

Abstract
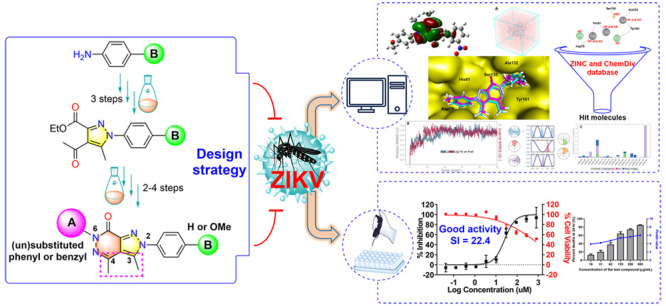
The Zika virus (ZIKV) is believed to cause birth defects, and no anti-ZIKV drugs have been approved by medical organizations to date. Starting from antimicrobial lead compounds with a pyrazolo[3,4-d]pyridazine-7-one scaffold, we synthesized 16 derivatives and screened their ability to interfere with ZIKV infection utilizing a cell-based phenotypic assay. Of these, five compounds showed significant inhibition of ZIKV with a selective index value greater than 4.6. In particular, compound 9b showed the best anti-ZIKV activity with a selectivity index of 22.4 (half-maximal effective concentration = 25.6 μM and 50% cytotoxic concentration = 572.4 μM). Through the brine shrimp lethality bioassay, 9b, 10b, 12, 17a, and 19a showed median lethal dose values in a range of 87.2–100.3 μg/mL. Compound 9b was also targeted to the NS2B-NS3 protease of ZIKV using molecular docking protocols, in which it acted as a noncompetitive inhibitor and strongly bound to five key amino acids (His51, Asp75, Ser135, Ala132, Tyr161). Utilizing the pharmacophore model of 9b, the top 20 hits were identified as prospective inhibitors of NS2B-NS3 protease, and six of them were confirmed for their stability with the protease via redocking and molecular dynamics simulations.
1. Introduction
ZIKV (Zika virus) is thought to cause birth defects as well as neurological problems.1 Currently, there are no medicines or vaccines against ZIKV or specific antiviral treatments for clinical ZIKV infection.1,2 Therefore, the development of new antivirus compounds that are effective, selective, and safe has been extremely necessary and urgent. In our previous research on bioactive heterocyclic compounds, berberine and benzimidazole derivatives were synthesized and exhibited a potential Zika antivirus with a selectivity index (SI) value greater than 10 and the half-maximal effective concentration (EC50) of 1.9–48.3 μM.3,4 In this study, we conducted works on heterocyclics and an assessment of their antiviral activity. It is found that bioorganic compounds containing heterocyclic amines (pyrazole or pyridazine) are particularly important in medicinal chemistry due to their diverse biological activities, and the relationships between their structures and pharmacological action have been studied for a long time.5−8 The heterocyclic amines possess potential biological properties including antimicrobial, analgesic, anticancer, antifeeding, antitubercular, antidiabetic, antifungal, antihypertensive, antiplatelet, anticonvulsant, anti-HIV, antiasthma, anti-inflammatory, antipyretic, and insecticidal activities.5−11 In particular, pyrazole-pyridazine compounds possess an extensive range of biological activities, and the fusion of pyrazole and pyridazine rings can give rise to five different bicyclic ring systems to help diversify and enhance the compound bioactivity (Figure 1A). For example, some pyrazolo-pyridazine derivatives showed antifungal12 and insecticidal activities.13 In medicinal chemistry, the pyrazolo-pyridazine compounds were also found to have anti-inflammatory14 and anticancer properties.7 These derivatives inhibited cyclin-dependent kinase,13 glycogen synthase kinase 3 (GSK-3),15 COX-2,16 and fibroblast growth factor receptor (FGFR)17 and were active against Leishmania amazonensis (Figure 1B).18
Figure 1.

Pyrazolo-pyridazine structures with different ring fusion patterns (A) and representative compounds containing a pyrazolo-pyridazine unit (B).
The types and preparations of the fused bicyclic ring structures such as pyrazolo[1,2-a]pyridazine, pyrazolo[3,4-d]pyridazinones, and pyrazolo[4,3-c]pyridazines have been reported by several synthesis methods.19−24 These compounds have shown diversity and abundance of pharmacology, especially antibacterial and antimicrobial activities.12,18,25,26 This also advocated an optimization of the synthesis of compounds containing the pyrazolo-pyridazine subunit with the repurposing of antiviral effects on ZIKV in this study. Zika virus NS2B-NS3 protease plays an essential role in viral replication.27 The viral protease is therefore considered as an ideal antiviral compound target. To facilitate the development of novel viral inhibitors, we also aimed to search for effective ZIKV inhibitors targeting NS2B-NS3 protease from the pyrazolo[3,4-d]pyridazine-7-one core with superior potency and selectivity.
In the course of our research on the design and synthesis of heterocyclic compounds bearing functional groups at the pyrazolo[3,4-d]pyridazine-7-one core, we have set up a versatile organic synthetic process in the laboratory. Besides, the evaluation of anti-ZIKV activity of the synthesized compounds was also carried out. The in vitro results revealed that synthesized pyrazolo[3,4-d]pyridazine-7-one compounds exhibit highly potent activities against ZIKV. Furthermore, docking, molecular dynamics, and pharmacophore model experiments have also been approached and proposed for the NS2B-NS3 protease of ZIKV in this study.
2. Results
2.1. Structural Design and Synthesis of Pyrazolo[3,4-d]pyridazine-7-one
In a series of previous studies, the pyrazolo[3,4-d]pyridazine-type compounds displayed effective antimicrobial and pharmacological properties (Figure 2).12,18,19,23,25,26 In this study, pyrazolo[3,4-d]pyridazine-7-one core structures have been utilized as a lead core to prepare different compounds with a direction of antiviral activity enhancement. Figure 2 shows an approach that makes chemical modifications of known antibacterial pyrazolo-pyridazines with an expectation of the formation of antiviral activity. The series of pyrazolo[3,4-d]pyridazine-7-one core compounds was designed and emphasized points on molecular modulation specifically modifying different substituents of phenyl or benzyl at N6 and their positions at N2. Additionally, the design and synthesis of heterocyclic compounds that bear functional groups at the pyrazolo[3,4-d]pyridazine scaffold were performed by a versatile process in the laboratory (Scheme 1). We synthesized the pyrazolo[3,4-d]pyridazine-7-one scaffold (7) from aniline derivatives as shown in Scheme 1. First, benzenediazonium chloride 2 was obtained by diazotizing aniline with NaNO2 in dilute HCl acid. In the next step, diazonium salt was used for the Japp-Klingemann reaction in situ by reacting with ethyl 2-chloroacetoacetate (3) to form an arylhydrazone.
Figure 2.

Rational design of the pyrazolo[3,4-d]pyridazin-7-one scaffold of lead compounds (A–D).
Scheme 1. General Procedure for the Synthesis of Compounds Containing the Pyrazolo[3,4-d]pyridazin-7-one Moiety.
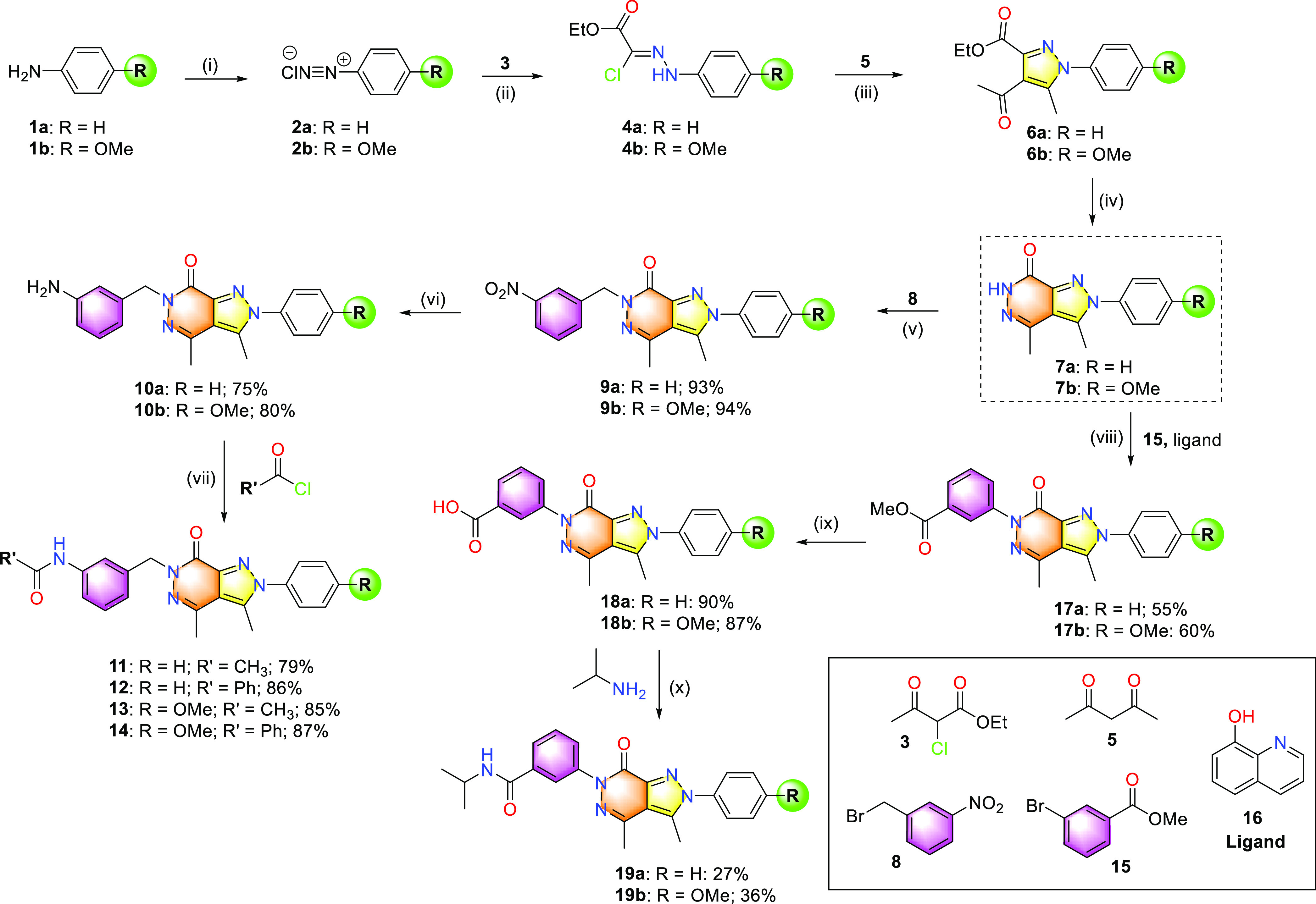
(i) Conc. HCl/H2O (1:1), NaNO2/H2O, 0 °C, 1 h; (ii) EtOH, NaOAc, 0 °C, 1 h; (iii) NaOEt, EtOH, rt, 1 h; (iv) hydrazine hydrate, EtOH, reflux, 1 h; (v) DMF, NaH, 0 °C, 30 min; (vi) SnCl2·H2O, EtOH, reflux, 1 h; (vii) MC, TEA, 0 °C, 1 h; (viii) CuI, K2CO3, DMSO, microwave, 150 °C; (ix) LiOH 1 M, rt, overnight; (x) DMF, HOBT, EDC·HCl, rt, −50 °C, overnight.
