

Abstract
Enzymes have been shaped by evolution over billions of years to catalyse the chemical reactions that support life on earth. Dispersed in the literature, or organised in online databases, knowledge about enzymes can be structured in distinct dimensions, either related to their quality as biological macromolecules, such as their sequence and structure, or related to their chemical functions, such as the catalytic site, kinetics, mechanism, and overall reaction. The evolution of enzymes can only be understood when each of these dimensions is considered. In addition, many of the properties of enzymes only make sense in the light of evolution. We start this review by outlining the main paradigms of enzyme evolution, including gene duplication and divergence, convergent evolution, and evolution by recombination of domains. In the second part, we overview the current collective knowledge about enzymes, as organised by different types of data and collected in several databases. We also highlight some increasingly powerful computational tools that can be used to close gaps in understanding, in particular for types of data that require laborious experimental protocols. We believe that recent advances in protein structure prediction will be a powerful catalyst for the prediction of binding, mechanism, and ultimately, chemical reactions. A comprehensive mapping of enzyme function and evolution may be attainable in the near future.
Keywords: biological databases, catalytic sites, enzyme evolution, enzyme mechanism, ligand binding, protein structure
Introduction
Lying at the interface between biology and chemistry, enzymes are complex subjects to study. To understand how they function, it is necessary to integrate diverse kinds of data, including amino-acid sequence [1], three-dimensional structure [2], knowledge about their catalytic residues [3] and co-factors [4], and the chemical reactions they catalyse [5,6]. Lastly, enzyme mechanisms [7], which are the sequence of bond changes and atom movements that happen in the active site during catalysis, present the fundamental explanation for how enzymes operate. Figure 1 provides an outline of the collective understanding of enzymes across these six dimensions and shows some of the databases containing this knowledge.
Figure 1. Six dimensions of enzymes, related databases and data diversity and biases.

*The list of databases and resources is not exhaustive, but it aims to be representative of the type and amount of data available.
In addition to this complexity, enzymes should be viewed as changing entities. They have been evolving for billions of years, as shaped by natural selection, and across millions of species. The study of evolution and the aforementioned dimensions cannot be detached from one another. While genetic mutations govern and constrain changes in the enzyme sequence (the first dimension in Figure 1), natural selection acts primarily at the level of the biological function (the chemical reaction for enzymes, the last dimension in Figure 1). Additionally, like the challenge of mapping genomes to phenomes [8], establishing a causal link between sequence changes and catalytic activity in enzymes requires examining the intermediate dimensions.
An ideal knowledge base of enzyme function and evolution would consist of multiple maps, each representing one of the dimensions mentioned in Figure 1, and it would explain how these different aspects of enzymes relate to each other and how they have changed through time. It would provide explanations, or predictions, for how changing one dimension would alter the others. This would allow us to understand the historical and natural process of change, enzyme evolution, and would be a significant contribution for enzyme engineering [9,10] and drug development [11,12].
How far are we from being able to build such a comprehensive resource? UniProt [1], the most extensively annotated dataset of protein sequence data, currently covers the complete genomes of hundreds of thousands of species, a substantial and partially representative collection of all life on earth. At the same time, data coverage on other aspects of proteins, and in particular enzymes, such as structure, ligand binding, mechanism, or the reaction, is not as extensive. Nevertheless, equipped with what is already known about enzyme function and evolution, and increasingly powerful and diverse methods, we believe it should be feasible to use sequence data as the seed to populate all the other dimensions, all the way through to the enzyme reaction. The recent advances in predicting structure from sequence [13] demonstrate that this seemingly utopian vision may be within reach.
Enzyme evolution
Like other biological components, enzymes are best understood in the light of evolution. Similarities between enzymes reveal their evolutionary relationships, with more similar sequences having diverged more recently. Whereas all organisms on earth share a common origin, the Last Universal Common Ancestor (LUCA) [14], proteins and enzymes can be grouped into several evolutionary families, which have, for the most part, separate evolutionary histories. Within families, enzymes present a remarkable degree of conservation, emphasising the significant selective pressure to preserve catalytic function. Certain structural folds and active sites, for example, are so well conserved that they can be traced back to LUCA [15]. Although estimations about the number and type of proteins in LUCA are uncertain [16,17], it is likely that a majority of them were enzymes. A recent consensus analysis, identified 199 enzymes among 366 possible ancestral proteins [18].
Since then, new enzymes, like all proteins, have mostly evolved by gene duplication and divergence [19]. Other genetic events, such as the fusion and swapping of domains, are rarer but also important, since they allow for larger evolutionary jumps and more significant changes of function [20]. Finally, de novo evolution of proteins from non-coding DNA is also possible, and not limited to the ancient past, as previously thought [21]. De novo proteins, are typically similar to small random peptidic chains, except for their improved solubility [22], and their physiological functions have been characterised for only a handful of cases, so they will not be discussed further in this review.
Enzyme evolution by gene duplication and divergence
Following the first observations of proteins sharing similar sequences, suggesting homology and common ancestry, Susumo Ohno proposed a neofunctionalization model based on gene duplication and divergence [23]. This model proposes that after a random duplication event, one of the gene copies can diverge without compromising organism fitness, and that these mutations may eventually result in the acquisition of a new function. The importance of duplication and divergence for neofunctionalisation has been reinforced since, but alternative models have been proposed [24] to address some limitations of Ohno's model. Most notably, these alternatives take into account that deleterious mutations typically accumulate faster than gain-of-function mutations [25], which often lead to complete loss of function and eventual deletion of the duplicated gene.
The IAD (Innovation–Amplification–Divergence) model, also called the Adaptive Radiation model [25,26], solves this apparent dilemma while also considering the significance of promiscuity for enzyme evolution. The sequence of events in the IAD model is depicted in Figure 2 and described below.
Figure 2. Models of divergent evolution and possible evolutionary relationships among extant enzymes.
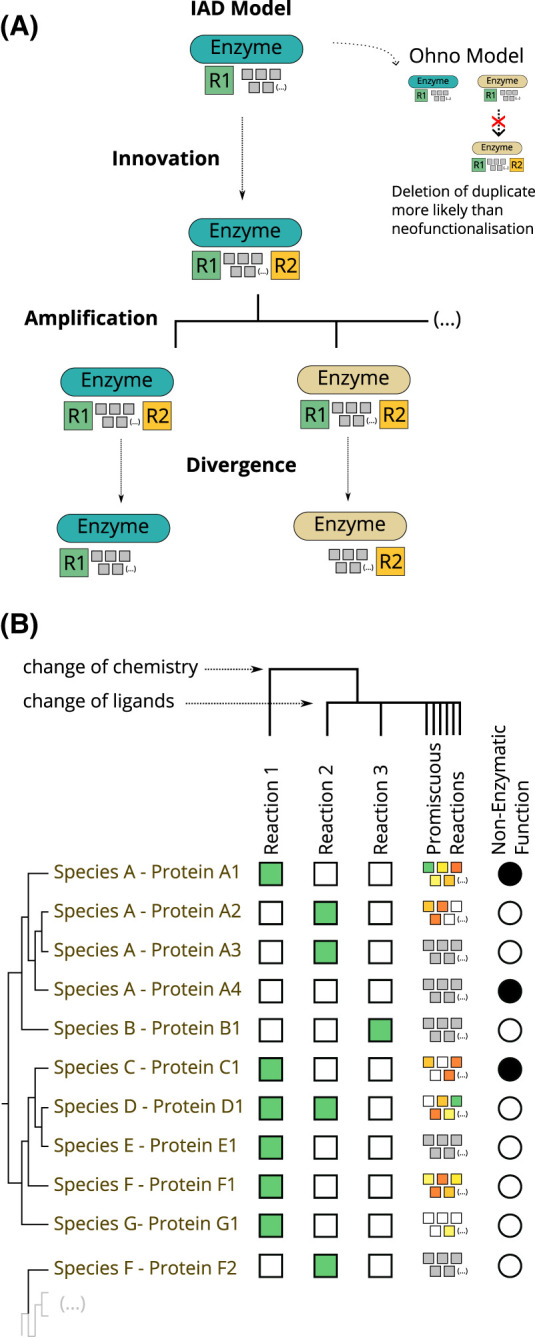
(A) The Innovation–Amplification–Divergence (IAD) model of enzyme evolution. Ohno's model is shown for comparison. A description of both models is given in the main text in the ‘Enzyme Evolution by Gene Duplication and Divergence' section. (B) Evolutionary and functional relationships between enzymes and non-enzymatic proteins. The colours of the squares are meant to indicate enzyme efficiency from green (high efficiency) to red (low efficiency). Grey squares indicate that the enzyme is likely promiscuous but was not tested for promiscuous reactions. Black circles indicate that the protein has a non-enzymatic reaction. The types of evolutionary relationships are discussed in the ‘Evolutionary and Functional Relationships Between Enzymes' section.
Innovation
During evolution, some mutations grant enzymes the ability to catalyse additional reactions that have no impact on fitness, since they are either too slow to affect metabolism or involve inaccessible substrates. These so-called promiscuous reactions are widespread [27] and they are considered a latent pool of innovation for evolution to use [27–29]. A promiscuous reaction might become important for fitness after a change in the organism's environment, such as the introduction of industrial chemicals in soils [30,31].
Amplification
The enzymatic efficiency for a new reaction is typically low and cannot be easily improved. Beneficial mutations for the new reaction often have negative effects for the old one (pleiotropy), resulting in an evolutionary impasse. Furthermore, distinct regulation of both reactions is impossible, unless they occur in different cells or tissues. The solution for these problems is the duplication or further amplification of the gene. In IAD, this amplification, defined as a selective expansion in the number of copies of a gene, is a favourable genetic event in itself, since it results in a larger number of enzymes and, ultimately, in an increase in the reaction turnover in the cell.
Divergence
After amplification, the copies of the gene are free to independently diverge. As some copies improve their catalytic efficiency towards one reaction, others are deleted from the genome as they no longer provide an advantage. Eventually, only two copies of the gene remain, each specialised in their chemical reaction and associated regulation.
