

Summary
Grain size is one of the important traits in wheat breeding programs aimed at improving yield, and cytokinins, mainly involved in cell division, have a positive impact on grain size. Here, we identified a novel wheat gene TaMADS‐GS encoding type I MADS‐box transcription factor, which regulates the cytokinins signalling pathway during early stages of grain development to modulate grain size and weight in wheat. TaMADS‐GS is exclusively expressed in grains at early stage of seed development and its knockout leads to delayed endosperm cellularization, smaller grain size and lower grain weight. TaMADS‐GS protein interacts with the Polycomb Repressive Complex 2 (PRC2) and leads to repression of genes encoding cytokinin oxidase/dehydrogenases (CKXs) stimulating cytokinins inactivation by mediating accumulation of the histone H3 trimethylation at lysine 27 (H3K27me3). Through the screening of a large wheat germplasm collection, an elite allele of the TaMADS‐GS exhibits higher ability to repress expression of genes inactivating cytokinins and a positive correlation with grain size and weight, thus representing a novel marker for breeding programs in wheat. Overall, these findings support the relevance of TaMADS‐GS as a key regulator of wheat grain size and weight.
Keywords: type I MADS‐box, cytokinin, grain size, H3K27me3, wheat
Introduction
Wheat (Triticum aestivum L.) is one of the most important staple crops, contributing 20% of calories and protein consumed worldwide (Pfeifer et al., 2014). Grain size and weight, depending on are prime breeding targets for increasing wheat yield. Both grain size and weight depend on seed development: a highly coordinated processes that relies on the concerted action of various regulatory pathways (Li and Li, 2016b; Liu et al., 2023). The genetic control of grain size and weight has been extensively investigated in many crops (Li et al., 2019; Li and Li, 2016a; Sun et al., 2013; Xiao et al., 2022). For example, in cereals the activity of genes promoting starch synthesis and accumulation during the middle‐to‐later stage of seed development is positively correlated with the grain size and weight (Cai et al., 2018; Xiao et al., 2022). Early endosperm development also plays a major role in determining grain size and weight. Arabidopsis (Arabidopsis thaliana) VQ motif protein HAIKU1 (IKU1), leucine‐rich repeat kinase HAIKU2 (IKU2), and the MINISEED3 (MINI3) member of the WRKY transcription factors (TFs) family act in the same pathway to promote early stages of seed development (Luo et al., 2005; Wang et al., 2010). Mutations affecting the function of these genes show precocious cellularization, leading to a premature arrest of seed growth, which ultimately results in seed size reduction. Similarly, mutation of the rice (Oryza sativa) OsbZIP76 (Basic region/leucine zipper motif) causes precocious endosperm cellularization and seed size reduction (Niu et al., 2020). In maize, VSK1 (varied‐kernel‐size) regulates mitosis and cytokinesis during early endosperm development and vks1 mutant displays a nonuniform small‐kernel phenotype (Huang et al., 2019).
The cytokinins (CKs) signalling pathway plays an essential role in regulating the early stages of seed development. CKs are required for cell division during early developmental stages and the increase of the CKs level leads to higher yield through regulation of grain sink potential (Emery et al., 2000; Jameson and Song, 2016; Rock and Quatrano, 1995; Yang et al., 2000, 2003). The cytokinin oxidase/dehydrogenases (CKXs) are enzymes that inactivate CKs and have been among the major genetic targets for yield improvement in wheat (Chen et al., 2020). For example, the wheat orthologues of the rice OsCKX2, TaCKX6‐D1 (subsequently renamed as TaCKX2.2.1‐3D) and TaCKX6a02 (subsequently renamed as TaCKX2.1‐3D), are associated with grain size, weight, and grain filling rate (Lu et al., 2015; Zhang et al., 2012). TaCKX4‐1 (subsequently renamed as TaCKX4‐3A) is positively associated with chlorophyll content and grain yield (Chang et al., 2015). Silencing of TaCKX2.2.1, TaCKX2.2.2 and TaCKX2.1 by RNA interference (RNAi) enhances grain weight (Jablonski et al., 2021). Similar indications were also reported for other plant species. The suppression of CKX gene expression in Arabidopsis, barley (Hordeum vulgare), cotton (Gossypium hirsutum), and rice improves seed quality and yield by increasing seed number and/or weight (Bartrina et al., 2011; Li et al., 2013, 2016; Zalewski et al., 2012; Zhao et al., 2015). However, the molecular mechanisms underlying the regulation of CKXs expression are still unknown.
Among transcription factors (TFs), type I MADS‐box play a key role in the regulation of female gametophyte and early endosperm development in Arabidopsis and grasses (Bemer et al., 2008; Colombo et al., 2008; Kang et al., 2008; Köhler et al., 2003a; Steffen et al., 2008). In general, a failure of the developmental program during early stages results in seed abortion or in the reduction of seed size. Arabidopsis type I MADS‐box gene PHERES1 (PHE1) is expressed transiently after fertilization in both embryo and endosperm and is epigenetically silenced by the Polycomb group gene MEDEA (MEA) (Köhler et al., 2003b). The seed abortion in the mea mutant is largely mediated by the deregulation of PHE1 expression. PHE1 regulates the expression of other TFs involved in seed development; however, the phe1 mutant has no abnormal seed phenotype, suggesting the presence of functional redundant paralogs (Wang et al., 2020). AGAMOUS‐LIKE 80 (AGL80) is another Arabidopsis type I MADS‐box that affects central cell development (Bemer et al., 2008). In the agl80 mutant, the central cell's nucleolus and vacuole fail to mature properly and endosperm development is not initiated after fertilization. During central cell development, the AGL80 protein physically interacts with the type I MADS‐box AGAMOUS‐LIKE 61 (AGL61) (Steffen et al., 2008). In agl61 mutant, polar nuclei fail to fuse and central cell morphology is abnormal and degenerates before fertilization (Bemer et al., 2008; Portereiko et al., 2006). Furthermore, the inactivation of the type I MADS‐box gene AGAMOUS‐LIKE 62 (AGL62) by the FERTILIZATION INDEPENDENT SEED (FIS) polycomb complex triggers endosperm cellularization during the syncytial phase of endosperm development (Kang et al., 2008). Rice type I MADS‐box genes also play important roles in the early stages of seed development, thus affecting grain size. Heat stress induces the repression of OsMADS82 and OsMADS87 expression, leading to early seed cellularization (Folsom et al., 2014). OsMADS78 and OsMADS79 are essential regulators of early seed developmental transition and their function affects both seed size and quality (Paul et al., 2020). In wheat, 128 genes encoding type I MADS‐box TFs have been identified and annotated, but the investigation of their functions is still limited and their role in seed development is unexplored (Raza et al., 2021). Even more importantly, in all plant species, knowledge about the crosstalk between distinct regulators of the early stage of seed development, such as type I MADS box and plant hormones, is largely lacking. Since hormones are involved in many pathways, affecting several aspects of plant development, their manipulation is expected to induce pleiotropic effects, also affecting undesired phenotypes aside from yield. Thus, to identify how hormones interact with other factors, such as TFs, to regulate early seed development is an essential step forward to specifically modulate grain size and weight, ultimately resulting in an increase in yield.
In this study, we report that two homoeologues, encoding a type I MADS‐box TF and named TaMADS‐GS‐A and ‐D, regulate grain size and weight in wheat by stabilizing the CKs signalling pathway. TaMADS‐GS genes are specifically expressed in seed, with higher transcript levels at early stage of seed development. Results achieved using Tamads‐gs knockout mutants and other assays show that TaMADS‐GS protein binds the putative proximal promoter of specific TaCKX genes and represses their expression, through physically interacting with components of Polycomb Repressive Complex 2 (PRC2) to induce the accumulation of the histone H3 trimethylated at lysine 27 (H3K27me3) in these genes. This pathway leads to the stabilization of CK levels, which positively correlates with seed size and weight. In addition, an elite TaMADS‐GS‐A haplotype was found by screening a large germplasm collection and showed to be positively associated with grain size and weight. These findings provide a novel gene related to wheat grain size and represent the first report of the molecular mechanisms underlying the crosstalk between a type I MADS box and the hormone CK to regulate the early stages of seed development.
Results
Identification of type I MADS‐box genes putatively involved in the regulation of early stages of seed development
A total of 128 putative proteins had been annotated as type I MADS‐box TFs in the wheat genome (Raza et al., 2021). Using publicly available wheat gene expression atlas data from RNA sequencing (RNA‐seq), including 36 sets of different tissues/organs and developmental stages (Ramírez‐González et al., 2018), we found a group of genes belonging to a common expression clade, where expression only occurs in seed tissues and is higher in the early stages of seed development (Figure S1A). Among them, TraesCS2A02G146400 and TraesCS2D02G150900 are homoeologues and, using reverse transcription quantitative PCR (RT‐qPCR), we found that, in seed harvested at 4 days after pollination (DAP), they showed the higher expression level compared to the other three genes (Figure S1B). Thus, we selected these two homoeologues for their functional analysis. TraesCS2A02G146400 accumulated at a higher level than TraesCS2D02G150900 and both homoeologues were expressed from 2‐ to 5‐DAP, with a sharp peak at 4‐DAP (Figure 1a). RNA in situ hybridization showed that both transcripts were detected in the endocarp, nucellar, nucellar projection, and endosperm of 4‐DAP seeds (Figure 1b, Figure S2).
Figure 1.
