

Abstract
Long recognized as an evolutionarily ancient cell type involved in tissue homeostasis and immune defense against pathogens, macrophages are being re-discovered as regulators of several diseases, including cancer. Tumor-associated macrophages (TAMs) represent the most abundant innate immune population in the tumor microenvironment (TME). Macrophages are professional phagocytic cells of the hematopoietic system specializing in the detection, phagocytosis and destruction of bacteria and other harmful micro-organisms, apoptotic cells and metabolic byproducts. In contrast to these healthy macrophage functions, TAMs support cancer cell growth and metastasis and mediate immunosuppressive effects on the adaptive immune cells of the TME. Cancer is one of the most potent insults on macrophage physiology, inducing changes that are intimately linked with disease progression. In this Review, we outline hallmarks of TAMs and discuss the emerging mechanisms that contribute to their pathophysiological adaptations and the vulnerabilities that provide attractive targets for therapeutic exploitation in cancer.
Macrophages are professional phagocytic cells specialized in the detection, phagocytosis and destruction of harmful organisms, apoptotic cells, insult-related debris, and metabolic byproducts, providing immediate defense1. After ingesting pathogens through phagocytosis, macrophages can directly present peptide antigens through the major histocompatibility complex class II (MHCII) to activate T helper cells. In contrast to dendritic cells (DCs), which present antigens in the lymph nodes and activate naive T cells, macrophages present antigens within tissues and cannot induce naive T cell activation2. Macrophages detect pathogen-associated molecular patterns (PAMPs), such as bacterial products, and damage-associated molecular patterns (DAMPs) produced in response to trauma, ischemia, or tissue damage. For this process, they use a system of pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs), which can bind specifically to pathogen components like bacterial lipopolysaccharides (LPS), RNA, DNA, or extracellular proteins, leading to the activation of signaling cascades and production of inflammatory mediators3. As a consequence, macrophages release soluble factors, such as cytokines, enzymes, or metabolites that affect other immune cell types. These steps can be subverted, resulting in a causal association of macrophages with diseases4.
In the adult host, macrophages originate mainly from blood monocytes produced from bone marrow (BM) myeloid progenitors, which leave the circulation to differentiate into macrophages in tissues. Macrophages generated during earlier stages of ontogeny, mainly from the yolk sac or fetal liver, also exist in various tissues5. These embryonic-derived macrophages persist throughout life as tissue-resident macrophages (TRMs) and participate in cancer evolution and metastasis. The adult bone marrow gives rise to Ly6C−(nonclassical) patrolling monocytes that detect pathogens and maintain vessel integrity6,7, and to Ly6C+ (classical) inflammatory monocytes, which are recruited to sites of infection, tissue injury and tumors. The Ly6C+ monocytes continuously replenish DCs in a process controlled by FLT3, generating monocyte-derived DCs (moDCs)5,8. Despite their distinct origins, the differentiation and expansion of all monocyte and macrophage lineages are regulated by CSF1R (a receptor of colony stimulating factor 1) and its ligands, IL-34 and CSF1 (refs.9,10). In some organs, such as kidney, liver, brain, and lung, macrophages originating from BM-derived monocytes co-exist with embryonically derived TRMs. Lineage-tracing studies have shown that microglia are primarily derived from the yolk-sac progenitors, whereas Kupffer cells of the liver have a mixed origin, from the yolk sac and fetal liver11,12. Major embryonically derived TRM populations are found in skin, spleen, pancreas, liver, brain, and lung13. In such tissues, the distinct origin of macrophages is of particular importance in the context of cancer, where TRMs and bone-marrow-derived TAMs differentially accumulate in primary versus metastatic tumors14,15.
