

Abstract
A key step of hepatitis B virus (HBV) replication is the selective packaging of pregenomic RNA (pgRNA) by core protein (Cp) dimers, forming a nucleocapsid where the reverse transcriptional viral DNA replication takes place. One approach in the development of new anti-HBV drugs is to disrupt the assembly of HBV nucleocapsids by misdirecting Cp dimers to assemble morphologically normal capsids devoid of pgRNA. In this study, we built upon our previous discovery of benzamide-derived HBV capsid assembly modulators by exploring fused bicyclic scaffolds with an exocyclic amide that is β, γ to the fused ring, and identified 1,2,3,4-tetrahydroquinoxaline derived phenyl ureas as a novel scaffold. Structure-activity relationship studies showed that a favorable hydrophobic substitution can be tolerated at the 2-position of the 1,2,3,4-tetrahydroquinoxaline core, and the resulting compound 88 demonstrated comparable or improved antiviral potencies in mouse and human hepatocyte-derived HBV-replicating cell lines compared to our previously reported benzamide compound, 38017 (8). In addition, a novel bis-urea series based on 1,2,3,4-tetrahydroquinoxaline was also found to inhibit HBV DNA replication with sub-micromolar EC50 values. The mode of action of these compounds is consistent with specific inhibition of pgRNA encapsidation into nucleocapsids in hepatocytes.
Keywords: 1,2,3,4-tetrahydroquinoxaline; phenyl ureas; hepatitis B virus; capsid assembly
Graphic Abstract
Introduction
Chronic Hepatitis B virus (HBV) infection remains a global public health challenge. The current standard of care medications, including nucleos(t)ide analogs (NAs) or viral DNA polymerase inhibitors and pegylated interferon-alpha (IFN-α), can efficiently suppress HBV replication, but rarely cure the viral infection.1 This is because the viral covalently closed circular DNA (cccDNA), the most stable viral genome replication intermediate, in the nuclei of infected hepatocytes is refractory to the current medications, and viral replication rebounds after the cessation of antiviral therapy.2 In addition, the current NA antiviral drugs are also unable to restore a functional antiviral immune response against HBV for durable immune control of HBV infection.3 It is anticipated that drugs with novel antiviral mechanisms are required, used either alone or in combination with the NAs and/or pegylated IFN-α, for a functional cure of chronic HBV infection.4–5
Selective packaging of viral pregenomic (pg) RNA and DNA polymerase complex by HBV core protein dimers forms a nucleocapsid where the reverse transcriptional HBV DNA replication takes place. Assembly of nucleocapsids is driven by hydrophobic interactions between the core protein dimer-dimer interface and can be disrupted by several chemotypes of small molecular core protein allosteric modulators (CpAMs).6–9 Since the discovery of two early chemotypes of CpAMs, phenylpropenamides (1, PPAs)10–12 and heteroaryldihydropyrimidines (2, HAPs)13–14, multiple types of molecules, such as sulfamoylbenzamides (3, SBAs),15 sulfamoylpyrroloamides (4, SPAs),16 glyoxamoylpyrroloxamides (5, GLPs),17–18 dibenzothiazepine derivatives (6, DBTs),19 and benzamides (7, 8, BAs),20 have been reported to modulate HBV capsid assembly (Fig. 1). The leads from HAP, SBA, and DBT families of CpAMs have been advanced to phase 2 clinical trials and demonstrated significant antiviral efficacy against HBV4.
Figure 1.
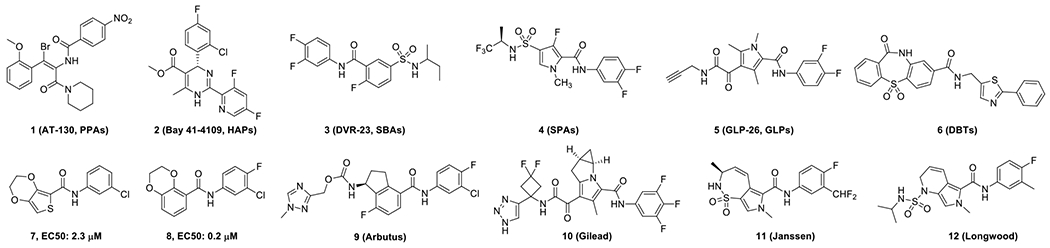
Representative HBV capsid assembly modulators.
The binding modes of five chemotypes of CpAMs – PPAs, HAPs, SBAs, Ciclopirox, and DBTs – with HBV core protein have been revealed either through X-ray crystallography21–25 or Cryo-EM technology26. Interestingly, the products of CpAM mis-directed core protein assembly are morphologically different even though they were found to bind at the same hydrophobic pocket between Cp dimer-dimer interfaces. A relatively large CpAM binding pocket allows for compounds with different structural scaffolds to adopt distinct binding poses and interact with distinct residues at the binding pocket. However, the detailed mechanism underlying the assembly of core protein dimers into structurally distinct products induced by different chemotypes of CpAMs at a whole-time scale remains to be elucidated. In addition to disrupting the assembly of nucleocapsids, CpAMs also induce the disassembly of in vitro assembled empty capsids and mature nucleocapsids containing double-stranded DNA in hepatocytes to modulate cccDNA synthesis, demonstrating the multifaceted effects a CpAM could play behind the final outcome.9 In agreement with their inhibition of nucleocapsid assembly, all CpAMs tested thus far have inhibited core protein dephosphorylation, which is associated with encapsidation of pgRNA and DNA polymerase complex. 27–28 Despite several types of CpAMs being available, discovery of new chemotypes should be helpful for probing other possible avenues of nucleocapsid assembly and disassembly and for identifying CpAMs with more therapeutically beneficial activities.
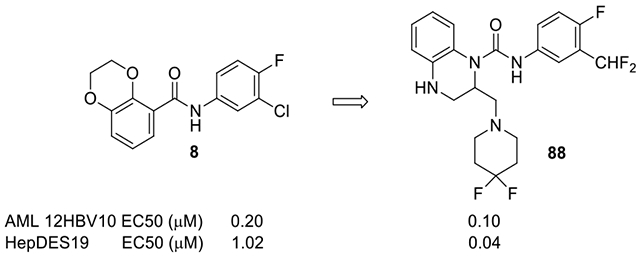
Based on the early high throughput screening campaign in our laboratory,15 we pioneered the identification of SBA modulators, which led to the discovery of a phase 1 clinical candidate, AB-423.29 In addition to the SBA compounds from that screening, we also observed another hit with a moderate EC50, 7, with a 2,3-dihydrothieno[3,4-b][1,4]dioxine scaffold, in which the fused 1,4-dioxane is β, γ to the exocyclic amide moiety. Preliminary structure-activity relationship (SAR) studies have progressed 7 to a new lead compound 38017 (8) with sub-micromolar EC50 in HBV AML12HBV10 cell line.20 Both compounds 7 and 8 possess unique bicyclic frameworks with the fused ring β, γ to exocyclic amide moieties, which are structurally different from previously known capsid modulators, and thus we continued the optimization of this novel series of CpAMs. During the course of our SAR optimization, several other types of CpAMs, 9-12 for example, with a common trait of bicyclic cores with fused rings β, γ to an exocyclic amide moiety were disclosed.30–34 Although different from ours, they supported the interest in this novel type of structure.
Our approach in continuing optimization was to upturn the phenyl in the 2,3-dihydrobenzo[1,4]dioxine in 8 and replace the oxygen atoms with either carbon or nitrogen atoms, thus connecting the aniline in the amide piece through either an amide or a urea group. This change allowed us to create and evaluate new molecules with different shapes and substitution potentials from 8. The resulting product 13 maintained the bicyclic core feature and served as a platform to introduce other functional groups for additional binding (Fig. 2). This acyl translocation has led to our discovery of 4-oxooctahydroquinoline-1(2H)-carboxamides as hepatitis B virus (HBV) capsid core protein assembly modulators.34 Herein, we report the identification of another bicyclic scaffold, 1,2,3,4-tetrahydroquinoxaline, which can be derivatized into phenyl ureas that affect the capsid core protein assembly efficiently.
Figure 2.
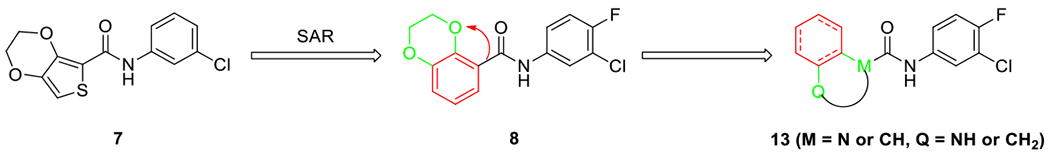
Strategy of new molecule design
Results and Discussion
Synthesis.
General synthetic routes are illustrated in Scheme 1 for the synthesis of urea 17, bis-urea 18, amide 23, and sulfamide 26. Ureas and bis-ureas were made from bicyclic amines through reactions with either isocyanates or phenyl carbamates35 (Scheme 1 A); amides were prepared from corresponding acids and anilines with a coupling reagent, EDCI (Scheme 1 B); a different linker, sulfonyl, was introduced through the reaction of an aniline with chlorosulfonic acid, then phosphorus pentachloride, and subsequently a bicyclic amine to provide the desired sulfamide36 for evaluation (Scheme 1 C).
Scheme 1.
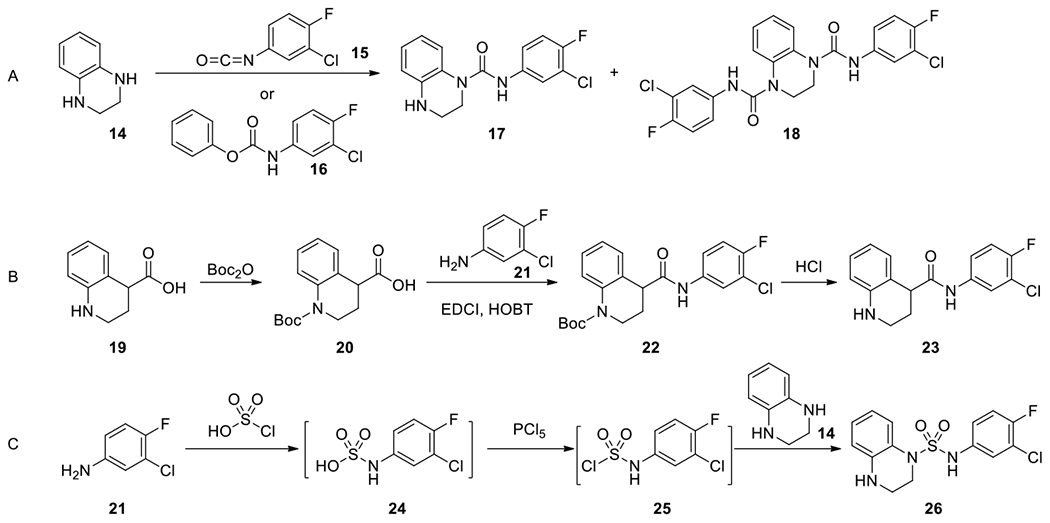
Illustrated synthesis of new compounds. Connection of an aniline to bicyclic ring through A, a urea linker; B, an amide linker; C, a sulfonamide linker.
Biological evaluation of new compounds.
The antiviral activity of compounds was tested by a dot blot hybridization assay in an immortalized mouse hepatocyte (AML12)-derived stable cell line (AML12HBV10) that supports a high level of HBV replication. This cell-based assay was the platform for our initial high throughput screening of HBV replication inhibitors, and it was used to determine EC50 and CC50 of new compounds and direct the SAR study. The antiviral activity of selected compounds was further confirmed in a human hepatoma-derived stable cell line supporting HBV replication (HepDES19). The mode of action of representative compounds on capsid assembly and pgRNA encapsidation was also investigated in hepatocytes by examination of viral RNA, encapsidated pgRNA, capsids, and viral DNA replication intermediates.
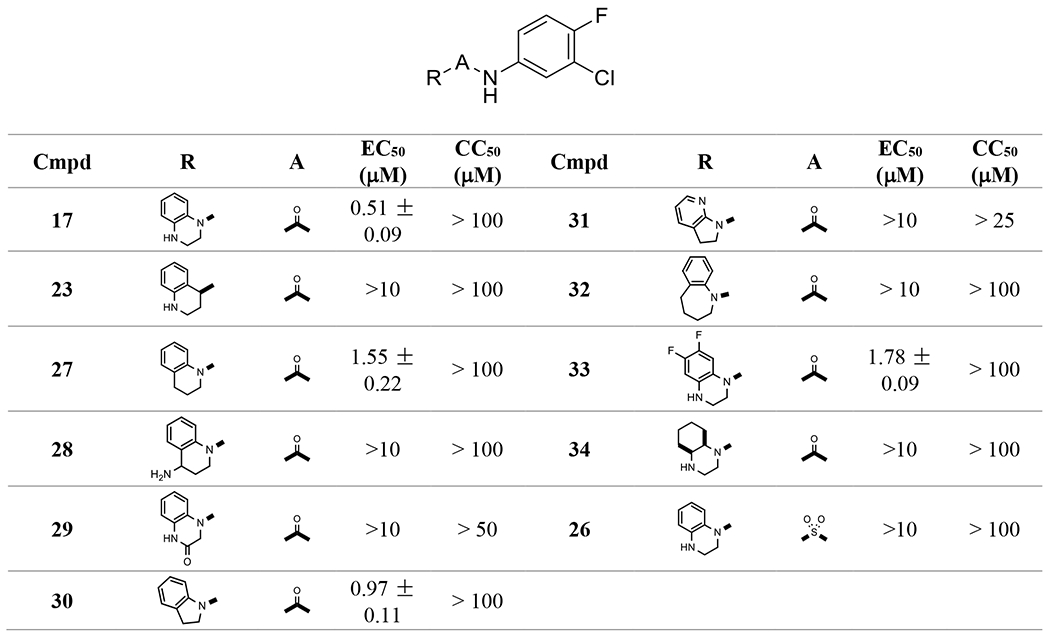
I. Bicyclic core
Bicyclic cores were first screened. The results are shown in Table 1. 1,2,3,4-tetrahydroquinoxaline in 17 gave an encouraging sub-micromolar EC50 (0.51 μM) and low cytotoxicity (CC50 > 100 μM). It is interesting to note that both nitrogen atoms are necessary for the activity. Replacement of 4-N with a CH2 in 27 reduced the activity by 3-fold, and substitution of 1-N with a CH resulted in the loss of activity in 23 (racemic). When the 4-N was placed outside the ring as in 28 (racemic), no activity was observed. Converting a basic 4-N to a neutral lactam in 29 led to an inactive compound. On the other hand, 30 with indoline maintained activity, but the benzo-fused 7-membered azepine in 32 diminished the activity. Saturation of the benzene ring as in 34 or replacement of the carbonyl with sulfonyl in 26 gave a loss of activity. The 1,2,3,4-tetrahydroquinoxaline is a pharmacophore that has been used in the preparation of bioactive compounds in other therapeutic fields, for instance as BET bromodomain inhibitors37 or DGAT1 inhibitors38. Considering the solubility benefit that 1,2,3,4-tetrahydroquinoxaline can bring when a salt is formed with its basic nitrogen at 4-position, as well as the hydrogen bond donating property of the NH at 4- position, which is different from the carbonyl in our 4-oxooctahydroquinoline-1(2H)-carboxamides34, we chose this scaffold to do further SAR and optimize the antiviral activity.
Table 1.
Central Pharmacophore evaluation and Amide Bond Structural Alteration. The bold bond designates the connecting bond.
|
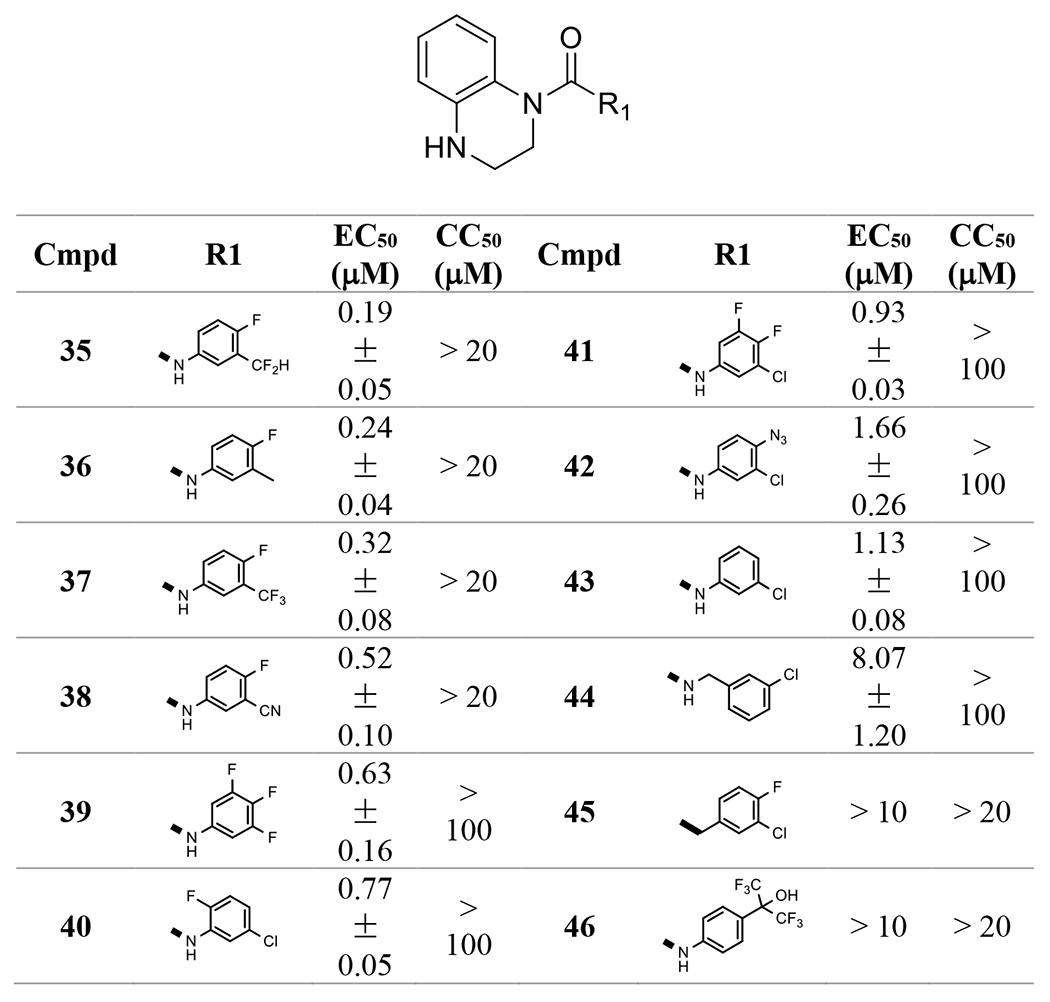
Next, we prepared ureas based on 1,2,3,4-tetrahydroquinoxaline with different anilines following the synthetic sequence described in Scheme 1A and evaluated these compounds for their anti-HBV activities and cytotoxicities (Table 2). The 3-(difluoromethyl)-4-fluoroaniline in 35 and 3-methyl-4-fluoroaniline in 36improved the potencies. However, using a benzylamine instead of aniline in 44 (3-chlorobenzylamine) reduced the activity, compared to the corresponding aniline 43. In addition, replacing the nitrogen of the aniline with a carbon in 45 resulted in a loss of activity, compared to the corresponding aniline 17. This suggests that an aniline is preferred at this position.