Expansion of enzyme evolutionary families
A family of related enzymes originated solely by duplication and divergence will have a unique common ancestor and can be organised in a well-ordered phylogenetic tree in a way that mimics the evolution of species and the tree of life. This hierarchy is complicated by domain recombination, so this is discussed separately below. While some enzyme families are very specific and tend to catalyse only one function across all the organisms where they are expressed, in other cases, the process of duplication and divergence leads to, over time, an increase in the number of members in protein families and also the number and variety of their functions. Some well-studied functionally diverse enzyme families include the haloacid dehalogenases [32], Glutathione Transferases [33], and the amidohydrolases [34].
Using structural classification systems like CATH [35] or SCOP [36] to identify distantly related homologues together with functional assignments, such as the enzyme commission (EC) [37], it is possible to categorise enzyme families in an encompassing scale. FunTree [38] is a resource that shows phylogenetic and functional relationships between enzymes based on CATH and EC, and has been used to study the general evolution patterns of enzymes. For example, among 379 enzyme families [39] for which the catalytic function can be assigned to a single structural domain, there are enzymes catalysing 2994 unique reactions, meaning that most types of reactions (at least according to the EC classification) have diverged from a common ancestor. The EC classification can also be used for a more detailed analysis. The EC hierarchy is composed by four levels: class, subclass, sub-subclass, and serial number. The three first levels are used to define the type of the reaction while the serial number specifies the substrates and products. Enzymes that have the same sub-subclass (the same first three EC numbers) and only differ in the last digit, catalyse essentially the same type of reaction on a different substrate. Most evolutionary changes observed in FunTree and similar datasets are at the fourth EC level. One can also use changes of EC class (the first number in the EC code) to find more radical changes of function, which for the mentioned 379 families account for <20% (18.6%) of the changes observed.
Illuminating as they may be, studies like these fall short of providing a casual explanation between the changes in the protein sequence and the observed functional changes (akin to limitations in genome-wide associations studies for the genome and phenome). To establish these causal links, a comprehensive analysis of the mutation's impact on the enzyme's structure, ligand binding, and catalytic mechanism is necessary. Evolutionary studies with this level of detail and across families are rare [40] because these analyses are difficult to automate, in particular when considering mechanistic data. However, as we discuss in the ‘Enzyme Mechanism' section, we have recently made some progress in systematising the knowledge about enzyme mechanisms into ‘rules of enzyme catalysis', which might be a future foundation for such studies.
Enzyme evolution by recombination of domains
The duplication models discussed above assume that an entire gene is duplicated, followed by independent changes in each copy. However, some genetic events can also lead to the insertion of genetic material from some genes into or next to other genes. Proteins domains are regions of the protein that fold independently and usually have a well-defined function. These domains serve as evolutionary units because genomic events that do not copy or move the entire domain are likely to disrupt its folding and function, rendering the resulting protein inactive. Throughout evolution, domains with distinct functions have been combined in different ways to create fully functional proteins [41,42].
The recombination of domains is also an important source of innovation in enzyme evolution. New domains can alter substrate specificity, regulate binding or catalytic activity, change the catalytic function, or simply add independent catalytic activities, resulting in multifunctional enzymes [43]. Furthermore, many enzymes that use co-factors evolve by combining the co-factor binding domains with other domains that bind the substrate or provide additional catalytic machinery. Notable examples include the Radical S-adenosylmethionine (SAM) superfamily [44], and FAD binding enzymes, such as Flavin dependent nitroreductases [45] and monooxygenases [20] where, interestingly, the sequence of the co-factor binding domains (but not the structure) is found interlaced with the sequence of the substrate binding domain.
Comprehensive and automated studies on the evolution of enzymes by domain recombination are challenging because the annotation of function in protein databases is traditionally given to the whole sequence. Preferably, one would want to know which function is contributed by each domain. The PDBe Knowledge Base and associated data sources [46], which provide residue-level annotations, might be a good starting point for future studies.
Convergent evolution
The same selective pressure keeping catalytic residues and mechanisms extremely well conserved during evolution, also leads to cases of convergent evolution, where enzymes evolve similar catalytic capabilities independently. The most clear-cut examples of convergent evolution in catalysis, are enzymes that have different structural folds but can catalyse the same overall reaction (sometimes called Non-homologous Isofunctional Enzymes) [47]. From a bioinformatics point of view, for annotated enzymes, these can be detected by searching for enzymes that have a different CATH code (or similar evolutionary classification), but the same EC number. In the FunTree study mentioned above [39], it was observed that 59% of EC reactions are catalysed by proteins belonging to at least two CATH superfamilies, suggesting that convergence of chemical function is surprisingly common.
Convergence can happen on different levels, as illustrated in the third panel of Figure 3. Complete convergence would be an example where the two enzymes catalyse exactly the same overall reaction (which implies having the same substrate and products) using an identical set of catalytic residues and reaction mechanism. Convergence only at the reaction level would mean that the enzymes catalyse the same reaction using a different mechanism and catalytic residues. It is also possible that enzymes catalyse a common catalytic step on different substrates or have converged to bind the same substrate or co-factor but catalyse a different reaction. Once more, a detailed account of the convergent evolution of enzymes would need to consider similarities at these different levels. This is even more important in the study of convergence than divergence because the lack of sequence similarity means that most of these relationships (such as mechanistic similarities) might be hidden in the data.
Figure 3. Some paradigms of enzyme evolution as defined by the similarities between the different dimensions of enzymes.
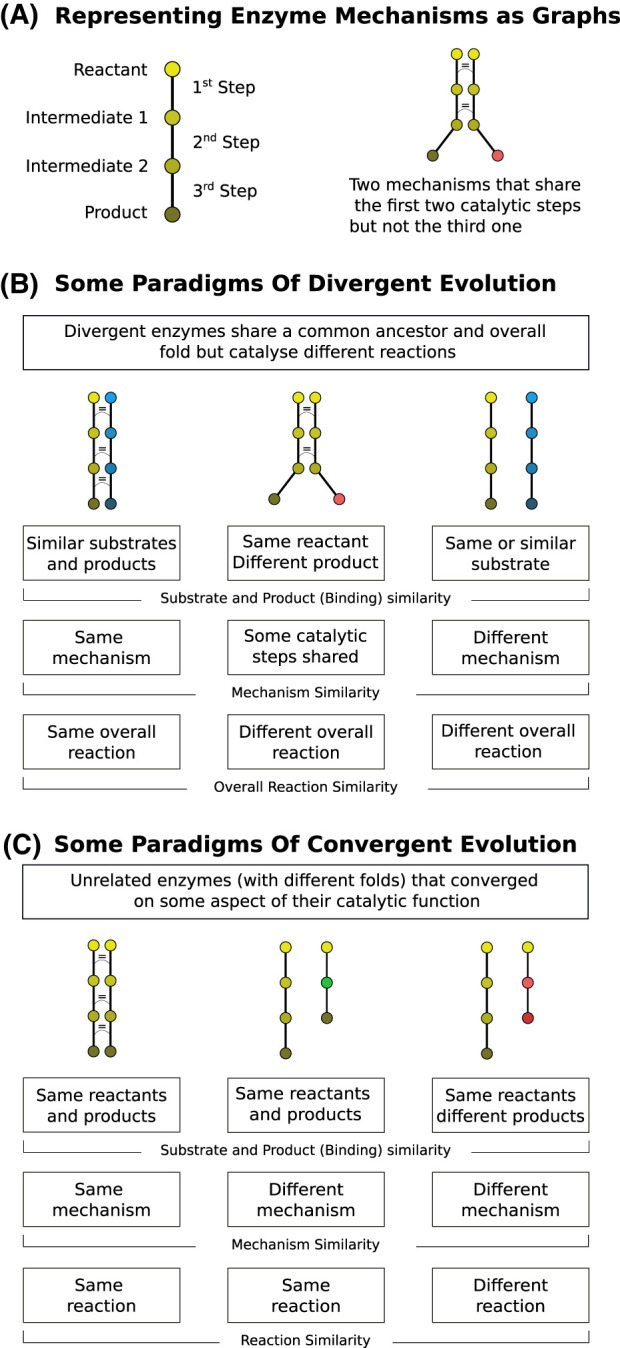
(A) Mechanisms are represented as graphs where nodes (circles) represent stable configurations of the active site along the mechanism (reactants, intermediates and products) while edges (lines) represent the catalytic steps. Two mechanisms can be compared by showing their graph representation side by side. An equal sign is used to represent steps with a high degree of similarity. Nodes with the same colour represent the same ligand (substrate or product) in both mechanisms. Transformations along the mechanism are indicated with a change of gradation to darker colours or complete change of colour when the transformation between the two enzymes are different. (B) Three paradigms (among other possibilities) of the divergent evolution of enzymes. Enzymes might evolve to (from left to right): catalyse the same reaction using the same mechanism on a different substrate; catalyse a different reaction on the same substrate by a partial change in the mechanism; catalyse a completely unrelated reaction on the same or different substrate. (C) Three paradigms (among other possibilities) of the convergent evolution of enzymes. Enzymes with unrelated ancestry might converge to (from left to right): catalyse the same reaction using a similar mechanism; catalyse the same reaction using another mechanism; bind the same substrate to perform unrelated reactions.
Functional and evolutionary relationships between enzymes
The complex interplay between the evolution of enzymes and the chemistry they catalyse leads to a rich tapestry of possible sequence-function(s) associations. For example, one enzyme can catalyse multiple reactions, differing in their substrates or overall chemical transformations, and the same reaction can be catalysed by related or unrelated enzymes. The second panel of Figure 2 shows some of the different possibilities and Figure 3 shows how these can be explained in terms of the underlying binding capabilities, catalytic machinery, and the reaction mechanisms.
All enzymes catalyse at least one chemical reaction that is important for the fitness of the organism. This might be called the enzyme's primary or native reaction. Some enzymes (protein D1 in Figure 2, for example) are able to catalyse more than one reaction (or the same reaction on different substrates) where the additional reactions are also important for fitness. This can be a well specified secondary reaction, or the case of broad-specificity enzymes, which are able to catalyse the same type of reaction across a range of substrates, as in the case of detoxifying enzymes.
