

Abstract
RNA biology has risen to prominence after a remarkable discovery of diverse functions of noncoding RNA (ncRNA). Most untranslated transcripts often exert their regulatory functions into RNA–RNA complexes via base pairing with complementary sequences in other RNAs. An interplay between RNAs is essential, as it possesses various functional roles in human cells, including genetic translation, RNA splicing, editing, ribosomal RNA maturation, RNA degradation and the regulation of metabolic pathways/riboswitches. Moreover, the pervasive transcription of the human genome allows for the discovery of novel genomic functions via RNA interactome investigation. The advancement of experimental procedures has resulted in an explosion of documented data, necessitating the development of efficient and precise computational tools and algorithms. This review provides an extensive update on RNA–RNA interaction (RRI) analysis via thermodynamic- and comparative-based RNA secondary structure prediction (RSP) and RNA–RNA interaction prediction (RIP) tools and their general functions. We also highlighted the current knowledge of RRIs and the limitations of RNA interactome mapping via experimental data. Then, the gap between RSP and RIP, the importance of RNA homologues, the relationship between pseudoknots, and RNA folding thermodynamics are discussed. It is hoped that these emerging prediction tools will deepen the understanding of RNA-associated interactions in human diseases and hasten treatment processes.
Keywords: RNA–RNA interaction prediction, RNA interactome, RNA structure prediction, computational tools
INTRODUCTION
More than 60 years ago, the central dogma of molecular biology was first introduced by Francis Crick as a model to describe the transfer of genetic information from DNA to protein [1]. Since then, several attempts have been made to interpret the composition of RNA subtypes in the human genome and their roles in protein synthesis [2, 3]. Typically, Watson–Crick base-pairing is known to maintain the genetic continuity of RNA replication, and encoded proteins are not involved as catalysts [1, 4]. The adaptability of RNA molecules has spawned the ‘RNA World’ hypothesis, in which RNA replication-based evolution takes precedence over DNA-centred evolution and protein synthesis [2, 5–7]. The ‘RNA World’ hypothesis depicts the possibility of storing genetic material via RNA alone and its ability to self-replicate as the primary source of catalytic mechanisms without the involvement of proteins [8–17]. Since the discovery of protein-encoding messenger RNA (mRNA) in the 1960s, it has received a great deal of attention due to its critical function in protein synthesis and is considered the inevitable intermediary necessity in producing proteins [18]. Nevertheless, high-throughput sequencing platforms create a paradigm shift, as over 90% of the human genome is transcribed into RNA [18, 19]. Of all, 2% of the RNA in the genome encodes proteins, while the remaining is easily transcribed into nonprotein-encoded RNA (also known as noncoding RNA or ncRNA) molecules [20–24]. In summary, advances in sequencing technology have enabled the discovery of ncRNAs, bringing RNA biology to the forefront and revealing the intricate role of ncRNAs in human cells [25–28].
Noncoding RNAs (ncRNAs) are RNA molecules that are not translated into proteins. Their length can be classified into three categories: (i) short (19 to 31 nucleotides), (ii) mid (20 to 200 nucleotides) and (iii) long (>200 nucleotides) [29]. Among them, microRNAs (miRNAs) are the most well-studied short ncRNAs, acting as supplementary posttranscriptional regulators and ‘buffers’ that maintain the robustness of biological systems [30, 31]. In contrast, long ncRNAs (lncRNAs) are the largest ncRNA subtype, with approximately 55 000 genes along the genome [32, 33].
Despite extensive functional studies, the molecular mechanisms of ncRNA-centric roles remain elusive and require advances in experimental biomedicine [34–36]. However, emerging RNA–RNA interaction (RRI) tools offer promise in reducing experimental efforts. Understanding these mechanisms requires investigating ncRNA interactions with cellular components such as proteins, DNA sites and other RNAs [37]. Remarkably, numerous classical ncRNAs communicate with other RNA subtypes, either directly via base pairing or indirectly via protein intermediates. Examples include transfer RNA-messenger RNA (tRNA–mRNA) interactions to translate genetic code; miRNA–mRNA interactions to stimulate mRNA degradation; and mRNA–protein interactions involving RNA splicing, editing and ribosomal RNA maturation [38–41]. These findings imply that RRIs portray a universal strategy utilized by many ncRNAs, and completely mapping these interactions could provide insight into ncRNA functions and mechanisms. The RNA interactome has emerged as a central component of many regulatory processes, prompting extensive research from both wet lab and computational researchers [42–45]. Nonetheless, mapping RRIs remains challenging, as current methods struggle to identify and differentiate between direct and indirect RRIs and may have limited resolution for specific RNA examination.
TYPES OF INTERACTIONS
RNA molecules are not just passive carriers of genetic information; they actively participate in various cellular processes through their interactions with other molecules [46]. Understanding these roles and interactions is crucial for advancing our knowledge of cellular biology. RNA molecules interact with other RNAs, proteins and DNA to carry out their functions.
RNA–DNA interactions are essential for several biological processes. One of the most well-known examples is transcription, where an RNA molecule is synthesised based on the DNA template. Another example is the process of reverse transcription in retroviruses, where viral RNA is reverse transcribed into DNA. For instance, in RNA interference (RNAi), small RNA molecules can bind to complementary sequences in mRNA molecules, leading to their degradation and thus preventing their translation into proteins [47]. Another example is the clustered regularly interspaced short palindromic repeats system, a bacterial defense mechanism that has been adapted for genome editing. In this Nobel-prize winner system, RNA molecules guide the Cas9 nuclease to specific locations in the DNA, allowing precise cuts to be made [48]. More recent studies have also highlighted the role of ncRNAs in regulating chromatin architecture via interaction with DNA or chromatin-associated proteins to modulate gene expression. Some ncRNAs function through the formation of R-loops with the complementary sequence from their transcribed loci and affect local gene expression [49].
RNA–protein interactions are fundamental to cellular processes and play a crucial role in the life cycle of an RNA molecule, from its synthesis and processing to its eventual function in protein synthesis. Proteins can bind to RNA to form ribonucleoprotein complexes, which are involved in various aspects of RNA metabolism, including splicing, polyadenylation, stability, transport, and translation [50]. The spliceosome, a large ribonucleoprotein complex, is responsible for removing introns from pre-mRNA, a process known as splicing, and is crucial for the maturation of mRNA molecules and their subsequent translation into proteins [51]. RNA–protein interactions also play a role in polyadenylation, the addition of a poly(A) tail to the 3′ end of an mRNA molecule that enhances the stability of the mRNA and facilitates its export from the nucleus and transport within the cell [52]. During protein translation, mRNA molecules interact with ribosomes, which are themselves ribonucleoprotein complexes, to synthesize proteins based on the sequence of the mRNA that determines the sequence of amino acids in the protein [53].
RNA also interacts with other RNA. For instance, RNA molecules can form complex secondary and tertiary structures through interactions with other RNA molecules, whereby these structures are critical for the function of many types of RNA, including ribosomal RNA (rRNA), transfer RNA (tRNA) and mRNA [54]. In the ribosome, which is a complex of rRNA and proteins, mRNA and tRNA interact to facilitate protein synthesis. The rRNA provides the structural framework for the ribosome and contributes to its catalytic activity [55]. RRIs also play a role in the regulation of gene expression. For instance, miRNAs can base-pair with target mRNAs to repress their translation or induce their degradation [56]. Dysregulation of RRIs can lead to various diseases. For example, mutations that affect the secondary structure of RNA can disrupt normal RRIs and lead to diseases such as cancer [57]. Understanding these interactions is crucial, as they play a significant role in cellular processes, and their dysregulation can lead to various diseases. Therefore, tools that can predict and analyse these interactions are of great importance in advancing our knowledge of cellular biology and developing therapeutic strategies.
Types of RNA–RNA interactions
There are two main types of interactions in RNA molecules, namely, cis-only and trans RRIs (Figure 1). The former is defined as the intramolecular base pairing between nucleotides within a single RNA molecule (Figure 1B) [58]. It permits canonical Watson–Crick base-pairing between {adenine (A) and uracil (U)} and {guanine (G) and cytosine (C)} and non-Watson–Crick/wobble base-pairing between {guanine (G) and uracil (U)} (formed by edge-to-edge hydrogen bonding interactions between the bases) (Figure 1A) [59–61]. The intramolecular RRI aids in the formation of short double-stranded helices and allows folding into specific 3D structures such as tRNA and mRNA, which form the basis for molecular recognition events [62, 63].
Figure 1.
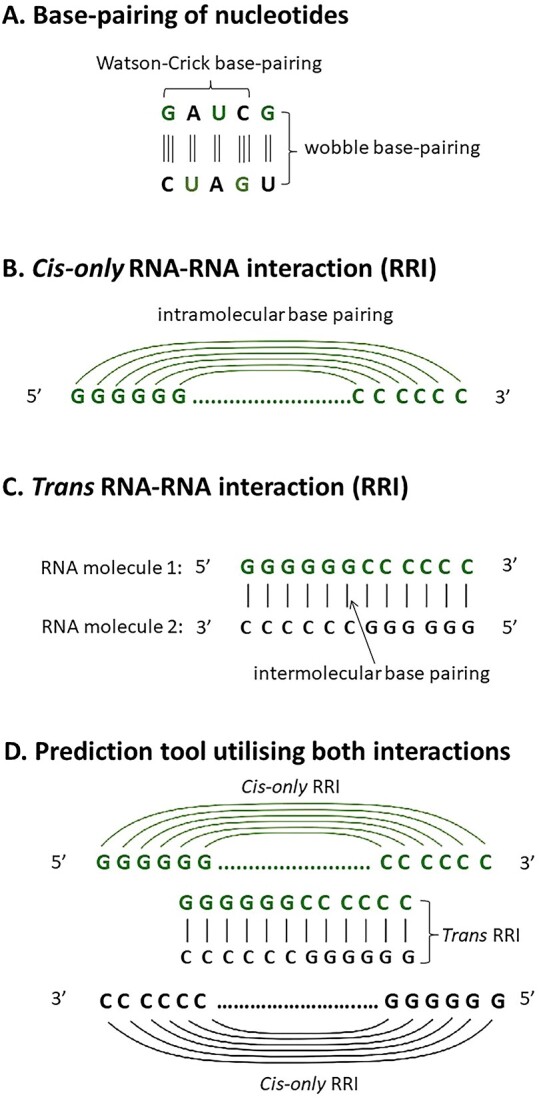
Potential interactions in RNA molecules. (A) Possible base-pairing of nucleotides. (B) cis-only RRI (intramolecular base-pairing) within a single RNA molecule. (C) Trans RRI (intermolecular base-pairing) between two identical RNA molecules. (D) Situation in concatenation-based prediction tool, where both RRI types are involved. Inter- and intramolecular base pairs are indicated by vertical pipe symbols and arches, respectively (adapted from [25]).
On the other hand, trans RRI is made up of two or more RNAs that interact intermolecularly via Watson–Crick base pairing, wobble base pairing or helical stacking (Figure 1C) [64, 65]. miRNAs, for example, can target the 3′ untranslated regions (3’ UTRs) of mRNAs [66–68], whereas spliceosomal small nuclear RNAs (snRNAs) recognize the intronic regions of pre-mRNAs [69, 70]. Duplex formation through base pairing of complementary nucleotides leads to naturally occurring RRIs. They are crucial for various processes, including RNA cleavage, RNA editing, RNA modification, RNA splicing, RNA translation, suppression of RNA translation and RNA degradation [71–75]. Additionally, base-pair interactions are crucial for semiconservative replication, energetically favourable arrangement of base pairs, and the formation of helical RNA structures [76]. Intramolecular interactions lead to the formation of RNA secondary structures, which is why researchers commonly refer to the prediction of cis-only RRIs as the method for RNA structure prediction (RSP). To summarise, intramolecular interactions form secondary RNA structures (cis-only RRI), while intermolecular interactions occur when two individual RNAs interact (trans RRI).
Predicting RRIs based solely on intra- or intermolecular interactions presents significant challenges due to the diverse conformations [77, 78] and conformational changes of RNA molecules [79–81]. Complexities also arise from the three-dimensional folding, secondary structures [82] and tertiary interactions of RNA molecules [83]. Therefore, focusing exclusively on one type of RNA interaction may result in the oversight of crucial interactions occurring across different regions of an RNA molecule [84]. Nonetheless, concatenating both intra- and intermolecular RNA interactions (Figure 1D) permits a more comprehensive analysis, capturing a broader range of interactions and revealing complex RNA networks. This integrated approach provides a more realistic representation of RRIs in biological systems and offers insights into their contribution to overall RNA architecture. Utilizing both types of interactions for prediction provides a more robust and holistic framework compared to relying on either one alone.
RRIs are modelled at various levels of complexity, depending on their common and distinguishing features, which are translated into sophisticated computational algorithms. Complexity refers to the intricacy and sophistication of the computational approach used to model RRIs. However, current RRI models cannot account for real-time biological and chemical information in the cellular environment, except at a coarser level of detail [85]. These models typically focus on sequence complementarity, thermodynamic stability, or structural motifs, which may not fully capture the intricacies of the cellular context [86]. Using RSP-like algorithm tools could facilitate RRI prediction (RIP) by providing reliable information on interacting nucleotide positions, revealing potential biological roles and regulatory mechanisms of mRNAs and ncRNAs [87]. In short, there is a need for RSP-like algorithms to better understand RNA sequences and their interactions in real time, improving RIP models and gaining deeper insights into their biological significance.
RNA–RNA INTERACTION MAPPING VIA EXPERIMENTAL DATA: LIMITATIONS AND TECHNIQUES
The secondary structure of ncRNA serves as a scaffold for the tertiary structure and facilitates catalytic and ligand binding interactions with various RNAs [33, 44, 88]. RIP tools use similar ideas and algorithms to predict RNA secondary structures. X-ray crystallography (single crystal X-ray diffraction (XRD)) and nuclear magnetic resonance (NMR) spectroscopy are the most accurate and robust conventional methods for detecting three-dimensional (3D) RNA structures [89, 90]. Although XRD provides high atomic resolution with no size limitations, crystallizing 3D RNA structures is challenging. Conversely, NMR excels when crystallization is impossible and provides solution state dynamics but has limitations on molecular weights (<50 kDa) [91]. Combining XRD and NMR results in a more accurate structure determination method, providing ncRNA structural information at a single base-pair resolution [92, 93]. Nonetheless, their widespread application is hampered by high experimental costs, low throughput, limited ncRNA resolution measurements and structure detection in vitro, difficulty in translating to in vivo conformation, and < 0.001% of ncRNAs identified from experimental data [94].
Numerous sequencing-based systems have been developed over the last decade for the experimental identification of RNA interactomes. However, current RRI mapping methods, such as RNA interactome analysis and sequencing (RIA-Seq) and RNA antisense purification (RAP)-Seq, do not directly assay RNA interactomes [95, 96]. Instead, they rely on anchored RNAs and molecular perturbations to identify interaction targets of specific RNAs [97]. This one-RNA-at-a-time approach makes it challenging to comprehensively identify all RRIs. Following this, several high-throughput techniques, including PARIS [98], SPLASH [99], LIGR-Seq [100] and MARIO [97], have been introduced. They map the entire RNA interactomes in vivo besides identifying interacting partners of specific target RNAs at a larger scale. Online databases such as RAID v2.0 [101], NPinter [102–104], RNAinter [105, 106] and RISE [107] organise and classify these RRIs based on curated data from various sources (bibliometrics, experimental data, etc.). Nevertheless, a complete picture of human RNA-associated interactions is lacking, with most observed interactions associated with ribosomal and small RNAs rather than ncRNAs. Tissue-specific expression patterns of RNAs require numerous repetitions of in vivo experiments to detect genome-wide RNA interactomes [20, 108]. Therefore, computational RIP methods remain indispensable compared to experimental approaches.
STATE-OF-THE-ART APPROACHES FOR RNA STRUCTURE AND INTERACTOME PREDICTION
Computational prediction methods are widely used for identifying RRIs. The discovery of the minimum free energy (MFE) structure of RNA sequences has garnered attention due to its association with RNA secondary structures and folding stability. The MFE of an RNA includes the sequence length, nucleotide content/composition and nucleotide order/arrangement [109]. Longer RNA sequences tend to be more stable due to stacking and hydrogen bond interactions [110]. The composition of nucleotides also influences RNA stability; G-C-rich sequences are more durable than A-U-rich sequences due to additional hydrogen bonds. The specific arrangement of nucleotides, including loop numbers and double helix conformations, contributes to folding structure stability [109].
This review aimed to summarise popular computational prediction tools for RIP based on two main strategies: deterministic dynamic programming (DDP) approach and comparative sequence analysis (homology), as illustrated in Figure 2A [85–87, 111–113]. This landscape reflects the growing interest and extensive research in the field of RIP. Figure 2B showcases the relationships between these two strategies.