Second, pyrazole-derived compound 6 bearing a carboxylate group and an acetyl group was prepared by condensation of an arylhydrazone with acetylacetone in the presence of NaOEt in ethanol. Third, the compound of 4-acetyl pyrazole 6 fairly readily reacted with hydrazine to produce pyrazolo[3,4-d]pyridazine-7-one scaffold (7) in high yield (>94%). A carefully detailed comparison of the spectral data recorded from compound 7a with compound 7b has shown their carbon skeleton similarity. The main differences in structure between 7a and 7b were the appearance of a methoxy group [δH 3.82 (s, 3H)] in the benzene moiety of 7b. The 1H NMR spectrum of compound 7a indicated the −NH– moiety of pyrazolo[3,4-d]pyridazine scaffold at δH 12.03 (s, 1H), N2-substituted phenyl moiety at δH 7.62 (s, 5H), and two methyl groups were attached to heterocyclic at C4 and C3 (δH 2.49, δH 2.60, respectively). In the 1H NMR spectrum of compound 7b, the signal at δH 11.96 indicated the −NH– moiety and the signals of two methyl groups revealed at 2.54 (s, 3H) and 2.45 (s, 3H). The N2-substituted aromatic ring system signals were observed at δH 7.12 (2H, d) and 7.49 (2H, d) (Supporting Information, Figures S1–S4).
Compound 7 was utilized as the raw material for synthesizing other compounds substituted for the benzyl group at the N6 site (Scheme 1). Compound 9 was obtained by a nucleophilic substitution reaction of 7 with 3-nitrobenzyl bromide in dimethylformamide (DMF). The nucleophilic substitution reaction between the pyrazolo-pyridazine derivative 7 and 3-nitrobenzyl bromide in DMF medium is very readily generating compound 9 in high yield (>93%). In the next step, the nitro group was converted to amine 10 by a reduction process using the reducing agent tin(II) chloride monohydrate in ethanol under reflux conditions. The amine functional group was coupled with acid chloride to form amides 11 to 14, respectively. They were prepared by the presence of triethanolamine (TEA) in methylene chloride (MC) at 0 °C, with good yields (>79%). Starting from the core structure of 7, the copper-catalyzed coupling reaction of aryl halide compound with nitrogen-containing nucleophile was applied for the synthesis of 17 by coupling between 7 and methyl 3-bromobenzoate (15) in the presence of CuI catalyst, K2CO3, dimethyl sulfoxide (DMSO), and 8-hydroxyquinoline ligand (16) under microwave conditions (yields of 55 to 60%). In the next steps, including ester group hydrolysis using LiOH and HCl, 18 was obtained with good yields (>87%). Finally, treatment of 18 with propane-2-amine gave the desired pyrazolo-pyridazinone 19 (yields of 27 to 36%). The carbodiimide 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) was used for solution-phase peptide couplings as its urea byproduct could be removed by washing in an aqueous workup. After the completion of the reaction, the products were purified by silica gel column chromatography and characterized by NMR and high-resolution mass spectrometry (HR-MS). Some synthesized compounds were also subjected to high-performance liquid chromatography (HPLC) and gas chromatography–flame ionization detector (GC-FID) analyses to test their purity. As a result, the selected compounds had a purity of more than 95% based on the area percentage (Supporting Information, Figures S5–S27).
2.2. Anti-ZIKV Activity of the Synthesized Pyrazolo[3,4-d]pyridazine-7-one
The present study aims to evaluate the antiviral activity of 16 pyrazolo[3,4-d]pyridazine-7-one compounds against the Zika virus strain MR766 of the Africa lineage, while also assessing their potential cytotoxic effects on Vero cells. Mycophenolic acid (MPA), an immunosuppressant, was used as a positive control.3,4 The African lineage strain ZIKV-MR766 was incubated with the compounds for 24 h prior to the Vero cell infection through the viral immunofluorescence assay; the results of anti-ZIKV bioassay are presented in Table 1, and the dose–response curves of the top five compounds were displayed in Figure 3. Five compounds (9b, 10b, 12, 17a, and 19a) showed activity against ZIKV with a selective index value greater than 4 (SI > 4), while the other nine compounds showed little to no effects on ZIKV (SI < 4). In particular, compound 9b exhibited a strong anti-ZIKV efficacy with an SI value of 22.4 (EC50 = 25.6 μM and 50% cytotoxic concentration (CC50) = 572.4 μM), and a potency of 2.6-fold to 4.6-fold than those of the other compounds. Two compounds (10b and 12) showed moderate activities with SI values greater than 6.9 (EC50 = 144.3 μM and 140.1 μM; CC50 ≥ 1000 μM and >1000 μM, respectively). Nevertheless, compounds 17a and 19a were weakly active against ZIKV (SI > 4.6) (EC50 = 218.1 and 87.2 μM; and CC50 ≥ 1000 and 435.6 μM, respectively).
Table 1. ZIKV Inhibitory Activity of the Synthesized Pyrazolo[3,4-d]pyridazine-7-ones against Zika Virus Strain MR766.
| Compd. | EC50a (μM) | CC50b (μM) | SIc |
|---|---|---|---|
| 7a | 697.5 ± 4.9 | 2606 ± 4.9 | 3.7 |
| 7b | 39.1 ± 1.8 | 114.6 ± 7.5 | 2.9 |
| 9a | NDe | ND | NAf |
| 9b | 25.6 ± 2.8 | 572.4 ± 5.1 | 22.4 |
| 10a | ND | ND | NA |
| 10b | 144.3 ± 3.9 | >1000 | >6.9 |
| 11 | 203.6 ± 3.2 | 361 ± 1.9 | 1.8 |
| 12 | 140.1 ± 13.7 | >1000 | >7.1 |
| 13 | 124.3 ± 4.6 | 245.4 ± 10.3 | 2.1 |
| 14 | 300.3 ± 6.5 | >1000 | >3.3 |
| 17a | 218.1 ± 3.8 | >1000 | >4.6 |
| 17b | 161.3 ± 9.1 | 368.5 ± 4.2 | 2.3 |
| 18a | >1000 | >1000 | NA |
| 18b | ND | ND | NA |
| 19a | 87.2 ± 7.3 | 435.6 ± 2.8 | 5.0 |
| 19b | 105.0 ± 4.6 | 299.8 ± 8.3 | 2.9 |
| MPAd | 0.94 | >1000 | >1063 |
EC50: median effective concentration.
CC50: 50% cytotoxic concentrations.
SI: Selectivity index (CC50/EC50).
MPA: mycophenolic acid (positive control).
ND: not determined.
NA: not applicable (compounds were tested, but CC50 and EC50 values could not be calculated). EC50 and CC50 values were calculated by analysis of 10-point dose–response curves in triplicate.
Figure 3.

Dose–response curves of compounds 9b, 10b, 12, 17a, and 19a against ZIKV and the reference inhibitor (MPA-mycophenolate acid). The percentage of ZIKV inhibition is shown in the black line. Percentage cell viability is shown in the red line. A compound shows good activity if the SI value is greater than 10 (SI of 10–6: activity; SI of 6–4: low activity; and SI < 4: no activity). A full antiviral data set for all compounds can be found in the Supporting Information, Table S1.
In addition, a cytotoxicity assay was conducted to determine their 50% cytotoxicity concentration (CC50) against virus-uninfected Vero cells. The results revealed that 7a, 9b, 10b, 11–14, 17a, 17b, 18a, 19a, and 19b exhibit low cytotoxicity, with CC50 values greater than 240 μM (Table 1). Furthermore, at a concentration of 10 μM, all of the test compounds maintained 90% of Vero cell viability. Notably, six compounds (7a, 9b, 10b, 12, 14, 17a, and 18a) displayed low cytotoxicity with CC50 values higher than 500 μM (Supporting Information, Table S1).
2.3. Toxicity Bioassay
One of the biggest concerns during the drug discovery process is the toxicity of the components, and any undesirable toxicity must be carefully monitored. Thus, in this study, we evaluated the toxicity of the compounds that have the highest anti-ZIKV efficacy by a brine shrimp lethality bioassay. The brine shrimp (Artemia salina) lethality assay was conducted on five compounds with strong anti-ZIKV efficacy (9b, 10b, 12, 17a, and 19a). Toxicity test results are presented in Table 2. The result is the lethality of compounds on brine shrimp with estimated median lethal dose (LD50) values that range from about 87.2 to 114.1 μg/mL. The tested compounds also showed concentration-dependent toxicity against brine shrimp. Besides, at concentrations greater than 500 μg/mL, all nauplii mortalities were higher than 80%. In particular, we found that these compounds have considerably lower toxicity than a berberine derivative, which was previously reported to be a ZIKV inhibitor4 (Supporting Information, Figure S28). Based on the toxicity index, these compounds were considered to be moderately toxic, indicating that they could be extended for further preclinical studies.
