2 weeks to mice with prolonged spared nerve injury (SNI)21,26 and sham controls, and, following 3 weeks of spontaneous withdrawal, we collected mPFC, NAc and ventral tegmental area (VTA) tissues for RNA-sequencing (RNA-seq) analysis. Overall, our study identifies adaptations in mouse reward circuits associated with oxycodone physical dependence and reveals that chronic neuropathic pain states affect sensory, affective and molecular signatures of opioid withdrawal. Guided by bioinformatic predictions from our RNA-seq analyses, we demonstrate that treatment with the HDAC1/HDAC2 inhibitor, Regenacy Brain Class I HDAC Inhibitor (RBC1HI), alleviates mechanical allodynia and prevents the expression of sensory and affective manifestations of oxycodone withdrawal, pointing to a new therapeutic approach for the management of chronic pain in opioid-dependent individuals.
Results
Oxycodone withdrawal produces thermal hyperalgesia and mechanical allodynia in spared nerve injury and sham groups of mice
We designed a new paradigm of oxycodone exposure to assess sensory and affective-like behaviors associated with spontaneous oxycodone withdrawal in mice with prolonged peripheral nerve injury and in sham controls (Fig. 1a). We first monitored sensory hypersensitivity in long-term SNI and sham groups of male mice with or without prolonged oxycodone exposure, followed by spontaneous drug withdrawal. Over 5 weeks, we assessed changes in body weight during oxycodone exposure and spontaneous withdrawal. No significant changes in weight were observed in long-term SNI (SNI-Sal) groups compared to sham-saline (Sham-Sal) controls. However, the weights of SNI and sham mice receiving chronic oxycodone injections (SNI-Oxy and Sham-Oxy, respectively) were significantly altered compared with Sham-Sal and SNI-Sal controls. Sham-Oxy and SNI-Oxy groups both showed decreased weights compared to Sham-Sal animals 2 d into oxycodone treatment. In addition, SNI-Oxy animals showed weight loss 4 d into withdrawal (Fig. 1b). Both SNI-Oxy and Sham-Oxy groups normalized their weight change by 17 d of drug withdrawal (Fig. 1b).
Fig. 1 |. Chronic oxycodone exposure and spontaneous oxycodone withdrawal produce weight loss and sensory deficits in long-term spared nerve injury and pain-free mice.
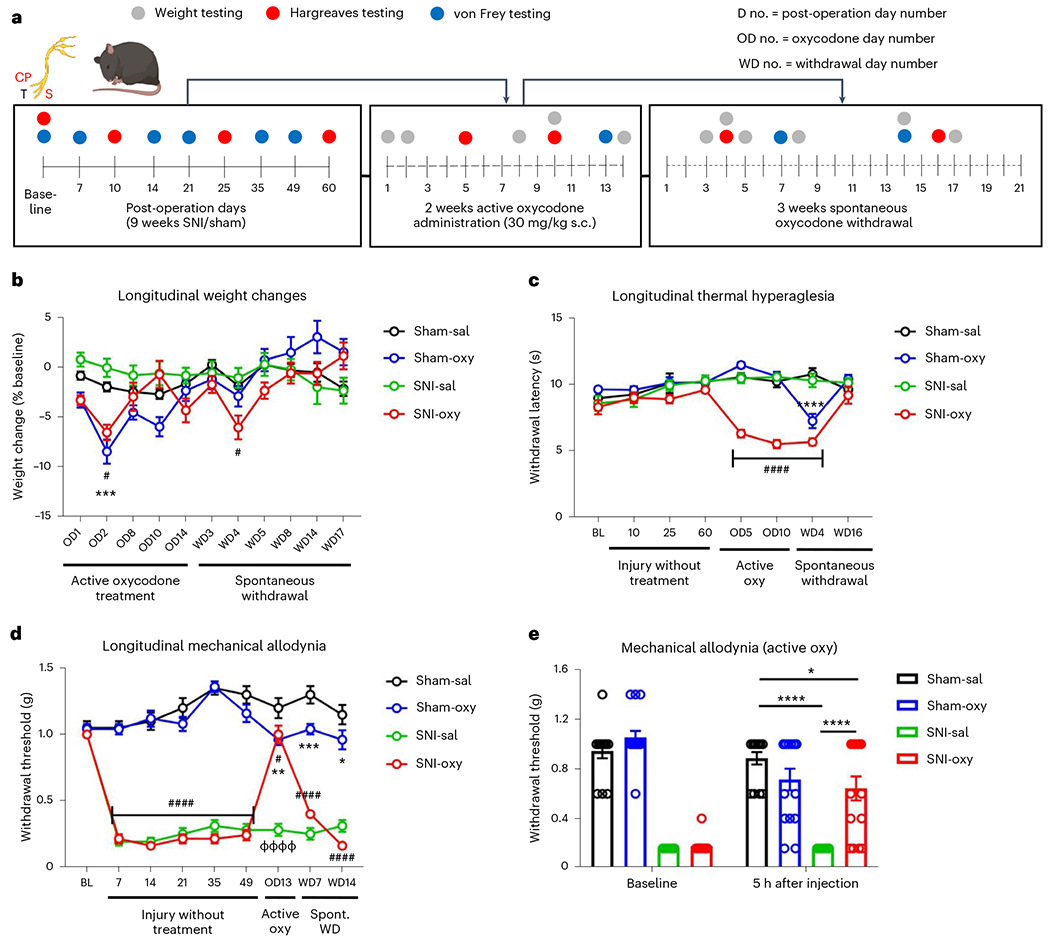
a, Schematic timeline depicting oxycodone exposure In male chronic SNI and sham mice. b, Weight was affected by oxycodone administration in both SNI and sham cohorts as compared with Sham-Sal-treated controls (Sham-Sal n = 11, Sham-Oxy n = 16; SNI-Sal n = 11, SNI-Oxy n = 14; repeated-measures two-way analysis of variance (ANOVA) interaction F30,480 = 6.653, P < 0.0001; Tukey’s multiple comparisons Sham-Oxy versus Sham-Sal OD2 q = 6.029, d.f. = 528, P = 0.0001; SNI-Oxy versus Sham-Sal OD2 q = 4.131, d.f. = 528, P = 0.019, WD4 q = 3.784, d.f. = 528, P = 0.0384). No significant weight change was observed between SNI controls (SNI-Sal) as compared to Sham-Sal. c, Oxycodone exposure produced significant and progressive thermal hyperalgesia in SNI mice (SNI-Oxy) at 5 d of oxycodone administration as compared to the Sham-Oxy group. Oxycodone-induced thermal hyperalgesia continued during spontaneous withdrawal in SNI-Oxy mice. Sham-Oxy mice displayed thermal hyperalgesia only during spontaneous withdrawal at early time points (WD4) (Sham-Sal n = 8, Sham-Oxy n = 10; SNI-Sal n = 8, SNI-Oxy n = 9; repeated-measures two-way ANOVA interaction F21,217 = 8.826, P < 0.0001; Tukey’s multiple comparisons. SNI-Oxy versus Sham-Sal OD5 q = 10.53, d.f. = 248, P < 0.0001, OD10 q = 11.64, d.f. = 248, P < 0.0001; Sham-Oxy versus Sham-Sal WD4 q = 8.943, d.f. = 248, P < 0.0001; SNI-Oxy versus Sham-Sal WD4 q = 12.63, d.f. = 248, P < 0.0001). d, Oxycodone-reduced mechanical thresholds in sham cohorts at 13 d of drug administration. During spontaneous withdrawal, Sham-Oxy mice displayed mild but significant mechanical allodynia up to 14 d after drug cessation as compared to Sham-Sal controls. SNI-Oxy mice showed significantly decreased mechanical allodynia during oxycodone administration (day 13) compared to all earlier time points under nerve injury, but thresholds returned to baseline levels during drug withdrawal (Sham-Sal n = 8, Sham-Oxy n = 10; SNI-Sal n = 8, SNI-Oxy n = 9; repeated-measures two-way ANOVA interaction F24,248 = 20.75, P < 0.0001; Tukey’s multiple comparisons, SNI-Oxy pretreatment days 7–49 P < 0.0001; Tukey’s multiple comparisons, OD13 SNI-Oxy versus Sham-Sal q = 4.07, d.f. = 279, P = 0.0223; SNI-Oxy versus SNI-Sal q = 14.65, d.f. = 279, P < 0.0001; Sham-Oxy versus Sham-Sal OD13 q = 5.003, d.f. = 279, P = 0.0027; Sham-Oxy versus Sham-Sal WD7 q = 5.42, d.f. = 279, P = 0.0009, WD14 q = 3.961, d.f. = 279, P = 0.0278). e, In a separate cohort, oxycodone alleviated SNI-induced mechanical allodynia 5 h after administration (13 d of daily treatment; Sham, Sal n = 14, Oxy, n = 15; SNI, Sal n = 14, Oxy n = 15; (F3,54 = 16.22, P < 0.0001; Sidak’s multiple comparisons, SNI-Sal versus Sham-Sal t = 8.717, d.f. = 108, P < 0.0001; SNI-Oxy versus SNI-Sal t = 5.897, d.f. = 108, P < 0.0001). Data indicate the mean ± s.e.m. Significance (P < 0.05) was calculated by means of two-way ANOVA with Tukey’s post hoc test for b–d and Sidak’s post hoc test for direct comparison of Sham-Oxy versus Sham-Sal (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001), SNI-Oxy versus Sham-Sal, (#P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001), SNI-Oxy versus SNI-Sal (ΦΦΦΦP < 0.0001). Oxy, oxycodone; Sal, saline; WD, withdrawal day; OD, oxycodone administration day.
We monitored thermal hypersensitivity using the Hargreaves assay. SNI-Oxy mice developed thermal hyperalgesia after 5 d of oxycodone injection, which was also observed on day 10 (Fig. 1c). Sham-Oxy groups showed no opioid-induced thermal hyperalgesia during the 2-week period of drug treatment; however, these animals displayed significant hyperalgesia at 4 d after the induction of spontaneous withdrawal (Fig. 1c). This effect was also observed with the SNI-Oxy group.
We next monitored the effect of chronic oxycodone administration on long-term mechanical allodynia associated with SNI. SNI reliably induced mechanical hypersensitivity through 49 d after surgery (Fig. 1d). We first monitored mechanical allodynia22,27 at 16 h after oxycodone injection on day 13 of drug treatment. At this time point after drug injection, SNI-Oxy mice showed significant, but partial, improvement from mechanical allodynia compared to pretreatment SNI thresholds, while the Sham-Oxy group demonstrated lower mechanical thresholds compared to all time points before Oxy treatment (Fig. 1d). During spontaneous withdrawal, we observed a persistent mechanical hypersensitivity in Sham-Oxy mice at 7 d and 14 d after oxycodone treatment discontinuation, whereas mice from the SNI-Oxy group returned to von Frey thresholds observed before drug treatment (Fig. 1d). We also observed significant, but partial, alleviation of mechanical allodynia at 13 d of oxycodone treatment in a separate SNI-Oxy group, when we tested mice 5 h after oxycodone injection (Fig. 1e). Overall, our findings suggest that chronic neuropathic pain states exacerbate symptoms of long-term oxycodone exposure, particularly during active treatment.
Oxycodone withdrawal produces deficits in social and emotional behavior
We next used a battery of behavioral paradigms to evaluate the effect of neuropathic pain states on emotional and motivational manifestations of chronic oxycodone administration and subsequent withdrawal (Fig. 2a). Peripheral nerve injury significantly reduced the percentage time spent on the bright side of the dark–light box 5 d after withdrawal, as it was lower in SNI-Sal compared to Sham-Sal mice and in SNI-Oxy compared to Sham-Oxy mice (Fig. 2b). In the marble-burying28 assay on day 9 after spontaneous oxycodone withdrawal, SNI-Oxy mice buried significantly fewer marbles than Sham-Oxy groups (Fig. 2c). Furthermore, as expected, SNI-Sal animals buried more marbles than their Sham-Sal counterparts. Interestingly, Sham-Oxy animals buried substantially more marbles than Sham-Sal animals, reinforcing the induction of anxiety-like states during spontaneous withdrawal alone. While at face value these findings would suggest that SNI-Oxy reduces anxiety levels, the combination of the two perturbations might also be interpreted as a reduced capacity to address environmental stressors, thus representing maladaptive indifference to these stressors. Voluntary running wheel activity29, which is considered a measure of anxiety and motivation, showed that Sham-Oxy animals ran significantly more than SNI-Oxy mice, suggesting high anxiety levels without nerve injury and decreased capacity with injury under oxycodone withdrawal (Fig. 2d). We observed a trend toward less running in SNI-Oxy compared to SNI-Sal mice, further suggesting reduced capacity in SNI-Oxy mice beyond injury-induced effects, and a trend toward more running in Sham-Oxy compared to Sham-Sal mice, suggesting withdrawal-induced anxiety states. We next assessed sociability using the two-choice preference model30 12 d after withdrawal. All groups showed no preference for either of the empty cages during the habituation phase of this test (Fig. 2e). In the sociability test, Sham-Sal and Sham-Oxy groups, as well as SNI-Sal mice, displayed preference for a social target, while SNI-Oxy mice showed no preference for a social target versus an empty cage, suggesting reduced sociability (Fig. 2f).
Fig. 2 |. Oxycodone withdrawal alters emotional and social behaviors.