Promiscuous reactions, on the other hand, are reactions that enzymes are able to catalyse but that are currently irrelevant for biological function and the fitness of the organism (there are other definitions of enzyme promiscuity, but we think this is the most useful) [27]. For example. these might be reactions that are too slow to have a metabolic impact, or reactions that involve substrates that do not exist in the organisms or their environment. Although not immediately important for fitness, promiscuity is crucial for the evolvability of enzymes, as explained above [29,48]. It is increasingly clear from substrate profiling studies that most, if not all, enzymes are promiscuous.
Enzymes might also perform non-catalytic functions. When the non-catalytic functions are independent from the catalytic activity, these are sometimes called moonlighting enzymes [49]. Conversely, pseudoenzymes (protein A4 in Figure 2) are proteins that do not have any catalytic function but are evolutionarily related to enzymes [50]. Typically, pseudoenzymes evolve from a catalytic ancestor that has lost its catalytic function [51].
Orthologous enzymes (proteins A1 and C1 in Figure 2, for example) are homologous proteins that have diverged following a speciation event and keep catalysing the same primary reaction. Paralogous enzymes (A1 and A2) arise from gene duplication within the same genome and evolve to catalyse different functions. Isozymes (A2 and A3) are enzymes in the same organism that catalyse the same reaction but might have differential regulation and expression, which justify the presence of a duplicate. Convergent evolution is at play when unrelated enzymes catalyse the same reaction (A2 and F2). The correct identification of all these evolutionary relationships is crucial to predicting the function of uncharacterised enzymes [52].
Evolution as an algorithm
As a search algorithm with the goal of finding catalytic proteins for a host of chemical reactions, enzyme evolution has several biological constrains, which limit the potential solutions it can find. Point mutations, the most common genetic event, restrict the size of potential changes to one residue position and to a small selection of the 20 amino acids (due to the genetic code, most amino acids changes are unreachable after mutating only one nucleotide). Series of mutations, which can be thought as a walk through the sequence space, cannot go through states where the activity of the enzymes is compromised or, depending on the selective pressure, even slightly lower. This means that the algorithm can get stuck in local maxima of fitness, and better maxima might be inaccessible because there is no favourable path to reach them (Evolution does not have foresight). One of the advantages of rational enzyme engineering is precisely the ability to make targeted jumps to parts of the sequence space that are unreachable to natural or even directed evolution.
The complexity of enzymes makes this a difficult search problem. Each of the dimensions discussed in this paper represent competing evolutionary goals, such as the maintenance of structural fold and stability or the enhancement of binding and the catalytic rate [45]. This results in an intricate evolutionary space where the effect of pleiotropic mutations and epistasis is significant [53]. It also explains why small changes and the overlap of catalytic functions (promiscuity) are so common, as opposed to large functional jumps, which will likely be deleterious. Finally, the presence of competing goals, coupled with the absence of a need for optimising beyond selective pressure, also justifies why evolution tends to produce enzymes that are ‘good enough’ rather than perfect.
Epistasis refers to the differential effect of mutations in one position being dependent on mutations in other positions of the same or different gene [54]. Epistasis is a reflection of the vast and multidimensional sequence/fitness space, where a particular mutation can only be called beneficial or deleterious (for enzyme activity, for example) in the context of a specific sequence, and where the role and importance of each position is not absolute but dependent on the environment. Pleiotropy refers to any genetic variants that affect more than one phenotype [53]. Pleiotropic mutations, in the context of enzyme evolution, are mutations that improve the ability of the enzyme to catalyse one reaction, while being detrimental to the other. These effects might be noticed at the level of the binding, the catalytic rate, or the overall mechanism. Pleiotropic mutations are an important reason for the necessity of the duplication of genes (together with independent regulation), because in most cases it is impossible to find a particular sequence that can effectively catalyse the two reactions of interest with optimal rates and expression.
Mapping the dimensions of enzyme catalysis
Information about the various dimensions of enzyme catalysis can be found scattered throughout the literature but also consolidated in several databases, each dedicated to specific types of data [46]. In addition to providing a centralised location for accessing information, databases offer the added benefits of normalisation and structured data models, which facilitate analysis and the re-use of data. Prediction methods, typically trained or tested against these data, allow researchers to use certain data points (like sequence) to fill the gaps in knowledge about other data (such as structure). Figure 1, together with the sections below, provide an outline of the collective understanding of enzymes across the six dimensions and shows some of the databases containing this knowledge. We also discuss how some of the missing data can be predicted from computational methods (summarised in Figure 4) and give a non-exhaustive account of some of these tools for each dimension.
Figure 4. Overview of existing computational methods used to predict the dimensions of enzymes based on known data [13,35,52,77–79,87,93,99,103,105,106,113,120,123,126–133].

*Ligand Binding refers both to the identification of ligand binding sites and the prediction of native ligands and their binding poses.
Sequence
Among the 6 dimensions shown in Figure 1, sequence is the one for which there are more experimentally determined data. This can be attributed to the increasing availability of sequencing methods, including recent advancements in metagenomic experiments, which enable the simultaneous sequencing of genomes from multiple species [55]. UniProtKB [1], a comprehensive database of protein sequences and associated biological knowledge, currently holds more than 246 million sequences belonging to more than 163 thousand proteomes (not considering redundant and excluded proteomes). At the same time, the MGnify database [56], which archives predicted protein sequences from the sequencing of metagenomic samples, contains more than 2.4 billion sequences.
The fraction of these proteins that has been experimentally characterised is minimal. Indeed, for most of the sequences in these databases, not even the existence of the protein has been confirmed, as protein sequences are solely predicted from genomic sequencing data. Swiss-Prot, a manually curated subset of UniProtKB that focus on well-studied organisms and proteins, contains 569 213 sequences (release 2023_1), or 0.23% of the total UniProt. Only 19.6% of the entries in Swiss-Prot contain evidence for the existence of the protein at the protein level, and another 9.8% at the transcription level. For TrEMBL, the non-curated portion of UniProt, these numbers are 0.08% and 0.55%, respectively. Finally, while the number of entries in Swiss-Prot has been essentially static for the past few years, TrEMBL and Mgnify have been growing exponentially, increasing the gap between the amount of raw sequence data and their functional characterisation.
Enzymes comprise 48.2% of Swiss-Prot (274 342), identified as all the entries associated with at least one EC number, while 15.5% (38.2 M) of TrEMBL entries are associated with an EC number. The enzyme coverage in Swiss-Prot and TrEMBL do not reflect the percentage of enzymes across life or even across the annotated species. On the one hand, there is an overrepresentation of enzymes in curated datasets such as Swiss-Prot, because enzymes have been more extensively studied in the past compared with other proteins. On the other hand, in non-curated datasets many proteins have not been assigned any function, including enzymes.
We have previously discussed the different ways enzymes are annotated in Swiss-Prot, and the data supporting these assignments, in the context of pseudoenzyme classification [57]. In that study, we verified that only 11% of Swiss-Prot entries contained experimental evidence related to the function of the protein, and of these, 46.9% (29 731) were labelled as enzymes. There is no better way to illustrate the challenge and the importance of computational methods to make correct functional assignments than contrast this number with the already mentioned number of non-redundant proteins in Mgnify, 2.4 billion, a difference of five orders of magnitude.
As discussed above, proteins (or domains) that evolve by duplication and divergence can be grouped in evolutionary families. Proteins in the same family can be identified by sequence similarity and typically perform the same or similar functions. These principles are the basis for several systems that try to organise the known protein sequence space. For example, the current version of Pfam [58] organises UniProt sequences into 19 632 related families of protein domains, based on matches to HMMs (hidden Markov models), representing each family. All sequences in a Pfam family share a common ancestor but they may not share the same function. Other notable classification systems of protein families include the CCD [59] and PantherDB [60]. PIRSF [61], rather than using domains, focus on the annotation of entire proteins. InterPro [62], which integrates these and other signatures in a single resource, assigns at least one signature to 84.1% of the sequences in UniProtKB. Evolutionary units smaller than the domain have also been identified and have been used to explain the evolutionary history and evolutionary relationships between domains [63].
Structure
Protein structures are available through the Protein Data Bank (PDB) [2], a structural archive of biological macromolecules. As of June 2023, the PDB archives 206 462 structures, most of them containing proteins (201 976). In contrast with most protein sequences in UniProt, which are determined in bulk by genome sequencing of entire organisms, methods to solve protein structures (X-ray crystallography, Electron Microscopy and NMR) are costly and time intensive and so, are typically used to characterise one system at a time. For this reason, the size and growth of PDB is more modest than UniProt. PDB is also more redundant, since many structures deposited in the PDB belong to the same protein. The current 201 976 protein structures in PDB correspond to only 62 433 UniProt sequences. PDB is also biased to better studied organisms and proteins. Almost one third of the PDB structures are of human proteins, for example, and more than two thirds are enzymes.
Structural families and structure prediction
Structural similarity can also be used to find evolutionary relationships among proteins and to define families and, because protein structure is much more conserved than sequence, it allows us to retrieve much older relationships. It is possible to recognise homologous proteins based on their overall fold even when their sequences have diverged beyond recognition. SCOP [36], CATH [35] and ECOD [64] are well known structural classification systems that group evolutionary related proteins together. Taking CATH as an example, all protein domains within the same superfamily (such as CATH:3.40.50.720 — NAD(P)-binding Rossmann-like Domain) share a common ancestor, and have emerged by duplication and divergence, either by speciation events (leading to the appearance of orthologs) or gene duplication (leading to the appearance of paralogs). As of June 2023, CATH categorises more than 536 000 domains, belonging to more than 186 000 PDB structures, into 6631 distinct superfamilies.
For many years, template-based methods, which can predict a protein's structure starting from the structure of a homologous protein, were the most efficient tool to bridge the gap between the number of known sequences and the number of available structures [65]. Recently, deep-learning methods, most notably Alphafold [13], which only requires an alignment of homologous sequences to the query, have been able to generate high quality structural models, even for proteins that do not have a known structural homologue. The alphafold database [66] currently provides structural predictions for most sequences in UniProt. ESMFold, another deep-learning structure prediction method, has been used to predict the structure of more than 700 million metagenomic sequences [67].
