

Abstract
Due to its high transmissibility, Omicron BA.1 ousted the Delta variant to become a dominating variant in late 2021 and was replaced by more transmissible Omicron BA.2 in March 2022. An important question is which new variants will dominate in the future. Topology-based deep learning models have had tremendous success in forecasting emerging variants in the past. However, topology is insensitive to homotopic shape evolution in virus-human protein-protein binding, which is crucial to viral evolution and transmission. This challenge is tackled with persistent Laplacian, which is able to capture both the topological change and homotopic shape evolution of data. Persistent Laplacian-based deep learning models are developed to systematically evaluate variant infectivity. Our comparative analysis of Alpha, Beta, Gamma, Delta, Lambda, Mu, and Omicron BA.1, BA.1.1, BA.2, BA.2.11, BA.2.12.1, BA.3, BA.4, and BA.5 unveils that Omicron BA.2.11, BA.2.12.1, BA.3, BA.4, and BA.5 are more contagious than BA.2. In particular, BA.4 and BA.5 are about 36% more infectious than BA.2 and are projected to become new dominant variants by natural selection. Moreover, the proposed models outperform the state-of-the-art methods on three major benchmark datasets for mutation-induced protein–protein binding free energy changes. Our key projection about BA4 and BA.5’s dominance made on May 1, 2022 (see arXiv:2205.00532) became a reality in late June 2022.
Keywords: SARS-CoV-2, Evolution, Infectivity, Deep learning, Persistent Laplacian
1. Introduction
The coronavirus disease, 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has lasted for more than years. The development of effective vaccines, monoclonal antibodies (mABs), and antiviral drugs have significantly improved our ability to bring COVID-19 pandemic under control. Nonetheless, the emerging SARS-CoV-2 variants have become a major threat to existing vaccines, monoclonal antibodies (mABs), and antiviral drugs.
The Omicron variant has mutations on various SARS-CoV-2 proteins, such as non-structure protein 3 (NSP3), NSP4, NSP5, NSP6, NSP12, NSP14, spike (S) protein, envelope protein, membrane protein, and nucleocapsid protein. Specifically, Omicron has three main lineages, BA.1 (B.1.1.529.1), BA.2 (B.1.1.529.2), and BA.3 (B.1.1.529.3), and many sub-lineages. Many new recombinants occurred, including XD, XE, and XF. XD and XE are recombination of Delta and BA.1, while XE is basically a BA.2 Omicron lineage carrying a piece of BA.1 at the front end of its genome. The S protein of XE is still BA.2.
The research community focuses its attention on the mutations at the S protein receptor-binding domain (RBD) due to the fact that the RBD facilitates the binding between the S protein and the host angiotensin-converting enzyme 2 (ACE2), which initiates the viral entry of a host cell and infection. It turns out that the binding strength between the S protein RBD and the ACE2 is proportional to the viral infectivity [1–5]. An artificial intelligence (AI) study revealed that natural selection is the governing mechanism for SARS-CoV-2 evolution [6]. Specifically, viral evolution selects those mutations that are able to strengthen the RBD-ACE2 binding. This mechanism led to the occurrence of many variants, such as Alpha, Beta, Gamma, Delta, Mu, etc. Natural selection in SARS-CoV-2 mutations was confirmed beyond doubt in April 2021 by the genotyping of over half a million viral genomes isolated from patients [7].
Additionally, antibodies are generated by the human immune response to infection or vaccination. A strong RBD-antibody binding would lock off RBD-ACE2 binding and directly neutralize the virus [8–10]. As such, mABs targeting the S protein, particularly the RBD, which are designed to treat viral infection. It was unveiled that viral evolution also selects those mutations that are able to weaken RBD-antibody binding, leading to vaccine breakthrough infections [11,12]. Therefore, a new virus with RBD mutations that make the virus more infectious and more capable of evading antibody protection would become the next dominant variant, which is the underlying principle for the successful forecasting of Omicron BA.2’s dominance [13].
In biophysics, the strength of protein–protein complex is measured by binding free energy (BFE). Mutation-induced BFE change is calculated by
| (1) |
where and are the BFE of wild type and mutant. A positive (negative) BFE change indicates the strengthening (weakening) of the protein–protein binding. Protein–protein BFE changes can be carried out in a variety of ways as shown in software packages FOLDX [14], SAAMBE [15], mCSM-AB [16], mCSM-PPI2 [17], BindProfX [18], etc. AI approaches take the advantage of existing data and often outperform other methods when experimental data become available. Due to the structural complexity and high dimensionality of protein–protein interactions (PPIs), methods that are able to effectively reduce the PPI structural complexity and dimensionality have demonstrated great advantages in predicting PPI BFE changes [19]. Advanced mathematics, particularly, persistent homology [20–25], offers tremendous abstraction of PPIs. Persistent homology is the main workhorse in popular topological data analysis (TDA) [26–29]. Element-specific persistent homology (EPH) has had tremendous success in computational biology [30,31] and worldwide competitions in computer-aided drug design [32].
Based on FPH, a topology-based network tree (TopNetTree) model was constructed from conventional neural network and decision trees for predicting PPI BFE changes [19]. In the past two years, this approach has been extended with SARS-CoV-2 related deep mutational data to predict the BFE changes RBD-ACE2 and RBD-antibody complexes up on RBD mutations [33,34]. Initially, in early 2020, TopNetTree model was applied to successfully predict that RBD residues 452 and 501 “have high chances to mutate into significantly more infectious COVID-19 strains” [6]. These RBD mutations later appeared in all major variants, Alpha, Beta, Delta, Gamma, Delta, Epsilon, Theta, Kappa, Lambada, Mu, and Omicron L452R/Q and N501Y mutations. In April 2021, the TopNetTree model predicted a list of 31 RBD antibody-escape mutations, including W353R, I401N, Y449D, Y449S, P491R, P491L, Q493P, etc. [7]. Notably, experimental results confirmed that mutations at RBD residues Y449, E484, Q493, S494, and Y505 enable the virus to escape antibodies [35]. It was revealed that variants found in the United Kingdom and South Africa in late 2020 would strengthen virus infectivity, which is consistent with the experimental results [36]. In summer 2021, a topology-based deep neural network trained with mAbs (TopNetmAb) was developed to forecast a list of most likely vaccine-escape RBD mutations, such as S494P, Q493L, K417N, F490S, F486L, R403K, E484K, L452R, K417T, F490L, E484Q, and A475S [34], and mutations S494P, K417N, E484K/Q, and L452R were designated as the variants of concern or variants of interest denounced by the Worldwide Health Organization (WHO). The correlation between the experimental deep mutational data [37] and AI-predicted RBD-mutation-induced BFE changes for all possible 3686 RBD mutations on the RBD-ACE2 complex is 0.7 [34]. In comparison, experimental deep mutational results for the same set of RBD mutations from 2 different labs only have a correlation of 0.67 [37,38]. TopNetmAb predictions of Omicron [39] and Omicron BA.2 [13] infectivity, vaccine breakthrough, and antibody resistance were nearly perfectly confirmed by experiments and pandemic evolution in the world. These mechanistic discovery and successful predictions may not be achievable via purely experimental means, indicate the indispensable role of AI for scientific discovery.