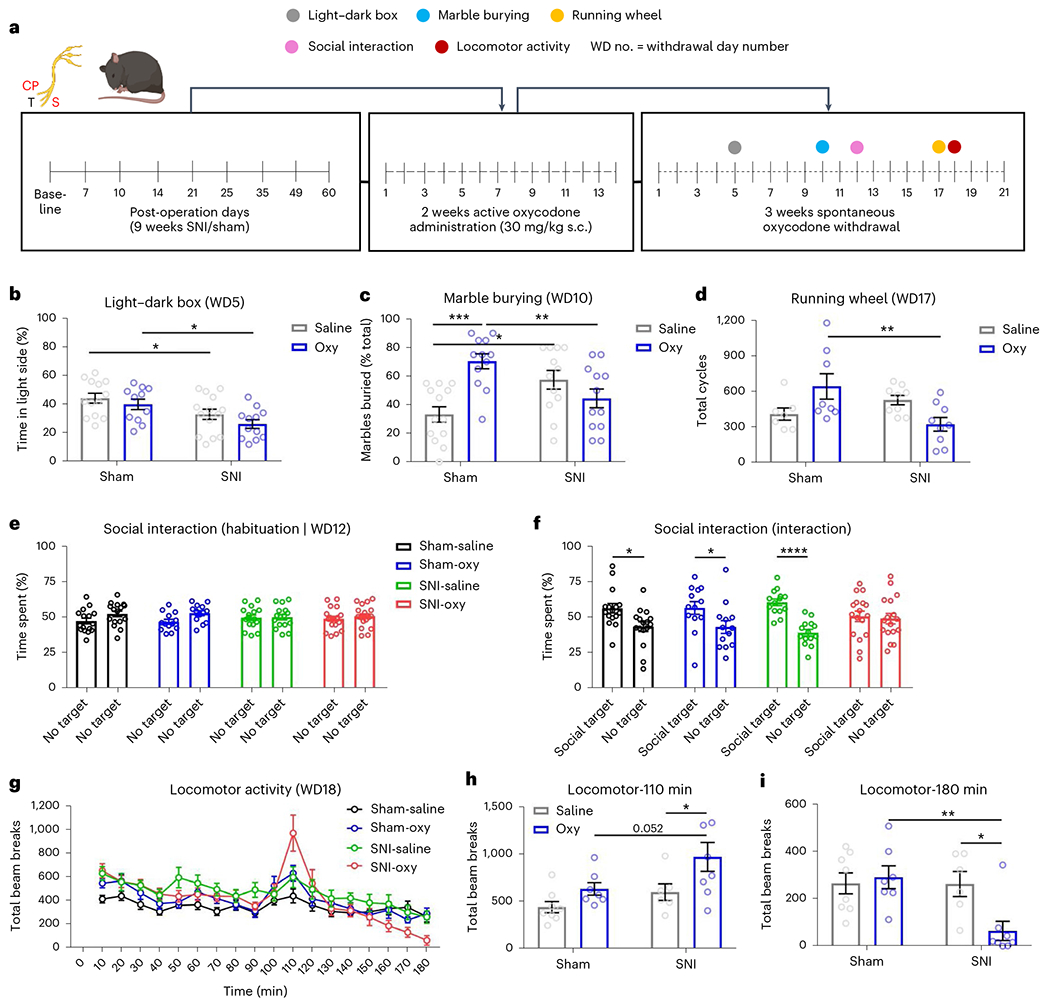
a, Schematic timeline depicting oxycodone exposure and behavioral assessment in male chronic SNI and sham mice. b, Five days after oxycodone withdrawal, anxiety-like behaviors were assessed using the light–dark assay. SNI-Oxy mice spent less time in the light chamber as compared to Sham-Sal controls. Oxycodone withdrawal decreased the time spent in the light chamber in the SNI-Oxy group compared to the Sham-Oxy group (Sham-Sal n = 13, Sham-Oxy n = 11; SNI-Sal n = 14, SNI-Oxy n = 12; two-way ANOVA interaction F1,46 = 0.1312, P = 0.7189; Sidak’s multiple comparisons SNI-Sal versus Sham-Sal t = 2.387, d.f. = 46, P = 0.0418; SNI-Oxy versus Sham-Oxy t = 2.696, d.f. = 46, P = 0.0195). c, In the marble-burying assay (day 10), Sham-Oxy mice and SNI-Sal mice buried more marbles as compared with Sham-Sal controls. The effect of oxycodone withdrawal was significant on the number of marbles buried between SNI-Oxy and Sham-Oxy mice. Sham-Oxy mice buried significantly more marbles compared to SNI-Oxy mice (Sham-Sal n = 13, Sham-Oxy n = 12; SNI-Sal n = 12, SNI-Oxy n = 12; two-way ANOVA; interaction, F1,45 = 17.89, P = 0.0001; Sidak’s multiple comparisons, SNI-Oxy versus Sham-Oxy t = 3.068, d.f. = 45, P = 0.0073; t = 2.912, d.f. = 45, P = 0.0111; SNI-Sal versus Sham-Sal t = 2.912, d.f. = 45, P = 0.0111; Sham-Oxy versus Sham-Sal t = 4.476, d.f. = 45, P = 0.0001). d, Running wheel performance was assessed at 17 d of oxycodone withdrawal. The effect of oxycodone on voluntary wheel running was significant between Sham-Oxy and SNI-Oxy groups of male mice (Sham-Sal n = 7, Sham-Oxy n = 8; SNI-Sal n = 9, SNI-Oxy n = 9; two-way ANOVA interaction F1,29 = 10.33, P = 0.0032; Sidak’s multiple comparisons, SNI-Oxy versus Sham-Oxy t = 3.38, d.f. = 29, P = 0.0042). e,f, Male mice were subjected to a sociability assay 12 d after oxycodone withdrawal. e, Mice from all cohorts showed no preference for either of the empty cages during the habituation phase as measured by the percentage of time spent with either object. f, During the sociability test, time spent by subject mice in the social target-containing field versus empty cage-containing side field was enhanced among all groups tested (Sham-Sal, Sham-Oxy and SNI-Sal) with the exception of the SNI-Oxy group (Sham-Sal n = 16, Sham-Oxy n = 13; SNI-Sal n = 16, SNI-Oxy n = 17; Sham two-way ANOVA interaction F1,56 = 0.01304, P = 0.9095; Sidak’s multiple comparisons, Sham-Sal t = 2.839, d.f. = 56, P = 0.0126; Sham-Oxy t = 2.331, d.f. = 56, P = 0.0462; SNI two-way ANOVA interaction, F1,58 = 9.51, P = 0.0031; Sidak’s multiple comparisons, SNI-Sal t = 4.489, d.f. = 58). g, At 18 d of oxycodone withdrawal, mice were habituated to the locomotor boxes for 80 min and then received a priming low dose of oxycodone (1 mg per kg body weight, s.c.). h,i, SNI-Oxy mice that were previously exposed to oxycodone demonstrated increased locomotor activity (Sham-Sal n = 8, Sham-Oxy n = 7; SNI-Sal n = 6, SNI-Oxy n = 8; two-way ANOVA interaction, F1,25 = 0.3945, P = 0.3945; Sidak’s multiple comparisons, SNI-Oxy versus SNI-Sal t = 2.486, d.f. = 25, P = 0.0395). Locomotor activity across conditions at 110 min (20 min after injection; h) and at 180 min (90 min after injection; i). Two-way ANOVA interaction, F1,25 = 5.842, P = 0.0233; Sidak’s multiple comparisons, SNI-Oxy versus SNI-Sal t = 2.959, d.f. = 25, P = 0.0133). Data represent the mean ± s.e.m. For two-way ANOVA multiple-comparisons testing between two groups, *P < 0.05 and **P < 0.01. Data represent the mean ± s.e.m.
Although we did not observe significant changes in the open field assay (Extended Data Fig. 1a), the elevated plus maze (EPM) assay showed an increase in the total time spent in the open arm in the SNI-Oxy group compared to the Sham-Oxy group (Extended Data Fig. 1b). No difference was observed between groups in the nesting assay (Extended Data Fig. 1c). Furthermore, Sham-Oxy, SNI-Sal and SNI-Oxy animals all displayed delayed latency to feed in a novel arena in the novelty-suppressed feeding assay31, with both oxycodone cohorts traveling a shorter distance than their saline counterparts (Extended Data Fig. 1d,e).
Finally, we show that the SNI-Oxy group was primed for locomotor activation in response to a low oxycodone dose (1 mg per kg body weight, subcutaneously (s.c.)) at 18 d of drug abstinence (Fig. 2g). Figure 2h shows locomotor activity at 110 min. This increased activity was followed by a low mobility phase only in the SNI-Oxy group, by minute 180 (Fig. 2i). Together, our findings reveal that chronic exposure to oxycodone in a prolonged nerve injury state leads to more severe physical and affective manifestations of drug withdrawal.
Oxycodone withdrawal in long-term spared nerve injury and sham states triggers broad transcriptomic patterns in the brain’s reward circuitry
We next sought to understand the transcriptional changes that correlate long-term peripheral nerve injury with oxycodone withdrawal in three brain regions implicated in chronic pain and opioid dependence: the NAc, mPFC and VTA. Tissues were collected from mice after 9 weeks of peripheral nerve injury, 14 d of oxycodone administration and 21 d of spontaneous withdrawal, and were then processed for RNA-seq21,32–34 (Fig. 3a). Differential expression (DE) analysis showed region-specific alterations in transcriptional profiles. The comparison of SNI-Oxy versus Sham-Sal groups resulted in 1,012 differentially expressed genes (DEGs) in the NAc, 1,116 genes in the mPFC and 533 genes in the VTA (Fig. 3b,e,h; nominal P < 0.05, log2fold change ≥ |0.5|). SNI-Sal triggered 1,457 DEGs in the NAc, 1,052 DEGs in the mPFC and 425 DEGs in the VTA as compared to Sham-Sal controls (Fig. 3b,e,h). The Sham-Oxy condition altered 2,609 genes in the NAc, 1,449 genes in the mPFC and 584 genes in the VTA as compared to Sham-Sal controls (Fig. 3b,e,h).
Fig. 3 |. Oxycodone withdrawal alters broad transcriptome patterns in brain reward regions of chronic neuropathic pain and pain-free mice.
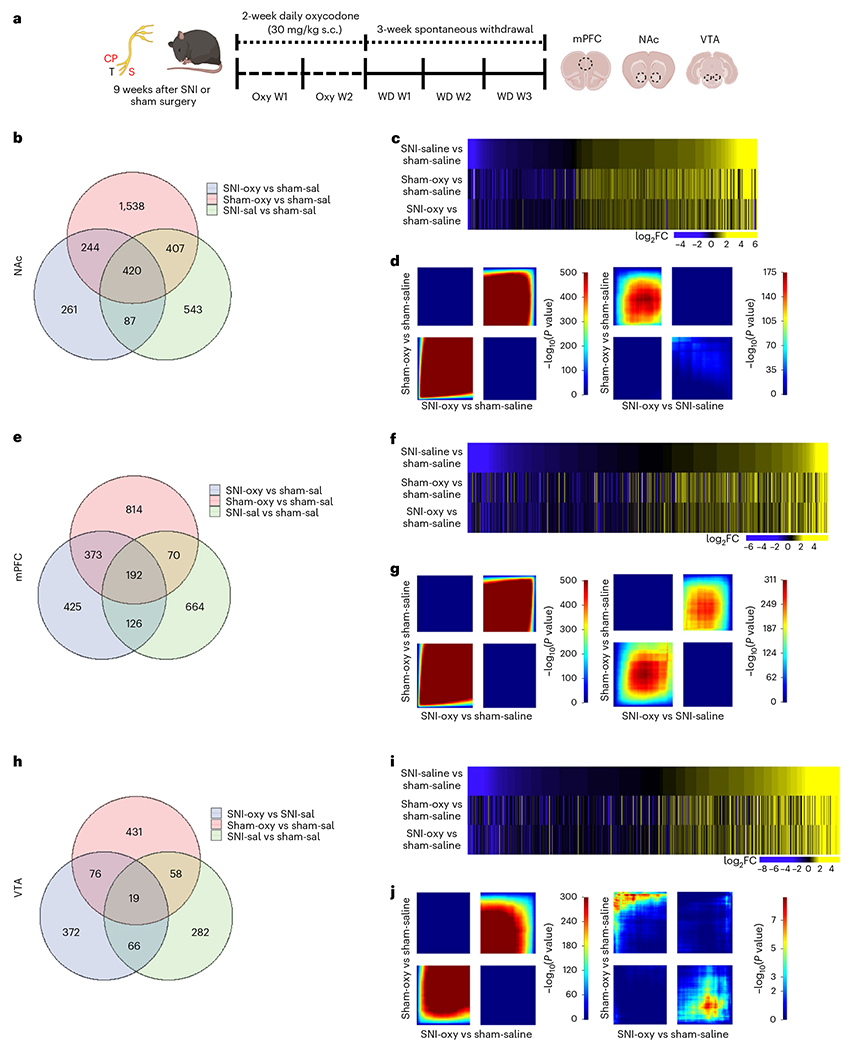
a, Schematic paradigm for tissue collection of brain regions for bulk RNA-seq studies from adult male C57BL/6 mice. b,e,h. Venn diagrams representing the number of DEGs altered by chronic pain states (SNI-Sal versus Sham-Sal), oxycodone withdrawal in sham (Sham-Oxy versus SNI-Sal) and oxycodone withdrawal under chronic nerve injury states (SNI-Oxy versus Sham-Sal). c,f,i. Representative union heat maps of DEGs in matched comparisons across the NAc, mPFC and VTA respectively; yellow indicates increasing log2 fold change of gene expression, and blue represents decreasing expression. d,g,j. Threshold-free comparison of DEGs by RRHO for the NAc, mPFC and VTA. Each pixel represents the overlap between the transcriptome of each comparison as noted, with the significance of overlap (−log10(P value)) of a hypergeometric test color coded. The lower-left quadrant includes co-upregulated genes, the upper-right quadrant includes co-downregulated genes, and the upper-left and lower-right quadrants include contra-regulated genes. Genes along each axis are sorted from most to least significantly regulated from the middle to outer corners.
To understand how various cell types contribute to these transcriptomic changes, we performed a deconvolution analysis of NAc RNA-seq data across all groups, as the NAc was the most transcriptionally dynamic region according to a single-cell RNA-seq dataset from Avey et al.35. This analysis revealed a significant reduction in the contribution of the neuron_1 cell subtype in Sham-Oxy mice compared to Sham-Sal mice, suggesting impaired neuronal function after spontaneous oxycodone withdrawal (Extended Data Fig. 2a). The SNI-Sal versus Sham-Sal comparison interestingly yielded a reduction in mural cell contribution in SNI-Sal mice, suggesting impaired vascular structure/function after prolonged peripheral nerve injury (Extended Data Fig. 2b). We also observed a trend toward a reduction in transcriptomic contributions from the neuron_1 and microglia subtypes in SNI-Oxy compared to Sham-Sal groups, but a trend toward upregulation in macrophage contribution, suggesting a combination of impaired neuronal and increased inflammatory cell function after withdrawal under a prolonged peripheral nerve injury state (Extended Data Fig. 2c). Overall, 420 DEGs (versus Sham-Sal) in the NAc, 192 DEGs in the mPFC and 19 DEGs in the VTA overlapped between Sham-Oxy, SNI-Sal and SNI-Oxy groups (Fig. 3b,e,h), suggesting a prominent role of the NAc in states of pain and opioid withdrawal with/without nerve injury. The highest number of DEGs was observed in the Sham-Oxy condition across all brain regions. Union heat maps sorted by log fold change, and compared with DEGs that were changed because of SNI (SNI-Sal versus Sham-Sal), showed a similar pattern of expression across Sham-Oxy vs Sham-Sal and SNI-Oxy vs SNI-Sal comparisons in the NAc, mPFC and VTA (Fig. 3c,f,i).
We next complemented these data analyses with a two-sided rank–rank hypergeometric overlap (RRHO) analysis to identify patterns and strengths of transcriptome-wide overlap in a threshold-free manner. RRHO analysis confirmed similar directional threshold-free regulation of genes between Sham-Oxy vs Sham-Sal and SNI-Oxy vs Sham-Sal comparisons (Fig. 3d,g,j) across all surveyed brain regions (NAc, mPFC and VTA). RRHO plots of the SNI-Oxy vs SNI-Sal comparison revealed an oppositely-regulated threshold-free transcriptional signature compared to the Sham-Oxy vs Sham-Sal condition in the NAc and VTA, while remaining similarly regulated in the mPFC (Fig. 3d,g,j). These data suggest that oxycodone withdrawal promotes broad transcriptional alterations under prolonged pain states that are unique for each brain region.
To better understand the molecular signature of DEGs affected by oxycodone withdrawal under SNI versus sham conditions, we performed Gene Ontology (GO) analysis for enriched biological processes and ingenuity pathway analysis (IPA). There was a significant DEG overlap between the SNI-Oxy versus SNI-Sal and Sham-Oxy versus Sham-Sal conditions in the NAc and mPFC (105 and 106, respectively; Fig. 4a,d). However, Oxy withdrawal caused substantially different transcriptomic profiles in nerve-injured groups compared to sham groups. Only 21 coexpressed DEGs between SNI-Oxy versus SNI-Sal and Sham-Oxy versus Sham-Sal were observed in the VTA (Fig. 4g). Figure 4a,d shows union heat maps of DEGs that are shared between the comparisons above, interestingly demonstrating opposite directional regulation of these genes in the NAc as opposed to unidirectional regulation in the mPFC. Figure 4g shows that shared DEGs between these conditions in the VTA did not have a clear directionality of regulation. Also, GO analysis of DEGs conserved between these treatment conditions in the NAc revealed an association with neuronal morphology, cAMP signaling and alcohol abuse, whereas in the mPFC, glutamate reception was a top hit (Fig. 4a,d). Of note, this analysis, which highlights the consistent effects of oxycodone withdrawal across injury states, was limited due to the few DEGs available when comparing these conditions. In the VTA (Fig. 4g), ADH1 and LRG1 appeared as conserved targets for predicted drug treatments.