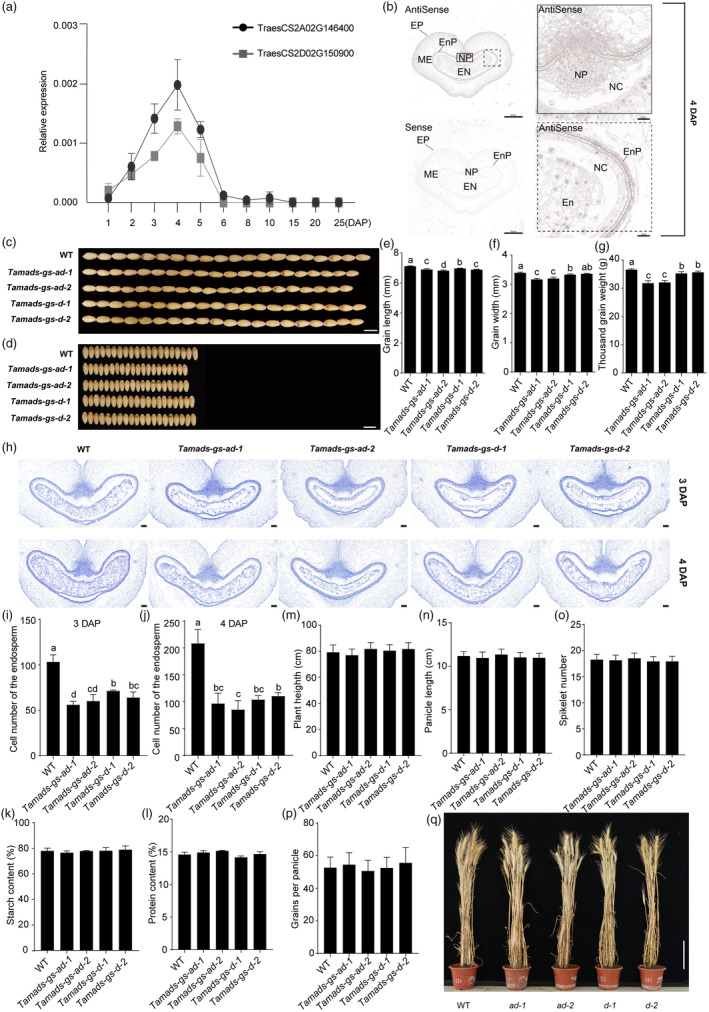
TaMADS‐GS expression in wheat seeds and phenotypes of wheat Tamads‐gs knockout lines. (a) Relative transcript levels of the two TaMADS‐GS homoeologs in the developing seeds at indicated days after pollination (DAP), as determined by RT‐qPCR. Data were normalized to wheat TaACTIN and reported as average value ± SD. (b) Representative picture of in situ hybridization assays performed using 4‐DAP seed and hybridized with antisense and sense TaMADS‐GS‐D transcript probes. EP, exocarp; ME, mesocarp; Enp, endocarp; NP, nucellar projection; NC, nucellar; En, endosperm. Scale bar in left and right sides of the panel represents 500 μm and 50 μm, respectively. (c–g) Grain width, grain length, and thousand‐grain weight measured in WT and Tamads‐gs knockout mutants. In c and d, the bar is 7 mm. Ten biological replicates were used for analysis, each with approximately 20 g seeds. Different letters above the graph bar indicate a statistically significant difference (P < 0.05) between different genotypes, while the presence of the same letter indicates the absence of statistically significant difference. (h) Representative cross sections of 3‐ and 4‐DAP seeds of the indicated genotypes. En, endosperm. Scale bar represents 100 μm. (i and j) The cell number of the endosperm in WT and Tamads‐gs knockout mutants. (k and l) The starch and protein content between WT and Tamads‐gs knockout mutants. (m–p) Plant height, panicle length, spikelet number and grains per panicle were measured in WT and Tamads‐gs knockout mutants. (q) Mature plants of WT and Tamads‐gs mutants. Scale bar, 16 cm. A total of 30 plants were selected to analyse plant height, panicle length, spikelet number and grains per panicle.
TaMADS‐GS knockout lines showed a lower grain size and weight, related to delayed endosperm cellularization
TraesCS2A02G146400 and TraesCS2D02G150900 knockout lines were generated in the wheat cultivar Fielder genetic background, using the CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 (CRISPR‐associated protein 9) technology. To target both homoeologues, two single guide RNAs (sgRNAs) were cloned in a unique vector (Figure S3). Four independent knockout lines were obtained and plants of all of them produced grains of smaller size compared to wild type (WT) (Figure 1c,d). Accordingly, by considering the “grain size” (GS) phenotype, TraesCS2A02G146400 and TraesCS2D02G150900 were named as TaMADS‐GS‐A and TaMADS‐GS‐D, respectively. In the two knockout lines Tamads‐gs‐ad‐1 and Tamads‐gs‐ad‐2, both homoeologues were mutated, while in the Tamads‐gs‐d‐1 and Tamads‐gs‐d‐2 lines, only the TaMADS‐GS‐D homoeologue was mutated. In all lines, mutations led to a frameshift and resulted in premature termination (Figure S3).
All four knockout lines showed a lower grain length and width and thousand‐grain weight (TGW) compared to the WT (Figure 1c–g). The Tamads‐gs‐ad double mutants showed a major grain length/width and TGW reduction compared to the Tamads‐gs‐d single mutants. Endosperm cellularization directly affects grain size in wheat and rice (Begcy and Walia, 2015; Niu et al., 2020) and the higher expression of TaMADS‐GS at an early stage of seed development prompts us to detect the alteration of endosperm cellularization in Tamads‐gs mutants. Histochemical analysis of seeds at 3‐ and 4‐DAP indicated that the four knockout lines had a slower rate of endosperm cellularization compared to the WT because, in WT and knockout lines, the complete cellularization was observed at 3‐ and 4‐DAP, respectively (Figure 1h). Cell number of endosperm from seeds of Tamads‐gs mutants at 3‐ and 4‐DAP was significantly lower than that in WT (Figure 1i,j). We measured the starch and protein content of mature seeds of WT and Tamads‐gs mutants, and no significant difference was observed among them, which indicated that TaMADS‐GS did not affect the synthesis of starch and protein (Figure 1k,l). In agreement with the seed‐specific expression of Tamads‐gs, no visible phenotypic differences, including plant height, panicle length, spikelet number, grains per panicle and flowering time, were detected in Tamads‐gs lines compared to WT (Figure 1m–q). Thus, alteration of Tamads‐gs activity does not result in undesired pleiotropic effects.
TaMADS‐GS affects the expression of genes involved in various physiological process
To identify the genes regulated by TaMADS‐GS, we focused on the single Tamads‐gs‐d knockout mutants because the observation of a phenotype in the Tamads‐gs‐d single mutants indicated that the function of TaMADS‐GS‐A and TaMADS‐GS‐D is not completely redundant. In addition, the major effect on seed size and weight observed in Tamads‐gs‐ad double compared to Tamads‐gs‐d single mutants suggest some kind of genetic interactions between the two homoeologues, thus making difficult the interpretation of results achieved from the double mutants, even if compared to results obtained from the single mutants. Transcriptome alteration was evaluated by RNA sequencing (RNA‐seq), using total RNA extracted from 4‐DAP seeds of WT and of the two independent knockout mutants Tamads‐gs‐d‐1 and ‐2. Results indicated that 5288 and 5808 genes were down‐ and up‐regulated in both mutants compared to WT, respectively (Data Set S1).
GO analysis of up‐regulated genes showed that TaMADS‐GS‐D negatively regulates the expression of genes involved in various physiological process, including photosynthesis, carbohydrate metabolism and signalling, cytokinin metabolic process, cell wall, and oxidative stresses (Figure S4A). GO analysis of down‐regulated genes also showed enrichment of various biological processes, including cell division, DNA repair, chromatin and RNA silencing, and microtubule‐based movement (Figure S4B). The downregulation of genes involved in cell division and DNA repair was in agreement with histological results, reporting a positive correlation between TaMADS‐GS‐D and cell number (Figure 1h). Among genes related to microtubule‐based movement, we found 94 kinesin‐like genes (Data Set S1), which promote cell growth and grain size in rice (Kitagawa et al., 2010; Ran et al., 2018). The enrichment of genes involved in chromatin‐ and RNA‐mediated silencing suggested that these mechanisms are somehow related to the TaMADS‐GS‐D regulation of grain size and weight.
TaMADS‐GS represses the expression of cytokinin oxidase/dehydrogenases (CKXs) genes
CK signalling pathway plays an essential role in regulating early stages of seed development (Emery et al., 2000; Jameson and Song, 2016; Rock and Quatrano, 1995; Yang et al., 2000, 2003), we focused on the differentially expressed genes involved in cytokinin metabolic process between Tamads‐gs and WT. Among them, nine TaCKX genes were up‐regulated in Tamads‐gs‐d mutant by RNA‐seq (Figure 2a) and the up‐regulation of eight out of nine TaCKX genes was confirmed by qRT‐PCR in both Tamads‐gs‐d single (Figure 2b) and Tamads‐gs‐ad double mutants (Figure S5). Since CKX enzymes are involved in irreversible CK inactivation (Vylíčilová et al., 2020) and trans‐zeatin (tZ) is considered the active CK form in wheat (Nguyen et al., 2020), the CK content was assessed by measuring tZ. Results showed that the tZ content was lower in the Tamads‐gs‐d single and Tamads‐gs‐ad double mutants compared to WT (Figure 2c). These observations are consistent with an increase of CKX enzymatic activity in the knockout mutants.
Figure 2.
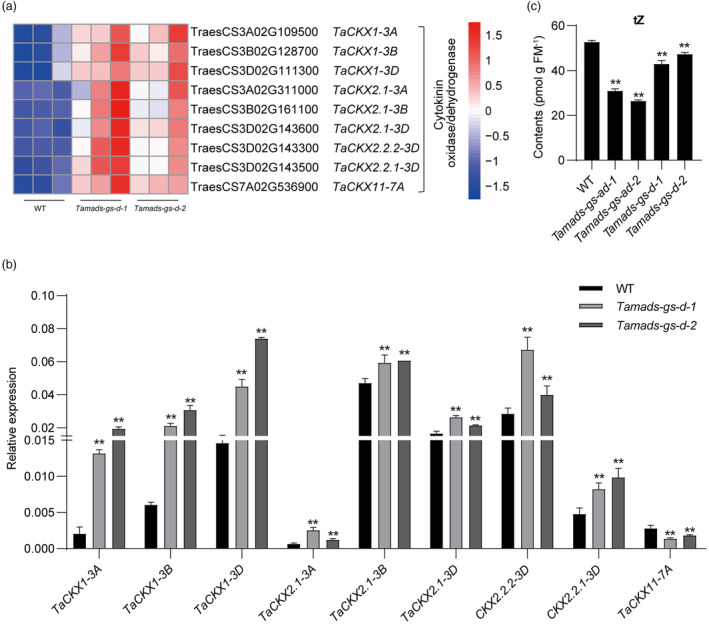
TaMADS‐GS regulates the expression of specific TaCKX genes and the level of active CKs. (a) TaCKX genes up‐regulated in both Tamads‐gs‐d mutants compared to WT, based on RNA‐seq results. (b) Results from RT‐qPCR for validation of different TaCKX transcripts levels in two Tamads‐gs‐d mutants compared to WT. Data were normalized using the TaACTIN gene and reported as average value ± SD. The single and double asterisks represent significant differences compared with WT and determined by Student's t test at P < 0.05 and P < 0.01, respectively. (c) Level of active tZ form of CKs measured in 4‐DAP seeds of WT and Tamads‐gs knockout mutants. Data are reported as average values of three replicates ±SD. Three biological replicates were performed for each sample. The double asterisks indicate a significant difference determined by Student's t‐test at P < 0.01.
MADS‐box proteins bind cis elements with a canonical sequence termed CArG motif (for C‐A/T‐rich‐G) (Dolan and Fields, 1991; Treisman, 1992). The CArG motif was found in the putative promoter (i.e. 2 kb upstream of the ATG) of five out of eight TaCKX genes that were up‐regulated in the Tamads‐gs mutants (Figures 2b and 3d), which prompted us to detect whether TaMADS‐GS could repress TaCKX genes expression. Firstly, transient transformation of wheat protoplasts was employed to verify that both TaMADS‐GS‐A and TaMADS‐GS‐D protein fused with the green fluorescent protein (GFP) localized in the nucleus, which were consistent with the two proteins being transcription factor (Figure S6). Secondly, the dual‐luciferase reporter assay was used to detect the ability of TaMADS‐GS to repress transcription. Results showed that the presence of TaMADS‐GS‐A or TaMADS‐GS‐D counteracted the VP16 transcriptional activation of the reporter gene (Figure 3a,b). Deletion analysis revealed that the MADS‐box domain of TaMADS‐GS was sufficient for the transcriptional inhibitory activity (Figure 3c).