Table 2.
Aniline derivative evaluation
|
To pursue additional bindings and thus improve potencies, we docked compounds 17 and 8 into the HAP pocket (Fig. 3) of the HBV capsid in the crystal structure 5D7Y with a grid centered on T128 of chain C and a 20Å box for the grid.39 Although the oxygen in the 1,4-dioxane of 8 is different from the NH in 17, as one is a hydrogen bond acceptor and the other is a hydrogen bond donor, it appears that both bicyclic rings were tolerated well in the HAP pocket. The 1-oxygen of 8 found leucine 140 in the B dimer as a hydrogen bond partner while the 4-NH of 17 formed a hydrogen bond with tyrosine 132 of the C dimer. Interestingly, there were some spaces available out of both 2- and 3- positions of 17 which could be filled up with functional groups.
Figure 3.
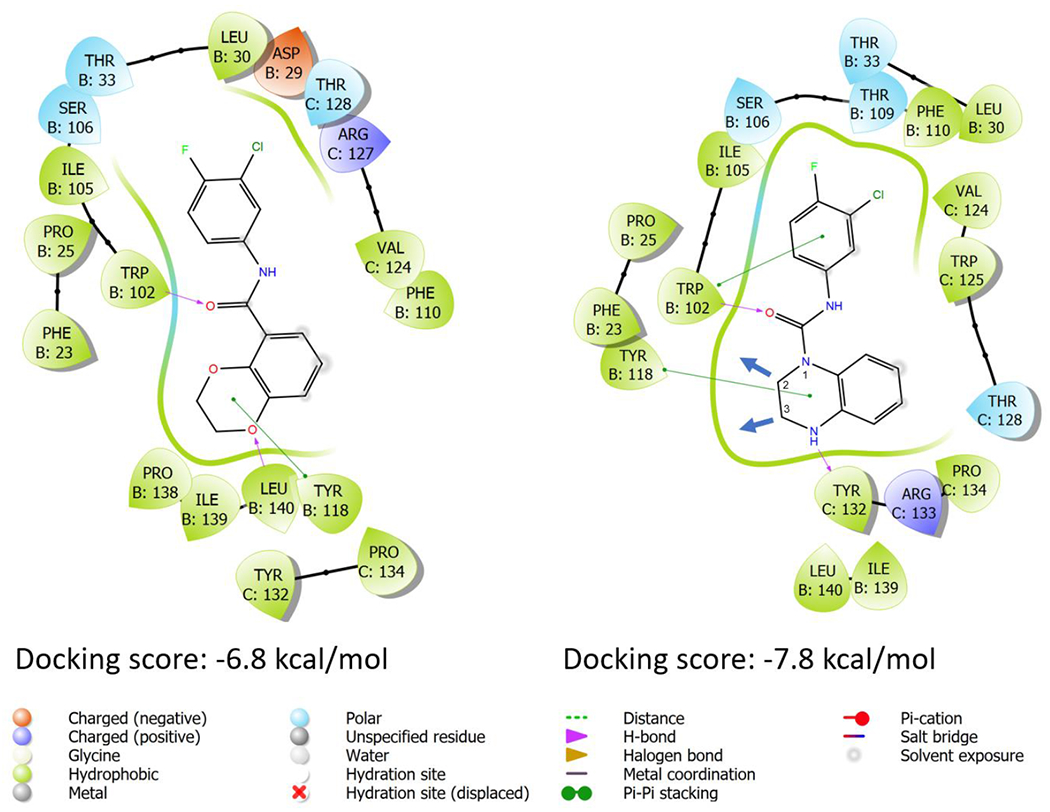
Docked 8 and 17 with the capsid residues in the dimer-dimer interface.
To evaluate the stability of the top compounds 8, 17, 88, 96 and 86 in the HAP pocket, a 10 ns replica exchange molecular dynamics simulation was run on the complexes. The MD simulations showed that all the complexes are well equilibrated as indicated by the convergence in the root mean square deviation (RMSD) plots (see supporting information) for both the protein and ligand. The ligand RMSD showed that all ligands stay in the HAP pocket throughout the simulation time. All the compounds maintained steady interaction with TRP102 occurring more than 80% of the simulation time, although for 88 the hydrogen bond with TRP102 is present less which is compensated by the other two other key interactions (PHE110 and THR128).
To explore these possibilities, we synthesized compounds with substitutions at 2- and 3- positions, respectively, and compared their antiviral activities. Compounds 51 and 69-70 were synthesized from commercial starting materials according to the procedure in Scheme 2.37 First, 2-isopropyl-1,2,3,4-tetrahydroquinoxaline (47) was treated with Boc2O. Regioselectivity was observed as the less hindered 4-Boc protected 48 was obtained as the major product. Further reactions with phosgene and aniline 21 provided 4-Boc protected urea 50, which was converted to urea 51 after acidic deprotection.
Scheme 2.
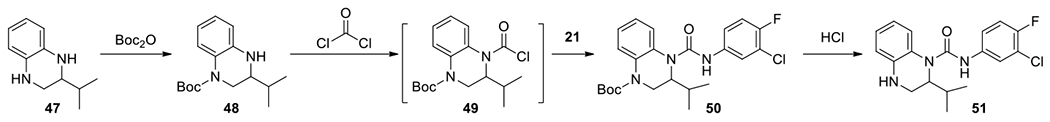
Exemplified synthesis of 2-substituted urea 51.
The Boc-protected isomer 48 was characterized by 1D and 2D NMR analysis in order to confirm the regioselectivity in presence of a substitution at 2-position. Interpretation of the 1H, 13C, HSQC, COSY, HMBC, NOESY spectra for 48 allowed for the assignment of the connectivity of the structure. Key HMBC correlations are depicted below in Fig. 4. A correlation between H-3 and C-4a, along with an observed correlation between H-2 and C-8a, together suggested the orientation of the two spin systems relative to one another. Additionally, a correlation between H-2 and the Boc carbonyl group strongly suggested that the Boc-group was attached to the nitrogen at position 1. Key correlations from the NOESY spectrum were observed between H-2′/3′/4′ (the Boc methyl protons) and H-8, as well between H-2″/3″ (the isopropyl methyl protons) and H-5. Together, these were consistent with the assignment of the Boc group to the 1-position. The appearance of these correlations, while of weak intensity, suggested that the tetrahydroquinoxazoline ring has some conformational freedom to allow the isopropyl groups to be close enough to H-5 to allow for observation of NOE’s. It should be noted that the magnitude of the chemical shift of H-8 (δH 7.49 ppm) is consistent with the presence of a de-shielding group nearby, such as a Boc carbonyl group.
Figure 4,
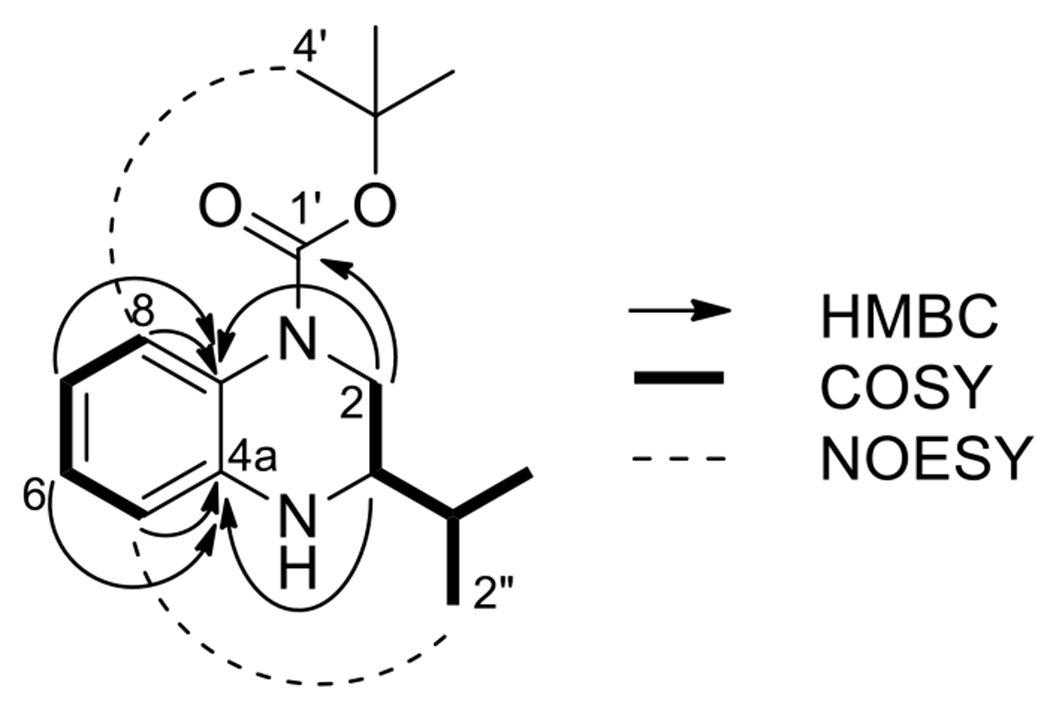
Determination of the structure of 48 via NMR analysis.
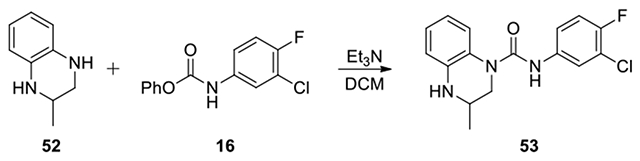
Due to the regioselectivity, 3-substituted 3,4-dihydroquinoxaline-1(2H)-carboxamides can be readily prepared as the major products by reacting the 3-substituted 1,2,3,4-tetrahydroquinoxalines with the active ester 16 directly (Scheme 3).
Scheme 3.
Synthesis of 3-substituted urea 53.
Both hydrophilic (e.g. morpholine)40 and hydrophobic (such as 3,3-difluropyrrolidine)41 substitutions out of the 6- position of the 1,4-dihydropyridine core of HAPs were found to be tolerated and beneficial to the activities in the optimization of the HAP scaffolds. Encouraged by these results, we also tried to install these substituents to the 1,2,3,4-tetrahydroquinoxaline scaffold. Compounds were initially synthesized through a linear route (Scheme 4). Starting from quinoxaline-2-carboxylic acid 54, a coupling reaction with an amine gave an amide 55, which was reduced to 1,2,3,4-tetrahydroquinoxaline 56 with a pendant substituent at position 2. Following regioselective Boc protection, reactions with phosgene and aniline 21 sequentially provided protected urea 60. Deprotection of Boc with HCl gave the final product 61. This route worked well, but it was linear and less productive, as it could only evaluate the SAR on anilines.
Scheme 4.
Linear synthesis of 2-substituted urea 61.
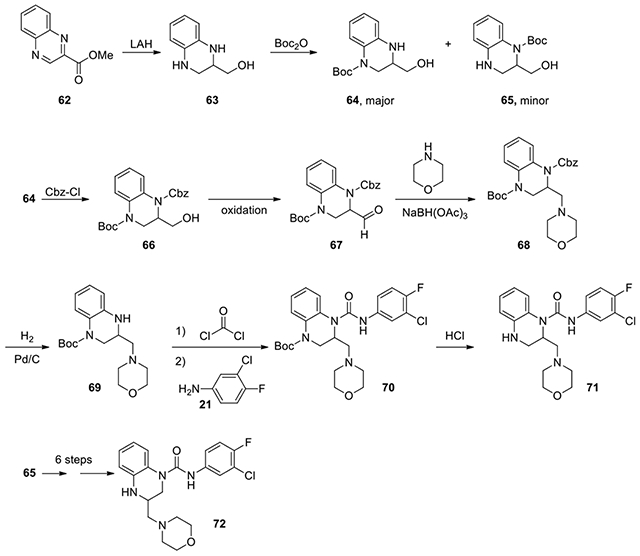
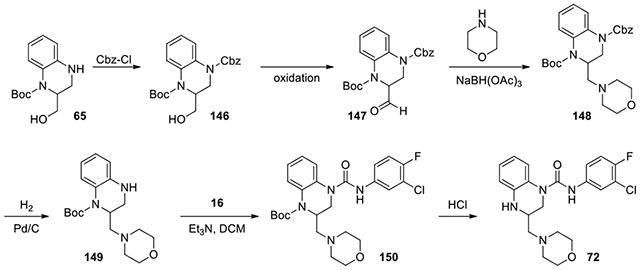
To improve productivity and make the synthesis versatile, allowing changes in both the anilines and substitutions at position 2, a common intermediate 67 was prepared (Scheme 5). Compounds 71 and 72 were prepared in 8 steps from methyl quinoxaline-2-carboxylate 62. Reduction with lithium aluminum hydride gave (1,2,3,4-tetrahydroquinoxalin-2-yl)methanol 63, which was treated with Boc2O leading to a major product 64 and a minor regioisomer 65. The free NH in 64 was protected with a Cbz group. The following Swern oxidation converted the alcohol in 66 to an aldehyde in 67. Reductive amination introduced a pendant group at position 2 of 68. Next, selective hydrogenation removed the Cbz protecting group at position 1, and the free NH in 69 was treated with isocyanate 15 to form the urea 70, which afforded compound 71 after the final deprotection with HCl to remove the Boc group. Compound 72, the regioisomer of 71, was made in a similar procedure starting from 65.
Scheme 5.
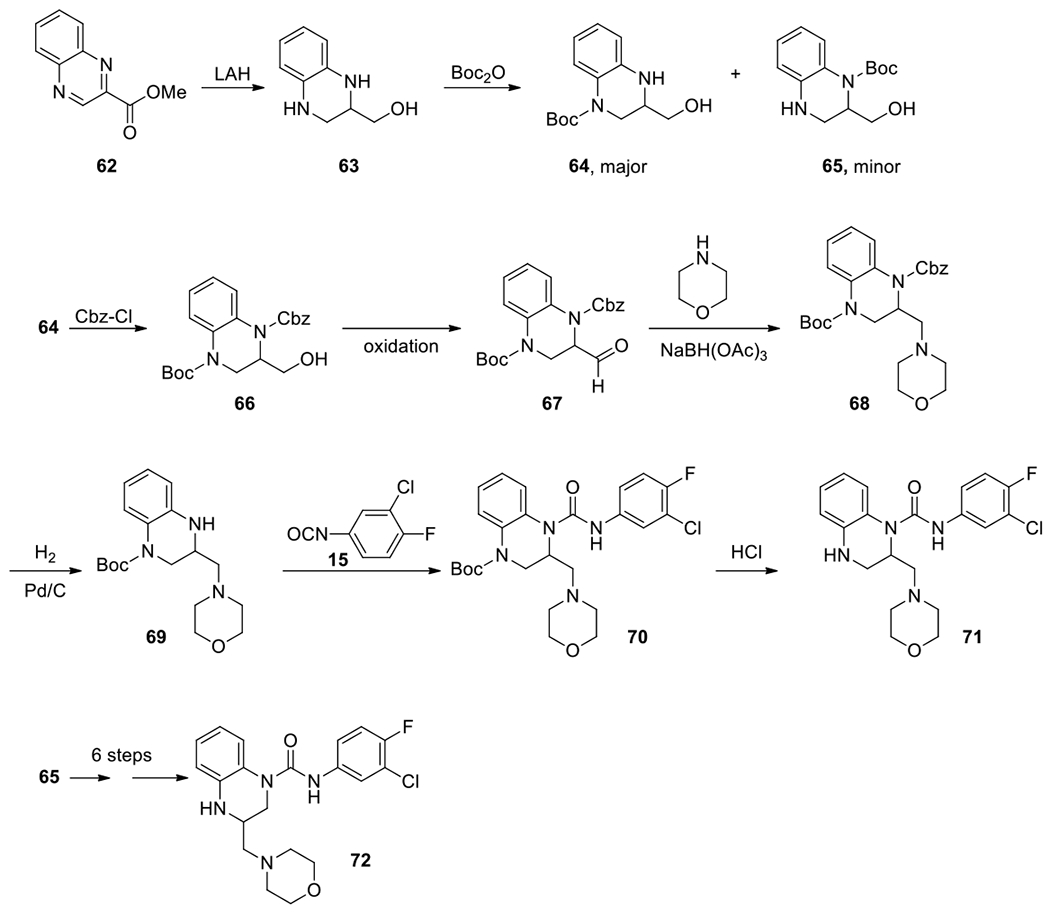
Synthesis of regioisomers at 2- and 3-positions.
Evaluation of these compounds demonstrated that substitutions at position 2 are much better tolerated than those at position 3 (73 vs 53, 74 vs 79). A phenyl group at the 2-position retained sub-micromolar activity (74). The most active compound was with a 4,4-difluoropiperidinyl-methyl group (61), which increased the potency more than two-fold. It is worth noting that a hydrophilic morpholine (71) reduced the activity significantly, which could suggest that hydrophobic moieties are preferred at this position. However, it is also true that hydrophobic 76 also causes a loss of activity, so it is possible that there are multiple factors at play, such as steric factors within the binding pocket.
With a favorable 4,4-difluoropiperidin-1-yl)methyl at position 2, we re-examined the aniline in 61 with anilines selected from a small set. The synthesis started from the aldehyde 67 as previously described. The intermediate 58 was prepared through reductive amination. Subsequent treatment with isocyanates made in situ provided a protected urea 81, which was converted to a desired product 82 after acidic deprotection (Scheme 6).
Scheme 6.
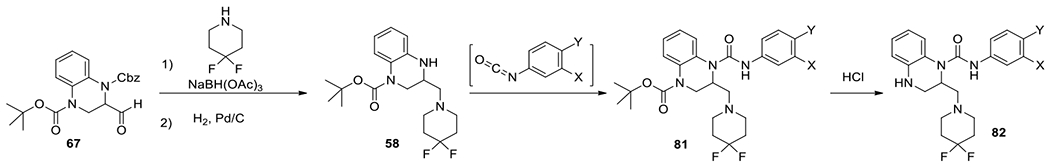
Synthesis of 2-((4,4-difluoropiperidin-1-yl)methyl)-dihydroquinoxaline-1(2H)-carboxamide.