Fig. 4 |. Predicted biological processes and transcriptional regulators altered by oxycodone withdrawal with chronic neuropathic pain and pain-free states.
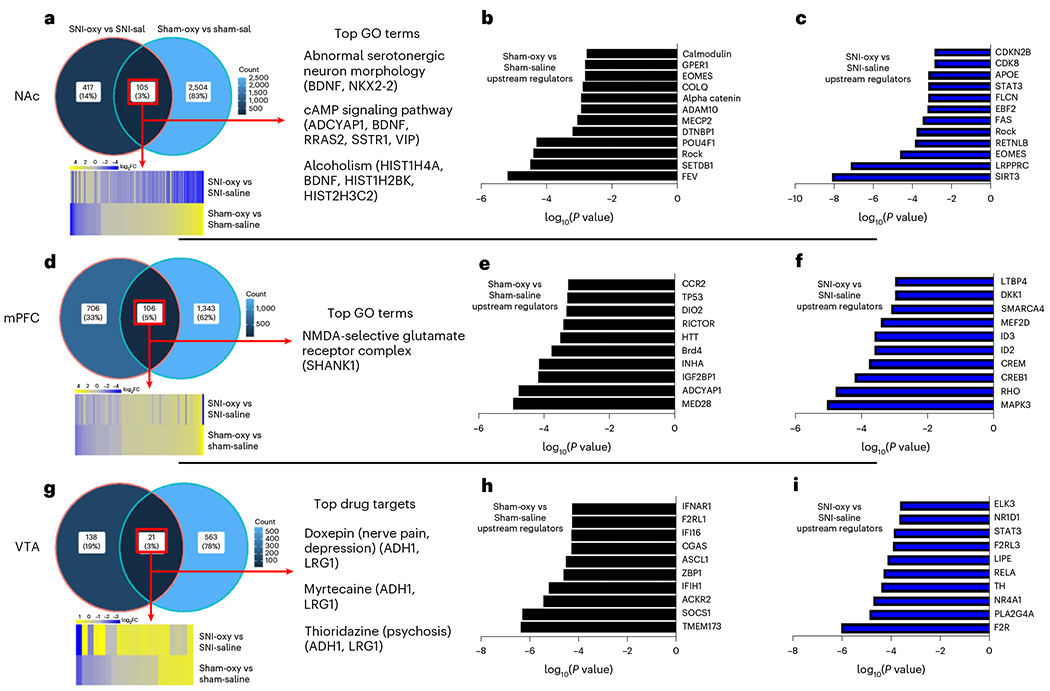
a,d,g, Venn diagrams depicting overlap of genes altered by oxycodone withdrawal under SNI-Oxy versus SNI-Sal and in Sham-Oxy versus Sham-Sal conditions in the NAc (a), mPFC (d) and VTA (g). These graphs also show respective union heat maps of commonly regulated DEGs between SNI-Oxy versus SNI-Sal and Sham-Oxy versus Sham-Sal conditions in the NAc, mPFC and VTA, as well as associated GO terms or predicted drug targets. b,c,e,f,h,i, Top predicted URs of Sham-Oxy versus Sham-Sal and SNI-Oxy versus SNI-Sal DEGs in the NAc (b and c), mPFC (e and f) and VTA (h and i). Top predicted URs of SNI-Oxy versus Sham-Sal control DEGs in NAc, mPFC and VTA.
Top predicted upstream regulators (URs) of transcriptional signatures were distinctly altered in several brain regions by oxycodone withdrawal in neuropathic conditions as well as in sham states. Within the NAc, FEV, SETDB1, DTNBP1 and COLQ were predicted URs for the Sham-Oxy versus Sham-Sal comparison (Fig. 4b), whereas SIRT3, LRPPRC and EOMES were predicted URs in SNI-Oxy versus SNI-Sal comparisons (Fig. 4c). The lack of overlap between top predicted URs emphasizes the distinct transcriptomic effects of oxycodone in nerve-injured versus uninjured conditions. However, URs from both conditions are implicated in epigenetic/transcriptional maintenance of the neuronal life cycle, such as FEV, SETDB1 and SIRT3. In the mPFC, ADCYAP1 and HTT were predicted URs in the Sham-Oxy versus Sham-Sal condition (Fig. 4e), whereas MAPK3, CREB1 and MEF2D (key regulators of activity in adult neurons) were predicted URs in the SNI-Oxy versus SNI-Sal comparison (Fig. 4f). Lastly, in the VTA, SOCS1 and CGAS were predicted URs in the Sham-Oxy versus Sham-Sal comparison (Fig. 4h), suggesting the engagement of interferon-related pathways by oxycodone withdrawal alone, whereas in the SNI-Oxy versus SNI-Sal comparison, RELA, STAT3 and ELK3 (which play a role in neuronal survival) were predicted URs (Fig. 4i).
We next performed an IPA canonical pathway analysis for the SNI-Oxy versus SNI-Sal comparison in the NAc, mPFC and VTA to better assess the molecular mechanisms affected by DEGs associated with withdrawal states under neuropathic pain. As seen in Table 1, conserved pathways across these brain regions included CREB signaling in neurons, G-protein-coupled receptor signaling, circadian rhythm signaling and osteoarthritis pathways.
Table 1 |.
Comparison of ingenuity pathway analysis top canonical pathways in mPFC, NAc and VTA tissues from SNI-Oxy and SNI-Sal animals
| Pathway | Brain region | –log(P value) | z-score | DEGs underlying pathway |
|---|---|---|---|---|
| CREB signaling in neurons | NAc | 3.61 | −2.4 | ADCY1, ADGRA1, ADGRD1, ADGRG6, ADRA2A, ATF2, CACNA1G, CYSLTR2, FGFR2, FPR3, GNB1L, GPR179, GPRC5D, HTR5A, LGR5, NMBR, NPFFR2, OXGR1, PTGFR, RRAS2, SSTR1, SSTR3 |
| mPFC | 2.04 | 0.471 | ADCY10, ADGRF4, ADGRL4, ADRB1, CACNG7, CREB5, F2RL3, FGFR2, GNAS, GNAT1, GNAT2, GNB1L, GNRHR, GPR182, GPR27, GPR3, GPR39, GPR6, GRPR, OPRM1, POLR2E, TBXA2R | |
| Hepatic fibrosis/hepatic stellate cell activation | NAc | 3 | – | ACTA2, CCN2, COL18A1, COL19A1, COL4A5, COL6A6, COL8A1, FGFR2, IL1RL2, TNFRSF1A |
| VTA | 3.42 | – | COL6A3, COL9A1, ICAM1, IL1RL2, MYH8, MYO1A | |
| Role of osteoclasts in rheumatoid arthritis signaling pathway | NAc | 2.92 | −1.155 | ADAM33, ATF2, COL18A1, COL19A1, COL4A5, COL6A6, COL8A1, FCGR2B, FRZB, IL1RL2, RRAS2, SFRP1, TNFRSF1A |
| VTA | 2.39 | −1.633 | CALCR, COL6A3, COL9A1, FOS, IL1RL2, IL7 | |
| G-protein-coupled receptor signaling | NAc | 2.77 | −2.558 | ADCY1, ADGRA1, ADGRD1, ADGRG6, ADRA2A, ATF2, CYSLTR2, FICD, FPR3, GNB1L, GPR179, GPRC5D, HTR5A, LGR5, NMBR, NPFFR2, OXGR1, PTGFR, RRAS2, SSTR1, SSTR3, TULP2 |
| mPFC | 2.43 | −1.177 | ADCY10, ADGRF4, ADGRL4, ADRB1, CNGA4, CREB5, DUSP1, EYA2, F2RL3, GNAS, GNAT1, GNAT2, GNB1L, GNRHR, GPR182, GPR27, GPR3, GPR39, GPR6, GRPR, LATS2, MAP2K3, OPRM1, PDE3A, SMPDL3B, TBXA2R | |
| VTA | 1.67 | −1.414 | BDKRB2, CALCR, FOS, GLP1R, HCAR1, MEF2C, PDE3A, RGS14 | |
| Regulation of cellular mechanics by calpain protease | NAc | 2.56 | – | CAPN11, CDK1, ITGA9, ITGAD, ITGAE, RRAS2 |
| VTA | 2.03 | – | ACTN2, CAPN11, ITGAD | |
| White adipose tissue browning pathway | NAc | 2.21 | −2.646 | ADCY1, ATF2, BDNF, CACNA1G, DIO2, FGFR2, PPARG |
| mPFC | 3.18 | 0 | ADCY10, BDNF, CACNG7, CEBPB, CREB5, FGFR2, GNAS, GUCY1A2, RXRG, THRA | |
| Wound healing signaling pathway | NAc | 2.18 | −0.632 | ACTA2, COL18A1, COL19A1, COL4A5, COL6A6, COL8A1, FGFR2, IL1RL2, RRAS2, TNFRSF1A |
| VTA | 2.1 | −1.342 | COL6A3, COL9A1, FOS, IL1RL2, LAMC2 | |
| Circadian rhythm signaling | NAc | 2 | – | ADCY1, ADCYAP1, ATF2, BDNF, CACNA1G, GNB1L, GUCY2G, NGF, RRAS2, VIP |
| mPFC | 1.95 | – | ADCY10, ADRB1, BDNF, CACNG7, CREB5, GNAS, GNB1L, GRPR, GUCY1A2, MAP2K3, MYC, RPS6KA5 | |
| Osteoarthritis pathway | NAc | 1.91 | 0 | ATF2, FRZB, IL1RL2, ITGA9, ITGAD, ITGAE, PPARG, RUNX2, TNFRSF1A |
| VTA | 2.97 | 0 | IL1RL2, ITGAD, MEF2C, PRKAG3, PTGS2, TCF7L2 | |
| Pulmonary fibrosis idiopathic signaling pathway | NAc | 1.85 | −0.905 | ACTA2, ATF2, CCN2, COL18A1, COL19A1, COL4A5, COL6A6, COL8A1, FGFR2, JARID2, RRAS2 |
| VTA | 1.66 | −1.342 | COL6A3, COL9A1, EGR1, FOS, TCF7L2 | |
| ILK signaling | mPFC | 2 | 0 | CDH1, CREB5, FLNC, LEF1, MYC, MYH4, MYO18B, MYO1H, RHOC, RPS6KA5 |
| VTA | 3.34 | −0.447 | ACTN2, FOS, KRT18, MYH8, MYO1A, PTGS2 |
DEGs underlying each pathway are bolded if they are conserved across brain regions within the same pathway. Statistics were generated in IPA.
Oxycodone withdrawal induces distinct transcriptional regulation in the NAc, mPFC and VTA in pain-free versus neuropathic pain states
Despite differences in explicit DEGs, an RRHO analysis strategy enabled us to look at overall concordance or lack thereof in gene expression in a threshold-free manner. As seen in Fig. 5a, when comparing Sham-Oxy versus Sham-Sal transcriptomic regulation across regions, the NAc and mPFC demonstrated high concordance–or similar directionality–of expression for the same genes. However, concordance was not observed when comparing the NAc or mPFC to VTA, suggesting different responses to oxycodone withdrawal across the mesolimbic system. Interestingly, when comparing SNI-Oxy to SNI-Sal transcriptomics across brain regions, the NAc and mPFC instead displayed mild counter-regulation of the same genes. The NAc and VTA increased in concordance, whereas the mPFC and VTA displayed more robust counter-regulation (Fig. 5b). Altogether, this analysis demonstrates highly similar responses between the NAc and mPFC during withdrawal in an injury-free state, yet a stronger transcriptomic relationship between the NAc/mPFC and VTA during withdrawal in injured states.
Fig. 5 |. Transcriptomic effects of oxycodone withdrawal are differentially expressed across reward-related brain regions with chronic SNI and Sham states.
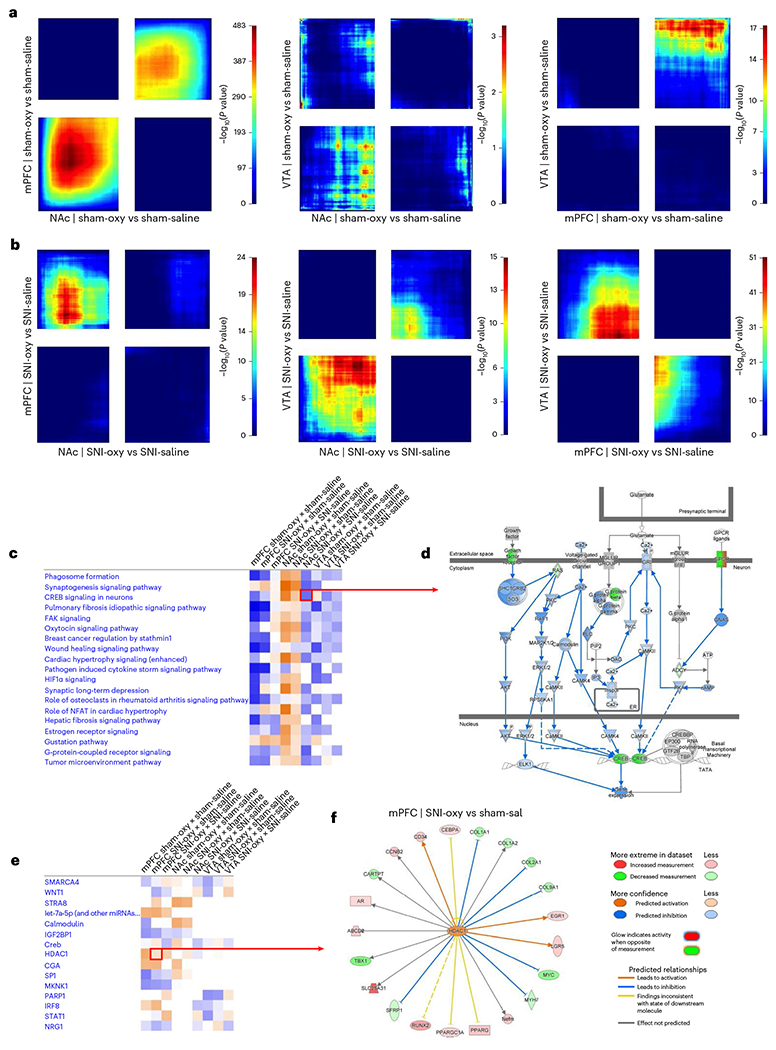
a,b, RRHO threshold-free comparisons of DEGs between the NAc, mPFC and VTA for oxycodone withdrawal with SNI and sham groups of adult male mice. Each pixel represents the overlap between the transcriptome of each comparison as noted, with the significance of overlap (−log10(P value)) of a hypergeometric test color coded. The lower-left quadrant includes co-upregulated genes, the upper-right quadrant includes co-downregulated genes, and the upper-left and lower-right quadrants include contra-regulated genes. Genes along each axis are sorted from most to least significantly regulated from the middle to outer corners. c, Top canonical pathways commonly regulated between Sham-Oxy versus Sham-Sal, SNI-Oxy versus Sham-Sal and SNI-Oxy versus SNI-Sal comparisons in the mPFC, NAc and VTA. d, Representation of significantly downregulated CREB signaling in neurons pathway in the NAc SNI-Oxy versus SNI-Sal condition. e, UR activity predictions across the aforementioned conditions/regions between oxycodone withdrawal under SNI and sham states. f, The genes predicted to be regulated by HDAC1 (UR) in the mPFC within the SNI-Oxy to Sham-Sal comparison.