Figure 3.
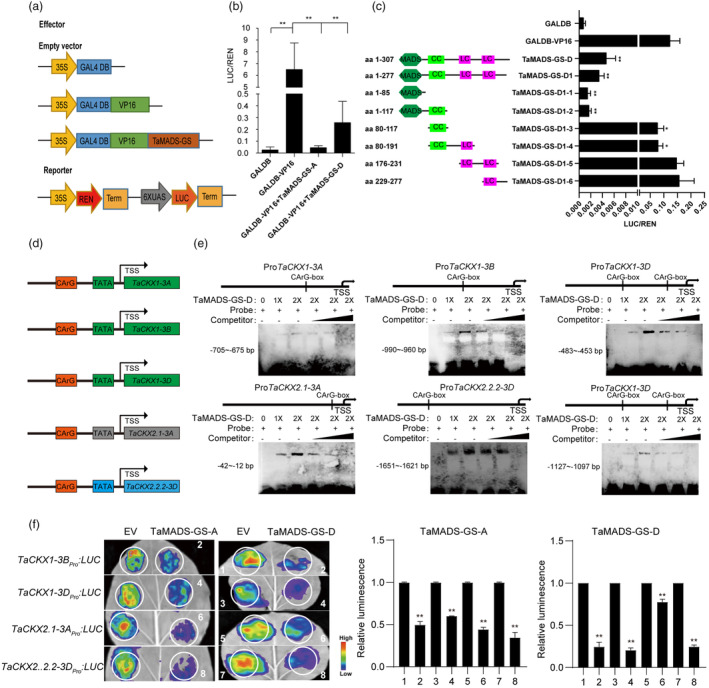
TaMADS‐GS protein represses transcription of specific TaCKX genes. (a) Schematization of constructs used for the dual‐luciferase transient activity assay. The 35S constitutive promoter of the Cauliflower mosaic virus in the reporter construct drives the expression of the Renilla luciferase (REN) gene and the REN signal was used as the control to measure the firefly luciferase (LUC)/Renilla luciferase (REN) ratio. (b) TaMADS‐GS acts as a transcriptional repressor, as determined by dual‐luciferase transient activity assay. Luciferase activity was strongly repressed by the GAL4DB‐VP16‐TaMADS‐GS‐D fusion protein compared to GAL4DB‐VP16. Average values of LUC/REN ratio ± SD (n = 10) are reported. The double asterisks indicate a significant difference determined by Student's t‐test at P < 0.01. (c) Transient transcriptional assays of the TaMADS‐GS‐D deletions schematized. The double asterisks indicate a significant difference determined by Student's t‐test at P < 0.01. (d) The CArG motif was found in the putative promoter of TaCKX genes. (e) Representative images of results achieved with EMSA assays. “+” indicates the addition the biotin‐labelled probe at different concentrations (1X or 2X). Unlabeled probe was used as competitor. (f) Representative image (left panel) and quantitative results (right panel) of the transient transcriptional assays. Heat scale from blue to red is reported and represent the measured LUC signal. Red means strong activation, and blue means weak activation. Average values ± SD (n = 3) are reported. The LUC signal detected when the reporter construct was co‐transformed with a construct expressing the GFP alone was set to 1, while the relative LUC value with respect to the reporter + GFP alone was calculated when the reporter construct was co‐transformed with the construct expressing the TaMADS‐GS‐D protein fused to GFP. The asterisk represents statistically significant differences compared between 1 and 2, 3 and 4, 5 and 6, 7 and 8, as determined by Student's t‐test at P < 0.01.
Electrophoretic mobility shift assay (EMSA) was performed and a dosage‐dependent TaMADS‐GS‐D binding was found for all five TaCKX promoters (Figure 3e). We selected four genes showing stronger TaMADS‐GS‐D binding to test the repression ability to TaCKX genes. The 2‐Kb proximal promoter of these TaCKX genes was cloned and placed upstream of the luciferase reporter gene; the resulting constructs, along with a plasmid expressing the TaMADS‐GS‐A or TaMADS‐GS‐D, were transiently co‐transformed in N. benthamiana leaves. The results showed that TaMADS‐GS repressed the expression of all tested promoters (Figure 3f). Altogether, these findings suggest that TaMADS‐GS binds and directly represses expression of TaCKX genes.
TaMADS‐GS regulates the enrichment of H3K27me3 on TaCKXs
H3K27me3 marks are associated with gene repression for tissues‐specific genes. Using the published epigenomic landscapes during wheat embryogenesis (Zhao et al., 2023), we found enrichment of H3K27me3 was increased during seed early development from 0‐ to 6‐DAP, accompanied by the decreased expression of TaCKX genes, which indicated that H3K27me3 was associated with repression of TaCKX genes. We would determine whether TaMADS‐GS regulates H3K27me3 modification on TaCKX genes for transcriptional repression during the endosperm cellularization process (Figure 4a).
Figure 4.
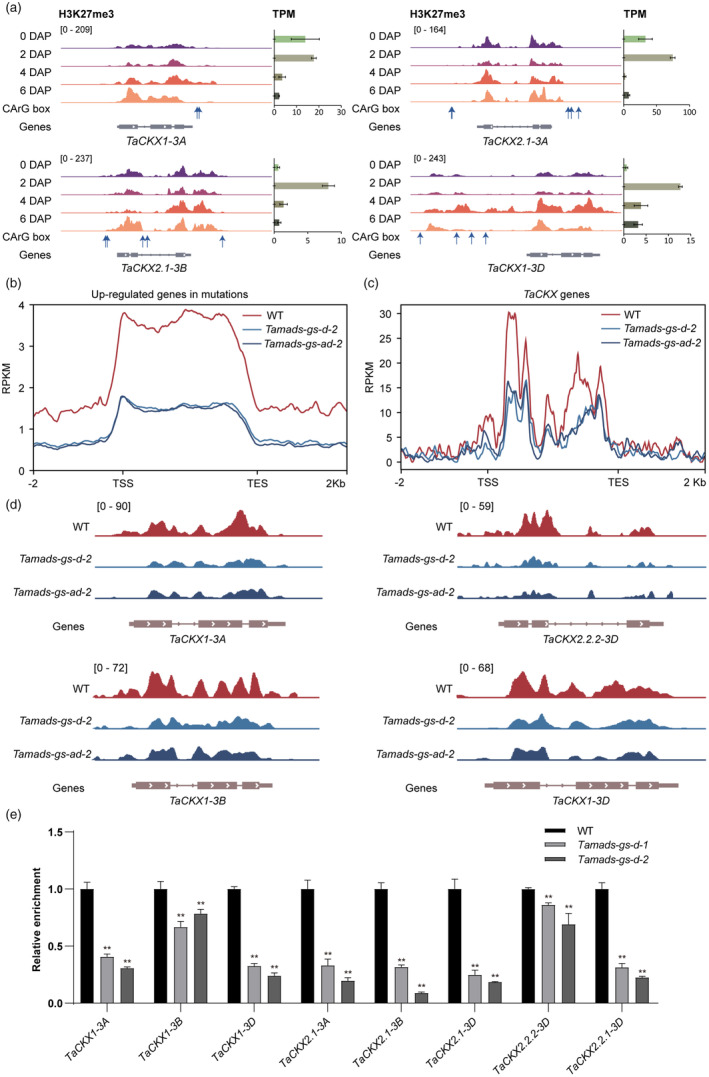
TaMADS‐GS regulates the enrichment of H3K27me3 on TaCKX. (a) The dynamic changes of H3K27me3 pattern at four TaCKX genes in seeds at 0, 2, 4 and 6 days after pollination. (b) H3K27me3 signals on the up‐regulated genes (RNA‐seq) in Tamads‐gs compared with WT peaks. (c) H3K27me3 signals on the TaCKXs in Tamads‐gs and WT. (d) Genome browser views of H3K27me3 levels on TaCKXs in Tamads‐gs compared with wild type. (e) Reduction of H3K27me3 level in TaCKX genes that are up‐regulated in the Tamads‐gs‐d mutants. ChIP‐qPCR was performed with three biological replicates and three technical replicates for each biological replication were performed. The average values of the mutants were standardized to the average value of the WT set to one. Standard deviation is reported. Double asterisk represents statistically significant differences compared to WT and determined by Student's t‐test at P < 0.01.
We performed the Cleavage Under Targets and Tagmentation (CUT&‐Tag) assay with anti‐H3K27me3 antibody to map the H3K27me3 on 4‐DAP seeds from WT, Tamads‐gs‐d‐2 and Tamads‐gs‐ad‐2 mutants. We identified 81 959 high confidence H3K27me3 peaks (18 333 genes) in Tamads‐gs‐d‐2, 61 466 peaks (13 146 genes) in Tamads‐gs‐ad‐2, and 76 432 peaks (16 452 genes) in WT. The peak number of Tamsds‐gs‐d‐2 was similar to that of the WT, but the peak number of Tamsds‐gs‐ad‐2 decreased by about 20% less than WT (Figure S7A). The peak distribution of the three samples were similar in the genome (Figure S7B).
For the 5808 up‐regulated genes in Tamsds‐gs mutants, the levels of H3K27me3 were decreased in both mutants relative to the WT (Figure 4b). This result indicated that TaMADS‐GS was associated with repression of genes with lower expression at early stage of seed development. Then we focused on the changes of H3K27me3 of eight TaCKX genes that were up‐regulated in the Tamads‐gs mutants, the result show that the H3K27me3 level in eight TaCKX genes is lower in the Tamads‐gs‐ad‐2 and Tamads‐gs‐d‐2 knock‐out lines, compared to WT (Figure 4c,d). Chromatin immunoprecipitation (ChIP) with antibody specific for H3K27me3 was performed using chromatin extracted from 4‐DAP seeds of the four independent Tamads‐gs mutants and WT. The immunoprecipitated DNA was used for quantitative PCR (qPCR) and the results showed that all eight TaCKX genes up‐regulated in Tamads‐gs mutants had a statistically significant reduction of the H3K27me3 level in the Tamads‐gs mutants compared to WT (Figures 4e, S7C). These findings indicate that TaMADS‐GS induces the accumulation of H3K27me3 in the TaCKX genes suggesting the mechanism related to the repression of their expression.
