

Abstract
Glycosaminoglycans (GAGs), such as hyaluronic acid (HA) and chondroitin sulfate (CS), have seen widespread adoptions as components of tissue engineering scaffolds due to their potent bioactive properties and ease of chemical modification. However, modification of the biopolymers will impair biological recognition of the GAG and reduce the bioactive properties of the material. In this work, we studied how the degree of thiolation of HA and CS, along with other key hydrogel design parameters, affected the physical and bioactive properties of the bulk hydrogel. Although properties, such as the HA molecular weight, did not have a major effect, increasing the degree of thiolation of both HA and CS decreased their biorecognition in experimental analogues for cell/matrix remodeling and binding. Furthermore, combining HA and CS into dual polymer network hydrogels also modulated the physical and bioactive properties, as seen with differences in gel stiffness, degradation rate, and encapsulated cell viability.
Keywords: Glycosaminoglycan, Hyaluronic Acid, Chondroitin Sulfate, Hydrogel, Tissue Engineering
Table of Contents Figure:
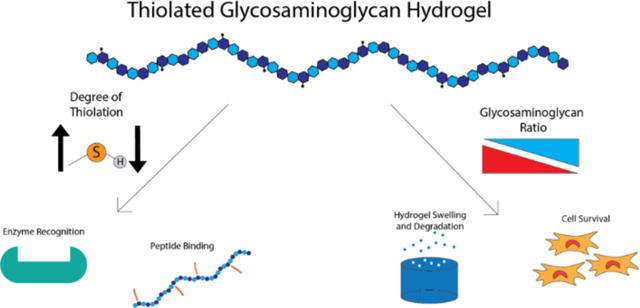
1. Introduction:
While the human body possesses natural healing capacity, situations can arise where the body is incapable of repairing the damaged tissue, such as in the case of critically sized tissue defects or disfunction of the natural repair pathways1,2. To address these medical needs, the field of tissue engineering has been developing methods to engineer replacement tissue. Tissue engineering approaches often employ cells seeded in a scaffold, such as a hydrogel, where the cells generate new tissue while the gel serves as a support structure. Synthetic polymers, such as poly(ethylene glycol) or poly(lactic acid), can provide scaffolds with very well-defined properties and characteristics; however, they lack the biological signals present in natural extracellular matrix3–6. On the other hand, natural polymers such as collagen and fibrin have also been used, but they lack the degree of chemical and mechanical tunability present in synthetic polymers3,7–11.
Over the past two decades, glycosaminoglycans (GAGs), including hyaluronic acid (HA) and chondroitin sulfate (CS), have seen increased use as components of tissue engineering scaffolds in a variety of fields, including bone12,13, cartilage14,15, skin16,17, vocal fold18, and nerve tissue regeneration19. HA and CS are naturally produced polymers, widely distributed within the extracellular matrix, and important for bodily function and cellular processes20,21. These GAGs possess potent bioactive properties including cell-directive signals22–24 and anti-inflammatory properties25, and they possess many chemically reactive hydroxyl and carboxylic acid groups. While these reactive moieties can exist as part of the bioactive motifs, they can also support chemical functionalization to allow for crosslinking and hydrogel formation. A wide variety of modifying agents including methacrylates26, thiols27, furans28, tyramines29, and various other species30 have been conjugated to HA and CS for gel formation. Although the addition of these groups can allow for increases in stiffness and compressive strength of the gel through increased crosslink density as well as provide handles for chemical addition of other bioactive factors, modification of the natural polymer structure may inhibit the ability of the cell to recognize and interact with native chemical moieties and thus reduce the number and potency of bioactive signals presented to the cells31,32. Among the previously reported studies, there is little consensus regarding the optimal degree of modification for HA and CS, and the degree of modification of these GAGs varies widely between papers.
In this study, we focused on designing CS and HA hydrogels that maximize the bioactive properties of the GAGs while maintaining the ability to crosslink the polymers into hydrogels and encapsulate cells. To do so, we synthesized and characterized hydrogels using thiolated HA and CS with increasing degrees of thiolation (DOT) and tested their biorecognition with two in vitro analogues of in vivo bioactivity. These tests included a hyaluronidase digestion assay, which represented how well encapsulated cells would be able to remodel their environment, and a GAG binding assay, which represented how well proteins, including receptors and growth factors, would be able to interact with the modified GAGs. Next, we examined the effect of HA molecular weight ranging from 40 kDa33 to 1.5 MDa19,34 and evaluated the viscoelastic properties of the resultant gels and effects of molecular weight on bioactivity. Finally, we fabricated and tested dual polymer network (DPN) hydrogels with the two GAGs to combine the chemical and biological signals of CS and HA. The ratio of CS to HA was tested to determine how it affected physical and bioactive properties of the hydrogel. Specifically, we fabricated CS/HA DPN hydrogels utilizing DOT and molecular weight that retained maximum biological activity, as determined using the hyaluronidase and peptide binding assays, to investigate how the combination of these two polymers affected hydrogel properties and cell survival.
2. Materials and Methods:
2.1: Preparation and Characterization of Thiolated Hyaluronic Acid and Chondroitin Sulfate
Thiolated HA (HA-SH) was prepared using a modified version of a previously reported protocol18. Hyaluronic acid (molecular weight (Mw): 10 kDa, 60 kDa, 100 kDa, 200 kDa, 500 kDa, Lifecore Biomedical) was first dissolved in 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) buffer with 0.2 wt% NaCl at a concentration of 2 mg/mL. To attach free thiol groups, dithio-bis(propionohydrazide) (DTP) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) were dissolved in the HA solution. The degree of thiolation was controlled through the amount of DTP added, and a 1:1 molar ratio of DTP to the desired percentage of modified HA carboxylic acid groups was used. EDC was added in a 2:1 molar ratio with respect to DTP. The reaction solution was titrated to a pH of 4.5 and was reacted overnight at room temperature. To cleave the disulfide bond of DTP, the HA solution was titrated to a pH of 8 and dithiothreitol (DTT) was added in 3:1 molar excess of the DTP. The DTT was allowed to react for 3 hours at room temperature before the solution was titrated to pH 4.5 to prevent the reformation of disulfide bonds. The polymer was then purified using a KrosFlo KR2i tangential flow filtration (TFF) unit (Repligen) using a 5 kDa molecular weight cut off column and a transmembrane pressure of 18 PSI. The solution was purified until a permeate volume of three times the reaction volume had been reached. After purification, the polymer was frozen and lyophilized until further use. Thiolated CS (CS-SH) (Mw: 40 kDa, Seikigaku Corporation) was synthesized using the same methods. Free thiol content of HA-SH and CS-SH were quantified using an Ellman’s assay, and cysteine was used to construct a standard curve. Degree of thiolation (DOT) was defined as the percentage of GAG carboxylic acid groups converted to free thiols. To validate the results from the Ellman’s assay, the DOT of low and high DOT HA-SH and CS-SH was determined using 1H nuclear magnetic resonance (NMR) spectroscopy. Samples were dissolved in D2O and run on a Bruker 800 MHz Avance III. DOT was determined by integrating the beta methylene peak on the thiol side chain and dividing by the integral of the of the N-acetyl methyl group of the N-acetylglucosamine monomer on both HA and CS27. DOT determined through NMR were within ± 5% of the Ellman’s assay (Figure S2, S3).