We next identified canonical pathways that are altered across brain regions for the Sham-Oxy versus Sham-Sal, SNI-Oxy versus Sham-Sal and SNI-Oxy versus SNI-Sal comparisons (Fig. 5c). Interestingly, in the mPFC and NAc, most pathways that were implicated in the Sham-Oxy versus Sham-Sal and SNI-Oxy versus Sham-Sal conditions were either much more mildly associated with or counter-regulated in the SNI-Oxy versus SNI-Sal condition, which highlights the unique transcriptomic signature of oxycodone under nerve injury. One such pathway is CREB signaling in neurons, which is uniquely downregulated in the NAc only in the SNI-Oxy versus SNI-Sal comparison and is crucial for the regulation of activity in adult neurons. When this pathway is expanded (Fig. 5d), inhibition of several cytoplasmic regulators of CREB activity was predicted to regulate and further contribute to the observed downregulation of CREB and G-protein signaling-related transcript isoforms. However, several top canonical pathways were conserved yet directionally counter-regulated between the mPFC/VTA and NAc under Sham-Oxy versus Sham-Sal and SNI-Oxy versus Sham-Sal comparisons. These included the synaptogenesis signaling pathway, CREB signaling in neurons, estrogen receptor signaling and G-protein-coupled receptor signaling.
We next identified top predicted URs across the aforementioned comparisons in the mPFC, NAc and VTA. Several previously well-defined transcriptional regulators were identified, including CREB1, TGFB1, BDNF and NF-κB. However, in searching for a transcriptional regulator that has been less well defined in the mesocorticolimbic system under neuropathic or withdrawal states, our UR analysis pointed to HDAC1. HDAC1 was not predicted to be altered in the VTA; it was predicted to be upregulated across comparisons in the mPFC and upregulated in NAc by the SNI-Oxy state (Fig. 5e). In the mPFC, SNI-Oxy versus Sham-Sal comparisons suggest an increase in HDAC1 activity, with transcripts associated with the extracellular matrix (Col1A1, Col2a1 and Col9a1) and transcriptional regulation (Myc and Egr1) undergoing gene expression changes that are concordant with published findings36–38 (Fig. 5f). In the NAc of Sham-Oxy mice, we observed differential expression of several genes implicated in drug dependence, addiction and pain, such as Mef2c, Bdnf and Sgk1 (refs. 39–42) all of which are HDAC1 targets, based on IPA UR predictions.
We validated the protein expression of HDAC1 in cells of the mPFC and NAc (Extended Data Fig. 3a,b), followed by RNAscope in situ hybridization to demonstrate Hdac1 transcript colocalization with common neuronal and microglial transcripts (Extended Data Fig. 3c–f).
A novel HDAC1/HDAC2 inhibitor reverses mechanical hypersensitivity and thermal hyperalgesia associated with chronic oxycodone exposure
As we identified the class I HDAC1 as a predicted UR of oxycodone withdrawal under injured and uninjured states in the NAc and mPFC, we hypothesized that inhibition of the deacetylase activity of HDAC1 might alleviate associated behavioral abnormalities. Class I HDACs comprise HDAC1, HDAC2 and HDAC3, and the evolutionarily more distant HDAC8 (ref. 43). HDAC1, HDAC2 and HDAC3 function as components of corepressor complexes that widely regulate gene transcription, while the role of the more distantly related HDAC8 in transcription is not clear. Of particular interest for the isoform-selective pharmacological interrogation of HDAC1–3 are ortho-aminoanilide-derived small-molecule inhibitors44. The novel inhibitor RBC1HI (Extended Data Fig. 4a) was identified in enzymatic assays performed with recombinant proteins as a potent inhibitor that showed a dose-dependent inhibition of class I HDACs, with highest specificity for HDAC1, followed by HDAC2, free HDAC3 and HDAC3 bound to NCOR2 in enzymatic assays performed with recombinant proteins (Extended Data Fig. 4b). However, because HDAC isoform selectivity depends in part on the binding of HDACs to other corepressor complex proteins, the true isoform specificity in any specific cell type is less well understood45. A recent study using the prototype HDAC1/HDAC2-selective benzamide-derived inhibitor Cpd-60, which potently targets free HDAC3, demonstrated that HDAC3 can associate with CoREST into a catalytically inactive complex following ligand binding45. We therefore evaluated the inhibitory activity of RBC1HI toward free HDAC3 and found only a modest increase in inhibitory activity, retaining high isoform selectivity over HDAC1 and HDAC2 (Extended Data Fig. 4b). Upon systemic administration, RBC1HI was rapidly cleared from the plasma but has a longer-lasting presence in brain parenchyma (Extended Data Fig. 4c,d), rendering the compound better suited for long-term studies than other class I HDAC inhibitors such as entinostat46 (MS-275). Injection of RBC1HI (3 mg per kg body weight, intraperitoneally (i.p.)) did not promote rewarding effects in the conditioned placed preference assay, unlike morphine (6 mg per kg body weight, s.c.; Extended Data Fig. 4e). Furthermore, RBC1HI did not affect ambulatory locomotor activity (Extended Data Fig. 4f).
Daily treatment with RBC1HI (3 mg per kg body weight, i.p.), initiated immediately after SNI/Sham surgery to assess the therapeutic effects of RBC1HI on acute nerve injury (Fig. 6a), partially reversed mechanical allodynia in male SNI mice (Fig. 6b). In a separate cohort, concurrent daily treatment with RBC1HI (3 mg per kg body weight, i.p.) and oxycodone prevented the development of thermal hyperalgesia associated with chronic oxycodone exposure in male SNI mice (Fig. 6c). It also prevented the development of thermal hyperalgesia during spontaneous withdrawal from oxycodone in both sham and SNI groups (Fig. 6c).
Fig. 6 |. Pharmacological inhibition of HDAC1/HDAC2 using RBC1HI ameliorates sensory hypersensitivity signs of opioid withdrawal.
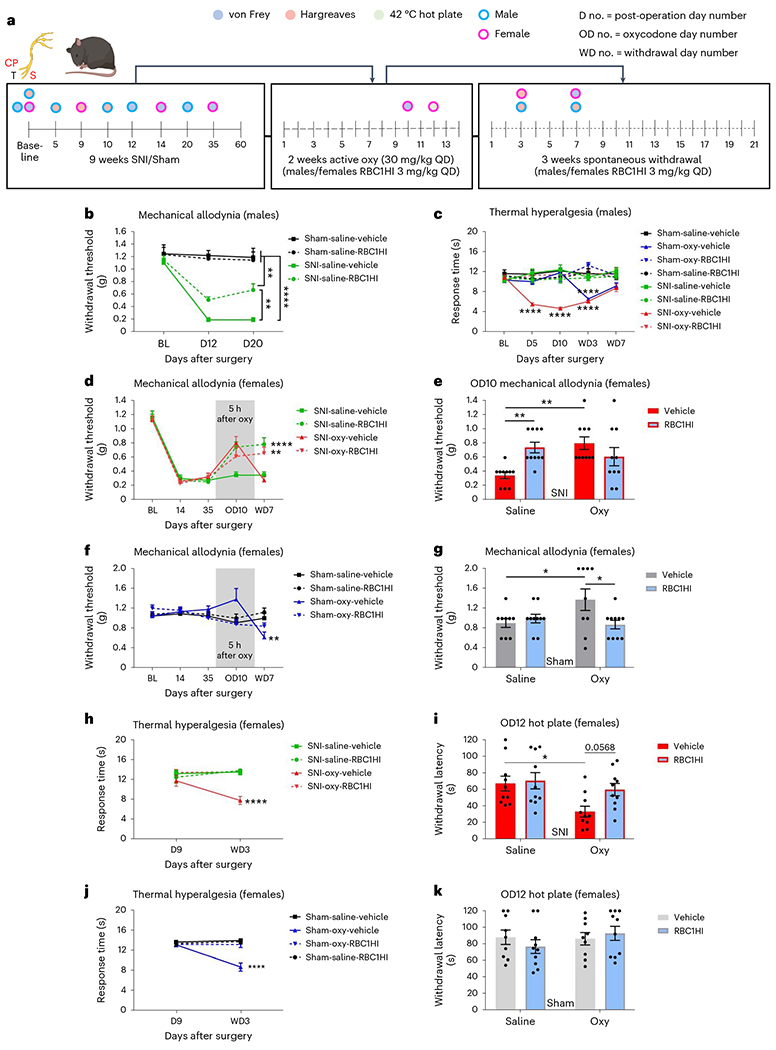
a, Schematic timeline of experimental design. b, RBC1HI promoted partial recovery from mechanical allodynia in groups of male mice with long-term SNI (Sham-Sal-Veh n = 7, Sham-Sal-RBC1HI n = 9; SNI-Sal-Veh n = 7, SNI-Sal-RBC1HI n = 10; repeated-measures two-way ANOVA interaction F6,58 = 5.858, P < 0.0001; Tukey’s multiple comparisons, day 20 SNI-Sal-Veh versus SNI-Sal-RBC1HI q = 5.12, d.f. = 87, P = 0.0027). c, Chronic treatment with 3 mg per kg body weight, RBC1HI prevented the development of oxycodone-induced hyperalgesia in male SNI groups exposed to chronic oxycodone. During spontaneous oxycodone withdrawal, inhibition HDAC1/HDAC2 prevented the induction of thermal hyperalgesia in both SNI and sham mice (Sham-Sal-Veh n = 7, Sham-Sal-RBC1HI n = 9; Sham-Oxy-Veh n = 8, Sham-Oxy-RBC1HI n = 10; SNI-Sal-Veh n = 7, SNI-Sal-RBC1HI n = 10; SNI-Oxy-Veh n = 8, SNI-Oxy-RBC1HI n = 7; repeated-measures two-way ANOVA interaction F28,232 = 5.01, P < 0.0001; Tukey’s multiple comparisons day 10 SNI-Oxy-Veh versus SNI-Oxy-RBC1HI q = 10.17, d.f. = 290, P < 0.0001; WD3 Sham-Oxy-Veh versus Sham-Oxy-RBC1HI q = 10.96, d.f. = 290, P < 0.0001; WD3 SNI-Oxy-Veh versus SNI-Oxy-RBC1HI q = 8.625, d.f. = 290, P < 0.0001). d,e, RBC1HI alleviated SNI-only and SNI-Oxy withdrawal-induced mechanical hypersensitivity on WD7. Five hours after Oxy administration on day 10, RBC1HI alleviated mechanical hypersensitivity in Sal-treated mice, but it did not affect the anti-allodynic response to Oxy, as Veh and RCB1H1 groups showed similar von Frey responses (SNI-Sal-Veh n = 10, SNI-Sal-RBC1HI n = 10, SNI-Oxy-Veh n = 10, SNI-Oxy-RBC1HI n = 10; repeated-measures two-way ANOVA interaction F12,144 = 4.531, P < 0.0001; Tukey’s multiple comparisons WD7 SNI-Oxy-RBC1HI versus SNI-Oxy-Veh q = 5.551, d.f. = 180, P = 0.0007; OD10 two-way ANOVA interaction F1,36 = 5.269, P = 0.0025; Sidak’s multiple comparisons SNI-Sal-Veh versus SNI-Sal-RBC1HI t = 3.112, d.f. = 36, P = 0.0072; SNI-Sal-Veh versus SNI-Oxy-Veh t = 3.589, d.f. = 36, P = 0.002). f,g, In sham mice, RCB1H1 prevented the analgesic effects of Oxy on mechanical thresholds and prevented the development of withdrawal-induced mechanical allodynia (Sham-Sal-Veh n = 9, Sham-Sal-RBC1HI n = 10, Sham-Oxy-Veh n = 9, Sham-Oxy-RBC1HI n = 10; two-way ANOVA interaction F1,34 = 5.269, P = 0.028; Sidak’s multiple comparisons OD10 Sham-Sal-Veh versus Sham-Oxy-Veh t = 2.517, d.f. = 34, P = 0.0332; OD10 Sham-Oxy-Veh versus Sham-Oxy-RBC1HI t = 2.754, d.f. = 34, P = 0.0187). h, RBC1HI pretreatment in this group of female mice also prevented the induction of thermal hyperalgesia after SNI with or without withdrawal in the Hargreaves assay (SNI-Sal-Veh n = 10, SNI-Sal-RBC1HI n = 10, SNI-Oxy-Veh n = 10, SNI-Oxy-RBC1HI n = 10; repeated-measures two-way ANOVA interaction F3,36 = 4.573, P = 0.0082; WD3 Tukey’s multiple comparisons SNI-Oxy-Veh versus SNI-Oxy-RBC1HI q = 7.742, d.f. = 72, P < 0.0001). i, When the same female Veh and RBC1HI SNI groups were tested in a 42 °C hot plate during active Oxy administration, RBC1HI ameliorated thermal hypersensitivity seen in SNI-Oxy mice (two-way ANOVA interaction F1,36 = 1.974, P = 0.1686; Sidak’s multiple comparisons OD12 SNI-Sal-Veh versus SNI-Oxy-Veh t = 2.899, d.f. = 36, P = 0.0126; OD12 SNI-Oxy-Veh versus SNI-Oxy-RBC1HI t = 2.277, d.f. = 36, P = 0.0568).j, RBC1HI effectively alleviated withdrawal-induced thermal hyperalgesia in Sham-Oxy animals (Sham-Sal-Veh n = 9, Sham-Sal-RBC1HI n = 10, Sham-Oxy-Veh n = 9, Sham-Oxy-RBC1HI n = 10; repeated-measures two-way ANOVA interaction F3,34 = 11.81, P < 0.0001; Tukey’s multiple comparisons Sham-Oxy-Veh versus Sham-Oxy-RBC1HI q = 9.016, d.f. = 68, P < 0.0001). k, There were no differences between Sham conditions in the 42 °C hot plate assay during active Oxy administration. Data indicate the mean ± s.e.m. Significance was calculated by means of two-way ANOVA with Bonferroni’s post hoc test; *P < 0.05, ***P < 0.001 and ****P < 0.0001. QD, once a day; Veh, vehicle.
We also assessed whether HDAC1/HDAC2 inhibition also had therapeutic-like actions in female mice. We initiated RBC1HI treatment at the same time as oxycodone administration. Indeed, RBC1HII exhibited anti-allodynic effects in SNI-Sal female mice 5 h after oxycodone administration (Fig. 6d,e) and also prevented mechanical allodynia by day 7 after spontaneous withdrawal (Fig. 6d). RBC1HI had no effects on mechanical thresholds in female Sham-Sal mice, yet interestingly it mitigated analgesia observed in Sham-Oxy females 5 h after administration (Fig. 6f,g). Similarly to our observations in male mice, administration of RBC1HI with oxycodone in female mice prevented the development of thermal hypersensitivity by day 3 after the onset of spontaneous withdrawal (Fig. 6h). We did not observe thermal hyperalgesia at 16 h after oxycodone injection in SNI-Oxy females in the Hargreaves assay. Therefore, we also assessed thermal hypersensitivity in a 42 °C hot plate assay and observed lower hot plate thresholds at day 12 of active oxycodone administration in SNI-Oxy animals; this effect seemed to be reversed by RBC1HI, although this effect did not reach statistical significance (Fig. 6i). RBC1HI also alleviated withdrawal-induced thermal hyperalgesia in Sham-Oxy female animals (Fig. 6j). We did not observe any thermal hypersensitivity in Sham-Oxy female mice during active oxycodone administration (Fig. 6k).