However, persistent homology and TDA provide only topological invariants, which may not be sufficient for representing PPI data. In particular, the shape of data arising from a family of homotopy geometries cannot be captured by persistent homology. For example, the geometry of each drum in an acoustic drum set is designed to offer a specific sound or frequency, but persistent homology is insensitive to the change in the sizes (or shapes) in the drum set. This challenge in TDA was addressed by the introduction of persistent Laplacian, or persistent spectral graph [40]. Persistent Laplacian manifests the full set of topological invariants and the homotopic shape evolution of data in its harmonic and non-harmonic spectra, respectively. Additional mathematical analysis [41] and a software package, i.e., HERMES [42], for persistent Laplacian have been reported in the literature. This method has been successfully applied to biological studies, including protein thermal stability [40], protein–ligand binding [43], and protein-protein binding problems [44].
In the present work, we introduce element-specific and site-specific persistent Laplacians to forecast emerging SARS-CoV-2 variants. We hypothesize that persistent Laplacians generate intrinsically low-dimensional representations of PPIs and dramatically reduce the dimensionality of PPI data, leading to a reliable high-throughput screening of emerging SARS-CoV-2 variants. To quantitatively validate this hypothesis, we integrate the harmonic and non-harmonic spectra of persistent Laplacians with efficient machine learning algorithms, i.e., gradient boosting tree (GBT) and deep neural network (Net), to predict PPI following mutations. The resulting topological and spectral-based machine learning models are validated on three major benchmark datasets, the AB-Bind database [45], SKEMPI dataset [46] and SKEMPI v2.0 dataset [47], giving rise to the state-of-the-art performance. Meanwhile, with additional training on SARS-CoV-2 related datasets, our models forecast emerging SARS-CoV-2 variants and recommend four Omicron subvariants, i.e., BA.2.11, BA.2.12.1, BA.4, and BA.5 for active surveillance. Our key projection of BA.4 and BA.5’s incoming dominance made in May 1, 2022 [48] had become reality in late June 2022.
2. Results
In this section, we first carry out the infectivity predictions on emerging SARS-CoV-2 variants. Next, three benchmark PPI datasets, i.e., the AB-Bind [45], SKEMPI [46], and SKEMPI 2.0 datasets [47] are employed to demonstrate the proposed persistent Laplacian-based AI models with ten-fold cross validations. Two evaluation metrics, Pearson correlation and the root-mean-square error (RMSE), are used to assess the quality of the present models. Lastly, we present the validation of our models on SARS-CoV-2-related datasets.
2.1. Emerging SARS-CoV-2 variants: Infectivity
Fig. 1 shows the RBD mutations of Omicron subvariants and their BFE changes of SARS-CoV-2 variants. A comparison is also given to other main SARS-CoV-2 variants Alpha, Beta, Gamma, Delta, Theta, Kappa, Lambda, and Mu variants. The Delta variant had the highest BFE change among the earlier variants and was the most infectious variant before the occurrence of the Omicron variant, which explains its dominance in 2021. Omicron BA.1, BA.2, and BA.3 have the common RBD mutations G339D, S373P, S375F, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, and Y505H. Omicron BA.1 has three distinct RBD mutations S371L, G446S, and G496S. Four distinct mutations, S371F, T376A, D405N, and R408S, were found for Omicron BA.2. Omicron BA.3 shares three mutations either with BA.1 or BA.2: S371F, D405N, and G446S. The AI-predicted BFE changes of BA.1, BA.2, and BA.3 are 2.60, 2.98, and 2.88 kcal/mol, respectively [13]. These values are significantly higher than those of other major SARS-CoV-2 variants as shown in Fig. 1. Note that Omicron BA.2 is the most infectious variant. It is about 20 and 4.2 times as infectious as the original SARS-CoV-2 and the Delta variant, respectively. The machine learning model also predicts that BA.2 is about 1.5 times as contagious as BA.1, which is highly consistent with experimental studies [50,51]. BA.2 has been the dominating variant since late March 2022 [13].
Fig. 1.
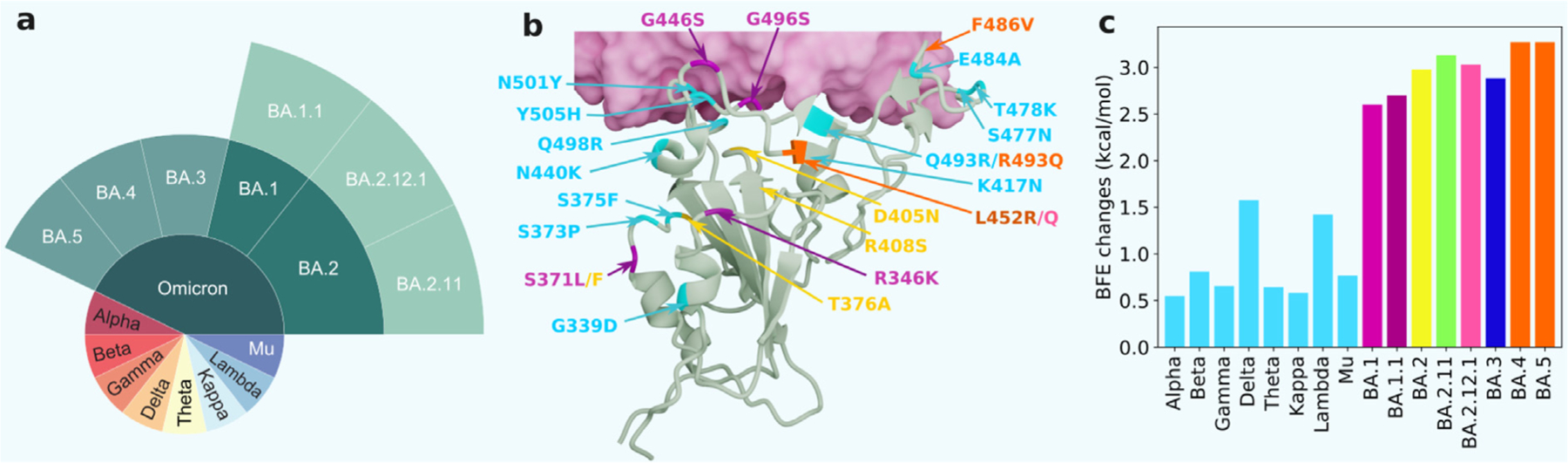
Illustration of major SARS-CoV-2 (sub)variants, the RBD mutations of Omicron subvariants at the RBD-ACE2 interface, and their mutation-induced BFE changes. a Prevailing SARS-CoV-2 variants and Omicron subvariants. b RBD mutations of Omicron subvariants at the RBD-ACE2 interface (PDB: 7T9L [49]). The shared 12 mutations are shown in cyan. BA.1 mutations are plotted with magenta. BA.2 mutations are marked in yellow. BA.4 and BA.5 mutations are labeled in orange. The rest colors can be matched from the right chart. c A comparison of predicted mutation-induced BFE changes for various SARS-CoV-2 variants and subvariants.