2.2: Fabrication of HA-SH and CS-SH Hydrogels
Hydrogels composed of 1.5 w/v% HA-SH and CS-SH were formed by crosslinking the polymer chains with poly(ethylene glycol) diacrylate (PEGDA) (Mw: 3.4 kDa, Alfa Aesar). HA-SH and CS-SH were dissolved at a concentration of 3 w/v% in 360 μL of phosphate buffered saline (PBS). For CS/HA DPN hydrogels, weight/ weight ratios of 10:0, 7:3, 5:5, 3:7, and 0:10 CS to HA were used with the total GAG content being kept at 1.5 w/v% (Table 1). For determination of the minimum DOT for gel formation, a stoichiometric quantity of PEGDA to fully react with all available free thiols was added. For experiments comparing properties of HA-SH and CS-SH with low and high thiolation degrees, the lower mass of PEGDA was used to crosslink both low and high DOT gels. To prevent spontaneous disulfide bridge formation in the high thiolation gels, N-ethyl maleimide (NEM) was added to the PEGDA fraction to cap free thiols in a 1:1 molar ratio of NEM to remaining free thiols after PEGDA crosslinking. For experiments involving CS/HA DPN gels, PEGDA content was held constant between all groups to maintain a constant crosslink density between groups, using the PEGDA required to fully crosslink the 10:0 group. For all experiments, PEGDA was dissolved in PBS and mixed with the HA-SH or CS-SH solution. To form the gels, the prepolymer solutions were titrated to pH 7.8, after which sufficient PBS was added to bring the concentration of the GAG to 1.5 w/v%. The hydrogels were then incubated overnight at 37 ºC in a humidified environment.
Table 1:
Concentrations of CS and HA used for CS/HA DPN hydrogels
| Ratio of CS/HA | 10:0 | 7:3 | 5:5 | 3:7 | 0:10 |
|---|---|---|---|---|---|
| Concentration of CS (mg/mL) | 15 | 10.5 | 7.5 | 4.5 | 0 |
| Concentration of HA (mg/mL) | 0 | 4.5 | 7.5 | 10.5 | 15 |
2.3: Mechanical Testing of GAG Hydrogels
To determine the stiffness of the HA-SH and CS-SH hydrogels, 150 μL hydrogels were polymerized directly onto Teflon coated microscope slides (Tekdon), with a hydrophilic area of 20 mm. After overnight incubation, stiffness of the hydrogels was determined using a Discovery Hybrid Rheometer (TA Instruments). An oscillation frequency sweep from 0.1 to 100 rad/s was performed using a 20 mm head with a constant stress of 1 Pa. Storage modulus and tan(δ) of the hydrogels were determined from the linear region of the sweep, if available. For determination of the minimum DOT for gel formation, 100 kDa HA-SH was used based off HA molecular weight experiments.
2.4: Enzymatic Degradation of GAG Hydrogels
For all degradation experiments, enzymatic degradation was conducted using a 50 U/mL solution of bovine testes hyaluronidase type I-S (Sigma Aldrich) in PBS. Degradation of hydrogels was conducted at 37 °C with the gels continuously agitated on a plate shaker.
2.4.1: Degradation of GAG homopolymer hydrogels
To determine the degradation rate of HA-SH of varying molecular weights, as well as HA-SH and CS-SH of low and high DOTs, 100 μL gels were made directly in 96-well plates and left to incubate overnight at 37 °C. At the start of the experiment, 100 μL fresh hyaluronidase solution was pipetted on top of the hydrogels then immediately removed to obtain a zero-hour time point. Fresh hyaluronidase solution was pipetted on top, with the supernatant removed and replaced with fresh hyaluronidase solution every two hours for a total of ten hours. To assess degradation of HA, a carbazole assay was performed to determine the concentration of free HA in the collected supernatants. To assess degradation of CS, a dimethylmethylene blue (DMMB) assay was performed to determine the concentration of free CS in the collected supernatants.
2.4.2: Degradation of CS/HA DPN hydrogels
To determine the degradation rate of CS/HA DPN hydrogels, 100 μL gels were made directly in the bottom of 0.5 mL microcentrifuge tubes, with the initial masses of the tubes recorded to calculate gel masses. After polymerization, hydrogels were first allowed to swell overnight with PBS. After overnight swelling, 300 μL of fresh hyaluronidase solution was added atop the gels and incubated for 24 hours at 37 °C. After 24 hours, the supernatant was discarded, the gels were blotted dry, and the mass of the gel was recorded. A total of seven time points were taken, and the hyaluronidase solution was replaced and the gel masses were recorded every 24 hours. After seven days, the gels were dialyzed against ultrapure water to remove salt at 4 °C overnight. Gels were then frozen and lyophilized, and the dry masses of the gels were recorded.
2.5: Peptide-Glycan Binding to GAG Hydrogels
2.5.1: Peptide-Glycan synthesis and characterization
The HA-binding peptide GAH (GAHWQFNALTVGSG) was synthesized with a C-terminal hydrazide for coupling to CS and was obtained from Chinese Peptide Company. The CS-binding peptide YKT (YKTNFRRYYRFGSG) was also synthesized with a C-terminal hydrazide for coupling to HA and was produced using a Liberty Blue peptide synthesizer with standard Fmoc solid phase peptide synthesis techniques. After synthesis, YKT was purified using reverse phase fast protein liquid chromatography, and the collected fractions were verified using matrix assisted time of flight mass spectrometry.
Synthesis of peptide-glycan conjugates was performed using a modified version of a previously reported method35. To synthesize GAH coupled to CS (CS-GAH) and YKT coupled to 100 kDa HA (HA-YKT), the GAGs were dissolved at 10 mg/mL in 0.1 M MES buffer with 8 M urea. One hundred molar excess of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride (DMTMM) was added with respect to the GAG. GAH was added in a 10:1 molar ratio of peptide to CS and YKT was added in a 30:1 ratio of peptide to HA. The solution was titrated to a pH of 4.5 and allowed to react for three days at room temperature. At the end of the reaction, the solutions were diluted 5X to stop the reaction and purified using TFF using the same parameters as the HA-SH and CS-SH synthesis. Peptide attachment was quantified by measuring the 280 nm absorbance of the peptide-glycan and comparing the results to a standard curve made of the free peptide.
2.5.2: Peptide-Glycan binding assay
To determine the binding ability of CS-GAH and HA-YKT to HA-SH and CS-SH, respectively, of low and high DOTs, 40 μL gels were made directly in a black opaque 96-well plate. After gelation, 10 μM solutions of CS-GAH and HA-YKT were pipetted atop the gels and left to incubate for thirty minutes at room temperature. After this time, the supernatant was removed, and the gels were washed three times with PBS to remove non adherent peptide-glycans. After washing, peptide-glycan attachment was quantified by measuring the intrinsic fluorescence of tryptophan and tyrosine residues of the gels to determine CS-GAH and HA-YKT binding, respectively. Intrinsic fluorescence of tryptophan was measured at an excitation of 295 nm and emission of 350, and the intrinsic fluorescence of tyrosine was measured at an excitation of 280 nm and emission of 305 nm.