TAMs are the most abundant immune population of the TME, representing ~50% of hematopoietic cells, and have heterogeneous properties spanning from anti-tumorigenic to pro-tumorigenic16. Antitumorigenic TAMs retain properties of antigen-presenting cells (APC), including high expression of MHCII, phagocytotic, and tumor-killing activity. Antitumorigenic TAMs secrete proinflammatory cytokines that support and activate adaptive immune cells17. In contrast, pro-tumorigenic TAMs are immunosuppressive and are characterized by low expression of MHCII and expression of inhibitory molecules such as PD-1, PD-L1, VISTA, B7-H4 and Tim3 (refs.18–24). Cues in the TME, including tumor-secreted soluble factors and metabolites, have been extensively studied and have been found to have an instrumental role in promoting the pro-tumorigenic features of TAMs, while suppressing the anti-tumorigenic features25. The inherent ability of TAMs to alter their properties, defined as plasticity, can cause significant changes that result in the generation of TAM subsets characterized by distinct abilities to support tumor growth and metastasis26,27. The distinct properties of TAMs are potential therapeutic targets (reviewed in ref.28).
Classical and patrolling monocytes
In tumor-bearing hosts, there is an increased output of classical Ly6C+ monocytes from BM myeloid progenitors, specifically common myeloid progenitors (CMPs) and granulocyte-macrophage progenitors (GMPs), which expand during cancer-mediated emergency myelopoiesis (Fig. 1a)29. Strong activation signals, such as those mediated by PAMPs, lead to a transient expansion and differentiation of myeloid progenitors to mature monocytes and granulocytes to protect the host. In contrast, during emergency myelopoiesis driven by continuous low-level stimulation mediated by cancer-derived growth factors, cytokines, and DAMPs, myeloid progenitors undergo modest expansion, with hindered differentiation leading to the accumulation of myeloid cells with immunosuppressive and tumor-promoting properties, named myeloid-derived suppressor cells (MDSCs). In mice, MDSCs consist of two major subsets, CD11b+Ly6ChiLy6G− monocytic