We have also examined other Omicron subvariants, namely, BA.1.1, BA.2.11, BA.2.12.1, BA.4, and BA.5. Compared with BA.1, BA.1.1 has one additional RBD mutation, i.e., R346K. BA.2.11 has one more RBD mutation, L452R, than BA.2 does. BA.2.12.1 has an extra RBD mutation, L452Q, compared with BA.2. BA.4 and BA.5 share the same set of RBD mutations but differ in ORF7b, nucleocapsid (N), and membrane (M) proteins. They have three additional RBD mutations, L452R, F486V, and R493Q compared with BA.2. Note that R493Q is a reversion to the wide type, Q493. It is interesting that L452R is one of Delta’s two RBD mutations. Additionally, mutations simultaneously occurred on two RBD residues, L452 and N501, which were singled out by our AI model in early 2020 [6].
Our AI-predicted BFE changes for BA.1.1, BA.2.11, BA.2.12.1, BA.4, and BA.5 are 2.70, 3.13, 3.03, 3.27, and 3.27 kcal/mol, respectively. It is noticed that BA.4 and BA.5 are predicted to be 1.36 times as infectious as BA.2 and have high potential to become new dominating SARS-CoV-2 variants.
2.2. The performance on the AB-Bind dataset
The AB-Bind dataset, including 1101 mutational data entries for experimentally determined BFE changes [45] is considered in the validation of the proposed models. Its 645 single mutations involving 29 antibody–antigen complexes are denoted as the AB-Bind S645 set. In the AB-Bind S645 set, about one-fifth of mutations strengthen the binding, while the rest are destabilizing mutations. In particular, 27 non-binders, which are mutants determined not to bind within the experimental sensitivity of the assay, are in the dataset. The mutation-induced binding free energy changes for these non-binders were set to −8 kcal/mol. For machine learning models, non-binders are outliers and can cause a very negative impact on model accuracy.
As shown in Table 1, our TopLapNetGBT and TopGBT models achieved the of 0.61 and 0.56 for the AB-Bind S645 set. In comparison, TopNet outperforms LapNet because TopNet includes auxiliary features, while LapNet has only Laplacian features. Note that our worst model (LapNet) still outperforms the other best model in the literature, while our best model is about 15% better than the other best model in the literature, indicating the predictive power of our topology and Laplacian-based machine learning models. Both GBTs and Nets models are quite sensitive to system errors as the model training is based on optimizing the mean-square error of the loss function. The BFE changes of 27 non-binders were defined to be −8 kcal/mol in the original dataset and did not follow the distribution of the whole dataset as the values is defined by For the TopLapGBT model, the RMSE of AB-Bind S645set is 1.13 kcal/mol and reduces to 0.82 kcal/mol when 27 non-binder samples are excluded. In this case, the of the TopLapNetGBT model is increased from 0.61 to 0.76 by excluding non-binder samples. The consensus results of GBT and Net have correlations of 0.58–0.59, which are lower than that of GBT but higher than that of Net. GBT models outperform Net models in the validation, showing that GBT performs better than Net on a small dataset.
Table 1.
Comparison of the Pearson correlation coefficients of various methods for the AB-bind S645 set.
| Method | Method | ||
|---|---|---|---|
| TopLapNetGBT | 0.61/0.76a | mCSM-AB | 0.53/0.56a |
| LapGBT | 0.60/0.71a | Discovery Studio | 0.45 |
| TopNetGBT | 0.59/0.76a | mCSM-PPI | 0.31 |
| LapNetGBT | 0.59/0.70a | FoldX | 0.34 |
| TopLapNet | 0.58/0.77a | STATIUM | 0.32 |
| TopLapGBT | 0.58/0.74a | DFIRE | 0.31 |
| LapNet | 0.58/0.70a | bASA | 0.22 |
| TopNet | 0.57/0.76a | dDFIRE | 0.19 |
| TopGBT | 0.56/0.73a | Rosetta | 0.16 |
2.3. The performance on the SKEMPI dataset
The SKEMPI dataset [46] has 3047 entries of BFE changes induced by mutations. This dataset is collected from the literature for protein-protein heterodimeric complexes with experimentally determined structures. It consists of single- and multi-mutations. Among them, 2317 single mutations out of 3047 entries are called the S2317 dataset. Recently, a subset of 1131 non-redundant interface single-mutations is selected and denoted as the S1131 set [18]. Table 2 shows the Pearson correlation coefficients on tenfold cross-validations of various models, including topology- and Laplacian-based models. The proposed topology- and Laplacian-based models are found to be more accurate than other existing methods. One may notice that for a larger training set, the consensus predictions of GBT and Net outperform GBT methods. Additionally, topology-based models contain topology features and auxiliary features, which include more biomolecular information than Laplacian-based models.
Table 2.
Comparison of the Pearson correlation coefficients of various methods for the S1131 set in the SKEMPI dataset.
| Method | Method | ||
|---|---|---|---|
| TopLapNetGBT | 0.87 | BindProfX | 0.738 |
| TopNetGBT | 0.87 | Profile-score+FoldX | 0.738 |
| TopLapNet | 0.86 | Profile-score | 0.675 |
| TopNet | 0.86 | SAAMBE | 0.624 |
| TopLapGBT | 0.86 | FoldX | 0.457 |
| TopGBT | 0.86 | BeAtMuSic | 0.272 |
| LapNetGBT | 0.81 | Dcomplex | 0.056 |
| LapNet | 0.81 | ||
| LapGBT | 0.78 |
Source: The results of other methods are adopted from Ref. [18].