2.6: MSC Viability in CS/HA DPN Hydrogels
2.6.1: Rabbit MSC isolation and culture
Rabbit MSCs were isolated from the bone marrow from the femurs of 6-month old New Zealand white rabbits. After euthanasia following an unrelated procedure, the discarded femurs were isolated. The neck of the femur was clipped off, and the bone marrow was rinsed out using warmed Dulbecco’s Modified Eagle Medium (DMEM) (Gibco) into 50 mL conical tubes. Erythrocytes were lysed through the addition of sterile deionized water into the tube, and the tubes were centrifuged to remove the dead cells. The resulting pellet was broken up and cells were plated on polystyrene tissue culture plates and incubated overnight in a humidified environment at 37 °C. The next day, the non-adherent cell population was aspirated, and the adherent MSC population was subsequently cultured using a medium consisting of low glucose DMEM with Glutamax supplement (Gibco), 10% fetal bovine serum (Gibco), 5% penicillin/streptomycin (Gibco), and 10 μg/ml basic fibroblast growth factor (Lonza). Cells were passaged at 70–80% confluence and used at passage two.
2.6.2: MSC Viability in CS/HA DPN Hydrogels
To assess cell viability as a function of CS/HA ratio on DPN hydrogels, MSCs were encapsulated in CS/HA DPN hydrogels at a cell concentration of 106 cells/mL. Ten μL gels were pipetted directly onto Ibidi μ-Slides and allowed to gel for one hour at 37 °C in a humidified environment. After polymerization, fresh cell medium was pipetted atop the gels, and the medium was changed every day. At time points of one and six days, the cell medium was aspirated and replaced with 4 μM of Calcein AM (Invitrogen) and 6 μM of ethidium homodimer (Invitrogen) in PBS to stain for live and dead cells, respectively. The cells were incubated for thirty minutes, after which the staining solution was removed and replaced with PBS. To image the cells, fluorescent images were taken using a Keyence BZ-X700 fluorescent microscope at a magnification of 10X. A Z-stack image of the gels was taken with a depth of 500 μm, and Z-stack images were combined using the full focus algorithm in the accompanying Keyence image analysis software. Each Z-stack image had a cross sectional area of 1.6 mm2, corresponding to 13% of the total hydrogel area. One image from the center of each biological replicate was taken. Live/dead counts were determined using the find maxima algorithm in ImageJ.
2.7: Statistics
Data are represented as means, and error bars correspond to standard deviation. For the comparison of two groups, statistical significance was determined using a T-Test. For comparing more than two groups, statistical significance was determined with single factor equal variance ANOVA, and differences between groups were determined using Tukey’s post hoc tests. Statistical analysis was performed using Graphpad Prism, and a probability value of 95% (P < 0.05) was used to determine statistical significance.
3. Results and Discussion:
3.1: Minimum Thiolation for Hydrogel Formation
To examine how degree of thiolation affected bioactivity of HA and CS, GAGs with varying percentages of thiolation, as determined by the percentages of available carboxylate groups that were converted to thiols, were synthesized. However, prior to bioactivity assays, the minimum DOT required for gelation had to be determined. Following reaction with DTP and EDC, HA with DOTs of 2.3%, 5.3%, 9.9%, 13.7%, 17.8%, and 24.0% were obtained (Table S1). Results from rheological testing showed that HA-SH with a DOT of 5.3% thiolation formed a gel (Figure 1) whereas, at a DOT of 2.3%, the HA-SH did not gel as denoted by a tan(δ) value greater than 1 (Figure 1c). Furthermore, at 5.3% thiolation, the gel exhibited an average G’ value of 16.5 Pa, which would not be robust enough for most tissue engineering applications. As expected, G’ values increased with increasing degrees of thiolation. The 17.8% DOT HA-SH produced robust gels with a modest DOT. This result, coupled with the data presented below showing a similar degree of thiolation (17.6%) was required for robust CS gelation, led to 17.8% DOT HA-SH being chosen for experiments investigating the impact of HA molecular weight on gel stability and as the low DOT for experiments investigating the impact of DOT on HA biological activity.
Figure 1:

Mechanical properties of HA-SH (MW: 100 kDa, 1.5 w/v%) and CS-SH (MW: 40 kDa, 1.5 w/v%) hydrogels increase as a function of DOT. (a) Storage modulus of HA-SH gels as a function of DOT. (b) Storage modulus of CS-SH gels as a function of DOT. (c) tan(δ) of HA-SH gels. (d) tan(δ) of CS-SH gels. * denotes statistical significance (P < 0.05) from all other groups. n.s. denotes no significance between groups (P > 0.05)
For CS-SH, it was found that a DOT higher than that required for HA-SH was needed to form a robust gel. Following reactions with DTP and EDC, CS with DOTs of 4.2%, 8.2%, 15.1%, 17.6%, 25.5%, 28.8%, and 35.1% were obtained (Table S2). Below 15.1%, CS-SH gels did not form, and 15.1% thiolated CS-SH formed a weak gel with a G’ of 43.2 Pa (Figure 1). To maintain parity with HA-SH in terms DOT, a CS-SH thiolation degree of 17.6% was chosen for future experiments. Given similar DOT levels, it was found that CS-SH hydrogels were weaker than HA-SH hydrogels (697.9 Pa for HA-SH and 304.4 Pa for CS-SH at a DOT of ~17.7%). The chosen DOT for HA-SH and CS-SH falls on the lower end of previously reported degrees of modification for both polymers, and other groups reported degrees of modification between 736,37 and 71%26,38. While other groups achieved hydrogel formation at degrees of modification lower than 17%, other factors such as reacting the GAG chains directly with one another36, the addition of other polymers39,40, or differences in the concentration of polymer used41 may account for the need for a DOT of ~17% to support gel formation in the study reported here. In this regard, the minimum DOT for gel formation is specific to a chosen crosslinking method and polymer concentration.
3.2: Effects of GAG DOT on Bioactivity
3.2.1: Effect of GAG DOT on Hyaluronidase Activity
To assess bioactivity of HA-SH hydrogels, one chosen metric was to measure hydrogel susceptibility to hyaluronidases as a measure of hyaluronidase recognition and degradation of HA as a function of DOT. This assay assessed the effect of thiolation on enzymatic recognition sites within the HA chain since fewer recognition sites would lead to decreased degradation. Hyaluronidase degradation was chosen as a proxy to understand how the degree of modification of the GAGs would affect the encapsulated cells’ ability to remodel the environment via secretion of hyaluronidase or how durable the material would be in a pro-inflammatory environment characterized by an increased level of hyaluronidases. To maintain consistent crosslink density, low DOT HA-SH (17.8%) and high DOT HA-SH (30.2%) were both crosslinked with the same amount of PEGDA needed to fully crosslink the 17.8% DOT HA-SH. Free thiols in the high DOT gels were capped with NEM to prevent disulfide bridge formation (Figure 3a). It has been previously demonstrated that maleimide groups react with thiols at a faster rate than acrylates; therefore, it is believed that the amount of free thiols available for crosslinking with PEGDA between the high and low DOT groups is roughly equivalent42. As such, any inefficiencies in crosslinking should be seen in both groups. This was further confirmed through rheological analysis of gels with high (with NEM) and low DOT, with the stiffness of the two groups being statistically equivalent (Figure S4).