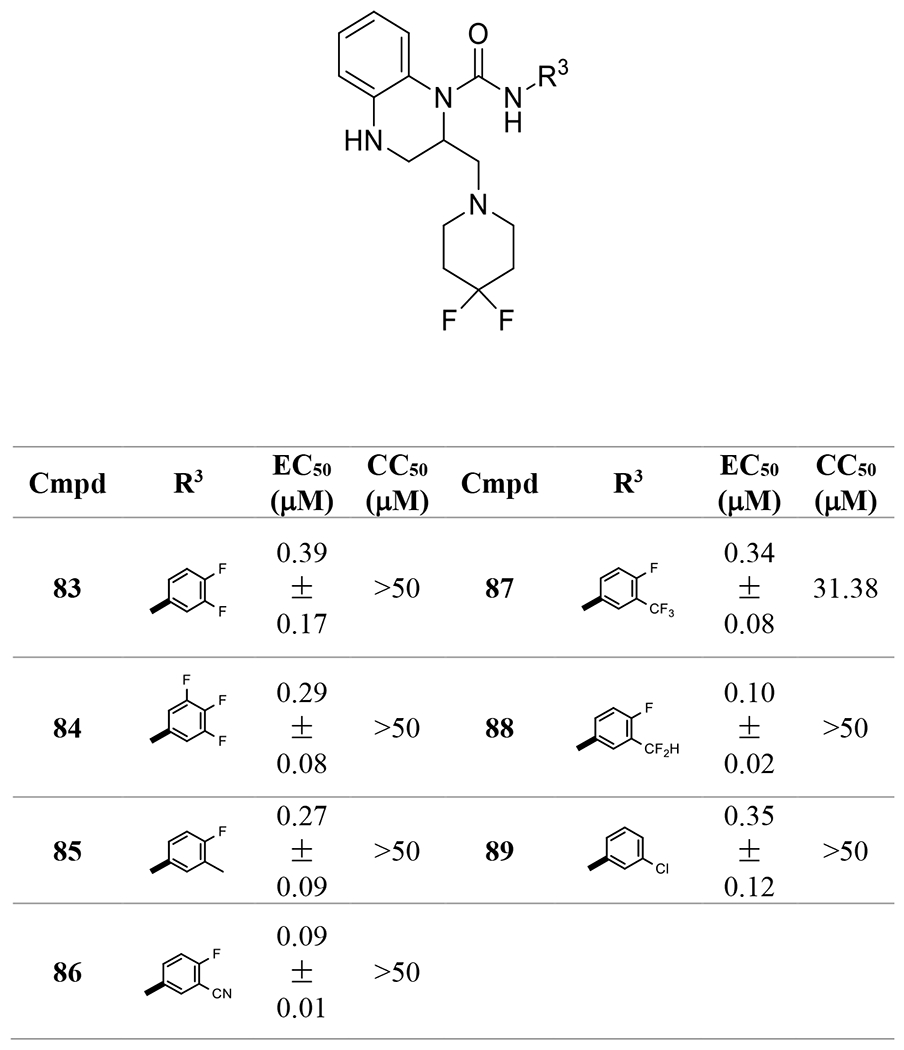
These compounds were also evaluated for their anti-HBV activities in the AML 12HBV10 cell line. Two anilines, 4-fluoro-3-cyanoaniline and 4-fluoro-3-(difluoromethyl)aniline in products 86 and 88, contributed to improved EC50s down to around 100 nM (Table 4). On the other hand, compounds 83-85, 87, and 89, with other anilines, had moderate sub-micromolar EC50s.
Table 4.
Aniline optimization with 2-substitution
|
II. Bis-ureas
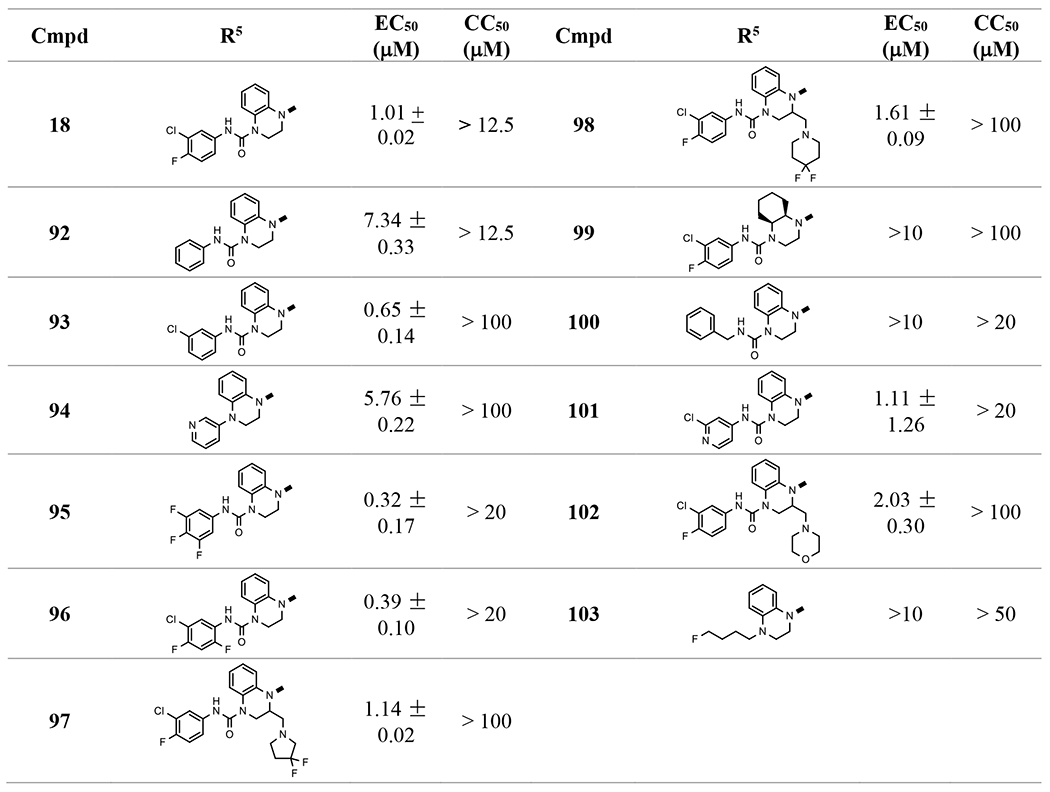
In the preparation of 1,2,3,4-tetrahydroquinoxaline based ureas (Scheme 1A), we also obtained bis-urea 18, which showed interesting low micromolar EC50 (1.01 μM, Table 5). 18 is a unique molecule and its structure appeared to be new as a capsid assembly modulator. Therefore, we investigated derivatives of this linear molecule. Anilines, benzylamines, and substituted amines were connected to the 4-N of 17, 61, or other intermediates prepared above through urea or alkyl moieties. The results showed that several bis-ureas, such as 95 and 96, had sub-micromolar activities. However, the pedant group at position 2 of the tetrahydroquinoxaline ring in 97 and 98, which we found to have a favorable effect in the mono-urea series, did not help to improve the activity significantly in this series, indicating there are different binding poses between these two chemical series.
Table 5.
Optimization of bis-ureas
|
III. Further analysis of selected compounds.
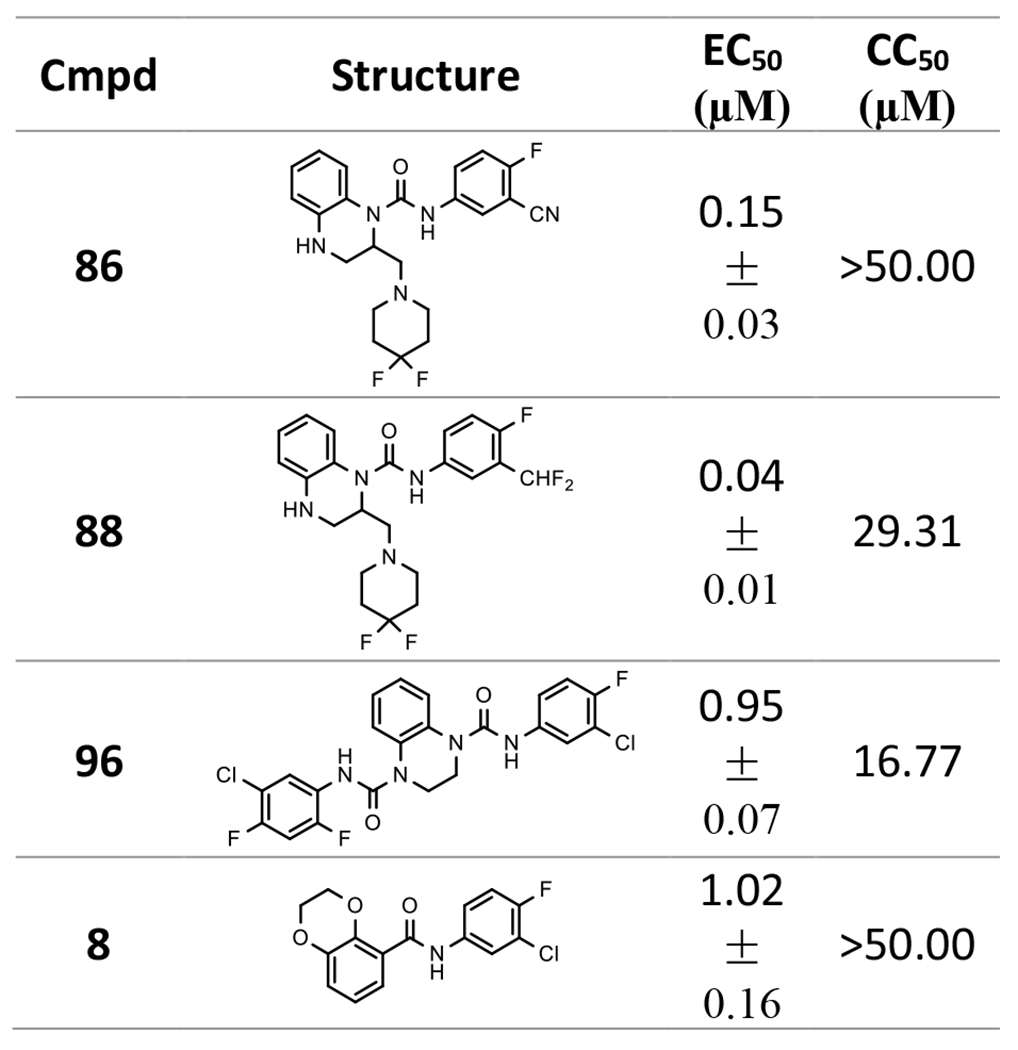
The anti-HBV activity of 86, 88, and 96 were further assessed in a human hepatoma-derived cell line HepDES19, along with 8 as a control. They demonstrated activities that inhibit HBV DNA replication in a concentration-dependent manner with EC50 values of 0.15, 0.04, and 0.95 μM respectively (Table 6), which are different from their EC50 values in AML12HBV10 cells (Tables 4–5), indicating HepDES19 has different sensitivities to these compounds.
Table 6.
Antiviral activity of selected compounds in HepDES19 cells
|
To ascertain that the newly synthesized compounds inhibit HBV replication by disruption of pgRNA encapsidation, two representative new compounds, 17 and 18, were tested in AML12HBV10 cells for their effects on capsid assembly, pgRNA encapsidation, and viral DNA synthesis, along with viral DNA polymerase inhibitor Entecavir (ETV), type I CpAM Bay 41-4109 (2), and an SBA chemotype of type II CpAM (DVR-23) (3)15 as controls. As anticipated, ETV treatment did not alter the levels of total viral RNA (Fig. 4A), core protein (Fig. 4B), encapsidated pgRNA (Fig. 4C), and capsids (Fig. 4D), but drastically reduced the amounts of capsid-associated viral DNA (Fig. 4D) and DNA replication intermediates (Fig. 4E). Also as expected, while Bay 41-4109 prevented the assembly of capsids (Fig. 4D), DVR-23 treatment induced the accumulation of capsids with faster electrophoresis mobility (Fig. 4D). Both Bay 41-4109 and DVR-23 significantly reduced the amounts of encapsidated pgRNA and as a consequence, viral DNA replication was inhibited (Fig. 4E). Similar to the pattern observed with DVR-23, treatment of AML12HBV10 cells with 17 and 18 did not alter the amounts of total viral RNA (Fig. 4A) and core protein (Fig. 4B), but 17 and 18 induced the assembly of capsids with faster electrophoresis mobility in a native agarose gel (Fig. 4D) and drastically reduced the amounts of encapsidated pgRNA (Fig. 4C) and viral DNA replication intermediates (Fig. 4E). Therefore, the mode of action of the newly synthesized compounds is consistent with type II CpAMs that induce the assembly of empty capsids devoid of viral pgRNA.
Conclusions
In summary, as part of our continuing efforts toward the discovery of novel benzamides with an additional fused ring β, γ to an amide bond as HBV capsid assembly modulators, we have screened a series of fused bicyclic scaffolds and translocated the connection of the amide bond from the benzene ring to the fused ring. The 1,2,3,4-tetrahydroquinoxaline core was identified as a good platform to build up phenyl ureas as bioisosteres of benzamides. SAR studies at three positions led to the discovery of compound 88, which was found to inhibit HBV potently in mouse and human hepatocytes (EC50 = 0.1 and 0.04 μM respectively). Moreover, novel bis-ureas based on the 1,2,3,4-tetrahydroquinoxaline motif were found to also be modulators against HBV capsid assembly at a sub-micromolar level. The modes of action for both of these two chemotypes of HBV capsid assembly modulators were found to fall into the typical type II CpAM, since they misdirect the Cp dimers to assemble empty capsids devoid of pgRNA and thus preclude the synthesis of viral DNA.
Materials and experimental details
Chemistry
All reagents and solvents used were purchased from commercial sources. Reactions were carried out under argon atmosphere. Flash column chromatography was performed on either a CombiFlash Rf+ or a CombiFlash Companion using the appropriate size Teledyne ISCO columns (20-40 microns or 40-60 microns) and prepacked silica gel-filled cartridges. Preparative high-performance liquid chromatography (HPLC) was performed using Gilson HPLC system with 331 and 332 pumps, a UV/VIS-155 detector, and a GX-271 liquid handler. Purifications were performed on a Phenomenex Luna LC Column (5 μm C18 100 Å, 150 x 21.2 mm). 1H NMR spectra were recorded on a 300 MHz INOVA VARIAN spectrometer. Chemical shifts values are given in ppm and referred against the internal standard of TMS (tetramethylsilane). The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet and dd, doublet of doublets. The coupling constants (J) are reported in Hertz (Hz). Mass Spectra were obtained on an Agilent 6120 mass spectrometer with electrospray ionization source (1200 Aligent LC-MS spectrometer, Positive). Mobile phase flow was 1.0 mL/min with a 3.0 min gradient from 20% aqueous media (0.1% formic acid) to 95% CH3CN (0.1% formic acid) and a 9.0 min total acquisition time. All the tested compounds possess a purity of at least 95%, which was determined by LC/MS Data recorded using an Agilent 1200 liquid chromatography and Agilent 6120 mass spectrometer, and further supported by clean NMR spectra. HRMS was were analyzed by direct infusion using constant flow of CH3CN:H2O / 9:1 on Bruker micrOTOF II mass spectrometer using ESI method in positive mode in the Mass Spectrometry and Proteomics Facility of University of Notre Dame.
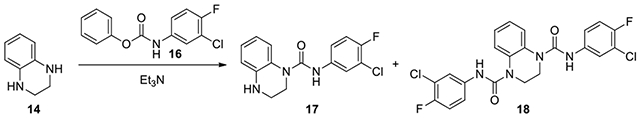
N-(3-chloro-4-fluorophenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (17):
A solution of 1,2,3,4-tetrahydroquinoxaline (182 mg, 1.36 mmol, 1.2 eq) and excess of Et3N (0.47 mL, 3.39 mmol, 3 eq) in DCM (4 mL) was added to a stirred solution of phenyl (3-chloro-4-fluorophenyl)carbamate (300 mg, 1.13 mmol, 1eq), which was synthesized by reacting 3-chloro-4-fluoroaniline with phenyl chloroformate.34–35 Upon completion, the reaction mixture was concentrated and the residue was purified by CombiFlash with a gradient of ethyl acetate in hexanes from 1:9 to 3:7 to afford 17 (309 mg, 89%): 1H NMR (300 MHz, CDCl3): δ (ppm) 7.55 (dd, J = 6.3, 2.7 Hz, 1H), 7.26-7.13 (m, 2H), 7.06-6.98 (m, 2H), 6.75-6.63 (m, 2H), 3.86-3.76 (m, 2H), 3.48-3.40 (m, 2H); Calculated for C15H13ClFN3O, 305.07; observed (M+H)+ 306.3; and minor 18 (10 mg).
N1,N4-bis(3-chloro-4-fluorophenyl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (18):
1H NMR (300 MHz, CDCl3): δ (ppm) 7.59-7.51 (m, 4H), 7.32-7.19 (m, 4H), 7.12-7.04 (m, 2H), 7.00 (s, 2H), 4.02 (s, 4H); Calculated for C22H16C12F2N4O2, 476.06; observed (M+H)+ 477.4.
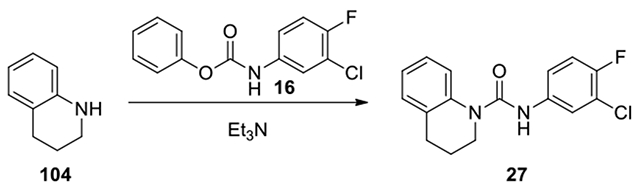
N-(3-chloro-4-fluorophenyl)-3,4-dihydroquinoline-1(2H)-carboxamide (27):
According to the procedure for the preparation of 17, 1,2,3,4-tetrahydroquinoline (104, 24 mg, 0.18 mmol) was treated with phenyl (3-chloro-4-fluorophenyl)carbamate (16, 40 mg, 0.15 mmol) to afford 27 (24 mg, 53%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.57-7.52 (m, 1H), 7.32-7.10 (m, 5H), 7.08-7.03 (m, 1H), 7.03-6.96 (s. broad, 1H), 3.85-3.77 (m, 2H), 2.84-2.75 (m, 2H), 2.06-1.94 (m, 2H); Calculated for C16H14ClFN2O, 304.08; observed (M+H)+ 305.1.
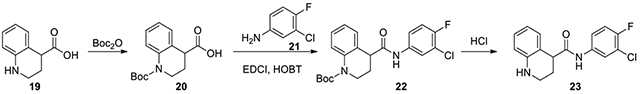
N-(3-chloro-4-fluorophenyl)-1,2,3,4-tetrahydroquinoline-4-carboxamide (23):
1.2.3.4-tctrahvdroquinolinc-4-carboxylic acid (19, 100 mg, 0.564 mmol) was dissolved in 2 mL THF and put on an ice bath at 0°C. 1 mL of 2N NaOH was added dropwise. After 10 minutes, di-tert-butyl dicarbonate (246 mg, 1.129 mmol) in 1 mL THF was added dropwise while vigorously stirring. The reaction was allowed to go to room temperature and left until completion determined with LC-MS. Then, several drops of 2N HCl was added until the pH was 5-6, and the mixture was extracted with EtOAc and DCM several times. The organic phases were combined and concentrated. The residue was purified on CombiFlash with a gradient of ethyl acetate in hexanes from 0:1 to 33:67 to provide the Boc protected acid 20. This acid was then dissolved in 3 mL DMF, and 3-chloro-4-fluoroaniline (21, 82 mg, 0.564 mmol), EDC·HCl (108 mg, 0.564 mmol), HOBt·H2O (86 mg, 0.564 mmol), and Et3N (2 eq) was added to the reaction, which was stirred overnight. It was then diluted with DCM and washed with H2O and brine. The organic phase was concentrated, and the residue was purified on CombiFlash with a gradient of ethyl acetate in hexanes from 0:1 to 2:8 to afford the amide 22. The Boc protected amide 22 was then dissolved in 1:1 MeOH to 4M HCl in dioxane. After 3.5 hours, the reaction was done and the mixture was concentrated. The residue was purified by HPLC with a gradient of acetonitrile in water from 25% to 100% to afford 23 (13 mg, 8%) after lyophilization. 1H NMR (300 MHz, DMSO): δ (ppm) 7.97 (dd, J = 6.9, 2.4 Hz, 1H), 7.56-7.49 (m, 1H), 7.38 (t, J = 9.3 Hz, 1H), 6.98-6.90 (m, 2H), 6.59-6.47 (m, 2H), 3.81-3.74 (m, 1H), 3.60-3.40 (m, 1H), 3.22-3.13 (m, 1H), 2.12-1.90 (m, 1H); Calculated for C16H14ClFN2O, 304.08; observed (M+H)+ 305.3.