Figure 2.

Foundation of RNA–RNA interaction prediction tools. (A) Two core strategies, namely, deterministic dynamic programming algorithm and comparative sequence analysis. (B) Venn diagram portraying the relationships between these strategies and emphasizing the overlap, demonstrating their interconnectedness.
Deterministic dynamic programming algorithm for individual RNA structure and RNA–RNA interaction prediction
The DDP algorithm is a popular and accurate type of RIP that relies on the thermodynamics model. It uses free energy minimization to predict RNA secondary structure based on a single sequence with a known function as an input [114]. DDP involves chemically altering nucleotides at Watson-Crick pairing sites in folded RNA using chemicals such as dimethyl sulfate and kethoxal. It is known as a "score-based method" that interprets the native RNA structure with a minimum/maximum total score of RNA folding prediction.
This approach relies on experimental approximations to account for the influence of sequence on stability for different RNA motifs. However, it does not account for pseudoknots, which are RNA structures formed by two nonnested base pairs. The nearest-neighbour model considers directly neighbouring bases and base pairs for each interaction [115, 116], utilizing loop-specific energy contributions to determine loop type- and context-specific contributions to the RNA structure [114, 117, 118].
Nussinov algorithm
The application of DDP in RSP ensures efficient computation [119, 120], producing consistent and identical results for identifying the lowest free energy structure. DDP simplifies complex RNA structures into simpler substructures through mathematical optimization and computer programming [119]. The DDP algorithm can be divided into several examples, as reported in Figure 2A. The Nussinov algorithm is the first DDP algorithm that efficiently predicts the optimal folding state of an RNA molecule by computing the maximum number of base-pairings [121]. However, it has several biases that can be noted as limitations. For instance, it (i) disregards differences in base-pairing strengths; the influence of loop sizes, base-pair stackings, loop context, multiloop, and pseudoknot formations on stability; (ii) lacks approximation-based prediction algorithms that cause the inability to predict pseudoknotted helices; (iii) does not consider folding kinetics, which does not apply to secondary RNA structures; (iv) exhibits asymmetry in the distribution of unpaired nucleotides, leading to destabilization of multibranch loops/helical junctions; (v) shows discontinuity in the formed base pairs; and (vi) is unable to create stem regions, thereby reducing its prediction accuracy [114, 122].
To address this, a minimum free energy (MFE) algorithm based on the Nussinov algorithm and nearest-neighbour model was proposed by Zuker in 1981 [123].
Minimum free energy algorithm
MFE algorithms, based on DPP, compute a series of complex free-energy parameters obtained from experimental methods. One example is the optical melting experiment that measures the thermodynamics of nucleotides. These algorithms breakdown a secondary RNA structure into substructures known as nearest-neighbour loops (Figure 3). The free energy of each nearest-neighbour loop is computed by adding its specific free energy parameters. The MFE approach can be categorised into four subclasses based on criteria, including intramolecular base pairs (internal structure), neglect of intramolecular structure, accessibility of the binding region, and the ability to predict the joint secondary structure of RNA duplexes [124].
Figure 3.
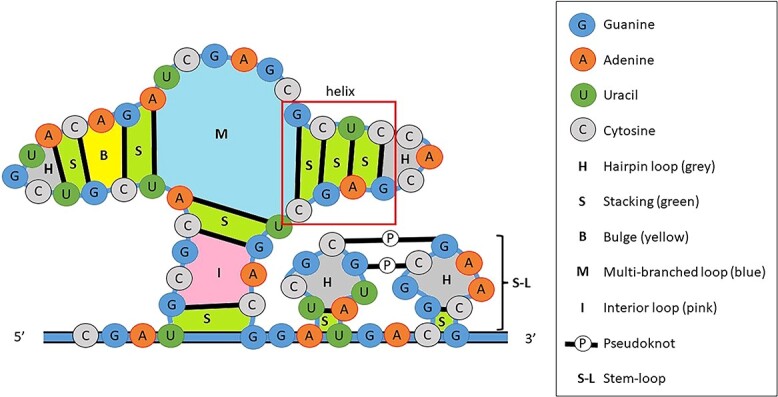
Loop decomposition of a nested RNA structure into hairpin loops (no enclosed base pairs), stackings (adjacent enclosed base pairs), bulges (only one side adjacent to enclosed base pair), multibranched loops (more than one directly enclosed base pair), interior loops (no stacked enclosed base pairs), pseudoknots (nucleotides in a loop pair with a region outside the helices that close the loop) and stem-loops (combination of the stem, double helix, and a loop) (adapted from [256]).
This review provides an overview of MFE algorithms derived from RSP and used in RIP tools to predict the RNA interactome in Tables 1–3 [42, 113, 114, 125]. It outlines the main prediction and output strategies employed by each algorithm. ‘Conservation’ indicates whether the prediction tools accept sequence alignments as input, which can help in identifying conserved regions within RNA molecules. ‘Suboptimal’ indicates whether the algorithms report suboptimal results in addition to a single MFE prediction. This feature allows the exploration of alternative RNA secondary structures with lower free energy but remain biologically relevant. The length of the interaction estimates the size of the predicted RNA–RNA helices, categorized as short (≤12 base pairs) or long (>12 base pairs). Additionally, the table distinguishes between local interactions and global predictions. ‘Local interactions’ involve single interactions with gaps and bulges, limited to a few base pairs. These predictions focus on aligning local regions with high similarity. In contrast, ‘global predictions’ span the entire RNA sequence, including multiple instances of local interactions separated by longer regions lacking intermolecular base pairs.
Table 1.
Interaction-only RIP tools based on MFE algorithms
| Characteristic | Interaction-only RIP Tool | Description | Input | Output | Applicable Species | Active (T)/Inactive (F) | ||
|---|---|---|---|---|---|---|---|---|
| Conservation | Suboptimal Prediction | Local Interaction Length | RNAduplex (RNA–RNA) [125] |
|
Two alignments of RNA sequences in CLUSTAL format with an equal number of sequences and the same order |
|
Any species (more accurate for humans and mice) | T |
| RNAaliduplex (RNA–RNA) [125] |
|
Multiple RNA sequences in CLUSTAL format (equal number of sequences and same order) |
|
Any species (more accurate for humans and mice) | T | |||
| No suboptimal | TargetRNA (sncRNA-mRNA) [127] |
|
A genomic sequence that may correspond to an sRNA gene |
|
Bacteria | T | ||
| No Conservation | Suboptimal Prediction | RIsearch (ncRNA-RNA) [129] |
|
RNA sequences in FASTA format |
|
Human | T | |
| GUUGle (miRNA) [130] |
|
A set of target sequences and a set of query sequences in various formats (nbrf/pir; codata; textual; unambiguous pure nucleotide; unambiguous pure RNA sequence; FASTA; FASTQ; XML; EMBL or ig), with a length threshold (k) |
|
All species | T | |||
| No Suboptimal |
Global interaction length | RNAhybrid (miRNA–mRNA) [126] |
|
Two RNA sequences, which will be handled simultaneously |
|
All species | T | |
G: guanine; lncRNA: long noncoding RNA; miRNA: microRNA; mRNA: messenger RNA; MSA: multiple sequence alignment; ncRNA: noncoding RNA; RIP: RNA–RNA interaction prediction; RRI: RNA–RNA interaction; RNA: ribonucleic acid; RSP: RNA structure prediction; sRNA: small RNA; U: uracil
Table 3.
Concatenation-based RIP tools based on MFE algorithms
| Characteristic | Concatenation-based RIP Tool | Description | Input | Output | Applicable Species |
Active (T)/ Inactive (F) |
|||
|---|---|---|---|---|---|---|---|---|---|
| Conservation | Suboptimal Prediction | Local Interaction Length | Nucleic Acid Package 4.0 (NUPACK 4.0) [148, 149] | • A growing software suite for the analysis and design of one or more species of interacting RNA strands. It enables analysis of nucleic acid sequences over complex and test tube ensembles containing arbitrary numbers of interacting strand species | At least 2 alignments of RNA sequences and allow specifications for the components, conditions of the RNA solution of interest, temperature, number of strand species, maximum complex size, strand sequences and strand concentrations | • Calculation of partition function, equilibrium base-pairing probabilities, MFE energy, proxy structure, suboptimal proxy structures, and Boltzmann sampled structures • Calculation of the partition function and MFE secondary structure for nonpseudoknot complexes of arbitrary numbers of interacting RNA strands • Calculation of the equilibrium concentrations for arbitrary species of complexes in a dilute solution • Calculation of equilibrium base-pairing observables for dilute solutions of interacting strand species via partition function and concentration information • Sequence design for >1 strand intended to adopt a nonpseudoknot target secondary structure at equilibrium |
All species | T | |
| No Conservation | No Suboptimal | UNAFold (Unified Nucleic Acid Folding and hybridization package) (DNAmelt & mFold) [146, 152] | • A tool with several closely related software applications available on the Worldwide Web for the prediction of the secondary structure of single-stranded nucleic acids; mFold has been replaced by UNAFold | One or 2 single-stranded RNA sequences in FASTA format with sequence name | • Prediction of RNA secondary structure (excluding pseudoknots) • Simulation of folding, hybridization, and melting pathways for one or two single-stranded NA sequences • Folding (secondary structure) prediction for single-stranded RNA via free energy minimisation, partition function calculations and stochastic sampling • Computation of entire melting profiles (plots), including melting temperatures (UV absorbance at 260 nm, heat capacity change (C(p)), and mole fractions of different molecular species |
All species | T | ||
| Suboptimal Prediction | Global Interaction Length | AccessFold (RNA–RNA, miRNA–mRNA, sRNA-mRNA) [262] | • A program for RIP with consideration for competing self-structure and allowing accessibility-based prediction as well as pairwise alignment | Two sequence files with sequence names for the first and second sequence | • Two approaches to evaluate accessibility: • Free energy density minimization • Pseudoenergy minimization • Minimization of the sum of free energy change and a pseudofree energy penalty for bimolecular pairing of nucleotides that are unlikely to be accessible for bimolecular structure • Prediction of binding sites that are split by unimolecular structures • Output is written to a CT file where the sequences are concatenated, with an intermolecular linker between them |
All species | T | ||
| PairFold [144] | • The first tool to predict suboptimal secondary structures of two interacting RNA strands and can handle complex joint structures | At least 2 sets of RNA sequences | • Prediction of the MFE pseudoknot-free secondary structure of two or more nucleic acid molecules via an extension of the Zuker and Stiegler algorithm [123] • Prediction of alternative low-energy suboptimal secondary structures for two NA molecules via suboptimal folding algorithm by Wuchty et al. [263] • Prediction of interactions between a probe and target RNA molecule or between pairs of strands in biomolecular nanostructures |
All species | |||||
| No Conservation | Suboptimal Prediction | Global Interaction Length | MultiFold [144] | • The first program to handle multiple RNA strands | At least 2 RNA sequences and accept MSA as input | • Standard thermodynamic parameters of the Turner group prediction of the MFE pseudoknot-free secondary structure of two or more nucleic acid molecules • Prediction of alternative low-energy (suboptimal) secondary structures for two nucleic acid molecules |
All species | ||
| RNAsoft (PairFold, CombFold, RNA designer, AveRNA, & HotKnots 2.0) (RNA–RNA) [143] | • A suite of RNA secondary structure prediction and design software tools, applicable for DNA sequences, and handles complex joint structures | Two RNA sequences with a description of a combinatorial set of RNA strands | • Summary of tools: • PairFold: Prediction of the MFE secondary structure formed by two input RNA molecules and interactions between a probe and target RNA molecule or between pairs of strands in biomolecular nanostructures • ComdFold: Prediction of the origin of a strand from a combinatorial set formed from RNA input strands and folding to a secondary structure with the lowest MFE • RNA designer: Designing an RNA sequence that folds to a given input secondary structure • AveRNA: Combination of the RNA secondary structures predicted by different algorithms to increase the overall accuracy • HotKnots 2.0: Prediction of short RNA secondary structures that are expected to form pseudoknots |
All species | |||||
| No Suboptimal | RNANUE (RNA–RNA) [147] | • A comprehensive and efficient analysis to detect RRIs from DDD (direct-duplex-detection) data | RNA sequencing files in a specific folder structure (the root folders must be specified for both treatment and control groups, and subfolders should represent arbitrary conditions that contain the read files) | • Split reads generation in SAM format • Clusters identification, including the IDs of the clusters, its length, size and genomic coordinates • Detection of RRIs, complementarity scores, and hybridization energies identification • MFE hybrid structure prediction and the probability in the ensemble of all possible interactions via RNAlib |
All species | T | |||
| RNAfold (RNA–RNA) [125] | • A web server which predicts secondary structures of single-stranded RNA sequences | RNA or DNA sequence in FASTA format with a limit of 7500 nt for partition function calculations and 10,000 nt for MFE-only predictions | • Interactive RNA secondary structure plot • RNA secondary structure plots with reliability annotation (partition function folding only) • Mountain plot (to predict and plot secondary structures) |
All species | T | ||||
| RNAcofold (RNA–RNA) [125, 145] | • A program like RNAfold, but allows users to specify two RNA sequences that can form a dimer structure, capable of interaction-only MFE-based method, and can handle complex joint structures | RNA sequences are read from stdin in the usual format | • Calculation of secondary structures of two RNAs with dimerization • Computation of the hybrid structure of two molecules • Computation of MFE structures, partition function (pf) and base pairing probability matrix (using the −p switch) • Computation of equilibrium concentrations for all five monomers and (homo/hetero)-dimer species, given input concentrations for the monomers (since dimer formation is concentration dependent) • Generation of PostScript structure plots and "dot plot" files containing the pair probabilities |
All species | T | ||||
| RNA–RNA interACTion prediction using Integer Programming (RactIP) (RNA–RNA) [163] | • A fast and accurate ML and probabilistic approach to predict RRI using integer programming, and handling complex joint structures | Two RNA sequences in FASTA format | • Integration of approximate information on an ensemble of equilibrium joint structures into the objective function of integer programming using posterior internal and external base-pairing probabilities • Prediction of RNA joint secondary structures under the general type of interaction including kissing hairpins • Prediction of the maximum expected accuracy (MEA) structure using integer programming (IP) with threshold cut via GNU Linear Programming Kit (GLPK) |
All species | T | ||||
CT: continuous tone; DNA: deoxyribonucleic acid; GNU: Gnu's Not Unix; MFE: minimum free energy; MSA: multiple sequence alignment; NA: nucleic acid; nt: number of nucleotides; RIP: RNA–RNA interaction prediction; RRI: RNA–RNA interaction; RNA: ribonucleic acid; UV: ultraviolet
Interaction-only approach
The first RIP method is known as the ‘interaction-only (IO)’ approach because it only considers intermolecular base pairs during computation and in the final predicted outcome [87]. The MFE derived from intermolecular base pairs between two RNA strands is called the hybridization energy. IO possesses fast algorithmic speed but lower accuracy, as it neglects intramolecular RNA structures that might disrupt and constrain certain intermolecular interactions. IO prediction tools compute the overall Gibbs free energy (ΔG) and determine the direction of RNA folding. The stable RNA structure is determined by minimizing free energy using thermodynamic data such as temperature and chemical composition. The goal is to find the structure with the lowest Gibbs free energy, indicating its most stable conformation under the given thermodynamic conditions. Examples include DuplexFold [126], targetRNA [127], RNAhybrid [126], RNAplex [128], RNAduplex, RNAaliduplex [125], RIsearch [129] and GUUGle [130] (Table 1).
The DuplexFold server predicts the lowest hybrid free energy conformation of two RNA sequences based on intermolecular base-pairing, whereas targetRNA identifies base-pair complementarity and calculates RRI scores using the MFE model for RNA duplexes [127]. Following targetRNA, RNAhybrid predicts eukaryotic miRNA target and prokaryotic sRNA target interactions [126]. Both targetRNA and RNAhybrid heavily rely on the energies of stacked back-to-back base pairs, interior loops, and bulges for their prediction. For more efficient computation and less complexity, the consideration of long interior loops is limited and excluded during the RIP process. Conversely, database-based RNAplex is explicitly designed to search for potential hybridization sites in a query RNA. It implements a slightly different energy model than RNAhybrid, shortening computational time and enabling target search on highly stable interactions.
Both RNAduplex and RNAaliduplex, included in the Vienna RNA 2.0 package, predict conserved RRI between two alignments [125]. In contrast, the RIsearch algorithm is designed to rapidly scan genome-wide ncRNA–RNA pairs. It incorporates a simplified Turner energy model to the Smith–Waterman–Gotoh algorithm, approximating the Turner nearest-neighbour energy model using the dinucleotide scoring matrix [129]. Interestingly, GUUGle stands out by not calculating Gibbs free energies to determine optimal interactions. Instead, it generates all ungapped interactions over a user-specified length, serving as an absolute baseline for predicted performance. Moreover, GUUGle is designed to reduce the search space for more complex algorithms [130]. Overall, all the IO methods predicted RRI solely based on intermolecular base pairs.