Figure 3:

HA-YKT Binding to CS-SH (MW: 40 kDa) and CS-GAH binding to HA-SH (MW: 100 kDa) as measured by intrinsic fluorescence of tyrosine and tryptophan Residues. * denotes statistical significance (P < 0.05) between groups
Following 10 hours of hyaluronidase degradation, it was found that the 30.2% DOT HA-SH degraded significantly less than the 17.8% DOT HA-SH, and these results suggest that fewer hyaluronidase recognition sites exist at a higher DOT (Figure 2b, c).
Figure 2:

Degradation of HA-SH (MW: 100 kDa, 1.5 w/v%) and CS-SH (MW: 40 kDa, 1.5 w/v%) hydrogels as a function of GAG DOT. (a) Schematic of NEM mediated capping of free thiols on HA-SH. (b) Degradation of HA-SH and CS-SH over time. (c) Cumulative Degradation of HA-SH and CS-SH at 10 hours. * denotes statistical significance (P < 0.05) between groups
Similarly, the bioactivity of CS-SH with respect to DOT was tested through hyaluronidase digestion since CS is also cleavable by hyaluronidase. Low DOT CS-SH (17.6%) and high DOT CS-SH (35.1%) were gelled with consistent crosslink density using the same methods as described for HA-SH. As expected, CS-SH generally degraded less than HA-SH due to the molecular differences between the two GAGs. Furthermore, similar to HA-SH, high DOT CS-SH also degraded less than low DOT CS-SH, and these results also suggest limited hyaluronidase recognition of CS due to increased modification (Figure 2b, c).
For both CS and HA, decreasing DOT resulted in increased susceptibility to hyaluronidase degradation. Increased susceptibility would not only allow encapsulated cells to more easily remodel their environment but also make the hydrogels more susceptible to degradation in the presence of increased concentrations of hyaluronidase, such as those found in pro-inflammatory environments43. As such, the optimal DOT of CS-SH and HA-SH will depend on the intended application and target environment of the hydrogel. While others have detailed how crosslink density affected the degradation rate of HA hydrogels, changes in degradation rate depended on both the degree of modification and the diffusivity of species within the hydrogel44,45. By capping free thiols, we were able keep crosslink density constant, removing a confounding factor with regard to hyaluronidase degradation.
3.2.2: Peptide-Glycan Interaction with Thiolated GAG Hydrogels
Another selected measure of bioactivity with regard to the DOT of the GAGs was the ability for biomolecules to interact with or bind to the thiolated GAGs, as occurs between proteins, such as growth factors, and GAGs within the extracellular matrix (ECM). As biomolecule analogs, the peptide-glycans CS-GAH and HA-YKT, which bind to HA and CS, respectively, served as proxies for protein and proteoglycans that interact with GAGs. These molecules were chosen as biomolecule analogs due to our previous experience developing and characterizing peptide-glycan constructs, with CS-GAH in particular previously described as an aggrecan mimetic that binds to HA35,46. Both GAH and YKT peptides were originally discovered from peptide arrays with the intention of maximizing interaction with HA and CS, respectively47,48. Following incubation with solubilized CS-GAH, which had an average of ten peptides bound to one CS molecule, it was found that significantly less CS-GAH bound to HA-SH of higher DOT compared to that of the lower DOT (Figure 3). Similarly, significantly less HA-YKT, which had an average 30 peptides bound to one HA molecule, bound to the high thiolation CS-SH compared to the lower thiolation CS-SH (Figure 3). These results are consistent with other previously reported protein binding studies. Kwon et al demonstrated decreased CD44 binding to HA as the HA was increasingly modified31. Although the HA and CS binding peptides are not derived from a specific protein, cationic charge motifs similar to those found in GAH and YKT peptides have been documented in the heparin binding domains of several proteins49. Because of these similarities in amino acid sequence and charge, the ability of these peptide-glycans to bind to HA-SH and CS-SH provides insight as to how heparin binding proteins might interact with the GAGs as a function of DOT. After six days of culture in HA-SH and CS-SH gels of high and low DOT, a small increase in MSC viability was found in the low DOT CS-SH gel compared to the high DOT gel, but no difference was found between the HA-SH gels of low and high DOT (figure S5). This difference in viability may be explained through differences in protein binding, as seen in the peptide-glycan binding assay.
3.3: Effect of HA Molecular Weight on Hydrogel Stiffness and Hyaluronidase Activity
To determine the effect of HA molecular weight on the enzymatic degradation of the resulting hydrogel, 10 kDa, 60 kDa, 100 kDa, 200 kDa, and 500 kDa HA-SH were synthesized with an approximate DOT of 17±0.4%. For this experiment, we hypothesized that hydrogels formed with higher molecular weight HA would be more resistant to degradation due to the increased number of crosslinks per polymer chain and reduced number of chain ends. As such, once one glycosidic bond was hydrolyzed by hyaluronidase, the probability of the cleaved chain remaining connected to the polymer network would be higher for polymers of higher molecular weight. To keep crosslink density consistent between all groups, the same amount of PEGDA was used to crosslink all HA-SH groups. Following 8 hours of enzymatic degradation with hyaluronidase, it was found that between 60 kDa and 500 kDa, there was little difference in the rate of enzymatic degradation, contrary to our initial hypothesis. However, the 10 kDa HA-SH did show significantly greater degradation than did the HA-SH of higher molecular weights (Figure 4a,b). To contextualize this difference in degradation between the different molecular weights of HA-SH, the stiffness of the intact gels was determined. It was found that between 60 kDa and 500 kDa, there was no significant difference in stiffness, but the gels made with 10 kDa HA-SH were significantly weaker than the other groups (Figure 4c). Thus, at an HA MW ≥60 kDA and a DOT of ~17%, the number of end groups does not significantly alter the stiffness of gels or the susceptibility to enzymatic degradation.
Figure 4:

Enzymatic Degradation of HA-SH Hydrogels (DOT: ~17±0.4%) with Respect to HA Molecular Weight. (a) Degradation of HA-SH gels of multiple molecular weights over time. (b) Cumulative degradation of HA-SH gels at eight hours. (c) Storage modulus of HA-SH with respect to HA molecular weight. Groups sharing letters are not statistically different (P > 0.05). * denotes statistical significance (P < 0.05) from all other groups. n.s. denotes no significance between groups ( P > 0.05).
Although the molecular weights of HA used in other reported hydrogel systems have varied between studies and ranged from 40 kDa50 to 1.5 MDa36, the results reported here suggest that this inconsistency between MW of HA studied should not have a large effect on the enzymatic degradation rates nor on the stiffness of HA gels. Therefore, when fabricating HA only or DPN hydrogels, molecular weights above 60 kDa may not be one of the governing factors in polymer design with regard to degradation or stiffness. Going forward, 100 kDa HA-SH was chosen for CS/HA DPN experiments as it readily dissolved at the high concentrations required for hydrogel synthesis compared to the 200 kDa and 500 kDa variants, which exhibited lower solubility and are challenging to work with due to higher solution viscosity.