2.4. The performance on the SKEMPI 2.0 dataset
The SKEMPI 2.0 [47] database is an updated version of the original SKEMPI database with new mutations from three other databases: AB-bind [45], PROXiMATE [52], and dbMPIKT [53]. This dataset has 7085 entries, including single-mutations and multi-mutations. To validate mCSM-PPI2, David et al. filtered only single-point mutations, selected 4169 variants in 319 different complexes, and denoted them as the S4169 set [17]. Additionally, set S8338 was derived from set S4169 by setting the BFE changes of the reverse mutations as the negative values of the original BFE changes induced by mutations. We present our tenfold cross-validation results on sets S4169 and S8338 in Table 3. For S4169, TopLapNetGBT has the most accurate result with of 0.82 and RMSE of 1.06 kcal/mol. Topology-based models, aided by auxiliary features, have correlations greater than 0.80 and RMSE from 1.04 kcal/mol to 1.10 kcal/mol. Purely Laplacian-based models also performed quite well, with the Pearson correlation of 0.76, which is the same as that of the mCSM-PPI2.
Table 3.
Comparison of the Pearson correlation coefficients of various methods for S4169 set and S8338 set in SKEMPI 2.0.
| S4169 | S8338 | ||
|---|---|---|---|
| Method | Method | ||
| TopLapNetGBT | 0.82 | TopLapNetGBT | 0.87 |
| TopNetGBT | 0.82 | TopLapNet | 0.87 |
| TopLapNet | 0.81 | TopNetGBT | 0.87 |
| TopLapGBT | 0.81 | TopNet | 0.86 |
| TopNet | 0.81 | TopLapGBT | 0.85 |
| TopGBT | 0.80 | TopGBT | 0.85 |
| LapNetGBT | 0.77 | LapNetGBT | 0.83 |
| mCSM-PPI2 | 0.76 | mCSM-PPI2 | 0.82 |
| LapNet | 0.76 | LapNet | 0.81 |
| LapGBT | 0.76 | LapGBT | 0.80 |
Source: Results of mCSM-PPI2 are from Ref. [17].
For the S8338 set, TopLapNetGBT has the highest Pearson correlation of 0.8702 and RMSE of 1.01 kcal/mol as shown in Table 3. TopLapNet has the most accurate results with of 0.8688 and RMSE of 0.984 kcal/mol. Topology models, aided by auxiliary features, have the in the range of (0.848, 0.870) and RMSE in the range of (1.070 kcal/mol, 0.984 kcal/mol). LapNet and LapGBT models have their values slightly lower than that of MCSM-PPI2, but the of their consensus (LapNetGBT) is higher than that of the mCSM-PPI2.
2.5. The performance on SARS-CoV-2 datasets
Training datasets have the utmost importance in implementing our machine learning model for SARS-CoV-2 applications. First, all the datasets mentioned above, including AB-bind,[45] PROXiMATE [52], dbMPIKT [53], SKEMPI [46], and SKEMPI 2.0 [47], are used in our model training. Additionally, SARS-CoV-2-related datasets are also employed to improve the prediction accuracy after a label transformation. These are deep mutational enrichment ratio data, including mutational scNeting data of ACE2 binding to the receptor-binding domain (RBD) of the S protein [54], mutational scNeting data of RBD binding to ACE2 [37,38], and mutational scNeting data of RBD binding to CTC-445.2 and of CTC-445.2 binding to the RBD [37]. Note that in our validation, our training datasets exclude the test dataset, which is a mutational scNeting data of RBD binding to ACE2. Here, these datasets provide more information on SARS-CoV-2 and can be used to calibrate the models to predict the real experimental results.
Here, we present a validation of our model BFE change prediction for mutations on S protein RBD compared to the experimental deep mutational enrichment data [37]. We compare between experimental deep mutational enrichment data and BFE change predictions on SARS-CoV-2 RBD binding to ACE2 in Fig. 2. Both BFE changes (Fig. 2 top) and enrichment ratios (Fig. 2 bottom) describe the binding affinity changes of the S protein RBD-ACE2 complex induced by mutations. It can be found that the predicted BFE changes are highly correlated to the enrichment ratio data. Pearson correlation is 0.69.
Fig. 2.

A comparison between experimental RBD deep mutation enrichment data and predicted BFE changes for SARS-CoV-2 RBD binding to ACE2 (6M0J) [37]. Top: machine learning predicted BFE changes for single-site mutants of the S protein RBD. Bottom: deep mutational scanning heatmap showing the average effect on the enrichment for single-site mutants of RBD when assayed by yeast display for binding to the S protein RBD [37].
3. Theories and methods
This section presents brief reviews of spectral graph theory, simplicial complex, and persistent Laplacian are presented. Machine learning and deep learning models are discussed in test datasets and validation settings.
3.1. Persistent Laplacians
3.1.1. Spectral graphs
Spectral graph theory studies the spectra of graph Laplacian matrices. It gives rise to the topological and spectral properties of underlying graphs or networks. Mathematically, a graph is an ordered pair , where is the vertex set with size and is the edge set. Denote the degree of each vertex , i.e., the number of edges that connects to . A specific Laplacian matrix can be given by
| (2) |
where “adjacent” is subject to a specific definition or connection rule.
Let order the eigenvalues of the graph Laplacian matrix as
| (3) |
The kernel dimension of is the multiplicity of 0 eigenvalues, indicating the number of connected components of , which is the topological property of the graph. The non-zero eigenvalues of contain the graph properties. In particular, is called the algebraic connectivity.
3.1.2. Simplicial complex
To construct a topological description of a graph, simplicial complex is used. For a set of points, , a -plane is well defined if the points are affinely independent, i.e., are linearly independent. Thus, one can have at most linearly independent vectors with at most affinely independent points in . An affine hull is the set of affine combinations, , and . Such an affine combination is a convex combination if all are non-negative. The convex hull is the set of convex combinations. A -simplex denoted as is the convex hull of affinely independent points. For example, 0-, 1-, 2-, and 3-simplex are vertexes, edges, triangles, and tetrahedrons. A simplicial complex is a collection of simplices in satisfying the following conditions such as the Cech complexes, Vietoris–Rips complexes, and alpha shapes. For example, the Vietoris–Rips complex of with radius consists of all subsets of radius at most as
| (4) |
For , its face is also in . The non-empty intersection of any two simplices is a face of them. The dimension of simplicial complex is defined as the maximum dimension of its simplex.