Table 2. Toxicity of the Selected Pyrazolo[3,4-d]pyridazine-7-one from Brine Shrimp Assay.
| 95%
fiducial limits |
|||
|---|---|---|---|
| Compd. | LD50 (μg/mL)a | Lower | Upper |
| 9b | 91.7 | 67.2 | 124.9 |
| 10b | 100.3 | 77.3 | 130.1 |
| 12 | 114.1 | 88.4 | 147.3 |
| 17a | 87.2 | 65.5 | 115.9 |
| 19a | 90.3 | 64.5 | 126.5 |
| Atropine SO4b | 737.8 | 477.4 | >1000 |
| Berberine chlorideb | 27.0 | 21.5 | 33.8 |
| ref. inhibitorc | 34.6 | 27.9 | 42.8 |
LD50: median lethal dose (μg/mL). LD values were estimated by probit analysis of 6-point dose (16–500 μg/mL).
Atropine sulfate monohydrate and berberine chloride were used as the positive controls. The solvent was used as the negative control (lower than 1% DMSO).
The ZIKV inhibitor (berberine derivative-4d) was described in our previous report, and it was also used as a standard.4
2.4. Docking Protocol
To study the interactions between ZIKV NS2B-NS3 protease (5H4I) and compounds 9b (most active) and 17a (least active), the Autodock program was used in a molecular docking study similar to docked berberine derivatives in our previous study.4 A comprehensive analysis of molecular interactions, hydrogen bonds, and hydrophobic interactions, particularly within the active site of the NS2B-NS3 protease, was performed for the selected compounds (Figure 4). The binding energies of both 9b and 7a were lower than those of the cocrystallized ligand (benzimidazole-1-ylmethanol), thus showing better binding affinity and stability than this ligand (−8.16, −7.60, and −6.53 kcal/mol, respectively) (Supporting Information, Figure S30). The benzyl function at N6 showed a more favorable orientation than the phenyl function in the hydrophobic region of the active site. This makes the aromatic ring of 9b sandwiched between the side chains of Ala132-Tyr161 with a distance shorter than that of 17b (3.58 and 4.35 Å for Ala132, and 5.35 and 5.40 Å for Tyr161, respectively). Compound 9b also showed a distance with Ser135 shorter than that of 17a (3.09 Å versus 3.54 Å, respectively) (Supporting Information, Figure S30). This may be the reason that 17b was less active than 9b. The substituted phenyl moieties (B) at N2 were found in the hydrophobic pocket and sandwiched with Asp75 and Asp83 (pi-anion). Both pi-pi-stacked and pi-pi T-shaped bonds were observed between the pyrazolo[3,4-d]pyridazine-7-one scaffold and His51 with a distance of about 4.25 to 5.33 Å. Additionally, 9b caused interactions with some key amino acids such as Thr134 (carbon–hydrogen bond), Gly151 (carbon–hydrogen bond), Trp50 (pialkyl), and Val72 (carbon–hydrogen bond). Compound 17a also displayed interactions with some amino acids (Tyr150 and Tyr130) in the binding pocket.
Figure 4.

Binding interaction between 9b (pink) and 17b (cyan) at the active site of NS2B-NS3.
2.5. Molecular Dynamic Simulation Study
A molecular dynamics simulation study was also performed to check the stability of the 9b-NS2B-NS3 complex during 50000 ps (or 50 ns) based on the molecular docking results (Figure 5A). The root-mean-square deviation (RMSD) plot indicated that the 9b-NS2B-NS3 complex reached its stable form after 10000 ps (or 10 ns), where the ligand is still intact with protein and not diffused away easily (Figure 5B). In addition, the molecular dynamic results displayed favored hydrophobic interactions with Tyr161 (94%), Phe84 (24%), and His51 (27%), hydrogen interaction with Ser135, and water-bridged interactions with Gly151 and Asn152 (Figure 5C). Figure 5D,E presents a ligand torsion histogram illustrating the dynamic changes in each rotatable bond throughout the simulation trajectory. These findings offer valuable insights into the conformational strain that preserves the ligand’s shape when bound to the protein.
Figure 5.

Molecular dynamics result for the 9b-NS2B-NS3 complex. (A) Protein at the center of the water box in simulation protocol using Desmond Academic license 2018–4. (B) The RMSD values for complex during the molecular dynamic simulation. (C) Protein interactions with the ligand 9b (hydrogen bonds, green; hydrophobic, violet; ionic, pink; and water bridges-blue). (D) Ligand torsion profile torsion and flexibility. (E) Ligand torsion angles.
2.6. HOMO and LUMO Analysis
The properties of the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) hold significant utility for chemists. Molecules characterized by higher HOMO values and lower LUMO values are prone to facile oxidation and reduction, respectively. Illustrated in Figure 6 are the HOMO and LUMO surfaces, revealing a concentration of HOMO orbitals primarily around the pyrazolo[3,4-d]pyridazine-7-one core. In both gas and water phases, compound 9b exhibits HOMO–LUMO energy gaps of 7.75 and 7.62 eV, respectively. These energy parameters signify the molecule’s remarkable stability and chemical hardness.
Figure 6.
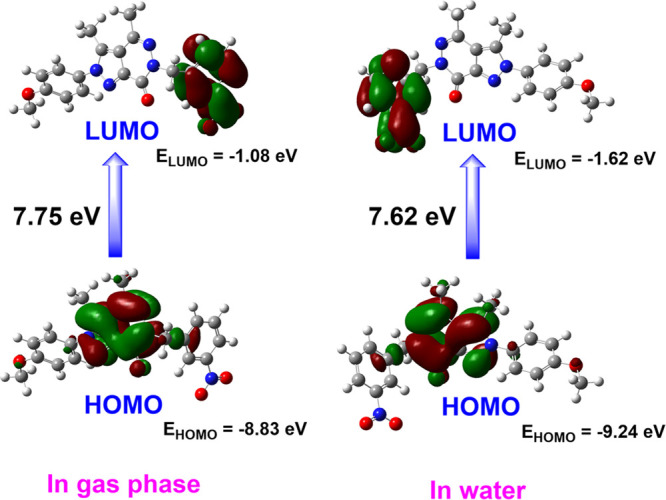
HOMO and LUMO orbitals of compound 9b in the gas phase and water.
2.7. Pharmacophore Model and Pharmacophore-Based Virtual Screening
To identify important pharmacological features necessary for the screening of promising ZIKV inhibitors, we have implemented pharmacological modeling based on the structure of NS2B-NS3 docked to inhibitor 9b using Pharmit online28 by loading the docked features. First, Pharmit identified 13 pharmacological features from compound 9b based on ligand–protease interactions (Figure 7A). Six sites involved in the interaction with key amino acids at the active site of NS2B-NS3 were selected to create a pharmacological model (Figure 7B). The pharmacophore model was chosen to contain one hydrogen acceptor from the hydroxyl group of Ser135, two hydrophobic regions engaging with Asp75 and Tyr161, and three featuring aromatic/hydrophobic regions interacting with His51 and Ala132. This pharmacophore model served for the virtual screening of compounds from databases or to optimize the modifications of lead compounds or inhibitors to obtain greater inhibitory efficacy. Two large databases, ZINC and ChemDiv, were used for virtual screening based on pharmacophore models in the present work. The ZINC database, containing more than 13 million molecules, yielded 1394 compounds that fit the model (RMSD < 1.0 Å) following screening, among which 536 molecules complied with Lipinski’s rule of five. Similarly, screening from the ChemDiv database (over 1 million molecules) resulted in 807 compounds that fit the model, among which 303 molecules followed Lipinski’s rule of five (Figure 7C). The two compounds ZINC1064562 (RMSD = 0.430 Å) and ChemDiv-L856–0019 (RMSD = 0.438 Å) exhibited the highest similarity to the model and were predicted to interact strongly with NS2B-NS3 protease, similar to compound 9b (Supporting Information, Table S2). The screened top three compounds of each database were used to perform molecular docking and molecular dynamic processes (Figure 8). Based on the results, it can be inferred that the six chosen compounds exhibit a favorable binding energy with the NS2-NS3 protease, as their predicted binding energies are lower than −6.9 kcal/mol. Notably, compound ChemDiv-L856–0019 displays even lower energies than 9b (−9.4 and −8.13 kcal/mol, respectively). Moreover, the stability of the ligand–receptor complex was assessed via molecular dynamics simulations over a period of 50 ns. The results indicate that the compounds maintain stability throughout the simulation; the RMSD values obtained were less than 4 Å. In summary, this investigation contributed to expanding the collection of compounds with the potential to effectively inhibit Zika virus.
Figure 7.

Pharmacophore model. (A) Pharmacophoric features of 9b (pink) at the active site of the NS2B-NS3 protease. (B) Pharmacophoric features of 9b were selected at the active site of NS2B-NS3 protease. (C) Pharmacophore-based virtual screening from the ZINC and ChemDiv database. HBA (hydrogen bond acceptor-organ sphere). HP (hydrophobic areas-green sphere). AR (aromatic ring-violet sphere).
Figure 8.

Redocking protocol and molecular dynamics of top three molecules virtual screened from ChemDiv and the ZINC database. HBA: hydrogen bond acceptor; HP: hydrophobic. Ar: aromatic ring.