3.4: Mechanical Properties of CS/HA DPN Hydrogels
After determining the effects of DOT on degradation and peptide-glycan binding, we sought to characterize hydrogels fabricated with a CS/HA DPN network. The goal of this study was to develop an understanding of how the combination of CS and HA, specifically the ratio in which the two GAGs were incorporated, affected the properties of the hydrogel. Two main considerations regarding the GAGs were taken into account. First, the molecular weight of the HA was more than double that of the CS, and therefore crosslinking the two polymers together at different ratios may affect the stiffness and degradation of the materials. Second, due to the sulfate groups present on CS but not HA, modulating the CS to HA ratio of the CS/HA DPN hydrogels would affect the internal charge of the hydrogels and could in turn affect swelling, stiffness, and degradation.
To test the mechanical properties of the CS/HA DPN hydrogels, gels with CS to HA w/w ratios of 10:0, 7:3, 5:5, 3:7, and 0:10 were fabricated with the same amount of PEGDA crosslinker to ensure that the crosslink density was consistent between groups. All groups containing any amount of CS had shear moduli that were not significantly different from one another (Figure 5). However, the group containing only HA had a significantly higher G’ than all groups containing CS, including the DPN group that was predominantly HA (Figure 5). These results suggest that inclusion of CS into the DPN reduces the modulus of the gel, but this reduction did not depend on CS concentration. Although it has been shown that inclusion of CS into collagen hydrogels can decrease gel stiffness through interactions with the protein, it has not been shown previously that CS has a similar effect on HA gels51. This observation warrants further future study to determine whether CS-HA interactions, or perhaps the presence of increased negative charge, are responsible for altering gel polymer structure and therefore the stiffness of these gels.
Figure 5:

Storage Modulus of CS/HA (CS MW: 40kDa, CS DOT: 17.6%; HA MW: 100 kDa, HA DOT: 17.8%; 1.5 w/v% GAG) DPN hydrogels at 1 rad/s. * denotes statistical significance (P < 0.05) from all other groups. n.s. denotes no significance between groups (P > 0.05)
3.5: CS/HA DPN Hydrogels Resist Enzymatic Degradation
The enzymatic degradation of CS/HA DPNs was tested to determine the effect of blending the two polymers on the degradation of the bulk hydrogel, given the differences in enzymatic degradation between CS-SH and HA-SH. Chemical analysis of the hydrogel supernatant could not be performed due to a lack of HA-specific assays. For example, the carbazole assay measures the presence of glucuronic acid from HA and CS indiscriminately, whereas HA enzyme-linked immunosorbent assays (ELISAs) are limited because of decreased recognition of the modified, short fragments of HA that are released upon degradation52. As a result, bulk degradation of the DPN hydrogels was assessed through recording the total mass of the swollen gels daily and measuring the change in mass of the polymer content within the gel at the end of seven days. As the gels degraded over the course of seven days, most gels experienced an initial increase in mass likely caused by decreased crosslink density and increased swelling capacity (Figure 6a,c). At longer times, a decrease in mass was observed likely coincident with sufficient polymer degradation to release free polymer fragments from the gels (Figure 6a,d). Although all groups reached a similar maximum increase in mass (~14–18%), the time point at which they did so roughly corresponded to their relative CS content. Groups with increased relative CS content reached their maximum mass earlier, and groups with increased relative HA content reached their peak later (Figure 6a).
Figure 6:

Swelling and Degradation Mechanics of CS/HA (CS MW: 40kDa, CS DOT: 17.6%; HA MW: 100 kDa, HA DOT: 17.8%; 1.5 w/v% GAG) DPN Hydrogels in the Presence of Hyaluronidase. (a) Change in total mass of gels over time. (b) Percent polymer degradation after seven days. (c) Percent change in total gel mass after four days of hyaluronidase treatment. (d) Percent change in total gel mass after seven days hyaluronidase treatment. Groups sharing letters are not statistically different (P > 0.05). * denotes statistical significance (P < 0.05) from all other groups. n.s. denotes no significance between groups (P > 0.05)
Based upon findings regarding the degradation of the CS-SH and HA-SH homopolymer gels, we expected the CS only gel to have the slowest degradation rate and that increased CS content in the DPN groups would result in slower degradation. However, after seven days, the percentage of the polymer degradation of the DPN hydrogels was significantly lower than both the CS and HA homopolymer gels (Figure 6b). Over the same period, there was no significant difference in polymer content loss between the DPN groups. Although initial homopolymer tests did show resistance to hyaluronidase by CS, that previous experiment did not explore the long-term degradation mechanics of the gels. As such, our initial hypothesis did not consider the swelling mechanics of DPN hydrogels as they degraded over a longer period of time. The CS hydrogel swelled to a greater degree and at a faster rate than all of the other groups, likely due to the high density of the negatively-charged sulfate groups on the CS backbone, and this swelling may have allowed for increased hyaluronidase infiltration and more rapid degradation. On the other hand, although the HA did not swell to the same degree as the CS gel, HA is more susceptible to hyaluronidase degradation and thus the HA gel degraded at a faster rate than the CS gel. Based on the observed degradation of the HA-only and CS-only gels, it is believed that DPN hydrogels degraded less due to a balance of decreased hyaluronidase-susceptibility of CS and the lower swelling capacity of HA. As such, given the multifaceted nature of exogenous enzyme degradation involving both enzyme substrate recognition and enzyme infiltration, DPN hydrogels demonstrated reduced polymer degradation over time.
3.6: CS/HA DPN Hydrogels Promote MSC Viability
Although it is known that MSCs are able to interact with HA through the CD44 receptor, it has not been shown directly that MSCs use CD44 to interact with CS. However, CD44 is able to interact with CS in a cell-free system, and these results suggest that MSCs can bind to CS through CD4453. To evaluate the ability of the CS/HA DPN hydrogels to support cell survival, rabbit MSCs were encapsulated and cultured in gels for periods of one and six days. At the end of these time points, live and dead cells were stained and counted to determine average cell viability with respect to the hydrogel formulation. After one day of culture, no significant difference was found in cell viability, and average cell viability of the MSCs ranged from 83% to 87% (Figure 7). After culturing for six days, it was found that the cell populations in the HA homopolymer gel decreased and exhibited an average viability of 60% (Figure 7). In contrast, cell populations cultured in groups containing any amount of CS showed significantly higher viability at six days, ranging from 76% to 83%, and no significant difference was found between groups incorporating CS (Figure 7).
Figure 7:

Live/dead assay of MSCs encapsulated in CS/HA (CS MW: 40kDa, CS DOT: 17.6%; HA MW: 100 kDa, HA DOT: 17.8%; 1.5 w/v% GAG) DPN Hydrogels. (a) MSCs in CS/HA DPN Hydrogels stained with Calcein AM (green) and Ethidium homodimer (red) at day six. Scale bar represents 100 μm (b) MSC viability at day one, n = 3. (c) MSC viability at day six, n = 3. (d) Change in MSC viability between day one and day six. * denotes statistical significance (P < 0.05) from all other groups. n.s. denotes no significance between groups (P > 0.05)
Although the MSCs should be able to bind and interact with the HA only gel via CD44, this formulation showed decreased cell viability compared to all CS groups. This finding suggests that the biological signals, in addition to CD44 interactions, provided by CS promote cell viability, but this signal does not promote cell viability in a dose-dependent manner. An alternative explanation to increased biological signaling coming directly from CS is that swelling due to increased CS content may have allowed enhanced nutrient diffusion and thus increased cell viability. Furthermore, the HA only gel was found to be significantly stiffer than all CS containing gels, with this increase in stiffness also possibly contributing to decreases in cell viability. Of note, all groups showed significant decreases in the percentage of viable cells between one day and six days of culture. This decrease may have been due to the lack of other supporting biological polymers, such as collagen, as similar decreases in cell viability over time in GAG-only gels have been reported elsewhere18,54. In those cases, cell viability increased following the inclusion of collagen into the polymer network. To promote cell viability in future studies, collagen or other ECM proteins can be included to provide support for encapsulated cells.