A -chain is a finite sum of simplices as with field of the coefficients for the sum, and the set of all chains in a group . The boundary operator maps defined as
| (5) |
where and stands for being omitted. A -chain is called -cycle if its boundary is zero. A chain complex is the sequence of chain groups connected by boundary operators
| (6) |
and the th homology group is defined by where and . The Betti numbers are defined by the ranks of th homology group . This, in practice, is counting holes in -dimension, such as reflects the number of connected components, gives the number of loops, and is the number of cavities. In a nutshell, the Betti number sequence reveals the intrinsic topological property of the system.
Recall that in graph theory, the degree of a vertex (0-simplex) is the number of edges that are adjacent to the vertex, denoted as . However, once we generalize this notion to -simplex, problem aroused since -simplex can have -simplices and -simplices adjacent to it at the same time. Therefore, the upper adjacency and lower adjacency are required to define the degree of a -simplex for [55,56].
Definition 3.1. Given two -simplices and of a simplicial complex . We say they are lower adjacent if they share a common -face, denoted as . The lower degree of -simplex is the number of nonempty -simplices in that are faces of , which is denoted as and is always .
Definition 3.2. Given two -simplices and of a simplicial complex . We say they are upper adjacent if they share a common -face, denoted as . The upper degree of -simplex is the number of -simplices in of which is a face, which is denoted .
Then, the degree of a -simplex is defined as:
| (7) |
3.1.3. Graph Laplacian
The graph Laplacian was introduced to enrich topological and geometric information of simplicial complexes via a filtration process. The preliminary concepts are about the oriented simplicial complex and -combinatorial Laplacian. More detail information can be found elsewhere [56–59]. The properties of the -combinatorial Laplacian matrix with its spectra are discussed in the following.
A -combinatorial Laplacian is defined based on oriented simplicial complexes, and its lower- and higher-dimensional simplexes can be employed to study a specifically oriented simplicial complex. An oriented simplicial complex is defined if all of its simplices are oriented. If and are upper adjacent with a common upper -simplex , they are similarly oriented if both have the same sign in and dissimilarly oriented if the signs are opposite. Additionally, if and are lower adjacent with a common lower -simplex , they are similarly oriented if has the same sign in and , and dissimilarly oriented if the signs are opposite. Similarly, -chains can be defined on the oriented simplicial complex , as well as -boundary operator.
The -combinatorial Laplacian is a linear operator for integer
| (8) |
where is the coboundary operator mapping . One property is preserved, which implies . The -combinatorial Laplacian matrix, denoted , is the matrix representation.
| (9) |
of operator , where and be the matrix representation of a -boundary operator and -coboundary operator, respectively, with respect to the standard basis for and with some assigned orderings. Then, the number of rows in corresponds to the number of -simplices and the number of columns shows the number of -simplices in , respectively. In addition, the upper and lower -combinatorial Laplacian matrices are denoted by and , respectively. Note that is the zero map which leads to being a zero matrix. Therefore, , with the (oriented) simplicial complex of dimension 1, which is actually a simple graph. Especially, 0-combinatorial Laplacian matrix is actually the Laplacian matrix defined in the spectral graph theory.
Given an oriented simplicial complex with , the entries of -combinatorial Laplacian matrices are given by [58]
| (10) |
| (11) |
3.1.4. Persistent spectral graphs
Persistent spectral graphs were introduced by integrating graph Laplacian and multiscale filtration [40]. Both topological and geometric information (i.e. connectivity and robustness of simple graphs) can be derived from analyzing the spectra of -combinatorial Laplacian matrix. However, this method is genuinely free of metrics or coordinates, which induced too little topological and geometric information that can be used to describe a single configuration. Therefore, persistent spectral graphs (PSG) is proposed to create a sequence of simplicial complexes induced by varying a filtration parameter, which is inspired by the idea of persistent homology and our earlier work in multiscale graphs. This section mainly introduce the construction of persistent spectral graphs.
First, a -combinatorial Laplacian matrix is symmetric and positive semi-definite. Therefore, its eigenvalues are all real and non-negative. The multiplicity of zero spectra (also called harmonic spectra) reveals the topological information, and the geometric information will be preserved in the non-harmonic spectra. More specifically, the multiplicity of zero spectra of is denoted by which is actually the th Betti number defined in the homology:
| (12) |
Naturally, persistent spectral theory creates a sequence of simplicial complexes induced by varying a filtration parameter [40]. A filtration of an oriented simplicial complex is a sequence of sub-complexes of
| (13) |
It induces a sequence of chain complexes Eq. (14) is given in Eq. (14).
 |
(14) |
For each sub-complexes , we define its corresponding chain group to be , and the -boundary operator will be denoted by . We say that if . then is an empty set and is a zero map. If , then
| (15) |
with being the -simplex, and being the -simplex for which its vertex is removed. Additionally, the adjoint operator is . The topological and spectral information of can be analyzed from along with the filtration parameter by diagonalizing the -combinatorial Laplacian matrix. We call the multiplicity of zero spectra of as its persistent Betti number , which counts the number of -dimensional holes in
| (16) |
Specifically, represents the number of connected components in reveals the number of one-dimensional loops or circles in , and shows the number of two-dimensional voids or cavities in . Moreover, the set of spectra of is given by:
| (17) |
where has dimension and spectra are arranged in ascending order. The smallest non-zero eigenvalue of is defined as . The -persistent -combinatorial Laplacian operator is defined by extending the boundary operator. Detailed descriptions can be found in Ref. [40].
3.2. Predictive models for mutation-induced protein–protein binding free energy changes
Since the harmonic spectra produced by the kernel of a persistent Laplacian contain exact topological information as that of persistent homology, we utilize a persistent homology software, GUDHI, to generate purely topological representations of PPIs in dimensions 0, 1, and 2. Additionally, persistent Laplacian spectra, including both harmonic and non-harmonic parts, are coded in Python. Machine learning and deep learning algorithms are implemented in Pytorch [60].
3.2.1. Persistent Laplacian representation of PPIs
To facilitate topological and shape analysis of PPIs via persistent Laplacians, we first composite the atoms in a protein–protein complex into various subsets.