Ligand binding
The active site is the region of the enzyme where the reaction takes place. It needs to fulfil two main roles for the catalytic activity to happen: to bind the required substrates and co-factors; and to provide the catalytic residues and surrounding environment that are conducive to catalysis. When it comes to binding, there are two levels of knowledge that we might have for a given enzyme. The first is to know which ligands bind the enzyme and what is their binding affinity. Binding databases typically include data on both natural substrates and enzyme inhibitors. BrendaDB [68], a database containing kinetic information of enzyme reactions, contains 176 610, 69 886, and 46 076 values of KM, IC50, and KI, respectively. BindingMOAD [69] and PDBbind [70], which focus on complexes that exist in the PDB, contain affinity data for 15 223 and 19 443 complexes, respectively. BindingDB [71] has more than 2.7 million data points extracted from both academic papers and patents.
The second type of knowledge about binding is related to where the ligand binds in the active site and what are the conformations the ligand and the enzyme adopt upon binding. These types of data are ultimately derived from the PDB, since many enzymes in the database include ligands in their active site, but other databases curate this information in different ways. These include the sc-PDB [72], BioLip [73], the already mentioned BindingMoad and the NLDB [74], which also includes predicted complexes.
We have previously analysed how well the PDB covers the binding of native ligands to enzyme structures against the known reactions in EC and KEGG [75]. We found that most enzymatic structures in the PDB have either no ligand in the active site or a ligand with low similarity to the native one. Only 26% of the enzyme structures in the PDB bind a molecule that is at least 70% similar to the cognate ligand. This coverage increases to 58.9% and 62.9% if we aggregate all the structures belonging to the same KEGG reaction, or EC number, respectively. Nonetheless, this still means that there is no adequate enzyme–ligand structure for more than one third of the reactions curated in these databases.
Protein–ligand prediction
While Alphafold and similar methods helped filled the gap in structural coverage, when compared with sequence, and fixed some of the experimental biases in PDB, it did not help with the lack of enzyme–ligand structures, since its predictions do not include ligands. Alphafill [76] alleviates this problem somewhat by finding ligands that bind structurally similar protein regions in PDB and transposing these ligands to the Alphafold structure, but this solution does not extend to ligands that do not exist in the PDB.
When it comes to predict binding to uncharacterised proteins, there are at least three subproblems to solve, the identification of ligand binding sites, the identification of the correct ligands and their binding pose, and the estimation of the binding affinity. There are numerous computer tools dedicated to answer one or more of these questions [77,78]. Template-based methods work by looking at similar sequences or structures that have been previously characterised. Knowledge about the phylogenetic relationships can also be useful here, since it is expected that orthologous enzymes will bind the same substrates while paralogues might differ. Machine learning methods are also trained on existing data and can use both sequence or structural features to identify binding sites and potential ligands. Simulation methods, most notably molecular docking [79], can be used to predict binding poses and affinities ab initio, starting from the protein structure.
Despite the abundance of tools to predict protein–ligand binding, this is still an open problem. An accurate and general solution to identify good ligands for a given protein would be useful not only for the study of evolution and enzyme function but would be revolutionary for drug discovery, so progress in this area is bound to continue.
Catalytic residues and co-factors
The catalytic residues are the amino acids in the active site of the enzyme that are responsible for accelerating the chemical reaction by lowering the energy of transition states or providing mechanistic paths that are not available elsewhere. The M-CSA (Mechanism and Catalytic Site Atlas) is the most comprehensive dataset of catalytic residues and includes curated annotations of the specific functions that the residues perform in each catalytic step. This data has been used in the past to better understand enzyme function and evolution. We recently did an overview of the frequency, roles and conservation of the catalytic residues across 648 enzyme families [3] and have also studied how mutations in the catalytic residues correlates with the evolution of pseudoenzymes [51,57]. The same dataset has also been used by others to answer biological questions [80,81], and to develop other data resources and methods [82–86], including most of the prediction methods discussed below.
Catalytic residues are extremely well conserved in evolution, even more so, in some examples, than the overall protein fold. For this reason, they are extremely important in the study of divergent evolution. Unlike the rest of the sequence, changes in catalytic residues are almost always associated with either a change or loss of catalytic function. Conversely, and unlike the overall protein sequence and structure, catalytic residues are also crucial to understand convergent evolution since the same active site composition and disposition can be found in unrelated enzymes.
We have recently reviewed the literature on studies and applications of 3D templates of catalytic residues [87], have analysed their flexibility in PDB structures [88], and their distribution in related and unrelated enzymes [89] using the M-CSA dataset. In related enzymes of both similar or divergent functions, active sites exhibit different degrees of structural variation, with the relative 3D disposition of catalytic residues being affected by their role in the mechanism and by binding of different substrates or products. With this geometric information we have generated several consensus templates representing compact clusters of catalytic residues. Recurring instances of these templates, which we have defined as the ‘3D modules of enzyme catalysis' [89], are typically associated with one or more functions and types of ligands and can themselves be used to better understand biological catalysis and evolution, and aid in enzyme design.
Co-factors are non-protein molecules that are required by many enzymes to perform their catalytic function. These molecules provide catalytic roles that cannot be performed by the canonical amino acids [90]. The evolutionary history of co-factors is interesting in its own right, since they are thought to be molecular fossils, catalysing reactions that can be traced back all the way to the origins of life. This argument has been initially made for nucleotide-like co-factors, which might be remnants of an RNA world [91], but has been extended to other organic and inorganic co-factors, which might have been the original catalysts in prebiotic geochemistry systems and later co-opted by RNA and protein-based enzymes [92]. Information about the roles of co-factors in enzyme mechanisms can be found in the M-CSA and the Co-factor database [4].
Prediction of catalytic residues
The identity of the catalytic residues of uncharacterised enzymes can be computationally inferred using both sequence and structural data. Due to their high conservation, a simple homology search and multi sequence alignment might be enough to identify potential catalytic residues, in particular, if the enzyme exists in distantly related species. In automated methods, conservation data is typically combined with other sequence-based features [93] and phylogenetic information [94,95]. Structurally, the clustering of catalytic residues in a well-defined pocket or cleft (the active site) and other features such as solvent accessibility, calculated pKa, and number of contacts, have been used by other methods [96,97]. It is also possible to create a network representation of the protein structure, which yields other descriptors such as closeness centrality that can also be used to distinguish catalytic residues [98]. Finally, some methods take an integrative approach by combining different types of data, typically with the help of machine learning algorithms to identify the best combination of features [99–103].
Enzyme mechanism
The enzyme mechanism comprises all the atomic movements and bond changes in the active site that are responsible for moving the catalytic reaction forward. It is a crucial piece of data to understand how enzymes work and how they have evolved. The M-CSA (Mechanism and Catalytic Site Atlas) database [7] contains detailed annotations of the individual catalytic steps of 734 enzymes mechanisms. This is a small number when compared with the other databases mentioned in this review, which reflects both the lower number of studies in the literature and the complex nature of the problem, which requires many different types of data coming from different types of experiments. Nevertheless, since many related enzymes share the same mechanism, the coverage across the protein space is more extensive than it initially appears. By assuming that homologous sequences with the same set of catalytic residues and catalysing the same reaction also share the same mechanisms, the annotations in M-CSA can be extended to more than 15 000 PDB structures and 70 000 Swiss-Prot sequences.
The literature is lacking in studies looking at the evolution of enzymes at the mechanistic level. Twenty years ago, Bartlett et al. [40] performed a manual analysis of 27 pairs of homologous enzymes with different functions, to learn how they differed in catalytic residues and mechanisms. The picture for this small subset of enzymes was diverse. While all enzyme pairs had at least some active site similarities, only 15 pairs exhibited mechanism similarity. In this last group, enzymes shared some catalytic steps and diverged at others, and these changes, typically at the start or end of the mechanism, were enough to completely change the overall catalysed reaction. Another study [104] charted the appearance of new catalytic steps over evolutionary time to find that half of the observed chemistry in enzymes was already present in LUCA, while the other half appeared progressively over time.
A large scale and complex analysis of the evolution of enzyme mechanisms has not been possible because until recently there was no way to automatically compare reaction mechanisms. We have recently generated a set of ‘catalytic rules' (see Figure 5) that are based on the catalytic steps annotated in M-CSA [105], which should be useful to automatically find similarities between the mechanisms of related and non-related enzymes.
Figure 5. Some rules of enzymatic catalysis.
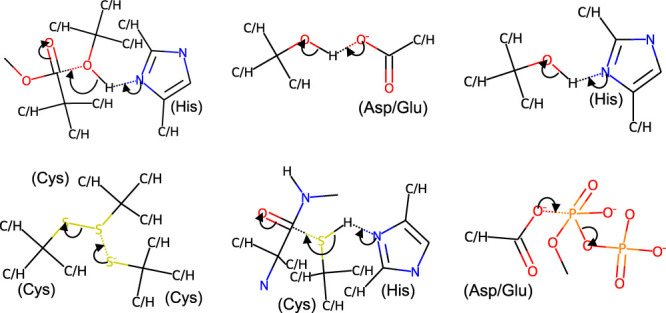
Each rule represents a type of chemical step observed in one or more reaction mechanisms annotated in the M-CSA (Mechanism and Catalytic Site Atlas). We have previously described the process of creating these rules and their possible usages for studying enzyme evolution [105,134].
Predicting the mechanism of enzymes
Simulation methods, such as QM/MM (Quantum Mechanism/Molecular Mechanics), are widely used to study the mechanism of enzymes in silico [106]. These methods provide a window to the active site by showing all catalytic events with atomic-level detail, something that is not accessible experimentally, due to the transient nature of the transition states and unstable intermediates. Although powerful, these methods are computationally expensive and difficult to setup, which limits their usage in large scale.
Homology can also be used to infer the mechanisms of enzymes but only if another enzyme with identical active site and function has already been characterised, in which case it can be assumed that both enzymes follow the same mechanism. To make use of the accumulated knowledge about enzyme mechanisms available in M-CSA, we have developed EzMechanism [105], a tool that can automatically generate mechanistic hypotheses for a given active site and chemical reaction. EzMechanism only takes into account local chemical similarities, so it also works for unrelated enzymes. Furthermore, it is able to compose mechanisms that have never been seen before. We are currently working on coupling the mechanistic hypotheses generated by EzMechanism to QM/MM calculations, with the objective of automatically describing their energetic profile.