3. Discussion
In this study, 16 pyrazolo[3,4-d]pyridazine-7-one compounds were rationally designed and tested against the Zika virus (MR766 Africa lineage). We also assessed their cytotoxicity in Vero cells. In a previous patent, pyrazolo-pyridazone compounds were found to inhibit hepatitis C virus (HCV) at concentrations ranging from 0.01 to 3.06 μM.29,30 Both hepatitis C virus and ZIKV belong to the genus Flavivirus and the family Flaviviridae. In our previous study, we also discovered benzimidazole derivatives as promising antiviral candidates for ZIKV treatment.3 Seven benzimidazole derivatives strongly inhibited the African ZIKV strain with EC50 values lower than 13 μM. In particular, these compounds all showed resistance to the HCV virus with a selectivity index (SI) greater than 3.5. Therefore, we hypothesized that pyrazolo-pyridazone compounds will have the potential for ZIKV inhibition. The pyrazolo[3,4-d]pyridazine compounds also displayed an effective antiviral activity against rotavirus Wa strain and adenovirus type 7.31 According to Akbas and Berbe,12 compound A, one of seven new pyrazolo[3,4-d]pyridazine-7-one derivatives that was prepared via a furandione derivative, had the highest antibacterial activity against Gram-negative and Gram-positive bacteria (four test bacteria) with MICs (minimum inhibitory concentrations) in the range of 0.31 to <0.0024 mg/mL. Besides, lead compound B was synthesized from reactions of pyrazole-3-carboxylic acid with some hydrazine compounds, which also showed inhibitory activity against six test bacteria, including Escherichia coli, Bacillus subtilis, Pseudomonas aeruginosa, Klebsiella pneumonia, Proteus vulgaris, and Xanthomonas compestris.19 In 2016, Mady et al. synthesized compound C, a pyrazolo[3,4-d]pyridazine-7-one linked to a diaryl sulfone moiety by using microwave irradiation; this compound also had effects on pathological bacterial strains such as E. coli O157 and S. typhimurium.25 Compound D displayed antibacterial activity against five bacteria owing to containing a pyrazole-pyridazine subunit, which was also stronger than those of the positive controls by Erythromycin, Rifampicin, and Amikacin.23 Based on the above-mentioned data, we designed a facile methodology for the design of pyrazolo[3,4-d]pyridazine-7-one linked to the methyl moiety at C3 and C4 from the original pyrazolo[3,4-d]pyridazine scaffold. The N2 and N6 positions of the pyrazolo[3,4-d]pyridazine-7-one core were introduced with benzyl or phenyl groups to form the respective compounds (Scheme 1 and Figure 2).
Sixteen compounds were synthesized, and then in vitro antiviral experiments were conducted to evaluate anti-ZIKV activity. The docking study, molecular dynamics, and pharmacophore model of the highly active compounds were also carried out subsequently. It is noteworthy that compound 9b was observed to be a potential new ZIKV inhibitor with an EC50 of 25.6 μM and a selectivity index value of greater than 22. In addition, compounds 10b, 12, 17a, and 19a also exhibited strong activity against Zika virus strain MR766 with SI values of 5.0 to >6.9 (Table 1 and Figure 3).
The brine shrimp lethality assay is a quick and simple biological test to estimate the lethal dose (LD) of purified compounds or plant extracts. Several studies suggest that these experiments are reasonably correlated with cytotoxic and antitumor properties.32−34 Berberine chloride and atropine sulfate were used as standard drugs in cytotoxicity activity. Berberine is relatively toxic parenterally and has been used orally with potential antineoplastic, radiosensitizing, anti-inflammatory, antilipidemic, and antidiabetic activities.35,36 Atropine was included in the World Health Organization’s list of Essential Medicines, which can be administered by intravenous, subcutaneous, intramuscular, intraosseous, oral, endotracheal, and ophthalmic methods.37 After 24 h of assessment, all the brine shrimp survived in the negative controls. In the positive controls treated with berberine, all the nauplii were dead at 125 μg/mL and the LD50 value was 27.0 μg/mL. In contrast, atropine sulfate showed mortality below 20% after 24 h (LD50 = 737.8 μg/mL) (Table 2 and Supporting Information, Figure S28). The brine shrimp’s bioassay for the lethality of selected compounds (9b, 10b, 12, 17a, and 19a) resulted in their LD50 values of 91.7, 100.3, 114.1, 87.2, and 90.3 μg/mL, respectively. Compared with our previously reported ZIKV inhibitors, the evaluated compounds possessed lower toxicity than those of the berberine derivatives (LD50 = 34.6 μg/mL).4 Therefore, the compounds in this study showed an acceptable toxicity range and could be candidates for the development of oral antiviral drugs and may have the potential to inhibit several cancer cell lines.
NS2B-NS3 protease plays a crucial role in the ZIKV activity due to it being responsible for site-specific cleavage of precursor polyprotein in ZIKV, and NS2B acts as a cofactor for NS2B-NS3 protease activation.38 Special, several NS2B-NS3 inhibitors were identified as bromocriptine (IC50 = 21.6 μM), novobiocin (IC50 = 14.2 μM), temoporfin (IC50 = 1.1 μM), and erythrosine B (IC50 = 1.7 μM).39−41 Choudhry et al. recently selected seven antiviral drug candidates, which were targeted to target NS2B-NS3 protease, using GOLD and AutoDock Vina software.39 Thirumoorthy et al. screened and identified three potential compounds derived from Andrographis paniculate for ZIKV inhibition by targeting NS2B-NS3.42 Therefore, NS2B-NS3 is the prioritized target for research in screening potential inhibitors against the Zika virus. In a recent work by us, a designed and synthesized berberine derivative was reported to be a potent inhibitor of Zika virus MR766 Africa lineage with an IC50 value of 5.3 μM and SI value of 15.3.4 This derivative caused a high affinity for binding to the NS2B-NS3 protease, which suggests a potential for its protease inhibition in the future. Using a virtual screening model to partially predict experimental results seemed to be the simplest and best option. First, the redocking was done with the reference ligand (cocrystallized ligand) into the active site of NS2B-NS3 to protocol authentication (Supporting Information, Figure S29) before we performed molecular docking for two selected compounds (9b and 17a). The obtained results displayed that the docked ligand completely overlapped with the cocrystallized ligand in protease root-mean-square deviation (RMSD value was 0.5936 Å). At the binding site, the docked ligand displayed interactions with some key amino acids as found in the crystal structure in the original PDB (5H4I).
The inhibitors’ binding mode with protease was previously reported, and His51, Asp75, and Ser135 were key amino acids that participated in H-bond and hydrophobic interactions.43,44 Especially, Ser135 played a vital role in the inhibition activity of the compound with this target enzyme. The benzimidazole ring of the cocrystallization ligand was sandwiched between the side chains of Ala132-Tyr161, so both of these amino acids were considered key residues.45 The docking protocol showed that both compounds interact with the key amino acids at the active site of NS2B-NS3 protease with binding energies lower than −7.6 kcal/mol. The 9b-NS2B-NS3 complex formed a catalytic triad at the active site characterized by the presence of the His51, Asp75, and Ser135 residues, resulting in a closed conformation of the oxyanion orifice.27,46 The inhibitor binding caused a change from the active to inactive state in the catalytic triad of the NS3 protease. However, 9b binding did not further induce significant conformational changes of protease (Figure 5), and this proposed a noncompetitive inhibitor binding on the ZIKV protease.27,46,47 The dynamic simulation for the NS2B-NS3 protease also revealed that compound 9b binds closely to five key amino acids (His51, Asp75, Ser135, Ala132, and Tyr161) at the active site of this protease. Unfortunately, in this study, we were unable to test NS2B-NS3 protease inhibition in vitro for providing more biological data. However, in this report, we attempted to use molecular docking and dynamic protocols to confirm 9b as a potential compound to target the NS2B-NS3 protease. This would provide insight into ZIKV inhibition of the analogues in the future.
From these ligand-protease interactions, we build the pharmacophore model from six different features using the Pharmit spectrum (one HBA feature, two HP features, and three features of both HP and AR). The arrangement of steric and electronic characteristics of the pharmacophore was necessary to guarantee the finest molecular interactions with the respective biological target or the discovery of new scaffolds binding the active site of the NS2B-NS3 protease. After pharmacophore-based virtual screening, 839 compounds that showed similarity to the pharmacophore model (RMSD values less than 1.0 Å) were selected (Figure 7). The top ten compounds were identified based on the pharmacophore model from two large databases, ZINC and ChemDiv (RMSD < 0.4 Å) (Supporting Information, Table S2). These compounds completely embodied the pharmacological features of the model, thereby enabling them to tightly bind to NS2B-NS3, and exhibited the ability to inhibit ZIKV. In particular, the redocking and molecular dynamics simulations of the top three compounds from each database revealed a significant binding affinity and stability with the NS2B-NS3 protease (Figure 8). Rehman et al. (2023) generated a pharmacophore model based on a five-featured pharmacophore hypothesis.48 The results identified three hit molecules as potential inhibitors of Zika NS2B-NS3 protease with binding free energies of −64.28, −55.15, and −50.16 kcal/mol, as predicted by Schrödinger’s GLIDE module. However, no follow-up in vitro evaluations has been conducted to verify these findings. The ZIKV NS3 protease pharmacophore model was utilized for virtual screening, resulting in the identification of 14 potential antiviral candidates by Pathak et al. (2020).49 Subsequently, these candidates were tested via in vitro assays, and the Food and Drug Administration (FDA)-approved drugs asunaprevir and simeprevir exhibited potent anti-ZIKV activities with EC50 values of 4.7 μM and 0.4 μM, respectively. Furthermore, both drugs were able to inhibit the viral protease, exhibiting IC50 values of 6.0 and 2.6 μM, respectively.
This study endeavors to shed new light on heterocyclic compounds with antiviral potential, paving the way for the future development of robust ZIKV inhibitors. Altogether, the study opens new investigation possibilities in the search for novel drugs composed of a pyrazolo[3,4-d]pyridazine-7-one core to combat ZIKV. The in vitro antiviral activity of our synthesized compounds against ZIKV and their docking inhibition on ZIKV NS2B-NS3 were reported for the first time. High-throughput virtual screening could be used to screen for prospective ZIKV inhibitors based on the predicted pharmacophore model, and it will also be a subject of future reports. Furthermore, in vitro and in vivo studies on enzymes and animals were also needed to confirm its pharmacological effects and antiviral efficacy on ZIKV. ZIKV infection could dramatically affect fetal neural development, especially in neural stem cells. Thus, the antiviral activity of 9b should be tested and validated in neural stem cells in the future.