MDSCs (M-MDSCs) and CD11b+Ly6CloLy6G+ polymorphonuclear MDSCs (PMN-MDSCs), which have a similar morphology and phenotype to normal monocytes and neutrophils, respectively. In humans, M-MDSCs are identified as CD11b+CD14–CD15+/CD66b+, and PMN-MDSCs as CD14+CD15−HLA-DRlo/– myelocytes (reviewed in ref.30). After egress from the BM, monocytes (or M-MDSCs) are recruited to the TME via chemokines of the CC and CXC families, such as CCL2, CCL5, and CXCL12 (Fig. 1a)31 that are produced by cancer cells early during tumorigenesis.
Fig. 1 |. In tumor-bearing hosts, tumor-released factors drive increased production and output of classical Ly6C+ monocytes and mDSCs from myeloid progenitors of the Bm.
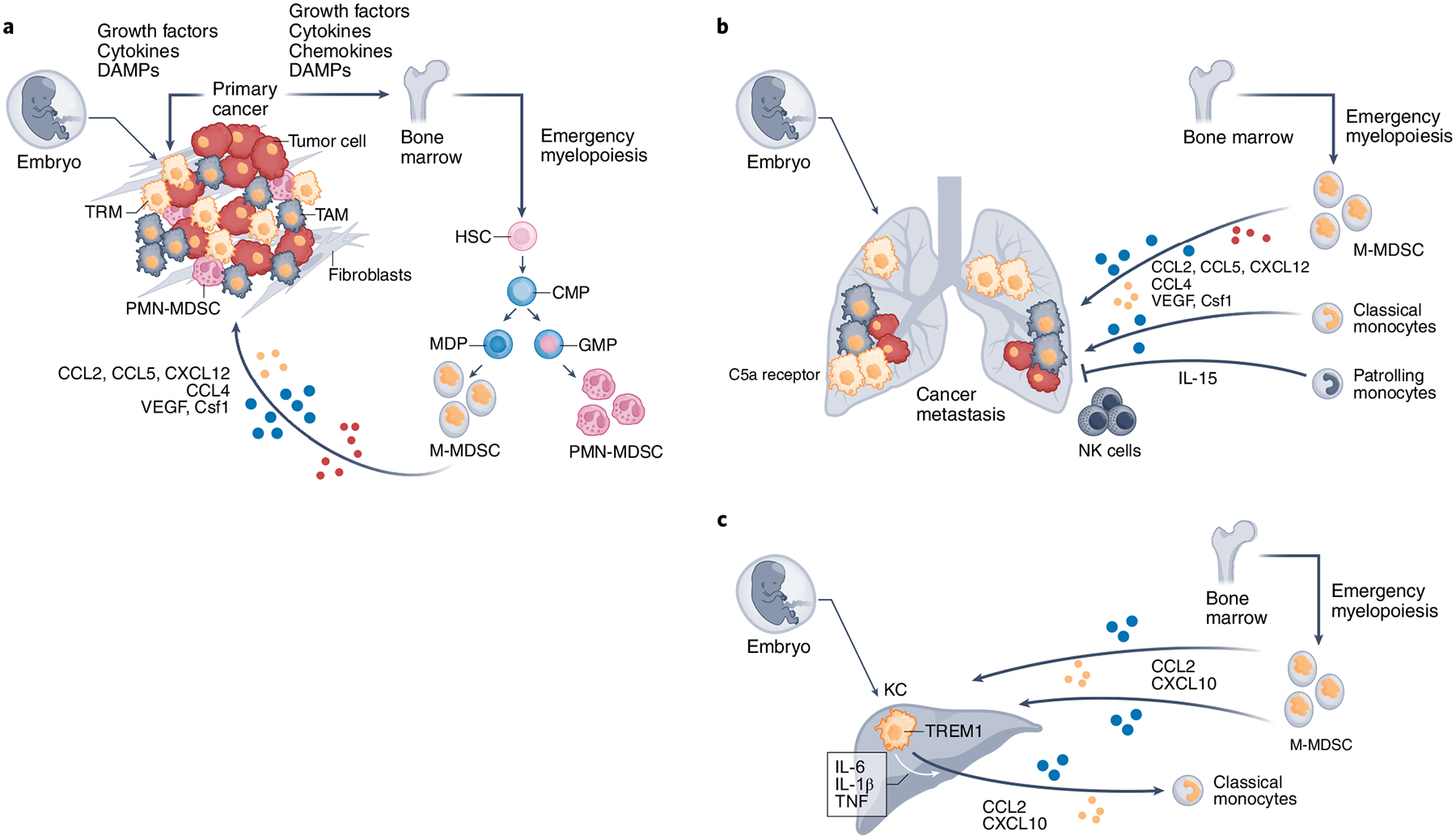
a–c, After egress, BM-derived monocytes, M-MDSCs, and PMN-MDSCs are recruited to the primary tumor (a) and metastasis sites (b,c) through chemotactic factors produced by cancer cells, tumor-associated fibroblasts, and TAMs. After localizing at the tumor, BM-derived monocytes and M-MDSCs differentiate to TAMs and promote cancer growth. In contrast to classical monocytes that give rise to TAMs, nonclassical patrolling monocytes seem to have a protective role against cancer progression because they accumulate at the sites of lung metastasis, produce IL-15, and orchestrate the recruitment and activation of NK cells, thereby inhibiting cancer invasion and growth. During cancer evolution, tissue-resident macrophages (TRMs) derived from embryonic hematopoietic organs, such as alveolar macrophages in the lung (b) and KCs in the liver (c) are the first to be subject to the effects of cancer-produced soluble factors, as well as other TME insults (a). They undergo early inflammatory changes, assist in the recruitment of BM-derived monocytes, and contribute to the generation of TAMs. In metastatic sites, TRMs might foster formation of the premetastatic niche (b,c). C5a receptor, complement receptor 5a. growth factors: M-CSF, gM-CSF; cytokines: IL-6, IL-1, IL-8; chemokines: CCL2, CCL5, CXCL12, CCL4; DAMPs: DNA, RNA, exosomes, uric acid, ATP, metabolites. HSC, hematopoietic stem cell; MDP, monocyte-DC progenitor.