4. Conclusion
In this study we demonstrated the effects of CS-SH and HA-SH polymer design on the physical and bioactive properties of hydrogels. The minimum DOT was determined for the formation of robust hydrogels. HA molecular weight ≥60 kDa was not found to be a large factor impacting the enzymatic degradation and hydrogel stiffness. Increasing the DOT of both HA-SH and CS-SH was found to decrease their bioactivity as determined by both their ability to be degraded by hyaluronidase and their ability to interact with HA and CS-binding peptides. These assays were performed as analogues for cellular interactions with their environment. CS-SH and HA-SH were incorporated together into DPN hydrogels with the eventual goal of stimulating encapsulated cells with the combined biological signals of the GAGs. DPN hydrogels resisted degradation by hyaluronidase to a greater degree than CS and HA homopolymer gels. Finally, the incorporation of CS in any amount into the DPN hydrogels demonstrated an increased ability to promote cell viability over the HA homopolymer gel.
Supplementary Material
Acknowledgements:
We would like to thank the Ripplinger lab at UC Davis for providing the rabbit hind limbs used in this experiment for MSC isolation. This work was funded by the NIH grant T32 HL086350.
Abbreviations:
- CS
chondroitin sulfate
- CS-SH
thiolated chondroitin sulfate
- DMEM
Dulbecco’s modified eagle media
- DMMB
dimethylmethylene blue
- DMTMM
4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride
- DOT
degree of thiolation
- DPN
dual polymer network
- DTP
dithio-bis(propionohydrazide)
- ELISA
enzyme-linked immunosorbent assays
- ECM
extracellular matrix
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- GAH
GAHWQFNALTVGSG
- GAG
Glycosaminoglycan
- HA
hyaluronic acid
- HA-SH
thiolated hyaluronic acid
- MES
2-(N-morpholino)ethanesulfonic acid
- MSC
mesenchymal stromal cell
- MW
molecular weight
- NEM
N-ethyl maleimide
- PEGDA
poly(ethylene glycol) diacrylate
- TFF
tangential flow filtration
- YKT
YKTNFRRYYRFGSG
Footnotes
Supporting Information:
• Quantification of free thiols by Ellman’s assay; 1H NMR spectra of HA-SH high DOT (30.4%), HA-SH low DOT (17.8%), CS-SH high DOT (34.8%), and CS-SH low DOT (17.6%), G’ of HA-SH high DOT, HA-SH low DOT, CS-SH high DOT, and CS-SH low DOT hydrogels; MSC viability in HA-SH high DOT, HA-SH low DOT, CS-SH high DOT, and CS-SH low DOT hydrogels at day one and day six.
6. References:
- (1).Roddy E; DeBaun MR; Daoud-Gray A; Yang YP; Gardner MJ Treatment of Critical-Sized Bone Defects: Clinical and Tissue Engineering Perspectives. Eur J Orthop Surg Traumatol 2018, 28 (3), 351–362. 10.1007/s00590-017-2063-0. [DOI] [PubMed] [Google Scholar]
- (2).Groeber F; Holeiter M; Hampel M; Hinderer S; Schenke-Layland K. Skin Tissue Engineering — In Vivo and in Vitro Applications. Adv. Drug Delivery Rev. 2011, 63 (4), 352–366. 10.1016/j.addr.2011.01.005. [DOI] [PubMed] [Google Scholar]
- (3).Spicer CD Hydrogel Scaffolds for Tissue Engineering: The Importance of Polymer Choice. Polym. Chem. 2020, 11 (2), 184–219. 10.1039/C9PY01021A. [DOI] [Google Scholar]
- (4).Lin L; Zhu J; Kottke-Marchant K; Marchant RE Biomimetic-Engineered Poly (Ethylene Glycol) Hydrogel for Smooth Muscle Cell Migration. Tissue Eng., Part A 2013, 20 (3–4), 864–873. 10.1089/ten.tea.2013.0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Zustiak SP; Durbal R; Leach JB Influence of Cell-Adhesive Peptide Ligands on Poly(Ethylene Glycol) Hydrogel Physical, Mechanical and Transport Properties. Acta Biomater. 2010, 6 (9), 3404–3414. 10.1016/j.actbio.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Flaig F; Ragot H; Simon A; Revet G; Kitsara M; Kitasato L; Hébraud A; Agbulut O; Schlatter G. Design of Functional Electrospun Scaffolds Based on Poly(Glycerol Sebacate) Elastomer and Poly(Lactic Acid) for Cardiac Tissue Engineering. ACS Biomater. Sci. Eng. 2020, 6 (4), 2388–2400. 10.1021/acsbiomaterials.0c00243. [DOI] [PubMed] [Google Scholar]
- (7).Bidault L; Deneufchatel M; Hindié M; Vancaeyzeele C; Fichet O; Larreta-Garde V. Fibrin-Based Interpenetrating Polymer Network Biomaterials with Tunable Biodegradability. Polymer 2015, 62, 19–27. 10.1016/j.polymer.2015.02.014. [DOI] [Google Scholar]
- (8).Ahmed TAE; Dare EV; Hincke M. Fibrin: A Versatile Scaffold for Tissue Engineering Applications. Tissue Eng., Part B 2008, 14 (2), 199–215. 10.1089/ten.teb.2007.0435. [DOI] [PubMed] [Google Scholar]
- (9).Ma K; Titan AL; Stafford M; Zheng C. hua; Levenston, M. E. Variations in Chondrogenesis of Human Bone Marrow-Derived Mesenchymal Stem Cells in Fibrin/Alginate Blended Hydrogels. Acta Biomater. 2012, 8 (10), 3754–3764. 10.1016/j.actbio.2012.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lotz C; Schmid FF; Oechsle E; Monaghan MG; Walles H; Groeber-Becker F. Cross-Linked Collagen Hydrogel Matrix Resisting Contraction To Facilitate Full-Thickness Skin Equivalents. ACS Appl. Mater. Interfaces 2017, 9 (24), 20417–20425. 10.1021/acsami.7b04017. [DOI] [PubMed] [Google Scholar]
- (11).Buitrago JO; Patel KD; El-Fiqi A; Lee J-H; Kundu B; Lee H-H; Kim H-W Silk Fibroin/Collagen Protein Hybrid Cell-Encapsulating Hydrogels with Tunable Gelation and Improved Physical and Biological Properties. Acta Biomater. 2018, 69, 218–233. 10.1016/j.actbio.2017.12.026. [DOI] [PubMed] [Google Scholar]