4. Conclusion
Our study described a discovery of the anti-ZIKV potential of 16 pyrazolo[3,4-d]pyridazine-7-one compounds and advocated 9b as an excellent candidate for further trials of ZIKV treatment. Compound 9b was a novel structural feature that produced anti-ZIKV activity comparable to the positive control MPA. Through the brine shrimp lethality bioassay, 9b, 10b, 12, 17a, and 19a showed acceptable toxicities with LD50 values in a range of 87.2–100.3 μg/mL. The docking dynamic simulation for the NS2B-NS3 protease also revealed that 9b acts as a noncompetitive inhibitor and binds closely to five key amino acids (His51, Asp75, Ser135, Ala132, Tyr161) at the active site of this protease. Using the pharmacophore model of 9b, a virtual screening was conducted to identify the top ZIKV inhibitors. Subsequently, molecular dynamics simulations were employed to evaluate the stability and binding affinity of the top six selected compounds with the NS2B-NS3 protease. More studies on in vitro and in vivo models are needed to confirm their therapeutic applications.
5. Materials and Methods
5.1. General Information
All reagents were purchased from commercial sources and used without further purification. Reactions were monitored by thin-layer chromatography (TLC) on 0.2 mm precoated silica gel 60 F254 plates (Merck), and compounds were visualized on TLC with UV light. The derivatives were synthesized and then purified by flash column chromatography using silica gel 45–63 μm (230–400 mesh) and 60 pore size. The NMR spectra were obtained from a Varian 300 MHz or Bruker 300 MHz spectrometer. The chemical shifts (δ) were reported in parts per million (ppm) relative to tetramethylsilane (TMS) or the internal solvent signal of deuterated solvents. Multiplicities were indicated by s (singlet), d (doublet), t (triplet), and m (multiplet).
5.2. Chemistry
Experimental Procedure of Compounds 2a–b
To a stirred solution of compound 1 (50 mmol, 1.0 equiv) in a mixture of cold H2O/conc. HCl (1:1; 20 mL) at 0 °C was slowly added a solution of NaNO2 (1.1 equiv) in H2O (10 mL). The mixture was stirred for 1 h at 0–5 °C, which gave the intermediate diazonium salt 2, and this compound was used without any further purification for the next reaction step.
Experimental Procedure of Compounds 4a–b
Then a stirred mixture of NaOAc (3.0 equiv) and compound 3 (1.1 equiv) in EtOH (150 mL) at 0 °C was added to the mentioned intermediate diazonium salt 2 portionwise. The mixture was stirred for 4 h and poured into 500 mL of water. The precipitate was filtered off, dried, and recrystallized from EtOH, giving compound 4.
Compound 4a
Yield: 94%. 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.53 (s, 1H, −NH−), 7.37–7.28 (m, 4H, ArH), 7.02–6.96 (m, 1H, ArH), 4.32–4.24 (m, 2H, −OCH2CH3), 1.32–1.27 (m, 3H, −OCH2CH3).
Compound 4b.
Yield: 54%. 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.43 (s, 1H, −NH−), 7.30–6.91 (m, 2H, ArH), 6.94–6.91 (m, 2H, ArH), 4.31–4.26 (m, 2H, −OCH2CH3), 3.72 (s, 3H, −OCH3), 1.31–1.26 (m, 3H, −OCH2CH3).
Experimental Procedure of Compound 6a–b
Compound 5, acetylacetone (1.1 equiv), was added to a stirred solution of 21% NaOEt/EtOH (1.1 equiv; diluted with 15 mL of anhydrous EtOH). The mixture was stirred at rt for 7 h, and then the finely powder compound 4 (20 mmol, 1.0 equiv) was added after being stirred for 4 h.
For Compound 6a
The mixture was quenched with water, extracted twice with EtOAc, washed with brine, concentrated, and purified by flash column chromatography (EtOAc/Hex, 1:4 to 1:3), which gave the desired product 6a in 93% yield as a yellow oil. 1H NMR (300 MHz, DMSO-d6, δ ppm): 7.62–7.54 (m, 5H, ArH), 4.38–4.30 (m, 2H, −OCH2CH3), 2.47 (s, 3H, −CH3), 2.37 (s, 3H, −COCH3), 1.33–1.28 (m, 3H, −OCH2CH3).
For Compound 6b
The mixture was added with cold water, and the precipitate was filtered off, dried, and recrystallized from EtOH, which gave desired product 6b in 86% yield as a pale yellow solid. 1H NMR (300 MHz, CDCl3, δ ppm): 7.33–7.26 (m, 2H, ArH), 7.00–6.98 (m, 2H, ArH), 4.49–4.42 (m, 2H, −OCH2CH3), 3.87 (s, 3H, −OCH3), 2.59 (s, 3H, −COCH3), 2.38 (s, 3H, −COCH3), 1.44–1.40 (m, 3H, −OCH2CH3).
Experimental Procedure of Compounds 7a–b
A mixture of compound 6 (5 mmol, 1.0 equiv) and excess hydrazine monohydrate in EtOH was refluxed for 2 h, then cooled, and kept at 0 °C for 20 min. The resulting precipitate was filtered off and dried to yield the desired product 7. Yield: 94% for 7a and 95% for 7b.
Compound 7a
1H NMR (300 MHz, DMSO-d6, δ ppm): 12.02 (s, 1H, −NH−), 7.63 (s, 5H, ArH), 2.61 (s, 3H, −CH3), 2.49 (s, 3H, −CH3). The molecular formula C13H12N4O was indicated by high-resolution electrospray ionization mass spectrometry (HR-ESI-MS) at m/z 241.1081 [M + H]+ (Calcd for [C13H12N4OH]+, 241.1084).
Compound 7b
1H NMR (300 MHz, DMSO-d6, δ ppm): 11.96 (s, 1H, −NH−), 7.49 (d, J = 8.8 Hz, 2H, ArH), 7.12 (d, J = 7.3 Hz, 2H, ArH), 3.82 (s, 3H, −OCH3), 2.54 (s, 3H, −CH3), 2.45 (s, 3H, −CH3). The molecular formula C14H14N4O2 was indicated by HR-ESI-MS at m/z 271.1189 [M + H]+ (Calcd for [C14H14N4O2H]+, 271.1190).
Experimental Procedure of Compounds 9a–9b
To a stirred solution of compound 7 (1.0 mmol, 1.0 equiv) in DMF (anhydrous, 5 mL) at 0 °C was added NaH (60% in oil). The mixture was stirred for 30 min, and then 8 was added. After the starting material disappeared (about 30 min), the reaction mixture was quenched with cold water and extracted with EtOAc twice. The combined organic phase was washed with water twice and brine, dried with anhydrous Na2SO4, filtered, and concentrated. The resulting residue was purified by flash column chromatography (EtOAc/Hex, 1:1 to 2:1), which gave the desired products 9a and 9b as white solids. Yield: 93% for 9a and 94% for 9b.
Compound 9a
1H NMR (300 MHz, CDCl3, δ ppm): 8.26 (s, 1H, ArH), 8.13 (d, J = 8.1 Hz, 1H, ArH), 7.81 (d, J = 7.0 Hz, 1H, ArH), 7.54–7.46 (m, 6H, ArH), 5.46 (s, 2H, −CH2−), 2.65 (s, 3H, −CH3), 2.57 (s, 3H, −CH3).
Compound 9b
1H NMR (300 MHz, DMSO-d6, δ ppm): 8.12–8.10 (m, 2H, ArH), 7.71 (d, J = 7.8 Hz, 1H, ArH), 7.61 (t, J = 8.1 Hz, 1H, ArH), 5.36 (s, 2H, −CH2−), 3.82 (s, 3H, −OCH3), 2.55 (s, 3H, −CH3), 2.49 (s, 3H, −CH3).
Experimental Procedure of Compounds 10a–b
To a stirred solution of compound 9 (0.5 mmol, 1.0 equiv) in EtOH (10 mL) at 80 °C was added SnCl2·H2O (5.0 equiv). The reaction mixture was stirred at 80 °C. After the starting material completely reacted (about 1 h), the reaction mixture was cooled to room temperature and neutralized with saturated NaHCO3 solution at 0 °C, and the resulting mixture was filtered through a diatomaceous earth layer. The filtrate was extracted twice with EtOAc, washed with brine, dried with anhydrous Na2SO4, filtered, concentrated, and purified by flash column chromatography (EtOAc/Hex, 4:1 to 5:1), which gave the desired products 3a–b as white solids. Yield: 75% for 10a and 94% for 10b.
Compound 10a
1H NMR (300 MHz, DMSO-d6, δ ppm): 7.60 (s, 5H, ArH), 6.90 (t, J = 7.6 Hz, 1H, ArH), 6.42–6.37 (m, 3H, ArH), 5.06 (s, 2H, −CH2−), 5.00 (s, 2H, −NH2), 2.59 (s, 3H, −CH3), 2.58 (s, 3H, −CH3).
Compound 10b
1H NMR (300 MHz, DMSO-d6, δ ppm): 7.52–7.48 (m, 2H, ArH), 7.15–7.11 (m, 2H, ArH), 6.90 (t, J = 7.7 Hz, 1H, ArH), 6.40–6.36 (m, 3H, ArH), 5.05 (s, 2H, −CH2−), 5.06 (s, 2H, −CH2−), 5.00 (s, 2H, −NH2), 3.82 (s, 3H, −OCH3), 2.55 (s, 3H, −CH3), 2.46 (s, 3H, −CH3).
Experimental Procedure of Compounds 11–14
To a stirred solution of compound 10 (0.2 mmol, 1.0 equiv) in DCM (5 mL) at 0 °C was added Et3N (3.0 equiv). Then acyl chlorides, CH3COCl or PhCOCl, (1.1 equiv) were added dropwise. The reaction mixture was stirred at 0 °C. After the starting material disappeared (30 min), the reaction mixture was added to cold water, extracted twice with DCM, washed with brine, dried with anhydrous Na2SO4, filtered, concentrated, and purified by flash column chromatography (EtOAc/Hex, 4:1 to 5:1), which gave the desired products 11 to 14 as white solids. Yield: 79 to 87%.