Accessibility-based approach
To overcome the shortcomings of IO prediction tools, the accessibility-based (AB) approach was introduced to predict intra- and intermolecular base pairs [87]. AB uses the McCaskill partition function algorithm to predict the pairing likelihood of single nucleotide sequences at each position of the input sequence data [131]. The stability of intermolecular interactions at specific positions is determined by calculating stacking base pairs and the likelihood of intramolecular base pairs being inaccessible within the RNA molecules. The energy needed to prevent interacting RNA segments from forming intramolecular base pairs is known as accessibility energy. Sfold [132], RNAup [133], IntaRNA [134, 135], RNAplex [128], RNApredator web server [136] (updated version of RNAplex), OligoWalk [137], BistaRNA [138], inRNAs [139], RIsearch2 [140], RIblast [141] and targetRNA2 [142] are examples of prediction tools that adopted the AB approach (Table 2).
Table 2.
Accessibility-based RIP tools based on MFE algorithms
| Characteristic | Accessibility-based RIP Tool | Description | Input | Output | Applicable Species |
Active (T)/ Inactive (F) |
||
|---|---|---|---|---|---|---|---|---|
| Conservation | Suboptimal Prediction | Local Interaction Length | LncRRIsearch (lncRNA–lncRNA and lncRNA–mRNA) [258] |
|
Longest mRNA and lncRNA transcript sequences excluding excluded transcripts in the pseudoautosomal region on the Y-chromosome |
|
Human, animal |
T |
| RIblast (sRNA and lncRNA TINCR) [141] |
|
A query RNA and a target RNA |
|
All species | T | |||
| TargetRNA2 (sRNA-mRNA) [142] |
|
Sequence of an sRNA in FASTA format and the name of a sequenced bacterial replicon |
|
Bacteria | T | |||
| RNAstructure (including ProbKnot, OligoWalk, bipartition, bifold, DuplexFold, Dynalign, Multilign, PARTS, TurboFold, etc.) (RNA–RNA) [259] |
|
Accept MSA as input, the name of a sequence file (SEQ, FASTA) or structure file (CT, DBN) containing the input sequence |
|
All species | T | |||
| Conservation | Suboptimal Prediction | Local Interaction Length | OligoWalk (siRNA-mRNA) [137] |
|
Only RNA oligomer is allowed, and 19 bases are recommended for siRNA design |
|
All species | T |
| RNAplex-aA and RNAplex-cA (RNA–RNA) [128] |
|
At least 1 FASTA file containing target and query RNA sequences or 2 CLUSTAL files as input |
|
Virus, animal | T | |||
| No Conservation | RNAplex-a and RNAplex-c (RNA–RNA) [128] |
|
||||||
| RIsearch2 (RIsearch and GUUGle) (RNA–RNA) [140] |
|
RNA sequences in FASTA format |
|
Multiple species | T | |||
| No Conservation | Suboptimal Prediction | Local Interaction Length | IntaRNA 2.0 (ncRNA-ncRNA) [134] |
|
At least 1 FASTA file containing target and query ncRNA sequences |
|
Bacteria | T |
| IntaRNA (ncRNA-ncRNA) [135] |
|
At least 1 set of noncoding RNA sequences in FASTA format with more than 1 but at most 100 sequences, each with a length ranging from 7 to 2000 nt |
|
Bacteria | T | |||
| No Suboptimal | InRNAs (RNA–RNA) [139] |
|
RNA pairs ranging from 20 to 60 nt |
|
All species | F | ||
| BistaRNA (mRNA of ncRNA) [138] |
|
mRNA sequences of specific ncRNA |
|
All species | T | |||
| RNAup (RNA–RNA) [133] |
|
One (accessibility) or 2 (interaction) RNA sequences in FASTA format with a limit of 5000 nt per sequence |
|
All species | T | |||
| No Conservation | No Suboptimal | Global Interaction Length | Sfold (siRNA and miRNA) [132, 261] |
|
RNA sequences in raw format, in FASTA format, or GenBank format (200 bases for an interactive job and 5000 bases for a batch job) |
|
All species | T |
| Suboptimal Prediction | RNApredator (RNA–RNA) [136] |
|
A single small RNA sequence consisting of lower or uppercase letters (A, T, C, G, U), where T is automatically converted into U (with confirmed genome) |
|
Bacteria | T | ||
A: Adenine; C: cytosine; G: guanine; lncRNA: long noncoding RNA; MFE: minimum free energy; miRNA: microRNA; mRNA: messenger RNA; MSA: multiple sequence alignment; NA: nucleic acid; ncRNA: noncoding RNA; NMR: nuclear magnetic resonance; nt: number of nucleotides; RIP: RNA–RNA interaction prediction; RRI: RNA–RNA interaction; RNA: ribonucleic acid; siRNA: short interfering RNA; srRNA: small regulatory RNA; sRNA: small RNA; T: thymine; TINCR: terminal differentiation-induced noncoding RNA; U: uracil
The Online Sfold tool predicts RNA secondary structure, target accessibility and hybridization energy [132]. It can compute the accessibility of binding regions and calculate the MFE of the RNA duplex via RNAup [133], IntaRNA 2.0 [134] and RNAplex [136]. However, RNAplex and RNAup cannot predict pseudoknots, while IntaRNA 2.0 is limited to interactions between single hairpin loops and excludes kissing hairpins (more complex pseudoknots/multiloops). OligoWalk predicts the hybridization of oligonucleotide binding by calculating the total free energy of an RNA sequence to the target sequence of a known structure [137]. BistaRNA and inRNAs provide insights into RNA accessibility and can predict multiple binding sites [138, 139]. Similarly, RNApredator is a fast accessibility-based prediction tool for single small RNA targets that uses a full nonpseudoknot partition function of interacting strands in a dilute solution [136].
RIsearch2 and RIblast are genome/transcriptome-wide scale RIP tools that implement the seed-and-extension approach to discover seed regions using suffix arrays and possess faster computational speed (64×) than other existing similar programs [141]. The seed regions are further refined using an energy model of the predicted RNA secondary structure [140]. On the other hand, TargetRNA2 is a tool for identifying targets of small regulatory RNAs (sRNAs) in bacteria via conserved regions, secondary structures, individual mRNA target secondary structures, and sRNA–mRNA hybridization energy. In RIP, TargetRNA2 suggests that the more conserved two sRNAs have in common, the more likely they are to interact with one another.
Concatenation-based approach
The third subclass of the MFE-based RIP tool involves both intermolecular and intramolecular base pairing of RNA. This approach is called concatenation-based, where two input sequences are concatenated and run through classical RSP algorithms to compute internal and external base pairs simultaneously [87]. Examples of concatenation-based tools include RNAsoft [143], PairFold [144], RNAfold [125], MultiFold [144], RNAcofold [125, 145], UNAFold (mfold/RNAfold) [146], RNAnue [147] and NUPACK [148, 149] (Table 3). However, they are limited due to the inability to predict pseudoknots accurately, where the base pairs are not well nested but overlap with each other.
In 2003, Andronescu et al. introduced an RNAsoft suite of programs to predict the secondary structure (PairFold), test combinatorial tag sets (CombFold) and design RNA strands (RNA Designer) [144, 150, 151]. PairFold is the first tool to predict suboptimal secondary structures of two interacting strands, and MultiFold is the first to handle multiple strands. Both programs use the standard thermodynamic parameters of Turner for RNA molecules [113, 132, 144]. RNAfold is a web tool that predicts the secondary structures of single-stranded RNA sequences [125]. Compared to RNAfold, RNAcofold allows the prediction of RNA secondary structures of single-stranded RNA sequences upon dimer formation [125, 145]. On the other hand, unified nucleic acid folding and hybridization package (UNAFold) is an amalgamation of mfold and DINAMelt. It predicts the pseudoknot-free RNA secondary structure of a single RNA sequence by simulating its folding, hybridization, and melting pathways. The prediction minimizes the global free energy using an improved algorithm by Zuker and Stiegler [125, 146, 151, 152]. RNAnue predicts inter- and intramolecular RRIs using complementary strands of double-stranded RNA information through direct-duplex-detection (DDD) methods [147].
Multiple sequence alignments and complex joint approach
Sequence alignment is a method to align DNA, RNA or protein sequences, predicting conserved regions that represent functional or evolutionary relationships between two sequences. Pairwise alignment determines the best-matching pattern of two sequences, whereas multiple sequence alignment involves multiple sequences simultaneously. Local alignment identifies local regions with the highest similarity level in sequences, whereas global alignment spans the entire sequence. RNAPLEX [128] and RNAduplex [125] are programmes that predict conserved RRIs using sequence alignments.
Another RIP tool of the MFE algorithm is known as the ‘complex joint’ (CJ), owing to MFE computation to identify the RRI between multiple RNA alignments. Unlike single RNA secondary structure-based RIP tools [33, 44], CJ can handle more complex joint structures with multiple interaction sites [153–158]. This capability is crucial, as ncRNAs often interact with target mRNAs in gene translation. Moreover, these relatively long regulatory antisense RNAs are not fully complementary to their target sequences. Instead, they rely on stable joint structures with mRNA via loop–loop interactions to facilitate regulatory functions [155]. Nevertheless, predicting these RNA secondary structure complexes with MSA is challenging (nondeterministic polynomial-time (NP)-hard problem), and only a few dedicated tools are available.
MultiRNAFold is a CJ-based package that includes three types of software: SimFold, PairFold and MultiFold [144]. It computes the MFE for predicting the secondary structure of interacting RNA molecules. Early attempts, such as PairFold [144] and RNAcofold [159], treated two interacting RNA sequences as a single sequence but faced challenges in predicting complex interactions such as kissing hairpins.
In 2007, Dirks et al. [160] introduced the NUPACK package, which efficiently computes the partition function of a single to multiple RNAs and concatenates input sequences in order, considering their symmetries and sequence heterogeneity. Similarly, BPPart, a revised algorithm of rip [157] and piRNA [154], computes the partition function for joint structures. The energy model is simplified by ignoring the entropy systems while retaining the thermodynamic information captured by more complex models [161]. The inRNAs algorithm predicts multiple binding sites in an RNA complex [139], while RIG utilizes multiple context-free grammars to model RRI [162]. Other CJ tools, such as IRIS [156], inteRNA [153] and piRNA [154], were previously available, but they are obsolete or no longer supported.
This review highlights that CJ methods are limited to relatively short RNA sequences to improve runtime performance. Although longer sequences cover a broader class of interacting RNA structures simultaneously, they are highly resource intensive and impractical for genome-wide scans. To overcome this challenge, Kato et al. [163] developed RactIP (RNA–RNA interaction prediction using integer programming), a novel method to increase the input RNA sequence length while optimizing runtime performance and prediction accuracy using the threshold cut technique.
Comparative sequence analysis for RNA structures and RNA–RNA interaction prediction
The structures of functional ncRNAs are crucial in understanding their functions and evolutionary conservation. Structural alignment compares a folded RNA to known reference ncRNAs, identifying similar regions called ‘conserved regions.’ Comparative sequence analysis allows the identification of these conserved regions. The alignment score represents the similarity in the ncRNA sequence and structure. Comparative analysis suggests that RNA-forming base pairs in RNA secondary structures tend to be more conserved and covary during evolution to maintain Watson–Crick and wobble pairings (compensatory mutations) [87, 164, 165]. This supports the theory that base pairs with fully conserved or retained structures from compensatory mutations are more functionally important than unconserved base pairs [87].
Multiple sequence alignment (MSA) is one of the oldest comparative studies used to detect common secondary structures from a set of homologous sequences. By including well-aligned and sufficiently divergent homologues, MSA provides valuable information for predicting evolutionarily conserved base pairs. This approach also significantly improves the accuracy of the RSP tool and overcomes shortcomings of the MFE-based approach, such as the difficulty in aligning RNA sequences with low similarity (<60%) and folding different primary sequences into the same secondary structures.
To date, comparative sequence analysis (homology) is more accurate than DPP approaches in RSP [166, 167]. This review highlights three major components of comparative sequence analysis (Figure 4A), including several examples of freely available homology-based tools in RIP, as tabulated in Tables 4–7 [164].
Figure 4.

Comparative RNA structure prediction (RSP) and RNA–RNA interaction prediction (RIP). (A) The three main approaches in comparative RSP: 1. Align-then-fold approach; 2. Fold-then-align approach and 3. Sankoff-type approach (alignment and folding simultaneously); (B) The two main approaches in comparative RIP are (i) interaction between two alignments via an individual RIP tool and (ii) interactions obtained from the joint output of multiple individual RIP tools (adapted from [257]).
Table 4.
Align-then-fold RSP tools based on comparative sequence analysis
| Characteristic | Align-then-fold RSP Tool | Description | Input | Output | Applicable Species |
Active (T)/Inactive (F) | ||
|---|---|---|---|---|---|---|---|---|
| Conservation | Suboptimal | Local Interaction Length | TurboFold II (part of RNAstructure) (RNA–RNA) [264] |
|
Homologous RNA sequences |
|
All species | T |
| No Suboptimal | RNAalifold (RNA–RNA) [172] |
|
Multiple RNA alignments in CLUSTAL W and FASTA format |
|
Virus, bacteria, human | T | ||
| Pfold [173] |
|
An alignment of up to 40 sequences and 500 positions in FASTA format with a phylogenetic tree relating the sequences |
|
Virus, bacteria, human | T | |||
| Global interaction length | PETfold (RNA–RNA) [174] |
|
One MSA in FASTA format |
|
Bacteria, virus | T | ||
CYK: Cocke–Younger–Kasami; MFE: minimum free energy; MSA: multiple sequence alignment; RNA: ribonucleic acid; RSP: RNA structure prediction; SCFG: stochastic context-free grammars
Table 7.
Fold-then-align RSP tools based on comparative sequence analysis
| Characteristic | Fold-then-align RSP Tool | Description | Input | Output | Applicable Species | Active (T)/Inactive (F) | ||
|---|---|---|---|---|---|---|---|---|
| Conservation | Suboptimal Prediction | Local Interaction Length | MARNA (surpassed by LOcARNA) [252, 253] |
|
RNA sequences in FASTA format (max 3 for RNAsubopt) |
|
Eukaryote | F |
| planACstar (RNA–RNA) [196] |
|
A set of alignments with several sequences per alignment |
|
Mammal | T | |||
| RNAspa (part of ViennaRNA package) (ncRNA) [270] |
|
A set of unaligned RNA sequences |
|
Virus, bacteria | T | |||
| RNAcast (RNA consensus abstract shape technique) (ncRNA) [193] |
|
At least 2 RNA sequences |
|
Virus | F | |||
| No Suboptimal | RNA Sampler (ncRNA-RNA) [198] |
|
Two RNA sequences |
|
Animal, eukaryote | T | ||
| Conservation | No Suboptimal | Global interaction length | MASTR (multiple alignment and structure prediction of ncRNAs) [197] |
|
At least 2 RNA sequences in FASTA format |
|
Human, eukaryote | T |
| LaRA 2 (ncRNA-RNA) [200, 272] |
|
At least 2 RNA sequences in FASTA format |
|
Bacteria, virus, eukaryote | T | |||
| T-Coffee (tree-based consistency objective function for alignment evaluation) [195] |
|
RNA, DNA and protein alignments from any source in FASTA format |
|
Parasite, bacteria, animal | T | |||
| Suboptimal Prediction | CMfinder (ncRNA) [199] |
|
Unaligned RNA sequences |
|
Bacteria, archaea | T | ||
| RNAforester (part of ViennaRNA package) [273] |
|
RNA secondary structures from stdin or RNA sequences and structures in FASTA format |
|
Bacteria, virus, eukaryote | T | |||
2D: 2-dimensional; ASCII: American standard code for information interchange; DNA: deoxyribonucleic acid; MSA: multiple sequence alignment; ncRNA: noncoding RNA; RNA: ribonucleic acid; T-Coffee: tree-based consistency objective function for alignment evaluation
Align-then-fold approach
The align-then-fold approach extends RSP to multiple sequences by aligning them based on similarity and then predicting the structure with the lowest free energy that is shared by the largest number of sequences [168]. This approach requires a conventional alignment tool (e.g., ClustalW [169, 170], MAFFT [171]), followed by RSP tools (e.g., RNAalifold [172], Pfold [173]). The RNAalifold web server is one of the most important and commonly used tools (combined with score-based methods) [172], whereas Pfold includes compensatory mutations for accurate secondary RSPs [173]. Meanwhile, PETfold combines thermodynamic and evolutionary perspectives into a single model [174]. In short, the align-then-fold method is efficient for sequences with high similarity (>60%) and is a computationally less expensive method than the Sankoff-type and fold-then-align methods.