- (12).Patterson J; Siew R; Herring SW; Lin ASP; Guldberg R; Stayton PS Hyaluronic Acid Hydrogels with Controlled Degradation Properties for Oriented Bone Regeneration. Biomaterials 2010, 31 (26), 6772–6781. 10.1016/j.biomaterials.2010.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhai P; Peng X; Li B; Liu Y; Sun H; Li X. The Application of Hyaluronic Acid in Bone Regeneration. Int. J. Biol. Macromol. 2020, 151, 1224–1239. 10.1016/j.ijbiomac.2019.10.169. [DOI] [PubMed] [Google Scholar]
- (14).Zhu M; Feng Q; Sun Y; Li G; Bian L. Effect of Cartilaginous Matrix Components on the Chondrogenesis and Hypertrophy of Mesenchymal Stem Cells in Hyaluronic Acid Hydrogels: Effect of Cartilaginous Matrix Components on Chondrogenesis and Hypertrophy of Human MSCs. J. Biomed. Mater. Res. 2017, 105 (8), 2292–2300. 10.1002/jbm.b.33760. [DOI] [PubMed] [Google Scholar]
- (15).Li H; Qi Z; Zheng S; Chang Y; Kong W; Fu C; Yu Z; Yang X; Pan S. The Application of Hyaluronic Acid-Based Hydrogels in Bone and Cartilage Tissue Engineering. Adv. Mater. Sci. Eng. 2019, 2019, e3027303. 10.1155/2019/3027303. [DOI] [Google Scholar]
- (16).Hu M; Sabelman EE; Cao Y; Chang J; Hentz VR Three-Dimensional Hyaluronic Acid Grafts Promote Healing and Reduce Scar Formation in Skin Incision Wounds. J. Biomed. Mater. Res., Part B 2003, 67B (1), 586–592. 10.1002/jbm.b.20001. [DOI] [PubMed] [Google Scholar]
- (17).Mahapatra C; Jin G-Z; Kim H-W Alginate-Hyaluronic Acid-Collagen Composite Hydrogel Favorable for the Culture of Chondrocytes and Their Phenotype Maintenance. Tissue Eng. Regen. Med. 2016, 13 (5), 538–546. 10.1007/s13770-016-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Walimbe T; Calve S; Panitch A; Sivasankar MP Incorporation of Types I and III Collagen in Tunable Hyaluronan Hydrogels for Vocal Fold Tissue Engineering. Acta Biomater. 2019, 87, 97–107. 10.1016/j.actbio.2019.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Suri S; Schmidt CE Cell-Laden Hydrogel Constructs of Hyaluronic Acid, Collagen, and Laminin for Neural Tissue Engineering. Tissue Eng., Part A 2010, 16 (5), 1703–1716. 10.1089/ten.tea.2009.0381. [DOI] [PubMed] [Google Scholar]
- (20).Fraser JRE; Laurent TC; Laurent UBG Hyaluronan: Its Nature, Distribution, Functions and Turnover. J. Intern. Med. 1997, 242 (1), 27–33. 10.1046/j.1365-2796.1997.00170.x. [DOI] [PubMed] [Google Scholar]
- (21).Mourão PAS Distribution of Chondroitin 4–Sulfate and Chondroitin 6–Sulfate in Human Articular and Growth Cartilage. Arthritis Rheum. 1988, 31 (8), 1028–1033. 10.1002/art.1780310814. [DOI] [PubMed] [Google Scholar]
- (22).Ishida O; Tanaka Y; Morimoto I; Takigawa M; Eto S. Chondrocytes Are Regulated by Cellular Adhesion Through CD44 and Hyaluronic Acid Pathway. J. Bone Miner. Res. 1997, 12 (10), 1657–1663. 10.1359/jbmr.1997.12.10.1657. [DOI] [PubMed] [Google Scholar]
- (23).Hamann KJ; Dowling TL; Neeley SP; Grant JA; Leff AR Hyaluronic Acid Enhances Cell Proliferation during Eosinopoiesis through the CD44 Surface Antigen. J. Immunol. 1995, 154 (8), 4073–4080. [PubMed] [Google Scholar]
- (24).Varghese S; Hwang NS; Canver AC; Theprungsirikul P; Lin DW; Elisseeff J. Chondroitin Sulfate Based Niches for Chondrogenic Differentiation of Mesenchymal Stem Cells. Matrix Biol. 2008, 27 (1), 12–21. 10.1016/j.matbio.2007.07.002. [DOI] [PubMed] [Google Scholar]
- (25).Ialenti A; Di Rosa M. Hyaluronic Acid Modulates Acute and Chronic Inflammation. Agents Actions 1994, 43 (1), 44–47. 10.1007/BF02005763. [DOI] [PubMed] [Google Scholar]
- (26).Fenn SL; Oldinski RA Visible Light Crosslinking of Methacrylated Hyaluronan Hydrogels for Injectable Tissue Repair. J. Biomed. Mater. Res. Part B Appl. Biomater. 2016, 104 (6), 1229–1236. 10.1002/jbm.b.33476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shu XZ; Liu Y; Luo Y; Roberts MC; Prestwich GD Disulfide Cross-Linked Hyaluronan Hydrogels. Biomacromolecules 2002, 3 (6), 1304–1311. 10.1021/bm025603c. [DOI] [PubMed] [Google Scholar]
- (28).Owen SC; Fisher SA; Tam RY; Nimmo CM; Shoichet MS Hyaluronic Acid Click Hydrogels Emulate the Extracellular Matrix. Langmuir 2013, 29 (24), 7393–7400. 10.1021/la305000w. [DOI] [PubMed] [Google Scholar]
- (29).Lee F; Eun Chung J; Kurisawa M. An Injectable Enzymatically Crosslinked Hyaluronic Acid – Tyramine Hydrogel System with Independent Tuning of Mechanical Strength and Gelation Rate. Soft Matter 2008, 4 (4), 880–887. 10.1039/B719557E. [DOI] [PubMed] [Google Scholar]
- (30).Mazumder MAJ; Fitzpatrick SD; Muirhead B; Sheardown H. Cell-Adhesive Thermogelling PNIPAAm/Hyaluronic Acid Cell Delivery Hydrogels for Potential Application as Minimally Invasive Retinal Therapeutics. J. Biomed. Mater. Res, Part A 2012, 100A (7), 1877–1887. 10.1002/jbm.a.34021. [DOI] [PubMed] [Google Scholar]
- (31).Kwon MY; Wang C; Galarraga JH; Puré E; Han L; Burdick JA Influence of Hyaluronic Acid Modification on CD44 Binding towards the Design of Hydrogel Biomaterials. Biomaterials 2019, 222, 119451. 10.1016/j.biomaterials.2019.119451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Eng D; Caplan M; Preul M; Panitch A. Hyaluronan Scaffolds: A Balance between Backbone Functionalization and Bioactivity. Acta Biomater. 2010, 6 (7), 2407–2414. 10.1016/j.actbio.2009.12.049. [DOI] [PubMed] [Google Scholar]
- (33).Erickson IE; Huang AH; Sengupta S; Kestle S; Burdick JA; Mauck RL Macromer Density Influences Mesenchymal Stem Cell Chondrogenesis and Maturation in Photocrosslinked Hyaluronic Acid Hydrogels. Osteoarthritis and Cartilage 2009, 17 (12), 1639–1648. 10.1016/j.joca.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yu F; Cao X; Zeng L; Zhang Q; Chen X. An Interpenetrating HA/G/CS Biomimic Hydrogel via Diels–Alder Click Chemistry for Cartilage Tissue Engineering. Carbohydrate Polymers 2013, 97 (1), 188–195. 10.1016/j.carbpol.2013.04.046. [DOI] [PubMed] [Google Scholar]