TaMADS‐GS interacts with TaFIE1 and TaCLF1
In plants, PRC2 mediates H3K27me3 deposition, an epigenetic mark of transcriptionally repressed chromatin to prevent the improper ectopic activation of gene expression (Hansen et al., 2008; Shen et al., 2021; Wang et al., 2022). Based on the presence of CArG box at the H3K27me3 enrichment areas of TaCKX genes (Figure 4a), we speculated that TaMADS‐GS may be involved in recruiting PRC2. Thus, physical interaction between TaMADS‐GS‐D and main members of the PRC2 complex, including Extra sex combs (ESC) and Enhancer of zeste [E (z)], were tested. In plants, the Drosophila ESC has been named as FERTILIZATION INDEPENDENT ENDOSPERM1 (FIE1). A total of seven TaFIE sequences were identified in wheat genome (Strejčková et al., 2020). Phylogenetic analysis showed that wheat TaFIE proteins are divided in two clades named as TaFIE1 and TaFIE2 (Figure S8A). Among them, homoeologues encoding TaFIE1 were highly expressed in early seed development (Figure S8B), thus, we picked one homoeologue TaFIE1‐7D1 to test its interaction with TaMADS‐GS‐D. The results indicated that TaFIE1‐7D1 can interact with TaMADS‐GS‐D by yeast two‐hybrid (Y2H) and LCI assays (Figure 5a,b). Nine putative wheat E(z) orthologues were found in wheat genome and they are divided into three phylogenetically distinct clades, named as TaCLF1, TaCLF2 and TaSWN (Figure S8C). Among these genes, three homoeologues encoding TaCLF1 were highly expressed during early seed development (Figure S8D), and we selected TaCLF1‐7D1.1 to test interaction with TaMADS‐GS‐D. We observed an interaction between TaCLF1‐7D1.1 and TaMADS‐GS‐D by Y2H and LCI assays (Figure 5c,d).
Figure 5.
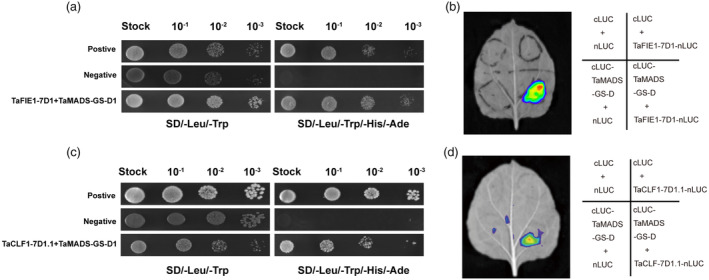
TaMADS‐GS protein interacts with distinct PRC2 components. (a) Representative image of the Y2H assay shows that TaMADS‐GS‐D1 interacts with TaFIE1. (b) Representative image of results achieved with LCI assay showing that TaMADS‐GS‐D interacts with TaFIE1. (c) Representative image of the Y2H assay showing that TaMADS‐GS‐D1 interacts with TaCLF1. (d) Representative image of results achieved with LCI assay showing that TaMADS‐GS‐D interacts with TaCLF1.
A specific TaMADS‐GS‐A haplotype positively correlates with wheat grain size and weight
Since using CRISPR/Cas9 method we were unable to achieve a knockout mutant only impairing TaMADS‐GS‐A function, we used natural genetic variation in the TaMADS‐GS‐A homoelogue to assess its association with grain size and weight. A wheat germplasm collection containing 233 accessions, including 129 modern cultivars and 104 landraces (Data Set S2) was analysed. No genetic polymorphism was found in the coding sequence of TaMADS‐GS‐D. This observation suggests that this homoeologue could not accumulate mutations due to its functional relevance, although it must be also considered that the D subgenome exhibits a low genetic diversity because, during the hybridization between Triticum turgidum (AABB) and Aegilops tauschii (DD) that formed the hexaploid Tritucum aestivum (AABBDD), only a small fraction of the D genome diversity was captured (Zhou et al., 2020). Conversely, four haplotypes were identified in the coding sequence of TaMADS‐GS‐A. They were named Hap1, Hap2, Hap3, and Hap4 and differ based on amino acid sequence variation at position 3, 16, and 126 (Figure 6a). Interestingly, 65% of landraces contained the Hap2 haplotype (Arg3‐Arg16‐Asn126), while 68% of modern cultivars possessed the Hap1 haplotype (Arg3‐Gln16‐Asn126) (Figure 6b). The Hap3 haplotype (Arg3‐Arg16‐Lys126) was present in a lower percentage and a similar distribution was found in landraces and modern cultivars (12 and 9%, respectively). The Hap4 haplotype (His3‐Arg16‐Asn126) was only detected in landraces at a frequency of 8% (Figure 6b). The finding that the Hap1 haplotype was enriched in modern cultivars characterized by higher yield compared to landraces supported a positive correlation between TaMADS‐GS‐A and yield. Accordingly, grain length, grain width, and TGW was higher in Hap1 (n = 104) compared to Hap2 (n = 97), Hap3 (n = 24), and Hap4 (n = 8) accessions (Figure 6c–e). In addition, luciferase‐based assays showed that the transient expression of the TaMADS‐GS‐A gene of the Hap1 haplotype repressed the expression of TaCKX1‐3B and TaCKX2.1‐3A promoters at a statistically significant higher level compared to other haplotypes (Figure 6f). Altogether, these results indicate that Hap1 haplotype represents an elite TaMADS‐GS‐A allele associated with larger grain size and weight and suggest that TaMADS‐GS‐A function, although not fully redundant, is similar to its TaMADS‐GS‐A homoeologue regarding the ability to repress TaCKX genes.
Figure 6.
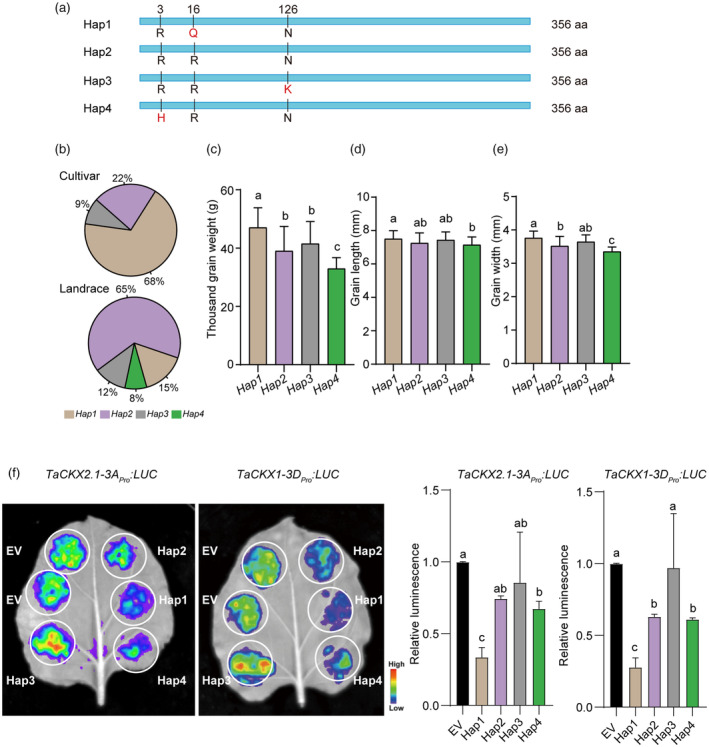
TaMADS‐GS‐A genetic haplotype is positively associated with grain size and weight. (a) Amino acid sequences alignment of four TaMADS‐GS‐A haplotypes (Hap1‐Hap4) indicating the detected polymorphisms. (b) Distribution of Hap1‐Hap4 haplotypes among 130 modern cultivars and 103 landrace accessions. (c–e) Multiple comparisons of the grain width, grain length and TGW of Hap1 (n = 104), Hap2 (n = 97), Hap3 (n = 24) and Hap4 (n = 8). Different letters above the graph bar indicate a statistically significant difference (P < 0.05) between different haplotypes, while the presence of the same letter indicates the absence of statistically significant difference. (f) Representative image (left panel) and quantitative results (right panel) with the transient transcriptional assays. Red means strong activation, and blue means weak activation. Error bars indicate SD (n = 3). Values by the different letters indicate a significant difference at P < 0.05.
Discussion
In this study, we used Tamads‐gs knockout mutants and identified specific genetic haplotypes to show that the two type I MADS‐Box TaMADS‐GS homoeologues positively regulate grain size and weight in wheat. Results from Tamads‐gs‐d knockout mutants indicated that TaMADS‐GS‐D represses the expression of specific TaCKX genes and induces the accumulation of H3K27me3 in these genes. Additional experiments confirmed the repressive activity of TaMADS‐GS‐D protein and showed that it binds the promoter of specific TaCKX genes and interacts with different components of PRC2. The TaMADS‐GS‐A protein also possesses some of these features. In addition, the TaMADS‐GS‐A Hap 1 haplotype with higher ability to repress TaCKX genes compared to other haplotypes, positively correlates with grain size and weight. These results suggest that both TaMADS‐GS‐A and ‐D homoeologues share similar mechanisms in regulating TaCKX gene expression. However, the observation of an evident phenotype in the knockout mutants, where only the TaMADS‐GS‐D homoeologue activity was impaired, as well as a larger reduction of grain size and weight in the Tamads‐gs‐ad double mutants compared to the Tamads‐gs‐d single mutants, indicate that the function of the two homoeologues is not fully redundant. In the future, the homozygous single mutant of Tamads‐gs‐a will be available to distinguish the contribution of two homoeologues.
Altogether, these findings prompt us to propose a novel molecular mechanism underlying seed size and weight regulation, which involves the crosstalk between a type I MADS box TF and CKs and occurs during the early stages of seed development (Figure 7). In wide type seed, TaMADS‐GS directly binds to the promoter of specific TaCKX genes and recruits PRC2, which, in turn, induces the accumulation of H3K27me3 in the gene body of these genes, leading to the repression of their expression. Since TaMADS‐GS is highly expressed in early stage of seed development (i.e. at 4‐DAP, during the endosperm cellularization phase), the lower expression of TaCKX genes avoid CKs inactivation during this key stage, where CKs are required for rapid endosperm nuclear and cell division (Bennett et al., 1973; Jameson and McWha, 1982). Conversely, in Tamads‐gs knockout mutants, the inactivation of this type I MADS‐box TF results in the up‐regulation of TaCKX genes, with the reduction of active CKs content that, in turn, delayed endosperm cellularization, a reduction of grain size and weight.
Figure 7.
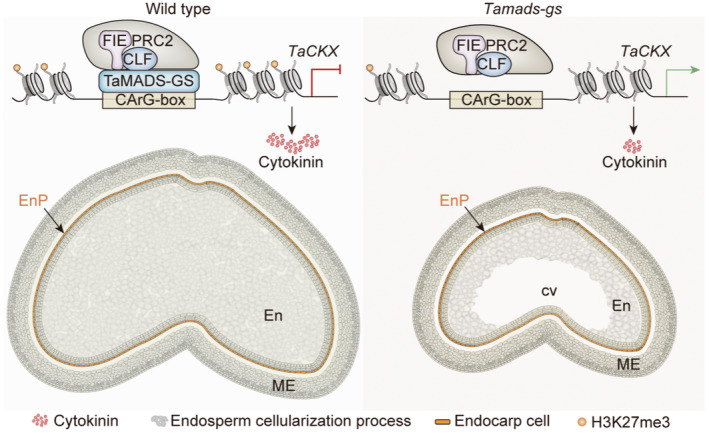
Model of TaMADS‐GS mediated regulation of grain size and weight by stabilizing the CKs signalling pathway. Schematization of the molecular mechanisms in WT and Tamads‐gs knockout mutant. See the text for details. ME, mesocarp; Enp, endocarp; En, endosperm.