Compound 11
1H NMR (300 MHz, DMSO-d6, δ ppm): 9.88 (s, 1H, −NH−), 7.60 (s, 5H, ArH), 7.56–7.50 (m, 1H, ArH), 7.37 (s, 1H, ArH), 7.20 (t, J = 7.9 Hz, 1H, ArH), 6.92 (d, J = 7.6 Hz, 1H, ArH), 5.18 (s, 2H, −CH2−), 2.58 (s, 3H, −CH3), 2.49 (s, 3H, −CH3), 1.96 (s, 3H, −COCH3). The molecular formula C22H21N5O2 was indicated by HR-ESI-MS at m/z 388.1772 [M + H]+ (Calcd for [C22H21N5O2H]+, 388.1768).
Compound 12
1H NMR (300 MHz, DMSO-d6, δ ppm): 10.22 (s, 1H, −NH−), 7.95–7.89 (m, 2H, ArH), 7.71 (d, J = 8.1 Hz, 1H, ArH), 7.64–7.44 (m, 9H, ArH), 7.30–7.25 (m, 1H, ArH), 7.01 (d, J = 7.6 Hz, 1H, >CH−), 5.23 (s, 2H, −CH2−), 2.59 (s, 3H, −CH3), 2.50 (s, 3H, −CH3). The molecular formula C27H23N5O2 was indicated by HR-ESI-MS at m/z 450.1932 [M + H]+ (Calcd for [C27H23N5O2H]+, 450.1925).
Compound 13
1H NMR (300 MHz, DMSO-d6, δ ppm): 9.89 (s, 1H, −NH−), 7.58–7.52 (m, 3H, ArH), 7.41 (s, 1H, ArH), 7.23 (t, J = 7.8 Hz, 1H, ArH), 7.17 (d, J = 8.5 Hz, 2H, ArH), 6.96 (d, J = 7.5 Hz, 1H, ArH), 5.21 (s, 2H, −CH2−), 3.87 (s, 3H, −OCH3), 2.59 (s, 3H, −CH3), 2.50 (s, 3H, −CH3), 2.00 (s, 3H, −COCH3). The molecular formula C23H23N5O3 was indicated by HR-ESI-MS at m/z 418.1885 [M + H]+ (Calcd for [C23H23N5O3H]+, 418.1874).
Compound 14: 1H NMR (300 MHz, DMSO-d6, δ ppm): 10.24 (s, 1H, −NH−), 7.93 (d, J = 7.4 Hz, 2H, ArH), 7.75 (d, J = 8.2 Hz, 1H, ArH), 7.66 (s, 1H, ArH), 7.60–7.48 (m, 5H, ArH), 7.31 (t, J = 7.9 Hz, 1H, ArH), 7.16 (d, J = 8.7 Hz, 2H, ArH), 7.04 (d, J = 7.6 Hz, 1H, ArH), 5.26 (s, 2H, −CH2−), 3.86 (s, 3H, −OCH3), 2.59 (s, 3H, −CH3), 2.53 (s, 3H, −CH3). The molecular formula C28H25N5O3 was indicated by HR-ESI-MS at m/z 480.2034 [M + H]+ (Calcd for [C28H25N5O3H]+, 480.2030).
Experimental Procedure of Compounds 17a–b
CuI (1.5 equiv) was added to a degassed mixture of compound 7 (1.0 mmol, 1.0 equiv), 15 (2.0 equiv), ligand 16 (0.15 equiv), and K2CO3 (1.5 equiv) in DMSO (anhydrous, 10 mL). The mixture was stirred at 150 °C under microwave conditions for 20 min. Then the reaction was quenched with cold water and added with EtOAc. The resulting suspension was filtered with a diatomaceous earth pad and washed several times with EtOAc. The organic phase was extracted twice with EtOAc, the combined EtOAc was washed 3 times with water, dried with Na2SO4, filtered, concentrated, and purified by flash column chromatography (EtOAc–Hex, 1:1 to 2:1), which gave the desired products 17a–b as white solids. Yield: 55% for 17a and 60% for 17b.
Compound 17a
1H NMR (300 MHz, DMSO-d6, δ (ppm): 8.16–8.09 (m, 1H, ArH), 7.94 (d, J = 7.7 Hz, 1H, ArH), 7.86 (d, J = 8.0 Hz, 1H, ArH), 7.63 (m, 6H, ArH), 3.86 (s, 3H, −COCH3), 2.63 (s, 3H, −CH3), 2.56 (s, 3H, −CH3).
Compound 17b
1H NMR (300 MHz, DMSO-d6, δ ppm): 8.14 (t, J = 1.9 Hz, 1H), 7.96–7.93 (m, 1H, ArH), 7.89–7.86 (m, 1H, ArH), 7.64 (t, J = 7.9 Hz, 1H, ArH), 7.57–7.53 (m, 2H, ArH), 7.19–7.13 (m, 2H, ArH), 3.88 (s, 3H, −OCH3), 3.85 (s, 3H, −COCH3), 2.61 (s, 3H, −CH3), 2.56 (s, 3H, −CH3).
Experimental Procedure of Compounds 18a–b
To a stirred solution of compound 17 (0.5 mmol, 1.0 equiv) in a mixture of tetrahydrofuran (THF)-MeOH (1:1, 10 mL) was added a solution of 1.0 M LiOH/H2O (excess). The reaction mixture was stirred at room temperature overnight. After the starting material disappeared, the mixture was concentrated to completely evaporate the solvents under a vacuum system. The resulting residue was acidified with 1.0 M HCl (aq) solution, then extracted twice with EtOAc, washed with brine, dried with Na2SO4, filtered, concentrated, and recrystallized, which gave the desired product 18a-b as white solids. Yield: 90% for 18a and 87% for 18b.
Compound 18a
1H NMR (300 MHz, DMSO-d6, δ ppm): 13.17 (br, 1H, −COOH), 8.13–8.09 (m, 1H, ArH), 7.93 (d, J = 7.6 Hz, 1H, ArH), 7.83 (d, J = 7.9 Hz, 1H, ArH), 7.69–7.57 (m, 6H, ArH), 2.65 (s, 3H, −CH3), 2.58 (s, 3H, −CH3).
Compound 18b
1H NMR (300 MHz, DMSO-d6, δ ppm): 13.17 (s, 1H, −COOH), 8.13 (s, 1H, ArH), 7.95 (d, J = 7.7 Hz, 1H, ArH), 7.85 (d, J = 7.9 Hz, 1H, ArH), 7.64 (d, J = 7.9 Hz, 1H, ArH), 7.59–7.55 (m, 2H, ArH), 7.18 (d, J = 8.4 Hz, 2H, ArH), 3.87 (s, 3H, −OCH3), 2.63 (s, 3H, −CH3), 2.58 (s, 3H, −CH3).
Experimental Procedure of Compounds 19a–b
To a stirred solution of compound 18 (0.5 mmol, 1.0 equiv), hydroxybenzotriazole (HOBT) (1.3 equiv) and EDC·HCl (1.3 equiv) in DMF (anhydrous, 5 mL) was added isopropylamine (excess). The mixture was stirred at room temperature to 50 °C overnight. After the starting material disappeared, the reaction product was added to cold water, extracted twice with EtOAc, washed twice with water, dried with Na2SO4, filtered, concentrated, and purified by flash column chromatography (EtOAc–Hex, 4:1 to EtOAc), which gave the desired product 19a-b as white solids. Yield: 27% for 19a and 36% for 19b.
Compound 19a
1H NMR (300 MHz, DMSO-d6, δ ppm): 8.33 (d, J = 7.8 Hz, 1H, ArH), 8.02 (d, J = 2.5 Hz, 1H, ArH), 7.88 (d, J = 7.7 Hz, 1H, ArH), 7.71–7.66 (m, 6H, ArH (5H), and −NH–(1H)), 7.57 (t, J = 7.8 Hz, 1H, ArH), 4.18–4.05 (m, 1H, −NH–CH–(CH3)2), 2.66 (s, 3H, −CH3), 2.60 (s, 3H, −CH3), 1.18 (d, J = 6.5 Hz, 6H, −CH–(CH3)2).
Compound 19b
1H NMR (300 MHz, DMSO-d6, δ ppm): 8.33 (d, J = 7.7 Hz, 1H, ArH), 8.02 (s, 1H, ArH), 7.88 (d, J = 7.7 Hz, 1H, ArH), 7.69 (d, J = 8.0 Hz, 1H, ArH), 7.59–7.54 (m, 3H, ArH (2H), and −NH–(1H)), 7.19 (d, J = 8.4 Hz, 2H, ArH), 4.18–4.07 (m, 1H, −NH–CH–(CH3)2), 3.88 (s, 3H, −OCH3), 2.64 (s, 3H, −CH3), 2.59 (s, 3H, −CH3), 1.18 (d, J = 6.4 Hz, 6H, −CH–(CH3)2).
5.3. Evaluation of Anti-ZIKV Activity
The experimental method against Zika virus was similar to that previously published.3,4 Vero cells (ATCC, Manassas, USA) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), 10% fetal bovine serum (FBS), 1% (penicillin and streptomycin), and 2 mM l-glutamine. Zika virus (MR766 Africa lineage, ATCC, USA, VR-1838) inoculated Aedes albopictus mosquito cells, clone C6/36. The culture was carried out for 14 days in EMEM, 1% FBS, 1% penicillin, and streptomycin, and then viral passages were kept at −80 °C for the experiments. Cells were pretreated with the test compounds at ten concentrations, then cells were infected (added 5 μL of ZIKV inoculum for each well) and continued to be incubated for 24 h. After 24 h, the cells were analyzed by immunofluorescence assay (should fix paraformaldehyde at 4%). Mycophenolate acid was used as a positive control in this study. The EC50 and CC50 values were calculated using Prism v7.0 software. The antiviral activity of tested compounds was evaluated by a 10-point DRC analysis in triplicate.