The recruitment and retention of BM-derived monocytes to metastatic sites are primarily regulated by the CCR2–CCL2 axis32,33 (Fig. 1b,c). As determined by studies in xenograph metastatic models, BM-derived human monocytes also migrate to tumor sites in a CCR2-dependent manner, where they differentiate to macrophages and promote cancer growth34. Although CCL2-mediated recruitment is dominant, factors such as the inducer of vascular growth, VEGFA, and Csf1 can also mediate recruitment of monocytes and conversion to TAMs35,36. CCL5 may stimulate monocyte production and recruitment to tumors37, whereas CCL4 (ref.38) produced by TAMs can recruit BM-derived monocytes.
Nonclassical ‘patrolling’ monocytes, identified as CX3CR1hiLy6C− in mice and CX3CR1hiCD14dimCD16+ in humans, are also generated in the BM. Patrolling monocytes have a protective role against cancer progression and mediate metastasis immunosurveillance39,40. In the tumor-bearing host, patrolling monocytes accumulate at the sites of metastasis and orchestrate IL-15-mediated recruitment and activation of NK cells, thereby inhibiting cancer invasion and growth (Fig. 1b)40. Mice lacking patrolling monocytes have impaired activation and accumulation of NK cells and develop multiple pulmonary metastases40. Although patrolling monocytes are initially protective, the effect of BM-derived TAMs gradually becomes dominant, resulting in progression of primary and metastatic tumors33,39. Under these conditions, inhibiting recruitment of BM-derived monocytes can reduce both primary tumor and metastatic burden34.
Tissue-resident macrophages
All healthy tissues harbor TRMs, which support defense, homeostasis, maintenance of tissue integrity, and wound healing5. During cancer evolution, TRMs are the first to be subject to the effects of cancer-produced soluble factors and other TME insults. They undergo early inflammatory changes, assist the recruitment of BM-derived monocytes and contribute to the generation of TAMs. The involvement of TRMs in tumor progression is distinct in various cancer types. For example, in a breast cancer mouse model, the numbers of TRMs progressively decreased over time, while the numbers of TAMs generated from BM-derived monocytes concomitantly increased. In this context, ablation of TRMs did not impact tumor growth, whereas ablation of circulating monocytes resulted in the reduction of tumor size41. In contrast, in a mouse model of pancreatic cancer, TRMs expanded during tumor progression and acquired a transcriptional profile favoring a profibrotic program typical of pancreatic adenocarcinoma, which was not disrupted by depletion of BM-derived macrophages but was reversed by depletion of TRMs42. In a lung cancer model, macrophages of both origins contributed to tumor growth and progression43. Thus, the role of TRMs in cancer growth seems to be organ-specific.
Conversion of Bm-derived monocytes to TAms
Recruitment of myeloid cells in tumors and subsequent conversion to TAMs requires integrin activation (Fig. 2). Cytokines and chemokines produced in the TME, including GM-CSF, IL-1β, SDF1α, VEGF, CSF1, and CCL2, promote emergency myelopoiesis, leukocyte trafficking to tumor, and extravasation by activating receptor-mediated signaling events that induce inside-out conformational changes of a4b1 integrin. Blockade of this pathway inhibits tumor inflammation and growth44. Specifically, IL-1β, SDF1α, and VEGF activate G-protein-coupled receptors (GPCRs), receptor tyrosine kinases (RTKs), and TLR/IL-1R, leading to activation of Ras and its downstream target phosphoinositide 3-kinase γ (PI3Kγ), the PI3K isoform predominantly expressed in myeloid cells45. PI3Kγ activates Bruton’s tyrosine kinase (BTK), phospholipase C (PLC), and RasGrp/CalDAG-GEFs, leading to the activation of Rap1 and its downstream effector RIAM45. The Rap1–RIAM module regulates reorganization of cytoskeletal actin, talin conformational changes, and integrin activation46,47, guiding leukocyte adhesion and migration48. Inhibition of this signaling cascade can decrease monocyte recruitment and TAM accumulation in tumors45. Activation of PI3Kγ mediates generation of TAMs by suppressing the activation of the transcription factor NF-κB and promoting c/EBPβ-mediated signaling49. However, the integrin aMb2 (CD11b/CD18), which is abundantly expressed in myelocytes does not appear to have any impact in the recruitment of monocytes to tumors and generation of TAMs50. Global deletion of CD11b in Itgam−/− mice did not compromise the ability of myeloid cells to accumulate in tumors, but CD11b-deficient myeloid cells or wild-type myeloid cells treated with a CD11b blocking antibody in vitro had an elevated expression of immunosuppressive genes and a simultaneous decrease of immunostimulatory genes50, suggesting that loss of aMb2 function might compromise the properties of myeloid cells by unidentified mechanisms.