Table 4 summarizes a comprehensive overview of align-then-fold RSP tools.
Sankoff-type approach
The Sankoff algorithm is the most rigorous and computationally expensive approach to align RNA structure [175]. It combines structural prediction and sequence comparison simultaneously, ensuring similarity between structures by considering base-pair input in both [175–177]. This approach yields more accurate predictions than methods that separate folding and alignment steps, but it requires additional computer memory [178]. The Sankoff-based tools include MARNA [179], Foldalign [180–182], Dynalign [183], Stemloc [184] and MXSCARNA [185] (Table 5). They employ the Sankoff algorithm to explore the structural space and calculate the optimal secondary structure considering both sequence and structure conservation [175–177]. Additionally, some variants use sequence-based heuristics to reduce computational complexity and align efficiently.
Table 5.
Sankoff-type (sequence-based heuristics) RSP tools based on comparative sequence analysis
| Characteristic | Sankoff-type (Sequence-based Heuristics) RSP Tool | Description | Input | Output | Applicable Species |
Active (T)/Inactive (F) | |||
|---|---|---|---|---|---|---|---|---|---|
| Conservation | Suboptimal Prediction | Local Interaction Length | Dynalign (part of RNAstructure) [267] |
|
Two RNA sequences (homologues) |
|
Bacteria, virus, eukaryote | T | |
| Multilign [268] |
|
At least two RNA sequence alignments |
|
Bacteria, virus, eukaryote | T | ||||
| MXSCARNA (multiplex stem candidate aligner for RNAs) [185] |
|
Two RNA sequences and accept MSA as input |
|
Bacteria, virus, eukaryote | T | ||||
| Global interaction length | Stemloc [184] |
|
Two RNA sequences (homologues), capable of pairwise alignment of multiple sequences |
|
Bacteria | T | |||
| No Suboptimal |
Local interaction length | CARNA (constraint-based alignment of RNA ensembles) [269] |
|
A set of RNA sequences in FASTA format and one dot plot per sequence in PostScript format |
|
Bacteria, eukaryote, virus | T | ||
| Global interaction length | Foldalign version 2.5 (ncRNA) [182] |
|
Two RNA sequences or entire sequences with lengths up to 10,000 nt and a maximum alignment length of 1000 nt |
|
Bacteria | T | |||
MFE: minimum free energy; ncRNA: noncoding RNA; nt: number of nucleotides; RNA: ribonucleic acid; RSP: RNA structure prediction; SCFG: stochastic context-free grammars
Another approach uses McCaskill's algorithm to calculate base-pair probabilities via dynamic programming (Table 6), such as PMcomp [186] and LocARNA [187], whereas FoldalignM [188] and Murlet [189] employ a different algorithm called ‘maximum expected accuracy’ (MEA). StrAl with PETcofold [190] combines Sankoff and McCaskill’s algorithm, using Sankoff for RSP and McCaskill’s algorithm for base-pair probability calculation. This approach reduces the structural search space, computational complexity, and runtime by utilizing a simplified energy model based on precalculated base-pair probabilities from McCaskill’s algorithm, rather than directly calculating loop energies as in the Sankoff approach. Notably, RNA alignment and folding is not part of the Sankoff algorithm but a separate algorithm integrating sequence alignment and RSP, providing a comprehensive analysis of both sequence and structure aspects. It combines subsequence alignment quality-based heuristics and the simplified energy model of PMcomp to simultaneously align and fold unaligned RNA sequences [184, 191].
Table 6.
Sankoff-type (base-pair probabilities) RSP tools based on comparative sequence analysis
| Characteristic | Sankoff-type (Base-pair) Probabilities) RSP Tool | Description | Input | Output | Applicable Species | Active (T)/Inactive (F) | |||
|---|---|---|---|---|---|---|---|---|---|
| Conservation | Suboptimal Prediction | Local interaction length | PMcomp [186] |
|
Two RNA sequences |
|
Bacteria, human, virus | F | |
| Global Interaction Length | FoldalignM (dependent on Vienna RNA package) (ncRNA) [188] |
|
Two or more RNA sequences or entire sequences and allow MSA as input |
|
Bacteria, human, virus | T | |||
| Murlet [189] |
|
RNA sequences in FASTA format with a maximum length of 300 nt |
|
Eukaryote | T | ||||
| No Suboptimal | LocARNA (local alignment of RNA)/LocARNA -P [187, 252] |
|
RNA sequences in FASTA format (recommendation for the analysis of RNAs ≤60% sequence identity, where alignments based on only sequence similarity are unreliable) |
|
Virus, bacteria, plant | T | |||
| Local interaction length | StrAl with PETcofold (ncRNA) [190] |
|
A set of alignments with several sequences per alignment |
|
Viruses, bacteria, eukaryote | F | |||
MSA: multiple sequence alignment; ncRNA: noncoding RNA; nt: number of nucleotides; RNA: ribonucleic acid; RSP: RNA structural prediction
Fold-then-align approach
The fold-then-align method involves first predicting the secondary structures of RNA sequences and then identifying the structure with the lowest free energy across all sequences. This method often employs MSA to improve conserved RSPs. Another approach explores a middle path, where individual secondary structures are identified for each sequence in sets, followed by postprocessing to determine the optimal structure shared by all sequences. However, the accuracy depends on the quality of input RNA structures and may be limited by the number of matched homologous sequences, leading to potential false positives. Consequently, the overall alignment quality is typically affected by individual RSP approaches [192]. RNAforester [193], RNAcast [193] and aliFreeFoldMulti [194] are examples of applications implementing the fold-then-align method (Table 7).
To improve accuracy despite limitations in alignment quality, Notredame and colleagues developed the T-Coffee tool by implementing a preprocessing procedure that generates a library of local and global pairwise alignments [195]. It creates a consensus MSA by combining signals from diverse heterogeneous sources, such as sequence and structure alignment programs. Other methods, including planACstar [196], MASTS [197] and RNA Sampler [198], use sampling techniques to refine alignment and folding structures. However, CMfinder [199] and LaRA [200] stand apart from conventional categories because CMfinder specifically detects new ncRNA families by combining RSP and covariance models, whereas LaRA focuses on the identification of local RNA alignments considering both sequence and secondary structure conservation. In short, thermodynamic-based methods work with single RNA sequences due to similar algorithms as RSP systems, while comparative sequence analysis methods require MSA to enhance the accuracy and performance of RSP or RIP.
Pairwise alignments
The conventional approach for comparative sequence analysis mainly focuses on RSP due to several challenges in detecting RIP. For instance, the limitation of prediction within in vitro settings, the prevalence of false-positive predictions due to the high magnitude of predicted RNA–RNA duplexes and potential interaction partners, and the impact of external factors (other interacting RNAs/small ligands/proteins in vivo). Comparative RIP identifies the role of an RNA regulator via direct base-pairing with its target RNA.
Two primary strategies for comparative RIP are shown in Figure 4B. Similar to comparative RSP, the first RIP method (individual RIP) predicts the interaction between two alignments rather than two distinct sequences. Hypothetically, strong sequence signals distinguish binding sites and interactions based on their conserved structural residues. It is commonly believed that homology can help deduce binding sites and interactions. Tools such as PETcofold [174] and RNAripalign [201] leverage this hypothesis. PETcofold is an extended version of PETfold capable of predicting conserved RRIs [174], whereas RNAripalign identifies RRIs based on sequence and structural conservation [201].
Richter and Backofen [202] proposed that interaction sites between RNAs may not always be strictly conserved, suggesting that conserved interactions can occur even without precise conserved interaction sites. However, their statements contradict most of the alignment-based hypotheses that assume strict conservation of interaction sites. Henceforth, a new method combining individual RIP tools without requiring a strict consensus is introduced. It generates more reliable results and uncovers conserved regulatory mechanisms across different systems. This second method outperforms individual RIP tools. RNAhybrid, published by Krüger and Rehmsmeier in 2006 [203], predicts homologous miRNAs on orthologous targets from various organisms. However, duplex energies predicted by RNAhybrid must be transformed into P values, as the former is strongly influenced by the GC content and frequency of dinucleotides of the selected organisms. As duplex prediction relies on base-pair stacking, maintaining the dinucleotide frequency is crucial, and mononucleotide shuffling would prevent the generation of random sequences that accurately represent the features of the nonrandom system. The joint P value is used to identify possible interactions between two RNA alignments [25]. Similarly, CopraRNA uses Hartung's method to compute a joint P value for a cluster of homologous RNA sequences [204, 205].
Table 8 provides a comprehensive summary of RIP tools focussing on pairwise alignment in comparative sequence analysis.
Table 8.
RIP tools based on pairwise alignment in comparative sequence analysis
| Characteristic | Comparative RIP Tool | Description | Input | Output | Applicable Species | Active (T)/Inactive (F) | |||
|---|---|---|---|---|---|---|---|---|---|
| Conservation | Suboptimal | Local Interaction Length | RNAripalign (part of rip) (RNA–RNA) [201] |
|
Two given MSA (allow incorporation of structure constraints as input parameters) |
|
Bacteria, virus, eukaryote | F | |
| No Suboptimal | CopraRNA (interaction calculated by IntaRNA) (sRNA-sRNA) [205, 274] |
|
At least 3 homologous sRNA sequences from 3 distinct organisms in FASTA format |
|
Human, bacteria, virus | T | |||
| Global interaction length | PETcofold (mRNA–RNA) [174] |
|
Two MSA with at least three shared sequence identifiers in FASTA format |
|
Bacteria, virus | T | |||
MFE: minimum free energy; mRNA: messenger RNA; MSA: multiple sequence alignment; ncRNA: noncoding RNA; RNA: ribonucleic acid; sRNA: small RNA
Pseudoknots: Loops and helical stems in RNA folding thermodynamics
RNAs contain an abundance of motifs, which are defined as discrete sequences or combinations of base juxtapositions. Structural motifs in RNA can form pseudoknots by base-pairing of single-stranded RNA regions in the hairpin loop with complementary nucleotides in the RNA chain [206]. The H-type pseudoknot is the most basic example, with a hairpin loop interacting with complementary nucleotides outside the loop [207]. Pseudoknots are critical components of RSP and RIP due to their involvement in translation readthrough mechanisms and are essential for identifying RNA complex functions [208]. Hinh et al. [209] also discovered a novel role of the ‘trans-pseudoknot’ RRI in the functional dimerization of human telomerase.
Additionally, the relationship between pseudoknots, RNA folding stability and conformational changes suggests that the interplay between loops and helical stems is essential in calculating RNA stability and folding thermodynamics [210–213]. Evaluating folding thermodynamics involves applying energy parameters to calculate the conformation energy and chain entropy, but this process can be computationally demanding and is limited to specific subclasses of pseudoknots [214].
For instance, using the DPP algorithm, Rivas and Eddy [215] developed an RSP tool called PKNOTS to fold optimal pseudoknotted RNAs (ranging from 100 to 200 nt), marking the beginning of prediction attempts on the secondary structure of RNA pseudoknots. PKNOTS can handle the broadest class of structures but is limited to small molecules due to its long running time [216]. Another DPP-based tool, HotKnots, offered faster prediction using a heuristic approach but could not guarantee the lowest free energy due to the vast conformational space and computational complexity. The search space is typically enormous, making an exhaustive search infeasible [216]. In short, existing DDP algorithms for pseudoknot prediction are both unreliable and inefficient.
Comparative methods are more reliable in predicting pseudoknot structures, but they are often selected in an ad hoc manner for specific purposes and require expert intervention [217]. The maximum weighted matching (MWM) algorithm can generate meaningful predictions, but it requires a large number of homologous sequences to detect strong covariance signals. However, the MWM algorithm is sensitive to noisy data such as misalignment, as it allows unrealistic interactions and may overlook the prevalence of helices as the most common structural elements in RNA structures [218, 219].
On the other hand, the iterated loop matching (ILM) algorithm combines both thermodynamic and comparative approaches to predict the secondary structure of RNA pseudoknots efficiently and reliably, even when only a few sequences are available. The ILM algorithm prioritises the formation of stable helices over computing a theoretically optimal structure, which proves to be beneficial by significantly enhancing the overall prediction accuracy. This advantage is particularly significant in situations where the available data are insufficient for a method such as MWM to generate reliable predictions using unrestricted models [220, 221].
Other examples of pseudoknot prediction tools are FlexStem and Kinefold. FlexStem constructed secondary RNA structures with pseudoknots by adding maximal stems based on the free energy model [222], whereas Kinefold used a long-term RNA folding simulation to predict pseudoknot structures with topological and geometrical constraints [223].
External pseudoknots or crossing interactions are formed when two interacting RNAs form pseudoknots. However, most of the thermodynamic-based tools disallowed the formation of pseudoknots and caused failure in predicting joint structures formed by nontrivial interactions between two RNAs. To address this problem, Eckart et al. developed NanoFolder, a program that predicts the base pairing of potential pseudoknots in RNA nanostructures. First, a simple energy model is used to calculate all possible helices, followed by a greedy algorithm to select the minimum free energy helices owing to their incorporation into the RNA complex [224]. Compared to NanoFolder, VfoldCPX uses a similar approach but a more advanced selection algorithm [225]. Meanwhile, IPknot could predict RNA secondary structures using a diverse set of pseudoknots from an individual sequence or MSA as an input [226]. Although comparative sequence analysis can predict pseudoknots, its accuracy is still limited. In brief, most of the computational methods predict the structure and RRI of pseudoknots using a thermodynamic-based approach, as reported in Table 9.
Table 9.
RSP and RIP tools involving pseudoknots
| Strategy | RSP and RSP tools involving pseudoknots | Description | Input | Output | Applicable Species | Active (T)/Inactive (F) |
|---|---|---|---|---|---|---|
| Thermodynamic-based approach | PknotsRG [275] |
|
A file containing one single RNA sequence in FASTA format |
|
Human, virus, bacteria | T |
| Thermodynamic-based approach | Kinefold (RNA–RNA) [223] |
|
A string of unmodified RNA/DNA bases (limit of 400 bases for renaturation fold and cotranscriptional fold) |
|
Virus, eukaryote | T |
| Thermodynamic-based approach | RNAMotif [276] |
|
A formal description of the permissible forms of the structure and the sequences contained within it |
|
Bacteria, virus | T |
| Thermodynamic-based approach | RCPred (RNA–RNA) [277] |
|
Multiple RNA secondary structures in the complex with possible interactions in each RNA pairs |
|
Bacteria, virus | T |
| Thermodynamic-based approach | Hyperfold (RNA–RNA) [278] |
|
RNA and DNA strand sequences (including temperature and concentration) |
|
Human | T |
| Thermodynamic-based approach | VfoldCPX (RNA–RNA) [225] |
|
Two RNA sequences including temperature (recommendation: 300 nt for RNA secondary structures without crossing base pairs, ≤150 nt for structures with H-type pseudoknots, and ≤ 120 nt for RNA secondary structures with pseudoknots and hairpin-hairpin kissed structures) |
|
Eukaryote | T |
| Thermodynamic-based approach (statistical mechanics) | Vfold (ncRNA-RNA) [279] |
|
RNA sequence in plain text form |
|
Human, virus | T |
| Thermodynamic-based approach (DDP heuristic algorithm) | HotKnots (RNA–RNA) [216] |
|
RNA sequences or sequence fragments |
|
Virus | T |
| Thermodynamic-based approach (DDP algorithm) | Pknots (RNA–RNA) [215] |
|
A single RNA sequence |
|
Bacteria, virus | T |
| Thermodynamic-based approach (heuristic algorithm) | FlexStem (RNA–RNA) [222] |
|
A ≥ 2 bp RNA secondary structure with a helical region or stem defined as an anti-parallel complementary strand |
|
Virus | T |
| Thermodynamic-based approach (empirical scoring function) | NanoFolder (RNA–RNA) [224] |
|
A set of RNA sequences combined with a descriptor for the desired target secondary structure |
|
Bacteria, human | T |
| Thermodynamic- or comparative-based approach (heuristic algorithm) | Iterated loop matching algorithm (RNA–RNA) [220] |
|
RNA homologous sequences |
|
Eukaryote | T |
| Thermodynamic- or comparative-based approach | ProbKnot (part of RNAstructure) (RNA–RNA) [280] |
|
A sequence file of DNA or RNA |
|
Human, virus | T |
| Comparative-based approach | IPknot (RNA–RNA) [226] |
|
A single sequence of RNA or MSA |
|
Virus, eukaryote | T |
2D: two-dimensional; 3D three-dimensional; bp: base pair; DNA: deoxyribonucleic acid; IP: integer programming; ILM: iterated loop matching; MEA: maximum expected accuracy; miRNA: microRNA; MSA: multiple sequence alignment; NA: nucleic acid; ncRNA: noncoding RNA; nt: number of nucleotides; RNA: ribonucleic acid
CHALLENGES IN RNA STRUCTURE AND RNA–RNA INTERACTION PREDICTION
With the rapid growth of biological data and technologies, there has been a surge in research for predicting structural RNA and RRI using computational approaches. However, researchers often overlook that the outputs from these tools do not reflect the actual RNA structure but rather assumption-based algorithms. In thermodynamic-based approaches, base pairs with higher free energies are occasionally ignored due to the lack of evidence in the literature. Representation of the ‘prediction/theoretical’ as the ‘true/actual’ RNA secondary structure or RRI results in the acceptance of an untested possibility without further investigation [82]. Moreover, the kinetic RNA structures that form during folding may serve as a crucial indicator of RNA functions [227]. For instance, riboswitches usually regulate metabolic functions via structural conformation instead of retaining a static native structure [228]. In addition, noncanonical base pairs also play a crucial role in forming tertiary RNA structures, necessitating their inclusion in the prediction process. Nevertheless, predicting both canonical and noncanonical base pairs remains a challenge. Noncanonical interactions must still be optimised as they may contain additional chemical probing information that facilitates RNA structure modelling and comprehension of functional RNA modules. In addition, predictions of RNA tertiary structure are less accurate in loop regions, where noncanonical pairs are required to evaluate structural details [229, 230].