Enzyme reaction
Thousands of biological reactions have been identified, particularly those associated with the primary metabolism, and have been categorised by different resources. The EC list [37], the most widely used classification system for enzyme reactions, currently includes 6743 EC numbers [107]. KEGG Reaction [108] and Rhea [5], two databases of biological enzyme reactions, contain annotations for 11 858 and 15 116 distinct chemical reactions, respectively. EnviPath [109], which focuses on reactions involved in the biotransformation of environmental contaminants, contains 4398 reactions. Of the 569 793 sequences currently annotated in Swiss-Prot, 274 744 are annotated with an EC number [1]. In TrEMBL, which comprises the 248 M unreviewed sequences of UniProt, and where most of the annotation attributions have been done by homology, there are almost 40 M sequences associated with an EC number.
Like protein sequence and structure, chemical reactions can also be grouped by similarity, and the number of these clusters give a better indication of the size of the chemical space than the total number of reactions. For example, many enzymatic reactions describe the same transformation performed on molecules that share a common chemical group [39]. Typically, these arise from cases of divergent evolution, where binding residues are mutated, while catalytic residues, and the overall mechanism remain conserved. However, unlike sequences and structures, similar enzyme reactions can also be the result of convergent evolution, as discussed in the ‘Convergent Evolution' section.
A possible measure of the true diversity of enzyme reactions is the third level of the EC classification. As explained in the ‘Expansion of Enzyme Evolutionary Families' section, reactions that only differ on the fourth level can be considered the same type of reaction applied to a different substrate. According to this measure, the EC currently describes 316 types of reactions, or sub-subclasses. The KEGG database groups their reactions in 65 reaction classes defined by a transformation pattern, which can also be understood as types of reaction. Both the EC hierarchy and the definition of KEGG reaction classes, are manually curated. An automated analysis using EC-BLAST on a set of 6000 fully specified and balanced reactions built from the KEGG reaction database, was able to create clusters based on similar bond changes and reaction centres. In this analysis, more than 700 clusters with more than one reaction were created [110].
While the data discussed above focus on the classification of reactions and the identity of substrates and products, other databases such as Brenda [68] and Sabio-RK [111] hold information about the kinetics of reactions. Brenda contains more than 85 000 kcat values while Sabio-RK contains more than 50 000 kinetic parameters, overall. Traditionally, using kinetic data for broad studies of enzyme function has been challenging because data has not been consistently provided in the literature. For example, in many papers of kinetic studies, the exact sequence of the protein being studied was either unknown, or not reported. Experimental conditions, which can greatly affect enzyme turn over, have also not been reported consistently. This problem has been addressed on recent years after the recognition of the importance of data standards. The STRENDA (Standards for Reporting Enzymology Data) guidelines, and their adoption by the main publications publishing enzymology studies, have been key to these advancements [112].
Prediction of enzyme reactions
Identifying the function of uncharacterised protein sequences and structures remains one of the most important outstanding goals in biology, and the number of existing tools to address this problem vast. As with the other dimensions, understanding conservation and neofunctionalization throughout evolution, is key to most of these prediction methods. Enzymes in the same family that contain the same conserved catalytic residues probably catalyse the same type of reaction, for example. The conservation of binding residues can further inform if the substrate specificities are the same. The existence of two orthologous enzymes in related species, and particularly when there are not paralogs, can also give a strong indication that both enzymes have the same function.
The CAFA (Critical Assessment of Functional Annotation) challenge is a competition aimed at evaluation existing computational tools for protein function prediction from sequence [113]. Methods are scored by how well they identify the most relevant GO terms for each sequence. In CAFA 3, the last challenge for which there is a report, the best methods at predicting the molecular function ontology were GoLabeler (now superseded by NetGo 3.0) [114] and CATH funfams. Both of these tools, as well as more recently developed methods, such as ProteinBert [115] and DeepGo [116], combine traditional sequence similarity methods with varied machine-learning approaches that are able to identify function-defining residues or motifs. Similar approaches do exist specific to enzymes, where the goal is to predict an EC number. A non-exhaustive list includes EzyPred [117], DEEPre [118], ECPred [119], and DeepEC [120]. Structural information is also considered by other methods, such as ProFunc [121], CO-FACTOR [122], and DeepFRI [123]. While using structure to predict function was traditionally less useful than using solely sequence, due to the limited availability of protein structures, this might now change with the ease of generating good structures for most sequences. Finally, information specific to the catalytic sites can also be used [124]. Methods that use templates of catalytic residues should be able to detect active site similarities in related but also unrelated enzymes when the catalytic residues converged to the same geometry [89].
By design, the methods discussed above are limited to identify chemical reactions that are already annotated in the classification system. Furthermore, most methods using sequence information together with machine learning lack interpretability, making the evaluation of assignments for specific enzymes tricky. Considering the methodologies described in the previous sections to predict the intermediate dimensions from sequence, it should be also possible, in principle, to predict the reactions of enzymes ab initio, in a way that is not limited to existing reactions. Alphafold and similar methods are already able to satisfactorily predict the structure of proteins from sequence. If predicting ligand binding and the enzyme mechanism becomes straightforward in the same manner, it will be possible to predict the reaction from sequence while establishing a clear causality chain across the six dimensions discussed here.
Conclusion
Theodosius Dobzhansky famously stated that ‘Nothing in Biology Makes Sense Except in the Light of Evolution'. This is clearly the case for enzymes, for which catalytic sites can be found conserved between bacteria and humans, and possible catalytic reactions can only arise by evolutionary paths that navigate the complex protein space while being nudged by epistatic and pleiotropic effects.
Most new catalytic reactions arise as a result of gene duplication and divergence. Changes in substrate specificity can be traced to changes in the binding residues and typically correspond to a last-digit modification of the EC number. Changes in the catalytic residues are much rarer but can lead to completely new chemical activities. Studies to understand the precise evolutionary processes of neofunctionalization are still lacking, in particular the role of mutations in the catalytic residues and changes in the enzyme mechanism. Ideally, we would like to classify all functional changes across several enzyme families according to the paradigms shown in Figure 3 (together with other possibilities).
Neutral drift and enzyme promiscuity have an important role in exploring the catalytic space without impacting fitness. Promiscuous functions can become adaptive after an environment or cellular change, and while initially inefficient, their activity can be improved after duplication and specialisation. The extent at which promiscuity is relevant for enzyme function and evolution has been recognised for several examples, but these kinds of data have not yet been used for large-scale computational studies, since available information and in its curation in databases is still limited [125].
The evolution of new enzymes through recombination of domains is another area that, in our opinion, would benefit from further studies. As explained above, domain recombination allows for big jumps in function and is particularly relevant for co-factor-containing enzymes. Studies focusing on the contribution of each domain for binding, catalytic residues, and the catalytic mechanism, would be helpful to understand domain recruitment during evolution.
In this review, we highlighted some databases with information about enzymes as well as some computational methods that can be used to close gaps in knowledge. These examples were meant to illustrate some of the available tools but are by no means exhaustive. Similarly, in the interest of brevity, other topics pertinent to enzyme function and evolution such as metabolic databases, enzyme engineering, in particular directed evolution and rational design, ancestral reconstruction, and the role of structural dynamics, have been excluded from the discussion.
One of the challenges of studying enzymes from the point of view of bioinformatics is related with the variety of the available data, which mirrors the complexity of enzymes as biological catalysts The integration of all these kinds of data, which we have organised here across six dimensions, is necessary to explain enzyme function and evolution, and crucial for efforts of enzyme design. Another challenge is the limited availability of data for certain dimensions, particularly when compared with the number of protein sequences. Our optimistic viewpoint is that by using different computational approaches, including template-based, machine learning, and simulation methods, it will be possible in the future to have a comprehensive knowledge base of enzyme function and evolution across these dimensions. In our opinion, the biggest obstacles to this vision are currently the prediction of protein–ligand binding and of the enzyme reaction mechanism.
Acknowledgements
This work was funded by the European Molecular Biology Laboratory (A.J.M.R., N.B., J.M.T.) and the EMBL International PhD Programme (I.G.R.).
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
CRediT Author Contribution
Antonio Ribeiro: Conceptualization, Writing — original draft, Writing — review and editing. Ioannis G. Riziotis: Conceptualization, Writing — review and editing. Neera Borkakoti: Conceptualization, Writing — review and editing. Janet M. Thornton: Conceptualization, Supervision, Funding acquisition, Project administration, Writing — review and editing.