Fig. 2 |. recruitment of Bm-derived monocytes in tumors and subsequent conversion to TAms requires activation of α4β1 integrin.
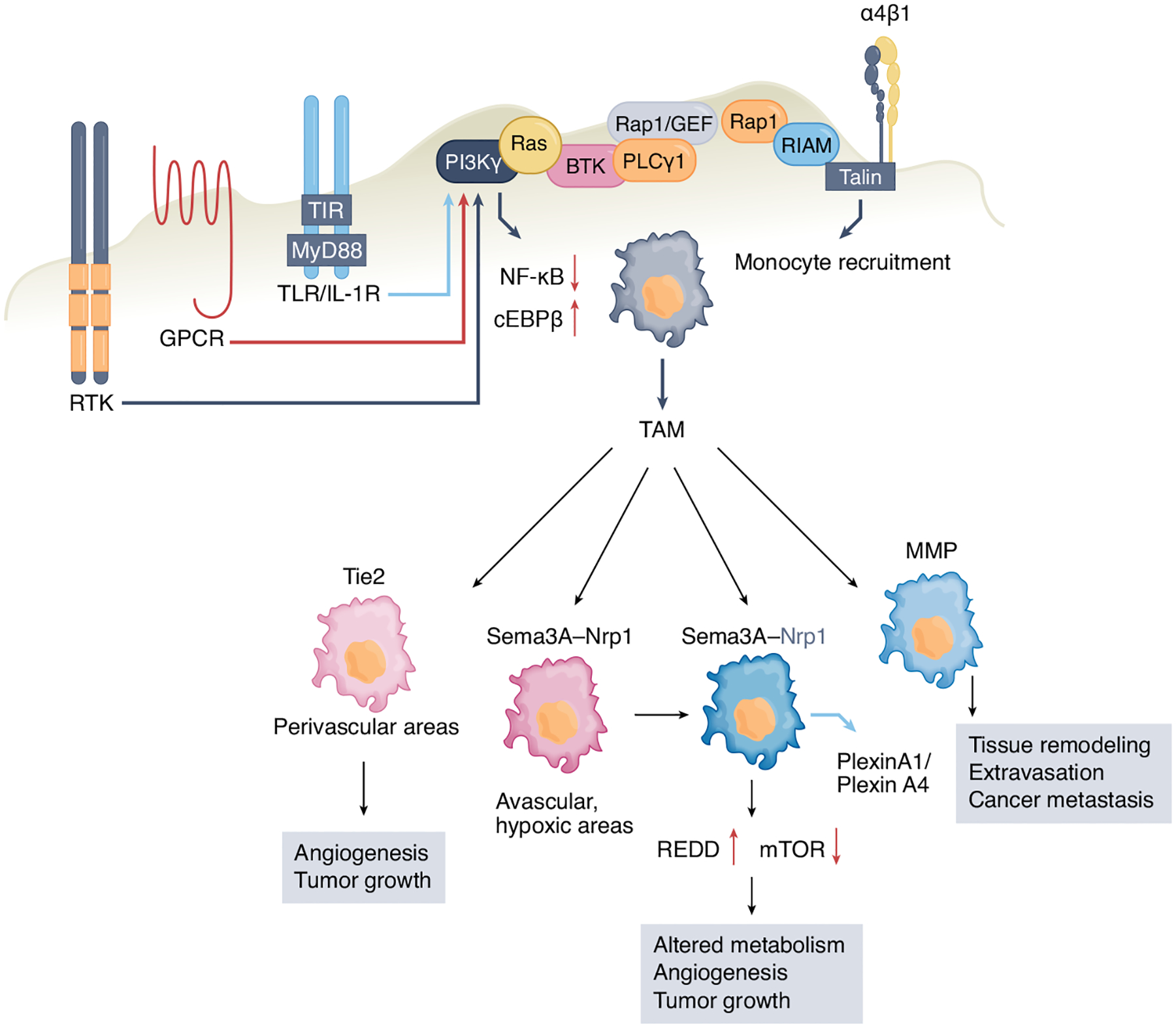
IL-1β, SDF1α, and VEgF activate receptor tyrosine kinases (RTK), g-protein-coupled receptors (gPCR), and TLR/IL-1R, leading to activation of Ras and its downstream target phosphoinositide 3-kinase γ (PI3Kγ), initiating a signaling cascade that leads to integrin activation. Activation of PI3Kγ inhibits signaling through the transcription factor NF-κB while promoting c/EBPβ-mediated signaling leading to the generation of immunosuppressive TAMs. Various subsets of TAMs within the same tumor have unique functional roles in supporting cancer growth. TAMs expressing the angiopoietin receptor Tie2 accumulate at the perivascular areas, where they support angiogenesis and tumor growth. TAMs are recruited to avascular, hypoxic tumor areas through the semaphorin 3A (Sema3A)–neuropilin-1 (Nrp1) pathway, where Nrp1 is downregulated and Sema3A entraps TAMs locally through plexinA1–plexinA4-mediated stop signals. Upregulation of REDD1, a negative regulator of mTOR, prevents glycolysis and promotes angiogenesis. Certain TAM subsets produce MMPs, which promote tissue remodeling, thereby facilitating monocyte and cancer cell migration, intravasation, and metastasis.
Although it is difficult to define when the recruited monocytes become TAMs, it is well-documented that tissue engagement changes the transcriptional profile of the recruited monocytes51. The precise mechanisms that drive this conversion are incompletely understood. It has been proposed that hypoxia-mediated enhancement of CD45 phosphatase activity in M-MDSCs alters activity of STAT3 and promotes TAM differentiation52. The enhanced phosphatase activity is induced by disruption of CD45 protein dimerization, an effect mediated by the sialic acid in the TME52. TAMs display distinct transcriptional profiles compared with circulating monocytes. Distinct transcriptional profiles have also been identified