Comparative-based techniques are limited by the need for a more extensive set of homologous sequences. Due to the limited knowledge of known RNA families, obtaining homologous sequences for all RNAs is unfeasible, resulting in a preference for score-based RSP with a single RNA sequence as input. The ‘predicted’ outputs should not be regarded as a substitute for comprehensive experimental RSP and RIP determination, as these algorithm-based prediction tools operate under the assumption that the nucleotides are likely to engage in secondary structure elements with the maximum predicted number of Watson-Crick base-pairings [117, 231, 232]. The automatic modelling methodology is another challenge in RSP and RIP tools. Due to limited experimental data, most currently available automated web servers only rely on RNA sequences as input with low accuracy. Therefore, integrating the experimental data into computational methods will be of assistance in enhancing the accuracy of RSP accuracy [79].
To improve the prediction accuracy of RIP and RSP tools, we concluded that five main challenges must be addressed as follows: (i) the limited number of examples with mapped interactions, (ii) limited focus on the kinetic RNA structures, (iii) the low specificity due to the restriction of single sequences, (iv) overreliance to ‘predicted’ output rather than experimental data and (v) the high cost for a search of complex types interactions provided a guaranteed maximum score is to be obtained.
ARTIFICIAL INTELLIGENCE: CURRENT TRENDS AND FUTURE DIRECTIONS
Artificial intelligence has emerged as a powerful approach to predicting RNA structure and function [233]. In previous years, numerous prediction methods have been developed with the primary goal of identifying RNA structures that are likely to exhibit an MFE state, such as proteins [234]. However, over the past two decades, machine learning (ML) has been proposed as an alternative methodology to enhance the accuracy and calculation speed of RIP and RSP tools [235]. It was previously overlooked due to limited accuracy resulting from small training datasets and the constraints of simplistic ML models [236]. Due to the recent surge in RNA sequence data and advancements in ML, particularly deep learning (DL), the latest ML-based approaches surpass existing traditional methods in both accuracy and applicability, providing an advantage in tackling complex questions in structural biology while dealing with large datasets. DL algorithms leverage reference structures to train scoring parameters for decomposed substructure analysis, making them a more efficient and scalable alternative to traditional experimental procedures [237].
RNA Interactome Scoper (RIscoper) is a ground-breaking AI tool based on natural language processing (NLP) that extracts RNA structure and interactions from published literature using an N-gram model [238]. NLP automates tasks by extracting useful information from unstructured text and converting it into a structured format for computational analysis. NLP techniques have substantially improved in recent years, demonstrating their effectiveness across various domains. These include literature-based discovery, aiding the analysis of high-throughput data such as gene expression and genome-wide association studies [239]. ML-based approaches, on the other hand, can be categorised into two major groups, each aligned with a distinct phase in the RSP and RIP process: ML-based scoring schemes and ML-driven prediction processes.
Score-based methods are the most widely used traditional computational methods and have dominated the field of RIP and RSP. Scoring methods assume that RNA structures must satisfy specific score-based criteria, which can vary depending on the RNA folding mechanism, making secondary RSP an optimization problem. Dynamic programming (DP) algorithms are commonly employed to discover the optimal structure by dividing it into smaller components with individual scores and require a sophisticated scoring scheme with numerous parameters. However, DP algorithms are often deemed inefficient for large inputs, as their running time increases rapidly with the input size based on RNA sequence length and may overlook unique base pairs and weak interactions [233]. Understanding the RNA folding mechanism through the score-based method is thus a formidable challenge, in contrast to data-driven ML methods that do not rely on such mechanisms.
In this review, we highlighted two categories of ML-based methods for RIP and RSP according to the subprocess, e.g. (i) score scheme based on ML (free energy parameter-refining approach, weighted approach, and probabilistic approach) and (ii) ML-driven prediction process (end-to-end approach and hybrid approach) (Table 10). All ML methods within these two categories trained their models through supervised learning, wherein model parameters were adjusted based on input–output pairs. RIP and RSP primarily employ features such as free energy parameters, RNA sequences, and sequence patterns as input, and the trained model outputs can be either classification labels or free energy values. The probabilistic approach based on ML is one of the earliest scoring schemes that used stochastic context-free grammars (SCFGs) to predict RNA structures and interactions. Datasets containing RNA sequences annotated with known secondary structures are used to estimate the probability parameters of the SCFG model [240].
Table 10.
Artificial intelligence-based RIP and RSP tools
| Strategy | AI-based RIP and RSP tool | Description | Input | Output | Applicable Species | Active (T)/Inactive (F) |
|---|---|---|---|---|---|---|
| N-gram statistics language model | RIscoper (RNA Interactome Scoper) (RNA–RNA) [238] |
|
Full texts or abstracts, with an online search tool connected to PubMed |
|
All species | T |
| Score scheme (free energy parameter-refining approach based on ML) | Constraint generation- RNAsoft [241] |
|
RNA sequence, all on one line; and RNA secondary structure in dot-parentheses format, all on one line |
|
All species | T |
| Score scheme (weighed approach based on ML) | ContextFold [243] |
|
One or more RNA sequences in FASTA format and accept MSA as input with optional structure constraints |
|
All species | T |
| Score scheme (a probabilistic approach based on ML) | Stochastic context-free grammars [168, 281, 282] |
|
An alignment of RNA sequences |
|
N/A | N/A |
| Predicting process based on ML (end-to-end approach) | SPOT-RNA [246] |
|
Single RNA sequence or batch of sequences |
|
Human | T |
| Predicting process based on ML (hybrid) | Deep learning method for state inference [284] |
|
Dataset of known input–output pairs |
|
Bacteria, animal, eukaryote, archaea | T |
| Predicting process based on ML (hybrid) | DMfold [247] |
|
Target RNA sequences with dot-bracket sequences as labels |
|
All species | T |
| Predicting process based on ML (hybrid) | MINT [248] |
|
A simple text file with a detailed description of the RNA or DNA structure in each conformation frame |
|
All species | T |
| Predicting process based on ML (hybrid) | CONTRAfold (CONditional TRAining for RNA Secondary Structure Prediction) (RNA–RNA) [242] |
|
Single RNA sequence |
|
All species | T |
2D: two-dimensional; 3D: 3-dimensional; AI: artificial intelligence; DNA: deoxyribonucleic acid; LSTM: long short-term memory; ML: machine learning; NA: nucleic acid; NMR: nuclear magnetic resonance; NNTM: nearest neighbor thermodynamic model; RIP: RNA–RNA interaction prediction; RNA: ribonucleic acid; RRI: RNA–RNA interaction; SCFG: stochastic context-free grammar; SHAPE: selective 2′-hydroxyl acylation analysed by primer extension
Andronescu and colleagues introduced the constraint generation (CG) method, a pioneering computational approach for estimating RNA-free energy parameters. This approach was designed to train on large datasets containing structural and thermodynamic information efficiently. By incorporating ML techniques, CG can predict and design RNA secondary structures with high accuracy [241]. Another notable tool, CONTRAfold, takes a different approach by using conditional log-linear models that generalise SCFGs through discriminative training and feature-rich scoring. This allows CONTRAfold to accurately predict RNA secondary structures based on probabilistic models [242]. ContextFold employs feature-rich scoring models that are trained extensively on large datasets [243]. This approach captures more complex relationships in the data, but there is a potential risk of overfitting, where the model becomes too specific to the training data and performs poorly on new, unseen data [244].
The ML-driven prediction process, on the other hand, adopts deep learning (DL) in predicting RNA structure [245]. SPOT-RNA, for instance, focuses on leveraging deep neural network learning to predict all base pairs, regardless of their association with local or nonlocal interactions. This approach leverages the power of DL to capture intricate patterns and features within RNA sequences [246]. To overcome limitations and enhance prediction accuracy, hybrid approaches have been introduced [233]. One example is the combination of thermodynamic and ML-based strategies, where the model of CONTRAfold and MFE (concatenation-based method and complex joint category) is used to predict RNA interactions [163, 242]. This hybrid method leverages the strengths of both thermodynamic principles and ML techniques to improve the accuracy of RIP. Nucleic Acid Package 4.0 (NUPACK 4.0), a hybrid tool, integrates ML-based and concatenation-based MFE methods for analysing and designing interacting RNA strands across multiple species. It enables the examination of RNA sequences in complex and test tube ensembles containing an arbitrary number of interacting strand species [148, 149].
For RSP, a method called DMfold has been proposed. DMfold combines deep learning and an improved base-pair maximization principle to predict RNA secondary structures with pseudoknots. By learning from similar RNA sequences instead of highly homogeneous sequences, DMfold reduces the requirement for auxiliary sequences and improves folding accuracy [247]. Motif identifier for nucleic acids trajectory (MINT) is an automatic tool to analyse 3D structures of RNA molecules, their molecular dynamics trajectories and other conformation changes [248]. On the other hand, CompaRNA utilizes a combination of 28 single-sequence methods and 13 comparative methods for continuous automated benchmarking [249, 250]. Although CompaRNA is primarily based on comparative sequence analysis rather than the ML method, it incorporates several ML-based tools, such as ContextFold and CONTRAfold, as part of its analysis pipeline [242, 243]. This demonstrates the synergy between comparative sequence analysis and machine learning, where ML algorithms complement evolutionary information and sequence conservation to improve predictions.
While ML techniques have significantly enhanced prediction methods in terms of accuracy, applicability, and processing speed, there remains a need for more sophisticated ML models to fully address the challenges of the RSP and RIP problems, particularly in predicting high-resolution structures [233]. Nevertheless, given the rapid expansion of RNA sequence data, the availability of high-performance hardware and continuous advancements in machine learning methods, there is a potential for the future development of cutting-edge RSP and RIP tools that could surpass traditional approaches in terms of both execution speed and accuracy.
SELECTING THE BEST APPROACH: PRACTICAL RECOMMENDATIONS
Choosing the most suitable method for RIP or RSP depends on the specific research objectives. For instance, if the primary goal is on RIP and identifying binding sites, the IO method may be the preferred option since it excels at detecting interaction regions and base-pairing sites. However, IO methods are not designed to provide detailed structural information about the individual molecules involved [87, 127, 142, 203]. On the other hand, the concatenation-based method is selected for predicting the MFE structure of an entire RNA molecule, considering potential intramolecular interactions and structural elements. These methods offer a comprehensive perspective on the folding behaviour of RNA and have the capability to capture complex structures and interactions. However, they are frequently computationally demanding, particularly when applied to large RNA molecules [87, 143].
Accessibility-based MFE algorithms, as employed in RNAup, IntaRNA, and RNAplex, have demonstrated superior performance in RSP and RIP when compared to the previous two types of tools [128, 133, 134]. In an analysis of a bacterial dataset by Umu and Gardner in 2017 [86], these algorithms showed their ability to distinguish nearly half of the native interactions from the background noise. This accomplishment is facilitated by the integration of well-designed negative controls such as dinucleotide shuffling, enabling the utilization of predicted MFE values and distinct scoring mechanisms to effectively discriminate native interactions from spurious ones [86, 251]. These accessibility algorithms are especially valuable for de novo predictions, particularly in scenarios where computational efficiency is essential, as is the case with IntaRNA and RNAplex, given that candidate target RNAs can be extensive, spanning thousands of nucleotides [128, 134, 135, 205]. RNAplex, in particular, excels at identifying correct interaction regions that might be embedded within larger RNA targets [128]. In essence, accessibility-based MFE algorithms excel IO and concatenation-based tools due to their consideration of RNA sequence structural accessibility and evaluation of base-pairing potential, improving the capability to discern real interactions from nonspecific interactions.
In the context of selecting RSP and RIP tools based on comparative sequence analysis, Pfold and RNAalifold generally exhibit strong performance, especially for well-aligned short sequences [172, 173]. However, it is worth noting that RNAalifold outperforms in terms of speed and is better suited for well-aligned, longer RNA sequences [172]. For datasets comprising short sequences (< 200 bases) with significant diversity, Dynalign is a suitable choice because it does not rely on sequence similarity, and its scoring function excludes sequence comparisons [183]. In other scenarios, a combination of RNAalifold and/or Pfold can be employed to fold similar RNA sequences [172, 173], while RNAforester and/or MARNA can be used to align these folded RNA molecules [252, 253]. Notably, most of the MSA algorithms do not favour transitions over transversions or employ ad hoc two-parameter methods to model these distinctions (e.g. ClustalW [170]). This can be relevant because structural RNA sequences often evolve rapidly through structure-neutral mutations, which tend to involve transitions rather than transversions [254, 255]. Therefore, multiple sequence algorithms that utilise more sophisticated yet accurate models of sequence evolution are likely to produce improved alignments for folding [164].
Table 11 offers a comprehensive overview of the advantages and limitations associated with MFE-based RSP and RIP tools. Additionally, Figure 5 presents a chronological depiction of the development timeline of RSP and RIP tools. Understanding this timeline is crucial for selecting the most appropriate tools based on research objectives and the evolution of available technologies.
Table 11.
Advantages and shortcomings of MFE-based RIP and RSP tools
| Type of RIP and RSP Tools | Advantages | Shortcomings | Ref. |
|---|---|---|---|
| Nussinov algorithm |
|
|
[114, 119–123, 285] |
| Interaction-only |
|
|
[87, 127, 142, 203] |
| Accessibility-based |
|
|
[86, 138, 139] |
| Concatenation-based |
|
|
[87, 143] |
MCC: Matthews correlation coefficient; MSA: multiple sequence alignment; RIP: RNA–RNA interaction prediction; RNA: ribonucleic acid; RRI: RNA–RNA interaction; RSP: RNA structure prediction
Figure 5.

Timeline of RNA structure prediction (RSP) and RNA–RNA interaction prediction (RIP) tools. (A) Chronological overview of RSP and RIP tools, highlighting the different approaches via minimum free energy and comparative sequence analysis; (B) tools involving pseudoknots and artificial intelligence.
CONCLUSION
In recent years, the intersection of structure-based RNA analysis and computational biology has garnered significant attention as researchers recognize the crucial role of RNA structures in RNA function. Despite the availability of large-scale RNA sequence data, the development of computational algorithms for RSP and RIP has faced challenges, including the complexity of RNA structures and limited training datasets. These challenges have been met with advancements in computational techniques, and the progress in RSP tools has provided a solid foundation for the development of RIP tools, enabling a deeper exploration of the intricate network of RRIs and their functional implications. This review aimed to provide a comprehensive overview of existing computational tools for both RSP and RIP, focusing on two main types of RRIs and the strategies employed to predict them. ML has also been integrated into RIP and RSP methodologies. However, it is important to note that ML-based methods cannot yet replace wet lab experiments and traditional computational approaches to obtain high-resolution RNA structures or accurate RIP. Nonetheless, the advent of deep learning technologies and high-performance hardware will foster a new generation of RIP and RSP tools with improved accuracy and running speed.
Key Points
Bridging the Gap: This comprehensive review features the connections between RSP and RIP, underscores the importance of RNA homologues, delves into the intricacies of pseudoknots and dissects the thermodynamics of RNA folding.
Informative Figures: Our review includes figures that elucidate RRI types, emphasise the two core strategies within RIP, simplify explanations of each strategy subtype, and present chronological timelines that trace the evolution of RSP and RIP tools.
Comprehensive Summary: A comprehensive summary of RSP and RIP tools, meticulously organised into detailed tables for each strategy type, is available. These tables encompass characteristics of the RSP and RIP tools, citations, concise definitions and functions, input and output specifications, applicable species, and status (active or inactive) for enhanced clarity.