The CArG motif was identified in the putative 2‐kb proximal promoter of five out of eight TaCKX genes that were up‐regulated in the Tamads‐gs‐d mutants. However, the reduction of H3K27me3 level was detected for all eight genes. This suggests that TaMADS‐GS‐D protein could bind some TaCKX genes and recruit PRC2 in other regions; for example in distally located cis‐regulatory elements with the CArG motif. In addition, RNA‐seq results indicated that TaMADS‐GS‐D also affects the expression of several genes distinct from TaCKX and not directly involved in the CK signalling pathway. The function of some of these genes is also associated with the cell division and/or grain size and weight. Among the up‐regulated genes in Tamads‐gs mutants compared to WT, some of them were not modified by H3K27me3, which indicated that these genes might be regulated by TaMADS‐GS trough additional pathways. Although it remains to be established what of these genes are regulated by direct TaMADS‐GS‐D binding to their promoters or by indirect effects, as well as whether PRC2 and H3K27me3 are also engaged in the regulation of these genes, this finding suggests that TaMADS‐GS‐D modulates grain size and weight not only acting on CKs stability but even through additional pathways.
So far, the majority of genes identified as regulators of grain size and weight in wheat affected middle and late stages of seed development (Jiang et al., 2011; Volpicella et al., 2016; Zhang et al., 2017). In addition, although the positive correlation between CKs and endosperm cellularization was already reported in rice (Yang et al., 2002), this study represents the first evidence that this connection also occurs in wheat. The delayed cellularization observed in Tamads‐gs knockout seed could be due to (i) the delay of the transition from syncytium to cellularization of the endosperm and/or (ii) the reduction of cellularization rate, because both process are related to CKs pathway. Thus, timely transition of endosperm from syncytial to cellularization ensures optimal nutritional quality of the seed (Aguirre et al., 2018). Although the further evidence supporting contribution of TaMADS‐GS to grain yield is still needed in the future, TaMADS‐GS represents a new candidate for regulating wheat grain size and weight and can be utilized during breeding process. For example, biotechnology‐based approaches could be used to increase active CKs form at first stages of endosperm development by specifically overexpressing TaMADS‐GS during these stages. Furthermore, the elite TaMADS‐GS‐A Hap 1 haplotype represents a novel genetic marker to be employed through classical breeding programs exploiting natural genetic variation.
Finally, our results are consistent with previous reports in wheat, Arabidopsis and other cereals indicating that increasing endogenous CK levels is one of strategies to increase yield (Cucinotta et al., 2018; Nguyen et al., 2020; Pineda Rodo et al., 2008; Shang et al., 2016). Thus, it is possible that similar mechanisms, connecting type I MADS‐box and CKs regulation of endosperm cellularization, are also conserved in other cereals. The information provided in this study will help in testing this hypothesis.
Materials and methods
Plant materials
The names and geographical origins of all wheat cultivars and landraces accessions used in this study are listed in Data Set S3. All wheat plants were grown in the experimental field of China Agricultural University in Beijing (40°08′15″N, 116°11′24″E) or in a greenhouse at a relative humidity of 75% and 26/20 °C day/night temperatures, with a light intensity of 3000 lux (Master GreenPower CG T 400W E40; Philips). Wild‐type and Tamads‐gs seeds were planted in a block containing five rows (1.5 m long) at a spacing of 20 cm in the field. Mature seeds from the plot were harvested and seeds from every six plants were mixed to represent a biological replicate, ten biological replicates were performed for phenotypic assessment. For gene expression analysis, whole seeds from 1‐ to 6‐DAP, 8‐DAP, 10‐DAP and the seeds without embryos of 15‐DAP, 20‐DAP and 25‐DAP were harvested from greenhouse. For each sample, seeds were collected from three different plants and mixed to represent a biological replicate and at least three replicates were performed. All samples were immediately frozen in liquid nitrogen and stored at −80 °C.
Phenotype analysis
The seed‐related phenotypes including TGW, grain length and width were determined using a camera‐assisted phenotyping system (Wanshen Detection Technology Co., Ltd, Hangzhou, China). Ten biological replicates were analysed, each with approximately 20 g seeds. A total of 30 plants were selected to analyse plant height, panicle length, spikelet number and grains per panicle. The cell number of endosperm was counted on the paraffin section by the microscope imaging system (DS‐U3, Nikon, Japan). Five replicates from five seeds were performed. Starch content was determined using Total Starch Assay Kit (Megazyme; Catalogue no.: KTSTA‐50A) based on the use of thermostable α‐amylase and amyloglucosidase as previously described (Gao et al., 2021). Three repeats were performed for each sample. Protein content was performed by near‐infrared spectrometry following protocols as previously described (Lin et al., 2014). Three repeats were performed for each sample.
RT‐qPCR
Total RNAs was extracted from seeds at the 1‐ to 6‐DAP, 8‐DAP, 10‐DAP, and from seeds without embryo of 15‐DAP, 20‐DAP and 25‐DAP using the TransZol Plant (TransGen Biotech, ET121‐01). First‐strand cDNAs were generated using a reverse transcription kit (Vazyme Biotech, R223‐01) according to the manufacturer's instructions. RT‐qPCR assays were performed using the SYBR Green PCR Master Mix (Vazyme Biotech, Q121‐02/03) with a CFX96 real‐time system (BioRad). The wheat TaACTIN (TraesCS5B02G124100) was used for standardization. The comparative CT (2−ΔCT) method (Schmittgen and Livak, 2008) was used to quantify the different mRNA levels. Three biological replicates and three technical replicates for each biological replicate were performed per gene. The results from one replicate are shown in the figures. Statistical test was performed by applying Student's t‐test at P < 0.05. The RT‐qPCR primers used in this study are listed in Data Set S3.
RNA in situ hybridization
For RNA in situ hybridization, the developing seeds of 4‐DAP harvested from greenhouse were fixed in FAA solution (50% (v/v) ethanol, 5% (v/v) glacial acetic acid and 4% (v/v) formaldehyde) at 4 °C for 24 h and embedded in paraffin. Pretreatment of sections, hybridization, and immunological detection were carried out as described previously (Sang et al., 2012). The sense and antisense RNA probes are listed in Data Set S3. Three replicates from three seeds were performed.
Histological analysis
Immature seeds at 3‐ and 4‐DAP were sampled from selected genotypes grown in the field. Seeds were fixed in FAA solution, subjected to vacuum pumping for 30 min, and embedded in paraffin. The wax block with tissue chips was cut on the paraffin slicer, achieving in slices with a thickness of 4 μm. Dewaxing was performed as follows: xylene I for 20 min, xylene II for 20 min, 100% ethanol I for 5 min, anhydrous ethanol II for 5 min, 100% ethanol II for 5 min, 75% ethanol for 5 min, rinsing with tap water. Plant tissue slices were treated with Toluidine Blue for 2–5 min and rinsed with tap water followed by the addition of xylene for 10 min and finally sealed with neutral gum. Detection was performed using a microscope imaging system (DS‐U3, Nikon, Japan). Five replicates from five seeds were performed.
Production of Tamads‐gs knockout mutants
Two sgRNAs were designed based on the first exon sequence of TaMADS‐GS‐A and TaMADS‐GS‐D, respectively, and using the E‐CRISP Design website (http://www.e‐crisp.org/E‐CRISP/designcrispr.html). Reverse complementary sgRNA sequences were synthesized, digested, and inserted into the expression cassette of the pBUE411 vector (Xing et al., 2014). The construct vector was transformed in wheat cultivar Fielder via Agrobacterium‐mediated (EHA105) transformation following the method previously described (Medvecká and Harwood, 2015).
RNA‐seq and data analysis
Total RNA was extracted from 4‐DAP WT, Tamads‐gs‐d‐1 and Tamads‐gs‐d‐2 seeds harvested from plants grown in the greenhouse using the TransZol (TransGen Biotech, ET121‐01) and following the manufacturers' instructions. Total RNA purity and concentration were examined using NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA), while RNA integrity and quantity were measured using the Agilent 2100 system (Agilent). For each genotype, three biological replicates were used. Used TruSeq RNA Sample Preparation Kit v2 (Illumina) according to the manufacturer's instructions to prepare RNA‐seq libraries and sequenced to generate 150‐bp paired‐end reads on the Illumina NovaSeq 6000 platform. Approximately 48–69 million reads were generated per sample. After screening and trimming, clean reads were aligned to wheat reference genome (IWGSC RefSeq v1.1) using STAR (Dobin et al., 2013). Approximately 39–53 million reads were aligned in the reference genome per sample. Aligned ratio is approximately 77.4%–86.1%. FeatureCounts was used to count the reads mapped to each gene (Love et al., 2014). After using a gene expression criterion of FPKM value ≥1 in at least one sample, DESeq2 v1.24.0 was used for differential expression gene analysis (Love et al., 2014) and genes with log2 (fold change) ≥ 1 and FDR <0.05 were identified as differentially expressed genes. GO enrichment analysis of differentially expressed genes (adjusted P‐value [false discovery rate] < 0.05) was implemented using ClusterProfiler (Yu et al., 2012).
CKs quantification
Endogenous CKs content was measured in 4‐DAP seeds harvested from plants grown in the field by Wuhan Greensword Creation Technology Co. Ltd. (http://www.greenswordcreation.com/index.html), according to a previously reported method based on LC–MS/MS analysis (Liu et al., 2010). Three biological replicates were tested. Statistical test was performed by applying Student's t‐test at P < 0.05.
Yeast two‐hybrid assay
Full‐length TaMADS‐GS‐D was cloned into pGBKT7 (BD) vector (Takara, Japan). However, we observed strong auto‐activation when we fused TaMADS‐GS‐D to the DNA‐binding domain of yeast GAL4 (Figure S9). Based on previous results indicating that the C‐terminal domain allows for transcriptional activation of some MADS‐domain proteins (Honma and Goto, 2001), we deleted the C‐terminal 29 amino acids of TaMADS‐GS‐D (TaMADS‐GS‐D1). Notably, this deletion abolished the self‐activation of the TaMADS‐GS‐D‐GAL4 fusion (Figure S9). For the pairwise yeast two‐hybrid interaction assays, the primers listed using PCR amplification in Data Set S3. Each amplified DNA fragment was cloned into the prey vector pGADT7 (Takara, Japan) and co‐transformed with bait vector expressing the TaMADS‐GS‐D1 genes fused with the BD domain. Yeast cells were spotted in stringent selection medium (synthetic dropout medium lacking Trp, Leu, His, and adenine (‐WLHA) or non‐selective medium lacking Trp and Leu (‐WL). Three replications were performed for each assay.
Firefly luciferase complementation imaging (LCI) assay
A split luciferase complementation assay with the pCAMBIA‐nLUC and pCAMBIA‐cLUC vectors was used to detect protein–protein interactions in N. benthamiana leaves (Chen et al., 2008). Firefly luciferase was divided into the N‐terminal (nLUC) and C‐terminal (cLUC) parts. The coding sequence of selected genes was cloned separately into pCAMBIA‐nLUC and pCAMBIA‐cLUC and co‐transformed in N. benthamiana leaves (Chen et al., 2008). LUC activity was analysed 48 h after infiltration, using the Night SHADE LB 985 (Berthold) system. Three replications were performed for each interaction.