5.4. Brine Shrimp Lethality Bioassay
The brine shrimp lethality bioassay was performed similar to that previously published by Mayer et al.32 First, the compounds were dissolved in small amounts of dimethyl sulfoxide (DMSO; Sigma, MA, USA), then serially diluted in simulated seawater to solutions of various concentrations (500, 250, 125, 62, 31, and 16 μg/mL). Second, randomly aspirate 20–35 naupliis into 5 mL of solution corresponding to each concentration. After 24 h, the number of surviving nauplii in each vial was counted and the brine shrimp’s lethality percentage was calculated. Finally, the LD50 values were determined by probit analysis. The procedures were independently repeated three times.
5.5. Docking Studies
All ligands were performed with the aid of the Gaussian 09 program.50 The geometries were fully optimized, making use of functional B3LYP in conjunction with the 6-31G(d,p) basis set for carbon and hydrogen atoms for compounds. The crystal structure of NS2B-NS3 in complex with a benzimidazol-1-ylmethanol inhibitor (PDB code: 5H4I) was retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB).27 All water molecules and small molecules were deleted by the Discovery Studio Visualizer. Autodock Tool version 4.2.6 was also used in the docking studies.49 Grid box parameters were set to cover all of the residues around the active site. More specifically, it was centered on the ligand and comprised 60 × 60 × 60 points with 0.375 Å in spacing. The Lamarckian Genetic Algorithm was used to explore the best conformational space for the ligand, with 100 docking runs for each ligand. The maximum numbers of generation and evaluation were set at 27,000 and 50,000,000, respectively. Other parameters were set as default. After the docking process was completed, 100 conformations of the ligands in complex with the receptor were obtained and ranked based on binding energy.
5.6. Molecular Dynamic and Pharmacophore Model
All calculations were performed with the Desmond package 2018–4 (academic license). The protein–ligand docked complex was solvated with the single-point charge water model, and the overall charge of the system was neutralized. The parameters were set by default by the software. The simulation system was relaxed by constant NPT ensemble conditions to generate simulation data for postsimulation analyses. The full system contained 24118 atoms, and the temperature value was defined as 300 K and stable atmospheric pressure (1 atm). Pharmacophore model using Pharmit online (https://pharmit.csb.pitt.edu/search.html).28
Acknowledgments
The authors would like to thank Can Tho University for its facilities support. This work was supported by the Ministry of Education and Training of Vietnam under grant No. B2019-TCT-37 (TCT-05) and Scientific research project of Can Tho University (T2023-19).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c06612.
Spectral data (1H NMR, HRMS spectra) and HPLC/GC-FID chromatograms for all compounds (Figures S1–S27); Biological assay data for all compounds (Table S1 and Figure S28); Molecular docking and pharmacophore model (Table S2 and Figures S29–S30) (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Pires L. C.; Dantas L. R.; Witkin S. S.; Bertozzi A. P. A. P.; Dezena R. de C. A. B.; Rodrigues M. M. D.; Gazeta R. E.; Passos S. D. Knowledge of Zika virus transmission and its prevention among high-risk pregnant women in Brazil. Viruses 2021, 13, 242. 10.3390/v13020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddique R.; Liu Y.; Nabi G.; Sajjad W.; Xue M.; Khan S. Zika virus potentiates the development of neurological defects and microcephaly: Challenges and control strategies. Front. Neurol. 2019, 10, 219. 10.3389/fneur.2019.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hue B. T. B.; Nguyen P. H.; De T. Q.; Van Hieu M.; Jo E.; Van Tuan N.; Thoa T. T.; Anh L. D.; Son N. H.; La Duc Thanh D.; Dupont-Rouzeyrol M.; Grailhe R.; Windisch M. P. Benzimidazole derivatives as novel zika virus inhibitors. ChemMedChem. 2020, 15, 1453. 10.1002/cmdc.202000124. [DOI] [PubMed] [Google Scholar]
- Nguyen C. Q.; Nguyen T. H. M.; Nguyen T. T. T.; Bui T. B. H.; Nguyen T. T.; Huynh N. T.; Le T. D.; Nguyen T. M. P.; Nguyen D. T.; Nguyen M. T.; Pham M. Q.; Tran Q. D.; Nguyen H. P. Designs, synthesis, docking studies, and biological evaluation of novel berberine derivatives targeting zika virus. J. Chem. 2021, 2021, e5567111 10.1155/2021/5567111. [DOI] [Google Scholar]
- Nagawade R. R.; Khanna V. V.; Bhagwat S. S.; Shinde D. B. Synthesis of new series of 1-aryl-1, 4-dihydro-4-oxo-6-methyl pyridazine-3-carboxylic acid as potential antibacterial agents. Eur. J. Med. Chem. 2005, 40, 1325. 10.1016/j.ejmech.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Ellis G. P.Synthesis of Fused Heterocycles ; Part (1), ; John Wiley & Sons, 2009; Vol. 47. [Google Scholar]
- Asif M. Pharmacologically potentials of hydrazonone containing compounds: A promising scaffold. Int. J. Adv. Chem. 2014, 2, 85. 10.14419/ijac.v2i2.2301. [DOI] [Google Scholar]
- Tang J.; Wang B.; Wu T.; Wan J.; Tu Z.; Njire M.; Wan B.; Franzblauc S. G.; Zhang T.; Lu X.; Ding K. Design, synthesis, and biological evaluation of pyrazolo [1, 5-a] pyridine-3-carboxamides as novel antitubercular agents. ACS Med. Chem. Lett. 2015, 6, 814. 10.1021/acsmedchemlett.5b00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelriheem N.; Zaki Y.; Abdelhamid A. Synthesis of some new pyrazolo [1, 5-a] pyrimidine, pyrazolo [5, 1-c] triazine, 1,3,4-thiadiazole and pyridine derivatives containing 1,2,3-triazole moiety. Chem. Cent. J. 2017, 11, 1. 10.1186/s13065-017-0282-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Gupta S.; Abadi L. F.; Gaikwad S.; Desai D.; Bhutani K. K. S.; Kulkarni S.; Singh I. P. Synthesis and in-vitro anti-HIV-1 evaluation of novel pyrazolo[4,3-c]pyridin-4-one derivatives. Eur. J. Med. Chem. 2019, 183, 111714. 10.1016/j.ejmech.2019.111714. [DOI] [PubMed] [Google Scholar]
- Arias-Gómez A.; Godoy A.; Portilla J. Functional Pyrazolo[1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses As an Antitumor Scaffold. Molecules 2021, 26, 2708. 10.3390/molecules26092708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbas E.; Berber I. Antibacterial and antifungal activities of new pyrazolo[3,4-d]pyridazin derivatives. Eur. J. Med. Chem. 2005, 40, 401. 10.1016/j.ejmech.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Braña M. F.; Cacho M.; García M. L.; Mayoral E. P.; López B.; de Pascual-Teresa B.; Ramos A.; Acero N.; Llinares F.; Muñoz-Mingarro D.; Lozach O.; Meijer L. Pyrazolo [3,4-c] pyridazines as novel and selective inhibitors of cyclin-dependent kinases. J. Med. Chem. 2005, 48, 6843. 10.1021/jm058013g. [DOI] [PubMed] [Google Scholar]
- Tewari A. K.; Mishra A. Synthesis and anti-inflammatory activities of N4, N5-disubstituted-3-methyl-1H-pyrazolo [3, 4-c] pyridazines. Bioorg. Med. Chem. 2001, 9, 715. 10.1016/S0968-0896(00)00285-6. [DOI] [PubMed] [Google Scholar]
- Tavares F. X.; Boucheron J. A.; Dickerson S. H.; Griffin R. J.; Preugschat F.; Thomson S. A.; Wang T. Y.; Zhou H.-Q. N-Phenyl-4-pyrazolo [1, 5-b] pyridazin-3-ylpyrimidin-2-amines as potent and selective inhibitors of glycogen synthase kinase 3 with good cellular efficacy. J. Med. Chem. 2004, 47, 4716. 10.1021/jm040063i. [DOI] [PubMed] [Google Scholar]
- Ghareb N.; Elshihawy H. A.; Abdel-Daim M. M.; Helal M. A. Novel pyrazoles and pyrazolo [1,2-a] pyridazines as selective COX-2 inhibitors; Ultrasound-assisted synthesis, biological evaluation, and DFT calculations. Bioor. Med. Chem. Lett. 2017, 27, 2377. 10.1016/j.bmcl.2017.04.020. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Dai Y.; Wu X.; Li F.; Liu B.; Li C.; Liu Q.; Zhou Y.; Wang B.; Zhu M.; Cui R.; Tan X.; Xiong Z.; Liu J.; Tan M.; Xu Y.; Geng M.; Jiang H.; Liu H.; Ai J.; Zheng M. Discovery and development of a series of pyrazolo [3, 4-d] pyridazinone compounds as the novel covalent fibroblast growth factor receptor inhibitors by the rational drug design. J. Med. Chem. 2019, 62, 7473. 10.1021/acs.jmedchem.9b00510. [DOI] [PubMed] [Google Scholar]
- Jacomini A. P.; Silva M. J. V.; Silva R. G. M.; Gonçalves D. S.; Volpato H.; Basso E. A.; Paula F. R.; Nakamura C. V.; Sarragiotto M. H.; Rosa F. A. Synthesis and evaluation against Leishmania amazonensis of novel pyrazolo [3,4-d] pyridazinone-N-acylhydrazone-(bi) thiophene hybrids. Eur. J. Med. Chem. 2016, 124, 340. 10.1016/j.ejmech.2016.08.048. [DOI] [PubMed] [Google Scholar]