Challenges and Future Directions: We highlight five primary challenges in RSP and RIP and elaborate on how the integration of artificial intelligence through machine learning and deep learning holds the potential to significantly enhance RSP and RIP.
Practical Recommendations: A dedicated section is included to offer valuable advice for the effective utilisation of RSP and RIP tools in various research applications.
ACKNOWLEDGEMENTS
The authors would like to thank Universiti Kebangsaan Malaysia and Ministry of Higher Education for the general supports.
Author Biographies
Francis Yew Fu Tieng recently graduated from the Universiti Kebangsaan Malaysia. His expertise lies in cancer genomics and transcriptomics, focusing on single-cell RNA-sequencing on circulating tumour cells. Additionally, he studies the interplay of RNA–RNA interactions, and identify the potential of natural products in alternative cancer treatment.
Muhammad-Redha Abdullah-Zawawi is a research fellow at the UKM Medical Molecular Biology Institute (UMBI), Universiti Kebangsaan Malaysia. His expertise includes bioinformatics and computational systems biology.
Nur Alyaa Afifah Md Shahri is a research officer at the UKM Medical Molecular Biology Institute (UMBI), Universiti Kebangsaan Malaysia. Her research focuses on exploring the significance of bioinformatics in molecular biology research and cancer genomics research.
Zeti-Azura Mohamed-Hussein leads the Bioinformatics & Computational Systems Biology (BCSB) group at Universiti Kebangsaan Malaysia. Her expertise includes bioinformatics approaches like sequence- and structure-based analysis, integrative omics analysis, comparative genomics, network analysis, and genomic variation studies.
Learn-Han Lee leads the Sunway Microbiomics Centre at Sunway University, Malaysia. He perceives teaching as a significant responsibility that enables him to fulfil his role as a scientist specializing in biochemistry, molecular biology, microbiology, and immunology.
Nurul-Syakima Ab Mutalib is a senior research fellow at the UKM Medical Molecular Biology Institute (UMBI), Universiti Kebangsaan Malaysia. Her expertise lies in cancer genomics, transcriptomics, and epigenomics, focusing on non-coding RNA as cancer biomarkers (microRNA). Additionally, she studies genetic variants in sudden cardiac death and identifies novel actinomycetes for potential antibacterial and anticancer drug discovery.
Contributor Information
Francis Yew Fu Tieng, UKM Medical Molecular Biology Institute (UMBI), Universiti Kebangsaan Malaysia (UKM), Kuala Lumpur 56000, Malaysia.
Muhammad-Redha Abdullah-Zawawi, UKM Medical Molecular Biology Institute (UMBI), Universiti Kebangsaan Malaysia (UKM), Kuala Lumpur 56000, Malaysia.
Nur Alyaa Afifah Md Shahri, UKM Medical Molecular Biology Institute (UMBI), Universiti Kebangsaan Malaysia (UKM), Kuala Lumpur 56000, Malaysia.
Zeti-Azura Mohamed-Hussein, Institute of Systems Biology (INBIOSIS), UKM, Selangor 43600, Malaysia; Department of Applied Physics, Faculty of Science and Technology, UKM, Selangor 43600, Malaysia.
Learn-Han Lee, Sunway Microbiomics Centre, School of Medical and Life Sciences, Sunway University, Sunway City 47500, Malaysia; Novel Bacteria and Drug Discovery Research Group, Microbiome and Bioresource Research Strength, Jeffrey Cheah School of Medicine and Health Sciences, Monash University of Malaysia, Selangor 47500, Malaysia.
Nurul-Syakima Ab Mutalib, UKM Medical Molecular Biology Institute (UMBI), Universiti Kebangsaan Malaysia (UKM), Kuala Lumpur 56000, Malaysia; Novel Bacteria and Drug Discovery Research Group, Microbiome and Bioresource Research Strength, Jeffrey Cheah School of Medicine and Health Sciences, Monash University of Malaysia, Selangor 47500, Malaysia; Faculty of Health Sciences, UKM, Kuala Lumpur 50300, Malaysia.
FUNDING
No funding was received.
DATA AVAILABILITY
Not applicable.
AUTHORS’ CONTRIBUTIONS
N.-S.A.M. and F.Y.F.T. conceptualise the idea. F.Y.F.T. prepared and wrote the original draft. F.Y.F.T. and M.-R.A.-Z. created all the figures. M.-R.A.-Z., N.-S.A.M. and F.Y.F.T. were involved in reviewing and editing the manuscript. N.S.A.M., M.-R.A.-Z., N.A.A.M.S., Z.-A.M.-H. and L.-H.L. provided critical scientific insights.
MATERIAL DISCLAIMER
The opinions expressed in Briefings in Bioinformatics are those of the authors and contributors and do not necessarily reflect those of the editors, the editorial board, Oxford University Press or the organization to which the authors are affiliated.
References
- 1. Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953;171:737–8. [DOI] [PubMed] [Google Scholar]
- 2. Crick FH. The origin of the genetic code. J Mol Biol 1968;38:367–79. [DOI] [PubMed] [Google Scholar]
- 3. Crick F. Central dogma of molecular biology. Nature 1970;227:561–3. [DOI] [PubMed] [Google Scholar]
- 4. Robertson MP, Joyce GF. The origins of the RNA world. Cold Spring Harb Perspect Biol 2012;4:a003608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orgel LE. Evolution of the genetic apparatus. J Mol Biol 1968;38:381–93. [DOI] [PubMed] [Google Scholar]
- 6. Woese CR, Dugre DH, Dugre SA, et al. On the fundamental nature and evolution of the genetic code. Cold Spring Harb Symp Quant Biol 1966;31:723–36. [DOI] [PubMed] [Google Scholar]
- 7. Woese CR, Dugre DH, Saxinger WC, Dugre SA. The molecular basis for the genetic code. Proc Natl Acad Sci U S A 1966;55:966–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ban N, Nissen P, Hansen J, et al. The complete atomic structure of the large ribosomal subunit at 2.4 a resolution. Science 2000;289:905–20. [DOI] [PubMed] [Google Scholar]
- 9. Gilbert W. Origin of life: the RNA world. Nature 1986;319:618–8. [Google Scholar]
- 10. Guerrier-Takada C, Gardiner K, Marsh T, et al. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983;35:849–57. [DOI] [PubMed] [Google Scholar]
- 11. Kruger K, Grabowski PJ, Zaug AJ, et al. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982;31:147–57. [DOI] [PubMed] [Google Scholar]
- 12. Lewin R. RNA catalysis gives fresh perspective on the origin of life: the old chicken-and-egg problem of the origin of life is illuminated in unexpected ways by recent results on the splicing of RNA precursors. Science 1986;231:545–6. [DOI] [PubMed] [Google Scholar]
- 13. Pace NR, Marsh TL. RNA catalysis and the origin of life. Orig Life Evol Biosph 1985;16:97–116. [DOI] [PubMed] [Google Scholar]
- 14. Shampo MA, Kyle RA, Steensma DP. Sidney Altman—Nobel laureate for work with RNA. Mayo Clin Proc 2012;87:e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharp PA. On the origin of RNA splicing and introns. Cell 1985;42:397–400. [DOI] [PubMed] [Google Scholar]
- 16. Wimberly BT, Brodersen DE, Clemons WM, et al. Structure of the 30S ribosomal subunit. Nature 2000;407:327–39. [DOI] [PubMed] [Google Scholar]
- 17. Yusupov MM, Yusupova GZ, Baucom A, et al. Crystal structure of the ribosome at 5.5 a resolution. Science 2001;292:883–96. [DOI] [PubMed] [Google Scholar]
- 18. Palazzo AF, Lee ES. Non-coding RNA: what is functional and what is junk? Front Genet 2015;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pertea M. The human transcriptome: an unfinished story. Genes (Basel) 2012;3:344–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011;25:1915–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carninci P, Kasukawa T, Katayama S, et al. The transcriptional landscape of the mammalian genome. Science 2005;309:1559–63. [DOI] [PubMed] [Google Scholar]
- 22. Guttman M, Amit I, Garber M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009;458:223–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Djebali S, Davis CA, Merkel A, et al. Landscape of transcription in human cells. Nature 2012;489:101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet 2013;9:e1003569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wright PR, Mann M, Backofen R. Structure and interaction prediction in prokaryotic RNA biology. Microbiol Spectr 2018;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell 2019;179:1033–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frías-Lasserre D, Villagra CA. The importance of ncRNAs as epigenetic mechanisms in phenotypic variation and organic evolution. Front Microbiol 2017;8:2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Micheel J, Safrastyan A, Wollny D. Advances in non-coding RNA sequencing. Noncoding RNA 2021;7:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ikemura T, Dahlberg JE. Small ribonucleic acids of Escherichia coli. I. Characterization by polyacrylamide gel electrophoresis and fingerprint analysis. J Biol Chem 1973;248:5024–32. [PubMed] [Google Scholar]
- 30. Lee RC, Feinbaum RL, Ambros V. The C. Elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993;75:843–54. [DOI] [PubMed] [Google Scholar]
- 31. Bhaskaran M, Mohan M. MicroRNAs: history, biogenesis, and their evolving role in animal development and disease. Vet Pathol 2014;51:759–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kung JTY, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics 2013;193:651–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zampetaki A, Albrecht A, Steinhofel K. Long non-coding RNA structure and function: is there a link? Front Physiol 2018;9:1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grote P, Wittler L, Währisch S, et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell 2013;24:206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Guttman M, Donaghey J, Carey BW, et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011;477:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sauvageau M, Goff LA, Lodato S, et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2013;2:e01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dawson WK, Bujnicki JM. Computational modeling of RNA 3D structures and interactions. Curr Opin Struct Biol 2016;37:22–8. [DOI] [PubMed] [Google Scholar]
- 38. Ambros V. The functions of animal microRNAs. Nature 2004;431:350–5. [DOI] [PubMed] [Google Scholar]
- 39. Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806–11. [DOI] [PubMed] [Google Scholar]
- 40. Holley RW, Apgar J, Everett GA, et al. Structure of a ribonucleic acid. Science 1965;147:1462–5. [DOI] [PubMed] [Google Scholar]
- 41. Holley RW, Everett GA, Madison JT, Zamir A. Nucleotide sequences in the yeast alanine transfer ribonucleic acid. J Biol Chem 1965;240:2122–8. [PubMed] [Google Scholar]
- 42. Dieterich C, Stadler PF. Computational biology of RNA interactions. Wiley Interdiscip Rev RNA 2013;4:107–20. [DOI] [PubMed] [Google Scholar]
- 43. Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 2013;153:654–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Graf J, Kretz M. From structure to function: route to understanding lncRNA mechanism. Bioessays 2020;42:e2000027. [DOI] [PubMed] [Google Scholar]
- 45. Kudla G, Granneman S, Hahn D, et al. Cross-linking, ligation, and sequencing of hybrids reveals RNA-RNA interactions in yeast. Proc Natl Acad Sci U S A 2011;108:10010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu Z, Chang HY. The RNA Base-pairing problem and base-pairing solutions. Cold Spring Harb Perspect Biol 2018;10:a034926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tsao N, Ashour ME, Mosammaparast N. How RNA impacts DNA repair. DNA Repair (Amst) 2023;131:103564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014;346:1258096. [DOI] [PubMed] [Google Scholar]
- 49. Khelifi G, Hussein SMI. A new view of genome organization through RNA directed interactions. Frontiers in Cell and Developmental Biology 2020;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Glisovic T, Bachorik JL, Yong J, Dreyfuss G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett 2008;582:1977–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Will CL, Lührmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol 2011;3:a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Passmore LA, Coller J. Roles of mRNA poly(a) tails in regulation of eukaryotic gene expression. Nat Rev Mol Cell Biol 2022;23:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Alberts B, Johnson A, Lewis J, et al. From RNA to protein. In: Molecular Biology of the Cell (4th ed). New York: Garland Science, 2002, pp. 132–3. [Google Scholar]
- 54. Assmann SM, Chou H-L, Bevilacqua PC. Rock, scissors, paper: how RNA structure informs function. Plant Cell 2023;35:1671–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Noller HF, Lancaster L, Zhou J, Mohan S. The ribosome moves: RNA mechanics and translocation. Nat Struct Mol Biol 2017;24:1021–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Macfarlane L-A, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics 2010;11:537–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Haruehanroengra P, Zheng YY, Zhou Y, et al. RNA modifications and cancer. RNA Biol 2020;17:1560–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Spencer M. The stereochemistry of deoxyribonucleic acid. II. Hydrogen-bonded pairs of bases. Acta Crystallogr 1959;12:66–71. [Google Scholar]
- 59. Luttermann C, Meyers G. The importance of inter- and intramolecular base pairing for translation reinitiation on a eukaryotic bicistronic mRNA. Genes Dev 2009;23:331–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Varani G, McClain, WH. The G·U wobble base pair. EMBO Rep 2000;1:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brenner S. Codon-anticodon pairing: the wobble hypothesis. In: Molecular Biology: A Selection of Papers. Massachusetts: Academic Press, 2012, pp. 370–7.
- 62. Murphy FV, Ramakrishnan V. Structure of a purine-purine wobble base pair in the decoding center of the ribosome. Nat Struct Mol Biol 2004;11:1251–2. [DOI] [PubMed] [Google Scholar]
- 63. Appasamy SD, Hamdani HY, Ramlan EI, Firdaus-Raih M. InterRNA: a database of base interactions in RNA structures. Nucleic Acids Res 2016;44:D266–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Treeck BV, Protter DSW, Matheny T, et al. RNA self-assembly contributes to stress granule formation and defining the stress granule transcriptome. PNAS 2018;115:2734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Van Treeck B, Parker R. Emerging roles for intermolecular RNA-RNA interactions in RNP assemblies. Cell 2018;174:791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lim LP, Lau NC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 2005;433:769–73. [DOI] [PubMed] [Google Scholar]
- 67. Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009;19:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Neph S, Vierstra J, Stergachis AB, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 2012;489:83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Buvoli M, Cobianchi F, Riva S. Interaction of hnRNP A1 with snRNPs and pre-mRNAs: evidence for a possible role of A1 RNA annealing activity in the first steps of spliceosome assembly. Nucleic Acids Res 1992;20:5017–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009;136:701–18. [DOI] [PubMed] [Google Scholar]
- 71. Seraphin B, Rosbash M. Identification of functional U1 snRNA-pre-mRNA complexes committed to spliceosome assembly and splicing. Cell 1989;59:349–58. [DOI] [PubMed] [Google Scholar]
- 72. Matera AG, Terns RM, Terns MP. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol 2007;8:209–20. [DOI] [PubMed] [Google Scholar]
- 73. Bachellerie JP, Cavaillé J, Hüttenhofer A. The expanding snoRNA world. Biochimie 2002;84:775–90. [DOI] [PubMed] [Google Scholar]
- 74. Kable ML, Seiwert SD, Heidmann S, Stuart K. RNA editing: a mechanism for gRNA-specified uridylate insertion into precursor mRNA. Science 1996;273:1189–95. [DOI] [PubMed] [Google Scholar]
- 75. Evans D, Marquez SM, Pace NR. RNase P: interface of the RNA and protein worlds. Trends Biochem Sci 2006;31:333–41. [DOI] [PubMed] [Google Scholar]
- 76. Alberts B, Johnson A, Lewis J, et al. The initiation and completion of DNA replication in chromosomes. In: Molecular Biology of the Cell (4th edition). New York: Garland Science, 2002, pp. 660–2. [Google Scholar]
- 77. Schneider B, Morávek Z, Berman HM. RNA conformational classes. Nucleic Acids Res 2004;32:1666–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schroeder SJ. Challenges and approaches to predicting RNA with multiple functional structures. RNA 2018;24:1615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shajani Z, Varani G. NMR studies of dynamics in RNA and DNA by 13C relaxation. Biopolymers 2007;86:348–59. [DOI] [PubMed] [Google Scholar]
- 80. Mustoe AM, Brooks CL, Al-Hashimi HM. Hierarchy of RNA functional dynamics. Annu Rev Biochem 2014;83:441–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Al-Hashimi HM. Beyond static structures of RNA by NMR: folding, refolding, and dynamics at atomic resolution. Biopolymers 2007;86:345–7. [DOI] [PubMed] [Google Scholar]
- 82. Vicens Q, Kieft JS. Thoughts on how to think (and talk) about RNA structure. Proc Natl Acad Sci 2022;119:e2112677119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhang J, Fei Y, Sun L, Zhang QC. Advances and opportunities in RNA structure experimental determination and computational modeling. Nat Methods 2022;19:1193–207. [DOI] [PubMed] [Google Scholar]
- 84. Dai X, Zhang S, Zaleta-Rivera K. RNA: interactions drive functionalities. Mol Biol Rep 2020;47:1413–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Meyer IM. Predicting novel RNA–RNA interactions. Curr Opin Struct Biol 2008;18:387–93. [DOI] [PubMed] [Google Scholar]
- 86. Umu SU, Gardner PP. A comprehensive benchmark of RNA–RNA interaction prediction tools for all domains of life. Bioinformatics 2017;33:988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lai D, Meyer IM. A comprehensive comparison of general RNA–RNA interaction prediction methods. Nucleic Acids Res 2016;44:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Matarrese MAG, Loppini A,Nicoletti M, et al. Assessment of tools for RNA secondary structure prediction and extraction: a final-user perspective. J Biomol Struct Dyn 2023;41:6917–36. [DOI] [PubMed] [Google Scholar]
- 89. Barnwal RP, Yang F, Varani G. Applications of NMR to structure determination of RNAs large and small. Arch Biochem Biophys 2017;628:42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Li B, Cao Y, Westhof E, Miao Z. Advances in RNA 3D structure Modeling using experimental data. Front Genet 2020;11:574485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Krishnan V, Rupp B. Macromolecular structure determination: comparison of X-ray crystallography and NMR spectroscopy. In: Encyclopedia of Life Sciences. New Jersey: John Wiley & Sons, 2012, a0002716.