- (35).Bernhard JC; Panitch A. Synthesis and Characterization of an Aggrecan Mimic. Acta Biomater. 2012, 8 (4), 1543–1550. 10.1016/j.actbio.2011.12.029. [DOI] [PubMed] [Google Scholar]
- (36).Poldervaart MT; Goversen B; de Ruijter M; Abbadessa A; Melchels FPW; Öner FC; Dhert WJA; Vermonden T; Alblas J. 3D Bioprinting of Methacrylated Hyaluronic Acid (MeHA) Hydrogel with Intrinsic Osteogenicity. PLoS One 2017, 12 (6), e0177628. 10.1371/journal.pone.0177628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fajardo AR; Fávaro SL; Rubira AF; Muniz EC Dual-Network Hydrogels Based on Chemically and Physically Crosslinked Chitosan/Chondroitin Sulfate. React. Funct. Polym. 2013, 73 (12), 1662–1671. 10.1016/j.reactfunctpolym.2013.10.003. [DOI] [Google Scholar]
- (38).Gao Y; Li B; Kong W; Yuan L; Guo L; Li C; Fan H; Fan Y; Zhang X. Injectable and Self-Crosslinkable Hydrogels Based on Collagen Type II and Activated Chondroitin Sulfate for Cell Delivery. Int. J. Biol. Macromol. 2018, 118, 2014–2020. 10.1016/j.ijbiomac.2018.07.079. [DOI] [PubMed] [Google Scholar]
- (39).Möller L; Krause A; Dahlmann J; Gruh I; Kirschning A; Dräger G. Preparation and Evaluation of Hydrogel-Composites from Methacrylated Hyaluronic Acid, Alginate, and Gelatin for Tissue Engineering. Int J Artif Organs 2011, 34 (2), 93–102. 10.5301/IJAO.2011.6397. [DOI] [PubMed] [Google Scholar]
- (40).Hozumi T; Kageyama T; Ohta S; Fukuda J; Ito T. Injectable Hydrogel with Slow Degradability Composed of Gelatin and Hyaluronic Acid Cross-Linked by Schiff’s Base Formation. Biomacromolecules 2018, 19 (2), 288–297. 10.1021/acs.biomac.7b01133. [DOI] [PubMed] [Google Scholar]
- (41).Tous E; Ifkovits JL; Koomalsingh KJ; Shuto T; Soeda T; Kondo N; Gorman JH; Gorman RC; Burdick JA Influence of Injectable Hyaluronic Acid Hydrogel Degradation Behavior on Infarction-Induced Ventricular Remodeling. Biomacromolecules 2011, 12 (11), 4127–4135. 10.1021/bm201198x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Gennari A; Wedgwood J; Lallana E; Francini N; Tirelli N. Thiol-Based Michael-Type Addition. A Systematic Evaluation of Its Controlling Factors. Tetrahedron 2020, 76 (47), 131637. 10.1016/j.tet.2020.131637. [DOI] [Google Scholar]
- (43).Yoshida M; Sai S; Marumo K; Tanaka T; Itano N; Kimata K; Fujii K. Expression Analysis of Three Isoforms of Hyaluronan Synthase and Hyaluronidase in the Synovium of Knees in Osteoarthritis and Rheumatoid Arthritis by Quantitative Real-Time Reverse Transcriptase Polymerase Chain Reaction. Arthritis Res Ther 2004, 6 (6), R514. 10.1186/ar1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Jeon O; Song SJ; Lee K-J; Park MH; Lee S-H; Hahn SK; Kim S; Kim B-S Mechanical Properties and Degradation Behaviors of Hyaluronic Acid Hydrogels Cross-Linked at Various Cross-Linking Densities. Carbohydr. Polym. 2007, 70 (3), 251–257. 10.1016/j.carbpol.2007.04.002. [DOI] [Google Scholar]
- (45).Nimmo CM; Owen SC; Shoichet MS Diels−Alder Click Cross-Linked Hyaluronic Acid Hydrogels for Tissue Engineering. Biomacromolecules 2011, 12 (3), 824–830. 10.1021/bm101446k. [DOI] [PubMed] [Google Scholar]
- (46).Sharma S; Panitch A; Neu CP Incorporation of an Aggrecan Mimic Prevents Proteolytic Degradation of Anisotropic Cartilage Analogs. Acta Biomater. 2013, 9 (1), 4618–4625. 10.1016/j.actbio.2012.08.041. [DOI] [PubMed] [Google Scholar]
- (47).Mummert ME; Mohamadzadeh M; Mummert DI; Mizumoto N; Takashima A. Development of a Peptide Inhibitor of Hyaluronan-Mediated Leukocyte Trafficking. J Exp Med 2000, 192 (6), 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Butterfield KC; Caplan M; Panitch A. Identification and Sequence Composition Characterization of Chondroitin Sulfate-Binding Peptides through Peptide Array Screening. Biochemistry 2010, 49 (7), 1549–1555. 10.1021/bi9021044. [DOI] [PubMed] [Google Scholar]
- (49).Heparin-Binding Domains in Vascular Biology | Arteriosclerosis, Thrombosis, and Vascular Biology 10.1161/01.ATV.0000137189.22999.3f (accessed 2021 −08 −19). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Gwon K; Kim E; Tae G. Heparin-Hyaluronic Acid Hydrogel in Support of Cellular Activities of 3D Encapsulated Adipose Derived Stem Cells. Acta Biomater. 2017, 49, 284–295. 10.1016/j.actbio.2016.12.001. [DOI] [PubMed] [Google Scholar]
- (51).Stuart K; Panitch A. Influence of Chondroitin Sulfate on Collagen Gel Structure and Mechanical Properties at Physiologically Relevant Levels. Biopolymers 2008, 89 (10), 841–851. 10.1002/bip.21024. [DOI] [PubMed] [Google Scholar]
- (52).Yuan H; Tank M; Alsofyani A; Shah N; Talati N; LoBello JC; Kim JR; Oonuki Y; de la Motte CA; Cowman MK Molecular Mass Dependence of Hyaluronan Detection by Sandwich ELISA-like Assay and Membrane Blotting Using Biotinylated Hyaluronan Binding Protein. Glycobiology 2013, 23 (11), 1270–1280. 10.1093/glycob/cwt064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Fujimoto T; Kawashima H; Tanaka T; Hirose M; Toyama-Sorimachi N; Matsuzawa Y; Miyasaka M. CD44 Binds a Chondroitin Sulfate Proteoglycan, Aggrecan. Int. Immunol. 2001, 13 (3), 359–366. 10.1093/intimm/13.3.359. [DOI] [PubMed] [Google Scholar]
- (54).Spearman BS; Agrawal NK; Rubiano A; Simmons CS; Mobini S; Schmidt CE Tunable Methacrylated Hyaluronic Acid-Based Hydrogels as Scaffolds for Soft Tissue Engineering Applications. J. Biomed. Mater. Res. Part A 2020, 108 (2), 279–291. 10.1002/jbm.a.36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.