Abbreviations
- EC
enzyme commission
- IAD
Innovation–Amplification–Divergence
- LUCA
Last Universal Common Ancestor
- PDB
Protein Data Bank
References
- 1.The UniProt Consortium. (2023) Uniprot: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531 10.1093/nar/gkac1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.wwPDB consortium. (2019) Protein data bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 47, D520–D528 10.1093/nar/gky949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ribeiro, A.J., Tyzack, J.D., Borkakoti, N., Holliday, G.L. and Thornton, J.M. (2020) A global analysis of function and conservation of catalytic residues in enzymes. J. Biol. Chem. 295, 314–324 10.1074/jbc.REV119.006289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fischer, J.D., Holliday, G.L. and Thornton, J.M. (2010) The CoFactor database: organic cofactors in enzyme catalysis. Bioinformatics 26, 2496–2497 10.1093/bioinformatics/btq442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bansal, P., Morgat, A., Axelsen, K.B., Muthukrishnan, V., Coudert, E., Aimo, L.et al. (2022) Rhea, the reaction knowledgebase in 2022. Nucleic Acids Res. 50, D693–D700 10.1093/nar/gkab1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. and Tanabe, M. (2015) KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 10.1093/nar/gkv1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ribeiro, A.J.M., Holliday, G.L., Furnham, N., Tyzack, J.D., Ferris, K. and Thornton, J.M. (2018) Mechanism and catalytic site atlas (M-CSA): a database of enzyme reaction mechanisms and active sites. Nucleic Acids Res. 46, D618–D623 10.1093/nar/gkx1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pigliucci, M. (2010) Genotype–phenotype mapping and the end of the ‘genes as blueprint’ metaphor. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 557–566 10.1098/rstb.2009.0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferreira, P., Fernandes, P.A. and Ramos, M.J. (2022) Modern computational methods for rational enzyme engineering. Chem. Catal. 2, 2481–2498 10.1016/j.checat.2022.09.036 [DOI] [Google Scholar]
- 10.Kiss, G., Çelebi-Ölçüm, N., Moretti, R., Baker, D. and Houk, K.N. (2013) Computational enzyme design. Angew. Chem. Int. Ed. Engl. 52, 5700–5725 10.1002/anie.201204077 [DOI] [PubMed] [Google Scholar]
- 11.Schramm, V.L. (2013) Transition states, analogues, and drug development. ACS Chem. Biol. 8, 71–81 10.1021/cb300631k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hopkins, A.L. and Groom, C.R. (2002) The druggable genome. Nat. Rev. Drug Discov. 1, 727–730 10.1038/nrd892 [DOI] [PubMed] [Google Scholar]
- 13.Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O.et al. (2021) Highly accurate protein structure prediction with alphaFold. Nature 596, 583–589 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woese, C. (1998) The universal ancestor. Proc. Natl Acad. Sci. U.S.A. 95, 6854–6859 10.1073/pnas.95.12.6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ranea, J.A.G., Sillero, A., Thornton, J.M. and Orengo, C.A. (2006) Protein superfamily evolution and the last universal common ancestor (LUCA). J. Mol. Evol. 63, 513–525 10.1007/s00239-005-0289-7 [DOI] [PubMed] [Google Scholar]
- 16.Berkemer, S.J. and McGlynn, S.E. (2020) A new analysis of archaea-bacteria domain separation: variable phylogenetic distance and the tempo of early evolution. Mol. Biol. Evol. 37, 2332–2340 10.1093/molbev/msaa089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weiss, M.C., Sousa, F.L., Mrnjavac, N., Neukirchen, S., Roettger, M., Nelson-Sathi, S.et al. (2016) The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 1, 16116 10.1038/nmicrobiol.2016.116 [DOI] [PubMed] [Google Scholar]
- 18.Gagler, D.C., Karas, B., Kempes, C.P., Malloy, J., Mierzejewski, V., Goldman, A.D.et al. (2022) Scaling laws in enzyme function reveal a new kind of biochemical universality. Proc. Natl Acad. Sci. U.S.A. 119, e2106655119 10.1073/pnas.2106655119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Copley, S.D. (2020) Evolution of new enzymes by gene duplication and divergence. FEBS J. 287, 1262–1283 10.1111/febs.15299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mascotti, M.L., Juri Ayub, M., Furnham, N., Thornton, J.M. and Laskowski, R.A. (2016) Chopping and changing: the evolution of the flavin-dependent monooxygenases. J. Mol. Biol. 428, 3131–3146 10.1016/j.jmb.2016.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bornberg-Bauer, E., Hlouchova, K. and Lange, A. (2021) Structure and function of naturally evolved de novo proteins. Curr. Opin. Struct. Biol. 68, 175–183 10.1016/j.sbi.2020.11.010 [DOI] [PubMed] [Google Scholar]
- 22.Heames, B., Buchel, F., Aubel, M., Tretyachenko, V., Loginov, D., Novák, P.et al. (2023) Experimental characterization of de novo proteins and their unevolved random-sequence counterparts. Nat. Ecol. Evol. 7, 570–580 10.1038/s41559-023-02010-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohno, S. (1970) Evolution by Gene Duplication, Springer Science & Business Media, New York [Google Scholar]
- 24.Innan, H. and Kondrashov, F. (2010) The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11, 97–108 10.1038/nrg2689 [DOI] [PubMed] [Google Scholar]
- 25.Bergthorsson, U., Andersson, D.I. and Roth, J.R. (2007) Ohno's dilemma: Evolution of new genes under continuous selection. Proc. Natl Acad. Sci. U.S.A. 104, 17004–17009 10.1073/pnas.0707158104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Francino, M.P. (2005) An adaptive radiation model for the origin of new gene functions. Nat. Genet. 37, 573–578 10.1038/ng1579 [DOI] [PubMed] [Google Scholar]
- 27.Copley, S.D. (2017) Shining a light on enzyme promiscuity. Curr. Opin. Struct. Biol. 47, 167–175 10.1016/j.sbi.2017.11.001 [DOI] [PubMed] [Google Scholar]
- 28.Tawfik, D.S. (2020) Enzyme promiscuity and evolution in light of cellular metabolism. FEBS J. 287, 1260–1261 10.1111/febs.15296 [DOI] [PubMed] [Google Scholar]
- 29.Pandya, C., Farelli, J.D., Dunaway-Mariano, D. and Allen, K.N. (2014) Enzyme promiscuity: engine of evolutionary innovation *. J. Biol. Chem. 289, 30229–30236 10.1074/jbc.R114.572990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seffernick, J.L. and Wackett, L.P. (2001) Rapid evolution of bacterial catabolic enzymes: a case study with atrazine chlorohydrolase. Biochemistry 40, 12747–12753 10.1021/bi011293r [DOI] [PubMed] [Google Scholar]
- 31.Renata, H., Wang, Z.J. and Arnold, F.H. (2015) Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem. Int. Ed. Engl. 54, 3351–3367 10.1002/anie.201409470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang, H., Pandya, C., Liu, C., Al-Obaidi, N.F., Wang, M., Zheng, L.et al. (2015) Panoramic view of a superfamily of phosphatases through substrate profiling. Proc. Natl Acad. Sci. U.S.A. 112, E1974–E1983 10.1073/pnas.1423570112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mashiyama, S.T., Malabanan, M.M., Akiva, E., Bhosle, R., Branch, M.C., Hillerich, B.et al. (2014) Large-scale determination of sequence, structure, and function relationships in cytosolic glutathione transferases across the biosphere. PLoS Biol. 12, e1001843 10.1371/journal.pbio.1001843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seibert, C.M. and Raushel, F.M. (2005) Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry 44, 6383–6391 10.1021/bi047326v [DOI] [PubMed] [Google Scholar]
- 35.Sillitoe, I., Bordin, N., Dawson, N., Waman, V.P., Ashford, P., Scholes, H.M.et al. (2021) CATH: increased structural coverage of functional space. Nucleic Acids Res. 49, D266–D273 10.1093/nar/gkaa1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandonia, J.-M., Guan, L., Lin, S., Yu, C., Fox, N.K. and Brenner, S.E. (2022) SCOPe: improvements to the structural classification of proteins – extended database to facilitate variant interpretation and machine learning. Nucleic Acids Res. 50, D553–D559 10.1093/nar/gkab1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonald, A.G. and Tipton, K.F. (2021) Enzyme nomenclature and classification: the state of the art. FEBS J. 290, 2214–2231 10.1111/febs.16274 [DOI] [PubMed] [Google Scholar]
- 38.Sillitoe, I. and Furnham, N. (2016) Funtree: advances in a resource for exploring and contextualising protein function evolution. Nucleic Acids Res. 44, D317–D323 10.1093/nar/gkv1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furnham, N., Dawson, N.L., Rahman, S.A., Thornton, J.M. and Orengo, C.A. (2016) Large-scale analysis exploring evolution of catalytic machineries and mechanisms in enzyme superfamilies. J. Mol. Biol. 428, 253–267 10.1016/j.jmb.2015.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bartlett, G.J., Borkakoti, N. and Thornton, J.M. (2003) Catalysing new reactions during evolution: economy of residues and mechanism. J. Mol. Biol. 331, 829–860 10.1016/s0022-2836(03)00734-4 [DOI] [PubMed] [Google Scholar]
- 41.Han, J.-H., Batey, S., Nickson, A.A., Teichmann, S.A. and Clarke, J. (2007) The folding and evolution of multidomain proteins. Nat. Rev. Mol. Cell Biol. 8, 319–330 10.1038/nrm2144 [DOI] [PubMed] [Google Scholar]
- 42.Vogel, C., Bashton, M., Kerrison, N.D., Chothia, C. and Teichmann, S.A. (2004) Structure, function and evolution of multidomain proteins. Curr. Opin. Struct. Biol. 14, 208–216 10.1016/j.sbi.2004.03.011 [DOI] [PubMed] [Google Scholar]
- 43.Bashton, M. and Chothia, C. (2007) The generation of new protein functions by the combination of domains. Structure 15, 85–99 10.1016/j.str.2006.11.009 [DOI] [PubMed] [Google Scholar]
- 44.Holliday, G.L., Akiva, E., Meng, E.C., Brown, S.D., Calhoun, S., Pieper, U.et al. (2018) Atlas of the radical SAM superfamily: divergent evolution of function using a “plug and play” domain. Methods Enzymol. 606, 1–71 10.1016/bs.mie.2018.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Akiva, E., Copp, J.N., Tokuriki, N. and Babbitt, P.C. (2017) Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily. Proc. Natl. Acad. Sci. U.S.A. 114, E9549–E9558 10.1073/pnas.1706849114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.PDBe-KB consortium. (2022) PDBe-KB: collaboratively defining the biological context of structural data. Nucleic Acids Res. 50, D534–D542 10.1093/nar/gkab988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Omelchenko, M.V., Galperin, M.Y., Wolf, Y.I. and Koonin, E.V. (2010) Non-homologous isofunctional enzymes: A systematic analysis of alternative solutions in enzyme evolution. Biol. Direct 5, 31 10.1186/1745-6150-5-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leveson-Gower, R.B., Mayer, C. and Roelfes, G. (2019) The importance of catalytic promiscuity for enzyme design and evolution. Nat. Rev. Chem. 3, 687–705 10.1038/s41570-019-0143-x [DOI] [Google Scholar]