: atoms of the mutation sites.
: atoms in the neighborhood of the mutation site within a cut-off distance .
: protein A atoms within of the binding site.
: protein B atoms within of the binding site.
- atoms in the system that has atoms of element type E. The distance matrix is specially designed such that it excludes the interactions between the atoms form the same set. For interactions between atoms and in set and/or set , the modified distance is defined as
where is the Euclidian distance between and .(18)
Molecular atoms of different can be constructed as points presented by as affinely independent points in simplicial complex. Persistent spectral graph is devised to track the multiscale topological and geometrical information over different scales along a filtration [40], resulting in significant important feature vectors for the machine learning method. Features generated by binned barcode vectorization can reflect the strength of atom bonds, van der Waals interactions, and can be easily incorporated into a machine learning model, which captures and discriminates local patterns. Using the atom subsets, for example and , simplicial complexes are constructed by only considering the edges from to for Vietoris–Rips complexes. Then from the Vietoris–Rips complex filtration, barcodes generated from persistent homology are enumerated by bar lengths in certain intervals with number 0 or 1. Meanwhile, for each complexes in the filtration, eigenvalues are calculated according to the graph Laplacian analysis. The statistics of eigenvalues such as sum, maximum, minimum, mean, and standard deviation are collected to have a normalized features for machine learning methods. Another method of vectorization is to get the statistics of bar lengths, birth values, and death values, such as sum, maximum, minimum, mean, and standard deviation. This method is applied to vectorize Betti-1 and Betti-2 barcodes obtained from alpha complex filtration based on the facts that higher-dimensional barcodes are sparser than barcodes.
3.2.2. Machine learning and deep learning algorithms
The features generated from the persistent spectral graph are tested by the gradient boosting tree (GBT) method and the deep neural network (Net) method. The validations are performed on the datasets discussed in the results section. The accurate prediction of the mutation-induced binding affinity changes of protein–protein complexes is very challenging. After effective feature-generations, a machine learning or deep learning model is also required for validations and real applications. The gradient boosting tree is a popular ensemble method for regression and classification problems. It builds a sequence of weak learners to correct training errors. By the assumption that the individual learners are likely to make different mistakes, the method combines weak learners to eliminate the overall error. Furthermore, a decision tree is added to the ensemble depending on the present prediction error on the training dataset. Therefore, this method is relatively robust against hyperparameter tuning and overfitting, especially for a moderate number of features. The GBT is shown for its robustness against overfitting, good performance for moderately small data sizes, and model interpretability. The present work uses the package provided by scikit-learn (v 0.23.0) [61]. The number of estimators and the learning rate are optimized for ensemble methods as 20 000 and 0.01, respectively. For each set, ten runs (with different random seeds) were done, and the average result is reported in this work. Considering a large number of features, the maximum number of features to consider is set to the square root of the given descriptor length for GBT methods to accelerate the training process. The parameter setting shows that the performance of the average of sufficient runs is decent.
A deep neural network is a network of neurons that maps an input feature layer to an output layer. The neural network mimics the human brain to solve problems with numerous neuron units with backpropagation to update weights on each layer. To reveal the facts of input features at different levels and abstract more properties, one can construct more layers and more neurons in each layer, known as a deep neural network. Optimization methods for feedforward neural networks and dropout methods are applied to prevent overfitting. The network layers and the number of neurons in each layer are determined by gird searches based on 10-fold cross-validations. Then, the hyperparameters of stochastic gradient descent (SGD) with momentum are set up based on the network structure. The network has 7 layers with 10 000 neurons in each layer. For SGD with momentum, the hyperparameters are momentum = 0.9 and weight_decay=0. The learning rate is 0.002 and the epoch is 400. The Net is implemented on Pytorch [60].
3.2.3. Predictive models
In our previous work, topology-based deep neural network trained with mAbs (TopNetmAb) was introduced to predict mAb binding free energy changes [34]. Persistent homology is the main workhorse for TopNetmAb, but auxiliary features inherited from our earlier TopNetTree [19] are utilized.
In this work, we construct a TopNet model from TopNetmAb by excluding mAb training data. A topology-based GBT model (TopBGT) is also developed in the present work by replacing Net in the TopNet model with GBT. Both TopNet and TopGBT include a set of auxiliary features inherited from our earlier TopNetTree [19] and TopNetmAb [34] to enhance their performance.
Additionally, to evaluate the performance of persistent Laplacian (Lap) for PPIs, we construct persistent Laplacian-based GBT (LapGBT) and persistent Laplacian-based deep neural network (LapNet). Note that, unlike TopNet and TopGBT, LapGBT and LapNet employ only persistent Laplacian features extracted from protein structures. Therefore, their performance depends purely on persistent Laplacian.
Moreover, TopLapGBT and TopLapNet are constructed by adding persistent Laplacian features to TopGBT and TopNet, respectively. Furthermore, the consensus of GBT and Net predictions are also used for validations, denoted as TopNetGBT and LapNetGBT, respectively. Finally, the consensus of TopLapNet and TopLapGBT is called TopLapNetGBT.
4. Conclusion
Due to natural selection, emerging SARS-CoV-2 variants are spreading worldwide with their increased transmissibility as a result of higher infectivity and/or stronger antibody resistance. The increase in antibody resistance also leads to vaccine breakthrough infections and jeopardizes the existing monoclonal antibody drugs. The spike protein plays the most important role in viral transmission because its receptor binding domain (RBD) binds to human ACE2 to facilitate the viral entry of host cells. Topological data analysis (TDA) of RBD-ACE2 binding free energy changes induced by RBD mutations enables the accurate forecasting of emerging SARS-CoV-2 variants [6,13,39,62].
However, the earlier TDA method is not sensitive to homotopic shape evolution during filtration, which is important for protein–protein interactions (PPIs). To overcome this obstacle, persistent Laplacian, which characterizes the topology and shape of data, is introduced in this work for analyzing PPIs. Paired with advanced machine learning and deep learning algorithms, the proposed persistent Laplacian method outperforms the state-of-art approaches in validation with mutation-induced binding free energy changes of PPIs using major benchmark datasets. An important forecasting from the present work is that Omicron subvariants BA.2.11, BA.212.1, BA.4, and BA.5 have a high potential to become new dominating variants in the world.