HDAC1/HDAC2 inhibition reversed emotional behavioral abnormalities associated with spontaneous oxycodone withdrawal
We next investigated the effect of HDAC1/HDAC2 inhibition on emotional behaviors after spontaneous oxycodone withdrawal in both SNI and sham male mice. Mice received RBC1HI once daily for 5 weeks concurrently with oxycodone exposure. We assessed various behaviors at time points matching experiments from Fig. 2. The treatment paradigm is depicted in Fig. 7a. We first assessed the effect of RBC1HI treatment on deficits in sociability and social novelty recognition in male groups of mice after 12 d of withdrawal. RBC1HI did not affect spatial exploration during the habituation phase of the paradigm (Fig. 7b and Extended Data Fig. 5a). RBC1HI treatment reversed the deficit in sociability observed in the SNI-Oxy group without affecting sociability in Sham-Sal or Sham-Oxy animals (Fig. 7c and Extended Data Fig. 5b). Interestingly, RBC1HI not only reversed a lack of social novelty recognition in SNI-Oxy mice under a withdrawal state, but also actually increased the time the mice spent with the novel mouse (Fig. 7d).
Fig. 7 |. RBC1HI treatment reverses affective symptoms of oxycodone withdrawal.
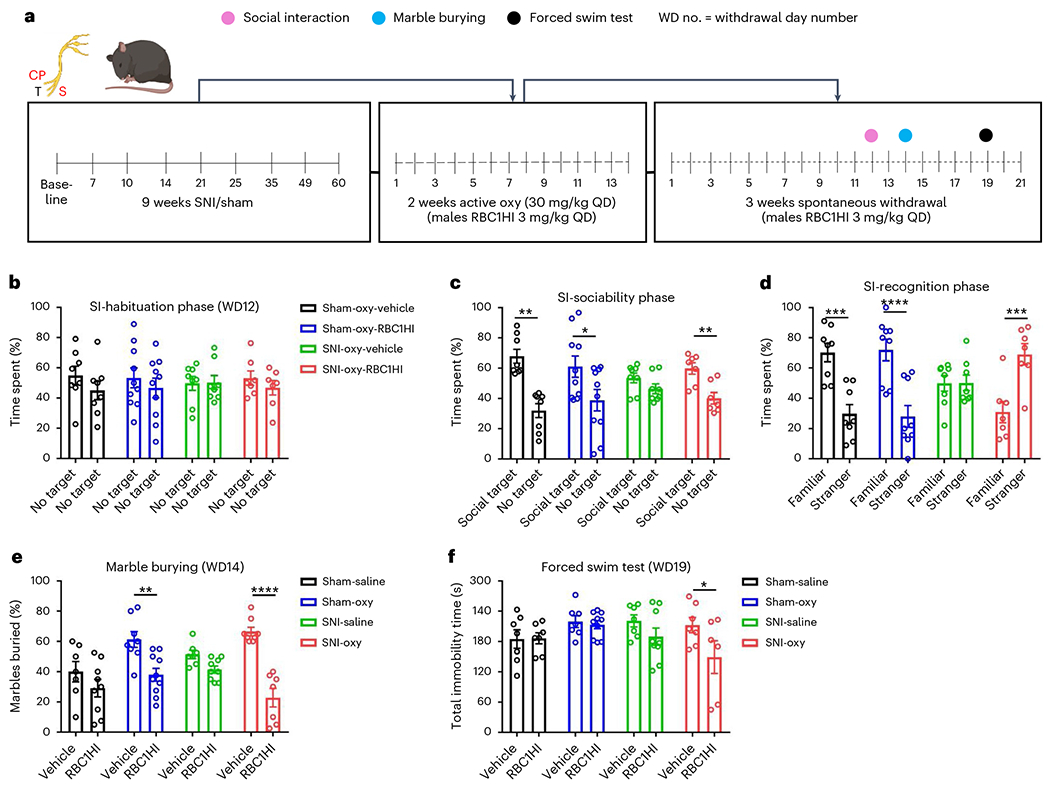
a, Schematic timeline of experimental design. b, Mice showed no preference for either empty cage during habituation for the social interaction assay (Sham-Oxy-Veh n = 8, Sham-Oxy-RBC1HI n = 10; SNI-Oxy-Veh n = 8, SNI-Oxy-RBC1HI n = 7; same for all phases). c, Sociability test: data are shown as the percentage of time spent by subject mice in the social target-containing field and empty cage-containing field. All groups of mice showed preference for the social target, except for the SNI-Oxy group (SNI two-way ANOVA interaction F1,26 = 3.209, P = 0.0849; Sidak’s multiple comparisons SNI-Oxy-RBC1HI social target versus no target t = 3.88, d.f. = 26, P = 0.0013). d, Social novelty recognition: data are shown as the percentage of time spent by subject mice in the stranger 1-containing field and stranger 2-containing field (SNI two-way ANOVA interaction F1,26 = 9.833, P = 0.0042; Sidak’s multiple comparisons SNI-Oxy-RBC1HI familiar versus stranger, t = 4.331, d.f. = 26, P = 0.0004). e, Treatment with RBC1HI decreased anxiety-like behaviors in the marble-burying assay 2 weeks after oxycodone withdrawal in both SNI and sham groups of male and Sham-Oxy female mice (Sham-Sal-Veh n = 7, Sham-Sal-RBC1HI n = 9; Sham-Oxy-Veh n = 8, Sham-Oxy-RBC1HI n = 10; SNI-Sal-Veh n = 7, SNI-Sal-RBC1HI n = 9; SNI-Oxy-Veh n = 8, SNI-Oxy-RBC1HI n = 7; SNI two-way ANOVA interaction F1,27 = 21.1, P < 0.0001; Sidak’s multiple comparisons SNI-Oxy-RBC1HI versus SNI-Oxy-Veh t = 8.324, d.f. = 27, P < 0.0001; Sham two-way ANOVA interaction F1,30 = 1.303, P = 0.2627; Sidak’s multiple comparisons Sham-Oxy-RBC1HI versus Sham-Oxy-Veh t = 3.119, d.f. = 30, P = 0.008). f, RBC1HI treatment reduced immobility time in SNI-Oxy mice (Sham-Sal-Veh n = 7, Sham-Sal-RBC1HI n = 7; Sham-Oxy-Veh n = 7, Sham-Oxy-RBC1HI n = 10; SNI-Sal-Veh n = 7, SNI-Sal-RBC1HI n = 9; SNI-Oxy-Veh n = 7, SNI-Oxy-RBC1HI n = 6; two-way ANOVA drug treatment F1,52 = 4.787, P = 0.0332; Sidak’s multiple comparisons SNI-Oxy-Veh versus SNI-Oxy-RBC1HI t = 2.627, d.f. = 52, P = 0.0444). Data indicate the mean ± s.e.m. Significance was calculated by means of two-way ANOVA with Bonferroni’s post hoc test; *P < 0.05, ***P < 0.001 and ****P < 0.0001.
In the marble-burying assay, RBC1HI treatment reversed the increased marble-burying in Sham-Oxy and SNI-Oxy mice (Fig. 7e), as well as in Sham-Oxy female mice (Extended Data Fig. 5c). In the forced swim assay47, RBC1HI pretreatment reduced immobility time in male SNI-Oxy mice on day 19 of withdrawal (Fig. 7f). Overall, our findings suggest that RBC1HI treatment efficiently alleviates sensory and affective signs of oxycodone withdrawal under chronic pain states.
Discussion
By establishing a new paradigm of prolonged oxycodone exposure, we effectively captured sensory, emotional and motivational abnormalities that characterize chronic opioid exposure and spontaneous withdrawal under neuropathic pain and pain-free states. We show that chronic oxycodone administration and withdrawal under neuropathic pain states promote maladaptive sensory and affective symptoms that are more severe than those observed in sham mice receiving the same oxycodone regimen. Bioinformatic analyses revealed unique transcriptional signatures in reward-related brain regions (NAc, mPFC and VTA) in response to long-term neuropathic pain and oxycodone withdrawal. Despite differences in gene expression adaptations, we identified several common biological pathways and URs associated with oxycodone withdrawal in both SNI and sham mice. Our study also demonstrates that inhibition of HDAC1/HDAC2 prevents behavioral deficits observed with oxycodone withdrawal especially under a state of prolonged neuropathic pain. No tolerance to the anti-allodynic and anti-hyperalgesic effects of RBC1HI was observed, as mice responded to the drug following several weeks of treatments (Fig. 6).
Within our paradigm, we captured the breadth of side effects associated with spontaneous withdrawal from prolonged exposure to opioids48–50. Previous studies in humans have also demonstrated adverse outcomes in neuropathic patients treated with chronic oxycodone6,51–53. In line with these studies, we found exacerbated behavioral deficits with opioid dependence under long-term neuropathic pain states. We showed that chronic oxycodone exposure produced opioid-induced hyperalgesia that persisted up to 7 d after discontinuation of drug treatment in both SNI and sham mice. Notably, chronic oxycodone treatment in SNI mice produced thermal hyperalgesia, which was observed as early as day 5 of treatment. Recent studies have documented that heroin and oxycodone withdrawal in rodents is manifested by deficits in emotional and social behaviors48,54. Although these studies used different treatment regimens and age groups, they both demonstrated the robust effects of opioid withdrawal on mouse behavior. In our study, we observed the emergence of emotional deficits at earlier time points in SNI mice. In assays measuring motivational or anxiety states, several changes were observed only in SNI-Oxy groups. For example, at early time points of oxycodone withdrawal, impaired sociability during withdrawal occurred only in long-term SNI mice. Relative to oxycodone withdrawal alone, the combination of withdrawal and SNI promoted anxiety-like behavior in the light–dark test55 but appeared to cause a loss of motivation to react to environmental stressors, as reflected by reduced marble burying and voluntary wheel running. Future studies applying operant tasks and conditioned place aversion protocols will further test how oxycodone affects motivational states in SNI and sham conditions.
The oxycodone regimen promoted sensory deficits over the 3-week withdrawal period in females as well, although other behaviors, such as oxycodone-induced hyperalgesia (16 h after injection) were only observed in male mice. We speculate that a different oxycodone dose or timing of behavioral assessment may be required to induce hyperalgesia in female mice. Notably, we were not able to collect data on sociability upon oxycodone withdrawal, because female mice showed a strong bias toward one side during the habituation phase of the behavioral tests. Furthermore, different drug regimens and assay time points may be required for studies with female mice, which we are currently exploring. Given the robust sex differences reported for pain and addiction mechanisms54,56,57, future studies will also involve modifications of the existing paradigms for a more extensive understanding of transcriptomic mechanisms in female cohorts of mice. The current work utilized bulk tissue sequencing analysis, which was sufficient to depict gene expression signatures among various treatments, but this methodology does not provide insight into cell-type-specific differential gene expression. Future work using single-nucleus RNA-seq may complement these findings and highlight neuronal and microglial subpopulations of interest.
While the molecular mechanisms underlying the actions of heroin and morphine in brain circuits mediating physical dependence and addiction have been investigated48,58–60, the mechanisms underlying oxycodone actions and the effect of chronic pain states on opioid responses are poorly understood. We have identified differences in transcriptional signatures in the NAc, mPFC and VTA after oxycodone withdrawal between chronic neuropathic and pain-free states. This differential pattern of expression reflects the diverse response of brain regions mediating sensory and affective signs of oxycodone withdrawal between neuropathic and pain-free conditions (Fig. 5). Previous studies highlighted key differences in oxycodone-induced gene expression in adult versus adolescent mice and the long-lasting effects of oxycodone on dopamine receptor expression across the NAc and VTA61,62.
Studies in humans have shown maladaptive increases in activity within brain reward pathway components in patients with chronic pain2,12,16. Animal studies have shown important roles of the NAc and VTA in altered motivational and emotional states under chronic pain13,21,63. The NAc is connected to several brain structures implicated in nociception, including the PFC64 and amygdalar nuclei2,24. Evidence from recent animal studies indicates that chronic pain states mainly affect the function of the indirect dopamine pathway, which primarily expresses dopamine D2 receptors65. Studies have also documented the importance of amygdala–NAc projections as well as mPFC–NAc projections in modulating chronic pain states24. We previously showed that a prolonged SNI states promote sensory and affective manifestations of chronic pain and induce differential expression of several genes and intracellular pathways implicated in mood disorders and nociceptive processing in the mPFC, NAc and periaqueductal gray21. Notably, several of these canonical pathways are also identified in the current study to be regulated by long-term SNI, such as G-protein-coupled receptor signaling, cAMP-mediated signaling and pregnenolone biosynthesis. Our findings also show that oxycodone exposure and withdrawal trigger gene signature patterns in the NAc and mPFC that are similar to those observed under SNI states. As expected, several genes associated with sensory hypersensitivity, pain perception and pain modulation appear as DEGs in these two studies. These genes include Bdnf, Capn11 and Tph2, among others. Likewise, we found opposite gene regulation between SNI and sham groups of mice with oxycodone withdrawal in genes known to be implicated in depression, anxiety and pain-like conditions.
Based on bioinformatic predictions, the histone modifier HDAC1, an epigenetic regulator with known actions in addiction66 and nociceptive processing67–69, plays a significant role in modulating gene expression associated with oxycodone withdrawal. The observation that systemic inhibition of class I HDACs effectively prevents sensory hyperalgesia and ameliorates emotional manifestations of oxycodone withdrawal may guide drug discovery efforts toward the development of compounds that can help patients with chronic pain who use prescription opioids transition to safer non-opioid medications. Studies causally link HDAC inhibitors to ‘epigenetic priming’70, suggesting that the drugs act in brain regions that are primed by a previous stressor such as chronic pain. We predict that class I HDAC inhibitors might be as efficacious for managing withdrawal from other opioid analgesics (for example, morphine and fentanyl), a subject that warrants future investigation.
Future work should also investigate the effect of class I HDACs or more selective HDAC1 inhibitors on the reinforcing actions of opioids and determine their potential for the treatment of addiction disorders. Because HDAC1 inhibitors67 and class I HDAC inhibitors attenuate mechanical hypersensitivity, it will be important to delineate the peripheral and central mechanisms by which they ameliorate sensory and affective signs of chronic pain. We expect that these inhibitors act at several sites within the nociceptive pathway, and at several regions that mediate the actions of opioids.
A major concern in pain therapeutics involves managing pain in patients who are dependent on or addicted to opioid analgesics, and this necessitates the development of novel treatment approaches that can be safely used in opioid-exposed patients. Indeed, several clinical studies document that pain patients who are dependent on opioids respond poorly to non-opioid analgesics71–74. This study may help to identify novel genes and pathways that can be targeted to prevent the development of physical dependence or opioid withdrawal in patients receiving long-term opioid treatment. Furthermore, inhibition of class I HDACs, and downstream pathways may provide a novel way to manage sensory hypersensitivity associated with chronic neuropathic pain conditions in opioid-naive as well as in opioid-dependent patients.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41593-023-01350-3.