- 49.Jeffery, C.J. (2014) An introduction to protein moonlighting. Biochem. Soc. Trans. 42, 1679–1683 10.1042/BST20140226 [DOI] [PubMed] [Google Scholar]
- 50.Eyers, P.A. and Murphy, J.M. (2016) The evolving world of pseudoenzymes: proteins, prejudice and zombies. BMC Biol. 14, 98 10.1186/s12915-016-0322-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ribeiro, A.J.M., Das, S., Dawson, N., Zaru, R., Orchard, S., Thornton, J.M.et al. (2019) Emerging concepts in pseudoenzyme classification, evolution, and signaling. Sci. Signal. 12, eaat9797 10.1126/scisignal.aat9797 [DOI] [PubMed] [Google Scholar]
- 52.Lee, D., Redfern, O. and Orengo, C. (2007) Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol. 8, 995–1005 10.1038/nrm2281 [DOI] [PubMed] [Google Scholar]
- 53.Soskine, M. and Tawfik, D.S. (2010) Mutational effects and the evolution of new protein functions. Nat. Rev. Genet. 11, 572–582 10.1038/nrg2808 [DOI] [PubMed] [Google Scholar]
- 54.Starr, T.N. and Thornton, J.W. (2016) Epistasis in protein evolution. Protein Sci. 25, 1204–1218 10.1002/pro.2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lobanov, V., Gobet, A. and Joyce, A. (2022) Ecosystem-specific microbiota and microbiome databases in the era of big data. Environ. Microbiome 17, 37 10.1186/s40793-022-00433-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richardson, L., Allen, B., Baldi, G., Beracochea, M., Bileschi, M.L., Burdett, T.et al. (2023) MGnify: the microbiome sequence data analysis resource in 2023. Nucleic Acids Res. 51, D753–D759 10.1093/nar/gkac1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ribeiro, A.J.M., Tyzack, J.D., Borkakoti, N. and Thornton, J.M. (2020) Identifying pseudoenzymes using functional annotation: pitfalls of common practice. FEBS J. 287, 4128–4140 10.1111/febs.15142 [DOI] [PubMed] [Google Scholar]
- 58.El-Gebali, S., Mistry, J., Bateman, A., Eddy, S.R., Luciani, A., Potter, S.C.et al. (2018) The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 10.1093/nar/gky995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang, J., Chitsaz, F., Derbyshire, M.K., Gonzales, N.R., Gwadz, M., Lu, S.et al. (2023) The conserved domain database in 2023. Nucleic Acids Res. 51, D384–D388 10.1093/nar/gkac1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thomas, P.D., Ebert, D., Muruganujan, A., Mushayahama, T., Albou, L.-P. and Mi, H. (2022) PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 31, 8–22 10.1002/pro.4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu, C.H., Nikolskaya, A., Huang, H., Yeh, L.-S.L., Natale, D.A., Vinayaka, C.R.et al. (2004) PIRSF: family classification system at the protein information resource. Nucleic Acids Res. 32, D112–D114 10.1093/nar/gkh097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paysan-Lafosse, T., Blum, M., Chuguransky, S., Grego, T., Pinto, B.L., Salazar, G.A.et al. (2023) Interpro in 2022. Nucleic Acids Res. 51, D418–D427 10.1093/nar/gkac993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Romero-Romero, S., Kordes, S., Michel, F. and Höcker, B. (2021) Evolution, folding, and design of TIM barrels and related proteins. Curr. Opin. Struct. Biol. 68, 94–104 10.1016/j.sbi.2020.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cheng, H., Schaeffer, R.D., Liao, Y., Kinch, L.N., Pei, J., Shi, S.et al. (2014) ECOD: an evolutionary classification of protein domains. PLoS Comput. Biol. 10, e1003926 10.1371/journal.pcbi.1003926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pearce, R. and Zhang, Y. (2021) Toward the solution of the protein structure prediction problem. J. Biol. Chem. 297, 100870 10.1016/j.jbc.2021.100870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G.et al. (2022) Alphafold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 10.1093/nar/gkab1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W.et al. (2023) Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 10.1126/science.ade2574 [DOI] [PubMed] [Google Scholar]
- 68.Chang, A., Jeske, L., Ulbrich, S., Hofmann, J., Koblitz, J., Schomburg, I.et al. (2021) BRENDA, the ELIXIR core data resource in 2021: new developments and updates. Nucleic Acids Res. 49, D498–D508 10.1093/nar/gkaa1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wagle, S., Smith, R.D., Dominic, A.J., DasGupta, D., Tripathi, S.K. and Carlson, H.A. (2023) Sunsetting binding MOAD with its last data update and the addition of 3D-ligand polypharmacology tools. Sci. Rep. 13, 3008 10.1038/s41598-023-29996-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu, Z., Li, Y., Han, L., Li, J., Liu, J., Zhao, Z.et al. (2015) PDB-wide collection of binding data: current status of the PDBbind database. Bioinformatics 31, 405–412 10.1093/bioinformatics/btu626 [DOI] [PubMed] [Google Scholar]
- 71.Gilson, M.K., Liu, T., Baitaluk, M., Nicola, G., Hwang, L. and Chong, J. (2016) BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 44, D1045–D1053 10.1093/nar/gkv1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Desaphy, J., Bret, G., Rognan, D. and Kellenberger, E. (2015) sc-PDB: a 3D-database of ligandable binding sites—10 years on. Nucleic Acids Res. 43, D399–D404 10.1093/nar/gku928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang, J., Roy, A. and Zhang, Y. (2013) Biolip: a semi-manually curated database for biologically relevant ligand–protein interactions. Nucleic Acids Res. 41, D1096–D1103 10.1093/nar/gks966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Murakami, Y., Omori, S. and Kinoshita, K. (2016) NLDB: a database for 3D protein–ligand interactions in enzymatic reactions. J. Struct. Funct. Genomics 17, 101–110 10.1007/s10969-016-9206-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tyzack, J.D., Fernando, L., Ribeiro, A.J., Borkakoti, N. and Thornton, J.M. (2018) Ranking enzyme structures in the PDB by bound ligand similarity to biological substrates. Structure 26, 565–571 10.1016/j.str.2018.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hekkelman, M.L., de Vries, I., Joosten, R.P. and Perrakis, A. (2023) Alphafill: enriching AlphaFold models with ligands and cofactors. Nat. Methods 20, 205–213 10.1038/s41592-022-01685-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhao, J., Cao, Y. and Zhang, L. (2020) Exploring the computational methods for protein-ligand binding site prediction. Comput. Struct. Biotechnol. J. 18, 417–426 10.1016/j.csbj.2020.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dhakal, A., McKay, C., Tanner, J.J. and Cheng, J. (2022) Artificial intelligence in the prediction of protein–ligand interactions: recent advances and future directions. Brief. Bioinform. 23, bbab476 10.1093/bib/bbab476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fan, J., Fu, A. and Zhang, L. (2019) Progress in molecular docking. Quant. Biol. 7, 83–89 10.1007/s40484-019-0172-y [DOI] [Google Scholar]
- 80.Babić, M., Janković, P., Marchesan, S., Mauša, G. and Kalafatovic, D. (2022) Esterase sequence composition patterns for the identification of catalytic triad microenvironment motifs. J. Chem. Inf. Model. 62, 6398–6410 10.1021/acs.jcim.2c00977 [DOI] [PubMed] [Google Scholar]
- 81.Pinney, M.M., Mokhtari, D.A., Akiva, E., Yabukarski, F., Sanchez, D.M., Liang, R.et al. (2021) Parallel molecular mechanisms for enzyme temperature adaptation. Science 371, eaay2784 10.1126/science.aay2784 [DOI] [PubMed] [Google Scholar]
- 82.Musil, M., Khan, R.T., Beier, A., Stourac, J., Konegger, H., Damborsky, J.et al. (2021) FireProtASR: a web server for fully automated ancestral sequence reconstruction. Brief. Bioinform. 22, bbaa337 10.1093/bib/bbaa337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moraes, J.P.A., Pappa, G.L., Pires, D.E.V. and Izidoro, S.C. (2017) GASS-WEB: a web server for identifying enzyme active sites based on genetic algorithms. Nucleic Acids Res. 45, W315–W319 10.1093/nar/gkx337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feehan, R., Franklin, M.W. and Slusky, J.S.G. (2021) Machine learning differentiates enzymatic and non-enzymatic metals in proteins. Nat. Commun. 12, 3712 10.1038/s41467-021-24070-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kelley, L.A., Mezulis, S., Yates, C.M., Wass, M.N. and Sternberg, M.J.E. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845 10.1038/nprot.2015.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Laskowski, R.A., Watson, J.D. and Thornton, J.M. (2005) Protein function prediction using local 3D templates. J. Mol. Biol. 351, 614–626 10.1016/j.jmb.2005.05.067 [DOI] [PubMed] [Google Scholar]
- 87.Riziotis, I.G. and Thornton, J.M. (2022) Capturing the geometry, function, and evolution of enzymes with 3D templates. Protein Sci. 31, e4363 10.1002/pro.4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Riziotis, I.G., Ribeiro, A.J.M., Borkakoti, N. and Thornton, J.M. (2022) Conformational variation in enzyme catalysis: a structural study on catalytic residues. J. Mol. Biol. 434, 167517 10.1016/j.jmb.2022.167517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Riziotis, I.G., Ribeiro, A.J.M., Borkakoti, N. and Thornton, J.M. (2023) The 3D modules of enzyme catalysis: deconstructing active sites into distinct functional entities. bioRxiv 435, 168254 10.1016/j.jmb.2023.168254 [DOI] [PubMed] [Google Scholar]
- 90.Fischer, J.D., Holliday, G.L., Rahman, S.A. and Thornton, J.M. (2010) The structures and physicochemical properties of organic cofactors in biocatalysis. J. Mol. Biol. 403, 803–824 10.1016/j.jmb.2010.09.018 [DOI] [PubMed] [Google Scholar]
- 91.White, H.B. (1976) Coenzymes as fossils of an earlier metabolic state. J. Mol. Evol. 7, 101–104 10.1007/BF01732468 [DOI] [PubMed] [Google Scholar]