- 92. Fürtig B, Richter C, Wöhnert J, Schwalbe H. NMR spectroscopy of RNA. Chembiochem 2003;4:936–62. [DOI] [PubMed] [Google Scholar]
- 93. Westhof E. Twenty years of RNA crystallography. RNA 2015;21:486–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Consortium TR . RNAcentral: a comprehensive database of non-coding RNA sequences. Nucleic Acids Res 2017;45:D128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kretz M, Siprashvili Z, Chu C, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013;493:231–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Engreitz JM, Sirokman K, McDonel P, et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell 2014;159:188–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Nguyen TC, Cao X, Yu P, et al. Mapping RNA–RNA interactome and RNA structure in vivo by MARIO. Nat Commun 2016;7:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lu Z, Zhang QC, Lee B, et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 2016;165:1267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Aw JGA, Shen Y, Wilm A, et al. In vivo mapping of eukaryotic RNA Interactomes reveals principles of higher-order organization and regulation. Mol Cell 2016;62:603–17. [DOI] [PubMed] [Google Scholar]
- 100. Sharma E, Sterne-Weiler T, O’Hanlon D, Blencowe BJ. Global mapping of human RNA-RNA interactions. Mol Cell 2016;62:618–26. [DOI] [PubMed] [Google Scholar]
- 101. Yi Y, Zhao Y, Li C, et al. RAID v2.0: an updated resource of RNA-associated interactions across organisms. Nucleic Acids Res 2017;45:D115–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Yuan J, Wu W, Xie C, et al. NPInter v2.0: an updated database of ncRNA interactions. Nucleic Acids Res 2014;42:D104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Teng X, Chen X, Xue H, et al. NPInter v4.0: an integrated database of ncRNA interactions. Nucleic Acids Res 2020;48:D160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wu T, Wang J, Liu C, et al. NPInter: the noncoding RNAs and protein related biomacromolecules interaction database. Nucleic Acids Res 2006;34:D150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Kang J, Tang Q, He J, et al. RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res 2021;50:D326–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lin Y, Liu T, Cui T, et al. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res 2020;48:D189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gong J, Shao D, Xu K, et al. RISE: a database of RNA interactome from sequencing experiments. Nucleic Acids Res 2018;46:D194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Iyer MK, Niknafs YS, Malik R, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 2015;47:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Trotta E. On the normalization of the minimum free energy of RNAs by sequence length. PloS One 2014;9:e113380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sykes MT, Levitt M. Simulations of RNA base pairs in a nanodroplet reveal solvation-dependent stability. Proc Natl Acad Sci 2007;104:12336–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Parsch J, Braverman JM, Stephan W. Comparative sequence analysis and patterns of covariation in RNA secondary structures. Genetics 2000;154:909–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Sato K, Akiyama M, Sakakibara Y. RNA secondary structure prediction using deep learning with thermodynamic integration. Nat Commun 2021;12:941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Zhang H, Zhang C, Li Z, et al. A new method of RNA secondary structure prediction based on convolutional neural network and dynamic programming. Front Genet 2019;10:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Mathews DH, Turner DH. Prediction of RNA secondary structure by free energy minimization. Curr Opin Struct Biol 2006;16:270–8. [DOI] [PubMed] [Google Scholar]
- 115. Tinoco I, Borer PN, Dengler B, et al. Improved estimation of secondary structure in ribonucleic acids. Nat New Biol 1973;246:40–1. [DOI] [PubMed] [Google Scholar]
- 116. Borer PN, Dengler B, Tinoco I, Uhlenbeck OC. Stability of ribonucleic acid double-stranded helices. J Mol Biol 1974;86:843–53. [DOI] [PubMed] [Google Scholar]
- 117. Turner DH, Mathews DH. NNDB: the nearest neighbor parameter database for predicting stability of nucleic acid secondary structure. Nucleic Acids Res 2010;38:D280–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 1999;288:911–40. [DOI] [PubMed] [Google Scholar]
- 119. Bellman R. The theory of dynamic programming. Bull Amer Math Soc 1954;60:503–15. [Google Scholar]
- 120. Bellman R. The structure of dynamic programming processes. In: Dynamic Programming (6th Ed). New Jersey: Princeton University Press, 1957, pp. 81–115.
- 121. Nussinov R, Pieczenik G, Griggs JR, Kleitman DJ. Algorithms for loop matchings. SIAM J Appl Math 1978;35:68–82. [Google Scholar]
- 122. Lyngsø RB. RNA secondary structure prediction by minimum free energy. In: Encyclopedia of Algorithms. Germany: SpringerLink, 2016, pp. 1846–50. [Google Scholar]
- 123. Zuker M, Stiegler P. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res 1981;9:133–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Backofen R, Hess WR. Computational prediction of sRNAs and their targets in bacteria. RNA Biol 2010;7:33–42. [DOI] [PubMed] [Google Scholar]
- 125. Lorenz R, Bernhart SH, Höner zu Siederdissen C, et al. ViennaRNA package 2.0. Algorithms for Molecular Biology 2011;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Rehmsmeier M, Steffen P, Hochsmann M, et al. Fast and effective prediction of microRNA/target duplexes. RNA 2004;10:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tjaden B. TargetRNA: a tool for predicting targets of small RNA action in bacteria. Nucleic Acids Res 2008;36:W109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tafer H, Hofacker IL. RNAplex: a fast tool for RNA–RNA interaction search. Bioinformatics 2008;24:2657–63. [DOI] [PubMed] [Google Scholar]
- 129. Wenzel A, Akbaşli E, Gorodkin J. RIsearch: fast RNA–RNA interaction search using a simplified nearest-neighbor energy model. Bioinformatics 2012;28:2738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Gerlach W, Giegerich R. GUUGle: a utility for fast exact matching under RNA complementary rules including G–U base pairing. Bioinformatics 2006;22:762–4. [DOI] [PubMed] [Google Scholar]
- 131. McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers 1990;29:1105–19. [DOI] [PubMed] [Google Scholar]
- 132. Ding Y, Chan CY, Lawrence CE. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res 2004;32:W135–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Mückstein U, Tafer H, Hackermüller J, et al. Thermodynamics of RNA-RNA binding. Bioinformatics 2006;22:1177–82. [DOI] [PubMed] [Google Scholar]
- 134. Mann M, Wright PR, Backofen R. IntaRNA 2.0: enhanced and customizable prediction of RNA–RNA interactions. Nucleic Acids Res 2017;45:W435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Busch A, Richter AS, Backofen R. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 2008;24:2849–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Eggenhofer F, Tafer H, Stadler PF, Hofacker IL. RNApredator: fast accessibility-based prediction of sRNA targets. Nucleic Acids Res 2011;39:W149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Lu ZJ, Mathews DH. OligoWalk: an online siRNA design tool utilizing hybridization thermodynamics. Nucleic Acids Res 2008;36:W104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Poolsap U, Kato Y, Sato K, et al. Using binding profiles to predict binding sites of target RNAs. J Bioinform Comput Biol 2011;09:697–713. [DOI] [PubMed] [Google Scholar]
- 139. Salari R, Backofen R, Sahinalp SC. Fast prediction of RNA-RNA interaction. Algorithms for Molecular Biology 2010;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Alkan F, Wenzel A, Palasca O, et al. RIsearch2: suffix array-based large-scale prediction of RNA-RNA interactions and siRNA off-targets. Nucleic Acids Res 2017;45:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Fukunaga T, Hamada M. RIblast: an ultrafast RNA-RNA interaction prediction system based on a seed-and-extension approach. Bioinformatics 2017;33:2666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kery MB, Feldman M, Livny J, Tjaden B. TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res 2014;42:W124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Andronescu M, Aguirre-Hernández R, Condon A, Hoos HH. RNAsoft: a suite of RNA secondary structure prediction and design software tools. Nucleic Acids Res 2003;31:3416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Andronescu M, Zhang ZC, Condon A. Secondary structure prediction of interacting RNA molecules. J Mol Biol 2005;345:987–1001. [DOI] [PubMed] [Google Scholar]
- 145. Lorenz R, Hofacker IL, Stadler PF. RNA folding with hard and soft constraints. Algorithms Mol Biol 2016;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Markham NR, Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol Biol 2008;453:3–31. [DOI] [PubMed] [Google Scholar]
- 147. Schäfer RA, Voß B. RNAnue: efficient data analysis for RNA–RNA interactomics. Nucleic Acids Res 2021;49:5493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Fornace ME, Porubsky NJ, Pierce NA. A unified dynamic programming framework for the analysis of interacting nucleic acid strands: enhanced models, scalability, and speed. ACS Synthetic Biology 2020;9:2665–78. [DOI] [PubMed] [Google Scholar]
- 149. Zadeh JN, Steenberg CD, Bois JS, et al. NUPACK: analysis and design of nucleic acid systems. J Comput Chem 2011;32:170–3. [DOI] [PubMed] [Google Scholar]
- 150. Hofacker IL, Stadler PF. Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics 2006;22:1172–6. [DOI] [PubMed] [Google Scholar]
- 151. Hofacker IL, Fontana W, Stadler PF, et al. Fast folding and comparison of RNA secondary structures. Monatsh Chem 1994;125:167–88. [Google Scholar]
- 152. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003;31:3406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Alkan C, Karakoç E, Nadeau JH, et al. RNA-RNA interaction prediction and antisense RNA target search. J Comput Biol 2006;13:267–82. [DOI] [PubMed] [Google Scholar]
- 154. Chitsaz H, Salari R, Sahinalp SC, Backofen R. A partition function algorithm for interacting nucleic acid strands. Bioinformatics 2009;25:i365–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Salari R, Backofen R, Sahinalp SC. Fast prediction of RNA-RNA interaction. Algorithms Mol Biol 2010;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Pervouchine DD. IRIS: intermolecular RNA interaction search. Genome Inform 2004;15:92–101. [PubMed] [Google Scholar]
- 157. Huang FWD, Qin J, Reidys CM, Stadler PF. Target prediction and a statistical sampling algorithm for RNA–RNA interaction. Bioinformatics 2010;26:175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Huang FWD, Qin J, Reidys CM, Stadler PF. Partition function and base pairing probabilities for RNA-RNA interaction prediction. Bioinformatics 2009;25:2646–54. [DOI] [PubMed] [Google Scholar]
- 159. Bernhart SH, Tafer H, Mückstein U, et al. Partition function and base pairing probabilities of RNA heterodimers. Algorithms Mol Biol 2006;1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Dirks RM, Bois JS, Schaeffer JM, et al. Thermodynamic analysis of interacting nucleic acid strands. SIAM Review 2007;49:65–88. [Google Scholar]
- 161. Ebrahimpour-Boroojeny A, Rajopadhye S, Chitsaz H. BPPart: RNA-RNA Interaction Partition Function in the Absence of Entropy. In: 21st International Workshop on Algorithms in Bioinformatics, virtual. Germany: Leibniz International Proceedings in Informatics, 2021, 201:14:1-14:24.
- 162. Kato Y, Akutsu T, Seki H. A grammatical approach to RNA–RNA interaction prediction. Pattern Recognition 2009;42:531–8. [Google Scholar]
- 163. Kato Y, Sato K, Hamada M, et al. RactIP: fast and accurate prediction of RNA-RNA interaction using integer programming. Bioinformatics 2010;26:i460–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Gardner PP, Giegerich R. A comprehensive comparison of comparative RNA structure prediction approaches. BMC Bioinformatics 2004;5:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Appasamy SD, Ramlan EI, Firdaus-Raih M. Comparative sequence and structure analysis reveals the conservation and diversity of nucleotide positions and their associated tertiary interactions in the riboswitches. PloS One 2013;8:e73984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Madison JT, Everett GA, Kung H. Nucleotide sequence of a yeast tyrosine transfer RNA. Science 1966;153:531–4. [DOI] [PubMed] [Google Scholar]
- 167. Gutell RR, Lee JC, Cannone JJ. The accuracy of ribosomal RNA comparative structure models. Curr Opin Struct Biol 2002;12:301–10. [DOI] [PubMed] [Google Scholar]
- 168. Knudsen B, Hein J. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics 1999;15:446–54. [DOI] [PubMed] [Google Scholar]
- 169. Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007;23:2947–8. [DOI] [PubMed] [Google Scholar]
- 170. Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics 2002;Chapter 2:Unit 2.3. [DOI] [PubMed] [Google Scholar]
- 171. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 2013;30:772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Bernhart SH, Hofacker IL, Will S, et al. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics 2008;9:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Knudsen B, Hein J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res 2003;31:3423–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Seemann SE, Menzel P, Backofen R, Gorodkin J. The PETfold and PETcofold web servers for intra- and intermolecular structures of multiple RNA sequences. Nucleic Acids Res 2011;39:W107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Sankoff D. Simultaneous solution of the RNA folding, alignment and protosequence problems. SIAM J Appl Math 1985;45:810–25. [Google Scholar]
- 176. Tahi F, Gouy M, Régnier M. Automatic RNA secondary structure prediction with a comparative approach. Comput Chem 2002;26:521–30. [DOI] [PubMed] [Google Scholar]
- 177. Tahi F, Engelen S, Regnier M. A fast algorithm for RNA secondary structure prediction including pseudoknots. Third IEEE Symposium on Bioinformatics and Bioengineering, Bethesda, MD, 2003. Proceedings. 2003; 11–7. IEEE Computer Society, Washington, DC, USA.