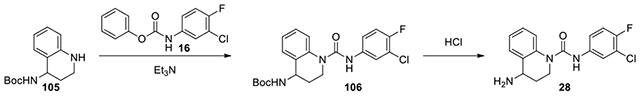
4-Amino-N-(3-chloro-4-fluorophenyl)-3,4-dihydroquinoline-1(2H)-carboxamide (28):
According to the procedure for the preparation of 17, tert-butyl (1,2,3,4-tetrahydroquinolin-4-yl)carbamate (105, 30 mg, 0.121 mmol) was treated with phenyl (3-chloro-4-fluorophenyl)carbamate (16, 35 mg, 0.133 mmol), to provide 106, which was then dissolved in 1:1 MeOH to 4M HCl in dioxane. After the reaction was complete determined by LC-MS, the mixture was concentrated and co-evaporated with MeOH thrice to remove excessive HCl and ether once to afford 28. 1H NMR (300 MHz, CD3OD): conformer mixture, δ (ppm) 7.66 (dd, J = 6.3, 2.4 Hz, 1H), 7.54-7.30 (m, 4H), 7.24-7.13 (m, 2H), 4.07-3.97 (m, 1H) 3.85-3.56 (m), 2.48-2.30 (m, 1H), 2.26-2.10 (m, 1H); Calculated for C16H15ClFN3O, 319.09; observed (M+H)+ 320.3.
N-(3-chloro-4-fluorophenyl)-3-oxo-3,4-dihydroquinoxaline-1(2H)-carboxamide (29):
According to the procedure for the preparation of compound 17, 3,4-dihydroquinoxalin-2(1H)-one (107, 134 mg, 0.906 mmol) was treated with phenyl (3-chloro-4-fluorophenyl)carbamate (16, 200 mg, 0.755 mmol) to afford 29 (115 mg, 48%). 1H NMR (300 MHz, DMSO): δ (ppm) 7.72 (dd, J = 6.7, 2.6 Hz, 1H), 7.46-7.38 (m, 2H), 7.39-7.29 (m, 1H), 7.16-7.08 (m, 1H), 7.06-6.98 (m, 2H), 4.26 (s, 2H); Calculated for C15H11ClFN3O2, 319.05; observed (M+H)+ 320.3.
N-(3-chloro-4-fluorophenyl)indoline-1-carboxamide (30):
According to the procedure for the preparation of compound 17, indoline (108, 18 mg, 0.15 mmol) was treated with phenyl (3-chloro-4-fluorophenyl)carbamate (16, 40 mg, 0.15 mmol) to afford 30 (45.3 mg, 100%) as a light yellow solid. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.90-7.84 (m, 1H), 7.65-7.59 (m, 1H), 7.30-7.19 (m, 3H), 7.12-7.04 (m, 1H), 7.02-6.95 (m, 1H), 6.45 (s, broad, 1H), 4.11-4.02 (m, 2H), 3.28-3.20 (m, 2H); Calculated for C15H12ClFN2O, 290.06; observed (M+H)+ 291.2.
N-(3-chloro-4-fluorophenyl)-2,3-dihydro-1H-pyrrolo[2,3-b]pyridine-1-carboxamide (31):
According to the procedure for the preparation of compound 17, 2,3-dihydro-1H-pyrrolo[2,3-b]pyridine (109, 18 mg, 0.15 mmol) was treated with phenyl (3-chloro-4-fluorophenyl)carbamate (16, 40 mg, 0.15 mmol) to afford 31 (30 mg, 69%) as a light yellow solid. 1H NMR (300 MHz, CDCl3): δ (ppm) 11.50 (s, 1H), 8.08-8.03 (m, 1H), 7.80-7.75 (m, 1H), 7.51-7.41 (m, 2H), 7.12-7.04 (m, 1H), 6.85-6.83 (m, 1H), 4.23-4.14 (m, 2H), 3.18-3.08 (m, 2H); Calculated for C14H11ClFN3O, 291.06; observed (M+H)+ 292.3.
N-(3-chloro-4-fluorophenyl)-2,3,4,5-tetrahydro-1H-benzo[b]azepine-1-carboxamide (32):
According to the procedure for the preparation of compound 17, 2,3,4,5-tetrahydro-1H-benzo[b]azepine (110, 26 mg, 0.18 mmol) was treated with phenyl (3-chloro-4-fluorophenyl)carbamate (16, 40 mg, 0.15 mmol) to afford 32 (37 mg, 77%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.50-7.45 (m, 1H), 7.37-7.24 (m, 4H), 7.14-7.06 (m, 1H), 7.02-6.96 (m, 1H), 6.19 (s, broad, 1H), 4.75-4.60 (m, 1H), 2.95-2.60 (m, 3H), 2.10-1.75 (m, 3H), 1.50-1.30 (m, 1H); Calculated for C17H16ClFN2O, 318.09; observed (M+H)+ 319.3.
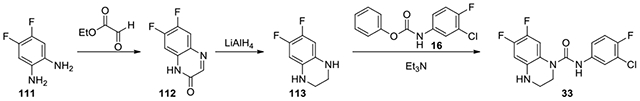
N-(3-chloro-4-fluorophenyl)-6,7-difluoro-3,4-dihydroquinoxaline-1(2H)-carboxamide (33):
50% ethyl glyoxylate in toluene (0.16 mL, 1.527 mmol) was added to 4,5-difluorobenzene-1,2-diamine (111, 200 mg, 1.388 mmol) in 20 mL MeOH. More ethyl glyoxylate (0.16 mL) was added after 3 hours, and the reaction was stirred overnight. After filtering, the solid (141 mg, 0.77 mmol) was dissolved in 8 mL THF and treated with 1M lithium aluminum hydride in THF (3.1 mL, 3.1 mmol). Upon completion, the reaction was quenched with EtOAc and MeOH, and diluted with ether. It was stirred with Na2SO4·10H2O overnight, and then filtered to afford 122 mg (0.719 mmol) intermediate 113. The intermediate was then treated with phenyl (3-chloro-4-fluorophenyl)carbamate (16, 229 mg, 0.863 mmol) according to the procedure for the preparation of compound 17,. After stirring overnight, the reaction was diluted with EtOAc and washed with 2N HCl twice, saturated NaHCO3, and brine. It was purified by HPLC with a gradient of acetonitrile in water from 50% to 90% to afford 33 (121 mg, 26%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.52 (dd, J = 6.6, 2.7 Hz, 1H), 7.21-7.14 (m, 1H), 7.11-7.01 (m, 2H), 6.47 (dd, J = 11.4, 7.2 Hz, 1H), 3.83-3.75 (m, 2H), 3.46-3.40 (m, 2H); Calculated for C15H11ClF3N3O, 341.05; observed (M+H)+ 342.3.
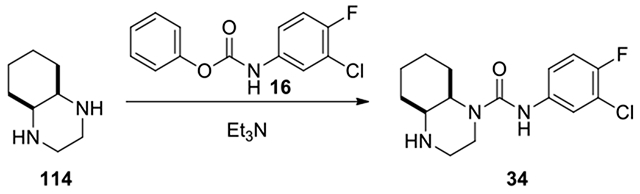
Cis-N-(3-chloro-4-fluorophenyl)octahydroquinoxaline-1(2H)-carboxamide (34):
According to the procedure for the preparation of compound 17, decahydroquinoxaline (114, 100 mg, 0.713 mmol) in DMF was treated with phenyl (3-chloro-4-fluorophenyl)carbamate (16, 189 mg, 0.713 mmol), to afford 34. 1H NMR (300 MHz, CD3OD): conformer mixture, δ (ppm) 7.59 (dd, J = 6.9, 2.7 Hz, 1H), 7.33-7.27 (m, 1H), 7.14 (t, J = 9.1 Hz, 1H), 4.45-4.35 (m, 1H), 4.25-4.15 (m, 1H), 3.76-3.70 (m, 1H), 3.70-3.63 (m, 1H), 3.60-3.53 (m, 2H), 3.53-3.32 (m), 3.25-3.13 (m), 2.10-1.85 (m, 4H), 1.80-1.58 (m, 2H), 1.58-1.36 (m, 2H); Calculated for C15H19ClFN3O, 311.12; observed (M+H)+ 312.4.
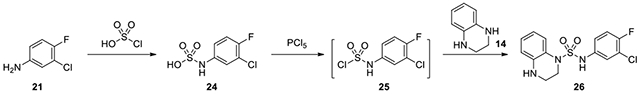
N-(3-chloro-4-fluorophenyl)-3,4-dihydroquinoxaline-1(2H)-sulfonamide (26):
Chlorosulfuric acid (46μL, 0.687 mmol) in 0.5 mL DCM was added dropwise to 3-chloro-4-fluoroaniline (21, 100 mg, 0.687 mmol) in 2 mL DCM at 0°C. After 30 minutes, the reaction was allowed to go to room temperature, and after 2 more hours, precipitate was filtered and dried on high vacuum. The solid was suspended in 3 mL toluene and phosphorus pentachloride (143 mg, 0.687 mmol) was added. The reaction was heated to 80°C for 6 hours, and then filtered. The filtrate was redissolved in pyridine and 1,2,3,4-tetrahydroquinoxaline was added. Upon completion, the reaction was diluted with EtOAc and washed with 2N HCl thrice, saturated NaHCO3 once, and brine once. The concentrated residue was purified on CombiFlash with a gradient of ethyl acetate in hexanes from 1:9 to 3:7 to afford 26. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.46-7.38 (m, 1H), 7.02-6.93 (m, 3H), 6.90-6.82 (m, 1H), 6.71-6.63 (m, 1H), 6.54-6.48 (m, 1H), 3.73-3.66 (m, 2H), 3.14-3.05 (m, 2H); Calculated for C14H13ClFN3O2S, 341.04; observed (M+H)+ 342.3.
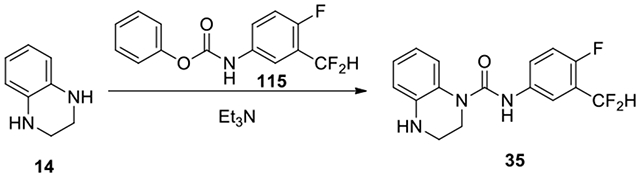
N-(3-(difluoromethyl)-4-fluorophenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (35):
3-(difluoromethyl)-4-fluoroaniline (39 mg, 0.24 mmol, 1 eq) was reacted with 1.2 eq of phenyl chloroformate in 1:1 EtOAc to saturated aqueous NaHCO3 overnight. The organic phase was concentrated and the resulting carbamate 115 was treated with 1,2,3,4-tetrahydroquinoxaline (14, 17 mg, 0.13 mmol) and Et3N (0.05 ml) in DCM overnight. The mixture was concentrated, and the residue was purified by HPLC with a gradient of acetonitrile in water from 50% to 59% to afford 35 (40.9 mg, 74%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.58-7.52 (m, 1H), 7.49-7.44 (m, 1H), 7.34-7.27 (m, 1H), 7.25-7.18 (m, 1H), 7.12-7.02 (m, 2H), 6.80-6.65 (m, 2H), 3.89-3.82 (m, 2H), 3.52-3.45 (m, 2H); Calculated for C14H16F3N3O, 321.11; observed (M+H)+ 322.5.
N-(4-fluoro-3-methylphenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (36):
According to the procedure for the preparation of compound 35, 4-fluoro-3-methylaniline (31 mg, 0.24 mmol) was treated with phenyl chloroformate and followed the same procedure to afford 36 (38.9 mg, 76%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.24-7.13 (m, 2H), 7.13-6.99 (m, 2H), 6.99-6.88 (m, 1H), 6.78-6.66 (m, 2H), 3.90-3.80 (m, 2H), 3.53-3.43 (m, 2H), 2.24 (s, 3H); Calculated for C16H16FN3O, 285.13; observed (M+H)+ 286.5.
N-(4-fluoro-3-(trifluoromethyl)phenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (37):
According to the procedure for the preparation of compound 35, 4-fluoro-3-(trifluoromethyl)aniline (44 mg, 0.24 mmol) was treated with phenyl chloroformate, and continued with the same procedure to afford 37 (32.1 mg, 79%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.62-7.50 (m, 2H), 7.24-7.02 (m, 3H), 6.81-6.68 (m, 2H), 3.90-3.82 (m, 2H), 3.54-3.43 (m, 2H); Calculated for C16H13F4N3O, 339.10; observed (M+H)+ 340.5.
N-(3-cyano-4-fluorophenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (38):
According to the procedure for the preparation of compound 35, 5-amino-2-fluorobenzonitrile (33 mg, 0.24 mmol) was treated with phenyl chloroformate and followed the same procedure to afford 38 (40.3 mg, 77%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.74-7.66 (m, 1H), 7.64-7.53 (m, 1H), 7.22-7.13 (m, 2H), 7.13-7.04 (m, 1H), 6.81-6.68 (m, 2H), 3.90-3.81 (m, 2H), 3.55-3.43 (m, 2H); Calculated for C16H13FN4O, 296.11; observed (M+H)+ 297.5.
N-(3,4,5-trifluorophenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (39):
According to the procedure for the preparation of compound 35, 3,4,5-trifuloroaniline (18 mg, 0.14 mmol) was treated with phenyl chloroformate, and continued with the same procedure and purified on CombiFlash with a gradient of ethyl acetate in hexanes from 1:9 to 3:7 to afford 39 (22.3 mg, 54%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.16-7.00 (m, 4H), 6.76-6.64 (m, 2H), 3.86-3.78 (m, 2H), 3.50-3.43 (m, 2H); Calculated for C15H12F3N3O, 307.09; observed (M+H)+ 308.3.
N-(5-chloro-2-fluorophenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (40):
To a solution of 1,2,3,4-tetrahydroquinoxaline (14, 67 mg, 0.50 mmol, 1 eq) and Et3N (1 eq) in DCM (2 mL), triphosgene (0.33 eq) was added and the mixture was stirred at 0°C for 1.5 h. Then the aniline (2 eq) was added, and the mixture was stirred at room temperature. After confirmation of disappearance of the starting materials, the mixture was evaporated to dryness. The residue was dilute with AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel, eluting with EtOAc /Hexane from 0:1 to 4:6 to give product 40. (14 mg, 9% yield, white solid). 1H NMR (300 MHz, Chloroform-d) δ 7.44 – 7.22 (m, 2H), 7.19 – 6.95 (m, 3H), 6.88 – 6.53 (m, 3H), 4.16 (s, 1H), 3.96 – 3.78 (m, 2H), 3.47 (td, J = 5.1, 2.7 Hz, 2H); Calculated for C15H13ClFN3O, 305.07; observed (M+H)+ 306.3.
N-(3-chloro-4,5-difluorophenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (41):
According to the procedure for the preparation of compound 35, 3-chloro-4,5-difluoroaniline (15 mg, 0.09 mmol) was treated with phenyl chloroformate, and continued with the same procedure and purified on CombiFlash with a gradient of ethyl acetate in hexanes from 0:1 to 25:75 to afford 41 (15.4 mg, 59%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.42-7.34 (m, 1H), 7.17-7.11 (m, 2H), 7.09-7.01 (m, 1H), 6.76-6.64 (m, 2H), 3.86-3.79 (m, 2H), 3.50-3.44 (m, 2H); Calculated for C15H12ClF2N3O, 323.06; observed (M+H)+ 324.3.
N-(4-azido-3-chlorophenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (42):
To a solution of 1,2,3,4-tetrahydroquinoxaline (14, 40 mg, 0.30 mmol, 1.5 eq) and Et3N (1.1 eq) in DCM (2 mL), the compound phenyl (4-azido-3-chlorophenyl)carbamate (121, 57 mg, 1 eq) was added and the mixture was stirred for 3 h. After confirmation of disappearance of the starting materials, the mixture was evaporated to dryness. The residue was diluted with AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4. and filtered. The filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel, eluting with EtOAc /Hexane from 0:1 to 2:8 to give product 42. (30 mg, 91% yield, yellow solid). 1H NMR (300 MHz, Chloroform-d) δ 7.52 (d, J = 2.4 Hz, 1H), 7.33 (dd, J = 8.7, 2.4 Hz, 1H), 7.23 (s, 1H), 7.17 (dd, J = 7.8, 1.5 Hz, 1H), 7.08 (d, J = 8.7 Hz, 1H), 7.05 – 6.98 (m, 2H), 6.79 – 6.69 (m, 1H), 6.67 (dd, J = 8.1, 1.2 Hz, 1H), 4.17 (s, 1H), 3.84 (dd, J = 5.7, 4.5 Hz, 2H), 3.47 (td, J = 5.1, 3.0 Hz, 2H); Calculated for C15H13ClN6O, 328.08; observed (M+H)+ 329.3.
N-(3-chlorophenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (43):
According to the procedure for the preparation of compound 35, 3-chloro-4,5-difluoroaniline (36.8 mg, 0.24 mmol) was treated with phenyl chloroformate, and continued the same procedure to afford 43 (40.6 mg, 59%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.48-7.41 (m, 1H), 7.23-7.16 (m, 3H), 7.10-7.00 (m, 2H), 6.78-6.66 (m 2H), 3.89-3.82 (m, 2H), 3.52-3.44 (m, 2H); Calculated for C15H14ClN3O, 287.08; observed (M+H)+ 288.4.
N-(3-chlorobenzyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (44):
To a solution of 1,2,3,4-tetrahydroquinoxaline (14, 67 mg, 0.50 mmol, 1 eq) and Et3N (1 eq) in DCM (2 mL), triphosgene (49 mg, 0.16 mmol, 0.33 eq) was added and the mixture was stirred at 0°C for 1.5 h. Then (3-chlorophenyl)methanamine (2 eq) was added and the mixture was stirred at room temperature. After confirmation of disappearance of the starting materials, the mixture was evaporated to dryness. The residue was diluted with AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4. and filtered. The filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel, eluting with EtOAc /Hexane from 0:1 to 3:7 to give product 44. (25 mg, 17% yield, white solid). 1H NMR (300 MHz, Chloroform- d) δ 7.31 – 7.21 (m, 3H), 7.20 – 7.12 (m, 2H), 7.07 (s, 1H), 7.05 – 6.93 (m, 1H), 6.84 – 6.69 (m, 2H), 5.86 (s, 1H), 4.43 (d, J = 5.1 Hz, 2H), 4.17 – 3.54 (m, 2H), 3.52 – 3.44 (m, 2H); Calculated for C16H16ClN3O, 301.10; observed (M+H)+ 302.3.
2-(3-chloro-4-fluorophenyl)-1-(3,4-dihydroquinoxalin-1(2H)-yl)ethanone (45):
2-(3-chloro-4-fluorophenyl)acetic acid (124, 20 mg, 0.11 mmol) was dissolved in thionyl chloride and refluxed at 75°C overnight. The solution was concentrated and co-evaporated with dry DCM thrice. The residue was redissolved in DCM, and 1,2,3,4-tetrahydroquinoxaline (14, 14 mg, 0.10 mmmol) and Et3N (0.05 ml) was added. Upon completion, the reaction was purified by HPLC with a gradient of acetonitrile in water from 45% to 55% to afford 45 (9.0 mg, 28%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.20-7.12 (m, 1H), 7.04-6.90 (m, 4H), 6.66-6.53 (m, 2H), 3.85-3.76 (m, 4H), 3.38-3.31 (m, 2H); Calculated for C16H14ClFN2O, 304.08; observed (M+H)+ 305.4.