Methods
Animals
Male and female C57BL/6J mice (8–9 weeks old) were obtained from Jackson Laboratories and maintained on a 12-h light–dark cycle with ad libitum access to food and water. All experiments were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee at Mount Sinai and of the Society for Neuroscience. All behavioral testing occurred during the animal’s light cycle. Experimenters were blinded to treatment groups, and the order of testing was counterbalanced during behavioral experiments. Different cohorts of animals were used for various behavioral assays and for RNA-seq studies. Behavioral analysis was performed in more than one cohort, except for locomotor assays.
Drug preparation
Oxycodone HCl (Sigma-Aldrich) was diluted in 0.9% sterile saline. RBC1HI (Regency Pharmaceuticals) was dissolved in 10% dimethyl-sulfoxide and 90%–5% dextrose in water, which was used as vehicle. Oxycodone was administered s.c. once a day at 30 mg per kg body weight for 2 weeks. RBC1HI was administered i.p. once a day at 3 mg per kg body weight. The 3-mg per kg body weight dose was selected as the lowest dose that suppressed SNI and oxycodone withdrawal-related behaviors in pilot studies.
Spared nerve injury surgery
SNI was performed on the left sciatic nerve, as described previously21,22,26. Briefly, using a stereomicroscope, a skin and muscle incision of the left hindlimb at mid-thigh level was performed to reach the sciatic nerve. The common peroneal and sural nerves were carefully ligated with 6.0-gauge silk sutures (Johnson & Johnson International) and transected, and 1–2-mm sections of these nerves were removed, while the tibial nerve was left intact. Skin and muscle were then closed with 4.0-gauge silk sutures (Johnson&Johnson International). The same procedure was followed in sham operated mice, but the nerves were left untouched.
Oxycodone spontaneous withdrawal protocol
To monitor oxycodone withdrawal in long-term neuropathic pain and pain-free states, we used a new oxycodone exposure paradigm. Mice received SNI or sham surgery and 9 weeks later were injected daily for 14 d with 30 mg per kg body weight oxycodone. Following this treatment, mice were allowed to spontaneously withdraw for 21 d. Sensory signs of spontaneous withdrawal were monitored starting the day after the last oxycodone injection. We monitored thermal and mechanical hyperalgesia during oxycodone injections and at acute time points of drug withdrawal. Locomotor activity, sociability, marble burying, voluntary running wheel and light–dark box activity were monitored during spontaneous withdrawal. For HDAC inhibitor studies, mice were injected with saline or RBC1HI (3 mg per kg body weight, i.p.) at the same time as oxycodone administration, throughout the period of oxycodone administration and up to 21 d after cessation of opioid treatment. Behaviors assessed after RBC1HI administration included von Frey and Hargreaves assays, marble burying, locomotor activity and sociability and social interaction.
von Frey assay
For the assessment of mechanical allodynia, we used von Frey filaments26,27 with ascending forces expressed in grams (Stoetling). Each filament was applied five times in a row against the ipsilateral hindpaw, with all mice receiving a filament application before returning for the next application to the first mouse. Hindpaw withdrawal or licking was marked as a positive allodynia response. A positive response in three of five repetitive stimuli was defined as the allodynia threshold.
Hargreaves test for thermal hyperalgesia
Mice were placed in Plexiglas boxes on top of a glass surface (IITC Life Science), and the latency to withdraw the injured hindpaw (left) was measured after a high-intensity heat beam (40%) was applied to the mid-plantar area27 (IITC Life Science). Two measurements were obtained with a 10-min interval, and the average was defined as the thermal nociceptive threshold. An intensity level of 40 and a cutoff time of 15 s was used to avoid potential tissue damage. Hindpaw withdrawal or licking was marked as a positive allodynia response.
Hot plate assay
A 42 °C hot plate was used to assess thermal hypersensitivity to a non-noxious stimulus. Briefly, an animal was placed on a hot plate in a plastic cylindrical enclosure. A cutoff time of 120 s was used, and the latency to respond was recorded upon seeing a positive response. This was defined as a hindpaw shake/lick or ajump.
Locomotor activity assay
Locomotor testing for the oxycodone priming experiment was performed using a beam break apparatus (Med Associates). Animals were allowed to habituate to the environment for 90 min, followed by an injection of either oxycodone (1 mg per kg body weight, s.c.) or vehicle. Animals were then monitored for another 90 min. Total beam breaks were taken for every animal in 10-min intervals.
Marble burying
The marble-burying test was conducted under red light conditions as previously described28. This assay was performed at the beginning of the second withdrawal week (day 9). After 1 h of acclimation to the testing room, mice were placed in a standard hamster cage filled with 15 cm of corn-cob bedding and topped with 20 glass marbles. After 30 min, the mice were removed and the number of marbles fully or partially buried (60% buried) was counted by two blinded observers and the percentage of marbles buried was calculated. Marbles that were covered by more than 60% bedding were counted as buried.
Voluntary wheel running
We used a wireless running wheel activity monitoring system (Low-Profile Wireless Running Wheel for Mouse, Med Associates). Mice were habituated for 2 d in their home cage with the running wheel apparatus29. On testing days, each mouse was monitored for 1 h. Mice that ran <100 cycles per hour were excluded from the study. The activity was calculated as the total number of revolutions during the testing period.
Dark–light chamber preference
An automated Med Associates place conditioning apparatus with equal-sized compartments was used for monitoring preference for a bright light versus a dark compartment. One side of the conditioned place preference chamber is illuminated by the overhead light located on the lid of the conditioned place preference apparatus. The other side is covered by black lid covers. We used identical mesh floors for both sides. Mice were placed in the central compartment and allowed to explore the apparatus for 5 min. Data were calculated as the percentage of time spent in each compartment and presented as the mean ± s.e.m.
Sociability and social novelty recognition test
Mice are placed in a two-choice field with an empty grid cage placed in each outer corner of the closed quadrants. For habituation, a subject mouse was allowed to freely explore the U-shaped two-choice field for 8 min. For the sociability and social interaction test, the subject mouse was allowed to explore the field containing a social target on one side and an inanimate grid cage on the other side for 8 min. For the social novelty recognition test, the field contains a social target 1 (an earlier stranger, which was used as the social target in the sociability test) on one side and a social target 2 (a new stranger) on the other side. The subject mouse was allowed to explore the field for 8 min. The mice were videotaped for 8 min and then scored using the EthoVision Tracking System for the total time spent in the quadrant with the social target and the inanimate grid cage and the novel social target30. Mice with strong bias for one side at habituation (over 65% on one side) were excluded from the experiment.
Forced swim test
The forced swim test was conducted following 1 h of habituation to the test room. Mice were placed individually in beakers containing 3 liters of water at 25 ± 1 °C for 6 min with ambient lighting. Immobility was recorded using a Canon HD camcorder (VIXIA HF R600). An independent experienced observer recorded total immobility times for 5.5 min, starting 30 s after the beginning of the assay and the latency to immobility47.
RNA extraction and RNA-seq library preparation
Twenty-one days after oxycodone treatment cessation, brains were removed rapidly, placed into ice-cold PBS, and sliced into 1-mm-thick coronal sections in a slice matrix. Bilateral punches were made from VTA (16 gauge), NAc (14 gauge) and mPFC (12 gauge) and flash-frozen in tubes on dry ice (n = 4–6 per group)21. Total RNA was isolated with TRIzol reagent (Invitrogen). All RNA samples were determined to have A260/280 values ≥ 1.8 (Nanodrop); samples for RNA-seq had RNA integrity number values > 9 (BioAnalyzer, Agilent). Approximately 500 ng of purified RNA was used to prepare libraries for sequencing using the TruSeq mRNA library prep kit (Illumina, RS-122-2001/2). NAc, mPFC and VTA samples were pooled (two animals per sample) before library preparation and were sequenced on an Illumina HiSeq 2000 apparatus with 125-nucleotide single-end reads at Beckman Coulter Genomics (currently GeneWiz). Samples were multiplexed to produce >30 million reads per sample.
Bioinformatic analysis
Read alignment, read counting and differential analysis were performed using HISAT2 (ref. 32), HT-Seq33 and the DESeq2 R package, respectively34. Differential analysis aimed at dissecting oxycodone withdrawal versus saline treatment effects in SNI and sham mice was performed using a 2 × 2 factorial design with the following formula: log(exp) ~ SNI + oxycodone + SNI: oxycodone. The differential lists were defined by a P-value cutoff < 0.05 and log2(fold change) <−0.5 or >0.5. Only terms with P value < 0.05 were reported. The ggVennDiagram R package was used for Venn diagram generation. The R package pheatmap was utilized to generate heatmaps for SNI-Sal vs Sham-Sal, Sham-Oxy vs. Sham-Sal, and SNI-Oxy vs Sham-Sal comparisons across the mPFC, NAc, and VTA. Pathway analysis was conducted using IPA. For pathway analysis, a cutoff of −log10(P value) > 1.3 was applied.
Rank–rank hypergeometric overlap RNA-seq analysis
Full threshold-free differential expression lists were ranked by the – log10(P value) multiplied by the sign of the fold change from the DESeq2 analysis. RRHO was used to evaluate the overlap of differential expression lists between oxycodone-withdrawing SNI and sham cohorts. A two-sided version of this analysis was used to test for coincidental and opposite enrichment. RRHO difference maps were produced for pairs of RRHO maps (SNI-Oxy versus Sham-Sal control compared to Sham-Oxy versus Sham-Sal control, and Sham-Oxy versus Sham-Sal control compared to SNI-Oxy versus SNI-Sal control).
Statistical analysis
Data were analyzed using GraphPad Prism 9 and the aforementioned R packages. No statistical methods were used to predetermine sample sizes but our sample sizes are similar to those reported in previous publications21,23,27,47. All behavioral (with the exception of assays assessing motor activity), RNAscope and immunofluorescence experiments were replicated more than one time. For the experiments monitoring behavioral responses over time, we used two-way repeated-measures ANOVAs followed by Tukey’s post hoc tests. For two-factor designs, we used two-way ANOVA followed by Sidak’s or Tukey’s post hoc test depending on the type of comparison. For data containing a single independent variable, we used unpaired two-tailed t-tests after an F test for variance. Results for behavioral and biochemical studies are expressed as the mean ± s.e.m. F and t values for each dataset and analysis are provided in the figure legends. Mice that demonstrated major sickness due to opioid withdrawal were removed from the study.
Extended methods
Anxiety and locomotor tests.
The EPM, open field and locomotor tests were performed under red light conditions, and mice were acclimatized to the room for 1 h before testing. The EPM consisted of two open arms and two closed arms situated opposite to each other (each measuring 12 × 50 cm). The number of entries and time spent in each arm were recorded and analyzed using Noldus EthoVision software. EPM was conducted at 11 d of oxycodone withdrawal. For the open field test, mice were allowed to freely explore the open field arena (50 × 50 × 20 cm) for 8 min. Time spent in the center versus border regions of the arena was recorded and analyzed using Noldus EthoVision. The open field test was conducted at 9d after oxycodone withdrawal. Analysis was performed by a blinded observer.
Nest building assay.
Mice were singly housed and provided cotton nesting material (Nestlet) and fresh bedding. After 12 h, mice were assessed for nest building. The nest quality was assessed with a complexity score from 1 (no nest) to 5 (complex nest with a wall surrounding the mouse)75. The nesting test was performed at 14 d of oxycodone withdrawal.
Novelty-suppressed feeding.
A modified novelty-suppressed feeding was performed after mice were single housed and subjected to food restriction overnight, before testing, to assess stress and motivation-like behavior76. On the day of testing, mice were habituated to the testing room for at least 1 h. Under red light conditions, mice were placed into a 40 × 40 × 20 cm arena with wood-chip bedding covering the floor and a single standard chow food pellet in the center of the arena. Mice were placed in the corner of the box, and the latency to eat was scored for up to 8 min. A video-tracking system (EthoVision, Noldus) measured locomotor activity. Data were analyzed as latency to eat in a novel arena. Total distance traveled was also assessed. ROUT test Q = 1% was used to identify outliers within the dataset, and animals that approached the foot pellet in under 100 s were disqualified.
RNA-seq deconvolution.
The tool CIBERSORTx (https://cibersortx.stanford.edu/index.php) was utilized to estimate the cell-type proportions in the Nac bulk RNA-seq dataset. First, a signature matrix file was constructed from the raw counts matrix of single-cell RNA-seq data from Avey et al.35. The signature matrix generated contained counts per million for genes specific to each cell type in the single-cell RNA-seq dataset. This matrix was further utilized for inferring the cell fractions in the bulk RNA-seq data; the bulk data being represented as a counts per million expression matrix. The S-mode (single-cell mode) batch correction was enabled to account for the technical differences between the bulk RNA-seq and single-cell RNA-seq unique molecular modifier-based data. Quantile normalization was disabled for the RNA-seq data, and the total number of permutations for P-value computation was set to 100. Cell-type proportions were estimated for each sample in the bulk RNA-seq data along with the P values indicating the statistical significance of the deconvolution.
RNAscope in situ hybridization.
The Fluorescent Multiplex V2 kit (Advanced Cell Diagnostics) was used for RNAscope fluorescence in situ hybridization. Specifically, we used the fresh frozen tissue protocol as detailed in the RNAscope Multiplex Fluorescent Reagent Kit v2 Assay User Manual. RNAscope probes were Hdac1 (Mm-Hdac1, 448071), Tubb3 for pan-neuronal labeling (Mm-Tubb3-C3, 423391-C3) and Aif1 for microglial labeling (Mm-Aif1-C3, 319141-C3). Opal dyes (Akoya Biosciences) used for secondary staining were Opal 690 for C1 and Opal 570 for C3. DAPI was used for nuclear staining. Images were taken on an LSM 880 confocal microscope (Zeiss, GER).
Immunofluorescence assays.
An anti-rabbit HDAC1 polyclonal antibody (Abcam 7028, 1:400 dilution) was used for immunofluorescence assays in PFC and NAc slices, as described77. Following overnight incubation with primary antibody, slices were washed three times with PBS solution and incubated for 2 h with donkey anti-rabbit 488 (Jackson labs, 1:300 dilution).
Sample collection, processing and storage for pharmacokinetic testing.
The animals were restrained manually, and approximately 110 μl blood at each time point was collected via retro-orbital puncture into pre-cold EDTA-K2 tubes at designated time points. Blood samples were centrifuged at 4 °C to obtain plasma within 15 min of sample collection. Whole brains were collected from the same animals for further analysis.
Biochemical histone deacetylase assay and pharmacokinetic profiling.
The HDAC binding assay was conducted as previously described78. Pharmacokinetic analysis was performed as previously described. Briefly, RBC1H1 was extracted from the brain and plasma by protein precipitation and analyzed using a high-performance liquid chromatography–tandem mass spectrometry method with electrospray ionization in positive mode as previously reported79.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Additional affective behavior assays after oxycodone withdrawal with or without SNI.