- 92.Goldman, A.D. and Kacar, B. (2021) Cofactors are remnants of life's origin and early evolution. J. Mol. Evol. 89, 127 10.1007/s00239-020-09988-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang, T., Zhang, H., Chen, K., Shen, S., Ruan, J. and Kurgan, L. (2008) Accurate sequence-based prediction of catalytic residues. Bioinformatics 24, 2329–2338 10.1093/bioinformatics/btn433 [DOI] [PubMed] [Google Scholar]
- 94.Dukka Bahadur, K.C. and Livesay, D.R. (2008) Improving position-specific predictions of protein functional sites using phylogenetic motifs. Bioinformatics 24, 2308–2316 10.1093/bioinformatics/btn454 [DOI] [PubMed] [Google Scholar]
- 95.Mihalek, I., Reš, I. and Lichtarge, O. (2004) A family of evolution–entropy hybrid methods for ranking protein residues by importance. J. Mol. Biol. 336, 1265–1282 10.1016/j.jmb.2003.12.078 [DOI] [PubMed] [Google Scholar]
- 96.Tang, Y.-R., Sheng, Z.-Y., Chen, Y.-Z. and Zhang, Z. (2008) An improved prediction of catalytic residues in enzyme structures. Protein Eng. Des. Sel. 21, 295–302 10.1093/protein/gzn003 [DOI] [PubMed] [Google Scholar]
- 97.Ondrechen, M.J., Clifton, J.G. and Ringe, D. (2001) THEMATICS: A simple computational predictor of enzyme function from structure. Proc. Natl Acad. Sci. U.S.A. 98, 12473–12478 10.1073/pnas.211436698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chea, E. and Livesay, D.R. (2007) How accurate and statistically robust are catalytic site predictions based on closeness centrality? BMC Bioinformatics 8, 153 10.1186/1471-2105-8-153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Song, J., Li, F., Takemoto, K., Haffari, G., Akutsu, T., Chou, K.-C.et al. (2018) PREvaIL, an integrative approach for inferring catalytic residues using sequence, structural, and network features in a machine-learning framework. J. Theor. Biol. 443, 125–137 10.1016/j.jtbi.2018.01.023 [DOI] [PubMed] [Google Scholar]
- 100.Dou, Y., Wang, J., Yang, J. and Zhang, C. (2012) L1pred: a sequence-based prediction tool for catalytic residues in enzymes with the L1-logreg classifier. PLoS ONE 7, e35666 10.1371/journal.pone.0035666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Petrova, N.V. and Wu, C.H. (2006) Prediction of catalytic residues using support vector machine with selected protein sequence and structural properties. BMC Bioinformatics 7, 312 10.1186/1471-2105-7-312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gutteridge, A., Bartlett, G.J. and Thornton, J.M. (2003) Using a neural network and spatial clustering to predict the location of active sites in enzymes. J. Mol. Biol. 330, 719–734 10.1016/S0022-2836(03)00515-1 [DOI] [PubMed] [Google Scholar]
- 103.Lopez, G., Maietta, P., Rodriguez, J.M., Valencia, A. and Tress, M.L. (2011) Firestar—advances in the prediction of functionally important residues. Nucleic Acids Res. 39, W235–W241 10.1093/nar/gkr437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nath, N., Mitchell, J.B.O. and Caetano-Anollés, G. (2014) The natural history of biocatalytic mechanisms. PLoS Comput. Biol. 10, e1003642 10.1371/journal.pcbi.1003642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ribeiro, A.J.M., Riziotis, I.G., Tyzack, J.D., Borkakoti, N. and Thornton, J.M. (2023) Ezmechanism: an automated tool to propose catalytic mechanisms of enzyme reactions. Nat. Methods 20, 1516–1522 10.1038/s41592-023-02006-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sousa, S.F., Ribeiro, A.J., Neves, R.P., Brás, N.F., Cerqueira, N.M., Fernandes, P.A.et al. (2017) Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. WIREs Comput. Mol. Sci. 7, e1281 10.1002/wcms.1281 [DOI] [Google Scholar]
- 107.Bairoch, A. (2000) The ENZYME database in 2000. Nucleic Acids Res. 28, 304–305 10.1093/nar/28.1.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kanehisa, M. and Goto, S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wicker, J., Lorsbach, T., Gütlein, M., Schmid, E., Latino, D., Kramer, S.et al. (2016) Envipath–The environmental contaminant biotransformation pathway resource. Nucleic Acids Res. 44, D502–D508 10.1093/nar/gkv1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rahman, S.A., Cuesta, S.M., Furnham, N., Holliday, G.L. and Thornton, J.M. (2014) EC-BLAST: a tool to automatically search and compare enzyme reactions. Nat. Methods 11, 171 10.1038/nmeth.2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wittig, U., Rey, M., Weidemann, A., Kania, R. and Müller, W. (2018) SABIO-RK: an updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Res. 46, D656–D660 10.1093/nar/gkx1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tipton, K.F., Armstrong, R.N., Bakker, B.M., Bairoch, A., Cornish-Bowden, A., Halling, P.J.et al. (2014) Standards for reporting enzyme data: the STRENDA consortium: what it aims to do and why it should be helpful. Perspect. Sci. 1, 131–137 10.1016/j.pisc.2014.02.012 [DOI] [Google Scholar]
- 113.Zhou, N., Jiang, Y., Bergquist, T.R., Lee, A.J., Kacsoh, B.Z., Crocker, A.W.et al. (2019) The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 20, 244 10.1186/s13059-019-1835-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang, S., You, R., Liu, Y., Xiong, Y. and Zhu, S. (2023) NetGO 3.0: protein language model improves large-scale functional annotations. Genomics Proteomics Bioinformatics 21, 349–358 10.1016/j.gpb.2023.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brandes, N., Ofer, D., Peleg, Y., Rappoport, N. and Linial, M. (2022) ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics 38, 2102–2110 10.1093/bioinformatics/btac020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kulmanov, M., Khan, M.A. and Hoehndorf, R. (2018) DeepGO: predicting protein functions from sequence and interactions using a deep ontology-aware classifier. Bioinformatics 34, 660–668 10.1093/bioinformatics/btx624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shen, H.-B. and Chou, K.-C. (2007) Ezypred: a top–down approach for predicting enzyme functional classes and subclasses. Biochem. Biophys. Res. Commun. 364, 53–59 10.1016/j.bbrc.2007.09.098 [DOI] [PubMed] [Google Scholar]
- 118.Li, Y., Wang, S., Umarov, R., Xie, B., Fan, M., Li, L.et al. (2018) DEEPre: sequence-based enzyme EC number prediction by deep learning. Bioinformatics 34, 760–769 10.1093/bioinformatics/btx680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dalkiran, A., Rifaioglu, A.S., Martin, M.J., Cetin-Atalay, R., Atalay, V. and Doğan, T. (2018) ECPred: a tool for the prediction of the enzymatic functions of protein sequences based on the EC nomenclature. BMC Bioinformatics 19, 334 10.1186/s12859-018-2368-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ryu, J.Y., Kim, H.U. and Lee, S.Y. (2019) Deep learning enables high-quality and high-throughput prediction of enzyme commission numbers. Proc. Natl Acad. Sci. U.S.A. 116, 13996–14001 10.1073/pnas.1821905116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Laskowski, R.A., Watson, J.D. and Thornton, J.M. (2005) Profunc: a server for predicting protein function from 3D structure. Nucleic Acids Res. 33, W89–W93 10.1093/nar/gki414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang, C., Freddolino, P.L. and Zhang, Y. (2017) COFACTOR: improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 45, W291–W299 10.1093/nar/gkx366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gligorijević, V., Renfrew, P.D., Kosciolek, T., Leman, J.K., Berenberg, D., Vatanen, T.et al. (2021) Structure-based protein function prediction using graph convolutional networks. Nat. Commun. 12, 3168 10.1038/s41467-021-23303-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nilmeier, J.P., Kirshner, D.A., Wong, S.E. and Lightstone, F.C. (2013) Rapid catalytic template searching as an enzyme function prediction procedure. PLoS ONE 8, e62535 10.1371/journal.pone.0062535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Velez Rueda, A.J., Palopoli, N., Zacarías, M., Sommese, L.M. and Parisi, G. (2019) Protmiscuity: a database of promiscuous proteins. Database (Oxford) 2019, baz103 10.1093/database/baz103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuhlman, B. and Bradley, P. (2019) Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 20, 681–697 10.1038/s41580-019-0163-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Evans, R., O'Neill, M., Pritzel, A., Antropova, N., Senior, A., Green, T.et al. (2021) Protein complex prediction with AlphaFold-Multimer. bioRxiv 10.1101/2021.10.04.463034 [DOI] [Google Scholar]
- 128.Das, S., Scholes, H.M., Sen, N. and Orengo, C. (2021) CATH functional families predict functional sites in proteins. Bioinformatics 37, 1099–1106 10.1093/bioinformatics/btaa937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shen, C., Ding, J., Wang, Z., Cao, D., Ding, X. and Hou, T. (2020) From machine learning to deep learning: advances in scoring functions for protein–ligand docking. WIREs Comput. Mol. Sci. 10, e1429 10.1002/wcms.1429 [DOI] [Google Scholar]
- 130.Sun, J., Wang, J., Xiong, D., Hu, J. and Liu, R. (2016) CRHunter: integrating multifaceted information to predict catalytic residues in enzymes. Sci. Rep. 6, 34044 10.1038/srep34044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Himo, F. and de Visser, S.P. (2022) Status report on the quantum chemical cluster approach for modeling enzyme reactions. Commun. Chem. 5, 29 10.1038/s42004-022-00642-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zou, Z., Tian, S., Gao, X. and Li, Y. (2019) mlDEEPre: multi-functional enzyme function prediction with hierarchical multi-label deep learning. Front. Genet. 9, 714 10.3389/fgene.2018.00714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yu, T., Cui, H., Li, J.C., Luo, Y., Jiang, G. and Zhao, H. (2023) Enzyme function prediction using contrastive learning. Science 379, 1358–1363 10.1126/science.adf2465 [DOI] [PubMed] [Google Scholar]
- 134.Ribeiro, A.J.M., Riziotis, I.G., Tyzack, J.D., Borkakoti, N. and Thornton, J.M. (2022) Using mechanism similarity to understand enzyme evolution. Biophys. Rev. 14, 1273–1280 10.1007/s12551-022-01022-9 [DOI] [PMC free article] [PubMed] [Google Scholar]