between TAMs and macrophages residing in adjacent healthy tissues51. In mouse models of breast cancer with lung metastatic disease, transcriptomics studies showed that newly recruited BM-derived monocytes convert into a precursor Ly6ChiCD11bhi cell population, which produces the chemokine ligand CCL3, and recruit metastasis-facilitating macrophages32.
It is possible that subsets of TAMs within the same tumor have unique functional roles in supporting cancer growth (Fig. 2). A subset of TAMs that express the angiopoietin receptor Tie2 accumulate in the perivascular areas, where they support angiogenesis, tumor growth, and tumor relapse after chemotherapy53,54. A distinct population of TAMs are recruited through the semaphorin 3A (Sema3A)–neuropilin-1 (Nrp1) pathway to avascular, hypoxic tumors areas, where they acquire pro-angiogenic and immunosuppressive properties. After accumulation in hypoxic niches, Nrp1 is downregulated and Sema3A entraps TAMs locally through plexinA1–plexinA4-mediated stop signals26. The pro-angiogenic properties of TAMs in the hypoxic niches also rely on their metabolism. REDD1, a negative regulator of mTOR that is upregulated in TAMs in hypoxic niches, prevents glycolysis and promotes the formation of abnormal blood vessels, contributing to tumor metastasis27. Other TAMs differentiate into metastasis-associated macrophages that are able to escort cancer cells at distal sites and facilitate their engraftment to form metastasis34. Conversely, IRF8, a transcription factor with a decisive role in monocyte and moDC lineage commitment, imprints a macrophage program that prevents metastasis55. Certain TAM subsets produce proteases such as metalloproteases (MMPs), which promote not only tissue remodeling, facilitating monocyte migration, but also cancer cell migration, intravasation, and relocation in distal sites, initiating metastatic foci56. The differential molecular properties of the spatially distinct subsets of TAMs might represent attractive targets for therapeutic intervention.