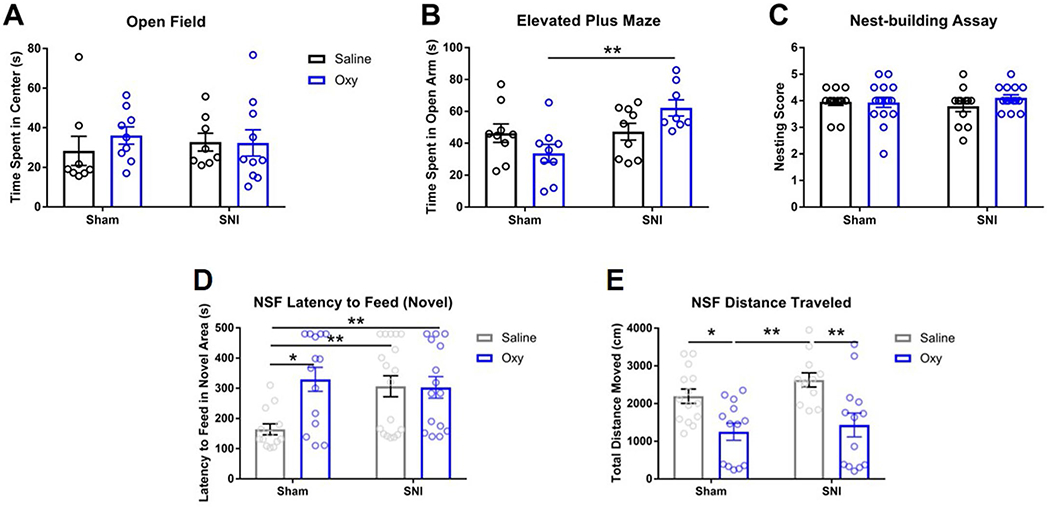
a. No significant difference was observed in the open-field assay (Sham-Sal n = 8, Sham-Oxy n = 9, SNI-Sal n = 8, SNI-Oxy n = 10). b. The time spent in the open arm of the elevated plus maze was significantly increased with oxycodone withdrawal in SNI animals when compared with Sham animals (Sham-Sal n = 9, Sham-Oxy n = 9, SNI-Sal n = 9, SNI-Oxy n = 8; two-way ANOVA interaction F1,31 = 6.299, p = 0.0175; Sidak’s multiple comparisons Oxy SNI vs Sham t = 3.62, df=31, p = 0.0021). c. No significant difference was observed in the nest building assay (Sham-Sal n = 13, Sham-Oxy n = 16, SNI-Sal n = 12, SNI-Oxy n = 14). d. Latency to eat in a novel environment after food deprivation was significantly affected in Sham-Oxy, SNI-Sal, and SNI-Oxy groups as compared with the Sham-Sal controls (Sham-Sal n = 13, Sham-Oxy n = 14, SNI-Sal n = 18, SNI-Oxy n = 15; two-way ANOVA interaction F1,56 = 6.052, p = 0.0170; Sidak’s m.c. SNI-Sal vs Sham-Sal t = 2.963, df=56, p = 0.0089; Sham-Oxy vs Sham-Sal t = 3.249, df=56, p = 0.0039; Tukey’s m.c. SNI-Oxy vs Sham-Sal q = 3.92, df=56, p = 0.0369). e. Total distance traveled during the NSF assay was significantly affected by oxycodone withdrawal in SNI-Oxy and Sham-Oxy (Sham-Sal n = 14, Sham-Oxy n = 13, SNI-Sal n = 12, SNI-Oxy n = 13; two-way ANOVA treatment F1,48 = 20.45, P < 0.001; Sidak’s m.c. Sham-Oxy vs Sham-Sal t = 2.88, df=48, p = 0.0118; SNI-Oxy vs SNI-Sal t = 3.505, df=48, p = 0.002). Values are represented as mean ± SEM *p < 0.05 and ** p < 0.001respectively.
Extended Data Fig. 2 |. NAc RNA-seq deconvolution across injury and withdrawal conditions.
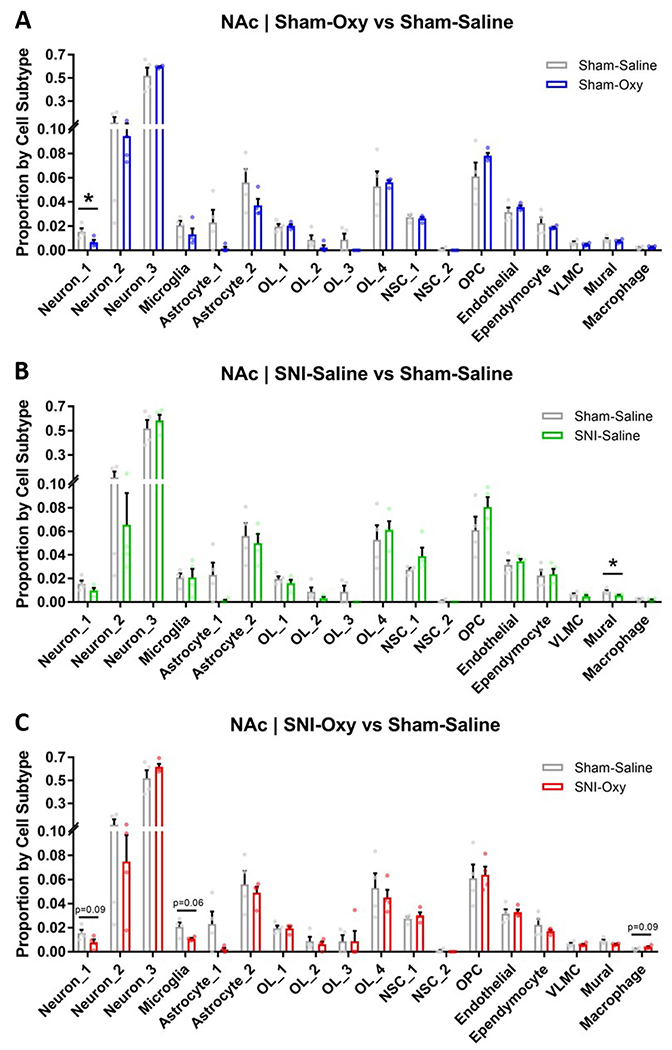
a–c. RNA-seq deconvolution of Sham-Oxy vs Sham-Saline, SNI-Saline vs Sham Saline, and SNI-Oxy vs Sham-Saline, respectively (n = 4 per group), demonstrating condition-specific alterations in transcriptomic representations of neuronal and vascular cell subtypes (Sham-Oxy vs Sham-Sal Neuron_1 unpaired t-test, t = 2.595, df=6, p = 0.0409; SNI-Sal vs Sham-Sal Mural unpaired t-test, t = 2.456, df=6, p = 0.0494, Neuron_1 unpaired t-test, t = 2.042, df=6, p = 0.0873, Microglia Welch’s unpaired t-test, t = 2.818, df=3.328, p = 0.0593; SNI-Oxy vs Sham-Sal unpaired t-test, t = 2.025, df=6, p = 0.0893). Values are represented as mean ± SEM *p < 0.05.
Extended Data Fig. 3 |. mPFC and NAc HDAC1 expression in neuronal and glial cells.
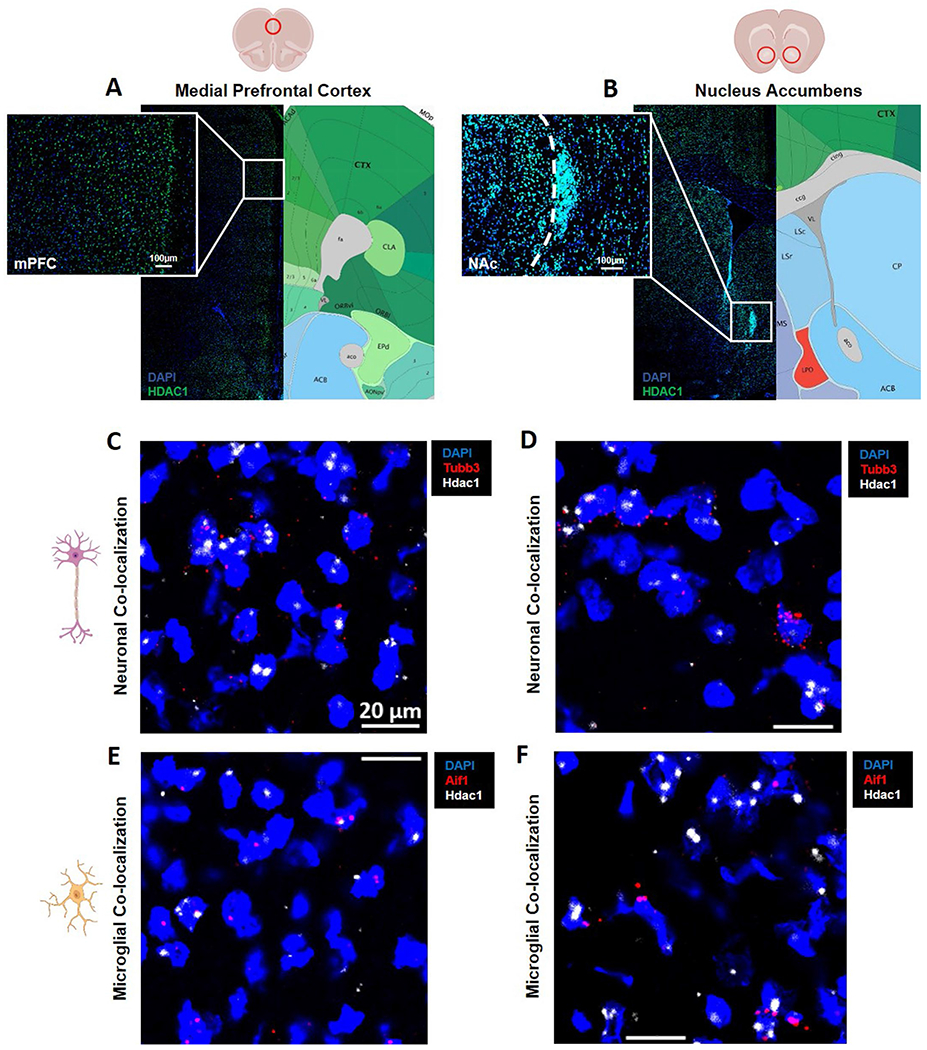
a, b. immunofluorescence staining of HDAC1 in the mPFC and NAc demonstrating high protein nuclear presence across most cells in these regions (naïve mice, n = 3 per group). c, d. RNAscope imaging of Hdac1 transcript colocalization with Tubb3, a pan-neuronal marker, in the mPFC and NAc, demonstrating neuronal expression. e, f. RNAscope imaging of Hdac1 transcript colocalization with Aif1, a microglial marker, demonstrating microglial expression.
Extended Data Fig. 4 |. Pharmacological and in vivo characterizations of RBC1HI.
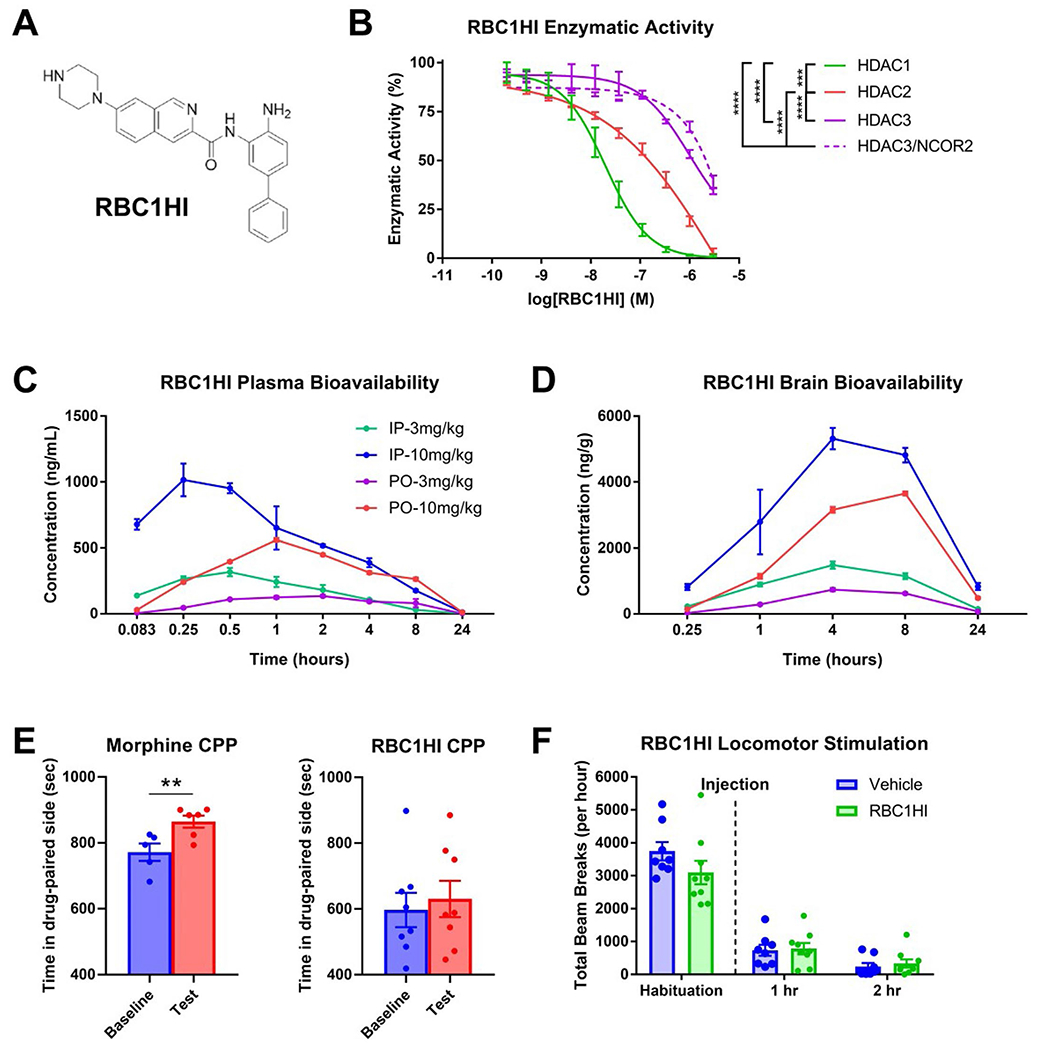
a. Molecular structure of RBC1HI. b. RBC1HI enzymatic inhibition of HDAC1,2,3 (‘HDAC3’=free versus HDAC3 bound to NCOR2) demonstrating highest specificity to HDAC1 (n = 3 replicates; (AUC one-way ANOVA: F3,8 = 191.1, p < 0.0001; Tukey’s m.c. HDAC1 vs HDAC2 q = 11.55, df=8, p = 0.0002; HDAC1 vs free HDAC3 q = 17.22, df=8, p < 0.0001; HDAC1 vs HDAC3/NCOR2 q = 16.21, df=8, p < 0.0001; HDAC2 vs free HDAC3 q = 17.22, df=8, p < 0.0001; HDAC2 vs HDAC3/NCOR2 q = 16.21, df=8, p < 0.0001). c. Plasma bioavailability of RBC1HI at 3 or 10 mg/kg and with intraperitoneal versus oral administration, demonstrating longer bioavailability with the intraperitoneal route. d. Brain bioavailability of RBC1HI with the aforementioned administration doses and routes, demonstrating prolonged drug presence with the intraperitoneal route. Brain to plasma concentration ratios over time. Dose-dependent circulating RBC1HI bioavailability as measured by plasma concentration and brain concentration over time (3 mg/kg i.p.; n = 3 male C57BL/6 mice per time point). e. RBC1HI (3 mg/kg intraperitoneal) did not promote place preference as seen with morphine (6 mg/kg subcutaneous). f. 3 mg/kg RBC1HI does not affect locomotor activity up to two hours after administration. Values are represented as mean ± SEM ****p < 0.0001.