- 178. Havgaard JH, Gorodkin J. RNA structural alignments, part I: Sankoff-based approaches for structural alignments. Methods Mol Biol 2014;1097:275–90. [DOI] [PubMed] [Google Scholar]
- 179. Höchsmann M, Töller T, Giegerich R, Kurtz S. Local similarity in RNA secondary structures. Proc IEEE Comput Soc Bioinform Conf 2003;2:159–68. [PubMed] [Google Scholar]
- 180. Gorodkin J, Heyer LJ, Stormo GD. Finding the most significant common sequence and structure motifs in a set of RNA sequences. Nucleic Acids Res 1997;25:3724–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Havgaard JH, Lyngsø RB, Stormo GD, et al. Pairwise local structural alignment of RNA sequences with sequence similarity less than 40%. Bioinformatics 2005;21:1815–24. [DOI] [PubMed] [Google Scholar]
- 182. Sundfeld D, Havgaard JH, de Melo ACMA, Gorodkin J. Foldalign 2.5: multithreaded implementation for pairwise structural RNA alignment. Bioinformatics 2016;32:1238–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Mathews DH, Turner DH. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J Mol Biol 2002;317:191–203. [DOI] [PubMed] [Google Scholar]
- 184. Bradley RK, Pachter L, Holmes I. Specific alignment of structured RNA: stochastic grammars and sequence annealing. Bioinformatics 2008;24:2677–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Tabei Y, Kiryu H, Kin T, Asai K. A fast structural multiple alignment method for long RNA sequences. BMC Bioinformatics 2008;9:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Hofacker IL, Bernhart SHF, Stadler PF. Alignment of RNA base pairing probability matrices. Bioinformatics 2004;20:2222–7. [DOI] [PubMed] [Google Scholar]
- 187. Will S, Reiche K, Hofacker IL, et al. Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering. PLoS Comput Biol 2007;3:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Torarinsson E, Havgaard JH, Gorodkin J. Multiple structural alignment and clustering of RNA sequences. Bioinformatics 2007;23:926–32. [DOI] [PubMed] [Google Scholar]
- 189. Kiryu H, Tabei Y, Kin T, Asai K. Murlet: a practical multiple alignment tool for structural RNA sequences. Bioinformatics 2007;23:1588–98. [DOI] [PubMed] [Google Scholar]
- 190. Dalli D, Wilm A, Mainz I, Steger G. StrAl: progressive alignment of non-coding RNA using base pairing probability vectors in quadratic time. Bioinformatics 2006;22:1593–9. [DOI] [PubMed] [Google Scholar]
- 191. Do CB, Foo C-S, Batzoglou S. A max-margin model for efficient simultaneous alignment and folding of RNA sequences. Bioinformatics 2008;24:i68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Allali J, Sagot M-F. A new distance for high level RNA secondary structure comparison. IEEE/ACM Trans Comput Biol Bioinform 2005;2:3–14. [DOI] [PubMed] [Google Scholar]
- 193. Reeder J, Giegerich R. Consensus shapes: an alternative to the Sankoff algorithm for RNA consensus structure prediction. Bioinformatics 2005;21:3516–23. [DOI] [PubMed] [Google Scholar]
- 194. Bossanyi M-A, Carpentier V, Glouzon J-PS, et al. aliFreeFoldMulti: alignment-free method to predict secondary structures of multiple RNA homologs. NAR Genomics and Bioinformatics 2020;2:lqaa086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 2000;302:205–17. [DOI] [PubMed] [Google Scholar]
- 196. Bremges A, Schirmer S, Giegerich R. Fine-tuning structural RNA alignments in the twilight zone. BMC Bioinformatics 2010;11:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Lindgreen S, Gardner PP, Krogh A. MASTR: multiple alignment and structure prediction of non-coding RNAs using simulated annealing. Bioinformatics 2007;23:3304–11. [DOI] [PubMed] [Google Scholar]
- 198. Xu X, Ji Y, Stormo GD. RNA sampler: a new sampling based algorithm for common RNA secondary structure prediction and structural alignment. Bioinformatics 2007;23:1883–91. [DOI] [PubMed] [Google Scholar]
- 199. Yao Z, Weinberg Z, Ruzzo WL. CMfinder—a covariance model based RNA motif finding algorithm. Bioinformatics 2006;22:445–52. [DOI] [PubMed] [Google Scholar]
- 200. Bauer M, Klau GW, Reinert K. Accurate multiple sequence-structure alignment of RNA sequences using combinatorial optimization. BMC Bioinformatics 2007;8:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Li AX, Marz M, Qin J, Reidys CM. RNA-RNA interaction prediction based on multiple sequence alignments. Bioinformatics 2011;27:456–63. [DOI] [PubMed] [Google Scholar]
- 202. Richter AS, Backofen R. Accessibility and conservation. RNA Biol 2012;9:954–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Krüger J, Rehmsmeier M. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res 2006;34:W451–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Hartung J. A note on combining dependent tests of significance. Biom J 1999;41:849–55. [Google Scholar]
- 205. Wright PR, Georg J, Mann M, et al. CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res 2014;42:W119–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Brierley I, Pennell S, Gilbert RJC. Viral RNA pseudoknots: versatile motifs in gene expression and replication. Nat Rev Microbiol 2007;5:598–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Puglisi JD, Wyatt JR, Tinoco I. Conformation of an RNA pseudoknot. J Mol Biol 1990;214:437–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Chiaruttini C, Milet M, Springer M. A long-range RNA-RNA interaction forms a pseudoknot required for translational control of the IF3-L35-L20 ribosomal protein operon in Escherichia coli. EMBO J 1996;15:4402–13. [PMC free article] [PubMed] [Google Scholar]
- 209. Ly H, Xu L, Rivera MA, et al. A role for a novel ‘trans-pseudoknot’ RNA–RNA interaction in the functional dimerization of human telomerase. Genes Dev 2003;17:1078–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Staple DW, Butcher SE. Pseudoknots: RNA structures with diverse functions. PLoS Biol 2005;3:e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Ten Dam E, Pleij K, Draper D. Structural and functional aspects of RNA pseudoknots. Biochemistry 1992;31:11665–76. [DOI] [PubMed] [Google Scholar]
- 212. Pleij CWA. Pseudoknots: a new motif in the RNA game. Trends Biochem Sci 1990;15:143–7. [DOI] [PubMed] [Google Scholar]
- 213. Pleij CWA, Bosch L. [21] RNA pseudoknot: structure, detection, and prediction. Methods Enzymol 1989;180:289–303. [DOI] [PubMed] [Google Scholar]
- 214. Nebel ME, Weinberg F. Algebraic and combinatorial properties of common RNA pseudoknot classes with applications. J Comput Biol 2012;19:1134–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Rivas E, Eddy SR. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J Mol Biol 1999;285:2053–68. [DOI] [PubMed] [Google Scholar]
- 216. Ren J, Rastegari B, Condon A, et al. HotKnots: heuristic prediction of RNA secondary structures including pseudoknots. RNA 2005;11:1494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Cary RB, Stormo GD. Graph-theoretic approach to RNA modeling using comparative data. Proc Int Conf Intell Syst Mol Biol 1995;3:75–80. [PubMed] [Google Scholar]
- 218. Page RDM. Comparative analysis of secondary structure of insect mitochondrial small subunit ribosomal RNA using maximum weighted matching. Nucleic Acids Res 2000;28:3839–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Tabaska JE, Cary RB, Gabow HN, Stormo GD. An RNA folding method capable of identifying pseudoknots and base triples. Bioinformatics 1998;14:691–9. [DOI] [PubMed] [Google Scholar]
- 220. Ruan J, Stormo GD, Zhang W. An iterated loop matching approach to the prediction of RNA secondary structures with pseudoknots. Bioinformatics 2003;20:58–66. [DOI] [PubMed] [Google Scholar]
- 221. Ruan J, Stormo GD, Zhang W. ILM: a web server for predicting RNA secondary structures with pseudoknots. Nucleic Acids Res 2004;32:W146–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Chen X, He S-M, Bu D, et al. FlexStem: improving predictions of RNA secondary structures with pseudoknots by reducing the search space. Bioinformatics 2008;24:1994–2001. [DOI] [PubMed] [Google Scholar]
- 223. Xayaphoummine A, Bucher T, Isambert H. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res 2005;33:W605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Bindewald E, Afonin K, Jaeger L, Shapiro BA. Multi-strand RNA secondary structure prediction and nanostructure design including pseudoknots. ACS Nano 2011;5:9542–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Xu X, Chen S-J. VfoldCPX server: predicting RNA-RNA complex structure and stability. PloS One 2016;11:e0163454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Sato K, Kato Y, Hamada M, et al. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 2011;27:i85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Gultyaev AP, van Batenburg FH, Pleij CW. The computer simulation of RNA folding pathways using a genetic algorithm. J Mol Biol 1995;250:37–51. [DOI] [PubMed] [Google Scholar]
- 228. Shi Y-Z, Wu Y-Y, Feng-Hua W, et al. RNA structure prediction: progress and perspective. Chinese Physics B 2014;23:078701. [Google Scholar]
- 229. Turner DH. Thermodynamics of base pairing. Curr Opin Struct Biol 1996;6:299–304. [DOI] [PubMed] [Google Scholar]
- 230. Antczak M, Zablocki M, Zok T, et al. RNAvista: a webserver to assess RNA secondary structures with non-canonical base pairs. Bioinformatics 2019;35:152–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Doshi KJ, Cannone JJ, Cobaugh CW, Gutell RR. Evaluation of the suitability of free-energy minimization using nearest-neighbor energy parameters for RNA secondary structure prediction. BMC Bioinformatics 2004;5:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. DiChiacchio L, Mathews DH. Predicting RNA-RNA interactions using RNAstructure. Methods Mol Biol 2016;1490:51–62. [DOI] [PubMed] [Google Scholar]
- 233. Zhao Q, Zhao Z, Fan X, et al. Review of machine learning methods for RNA secondary structure prediction. PLoS Comput Biol 2021;17:e21009291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Sato K, Hamada M. Recent trends in RNA informatics: a review of machine learning and deep learning for RNA secondary structure prediction and RNA drug discovery. Brief Bioinform 2023;24:bbad186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Justyna M, Antczak M, Szachniuk M. Machine learning for RNA 2D structure prediction benchmarked on experimental data. Brief Bioinform 2023;24:bbad153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Le Quy T, Roy A, Iosifidis V, et al. A survey on datasets for fairness-aware machine learning. WIREs Data Mining Knowledge Discov 2022;12:e1452. [Google Scholar]
- 237. Townshend RJL, Eismann S, Watkins AM, et al. Geometric deep learning of RNA structure. Science 2021;373:1047–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Zhang Y, Liu T, Chen L, et al. RIscoper: a tool for RNA–RNA interaction extraction from the literature. Bioinformatics 2019;35:3199–202. [DOI] [PubMed] [Google Scholar]
- 239. Chen Q, Leaman R, Allot A, et al. Artificial intelligence in action: addressing the COVID-19 pandemic with natural language processing. Ann Rev Biomed Data Sci 2021;4:313–39. [DOI] [PubMed] [Google Scholar]
- 240. JWJ A, Tataru P, Staines J, et al. Evolving stochastic context-free grammars for RNA secondary structure prediction. BMC Bioinformatics 2012;13:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Andronescu M, Condon A, Hoos HH, et al. Efficient parameter estimation for RNA secondary structure prediction. Bioinformatics 2007;23:i19–28. [DOI] [PubMed] [Google Scholar]
- 242. Do CB, Woods DA, Batzoglou S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 2006;22:e90–8. [DOI] [PubMed] [Google Scholar]
- 243. Zakov S, Goldberg Y, Elhadad M, Ziv-ukelson M. Rich parameterization improves RNA structure prediction. J Comput Biol 2011;18:1525–42. [DOI] [PubMed] [Google Scholar]
- 244. Rivas E, Lang R, Eddy SR. A range of complex probabilistic models for RNA secondary structure prediction that includes the nearest-neighbor model and more. RNA (New York, NY) 2012;18:193–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Yu H, Qi Y, Ding Y. Deep learning in RNA structure studies. Front Mol Biosci 2022;9:869601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Singh J, Hanson J, Paliwal K, Zhou Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat Commun 2019;10:5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Wang L, Liu Y, Zhong X, et al. DMfold: a novel method to predict RNA secondary structure with pseudoknots based on deep learning and improved base pair maximization principle. Front Genet 2019;10:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Górska A, Jasiński M, Trylska J. MINT: software to identify motifs and short-range interactions in trajectories of nucleic acids. Nucleic Acids Res 2015;43:e114–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Puton T, Kozlowski LP, Rother KM, Bujnicki JM. CompaRNA: a server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res 2014;42:5403–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Puton T, Kozlowski LP, Rother KM, Bujnicki JM. CompaRNA: a server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res 2013;41:4307–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Umu SU, Poole AM, Dobson RC, et al. Avoidance of stochastic RNA interactions can be harnessed to control protein expression levels in bacteria and archaea. Elife 5:e13479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Will S, Joshi T, Hofacker IL, et al. LocARNA-P: accurate boundary prediction and improved detection of structural RNAs. RNA 2012;18:900–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Siebert S, Backofen R. MARNA: multiple alignment and consensus structure prediction of RNAs based on sequence structure comparisons. Bioinformatics 2005;21:3352–9. [DOI] [PubMed] [Google Scholar]
- 254. Higgs PG. RNA secondary structure: physical and computational aspects. Q Rev Biophys 2000;33:199–253. [DOI] [PubMed] [Google Scholar]
- 255. Klein RJ, Eddy SR. RSEARCH: finding homologs of single structured RNA sequences. BMC Bioinformatics 2003;4:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Mamuye A, Merelli E, Tesei L. A graph grammar for modelling RNA folding. Electron Proc Theor Comput Sci 2016;231:31–41. [Google Scholar]
- 257. Pervouchine DD. Towards long-range RNA structure prediction in eukaryotic genes. Genes 2018;9:302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Fukunaga T, Iwakiri J, Ono Y, Hamada M. LncRRIsearch: a web server for lncRNA-RNA interaction prediction integrated with tissue-specific expression and subcellular localization data. Front Genet 2019;10:462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Reuter JS, Mathews DH. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 2010;11:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Reynolds A, Leake D, Boese Q, et al. Rational siRNA design for RNA interference. Nat Biotechnol 2004;22:326–30. [DOI] [PubMed] [Google Scholar]
- 261. Rennie W, Kanoria S, Liu C, et al. Sfold Tools for MicroRNA Target Prediction. Methods Mol Biol 2019;1970:31–42. [DOI] [PubMed] [Google Scholar]
- 262. DiChiacchio L, Sloma MF, Mathews DH. AccessFold: predicting RNA–RNA interactions with consideration for competing self-structure. Bioinformatics 2016;32:1033–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Wuchty S, Fontana W, Hofacker IL, Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers 1999;49:145–65. [DOI] [PubMed] [Google Scholar]
- 264. Tan Z, Fu Y, Sharma G, Mathews DH. TurboFold II: RNA structural alignment and secondary structure prediction informed by multiple homologs. Nucleic Acids Res 2017;45:11570–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 1981;17:368–76. [DOI] [PubMed] [Google Scholar]
- 266. Durbin R, Eddy SR, Krogh A, et al. Chapter 9: Transformational grammars. In: Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acidssource. New York, USA: Cambridge University Press, 1998, pp. 233–59.
- 267. Fu Y, Sharma G, Mathews DH. Dynalign II: common secondary structure prediction for RNA homologs with domain insertions. Nucleic Acids Res 2014;42:13939–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Xu Z, Mathews DH. Multilign: an algorithm to predict secondary structures conserved in multiple RNA sequences. Bioinformatics 2011;27:626–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Sorescu DA, Möhl M, Mann M, et al. CARNA—alignment of RNA structure ensembles. Nucleic Acids Res 2012;40:W49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Horesh Y, Doniger T, Michaeli S, Unger R. RNAspa: a shortest path approach for comparative prediction of the secondary structure of ncRNA molecules. BMC Bioinformatics 2007;8:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Ji Y, Xu X, Stormo GD. A graph theoretical approach for predicting common RNA secondary structure motifs including pseudoknots in unaligned sequences. Bioinformatics 2004;20:1591–602. [DOI] [PubMed] [Google Scholar]
- 272. Winkler J, Urgese G, Ficarra E, Reinert K. LaRA 2: parallel and vectorized program for sequence–structure alignment of RNA sequences. BMC Bioinformatics 2022;23:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Hochsmann M, Toller T, Giegerich R, et al. Local similarity in RNA secondary structures. In: Computational Systems Bioinformatics. Proceedings of the 2003 IEEE Bioinformatics Conference. Stanford, CA, 2003, pp. 159–68. The Institute of Electrical and Electronics Engineers, New York, USA. [PubMed]
- 274. Raden M, Ali SM, Alkhnbashi OS, et al. Freiburg RNA tools: a central online resource for RNA-focused research and teaching. Nucleic Acids Res 2018;46:W25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Reeder J, Steffen P, Giegerich R. pknotsRG: RNA pseudoknot folding including near-optimal structures and sliding windows. Nucleic Acids Res 2007;35:W320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Macke TJ, Ecker DJ, Gutell RR, et al. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res 2001;29:4724–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Legendre A, Angel E, Tahi F. RCPred: RNA complex prediction as a constrained maximum weight clique problem. BMC Bioinformatics 2019;20:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Bindewald E, Afonin KA, Viard M, et al. Multistrand structure prediction of nucleic acid assemblies and design of RNA switches. Nano Lett 2016;16:1726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Xu X, Zhao P, Chen S-J. Vfold: a web server for RNA structure and folding thermodynamics prediction. PloS One 2014;9:e107504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Bellaousov S, Mathews DH. ProbKnot: fast prediction of RNA secondary structure including pseudoknots. RNA 2010;16:1870–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Krogh A, Brown M, Mian IS, et al. Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol 1994;235:1501–31. [DOI] [PubMed] [Google Scholar]
- 282. Sükösd Z, Andersen ES, Lyngsø R. SCFGs in RNA secondary structure prediction RNA secondary structure prediction: a hands-on approach. Methods Mol Biol 2014;1097:143–62. [DOI] [PubMed] [Google Scholar]
- 283. Darty K, Denise A, Ponty Y. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009;25:1974–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Willmott D, Murrugarra D, Ye Q. Improving RNA secondary structure prediction via state inference with deep recurrent neural networks. Comput Mathemat Biophys 2020;8:36–50. [Google Scholar]
- 285. Deschenes A, Wiese KC, Poonian J. Comparison of dynamic programming and evolutionary algorithms for RNA secondary structure prediction. In: 2004 IEEE International Geoscience and Remote Sensing (IEEE Cat. No.04CH37612). Anchorage, AK, USA, 2004, 214–22. The Institute of Electrical and Electronics Engineers, New York, USA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.