Subcellular localization
TaMADS‐GS‐A and TaMADS‐GS‐D cDNA were amplified using specific primers and cloned upstream and in frame of the GFP gene in the pCAMBIA1300 vector (Chen et al., 2022). The fusion construct pMAS::TaMADS‐GS‐A/D‐GFP and the pMAS::GFP control were used for transient expression assays after PEG‐mediated transfection in wheat mesophyll protoplasts using a previously described method (Shan et al., 2014; Yoo et al., 2007). Transformed cells were cultured at 28 °C for 14 h and the GFP signal was detected with a confocal microscope (Zeiss LSM‐880). Three replicates were performed.
EMSA
The full‐length CDS of TaMADS‐GS‐D was cloned into the pGEX6P‐1 vector (Solarbio, P0300) and in frame with the GST gene. The GST‐tagged TaMADS‐GS‐D protein was expressed in Escherichia coli (BL21 DE3). 0.1 mm isopropyl‐β‐D‐thiogalactopyranoside (IPTG) was used to induce recombinant protein expression in Luria Bertani (LB) buffer overnight at 16 °C. The harvested cells in phosphate‐buffered saline (137 mm NaCl, 10 mm Na2HPO4, 2.7 mm KCl, 2 mm KH2PO4, 1 mm phenylmethanesulfonyl fluoride and 1/4 of a tablet of protease inhibitor cocktail) were sonicated for 1 h, and centrifuged at 13 000 g for 30 min. The supernatant was mixed with GST MAG AgaroseBeads (Cat No. 20508ES50; Yeasen, Shanghai, China) and rocked for 2 h at 4 °C. The fusion protein was eluted from the beads with 50 mm Tris–HCl (pH 8.0) containing 10 mm reduced glutathione.
Oligonucleotide probes of 30 nucleotides were synthesized and labelled with biotin at their 5′‐end (Invitrogen). Double‐stranded probes were achieved by adding complementary oligonucleotides and cooling from 100 °C to room temperature. The sequences of the probes are listed in Data Set S3. The EMSA assays were carried out using the Light Shift Chemiluminescent EMSA Kit (no. 20148; Thermo Fisher Scientific) following manufacturer instructions (Liu et al., 2021). Three replicates were performed.
Transcriptional repression assay
The transcriptional repression assay was based on the dual‐luciferase transient expression in wheat mesophyll protoplasts, which was performed as previously described (Zhou et al., 2021). The TaMADS‐GS‐A or TaMADS‐GS‐D coding sequence was cloned into the vector and in frame with GAL4‐DB and VP16, to generate the effector constructs GAL4DB‐VP16‐TaMADS‐GS‐A or GAL4DB‐VP16‐TaMADS‐GS‐D, respectively. For the reporter construct, a promoter with 6x GAL4 UAS sequence and a TATA box was introduced into pGreenII 0800‐LUC. The reporter and effector constructs were co‐transformed into wheat mesophyll protoplasts, using the previously reported method (Zhou et al., 2021). The empty effector construct was used as control.
Transcriptional luciferase assays through transient transformation of N. benthamiana leaves were performed as previously described (Liu et al., 2021). The 2‐kb putative promoter sequence upstream of the start codon ATG of desired genes was cloned into the plant binary vector pGWB35 (Nakamura et al., 2013) and upstream of the luciferase reporter gene LUC, using the Gateway kit (Invitrogen) to generate the reporter constructs. The effector construct was generated by cloning the TaMADS‐GS‐A or TaMADS‐GS‐D kit into the pCAMBIA1300 expression vector (Liu et al., 2021). The reporters were separately co‐transformed with the effector in N. benthamiana leaves through Agrobacterium‐mediated transient expression (Liu et al., 2021). The luminescence activities were captured and quantified 48 h after infiltration, using NightSHADE LB 985 (Berthold) with Indigo software. In each experiment, three independent N. benthamiana leaves were infiltrated and analysed and three technical replicates were used for quantification. Statistical test was performed by applying Student's t‐test at P < 0.05.
Phylogenetic analysis
Predicted amino acid sequences of different components of wheat PRC2 were reported by Strejčková et al. (2020). Predicted amino acid sequences of PRC2 proteins of Arabidopsis, rice, barley and maize were reported by Tonosaki and Kinoshita (2015). The amino acid sequences of PRC2 domain were aligned with ClustalW (Kumar et al., 2016), and a phylogenetic tree was constructed using the neighbour‐joining method with MEGA 7 software with default parameters (Kumar et al., 2016). The evolutionary distances were calculated using the Poisson model (Hayat and Higgins, 2014). The phylogeny test was computed using bootstrap method with 10 000 replications (Saitou and Nei, 1987).
CUT&‐tag and data analysis
Cleavage Under Targets and Tagmentation (CUT&‐Tag) were performed using seeds at 4‐DAP from two Tamads‐gs mutants (Tamads‐gs‐d‐1 and Tamads‐gs‐d‐2) and wild‐type plants. Two biological replicates were performed independently. The CUT&‐Tag experiment was performed exactly following protocols as previously described (Zhao et al., 2023). Finally, the purified PCR products were sequenced using an Illumina NovaSeq platform.
For next‐generation data analysis, the pipeline was largely based on the previous study (Zhao et al., 2023). The raw fastq data was filtered using fastp (v0.20.0) (Chen et al., 2018). The filtered reads were mapped to wheat reference genome‐Ref‐Seq v1.0 (IWGSC 2018; https://urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Assemblies/v1.0/) using BWA‐MEM (v0.7.17) (Li and Durbin, 2009). MACS2 (v2.1.2) was used for peak calling. To reduce influence of sequencing depth and sample differences, a “reads‐in peaks normalization” method (Corces et al., 2018) was used to obtain high‐quality peaks. The peaks were further assigned to genes using ChIPseeker (Yu et al., 2015). For Cut&Tag signal comparison, ChIPseqSpikeInFree (Jin et al., 2020) (v1.2.4) was first applied to calculate scale factors, and the bam files were converted to bigwig files via deepTools (3.3.0) according to the scale factors (Ramírez et al., 2014).
ChIP‐qPCR
The ChIP assays were performed as previously described (Yang et al., 2016). Briefly, about 1.5 g of 4‐DAP seeds harvested from plants grown in the field were cross‐linked with 1% formaldehyde in GB buffer (0.4 M sucrose, 10 mm Tris–HCl, pH 8.0, and 1 mm EDTA) for 10 min under a vacuum. Chromatin was extracted and fragmented to 200–500 bp by sonication (15 min sonication, 20/40 s on/off, at high energy level) with a Bioruptor Plus System (Qsonica). Ten microliter of the anti‐H3 trimethyl‐Lys 27 antibody (CST C36B11) was used for immunoprecipitation of the protein‐DNA complexes. The immunoprecipitated was dissolved in 30 μL pH 7.5, 10 mm Tris–HCl and a fraction was used as the DNA template for qPCR analysis. De‐cross‐linked DNA from the chromatin fraction before incubation with the antibody was used as control (input sample). The qPCR values were standardized to the input sample (Zheng et al., 2019). Three biological replicates and three technical replicates for each biological replicate were performed per gene. Statistical test was performed by applying Student's t‐test at P < 0.05. Primer sequences are listed in Data Set S3.
Accession numbers
RNA sequencing data are available at the Sequence Read Archive (SRA) under accession no. PRJNA905220. CUT&‐Tag data are available at the Sequence Read Archive (SRA) under accession no. PRJNA956711.
Author contributions
YY and QS conceived the project; JZ performed the experiments; ZZ, LZ performed bioinformatics analysis; RZ, CY, XZ, SC, and QC provided technological assistance; VR, JX, MX, JD, WG, ZH, JL, HP, and ZN provided theoretical contributions to the project; JZ, YY and QS analysed the data and wrote the article.
Conflicts of interest
The authors declare no conflicts of interest.
Supporting information
Figure S1 Spatial and temporal expression of type I TaMADS‐box genes.
Figure S2 Representative picture of in situ hybridization assays performed using 4‐DAP seed and hybridized with antisense and sense TaMADS‐GS‐A transcript probes.
Figure S3 Strategy used to produce the Tamads‐gs knockout lines.
Figure S4 GO analysis of differentially expressed genes in 4‐DAP seed of WT and Tamads‐gs‐d knockout mutants.
Figure S5 Relative transcript levels of different TaCKX genes in Tamads‐gs‐ad mutants and WT.
Figure S6 Subcellular localization of TaMADS‐GS.
Figure S7 The results of CUT&‐Tag and ChIP‐qPCR analysis.
Figure S8 Phylogenetic analysis and spatial and temporal expression of PRC2 proteins.
Figure S9 Full‐length TaMADS‐GS‐D cDNA fused with the GAL4 BD domain can self‐activate the expression of the reporter genes in Y2H assays.
Data Set S1 List of differentially expressed transcripts in the Tamads‐gs mutant and wild type.
Data Set S2 Information of 233 wheat accessions used for haplotype analysis.
Data Set S3 Primers used for gene expression and vector construction.
Acknowledgements
We thank Dr Yusheng Zhao (Chinese Academy of Sciences) for helpful discussions and comments on the text; Dr Rentao Song (China Agricultural University) for providing the transient expression vectors (pGreenII 0800‐LUC). This work was supported by the National Key Research and Development Program of China (2022YFD1200203), National Natural Science Foundation of China (Grant no. 32125030), Hainan Yazhou Bay Seed Lab (no. B21HJ0502), Major Program of National Agricultural Science and Technology of China (NK20220607), Frontiers Science Center for Molecular Design Breeding (no. 2022TC149) and National Natural Science Foundation of China (Grant no. 32001540). Pinduoduo‐China Agricultural University Research Fund (PC2023A01003).
Contributor Information
Qixin Sun, Email: qxsun@cau.edu.cn.
Yingyin Yao, Email: yingyin@cau.edu.cn.