N-(4-(1,1,1,3,3,3-hexafluoro-2-hydroxypropan-2-yl)phenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (46):
According to the procedure for the preparation of compound 35, 2-(4-aminophenyl)-1,1,1,3,3,3-hexafluoropropan-2-ol (126, 20 mg, 0.08 mmol, 1 eq) was treated with phenyl chloroformate (9 μl, 0.08 mmol, 1 eq), and continued with the same procedure to afford 46 (26.9 mg, 75%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.66-7.56 (m, 2H), 7.54-7.44 (m, 2H), 7.26-7.18 (m, 1H), 7.00-6.90 (m, 1H), 6.79-6.66 (m, 2H), 3.82-3.74 (m, 2H), 3.47-3.38 (m, 2H); Calculated for C18H15F6N3O2, 419.11; observed (M+H)+ 420.5.
N-(3-chloro-4-fluorophenyl)-2-isopropyl-3,4-dihydroquinoxaline-1(2H)-carboxamide (51):
According to the procedure for the preparation of compound 73, 2-isopropyl-1,2,3,4-tetrahydroquinoxaline (47, 48 mg, 0.27 mmol) was treated di-tert-butyl dicarbonate (68.4 mg, 0.31 mmol), and continued with the same procedure to afford 51 (12.3 mg, 13%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.62 (dd, J = 6.3, 2.4 Hz, 1H), 7.28-7.22 (m, 1H), 7.16-7.06 (m, 2H), 7.00-6.92 (m, 1H), 6.70-6.61 (m, 2H), 4.16-4.08 (m, 1H), 3.62-3.52 (m, 1H), 3.28-3.20 (m, 1H), 1.62-1.45 (m, 1H), 1.00 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H); Calculated for C18H19ClFN3O, 347.12; observed (M+H)+ 348.4.
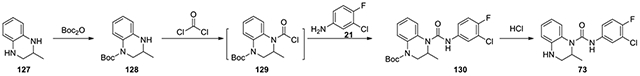
N-(3-chloro-4-fluorophenyl)-2-methyl-3,4-dihydroquinoxaline-1(2H)-carboxamide (73):
di-tert-butyl dicarbonate (352.7 mg, 1.62 mmol, 1.15 eq) was added to 2-methyl-1,2,3,4-tetrahydroquinoxaline (127, 208 mg, 1.40 mmol, 1 eq) in DCM. Upon completion, the reaction was purified by CombiFlash with a gradient of ethyl acetate in hexanes from 0:1 to 1:9 to afford a Boc-protected intermediate 128 (298 mg, 85%). The intermediate 128 (60 mg, 0.242 mmol, 1 eq) and DIPEA (0.13 mL, 0.726 mmol, 3 eq) was dissolved in 2 mL toluene. 15% phosgene in toluene (0.52 mL, 0.726 mmol) was added, and the reaction was stirred for 5.5 hours. The reaction was stirred under vacuum for an hour to get rid of excess phosgene. 3-Chloro-4-fluoroaniline and DIPEA in 1 mL toluene was added dropwise and stirred for several days. Then, it was diluted in EtOAc and washed with 2N HCl, NaHCO3, and brine. The residue was purified by CombiFlash with ethyl acetate in hexanes from 1:9 to 3:7 to provide 130 as an oil, which was dissolved in 1:1 MeOH to 4M HCl in dioxane. After 3 hours, it was concentrated and purified by HPLC with acetonitrile : water from 50% to 80% to afford 73 (63.5 mg, 82%). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.62 (dd, J = 6.6, 2.7 Hz, 1H), 7.30-7.23 (m, 1H), 7.16-7.07 (m, 2H), 6.98-6.91 (m, 1H), 6.72-6.61 (m, 2H), 4.75-4.66 (m, 1H), 3.39-3.32 (m, 1H), 3.28-3.22 (m, 1H), 1.09 (d, J = 6.9 Hz, 3H); Calculated for C16H15ClFN3O, 319.09; observed (M+H)+ 320.4.
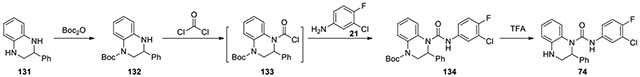
N-(3-chloro-4-fluorophenyl)-2-phenyl-3,4-dihydroquinoxaline-1(2H)-carboxamide (74):
According to the procedures for the preparation of compound 73, 2-phenyl-1,2,3,4-tetrahydroquinoxaline (131, 100 mg, 0.48 mmol) was treated with the same procedure to afford Boc-protected 134 (41.4 mg, 18%). To a solution of Boc-protected urea 134 (27 mg) in 2 mL of DCM, trifluoroacetic acid (2 ml) was added and the mixture was stirred at r.t. for 2 h. After confirmation of disappearance of the starting materials, the reaction was quenched with saturated aqueous NaHCO3 solution at 0 °C. The organic layer was washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by CombiFlash with ethyl acetate in hexanes from 0:1 to 2:8 to give product 74 as a white solid (20.7 mg, 97%). 1H NMR (300 MHz, Chloroform-d) δ 7.61 (dd, J = 6.6, 2.7 Hz, 1H), 7.39 – 7.23 (m, 3H), 7.28 – 7.12 (m, 4H), 7.06 (t, J = 8.7 Hz, 1H), 6.96 (ddd, J = 7.8, 7.2, 1.5 Hz, 1H), 6.79 – 6.64 (m, 1H), 6.59 (dd, J = 8.1, 1.5 Hz, 1H), 5.97 (d, J = 4.2 Hz, 1H), 3.96 (dd, J = 12.3, 2.1 Hz, 1H), 3.71 (dd, J = 12.3, 4.5 Hz, 1H), 3.06 (s, 1H); Calculated for C21H17ClFN3O, 381.10; observed (M+H)+ 382.4.
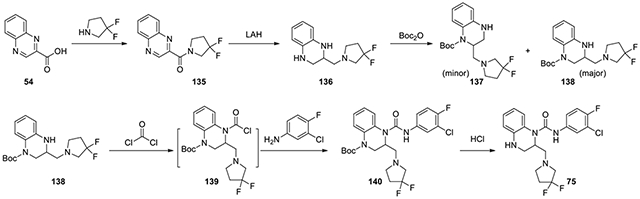
N-(3-chloro-4-fluorophenyl)-2-((3,3-difluoropyrrolidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (75):
1.1 eq 3,3-difluoropyrrolidine (315 mg, 2.2 mmol), 1.2 eq EDC·HCl (458 mg, 2.4 mmol), 1.2 eq HOBt·H2O (367 mg, 2.4 mmol), and DIPEA (0.76 ml, 2.2 eq) was added to quinoxaline-2-carboxylic acid 54 (348 mg, 2.0 mmol, 1 eq) in DMF. After stirring overnight, it was purified by CombiFlash with ethyl acetate in hexanes from 0:1 to 5:5 to provide the amide 135 (286 mg, 54%). The amide 135 (177.0 mg, 0.67 mmol, 1 eq) was dissolved in THF. Lithium aluminium hydride in THF (2.70 ml, 1.0 M, 2.7 mmol) was added and the solution was refluxed at 70°C overnight. Then it was quenched with a few drops of EtOAc, diluted with ether, and treated with Na2SO4·10H2O for 2 hours. The mixture was filtered and the filtrate was concentrated and then redissolved in DCM. Di-tert-butyl dicarbonate (160.8 mg, 0.74 mmol) in DCM was added dropwise. Upon completion, the reaction was purified by CombiFlash with ethyl acetate in hexanes from 0:1 to 4:6 to afford the two regiosomers, 137 and 138. The main isomer 138, tert-butyl 3-((3,3-difluoropyrrolidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate (90 mg, 0.25 mmol), was treated with excess DIPEA (0.13 ml, 0.76 mmol) and 15% phosgene in toluene (0.55 ml, 0.76 mmol) , and the reaction was stirred for 5.5 hours. The reaction was concentrated under vacuum for an hour to get rid of excess phosgene. The residue 139 was dissolved in toluene (1 ml) and treated with 3-chloro-4-fluoroaniline (74 mg, 0.51 mmol) and DIPEA (3 eq) in 1 mL toluene dropwise. The mixture was stirred for over a weekend. Then, it was diluted in EtOAc and washed with 2N HCl, NaHCO3, and brine. The organic phase was concentrated, and the residue was purified by CombiFlash with ethyl acetate and hexanes from 1:9 to 3:7 to give the Boc protected 140, which was dissolved in 1:1 MeOH to 4M HCl in dioxane. After 3 hours, it was purified by HPLC with acetonitrile and water from 40% to 80% to afford 75 (5.2 mg). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.76-7.70 (m, 1H), 7.37-7.28 (m, 1H), 7.28-7.22 (m, 1H), 7.18-7.10 (m, 1H), 7.02-6.94 (m, 1H), 6.76-6.68 (m, 2H), 4.01-3.89 (m, 1H), 3.76-3.64 (m, 1H), 3.47-3.17 (m, 7H), 2.71-2.54 (m, 2H); Calculated for C20H20ClF3N4O, 424.13; observed (M+H)+ 425.5.
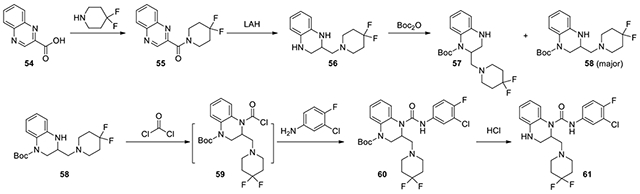
N-(3-chloro-4-fluorophenyl)-2-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (61):
According to the procedure for the preparation of compound 75, starting from 4,4-difluoropiperidine (345 mg, 2.2 mmol) and quinoxaline-2-carboxylic acid (54, 348 mg, 2.0 mmol) through the same amide formation, reduction, and protection procedure to afford two Boc-containing regioisomers 57 and 58. The main isomer 58, tert-butyl 3-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate, was used following the same procedure for introduction of the urea bond to afford 61 (35.6 mg). 1H NMR (300 MHz, CD3OD): δ (ppm) 7.76-7.68 (m, 1H), 7.37-7.23 (m, 2H), 7.18-6.96 (m, 2H), 6.86-6.79 (m, 1H), 6.79-6.70 (m, 1H), 5.09-4.97 (m, 1H), 4.78-4.63 (m, 2H), 3.66-3.12 (m, 6H), 2.50-2.24 (m, 4H); Calculated for C21H22ClF3N4O, 438.14; observed (M+H)+ 439.5.
N-(3-chloro-4-fluorophenyl)-2-(morpholinomethyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (71):
lithium aluminum hydride (6.0 ml, 2.4 M, 14.4 mmol, 3.3 eq) was added to methyl quinoxaline-2-carboxylate (820 mg, 4.4 mmol, 1 eq) in THF at room temperature. After 4 hours, it was diluted with ether, cooled to 0°C, quenched with Na2SO4·10H2O, and stirred overnight. After filtration, the filtrate was concentrated and redissolved in DCM and treated with di-tert-butyl decarbonate (1.16 g, 5.28 mmol). After the reaction was completed as determined by LC-MS, the reaction mixture was concentrated and the residue was purified by CombiFlash with ethyl acetate and dichloromethane from 0:1 to 2:8 to obtain the first isomer 64 (663.5 mg, 57%) and to 3:7 to afford the second isomer 65 (130 mg, 11%). The main isomer 64 (first), tert-butyl 3-(hydroxymethyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate (663.5 mg, 2.51 mmol), was dissolved in saturated NaHCO3 and EtOAc, along with 1.25 eq benzyl chloroformate (0.46 ml, 3.14 mmol). After stirring overnight, the organic phase was separated and concentrated. The residue was purified by CombiFlash with ethyl acetate and dichloromethane from 0:1 to 2:8 to give the protected intermediate 66 (345 mg, lost some due to a fraction spill). The alcohol 66 was dissolved in DCM and added dropwise to a flask of 3 eq DMSO and 2 eq oxalyl chloride in DCM that had been stirring at −78°C for 20 minutes. After 4 hours, 4 eq Et3N was added and the reaction was allowed to go to room temperature. After stirring overnight, it was quenched with saturated NH4Cl and extracted with DCM twice and concentrated. The residue was purified on silica gel with ethyl acetate and hexanes from 0:1 to 2:8 to afford the aldehyde intermediate, 1-benzyl 4-tert-butyl 2-formyl-2,3-dihydroquinoxaline-1,4-dicarboxylate 67 (290 mg, 84%). The aldehyde 67 (50 mg, 0.13 mmol, 1 eq) was dissolved in DCE and added dropwise to morpholine (16.5 mg, 0.19 mmol, 1.5 eq) and sodium triacetoxyborohydride (55.1 mg, 0.26 mmol, 2 eq) in DCE, and left stirring overnight. The reaction was quenched with saturated NaHCO3 and stirred vigorously for 2 hours. The organic phase was isolated and concentrated. The residue was purified by CombiFlash with ethyl acetate and dichloromethane from 0:1 to 2:8 to give 68 as a clear oil (52 mg, 86%), which then underwent hydrogenation (2 atm) in EtOH for 2 hours with Pd/C (10%, 10 mg) to remove the Cbz group. The reaction mixture was filtered through a pad of celite and concentrated. The residue was dissolved in 2 mL toluene with excess DIPEA (57μl, 0.33 mmol) and 15% phosgene in toluene (0.24 ml, 0.33 mmol) was added. The reaction was stirred for 5.5 hours, and then stirred under vacuum for an hour to get rid of excess phosgene, and then dissolved in DCM (2 ml). 3-chloro-4-fluoroaniline (32 mg, 0.22 mmol)and DIPEA (57μl, 0.33 mmol) in 1 mL DCM was added dropwise, and stirred for 24 hours. The mixture was diluted in EtOAc and washed with 2N HCl, NaHCO3, and brine. The organic phase was isolated and concentrated. The residue was purified by HPLC with acetonitrile and water from 1:9 to 9:1 to afford Boc protected 70 which was dissolved in 1:1 MeOH to 4M HCl in dioxane. After 3 hours, it was concentrated to afford 71 as a tan solid. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.66-7.70 (m, 1H), 7.32-7.19 (m, 2H), 7.15-7.00 (m, 2H), 6.92-6.82 (m, 2H), 4.26-4.12 (m, 2H), 4.06-3.90 (m, 2H), 3.80-3.68 (m, 3H), 3.68-3.58 (m, 1H), 3.46-3.36 (m, 2H), 3.36-3.25 (m, 1H), 3.18-2.90 (m, 2H); Calculated for C20H22ClFN4O2, 404.14; observed (M+H)+ 405.5.
N-(3-chloro-4-fluorophenyl)-2-((4,4-dimethylpiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (76):
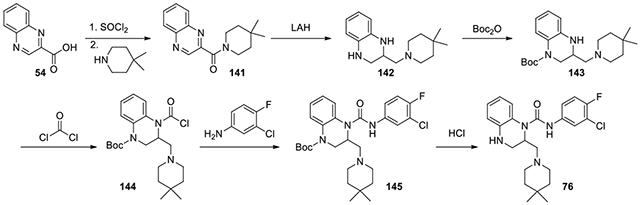
Quinoxaline-2-carboxylic acid (54, 200 mg, 1.15 mmol) was refluxed in thionyl chloride at 75°C overnight to afford quinoxaline-2-carbonyl chloride after concentration. The residue was then treated with 4,4-dimethylpiperidine (189 mg, 1.26 mmol) and Et3N (0.5 ml) in DCM. After stirring overnight, it was purified by CombiFlash with ethyl acetate and hexanes from 0:1 to 6:4 to afford the amide 141 (231.3 mg, 75%). Starting from this amide, following to the procedure for the preparation of compound 75, to afford the two Boc-containing regioisomers. The main isomer, tert-butyl 3-((4,4-dimethylpiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate 143 (155 mg, 0.43 mmol), was used following the same procedure for introduction of the urea bond to afford 76. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.62-7.56 (m, 1H), 7.25-7.20 (m, 1H), 7.20-7.13 (m, 1H), 7.13-7.03 (m, 2H), 6.83-6.70 (m, 2H), 3.80-3.65 (m, 1H), 3.60-3.41 (m, 1H), 3.45-3.37 (m, 1H), 3.36-3.22 (m, 2H), 3.21-3.10 (m, 2H), 3.10-2.97 (m, 1H), 2.97-2.86 (m, 1H), 2.03-1.83 (m, 2H), 1.62-1.43 (m, 2H), 1.05 (s, 6H); Calculated for C23H28ClFN4O, 430.19; observed (M+H)+ 431.6.
N-(3-chloro-4-fluorophenyl)-3-methyl-3,4-dihydroquinoxaline-1(2H)-carboxamide (53):
To a solution of 3-methyl-3,4-dihydroquinoxaline (52, 15 mg, 0.10 mmol) and Et3N (1.1 eq) in DCM (2 mL), phenyl (3-chloro-4-fluorophenyl)carbamate (16, 39.8 mg, 0.15 mmol) was added and the mixture was stirred for 3 h. After confirmation of disappearance of the starting materials, the mixture was evaporated to dryness. The residue was poured into AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel, eluting with EtOAc /hexane from 0:1 to 2:8 to give product 53 (9 mg, 56% yield, white solid). 1H NMR (300 MHz, Chloroform-d) δ 7.46 (dd, J = 6.3, 2.7 Hz, 1H), 7.26 – 7.14 (m, 2H), 7.13 – 6.98 (m, 2H), 6.88 – 6.68 (m, 2H), 6.62 (s, 1H), 4.32 (dd, J = 12.9, 3.6 Hz, 1H), 3.84 – 3.39 (m, 1H), 3.09 (dd, J = 12.9, 8.7 Hz, 1H), 1.29 (d, J = 6.3 Hz, 3H); Calculated for C16H15ClFN3O, 319.09; observed (M+H)+ 320.3.
N-(3-chloro-4-fluorophenyl)-3-(morpholinomethyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (72):
According to the procedure for the preparation of compound 71 and 53, the minor isomer, tert-butyl 2-(hydroxymethyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate 65 (142 mg, 0.36 mmol), was used to afford product 72 (127 mg) as a pink solid. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.59-7.54 (m, 1H), 7.20-7.12 (m, 2H), 7.08-7.00 (m, 2H), 6.77-6.68 (m, 2H), 4.28-4.10 (m, 3H), 4.02-3.86 (m, 6H), 3.38-3.35 (m, 1H), 3.27-3.19 (m, 3H); Calculated for C20H22ClFN4O2, 404.14; observed (M+H)+ 405.5.