- Bildirici İ.; Şener A.; Atalan E.; Battal A.; Genç H. Synthesis and antibacterial activity of 4-benzoyl-1-(4-carboxy-phenyl)-5-phenyl-1 H-pyrazole-3-carboxylic acid and derivatives. Med. Chem. Res. 2009, 18, 327. 10.1007/s00044-008-9130-3. [DOI] [Google Scholar]
- Fayed A.; Al-Harbi N.; Amr A. E.-G.; Kalmoush A.; Shadid K.; Flefel E. Synthesis, reactions, and pharmacological evaluations of some novel pyridazolopyridiazine candidates. J. Heterocycl. Chem. 2014, 51, 1770. 10.1002/jhet.1881. [DOI] [Google Scholar]
- Deeb A.; Abdelgawad A. A. M. Utility of 3-amino-1 H-pyrazolo [3, 4-c] pyridazine in heterocyclic synthesis. Chem. Heterocycl. Compd. 2015, 51, 785. 10.1007/s10593-015-1776-4. [DOI] [Google Scholar]
- Ghorbani-Vaghei R.; Mahmoodi J.; Maghbooli Y. Preparation and characterization of nanomagnetic piperidinium benzene-1, 3-disulfonate ionic liquid as a novel, green and heterogeneous catalyst and its use in the synthesis of 1H-pyrazolo [1,2-b] phthalazine-5, 10-diones and 1H-pyrazolo [1,2-a] pyridazine-5, 8-diones under solvent-free conditions. Appl. Organomet. Chem. 2017, 31, e3717 10.1002/aoc.3717. [DOI] [Google Scholar]
- Çetin A.; Bildirici İ. A study on synthesis and antimicrobial activity of 4-acyl-pyrazoles. J. Saudi Chem. Soc. 2018, 22, 279. 10.1016/j.jscs.2016.05.008. [DOI] [Google Scholar]
- Kumbar M. N.; Shaikh S. K. J.; Kamble R. R.; Bayannavar P. K.; Kamble A. A.; Hunnur R. K. Serendipitous formation of 2H-pyrazolo [3, 4-d] pyridazin-7 (6H)-ones from 3-arylsydnones. ACS Omega 2019, 4, 4955. 10.1021/acsomega.8b02013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mady M. F.; Saleh T. S.; El-Kateb A. A.; Abd El-Rahman N. M.; Abd El-Moez S. I. Microwave-assisted synthesis of novel pyrazole and pyrazolo [3, 4-d] pyridazine derivatives incorporating diaryl sulfone moiety as potential antimicrobial agents. Res. Chem. Intermed. 2016, 42, 753. 10.1007/s11164-015-2054-x. [DOI] [Google Scholar]
- Jacomini A. P.; da Silva M. J. V.; Poletto J.; Ribeiro G. M.; Yokoyama J. T. C.; Bidóia D. L.; Paula F. R.; Nakamura C. V.; Sarragiotto M. H.; Rosa F. A. Potential antileishmanial activity of 4-N-acylhydrazone pyrazolo [3,4-d] pyridazin-7-ones: Synthesis, in vitro biological evaluations and computational studies. J. Braz. Chem. Soc. 2018, 29, 2657. 10.21577/0103-5053.20180134. [DOI] [Google Scholar]
- Zhang Z.; Li Y.; Loh Y. R.; Phoo W. W.; Hung A. W.; Kang C.; Luo D. Crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science 2016, 354, 1597. 10.1126/science.aai9309. [DOI] [PubMed] [Google Scholar]
- Sunseri J.; Koes D. R. Pharmit: interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442. 10.1093/nar/gkw287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadow J. F.; Pracitto R. United States, US8324212B2, 2012.
- Elfiky A. A. Zika viral polymerase inhibition using anti-HCV drugs both in market and under clinical trials. J. Med. Virol. 2016, 88, 2044. 10.1002/jmv.24678. [DOI] [PubMed] [Google Scholar]
- Hamdy N. A.; El-Senousy W. M. Synthesis and antiviral evalution of some novel pyrazoles and pyrazolo[3,4-d]pyridazines bearing 5,6,7,8-tetrahydronaphthalene. Acta Pol. Pharm. 2012, 70, 99–110. [PubMed] [Google Scholar]
- Meyer B. N.; Ferrigni N. R.; Putnam J. E.; Jacobsen L. B.; Nichols D. E.; McLaughlin J. L. Brine shrimp: a convenient general bioassay for active plant constituents. Planta Med. 1982, 45, 31. 10.1055/s-2007-971236. [DOI] [PubMed] [Google Scholar]
- Ali M. I.; Liton M. A. K. Cytotoxicity, 2D-and 3D-QSAR study of some halogen containing hydroxy and amino substituted aromatic compounds. World J. Org. Chem. 2015, 3 (1), 16–26. 10.12691/wjoc-3-1-4. [DOI] [Google Scholar]
- Rayees Ahmad M.; Girija Sastry V.; Bano N.; Anwar S. Synthesis of novel chalcone derivatives by conventional and microwave irradiation methods and their pharmacological activities. Arabian J. Chem. 2016, 9, S931. 10.1016/j.arabjc.2011.09.002. [DOI] [Google Scholar]
- Definition of berberine chloride - NCI Drug Dictionary - NCI, https://www.cancer.gov/publications/dictionaries/cancer-drug/def/berberine-chloride, (accessed 2022-08-24).
- Berberine , https://go.drugbank.com/drugs/DB04115, (accessed 2022-08-24).
- Atropine , https://go.drugbank.com/drugs/DB00572, (accessed 24, 2022-08-24).
- Yadav R.; Selvaraj C.; Aarthy M.; Kumar P.; Kumar A.; Singh S. K.; Giri R. Investigating into the molecular interactions of flavonoids targeting NS2B-NS3 protease from ZIKA virus through in-silico approaches. J. Biomol. Struct. Dyn. 2021, 39, 272. 10.1080/07391102.2019.1709546. [DOI] [PubMed] [Google Scholar]
- Choudhry H.; Alzahrani F. A.; Hassan M. A.; Alghamdi A.; Abdulaal W. H.; Bakhrebah M. A.; Zamzami M. A.; Helmi N.; Bokhari F. F.; Zeyadi M.; Baothman O. A.; Kamal M. A.; Warsi M. K.; Ali A.; Jarullah B.; Jamal M. S. Zika virus targeting by screening inhibitors against NS2B/NS3 protease. Biomed. Res. Int. 2019, 2019, e3947245 10.1155/2019/3947245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Liang R.; Gao Y.; Li Y.; Deng X.; Xiang R.; Zhang Y.; Ying T.; Jiang S.; Yu F. Development of small-molecule inhibitors against zika virus infection. Front. Microbiol. 2019, 10, 2725. 10.3389/fmicb.2019.02725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss S.; Nitsche C. Inhibitors of the Zika virus protease NS2B-NS3. Bioor. Med. Chem. Lett. 2020, 30, 126965. 10.1016/j.bmcl.2020.126965. [DOI] [PubMed] [Google Scholar]
- Thirumoorthy G.; Tarachand S. P.; Nagella P.; Veerappa Lakshmaiah V. Identification of potential ZIKV NS2B-NS3 protease inhibitors from Andrographis paniculata: An in silico approach. J. Biomol. Struct. Dyn. 2022, 40, 11203. 10.1080/07391102.2021.1956592. [DOI] [PubMed] [Google Scholar]
- Coronado M. A.; Eberle R. J.; Bleffert N.; Feuerstein S.; Olivier D. S.; de Moraes F. R.; Willbold D.; Arni R. K. Zika virus NS2B/NS3 proteinase: A new target for an old drug-Suramin a lead compound for NS2B/NS3 proteinase inhibition. Antivir. Res. 2018, 160, 118. 10.1016/j.antiviral.2018.10.019. [DOI] [PubMed] [Google Scholar]
- Wu H.; Bock S.; Snitko M.; Berger T.; Weidner T.; Holloway S.; Kanitz M.; Diederich W. E.; Steuber H.; Walter C.; Hofmann D.; Weißbrich B.; Spannaus R.; Acosta E. G.; Bartenschlager R.; Engels B.; Schirmeister T.; Bodem J. Novel dengue virus NS2B/NS3 protease inhibitors. J. Bodem, Antimicrob. Agents Chemother. 2015, 59, 1100. 10.1128/AAC.03543-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman H. M.; Sajjad M.; Ali M. A.; Gul R.; Irfan M.; Naveed M.; Bhinder M. A.; Ghani M. U.; Hussain N.; Said A. S. A.; Al Haddad A. H. I.; Saleem M. Identification of NS2B-NS3 protease inhibitors for therapeutic application in zikv infection: A pharmacophore-based high-throughput virtual screening and MD simulations approaches. Vaccines 2023, 11, 131. 10.3390/vaccines11010131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J.; Lee H.; Kotak A.; Johnson M. E. MD simulations reveal alternate conformations of the oxyanion hole in the Zika virus NS2B/NS3 protease. Proteins: Struct. Funct. Bioinform. 2020, 88, 345. 10.1002/prot.25809. [DOI] [PubMed] [Google Scholar]
- de Paula Junior V. F.; van Tilburg M. F.; Morais P. A.; Júnior F. F. M.; Lima E. G.; Oliveira V. T. D. S.; Guedes M. I. F.; Caetano E. W. S.; Freire V. N. Quantum biochemistry and MM-PBSA description of the ZIKV NS2B-NS3 protease: Insights into the binding interactions beyond the catalytic triad pocket. Int. J. Mol. Sci. 2022, 23, 10088. 10.3390/ijms231710088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak N.; Kuo Y.-P.; Chang T.-Y.; Huang C.-T.; Hung H.-C.; Hsu J. T.-A.; Yu G.-Y.; Yang J.-M. Zika virus NS3 protease pharmacophore anchor model and drug discovery. Sci. Rep. 2020, 10, 8929. 10.1038/s41598-020-65489-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch M.; Trucks G.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.. Gaussian 09 , (Revision A.02); Gaussian Inc.: Wallingford, CT, 2016.
- Morris G. M.; Goodsell D. S.; Halliday R. S.; Huey R.; Hart W. E.; Belew R. K.; Olson A. J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639.. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.