References
- Aguirre, M. , Kiegle, E. , Leo, G. and Ezquer, I. (2018) Carbohydrate reserves and seed development: an overview. Plant Reprod. 31, 263–290. [DOI] [PubMed] [Google Scholar]
- Bartrina, I. , Otto, E. , Strnad, M. , Werner, T. and Schmülling, T. (2011) Cytokinin regulates the activity of reproductive meristems, flower organ size, ovule formation, and thus seed yield in Arabidopsis thaliana . Plant Cell 23, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begcy, K. and Walia, H. (2015) Drought stress delays endosperm development and misregulates genes associated with cytoskeleton organization and grain quality proteins in developing wheat seeds. Plant Sci. 240, 109–119. [DOI] [PubMed] [Google Scholar]
- Bemer, M. , Wolters‐Arts, M. , Grossniklaus, U. and Angenent, G.C. (2008) The MADS domain protein DIANA acts together with AGAMOUS‐LIKE80 to specify the central cell in Arabidopsis ovules. Plant Cell 20, 2088–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M.D. , Rao, M. , Smith, J. and Bayliss, M. (1973) Cell development in the anther, the ovule, and the young seed of Triticum aestivum L. var. Chinese Spring. Philos. Trans. R. Soc. London. B, Biol. Sci. 266, 39–81. [Google Scholar]
- Cai, Y. , Zhang, W. , Jin, J. , Yang, X. , You, X. , Yan, H. , Wang, L. et al. (2018) OsPKpα1 encodes a plastidic pyruvate kinase that affects starch biosynthesis in the rice endosperm. J. Integr. Plant Biol. 60, 1097–1118. [DOI] [PubMed] [Google Scholar]
- Chang, C. , Lu, J. , Zhang, H.P. , Ma, C.X. and Sun, G. (2015) Copy number variation of cytokinin oxidase gene Tackx4 associated with grain weight and chlorophyll content of flag leaf in common wheat. PloS One 10, e0145970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , Zou, Y. , Shang, Y. , Lin, H. , Wang, Y. , Cai, R. , Tang, X. et al. (2008) Firefly luciferase complementation imaging assay for protein‐protein interactions in plants. Plant Physiol. 146, 368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, S. , Zhou, Y. , Chen, Y. and Gu, J. (2018) fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics (Oxford, England) 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, L. , Zhao, J. , Song, J. and Jameson, P.E. (2020) Cytokinin dehydrogenase: a genetic target for yield improvement in wheat. Plant Biotechnol. J. 18, 614–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Q. , Yang, C. , Zhang, Z. , Wang, Z. , Chen, Y. , Rossi, V. , Chen, W. et al. (2022) Unprocessed wheat γ‐gliadin reduces gluten accumulation associated with the endoplasmic reticulum stress and elevated cell death. New Phytol. 236, 146–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, M. , Masiero, S. , Vanzulli, S. , Lardelli, P. , Kater, M.M. and Colombo, L. (2008) AGL23, a type I MADS‐box gene that controls female gametophyte and embryo development in Arabidopsis. Plant J. 54, 1037–1048. [DOI] [PubMed] [Google Scholar]
- Corces, M.R. , Granja, J.M. , Shams, S. , Louie, B.H. , Seoane, J.A. , Zhou, W. , Silva, T.C. et al. (2018) The chromatin accessibility landscape of primary human cancers. Science (New York, N.Y.) 362, eaav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucinotta, M. , Manrique, S. , Cuesta, C. , Benkova, E. , Novak, O. and Colombo, L. (2018) CUP‐SHAPED COTYLEDON1 (CUC1) and CUC2 regulate cytokinin homeostasis to determine ovule number in Arabidopsis. J. Exp. Bot. 69, 5169–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin, A. , Davis, C.A. , Schlesinger, F. , Drenkow, J. , Zaleski, C. , Jha, S. , Batut, P. et al. (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics (Oxford, England) 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan, J.W. and Fields, S. (1991) Cell‐type‐specific transcription in yeast. Biochim. Biophys. Acta‐Gene Struct. Exp. 1088, 155–169. [DOI] [PubMed] [Google Scholar]
- Emery, R.J. , Ma, Q. and Atkins, C.A. (2000) The forms and sources of cytokinins in developing white lupine seeds and fruits. Plant Physiol. 123, 1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folsom, J.J. , Begcy, K. , Hao, X. , Wang, D. and Walia, H. (2014) Rice fertilization‐Independent Endosperm1 regulates seed size under heat stress by controlling early endosperm development. Plant Physiol. 165, 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, Y. , An, K. , Guo, W. , Chen, Y. , Zhang, R. , Zhang, X. , Chang, S. et al. (2021) The endosperm‐specific transcription factor TaNAC019 regulates glutenin and starch accumulation and its elite allele improves wheat grain quality. Plant Cell 33, 603–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, K.H. , Bracken, A.P. , Pasini, D. , Dietrich, N. , Gehani, S.S. , Monrad, A. , Rappsilber, J. et al. (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 10, 1291–1300. [DOI] [PubMed] [Google Scholar]
- Hayat, M.J. and Higgins, M. (2014) Understanding poisson regression. J. Nurs. Educ. 53, 207–215. [DOI] [PubMed] [Google Scholar]
- Honma, T. and Goto, K. (2001) Complexes of MADS‐box proteins are sufficient to convert leaves into floral organs. Nature 409, 525–529. [DOI] [PubMed] [Google Scholar]
- Huang, Y. , Wang, H. , Huang, X. , Wang, Q. , Wang, J. , An, D. , Li, J. et al. (2019) Maize VKS1 regulates mitosis and cytokinesis during early endosperm development. Plant Cell 31, 1238–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski, B. , Szala, K. , Przyborowski, M. , Bajguz, A. , Chmur, M. , Gasparis, S. , Orczyk, W. et al. (2021) TaCKX2.2 genes coordinate expression of other TaCKX family members, regulate phytohormone content and yield‐related traits of wheat. Int. J. Mol. Sci. 22, 4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jameson, P. and McWha, J.A. (1982) Cytokinins and changes in their activity during the development of grains of wheat (Triticum aestivum L.). J. Plant Physiol. 106, 27. [Google Scholar]
- Jameson, P.E. and Song, J. (2016) Cytokinin: a key driver of seed yield. J. Exp. Bot. 67, 593–606. [DOI] [PubMed] [Google Scholar]
- Jiang, Q. , Hou, J. , Hao, C. , Wang, L. , Ge, H. , Dong, Y. and Zhang, X. (2011) The wheat (T. aestivum) sucrose synthase 2 gene (TaSus2) active in endosperm development is associated with yield traits. Funct. Integr. Genomics 11, 49–61. [DOI] [PubMed] [Google Scholar]
- Jin, H. , Kasper, L.H. , Larson, J.D. , Wu, G. , Baker, S.J. , Zhang, J. and Fan, Y. (2020) ChIPseqSpikeInFree: a ChIP‐seq normalization approach to reveal global changes in histone modifications without spike‐in. Bioinformatics (Oxford, England) 36, 1270–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, I.H. , Steffen, J.G. , Portereiko, M.F. , Lloyd, A. and Drews, G.N. (2008) The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell 20, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa, K. , Kurinami, S. , Oki, K. , Abe, Y. , Ando, T. , Kono, I. , Yano, M. et al. (2010) A novel kinesin 13 protein regulating rice seed length. Plant Cell Physiol. 51, 1315–1329. [DOI] [PubMed] [Google Scholar]
- Köhler, C. , Hennig, L. , Bouveret, R. , Gheyselinck, J. , Grossniklaus, U. and Gruissem, W. (2003a) Arabidopsis MSI1 is a component of the MEA/FIE Polycomb group complex and required for seed development. EMBO J. 22, 4804–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, C. , Hennig, L. , Spillane, C. , Pien, S. , Gruissem, W. and Grossniklaus, U. (2003b) The Polycomb‐group protein MEDEA regulates seed development by controlling expression of the MADS‐box gene PHERES1. Genes Dev. 17, 1540–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. and Tamura, K. (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics (Oxford, England) 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N. and Li, Y. (2016a) Signaling pathways of seed size control in plants. Curr. Opin. Plant Biol. 33, 23–32. [DOI] [PubMed] [Google Scholar]
- Li, S. , Zhao, B. , Yuan, D. , Duan, M. , Qian, Q. , Tang, L. , Wang, B. et al. (2013) Rice zinc finger protein DST enhances grain production through controlling Gn1a/OsCKX2 expression. Proc. Natl Acad. Sci. USA 110, 3167–3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Li, X. , Zhou, Z. , Wu, P. , Fang, M. , Pan, X. , Lin, Q. et al. (2016) Reassessment of the four yield‐related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front. Plant Sci. 7, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N. , Xu, R. and Li, Y. (2019) Molecular networks of seed size control in plants. Annu. Rev. Plant Biol. 70, 435–463. [DOI] [PubMed] [Google Scholar]
- Lin, C. , Chen, X. , Jian, L. , Shi, C. , Jin, X. and Zhang, G. (2014) Determination of grain protein content by near‐infrared spectrometry and multivariate calibration in barley. Food Chem. 162, 10–15. [DOI] [PubMed] [Google Scholar]
- Liu, Z. , Wei, F. and Feng, Y.‐Q. (2010) Determination of cytokinins in plant samples by polymer monolith microextraction coupled with hydrophilic interaction chromatography‐tandem mass spectrometry. Anal. Methods 2, 1676–1685. [Google Scholar]
- Liu, J. , Chen, Z. , Wang, Z. , Zhang, Z. , Xie, X. , Wang, Z. , Chai, L. et al. (2021) Ectopic expression of VRT‐A2 underlies the origin of Triticum polonicum and Triticum petropavlovskyi with long outer glumes and grains. Mol. Plant 14, 1472–1488. [DOI] [PubMed] [Google Scholar]
- Liu, H. , Luo, Q. , Tan, C. , Song, J. , Zhang, T. and Men, S. (2023) Biosynthesis‐ and transport‐mediated dynamic auxin distribution during seed development controls seed size in Arabidopsis. Plant J. 113, 1259–1277. [DOI] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, J. , Chang, C. , Zhang, H.P. , Wang, S.X. , Sun, G. , Xiao, S.H. and Ma, C.X. (2015) Identification of a novel allele of TaCKX6a02 associated with grain size, filling rate and weight of common wheat. PloS One 10, e0144765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, M. , Dennis, E.S. , Berger, F. , Peacock, W.J. and Chaudhury, A. (2005) MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine‐rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc. Natl Acad. Sci. USA 102, 17531–17536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvecká, E. and Harwood, W.A. (2015) Wheat (Triticum aestivum L.) transformation using mature embryos. Methods Mol. Biol. (Clifton, N.J.) 1223, 199–209. [DOI] [PubMed] [Google Scholar]
- Nakamura, H. , Xue, Y.L. , Miyakawa, T. , Hou, F. , Qin, H.M. , Fukui, K. , Shi, X. et al. (2013) Molecular mechanism of strigolactone perception by DWARF14. Nat. Commun. 4, 2613. [DOI] [PubMed] [Google Scholar]
- Nguyen, H.N. , Perry, L. , Kisiala, A. , Olechowski, H. and Emery, R.J.N. (2020) Cytokinin activity during early kernel development corresponds positively with yield potential and later stage ABA accumulation in field‐grown wheat (Triticum aestivum L.). Planta 252, 76. [DOI] [PubMed] [Google Scholar]