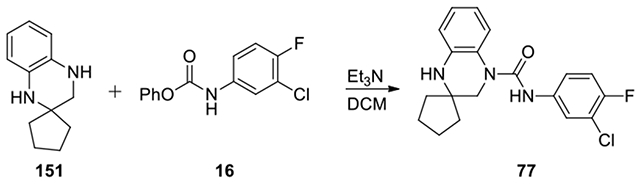
N-(3-chloro-4-fluorophenyl)-1’H-spiro[cyclopentane-1,2’-quinoxaline]-4’(3’H)-carboxamide (77):
To a solution of 3’,4’-dihydro-1’H-spiro[cyclopentane-1,2’-quinoxaline] (151, 32 mg, 0.17 mmol) and Et3N (1.1 eq) in DCM (2 mL), phenyl (3-chloro-4-fluorophenyl)carbamate (16, 67 mg, 0.26 mmol) was added and the mixture was stirred for 3 h. After confirmation of disappearance of the starting materials, the mixture was evaporated to dryness. The residue was diluted with AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel, eluting with EtOAc /hexane from 0:1 to 1:3 to give product 77. (16 mg, 26% yield, white solid). 1H NMR (300 MHz, Chloroform-d) δ 7.58 (dd, J = 6.5, 2.7 Hz, 1H), 7.40 – 7.14 (m, 3H), 7.12 – 6.91 (m, 2H), 6.69 (ddd, J = 8.1, 7.2, 1.4 Hz, 1H), 6.62 (dd, J = 8.1, 1.5 Hz, 1H), 4.34 – 3.89 (m, 1H), 3.70 (s, 2H), 1.85 – 1.63 (m, 6H), 1.59 – 1.38 (m, 2H); Calculated for C19H19ClFN3O, 359.12; observed (M+H)+ 360.4.
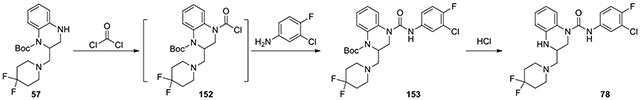
N-(3-chloro-4-fluorophenyl)-3-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (78):
According to the procedure for the preparation of compound 61, the minor isomer, tert-butyl 2-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate 57 (25.4 mg, 0.069 mmol), was used to afford product 78 (16.0 mg) as a white solid. 1H NMR (300 MHz, CD3OD): δ (ppm) 7.65 (dd, J = 6.6, 2.7 Hz, 1H), 7.32-7.22 (m, 2H), 7.15 (t, J = 9.0 Hz, 1H), 7.05-6.96 (m, 1H), 6.84-6.72 (m, 2H), 4.38-4.30 (m, 1H), 4.22-4.12 (m, 1H), 3.65-3.38 (m, 3H), 3.26-3.00 (m, 4H), 2.46-2.24 (m, 4H); Calculated for C21H22ClF3N4O, 438.14; observed (M+H)+ 439.5.
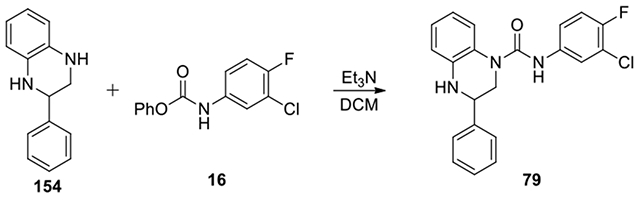
N-(3-chloro-4-fluorophenyl)-3-phenyl-3,4-dihydroquinoxaline-1(2H)-carboxamide (79):
To a solution of 2-phenyl-1,2,3,4-tetrahydroquinoxaline (154, 21 mg, 0.10 mmol) and Et3N (1.1 eq) in DCM (2 mL), phenyl (3-chloro-4-fluorophenyl)carbamate 16 (39.8 mg, 0.15 mmol) was added and the mixture was stirred for 3 h. After confirmation of disappearance of the starting materials, the mixture was evaporated to dryness. The residue was diluted with AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel, eluting with EtOAc /hexane from 0:1 to 2:8 to give product 79 (24 mg, 62% yield, orange solid). 1H NMR (300 MHz, Chloroform-d) δ 7.51 (dd, J = 6.6, 2.7 Hz, 1H), 7.46 – 7.27 (m, 5H), 7.25 – 6.94 (m, 5H), 6.84 – 6.48 (m, 2H), 4.84 – 4.46 (m, 2H), 4.40 (s, 1H), 3.24 (dd, J = 12.9, 8.4 Hz, 1H); Calculated for C21H17ClFN3O, 381.10; observed (M+H)+ 382.4.
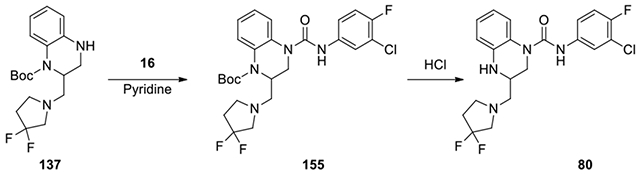
N-(3-chloro-4-fluorophenyl)-3-((3,3-difluoropyrrolidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (80):
According to the procedure for the preparation of compound 53, the minor isomer, tert-butyl 2-((3,3-difluoropyrrolidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate 137 (23.8 mg, 0.067 mmol), was used to afford 80 (3.5 mg) as a clear oil. 1H NMR (300 MHz, CD3OD): δ (ppm) 7.66 (dd, J = 6.3, 2.4 Hz, 1H), 7.33-7.22 (m, 2H), 7.15 (t, J = 9.3 Hz, 1H), 7.04-6.96 (m, 1H), 6.83-6.70 (m, 2H), 4.26-4.17 (m, 1H), 4.02.-3.92 (m, 1H), 3.81-3.68 (m, 2H), 3.58-3.48 (m, 2H), 3.24-3.00 (m, 4H), 2.64-2.48 (m, 2H); Calculated for C20H20ClF3N4O, 424.13; observed (M+H)+ 425.5.
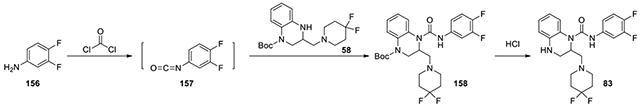
N-(3,4-difluorophenyl)-2-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (83):
1.5 eq 3,4-difluoroaniline (7.9 mg, 0.06 mmol) was dissolved in chloroform with DIPEA (0.04 ml, 0.24 mmol) and 3 eq of 15% phosgene in toluene (0.088 ml, 0.12 mmol). After 4 hours, it was concentrated, and the residue was redissolved in DCM. The solution was added to the intermediate tert-butyl 3-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate (58, 15 mg, 0.04 mmol, 1 eq) and 3 eq of DIPEA (0.02 ml) in DCM and the mixture was stirred at room temperature for 24 hrs. The mixture was concentrated, and the residue was dissolved in THF (0.5ml) and loaded on the silica gel column (ISCO, 12g) and eluted with ethyl acetate and dichloromethane from 0:1 to 2:8 to give the Boc-protected 158, which was stirred in 1:1 MeOH to 4M HCl in dioxane (1 ml: 1ml) for 4 hours. The solution was concentrated and then was purified by HPLC with acetonitrile and water from 30% to 51% to afford 83 (3.4 mg). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.50-7.40 (m, 1H), 7.18-7.00 (m, 3H), 6.98-6.90 (m, 1H), 6.83-6.68 (m, 2H), 5.28-5.18 (m, 1H), 3.55-3.46 (m, 2H), 3.46-3.33 (m, 3H), 3.33-3.23 (m, 1H), 2.99-2.89 (m, 1H), 2.50-2.32 (m, 5H); Calculated for C21H22F4N4O, 422.17; observed (M+H)+ 423.7.
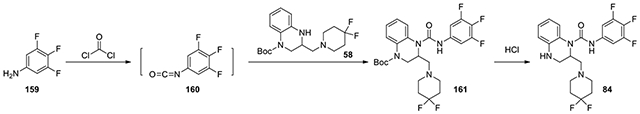
2-((4,4-difluoropiperidin-1-yl)methyl)-N-(3,4,5-trifluorophenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (84):
According to the procedure for the preparation of compound 83, 3,4,5-trifluoroaniline (159, 8.8 mg, 0.06 mmol) was treated with phosgene, and then added to tert-butyl 3-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate (58, 15 mg, 0.04 mmol, 1 eq). The following steps of the same procedure was followed to afford 84 (8.2 mg). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.18-7.02 (m, 4H), 6.82-6.65 (m, 2H), 5.23-5.12 (m, 1H), 3.56-3.20 (m, 7H), 2.96-2.82 (m, 1H), 2.46-2.27 (m, 4H); Calculated for C21H21F5N4O, 440.16; observed (M+H)+ 441.7.
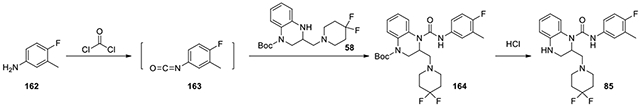
2-((4,4-difluoropiperidin-1-yl)methyl)-N-(4-fluoro-3-methylphenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide 85):
According to the procedure for the preparation of compound 83, 4-fluoro-3-methylaniline (162, 7.5 mg, 0.06 mmol) was treated with phosgene, and then added to tert-butyl 3-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate (58, 15 mg, 0.04 mmol, 1 eq). The following steps of the same procedure was followed to afford 85 (7.2 mg). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.22-7.00 (m, 4H), 6.96-6.84 (m, 1H), 6.82-6.65 (m, 2H), 5.27-5.16 (m, 1H), 3.54-3.22 (m, 5H), 2.98-2.84 (m, 1H), 2.46-2.26 (m, 6H), 2.22 (s, 3H); Calculated for C22H25F3N4O, 418.20; observed (M+H)+ 419.7.
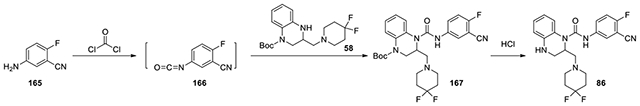
N-(3-cyano-4-fluorophenyl)-2-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (86):
According to the procedure for the preparation of compound 83, 5-amino-2-fluorobenzonitrile (165, 8.2 mg, 0.06 mmol) was treated with phosgene, and then added to tert-butyl 3-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate (58, 15 mg, 0.04 mmol, 1 eq). The following steps of the same procedure was followed to afford 86 (10.1 mg). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.80-7.72 (m, 1H), 7.64-7.55 (m, 1H), 7.22-7.06 (m, 3H), 6.85-6.76 (m, 1H), 6.76-6.68 (m, 1H), 5.30-5.20 (m, 1H), 3.60-3.47 (m, 2H), 3.47-3.33 (m, 3H), 3.30-3.20 (m, 1H), 3.04-2.92 (m, 1H), 2.50-2.33 (m, 5H); Calculated for C22H22F3N5O, 429.18; observed (M+H)+ 430.7.
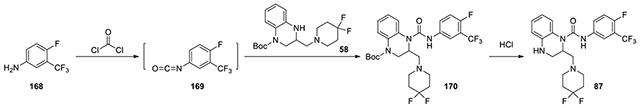
2-((4,4-difluoropiperidin-1-yl)methyl)-N-(4-fluoro-3-(trifluoromethyl)phenyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (87):
According to the procedure for the preparation of compound 83, 4-fluoro-3-(trifluoromethyl)aniline (168, 10.7 mg, 0.06 mmol) was treated with phosgene, and then added to tert-butyl 3-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate (58, 15 mg, 0.04 mmol, 1 eq). The following steps of the same procedure was followed to afford 87 (12.9 mg). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.66-7.54 (m, 2H), 7.21-7.05 (m, 3H), 6.84-6.76 (m, 1H), 6.76-6.68 (m, 1H), 5.28-5.20 (m, 1H), 3.56-3.46 (m, 2H), 3.46-3.33 (m, 3H), 3.33-3.23 (m, 1H), 3.02-2.92 (m, 1H), 2.49-2.31 (m, 5H); Calculated for C22H22F6N4O, 472.17; observed (M+H)+ 473.8.
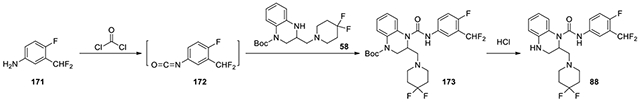
N-(3-(difluoromethyl)-4-fluorophenyl)-2-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (88):
According to the procedure for the preparation of compound 83, 3-(difluoromethyl)-4-fluoroaniline (171, 9.7 mg, 0.06 mmol) was treated with phosgene, and then added to tert-butyl 3-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate (58, 15 mg, 0.04 mmol, 1 eq). The following steps of the same procedure was followed to afford 88. 1H NMR (400 MHz, CDCl3): δ 7.55 (m, 1 H), 7.51 (m, 1 H), 7.36 (s, 1 H), 7.19 (dd, J1 = 8.0 Hz, J2 = 1.1 Hz, 1 H), 7.12 (m, 1 H), 7.10 (m, 1 H), 6.85 (t, J = 54.9 Hz, 1 H), 6.83 (ddd, J1 = 8.1 Hz, J2 = 7.0 Hz, J3 = 1.3 Hz,1 H), 6.72 (dd, J1 = 8.0 Hz, J2 = 1.4 Hz, 1 H), 5.29 (m, 1 H), 3.51 (bs, 4 H), 3.50 (dd, J1 = 12.4 Hz, J2 = 1.6 Hz, 1 H), 3.43 (dd, J1 = 12.5 Hz, J2 = 3.8 Hz, 1 H), 3.29 (dd, J1 = 13.8 Hz, J2 = 3.0 Hz, 1 H), 3.09 (dd, J1 = 13.7 Hz, J2 = 9.2 Hz, 1 H), 2.40 (bs, 4 H); 13C NMR (100 MHz, CDCl3): δ 161.4 (d, J = 39 Hz), 156.7 (d, J = 249 Hz), 153.4, 136.3, 134.0, 134.0, 127.5, 124.5, 124.4, 118.8, 118.5 (d, J = 23 Hz), 117.0 (t, J = 289 Hz), 116.6 (d, J = 22 Hz), 116.2, 110.3 (td, J1 = 238, J2 = 4.7), 56.4, 49.9 (2C), 44.2, 43.6, 31.2 (2C, t, J = 26 Hz). HRMS, Calculated for C22H23F5N4O (M+H)+, 455.1865; observed (M+H)+ 455.1861.
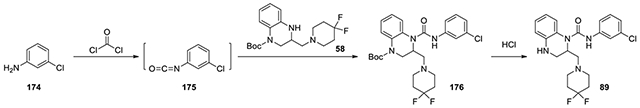
N-(3-chlorophenyl)-2-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (89):
According to the procedure for the preparation of compound 83, 3-chloroaniline (174, 8.7 mg, 0.06 mmol) was treated with phosgene, and then added to tert-butyl 3-((4,4-difluoropiperidin-1-yl)methyl)-3,4-dihydroquinoxaline-1(2H)-carboxylate (58, 15 mg, 0.04 mmol, 1 eq). The following steps of the same procedure was followed to afford 89. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.51-7.45 (m, 1H), 7.25-7.19 (m, 1H), 7.19-7.00 (m, 3H), 6.83-6.68 (m, 2H), 5.28-5.19 (m, 1H), 3.56-3.46 (m, 2H), 3.46-3.25 (m, 5H), 2.96-2.85 (m, 1H), 2.48-2.30 (m, 4H); Calculated for C21H23ClF2N4O, 420.15; observed (M+H)+ 421.7.
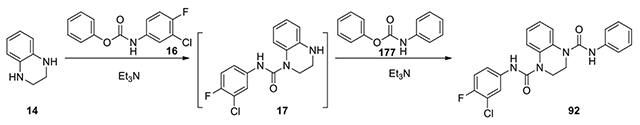
N1-(3-chloro-4-fluorophenyl)-N4-phenyl-2,3-dihydroquinoxaline-1,4-dicarboxamide (92):
To a solution of 1,2,3,4-tetrahydroquinoxaline (14, 67 mg, 0.5 mmol, 1 eq) and Et3N (0.15 ml, 2.2 eq) in DCM (2 mL), (3-chloro-4-fluorophenyl)carbamate (16, 132.8 mg, 0.5 mmol, 1 eq) was added and the mixture was stirred for 3 h. After confirmation of disappearance of the starting materials, the mixture was further treated with phenyl phenylcarbamate (177, 106.6 mg, 0.5 mmol, 1 eq). The solution was stirred under reflux for 2 days. The mixture was concentrated, and the residue was poured into AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was evaporated to dryness. The residue was purified by column chromatography on silica gel, eluting with EtOAc/Hexane from 25% to 40% to give product 92. (64 mg, 30% yield, white solid). 1H NMR (300 MHz, Chloroform-d) δ 7.63 – 7.49 (m, 3H), 7.40 (dd, J = 8.7, 1.2 Hz, 2H), 7.36 – 7.28 (m, 2H), 7.29 – 7.16 (m, 3H), 7.13 – 7.01 (m, 2H), 6.94 (s, 2H), 4.03 (s, 4H); Calculated for C22H18ClFN4O2, 424.11; observed (M+H)+ 425.4.
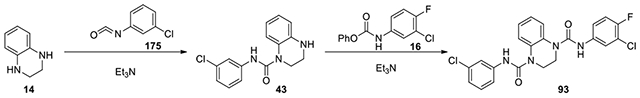
N1-(3-chloro-4-fluorophenyl)-N4-(3-chlorophenyl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (93):
Similar to the procedure for the preparation of compound 92, 1,2,3,4-tetrahydroquinoxaline (14, 30 mg, 0.22 mmol, 1 eq) was treated with 3-chlorophenyl isocyanate (175, 27 μl, 0.22 mmol, 1 eq) and (3-cliloro-4-fluorophenyl)carbamate (30 mg, 0.11 mmol) subsequently. The mixture was purified by HPLC with a gradient of acetonitrile in water from 45% to 100% to afford 93 (5.2 mg) as a white solid. 1H NMR (300 MHz, CDCl3): δ (ppm)7.58-7.52 (m, 3H), 7.51-7.48 (m, 2H), 7.30-7.27 (m, 2H), 7.26-7.19 (m, 3H), 7.24-7.19 (m, 2H), 4.02 (s, 4H); Calculated for C22H17Cl2FN4O2, 458.07; observed (M+H)+ 459.4.
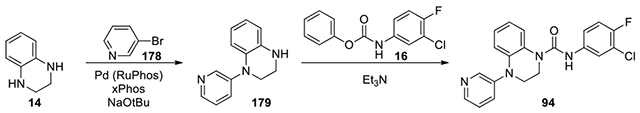
N1-(3-chloro-4-fluorophenyl)-N4-(pyridine-3-yl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (94):
To a sealed tube was charged 1,2,3,4-tetrahydroquinoxaline (14, 134 mg, 1.0 mmol, 1 eq), 3-bromopyrine (178, 40mg, 0.25 mmol), Pd(RuPhos) (15 mg), xPhos (28.6 mg), NaOtBu (115 mg), and THF (2 ml). After degassed, the mixture was stirred at 85 °C under Ar for overnight. The mixture was concentrated and purified on HPLC with a gradient of acetonitrile in water from 10% to 100% to provide the intermediate 179 (44 mg, 83%), which was treated with 3-chloro-4-fluorophenyl)carbamate (16, 30 mg, 0.11 mmol) following the procedure used for preparation of 92. The residue was purified by HPLC with a gradient of acetonitrile in water from 30% to 100% to give product 94 (23 mg, 29% yield, yellow solid). 1H NMR (300 MHz, Chloroform-d) δ 8.87 (s, 1H), 8.31 (d, J = 5.1 Hz, 1H), 7.96 (d, J = 8.4 Hz 1H), 7.68 (dd, J = 8.7, 5.1 Hz, 1H), 7.59 (dd, J = 6.3, 2.7 Hz, 1H), 7.43 (dd, J = 7.8, 1.5 Hz, 1H), 7.35 – 7.27 (m, 1H), 7.26 – 7.12 (m, 4H), 7.07 (t, J = 8.7 Hz, 1H), 4.12 (t, J = 6.0 Hz, 2H), 3.93 (t, J = 6.0 Hz, 2H); Calculated for C20H16ClFN4O, 382.10; observed (M+H)+ 383.4.