Extended Data Fig. 5 |. Additional affective behaviors after treatment with RBC1HI.
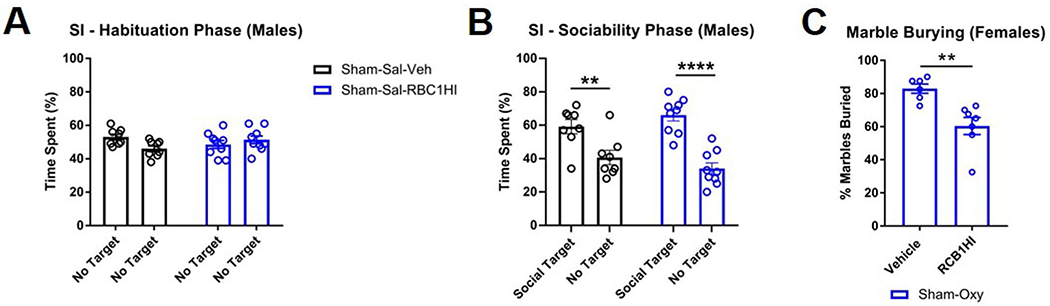
a. Habituation phase of Sham-Sal male mice after receiving Veh or RBC1HI, demonstrating equal interaction times for two empty cups (Sham-Sal-Veh n = 9, Sham-Sal-RBC1HI n = 10). b. RBC1HI and Veh animals both showed a preference for social targets in the sociability phase of the social interaction assay (Sham-Sal-Veh n = 8, Sham-Sal-RBC1HI n = 9). c. Female mice undergoing spontaneous withdrawal from oxycodone bury fewer marbles after longitudinal RBC1HI treatment (Sham-Oxy-Veh n = 6, Sham-Oxy-RBC1HI n = 7; unpaired t-test t = 3.663, df=11, p = 0.0037). Values are represented as meant ± SEM **p < 0.01, ****p < 0.0001.
Acknowledgements
This study was supported by National Institute of Neurological Disorders and Stroke NS086444 (to V.Z.), R01 NS111351, (to V.Z.), R01NS086444S1 (to R.S.) and National Institute on Drug Abuse P01 DA047233 (to E.J.N. and V.Z.), T32 5T32DA007135-34 (to K.D.P.) and R01NS08644S1 (to K.D.P).
Footnotes
Competing interests
The authors declare no competing interests. V.Z. and K.D.P. are inventors on a provisional patent regarding RBC1HI as part of an agreement between the Icahn School of Medicine at Mount Sinai and Regenacy Pharmaceuticals.
Extended data is available for this paper at https://doi.org/10.1038/s41593-023-01350-3.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41593-023-01350-3.
Data availability
RNA-seq data are available on the NCBI Gene Expression Omnibus under accession GSE223541. Other data from this study can be made available upon request.
References
- 1.Darcq E & Kieffer BL Opioid receptors: drivers to addiction? Nat. Rev. Neurosci 19, 499–514 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Serafini RA, Pryce KD & Zachariou V The mesolimbic dopamine system in chronic pain and associated affective comorbidities. Biol. Psychiatry 87, 64–73 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finnerup NB et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 14, 162–173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dworkin RH et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain 132, 237–251 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Cruccu G Treatment of painful neuropathy. Curr. Opin. Neurol 20, 531–535 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Hooten WM, Lamer TJ & Twyner C Opioid-induced hyperalgesia in community-dwelling adults with chronic pain. Pain 156, 1145–1152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volkow ND, Michaelides M & Baler R The neuroscience of drug reward and addiction. Physiol. Rev 99, 2115–2140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y et al. Chronic oxycodone self-administration altered reward-related genes in the ventral and dorsal striatum of C57BL/6J mice: an RNA-seq analysis. Neuroscience 393, 333–349 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Koob GF & Volkow ND Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3, 760–773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baliki MN, Chang PC, Baria AT, Centeno MV & Apkarian AV Resting-sate functional reorganization of the rat limbic system following neuropathic injury. Sci. Rep 4, 6186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apkarian VA, Hashmi JA & Baliki MN Pain and the brain: specificity and plasticity of the brain in clinical chronic pain. Pain 152, S49–S64 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seminowicz DA et al. Pain-related nucleus accumbens function: modulation by reward and sleep disruption. Pain 160, 1196–1207 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartz N et al. Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science 345, 535–542 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Browne CJ, Godino A, Salery M & Nestler EJ Epigenetic mechanisms of opioid addiction. Biol. Psychiatry 87, 22–33 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jullie D, Gondin AB, von Zastrow M & Canals M Opioid pharmacology under the microscope. Mol. Pharm 98, 425–432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baliki MN, Geha PY, Fields HL & Apkarian AV Predicting value of pain and analgesia: nucleus accumbens response to noxious stimuli changes in the presence of chronic pain. Neuron 66, 149–160 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Darnall B To treat pain, study people in all their complexity. Nature 557, 7 (2018). [DOI] [PubMed] [Google Scholar]
- 18.McPherson ML et al. Safe and appropriate use of methadone in hospice and palliative care: expert consensus white paper. J. Pain Symptom Manag 57, 635–645 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Bertin C et al. Analgesic opioid use disorders in patients with chronic non-cancer pain: a holistic approach for tailored management. Neurosci. Biobehav. Rev 121, 160–174 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Delorme J et al. Prevalence and characteristics of chronic pain in buprenorphine and methadone-maintained patients. Front. Psychiatry 12, 641430 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Descalzi G et al. Neuropathic pain promotes adaptive changes in gene expression in brain networks involved in stress and depression. Sci. Signal 10, eaaj1549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mitsi V et al. RGS9-2–controlled adaptations in the striatum determine the onset of action and efficacy of antidepressants in neuropathic pain states. Proc. Natl Acad. Sci. USA 112, E5088–E5097 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitsi V & Zachariou V Modulation of pain, nociception, and analgesia by the brain reward center. Neuroscience 338, 81–92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Corder G et al. An amygdalar neural ensemble that encodes the unpleasantness of pain. Science 363, 276–281 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren W et al. Adaptive alterations in the mesoaccumbal network after peripheral nerve injury. Pain 162, 895–906 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shields SD, Eckert WA 3rd & Basbaum AI Spared nerve injury model of neuropathic pain in the mouse: a behavioral and anatomic analysis. J. Pain 4, 465–470 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Avrampou K et al. RGS4 maintains chronic pain symptoms in rodent models. J. Neurosci 39, 8291–8304 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson EM et al. Knockdown of the histone di-methyltransferase G9a in nucleus accumbens shell decreases cocaine self-administration, stress-induced reinstatement, and anxiety. Neuropsychopharmacology 44, 1370–1376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cobos EJ et al. Inflammation-induced decrease in voluntary wheel running in mice: a nonreflexive test for evaluating inflammatory pain and analgesia. Pain 153, 876–884 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee E-H, Park J-Y, Lee Y & Han P-L Sociability and social novelty preference tests using a U-shaped two-choice field. Bio. Protoc 8, e2853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lustberg D et al. Noradrenergic circuits in the forebrain control affective responses to novelty. Psychopharmacol. 237, 3337–3355 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D, Paggi JM, Park C, Bennett C & Salzberg SL Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anders S, Pyl PT & Huber W HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Avey D et al. Single-cell RNA-seq uncovers a robust transcriptional response to morphine by glia. Cell Rep. 24, 3619–3629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray MH, Williams BR, Kuppe MK, Bryant CD & Logan RW A glitch in the matrix: the role of extracellular matrix remodeling in opioid use disorder. Front. Integr. Neurosci 16, 899637 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flaisher-Grinberg S, Persaud SD, Loh HH & Wei LN Stress-induced epigenetic regulation of κ-opioid receptor gene involves transcription factor c-Myc. Proc. Natl Acad. Sci. USA 109, 9167–9172 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Perez D, Ferenczi S, Kovacs KJ & Milanes MV Distinct regulation pattern of Egr-1, BDNF and Arc during morphine-withdrawal conditioned place aversion paradigm: role of glucocorticoids. Behav. Brain Res 360, 244–254 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Dietrich JB, Takemori H, Grosch-Dirrig S, Bertorello A & Zwiller J Cocaine induces the expression of MEF2C transcription factor in rat striatum through activation of SIK1 and phosphorylation of the histone deacetylase HDAC5. Synapse 66, 61–70 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Peng HY et al. Spinal SGK1/GRASP-1/Rab4 is involved in complete Freund’s adjuvant-induced inflammatory pain via regulating dorsal horn GluR1-containing AMPA receptor trafficking in rats. Pain 153, 2380–2392 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Bawor M et al. Contribution of BDNF and DRD2 genetic polymorphisms to continued opioid use in patients receiving methadone treatment for opioid use disorder: an observational study. Addict. Sci. Clin. Pract 10, 19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sarkar S, Jain R, Kethawath SM, Gupta R & Kumar M Serum BDNF levels in patients with opioid dependence during the early withdrawal period: a case control study. Neurosci. Lett 681, 100–104 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Gregoretti IV, Lee YM & Goodson HV Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol 338, 17–31 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Payne NC, Maksoud S, Tannous BA & Mazitschek R A direct high-throughput protein quantification strategy facilitates discovery and characterization of a celastrol-derived BRD4 degrader. Cell Chem. Biol 29, 1333–1340 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Payne NC & Mazitschek R Resolving the deceptive isoform and complex selectivity of HDAC1/2 inhibitors. Cell Chem. Biol 29, 1140–1152 (2022). [DOI] [PubMed] [Google Scholar]
- 46.Simonini MV et al. The benzamide MS-275 is a potent, long-lasting brain region-selective inhibitor of histone deacetylases. Proc. Natl Acad. Sci. USA 103, 1587–1592 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stratinaki M et al. Regulator of G-protein signaling 4 is a crucial modulator of antidepressant drug action in depression and neuropathic pain models. Proc. Natl Acad. Sci. USA 110, 8254–8259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lutz PE et al. Distinct mu, delta, and kappa opioid receptor mechanisms underlie low sociability and depressive-like behaviors during heroin abstinence. Neuropsychopharmacology 39, 2694–2705 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Green-Fulgham SM et al. Oxycodone, fentanyl, and morphine amplify established neuropathic pain in male rats. Pain 160, 2634–2640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y et al. Behavioral and neurochemical changes induced by oxycodone differ between adolescent and adult mice. Neuropsychopharmacology 34, 912–922 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffman EM, Watson JC, St Sauver J, Staff NP & Klein CJ Association of long-term opioid therapy with functional status, adverse outcomes, and mortality among patients with polyneuropathy. JAMA Neurol. 74, 773–779 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Comer SD, Sullivan MA, Vosburg SK, Kowalczyk WJ & Houser J Abuse liability of oxycodone as a function of pain and drug use history. Drug Alcohol Depend. 109, 130–138 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darnall BD et al. Patient-centered prescription opioid tapering in community outpatients with chronic pain. JAMA Intern. Med 178, 707–708 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bravo IM et al. Divergent behavioral responses in protracted opioid withdrawal in male and female C57BL/6J mice. Eur. J. Neurosci 51, 742–754 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young R & Johnson DN Comparison of routes of administration and time course effects of zacopride and buspirone in mice using an automated light/dark test. Pharm. Biochem Behav 40, 733–737 (1991). [DOI] [PubMed] [Google Scholar]
- 56.Fulenwider HD et al. Sex differences in oral oxycodone self-administration and stress-primed reinstatement in rats. Addict. Biol 25, e12822 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Collins D, Reed B, Zhang Y & Kreek MJ Sex differences in responsiveness to the prescription opioid oxycodone in mice. Pharm. Biochem. Behav 148, 99–105 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Lane DA et al. Region-specific changes in the subcellular distribution of AMPA receptor GluR1 subunit in the rat ventral tegmental area after acute or chronic morphine administration. J. Neurosci 28, 9670–9681 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Raehal KM et al. Morphine-induced physiological and behavioral responses in mice lacking G protein-coupled receptor kinase 6. Drug Alcohol Depend. 104, 187–196 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matthes HW et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383, 819–823 (1996). [DOI] [PubMed] [Google Scholar]
- 61.Carpenter MD, Manners MT, Heller EA & Blendy JA Adolescent oxycodone exposure inhibits withdrawal-induced expression of genes associated with the dopamine transmission. Addict. Biol 26, e12994 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanchez V, Carpenter MD, Yohn NL & Blendy JA Long-lasting effects of adolescent oxycodone exposure on reward-related behavior and gene expression in mice. Psychopharmacol. 233, 3991–4002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Markovic T et al. Pain induces adaptations in ventral tegmental area dopamine neurons to drive anhedonia-like behavior. Nat. Neurosci 24, 1601–1613 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou H et al. Inhibition of the prefrontal projection to the nucleus accumbens enhances pain sensitivity and affect. Front. Cell Neurosci 12, 240 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ren W et al. The indirect pathway of the nucleus accumbens shell amplifies neuropathic pain. Nat. Neurosci 19, 220–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kennedy PJ et al. Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation. Nat. Neurosci 16, 434–440 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanna MD, Guandalini L, Romanelli MN & Galeotti N The new HDAC1 inhibitor LG325 ameliorates neuropathic pain in a mouse model. Pharm. Biochem Behav 160, 70–75 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Borgonetti V & Galeotti N Combined inhibition of histone deacetylases and BET family proteins as epigenetic therapy for nerve injury-induced neuropathic pain. Pharm. Res 165, 105431 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Sanna MD & Galeotti N The HDAC1/c-JUN complex is essential in the promotion of nerve injury-induced neuropathic pain through JNK signaling. Eur. J. Pharm 825, 99–106 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Graff J & Tsai LH Histone acetylation: molecular mnemonics on the chromatin. Nat. Rev. Neurosci 14, 97–111 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Quinlan J, Willson H & Grange K Hopes and fears before opioid tapering: a quantitative and qualitative study of patients with chronic pain and long-term opioids. Br. J. Pain 15, 120–128 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jantarada C, Silva C & Guimaraes-Pereira L Prevalence of problematic use of opioids in patients with chronic noncancer pain: a systematic review with meta-analysis. Pain. Pract 21, 715–729 (2021). [DOI] [PubMed] [Google Scholar]
- 73.Smit T et al. Anxiety sensitivity and pain intensity independently predict opioid misuse and dependence in chronic pain patients. Psychiatry Res. 294, 113523 (2020). [DOI] [PubMed] [Google Scholar]
- 74.Throckmorton DC, Gottlieb S & Woodcock J The FDA and the next wave of drug abuse—proactive pharmacovigilance. N. Engl. J. Med 379, 205–207 (2018). [DOI] [PubMed] [Google Scholar]
- 75.Deacon RM Assessing nest building in mice. Nat. Protoc 1, 1117–1119 (2006). [DOI] [PubMed] [Google Scholar]
- 76.Pena CJ et al. Early life stress alters transcriptomic patterning across reward circuitry in male and female mice. Nat. Commun 10, 5098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gaspari S et al. Suppression of RGSz1 function optimizes the actions of opioid analgesics by mechanisms that involve the Wnt/β-catenin pathway. Proc. Natl Acad. Sci. USA 115, E2085–E2094 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bradner JE et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc. Natl Acad. Sci. USA 107, 12617–12622 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jochems J et al. Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology 39, 389–400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
RNA-seq data are available on the NCBI Gene Expression Omnibus under accession GSE223541. Other data from this study can be made available upon request.