- Niu, B. , Deng, H. , Li, T. , Sharma, S. , Yun, Q. , Li, Q. , Zhiguo, E. et al. (2020) OsbZIP76 interacts with OsNF‐YBs and regulates endosperm cellularization in rice (Oryza sativa). J. Integr. Plant Biol. 62, 1983–1996. [DOI] [PubMed] [Google Scholar]
- Paul, P. , Dhatt, B.K. , Miller, M. , Folsom, J.J. , Wang, Z. , Krassovskaya, I. , Liu, K. et al. (2020) MADS78 and MADS79 are essential regulators of early seed development in rice. Plant Physiol. 182, 933–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer, M. , Kugler, K.G. , Sandve, S.R. , Zhan, B. , Rudi, H. , Hvidsten, T.R. , Mayer, K.F. et al. (2014) Genome interplay in the grain transcriptome of hexaploid bread wheat. Science (New York, N.Y.) 345, 1250091. [DOI] [PubMed] [Google Scholar]
- Pineda Rodo, A. , Brugière, N. , Vankova, R. , Malbeck, J. , Olson, J.M. , Haines, S.C. , Martin, R.C. et al. (2008) Over‐expression of a zeatin O‐glucosylation gene in maize leads to growth retardation and tasselseed formation. J. Exp. Bot. 59, 2673–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portereiko, M.F. , Lloyd, A. , Steffen, J.G. , Punwani, J.A. , Otsuga, D. and Drews, G.N. (2006) AGL80 is required for central cell and endosperm development in Arabidopsis. Plant Cell 18, 1862–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez, F. , Dündar, F. , Diehl, S. , Grüning, B.A. and Manke, T. (2014) deepTools: a flexible platform for exploring deep‐sequencing data. Nucleic Acids Res. 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez‐González, R.H. , Borrill, P. , Lang, D. , Harrington, S.A. , Brinton, J. , Venturini, L. , Davey, M. et al. (2018) The transcriptional landscape of polyploid wheat. Science (New York, N.Y.) 361, eaar6089. [DOI] [PubMed] [Google Scholar]
- Ran, Q. , Akhter, D. , Chengcong, Y. , Nath, U.K. , Eshag, J. , Xiaoli, J. and Chunhai, S. (2018) SRG1, encoding a kinesin‐4 protein, is an important factor for determining grain shape in rice. Rice Sci. 25, 297–307. [Google Scholar]
- Raza, Q. , Riaz, A. , Atif, R.M. , Hussain, B. , Rana, I.A. , Ali, Z. , Budak, H. et al. (2021) Genome‐wide diversity of MADS‐box genes in bread wheat is associated with its rapid global adaptability. Front. Genet. 12, 818880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock, C.D. and Quatrano, R.S. (1995) The role of hormones during seed development. In Plant Hormones, Physiology, Biochemistry and Molecular Biology, pp 671–697. Dordrecht: Springer, Netherlands. [Google Scholar]
- Saitou, N. and Nei, M. (1987) The neighbor‐joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425. [DOI] [PubMed] [Google Scholar]
- Sang, X. , Li, Y. , Luo, Z. , Ren, D. , Fang, L. , Wang, N. , Zhao, F. et al. (2012) CHIMERIC FLORAL ORGANS1, encoding a monocot‐specific MADS box protein, regulates floral organ identity in rice. Plant Physiol. 160, 788–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen, T.D. and Livak, K.J. (2008) Analyzing real‐time PCR data by the comparative C (T) method. Nat. Protoc. 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Shan, Q. , Wang, Y. , Li, J. and Gao, C. (2014) Genome editing in rice and wheat using the CRISPR/Cas system. Nat. Protoc. 9, 2395–2410. [DOI] [PubMed] [Google Scholar]
- Shang, X.L. , Xie, R.R. , Tian, H. , Wang, Q.L. and Guo, F.Q. (2016) Putative zeatin O‐glucosyltransferase OscZOG1 regulates root and shoot development and formation of agronomic traits in rice. J. Integr. Plant Biol. 58, 627–641. [DOI] [PubMed] [Google Scholar]
- Shen, Q. , Lin, Y. , Li, Y. and Wang, G. (2021) Dynamics of H3K27me3 modification on plant adaptation to environmental cues. Plants (Basel, Switzerland) 10, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen, J.G. , Kang, I.H. , Portereiko, M.F. , Lloyd, A. and Drews, G.N. (2008) AGL61 interacts with AGL80 and is required for central cell development in Arabidopsis. Plant Physiol. 148, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strejčková, B. , Čegan, R. , Pecinka, A. , Milec, Z. and Šafář, J. (2020) Identification of polycomb repressive complex 1 and 2 core components in hexaploid bread wheat. BMC Plant Biol. 20, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, L. , Li, X. , Fu, Y. , Zhu, Z. , Tan, L. , Liu, F. , Sun, X. et al. (2013) GS6, a member of the GRAS gene family, negatively regulates grain size in rice. J. Integr. Plant Biol. 55, 938–949. [DOI] [PubMed] [Google Scholar]
- Tonosaki, K. and Kinoshita, T. (2015) Possible roles for polycomb repressive complex 2 in cereal endosperm. Front. Plant Sci. 6, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman, R. (1992) The serum response element. Trends Biochem. Sci. 17, 423–426. [DOI] [PubMed] [Google Scholar]
- Volpicella, M. , Fanizza, I. , Leoni, C. , Gadaleta, A. , Nigro, D. , Gattulli, B. , Mangini, G. et al. (2016) Identification and characterization of the sucrose synthase 2 gene (Sus2) in durum wheat. Front. Plant Sci. 7, 266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vylíčilová, H. , Bryksová, M. , Matušková, V. , Doležal, K. , Plíhalová, L. and Strnad, M. (2020) Naturally occurring and artificial N9‐cytokinin conjugates: from synthesis to biological activity and back. Biomolecules 10, 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, A. , Garcia, D. , Zhang, H. , Feng, K. , Chaudhury, A. , Berger, F. , Peacock, W.J. et al. (2010) The VQ motif protein IKU1 regulates endosperm growth and seed size in Arabidopsis. Plant J. 63, 670–679. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Jiang, H. and Wang, G. (2020) PHERES1 controls endosperm gene imprinting and seed development. Trends Plant Sci. 25, 517–519. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Tong, X. , Tang, L. , Wang, Y. , Zhao, J. , Li, Z. , Liu, X. et al. (2022) RLB (RICE LATERAL BRANCH) recruits PRC2‐mediated H3K27 tri‐methylation on OsCKX4 to regulate lateral branching. Plant Physiol. 188, 460–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, J. , Liu, B. , Yao, Y. , Guo, Z. , Jia, H. , Kong, L. , Zhang, A. et al. (2022) Wheat genomic study for genetic improvement of traits in China. Sci. China Life Sci. 65, 1718–1775. [DOI] [PubMed] [Google Scholar]
- Xing, H.L. , Dong, L. , Wang, Z.P. , Zhang, H.Y. , Han, C.Y. , Liu, B. , Wang, X.C. et al. (2014) A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 14, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Peng, S. , Visperas, R.M. , Sanico, A.L. , Zhu, Q. and Gu, S. (2000) Grain filling pattern and cytokinin content in the grains and roots of rice plants. Plant Growth Regul. 30, 261–270. [Google Scholar]
- Yang, J. , Zhang, J. , Huang, Z. , Wang, Z. , Zhu, Q. and Liu, L. (2002) Correlation of cytokinin levels in the endosperms and roots with cell number and cell division activity during endosperm development in rice. Ann. Bot. 90, 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Zhang, J. , Wang, Z. and Zhu, Q. (2003) Hormones in the grains in relation to sink strength and postanthesis development of spikelets in rice. Plant Growth Regul. 41, 185–195. [Google Scholar]
- Yang, H. , Liu, X. , Xin, M. , Du, J. , Hu, Z. , Peng, H. , Rossi, V. et al. (2016) Genome‐wide mapping of targets of maize histone deacetylase HDA101 reveals its function and regulatory mechanism during seed development. Plant Cell 28, 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo, S.‐D. , Cho, Y.‐H. and Sheen, J. (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat. Protoc. 2, 1565–1572. [DOI] [PubMed] [Google Scholar]
- Yu, G. , Wang, L.G. , Han, Y. and He, Q.Y. (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. Omics: J. Integr. Biol. 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, G. , Wang, L.G. and He, Q.Y. (2015) ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics (Oxford, England) 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zalewski, W. , Orczyk, W. , Gasparis, S. and Nadolska‐Orczyk, A. (2012) HvCKX2 gene silencing by biolistic or Agrobacterium‐mediated transformation in barley leads to different phenotypes. BMC Plant Biol. 12, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Zhao, Y.L. , Gao, L.F. , Zhao, G.Y. , Zhou, R.H. , Zhang, B.S. and Jia, J.Z. (2012) TaCKX6‐D1, the ortholog of rice OsCKX2, is associated with grain weight in hexaploid wheat. New Phytol. 195, 574–584. [DOI] [PubMed] [Google Scholar]
- Zhang, P. , He, Z. , Tian, X. , Gao, F. , Liu, J. , Wen, W. , Fu, L. et al. (2017) Cloning of TaTPP‐6AL1 associated with grain weight in bread wheat and development of functional marker. Mol. Breed. 37, 1–8.28127252 [Google Scholar]
- Zhao, J. , Bai, W. , Zeng, Q. , Song, S. , Zhang, M. , Li, X. , Hou, L. et al. (2015) Moderately enhancing cytokinin level by down‐regulation of GhCKX expression in cotton concurrently increases fiber and seed yield. Mol. Breed. 35, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , Yang, Y. , Chen, J. , Lin, X. , Zhang, H. , Wang, H. , Wang, H. et al. (2023) Dynamic chromatin regulatory programs during embryogenesis of hexaploid wheat. Genome Biol. 24, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, M. , Liu, X. , Lin, J. , Liu, X. , Wang, Z. , Xin, M. , Yao, Y. et al. (2019) Histone acetyltransferase GCN5 contributes to cell wall integrity and salt stress tolerance by altering the expression of cellulose synthesis genes. Plant J. 97, 587–602. [DOI] [PubMed] [Google Scholar]
- Zhou, Y. , Zhao, X. , Li, Y. , Xu, J. , Bi, A. , Kang, L. , Xu, D. et al. (2020) Triticum population sequencing provides insights into wheat adaptation. Nat. Genet. 52, 1412–1422. [DOI] [PubMed] [Google Scholar]
- Zhou, L. , Zhu, C. , Fang, X. , Liu, H. , Zhong, S. , Li, Y. , Liu, J. et al. (2021) Gene duplication drove the loss of awn in sorghum. Mol. Plant 14, 1831–1845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Spatial and temporal expression of type I TaMADS‐box genes.
Figure S2 Representative picture of in situ hybridization assays performed using 4‐DAP seed and hybridized with antisense and sense TaMADS‐GS‐A transcript probes.
Figure S3 Strategy used to produce the Tamads‐gs knockout lines.
Figure S4 GO analysis of differentially expressed genes in 4‐DAP seed of WT and Tamads‐gs‐d knockout mutants.
Figure S5 Relative transcript levels of different TaCKX genes in Tamads‐gs‐ad mutants and WT.
Figure S6 Subcellular localization of TaMADS‐GS.
Figure S7 The results of CUT&‐Tag and ChIP‐qPCR analysis.
Figure S8 Phylogenetic analysis and spatial and temporal expression of PRC2 proteins.
Figure S9 Full‐length TaMADS‐GS‐D cDNA fused with the GAL4 BD domain can self‐activate the expression of the reporter genes in Y2H assays.
Data Set S1 List of differentially expressed transcripts in the Tamads‐gs mutant and wild type.
Data Set S2 Information of 233 wheat accessions used for haplotype analysis.
Data Set S3 Primers used for gene expression and vector construction.