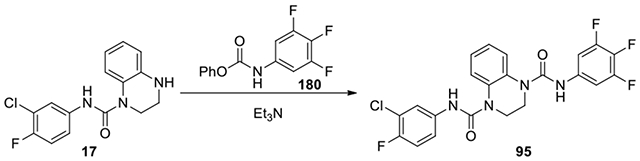
N1-(3-chloro-4-fluorophenyl)-N4-(3,4,5-trifluorophenyl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (95):
3,4,5-tifluoroaniline (27mg, 0.19 mmol, 1 eq) was treated with phenyl chloroformate (50.5 mg, 0.19 mmol) in 1:1 EtOAc to saturated NaHCO3. After 2 hours, the organic phase was isolated and concentrated. The residual 180 was dissolved in DCM, and treated with 1 eq compound 17 (50.5 mg) and Et3N (2 eq). After stirring overnight, it was concentrated, and the residue was purified by HPLC with a gradient of acetonitrile in water from 10% to 90% to afford 95 (29.3 mg 33%) as a white solid. 1H NMR (300 MHz, CD3OD): δ (ppm) 7.68-7.60 (m, 1H), 7.60-7.47 (m, 2H), 7.36-7.23 (m, 3H), 7.23-7.11 (m, 3H), 3.97 (s, 4H); Calculated for C22H15ClF4N4O2, 478.08; observed (M+H)+ 479.6.
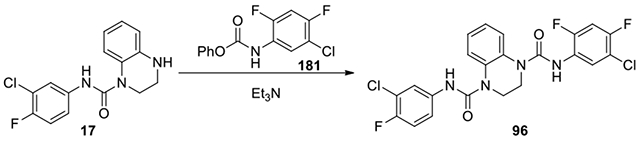
N1-(5-chloro-2,4-difluorophenyl)-N4-(3-chloro-4-fluorophenyl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (96):
According to the procedure for the preparation of compound 95, 5-chloro-2,4-difluoroaniline (30 mg, 0.19 mmol, 1 eq) was treated with phenyl chloroformate, and then added to compound 17, to afford 96 (6.4 mg, 7%) as a light purple solid. 1H NMR (300 MHz, CD3OD): δ (ppm) 7.94-7.85 (m, 1H), 7.68-7.50 (m, 3H), 7.36-7.28 (m, 1H), 7.28-7.11 (m, 4H), 3.98 (s, 4H); Calculated for C22H15Cl2F3N4O2, 494.05; observed (M+H)+ 495.5.
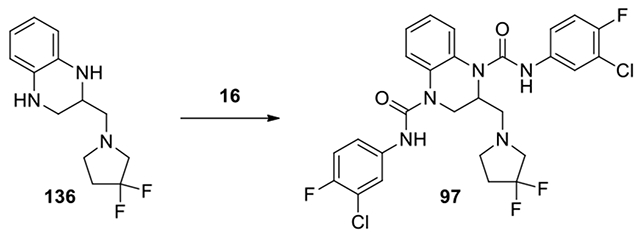
N1,N4-bis(3-chloro-4-fluorophenyl)-2-((3,3-difluoropyrrolidin-1-yl)methyl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (97):
To a solution of amine 136 (192 mg, 0.76 mmol) and DBU (1.1 eq) in DCM (2 mL), compound 16 was added and the mixture was stirred at 40°C for 3 d. The mixture was evaporated to dryness. The residue was poured into AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was evaporated to dryness. The residue was purified by HPLC, eluting with acetonitrile in water from 40% to 80% to give product 56-8. (0.1 mg, 0.02% yield, white solid). Calculated for C27H23Cl2F4N5O2, 595.1; observed (M+H)+ = 596.6. 1H NMR (300 MHz, Chloroform-d) δ 8.08 (s, 1H), 7.68 (s, 1H), 7.63 (dd, J = 6.6, 2.7 Hz, 1H), 7.55 – 7.42 (m, 2H), 7.32 – 7.27 (m, 1H), 7.27 – 7.24 (m, 3H), 7.22 – 7.16 (m, 1H), 7.15 – 6.99 (m, 2H), 4.99 (d, J = 8.0 Hz, 1H), 4.40 (dd, J = 13.8, 6.3 Hz, 1H), 3.78 (s, 2H), 3.75 – 3.61 (m, 2H), 3.58 – 3.29 (m, 2H), 3.09 – 2.92 (m, 1H), 2.59 (tt, J = 13.8, 7.2 Hz, 2H).
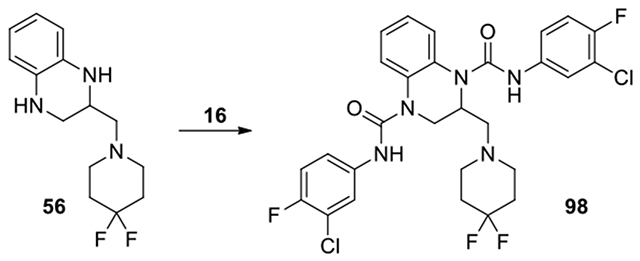
N1,N4-bis(3-chloro-4-fluorophenyl)-2-((4,4-difluoropiperidin-1-yl)methyl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (98):
To a solution of amine 56 (116 mg, 0.43 mmol) and DBU (1.1 eq) in DCM (2 mL), compound 16 was added and the mixture was stirred at 40°C for 3 d. The mixture was evaporated to dryness. The residue was poured into AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was evaporated to dryness. The residue was purified by HPLC, eluting with acetonitrile in water from 40% to 75% gave product 98. (1.7 mg, 0.6% yield, white solid). 1H NMR (300 MHz, Chloroform-d) δ 8.04 (s, 1H), 7.67 (dd, J = 6.6, 2.7 Hz, 1H), 7.56 – 7.50 (m, 2H), 7.47 – 7.38 (m, 1H), 7.37 – 7.28 (m, 2H), 7.27 – 7.25 (m, 2H), 7.23 ∓ 7.15 (m, 1H), 7.09 (td, J = 8.7, 6.0 Hz, 2H), 5.17 (dt, J = 10.2, 4.8 Hz, 1H), 4.20 (dd, J = 13.8, 5.7 Hz, 1H), 3.92 (dd, J = 13.8, 5.4 Hz, 1H), 3.57 (dd, J = 13.2, 8.1 Hz, 1H), 3.02 (dd, J = 13.2, 4.5 Hz, 1H), 2.68 – 1.81 (m, 8H). Calculated for C28H25Cl2F4N5O2, 609.13; observed (M+H)+ = 610.6.
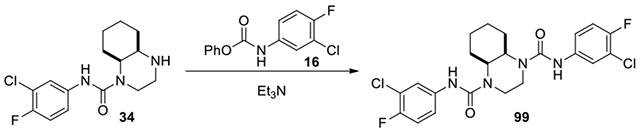
Cis-N1,N4-bis(3-chloro-4-fluorophenyl)octahydroquinoxaline-1,4-dicarboxamide (99):
Compound 34 (20 mg, 0.064 mmol) and phenyl (3-chloro-4-fluorophenyl)carbamate (17 mg, 0.064 mmol) were dissolved in DCM with excess Et3N. The mixture was stirred at rt for 3 days. The mixture was diluted with EtOAc and washed with sat. NaHCO3 and brine. The organic phase was isolated and concentrated. The residue was purified on CombiFlash with a gradient of ethyl acetate in hexanes from 50% to 100% to afford 99 (10.3 mg, 33%) as a clear oil. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.56 (dd, J = 6.6, 2.7 Hz, 2H), 7.22-7.15 (m, 2H), 7.04 (t,J = 9.0 Hz, 2H), 4.28-4.19 (m, 2H), 4.06-3.94 (m, 2H), 3.68-3.55 (m, 2H), 2.10-1.94 (m, 2H), 1.84-1.72 (m, 2H), 1.66-1.38 (m, 4H); Calculated for C22H22Cl2F2N4O2, 482.11; observed (M+H)+ 483.4.
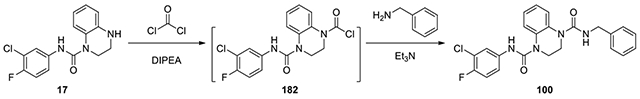
N1-benzyl-N4-(3-chloro-4-fluorophenyl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (100):
Compound 17 (50.8 mg, 0.25 mmol) and excess of DIPEA (0.13 ml, 0.75 mmol) was dissolved in toluene (1ml) under argon. 3 eq of 15% phosgene in toluene (0.53 ml, 0.75 mmol) was added and the reaction was stirred overnight. The solution was concentrated, and the residue was redissolved in DCM (1 ml), and then added to benzylamine (29 mg, 0.27 mmol) and excess of Et3N (0.1 ml) in DCM (1 ml). The mixture was stirred overnight and concentrated. The residue was purified by HPLC with a gradient of acetonitrile in water from 50% to 60% to afford 100 (54.2 mg 45%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.53-7.47 (m, 2H), 7.42-7.38 (m, 1H), 7.31-7.13 (m, 6H), 7.13-7.05 (m, 2H), 7.00 (t, J = 8.7 Hz, 1H), 4.40 (s, 2H), 3.92-3.79 (m, 4H); Calculated for C23H20ClFN4O2, 438.13; observed (M+H)+ 439.6.
N1-(3-chloro-4-fluorophenyl)-N4-(2-chloropyridin-4-yl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (101):
According to the procedure for the preparation of compound 100, the intermediate was treated with 2-chloropyridin-4-amine (35 mg, 0.27 mmol) afforded 101 (6.7 mg, 5.8%). 1H NMR (300 MHz, CDCl3): δ (ppm) 8.12 (d, J = 6.0 Hz, 1H), 7.54-7.46 (m, 3H), 7.43-7.38 (m, 1H) 7.30-7.26 (m, 1H), 7.24-7.16 (m, 3H), 7.03 (t, J = 8.7 Hz, 1H), 3.96-3.92 (m, 4H); Calculated for C21H16Cl2FN5O2, 459.07; observed (M+H)+ 460.6.
N1,N4-bis(3-chloro-4-fluorophenyl)-2-(morpholinomethyl)-2,3-dihydroquinoxaline-1,4-dicarboxamide (102):
According to the procedure for the preparation of compound 75 starting from quinoxaline-2-carboxylic acid (184 mg), The intermediate 184 was treated with compound 16 (200 mg) and the mixture was stirred at 40°C for 3 d. The mixture was evaporated to dryness. The residue was diluted with AcOEt (10 mL). The organic solution was washed with H2O and brine, dried over anhydrous Na2SO4, and filtered. The filtrate was evaporated to dryness. The residue was purified by HPLC, eluting with CH3CN/water from 10% to 90% to give product 102 (7 mg, 1.5% yield, white solid). 1H NMR (300 MHz, Chloroform-d) δ 8.14 (s, 1H), 7.68 (dd, J = 6.5, 2.7 Hz, 1H), 7.62 (s, 1H), 7.54 (dd, J = 6.6, 2.7 Hz, 2H), 7.47 – 7.37 (m, 1H), 7.37 – 7.28 (m, 1H), 7.25 – 7.14 (m, 3H), 7.09 (td, J = 8.7, 6.0 Hz, 2H), 5.21 (dd, J = 8.4, 4.8 Hz, 1H), 4.18 (dd, J = 13.5, 5.4 Hz, 1H), 4.06 – 3.88 (m, 4H), 3.56 (dd, J = 13.2, 8.1 Hz, 1H), 3.04 (t, J = 2.4 Hz, 1H), 2.99 (d, J = 4.5 Hz, 1H), 2.79 (q, J = 7.2 Hz, 2H), 2.35 (t, J = 7.5 Hz, 1H), 2.29 – 2.12 ((t, J = 7.5 Hz, 1H); Calculated for C27H25Cl2F2N5O3, 575.13; observed (M+H)+ 576.6.
N-(3-chloro-4-fluorophenyl)-4-(4-fluorobutyl)-3,4-dihydroquinoxaline-1(2H)-carboxamide (103):
Potassium carbonate (36 mg, 0.26 mmol) and 1-bromo-4-fluorobutane (14 μL, 0.13 mmol) was added to compound 17 (40 mg, 0.13 mmol) in acetonitrile. The reaction was heated to 85°C over 2 days. Upon completion, it was diluted with EtOAc and washed with 2N HCl thrice, saturated NaHCO3, and brine. It was purified by CombiFlash at 40% EtOAc in hexanes to afford 103 (2.1 mg, 4%). 1H NMR (300 MHz, CDCl3): δ (ppm) 7.06-6.97 (m, 2H), 6.94-6.82 (m, 2H), 6.75-6.68 (m, 1H), 6.54-6.47 (m, 1H), 6.40-6.35 (m, 1H), 4.56-4.51 (m, 1H), 4.40-4.35 (m, 1H), 3.76-3.66 (m, 2H), 3.62-3.56 (m, 2H), 3.47-3.41 (m, 2H), 0.93-0.77 (m, 4H); Calculated for C19H20ClF2N3O, 379.13; observed (M+H)+ 380.4.
Biological evaluation
Materials
AML12HBV10 and HepDES19 cells are immortalized mouse hepatocyte (AML12)- and human hepatoma cell (HepG2)-derived stable cell lines supporting the replication of a stably-transfected envelope protein-deficient HBV genome in a tetracycline-inducible manner 8–9. These cell lines were maintained in DMEM/F12 medium (Corning) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml tetracycline and 200 μg/ml G-418. When cultured in medium without tetracycline, HBV pg RNA transcription will be activated and viral DNA replication occurs subsequently. DVR-23 was synthesized in house. Bay 41-4109 is a gift from Dr. Lai Wei at Peking University, Beijing China. Entecavir is a gift from Dr. William S. Mason at Fox Chase Cancer Center, Philadelphia.
Antiviral and cytotoxicity assays in AML12HBV10 cells
The cells were seeded into 96-well plates at a density of 2 x 104 cells per well and cultured in the absence of tetracycline. One day after seeding, cells were mock-treated or treated with a serial 2-fold dilution of compound with DMEM/F12 medium (Corning), ranging from 10 μM to 0.08 μM, for 48 h and lysed by addition of 100 μl per well of lysis buffer containing 10 mM Tris-HCl (pH 7.5), 1 mM EDTA, 100 mM NaCl and 1% NP-40. Half of the lysate was added to DNA denaturing solution containing 1.5M NaCl and 1M NaOH. After 5 minutes of incubation at room temperature, 100μl of neutralization solution containing 1.5M NaCl, 1M Tris-HCl (pH 7.4) was added. Using a 96-well dot-blot manifold (Bio-Rad), the lysates were applied to a Hybond-N+ membrane (Amersham). HBV DNA in the cell lysates was detected by hybridization with alpha-32P-UTP-labelled (800 Ci/mmol, PerkinElmer) riboprobe specific for HBV minus strand DNA. After overnight incubation, membrane was washed twice, one hour each, with buffer containing 0.1X SSC and 0.1%SDS at 65°C, and exposed to a phosphoimager screen (GE Healthcare). Quantification done by QuantityOne software was used to determine the concentration that reduces the amount of HBV DNA by 50% (EC50). To determine the cytotoxicity, the cells were treated with a serial 2-fold dilution of compound, ranging from 50 μM to 1.56 μM, for 48 h under the same culture condition for the antiviral assay. The cell viability was inspected under microscopy and quantified by a MTT assay (Sigma) and expressed as the concentration of compound that reduced the viability of the cells by 50% (CC50).
Antiviral and cytotoxicity assays in HepDES19 cells
For the antiviral activity assay, HepDES19 cells were seeded into 24-well plates and cultured in the absence of tetracycline for 2 days. The cells were then mock-treated or treated with a serial 2-fold dilution of compound for an additional 4 days. Cytoplasmic HBV core DNA were extracted and quantified by a qPCR assay as previously described.15,20 The antiviral activity (EC50) was determined from triplicate biological experiments by a regression method of GraphPad Prism. To determine the cytotoxicity, HepDES19 cells seeded in 96 well plates were treated with a serial 3-fold dilution of compound, ranging from 50 μM to 0.39 μM, for 6 days under the same culture condition for the antiviral assay. The cell viability was inspected under microscopy and CC50 values were determined by a MTT assay.
Mode of action study in AML12HBV10 cells.
AML12HBV10 cells were seeded into 24-well plates and cultured in the presence or absence of tetracycline for 24 h and then mock-treated or treated with the indicated compounds at desired concentrations for an additional 48 h. The amounts of intracellular total viral RNA, core protein, capsids, encapsidated pgRNA and viral DNA replication intermediates were determined with the methods described previously.15,20
Supplementary Material
Figure 5, Determination of HBV replication step inhibited by compound 17 and 18.
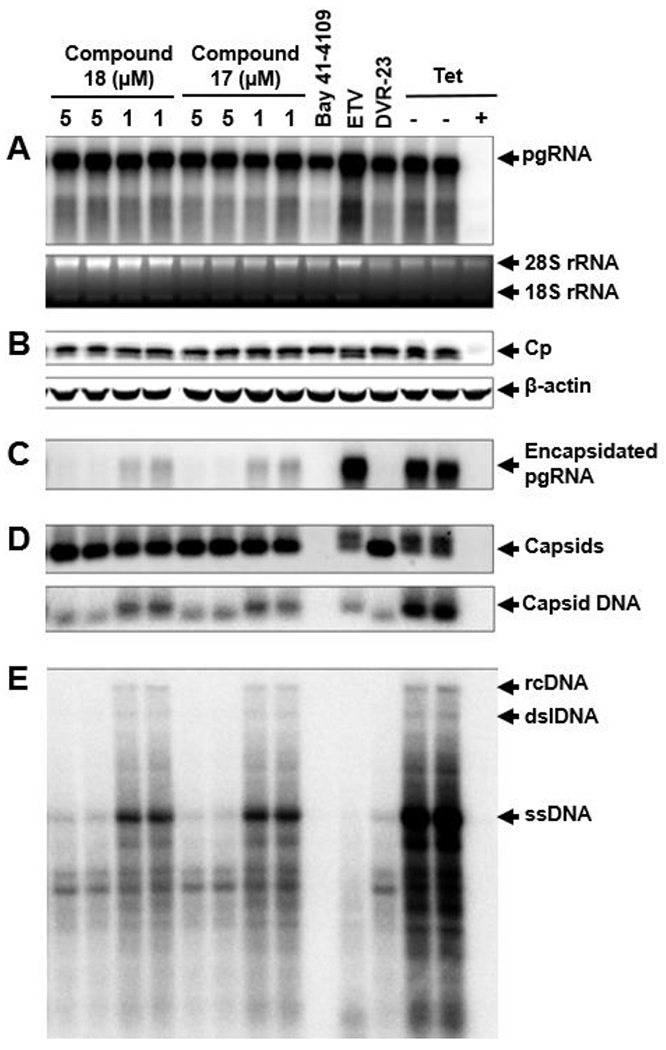
AML12HBV10 cells were cultured in the presence of tetracycline (tet) to inhibit pgRNA transcription and serve as negative control, or in the absence of tet to induce pgRNA transcription and subsequent HBV DNA replication in cytoplasmic capsids. The cells were mock-treated (DMSO) or treated with ETV (1 μM), Bay 41-4109 (2 μM), DVR-23 (5 μM), Compound 17 (1 or 5 μM), or 18 (1 or 5 μM) for 2 days, starting at 24 h post the removal of tet. Intracellular total HBV RNA (A) and encapsidated pgRNA (C) were detected by Northern blot hybridization. Ribosomal RNA (rRNA) served as loading control. HBV core protein (Cp) was detected by Western blot assay with an antibody against C-terminal 14 amino acid residues of core protein (Cp). β-actin served as loading control (B). HBV capsids and core associated DNA were detected by native agarose gel electrophoresis-based particle gel assay (D). Core associated HBV DNA replication intermediates were analyzed by Southern blot assay (E). rcDNA, relaxed circular DNA. dslDNA, double-stranded linear DNA. ssDNA, single-strand DNA.