Acknowledgments
This work was supported in part by NIH, USA grants R01GM126189 and R01AI164266, National Science Foundation, USA grants DMS-2052983, DMS-1761320, and IIS-1900473, NASA, USA grant 80NSSC21M0023, Michigan Economic Development Corporation, USA, Michigan State University Foundation, USA, Bristol-Myers Squibb, USA 65109, and Pfizer, USA.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Li W, Shi Z, Yu M, Ren W, Smith C, Epstein JH, Wang H, Crameri G, Hu Z, Zhang H, et al. , Bats are natural reservoirs of SARS-like coronaviruses, Science 310 (5748) (2005) 676–679. [DOI] [PubMed] [Google Scholar]
- [2].Qu X-X, Hao P, Song X-J, Jiang S-M, Liu Y-X, Wang P-G, Rao X, Song H-D, Wang S-Y, Zuo Y, et al. , Identification of two critical amino acid residues of the severe acute respiratory syndrome coronavirus spike protein for its variation in zoonotic tropism transition via a double substitution strategy, J. Biol. Chem 280 (33) (2005) 29588–29595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Song H-D, Tu C-C, Zhang G-W, Wang S-Y, Zheng K, Lei L-C, Chen Q-X, Gao Y-W, Zhou H-Q, Xiang H, et al. , Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human, Proc. Natl. Acad. Sci 102 (7) (2005) 2430–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, et al. , SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor, Cell 181 (2) (2020) 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D, Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein, Cell (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chen J, Wang R, Wang M, Wei G-W, Mutations strengthened SARS-CoV-2 infectivity, J. Mol. Biol 432 (19) (2020) 5212–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang R, Chen J, Gao K, Wei G-W, Vaccine-escape and fast-growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID-19-devastated countries, Genomics 113 (4) (2021) 2158–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wang C, Li W, Drabek D, Okba NM, van Haperen R, Osterhaus AD, van Kuppeveld FJ, Haagmans BL, Grosveld F, Bosch B-J, A human monoclonal antibody blocking SARS-CoV-2 infection, Nature Commun. 11 (1) (2020) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Yu F, Xiang R, Deng X, Wang L, Yu Z, Tian S, Liang R, Li Y, Ying T, Jiang S, Receptor-binding domain-specific human neutralizing monoclonal antibodies against SARS-CoV and SARS-CoV-2, Signal Transduct. Target. Ther 5 (1) (2020) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Li C, Tian X, Jia X, Wan J, Lu L, Jiang S, Lan F, Lu Y, Wu Y, Ying T, The impact of receptor-binding domain natural mutations on antibody recognition of SARS-CoV-2, Signal Transduct. Target. Ther 6 (1) (2021) 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang R, Chen J, Wei G-W, Mechanisms of sars-cov-2 evolution revealing vaccine-resistant mutations in europe and america, J. Phys. Chem. Lett 12 (2021) 11850–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang R, Chen J, Hozumi Y, Yin C, Wei G-W, Emerging vaccine-breakthrough SARS-CoV-2 variants, ACS Infect. Dis 8 (3) (2022) 546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Chen J, Wei G-W, Omicron ba. 2 (b.1.1. 529.2) high potential for becoming the next dominant variant, J. Phys. Chem. Lett 13 (2022) 3840–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Guerois R, Nielsen JE, Serrano L, Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations, J. Mol. Biol 320 (2) (2002) 369–387. [DOI] [PubMed] [Google Scholar]
- [15].Petukh M, Dai L, Alexov E, Saambe: webserver to predict the charge of binding free energy caused by amino acids mutations, Int. J. Mol. Sci 17 (4) (2016) 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pires DE, Ascher DB, Mcsm-ab: a web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures, Nucleic Acids Res. 44 (W1) (2016) W469–W473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rodrigues CH, Myung Y, Pires DE, Ascher DB, Mcsm-ppi2: predicting the effects of mutations on protein-protein interactions, Nucleic Acids Res. 47 (W1) (2019) W338–W344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Xiong P, Zhang C, Zheng W, Zhang Y, Bindprofx: assessing mutation-induced binding affinity change by protein interface profiles with pseudo-counts, J. Mol. Biol 429 (3) (2017) 426–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang M, Cang Z, Wei G-W, A topology-based network tree for the prediction of protein-protein binding affinity changes following mutation, Nat. Mach. Intell 2 (2) (2020) 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Frosini P, Measuring shapes by size functions, in: Intelligent Robots and Computer Vision X: Algorithms and Techniques, Vol. 1607, International Society for Optics and Photonics, 1992, pp. 122–133. [Google Scholar]
- [21].Edelsbrunner H, Letscher D, Zomorodian A, Topological persistence and simplification, in: Proceedings 41st Annual Symposium on Foundations of Computer Science, IEEE, 2000, pp. 454–463. [Google Scholar]
- [22].Zomorodian A, Carlsson G, Computing persistent homology, Discrete Comput. Geom 33 (2) (2005) 249–274. [Google Scholar]
- [23].Carlsson G, Topology and data, Bull. Amer. Math. Soc 46 (2) (2009) 255–308. [Google Scholar]
- [24].Mischaikow K, Nanda V, Morse theory for filtrations and efficient computation of persistent homology, Discrete Comput. Geom 50 (2) (2013) 330–353. [Google Scholar]
- [25].Xia KL, Wei GW, Persistent homology analysis of protein structure, flexibility and folding, Int. J. Numer. Methods Biomed. Eng 30 (2014) 814–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].De Silva V, Ghrist R, Coverage in sensor networks via persistent homology, Algebr. Geom. Topol 7 (1) (2007) 339–358. [Google Scholar]
- [27].Yao Y, Sun J, Huang XH, Bowman GR, Singh G, Lesnick M, Guibas LJ, Pande VS, Carlsson G, Topological methods for exploring low-density states in biomolecular folding pathways, J. Chem. Phys 130 (2009) 144115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bubenik P, Scott JA, Categorification of persistent homology, Discrete Comput. Geom 51 (3) (2014) 600–627. [Google Scholar]
- [29].Dey TK, Fan F, Wang Y, Computing topological persistence for simplicial maps, in: Proceedings of the Thirtieth Annual Symposium on Computational Geometry, ACM, 2014, p. 345. [Google Scholar]