Scheme 7.
Synthesis of mixed bis-ureas.
Table 3.
2- Vs 3-substitution
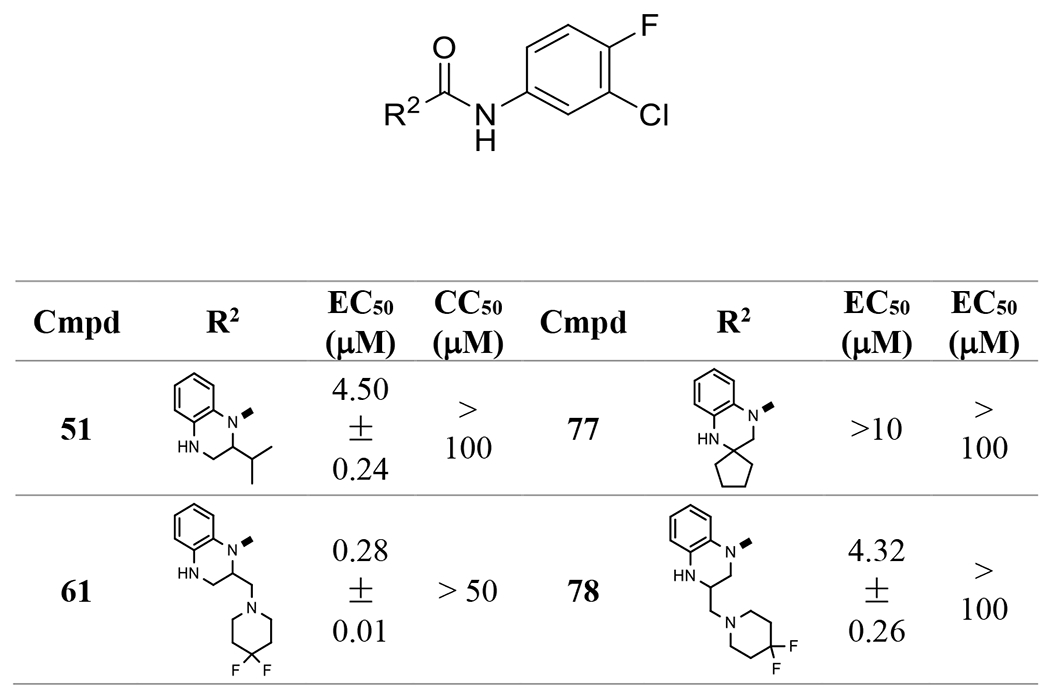
|
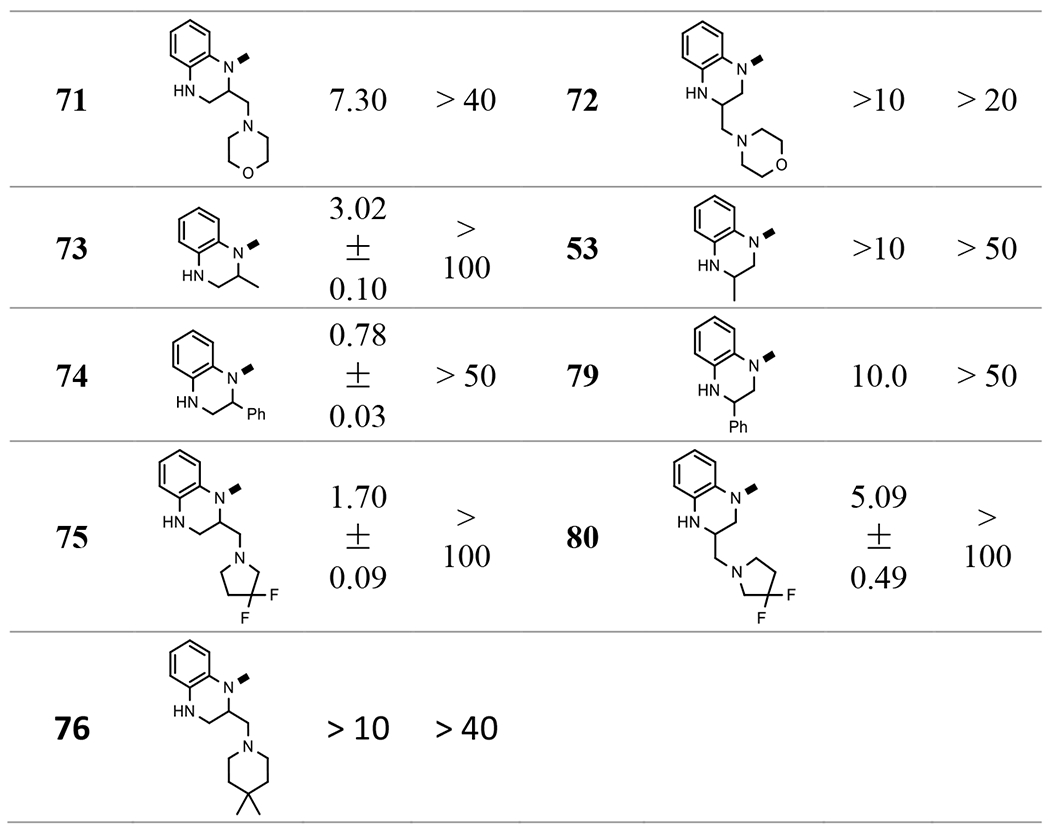
|
Highlights.
Phenyl ureas of 1,2,3,4-tetrahydroquinoxaline modulate HBV pgRNA encapsidation
Bis-ureas based on 1,2,3,4-tetrahydroquinoxaline core inhibit HBV DNA replication
Both types of compounds induce the assembly of empty capsids devoid of viral pgRNA
Acknowledgments
This work was supported by grants from the National Institutes of Health, USA (AI113267) and appreciation of The Commonwealth of Pennsylvania through the Hepatitis B Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Liaw Y-F; Chu C-M, Hepatitis B virus infection, Lancet (2009), 373(9663), 582–592. [DOI] [PubMed] [Google Scholar]
- 2.Xia Y; Guo H, Hepatitis B virus cccDNA: Formation, regulation and therapeutic potential, Antiviral Res (2020), 180, 104824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez MG; Villeret F; Testoni B; Zoulim F, Can we cure hepatitis B virus with novel direct-acting antivirals?, Liver Int (2020), 40(S1), 27–34. [DOI] [PubMed] [Google Scholar]
- 4.Fanning GC; Zoulim F; Hou J; Bertoletti A, Therapeutic strategies for hepatitis B virus infection: towards a cure, Nat Rev Drug Discov. 2019. Nov;18(11):827–844. [DOI] [PubMed] [Google Scholar]
- 5.Hu J; Cheng J; Tang L; Hu Z; Luo Y; Li Y; Zhou T; Chang J; Guo J-T, Virological Basis for the Cure of Chronic Hepatitis B, ACS infect dis (2019), 5(5), 659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wynne SA; Crowther RA; Leslie AGW The crystal structure of the human hepatitis B virus capsid. Mol. Cell 1999, 3, 771–780. [DOI] [PubMed] [Google Scholar]
- 7.Ceres P; Zlotnick A, Weak Protein-Protein Interactions Are Sufficient To Drive Assembly of Hepatitis B Virus Capsids,. Biochemistry (2002), 41(39), 11525–11531. [DOI] [PubMed] [Google Scholar]
- 8.Nijampatnam B; Liotta DC, Recent advances in the development of HBV capsid assembly modulators, Curr Opin Chem Biol 2019, 50:73–79. [DOI] [PubMed] [Google Scholar]
- 9.Viswanathan U; Mani N; Hu Z; Ban H; Du Y; Hu J; Chang J; Guo J-T, Targeting the multifunctional HBV core protein as a potential cure for chronic hepatitis B, Antiviral Res 182 (2020) 104917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.King RW; Ladner SK; Miller TJ; Zaifert K; Perni RB; Conway SC; Otto MJ Inhibition of human hepatitis B virus replication by AT-61, a phenylpropenamide derivative, alone and in combination with (−)beta-L-2’,3’-dideoxy-3’-thiacytidine [published correction appears in Antimicrob Agents Chemother 1999 Mar;43(3):726], Antimicrob Agents Chemother. 1998;42(12):3179–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perni RB; Conway SC; Ladner SK; Zaifert K; Otto MJ; King RW 2000. Phenylpropenamide derivatives as inhibitors of hepatitis B virus replication. Bioorg. Med. Chem. Lett 10:2687–2690. [DOI] [PubMed] [Google Scholar]
- 12.Wang P; Naduthambi D; Mosley RT; Niu C; Furman PA; Otto MJ; and Sofia MJ (2011) Phenylpropenamide derivatives: anti-hepatitis B virus activity of the Z isomer, SAR and the search for novel analogs. Bioorg. Med. Chem. Lett 21 (15), 4642–4647. [DOI] [PubMed] [Google Scholar]
- 13.Weber O; Schlemmer KH; Hartmann E; Hagelschuer I; Paessens A; Graef E; Deres K; Goldmann S; Niewoehner U; Stoltefuss J; Haebich D; Ruebsamen-Waigmann H; Wohlfeil S Inhibition of human hepatitis B virus (HBV) by a novel non-nucleosidic compound in a transgenic mouse model. Antiviral Res. 2002;54(2):69–78. [DOI] [PubMed] [Google Scholar]
- 14.Deres K; Schröder CH; Paessens A; Goldmann S; Hacker HJ; Weber O; Krämer T; Niewöhner U; Pleiss U; Stoltefuss J; Graef E; Koletzki D; Masantschek RN; Reimann A; Jaeger R; Gross R; Beckermann B; Schlemmer KH; Haebich D; Rübsamen-Waigmann H Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299(5608):893–896. [DOI] [PubMed] [Google Scholar]
- 15.Campagna MR; Liu F; Mao R; Mills C; Cai D; Guo F; Zhao X; Ye H; Cuconati A; Guo H; Chang J; Xu X; Block TM; Guo J-T Sulfamoylbenzamide derivatives inhibit the assembly of hepatitis B virus nucleocapsids. J Virol. 2013;87(12):6931–6942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vandyck K; Hache GYP; Last SJ; McGowan DC; Rombouts G; Verschueren WG; Raboisson PJ-MB Sulphamoylpyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis B, WO2014184350A1.
- 17.Vandyck K; Kesteleyn B; Rudolf R; Pieters S; Maria A; Rombouts G; Verschueren WG; Raboisson PJ-MB Glyoxamide substituted pyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis B, WO2015011281
- 18.Amblard F; Boucle S; Bassit L; Cox Bryan; Sari Ozkan; Tao Sijia; Chen Zhe; Ozturk Tugba; Verma Kiran; Russell Olivia; Rat Virgile; de Rocquigny Hugues; Fiquet Oriane; Boussand Maud; Di Santo James; Strick-Marchand Helene; Schinazi Raymond F. Novel Hepatitis B Virus Capsid Assembly Modulator Induces Potent Antiviral Responses In Vitro and in Humanized Mice. Antimicrob Agents Chemother. 2020;64(2):e01701–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turner W; Arnold LD; Maag H; Li L; Bures M; Haydar S; Francis S Hepatitis B core protein modulators, WO2017048950.
- 20.Wu S; Zhao Q; Zhang P; Kulp J; Hu L; Hwang N; Zhang J; Block TM; Xu X; Du Y; Chang J; Guo JT Discovery and mechanistic study of benzamide derivatives that modulate hepatitis B virus capsid assembly. J Virol. 2017;91(16):e00519–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katen SP; Tan Z; Chirapu SR; Finn MG; Zlotnick A Assembly-directed antivirals differentially bind quasiequivalent pockets to modify hepatitis B virus capsid tertiary and quaternary structure, (2013) Structure 21: 1406–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bourne CR; Finn MG; Zlotnick A (2006) Global structural changes in hepatitis B virus capsids induced by the assembly effector HAP1. J Virol 80(22):11055–11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klumpp K; Lam AM; Lukacs C; Vogel R; Ren S; Espiritu C; Baydo R; Atkins K; Abendroth J; Liao G; Efimov A; Hartman G; Flores OA High-resolution crystal structure of a hepatitis B virus replication inhibitor bound to the viral core protein. Proc Natl Acad Sci U S A. 2015; 112(49):15196–15201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Z; Hu T; Zhou X; Wildum S; Garcia-Alcalde F; Xu Z; Wu D; Mao Y; Tian X; Zhou Y; Shen F; Zhang Z; Tang G; Najera I; Yang G; Shen HC; Young JAT; Qin N Heteroaryldihydropyrimidine (HAP) and Sulfamoylbenzamide (SBA) Inhibit Hepatitis B Virus Replication by Different Molecular Mechanisms. Sci Rep. 2017;7:42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang JA; Kim S; Park M; Park H-J; Kim J-H; Park S; Hwang J-R; Kim Y-C; Kim YJ; Cho Y; Jin MS; Park S-G Ciclopirox inhibits Hepatitis B Virus secretion by blocking capsid assembly. Nat Commun. 2019;10(1):2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlicksup CJ; Laughlin P; Dunkelbarger S; Wang JC; Zlotnick A Local stabilization of subunit-subunit contacts causes global destabilization of Hepatitis B virus capsids, ACS Chem. Biol 2020, 15, 6, 1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao Q; Hu Z; Cheng J; Wu S; Luo Y; Chang J; Hu J; Guo J-T Hepatitis B Virus Core Protein Dephosphorylation Occurs during Pregenomic RNA Encapsidation, (2018), J Virol 92:e02139–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ludgate L; Adams C; Hu J, Phosphorylation state-dependent interactions of hepadnavirus core protein with host factors, PloS one (2011), 6(12), e29566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mani N; Cole AG; Phelps JR; Ardzinski A; Cobarrubias KD; Cuconati A; Dorsey BD; Evangelista E; Fan K; Guo F; Guo H; Guo J-T; Harasym TO; Kadhim S; Kultgen SG; Lee ACH; Li AHL; Long Q; Majeski SA; Mao R; McClintock KD; Reid SP; Rijnbrand R; Snead NM; Steuer HMM; Stever K; Tang S; Wang X; Zhao Q; Sofia MJ Preclinical Profile of AB-423, an Inhibitor of Hepatitis B Virus Pregenomic RNA Encapsidation, Antimicrob Agents Chemother. May 2018, 62 (6) e00082–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mani N; Cole AG; Phelps JR; Ardzinski A; Burns R; Chiu T; Cuconati A; Dorsey BD; Evangelista E; Fan K; Guo F; Harasym TO; Kadhim S; Kowalski R; Kultgen SG; Lee ACH; Li AH; Majeski SA; Miller A; Pasetka C; Reid SP; Rijnbrand R; Micolochick SHM; Stever K; Tang S; Teng X; Wang X; Sofia MJ Preclinical characterization of AB-506, an inhibitor of HBV replication targeting the viral core protein. Antiviral Res. 2022. Jan;197:105211. [DOI] [PubMed] [Google Scholar]
- 31.Du J; Kaplan JA, Kirschberg TA, Kobayashi T, Lazerwith SE, Lee RA, Medley JW, Mitchell ML, Morganelli PA, Pyun H-J, Shevick SL, Squires NH, Watkins WJ, Substituted pyrrolizine compounds and uses thereof. WO2018039531. 2018. March 01.
- 32.Vendeville SMH; Last SJ; Demin SD; Grosse SC; Haché GYP; Hu L; Pieters SMA; Rombouts G; Vandyck K; Verschueren WG; Raboisson PJ-MB Cyclized sulfamoylarylamide derivatives and the use thereof as medicaments for the treatment of hepatitis B. WO2017001655.
- 33.Wang Z; Fan G; Wang X; Thiourea and urea compound and use thereof. WO2018133845.
- 34.Hwang N, Ban H, Wu S, McGuire K, Hernandez E, Cheng J, Zhao Q, Suresh M, Blass B, Viswanathan U, Kulp J, Chang J, Clement J, Menne S, Guo JT, Du Y. 4-Oxooctahydroquinoline-1(2H)-carboxamides as hepatitis B virus (HBV) capsid core protein assembly modulators. Bioorg Med Chem Lett. 2021. Dec 31;58:128518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwang N, Ban H, Chen J, Ma J, Liu H, Lam P, Kulp J, Menne S, Chang J, Guo JT, Du Y. Synthesis of 4-oxotetrahydropyrimidine-1(2H)-carboxamides derivatives as capsid assembly modulators of hepatitis B virus. Med Chem Res. 2021;1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aebi J; Bur D; Chucholowski A; Dehmlow H; Kitas EA; Obst U; Wessel HP Preparation of pyrrolidinethiols and analogs as metalloprotease inhibitors, WO2002008185 A1. Page 114
- 37.Bair KW; Herbertz T; Kauffman GS; Kayser-Bricker KJ; Luke GP; Martin MW; Millan DS; Schiller SER; Talbot AC; Tebbe MJ, Benzopiperazine compositions as BET bromodomain inhibitors and their preparation, WO2015074081 A1.
- 38.Zhou G; Ting PC; Wishart G; Zorn N; Aslanian RG; Lin M; Smith M; Walker SS; Cook J, Van Heek M; Lachowicz J Discovery of novel quinoline carboxylic acid series as DGAT1 inhibitors. Bioorg Med Chem Lett. 2014;24(7):1790–1794. [DOI] [PubMed] [Google Scholar]
- 39.Schrödinger Release 2021-3: Maestro, Schrödinger, LLC, New York, NY, 2021. [Google Scholar]
- 40.Ren Q; Liu X; Luo Z; Li J; Wang C; Goldmann S; Zhang J; Zhang Y Discovery of hepatitis B virus capsid assembly inhibitors leading to a heteroaryldihydropyrimidine based clinical candidate (GLS4). Bioorg Med Chem. 2017. Feb 1;25(3):1042–1056. [DOI] [PubMed] [Google Scholar]
- 41.Qiu Z; Lin X; Zhang W; Zhou M; Guo L; Kocer B; Wu G; Zhang Z; Liu H; Shi H; Kou B; Hu T; Hu Y; Huang M; Yan SF; Xu Z; Zhou Z; Qin N; Wang YF; Ren S; Qiu H; Zhang Y; Zhang Y; Wu X; Sun K; Zhong S; Xie J; Ottaviani G; Zhou Y; Zhu L; Tian X; Shi L; Shen F; Mao Y; Zhou X; Gao L; Young JAT; Wu JZ; Yang G; Mayweg AV; Shen HC; Tang G; Zhu W Discovery and Pre-Clinical Characterization of Third-Generation 4-H Heteroaryldihydropyrimidine (HAP) Analogues as Hepatitis B Virus (HBV) Capsid Inhibitors. J Med Chem. 2017. Apr 27;60(8):3352–3371. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.