- [30].Cang Z, Wei G-W, Topologynet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions, PLoS Comput. Biol 13 (7) (2017) e1005690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cang Z, Mu L, Wei G-W, Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening, PLoS Comput. Biol 14 (1) (2018) e1005929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nguyen DD, Cang Z, Wu K, Wang M, Cao Y, Wei G-W, Mathematical deep learning for pose and binding affinity prediction and ranking in D3R Grand challenges, J. Comput. Aided Mol. Des 33 (1) (2019) 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen J, Gao K, Wang R, Wei G-W, Prediction and mitigation of mutation threats to COVID-19 vaccines and antibody therapies, Chem. Sci 12 (20) (2021) 6929–6948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen J, Gao K, Wang R, Wei G-W, Revealing the threat of emerging SARS-CoV-2 mutations to antibody therapies, J. Mol. Biol 433 (7744) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Alenquer M, Ferreira F, Lousa D, Valério M, Medina-Lopes M, Bergman M-L, Gonçalves J, Demengeot J, Leite RB, Lilue J, et al. , Signatures in sars-cov-2 spike protein conferring escape to neutralizing antibodies, PLoS Pathog. 17 (8) (2021) e1009772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dupont L, Snell LB, Graham C, Seow J, Merrick B, Lechmere T, Maguire TJ, Hallett SR, Pickering S, Charalampous T, et al. , Neutralizing antibody activity in convalescent sera from infection in humans with sars-cov-2 and variants of concern, Nat. Microbiol (2021) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Linsky TW, Vergara R, Codina N, Nelson JW, Walker MJ, Su W, O Barnes C, Hsiang T-Y, Esser-Nobis K, Yu K, et al. , De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2, Science 370 (6521) (2020) 1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KH, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC, et al. , Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding, Cell 182 (5) (2020) 1295–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen J, Wang R, Gilby NB, Wei G-W, Omicron variant (b, 1.1. 529) infectivity, vaccine breakthrough, and antibody resistance, J. Chem. Inf. Model 62 (2) (2022) 412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wang R, Nguyen DD, Wei G-W, Persistent spectral graph, Int. J. Numer. Methods Biomed. Eng 36 (9) (2020) e3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mémoli F, Wan Z, Wang Y, Persistent laplacians: Properties, algorithms and implications, 2020, arXiv preprint arXiv:2012.02808. [Google Scholar]
- [42].Wang R, Zhao R, Ribando-Gros E, Chen J, Tong Y, Wei G-W, Hermes: Persistent spectral graph software, Found. Data Sci. (Springfield, Mo.) 3 (1) (2021) 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Meng Z, Xia K, Persistent spectral-based machine learning (perspect ml) for protein-ligand binding affinity prediction, Sci. Adv 7 (19) (2021) eabc5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wee J, Xia K, Persistent spectral based ensemble learning (perspect-el) for protein-protein binding affinity prediction, Brief. Bioinform 23 (2) (2022). [DOI] [PubMed] [Google Scholar]
- [45].Sirin S, Apgar JR, Bennett EM, Keating AE, AB-Bind: antibody binding mutational database for computational affinity predictions, Prot. Sci 25 (2) (2016) 393–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Moal IH, Fernández-Recio J, SKEMPI: a structural kinetic and energetic database of mutant protein interactions and its use in empirical models, Bioinformatics 28 (20) (2012) 2600–2607. [DOI] [PubMed] [Google Scholar]
- [47].Jankauskaitė J, Jiménez-García B, Dapkūnas J, Fernández-Recio J, Moal IH, SKEMPI 2.0: an updated benchmark of changes in protein-protein binding energy, kinetics and thermodynamics upon mutation, Bioinformatics 35 (3) (2019) 462–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chen J, Qiu Y, Wang R, Wei G-W, Persistent laplacian projected omicron ba. 4 and ba. 5 to become new dominating variants, 2022, arXiv preprint arXiv:2205.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Mannar D, Saville JW, Zhu X, Srivastava SS, Berezuk AM, Tuttle K, Marquez C, Sekirov I, Subramaniam S, Sars-cov-2 omicron variant: Ace2 binding, cryo-em structure of spike protein-ace2 complex and antibody evasion, BioRxiv (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].BA2 reinfection, 0000. https://www.timesofisrael.com/several-cases-of-omicron-reinfection-said-detected-in-israel-with-new-ba2-strain/.
- [51].Lyngse FP, Kirkeby CT, Denwood M, Christiansen LE, Mølbak K, Møller CH, Skov RL, Krause TG, Rasmussen M, Sieber RN, et al. , Transmission of sars-cov-2 omicron voc subvariants ba. 1 and ba. 2: Evidence from danish households, MedRxiv (2022) [Google Scholar]
- [52].Jemimah S, Yugandhar K, Michael Gromiha M, Proximate: a database of mutant protein-protein complex thermodynamics and kinetics, Bioinformatics 33 (17) (2017) 2787–2788. [DOI] [PubMed] [Google Scholar]
- [53].Liu Q, Chen P, Wang B, Zhang J, Li J, Dbmpikt: a database of kinetic and thermodynamic mutant protein interactions, BMC Bioinformatics 19 (1) (2018) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Procko E, The sequence of human ace2 is suboptimal for binding the s spike protein of sars coronavirus 2, BioRxiv (2020). [Google Scholar]
- [55].Serrano DH, Gómez DS, Centrality measures in simplicial complexes: applications of tda to network science, 2019, arXiv preprint arXiv:1908.02967. [Google Scholar]
- [56].Maletić S, Rajković M, Consensus formation on a simplicial complex of opinions, Phys. A 397 (March) (2014) 111–120. [Google Scholar]
- [57].Hernández Serrano D, Sánchez Gómez D, Higher order degree in simplicial complexes, multi combinatorial laplacian and applications of tda to complex networks, 2021, arXiv preprint arXiv:1908.02583. [Google Scholar]
- [58].Goldberg TE, Combinatorial Laplacians of Simplicial Complexes Senior Thesis, Bard College, 2002. [Google Scholar]
- [59].Horak D, Jost J, Spectra of combinatorial laplace operators on simplicial complexes, Adv. Math 244 (2013) 303–336. [Google Scholar]
- [60].Paszke A, Gross S, Massa F, Lerer A, Bradbury J, Chanan G, Killeen T, Lin Z, Gimelshein N, Antiga L, et al. , Pytorch: An imperative style, high-performance deep learning library, Adv. Neural Inf. Process. Syst 32 (2019). [Google Scholar]
- [61].Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. , Scikit-learn: Machine learning in python, J. Mach. Learn. Res 12 (2011) 2825–2830. [Google Scholar]
- [62].Wang R, Chen J, Hozumi Y, Yin C, Wei G-W, Emerging vaccine-breakthrough SARS-CoV-2 variants, ACS Infect. Dis 8 (3) (2021) 546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]