Bm-derived macrophages and Trms in primary and metastatic cancer
Since TRMs colonize tissue-specific niches during embryonic development and have self-renewal capacity5,13, they are present in metastases-targeted organs prior to cancer growth and might mediate local tissue alterations that facilitate metastasis. This hypothesis was investigated in depth in a metastatic breast cancer mouse model by looking at the alveolar macrophages57, the TRMs of the lung5. Alveolar macrophages accumulated in premetastatic lungs through complement C5a receptor-mediated proliferation, reduced the number and maturation of lung dendritic cells, suppressed type 1 helper T (TH1) responses and enhanced lung metastases (Fig. 1b). Depletion of alveolar macrophages reversed immunosuppression, strengthened local TH1 cell responses, and reduced metastatic burden. Thus, TRMs might foster the formation of the premetastatic niche and support development of metastatic disease57. Consistent with this notion, in three different lung cancer models it was found that BM-derived macrophages facilitated metastatic tumor spreading, whereas TRMs supported proliferation of cancer cells at the primary tumor site43.
In liver Kupffer cells (KCs), the local TRMs, TREM-1-mediated activation results in secretion of proinflammatory cytokines, including IL-6, IL-1β, TNF, CCL2, and CXCL10, leading to inflammatory liver injury and subsequent carcinogenesis (Fig. 1c)58. CCL2 is highly expressed in individuals with hepatocellular carcinoma (HCC), whereas blockade of the CCL2–CCR2 pathway in a mouse model of HCC prevented TAM accumulation and tumor growth, suggesting a role for KCs in regulating the immunological TME profile in HCC59. In addition, KCs might protect against metastasis of colorectal carcinoma (CRC) to the liver, as indicated by the increased number of metastases in livers of KC-deficient mice60.
Some of the most informative studies regarding the contribution of TRMs and BM-derived macrophages in primary versus metastatic cancer growth have been generated in the context of brain cancer. In the homeostatic central nervous system (CNS), innate immunity is solely accomplished by parenchymal microglia, and to a lesser degree by border-associated macrophages (BAMs). Microglia are a unique population of myeloid mononuclear phagocytic cells that originate during embryogenesis from erythromyeloid progenitors in the yolk sac11,61 and are divergent from BAMs (reviewed in ref.62). Microglia play a dynamic role in brain wiring during CNS development by phagocytosing apoptotic neurons and by selectively remodeling synapses. Microglia accomplish these tasks by surveying the environment, sensing changes in brain function and physiology, and responding accordingly63. Genes representing the microglial sensing system are well-defined64,65, and responses of microglia to pathologies vary widely, depending on their location within the CNS. High-resolution single-cell RNA-sequencing studies of mouse and human microglia indicated the existence of highly heterogeneous populations of microglia under normalcy66.
Primary brain cancers (which originate within the CNS) are composed of histopathologically and molecularly distinct neoplasms67, and their tumor immune microenvironment demonstrates remarkable heterogeneity68. Glioblastoma, the most common primary malignant brain cancer, is molecularly well characterized. Glioblastoma is considered highly immunosuppressive, with minimal cytotoxic lymphoid infiltration and the presence of considerable numbers of suppressive myeloid cells, such as macrophages, microglia, and MDSCs. Microglia, BAMs, and BM-derived TAMs constitute up to 40% of the tumor cellular composition. Microglia and monocyte-derived TAMs are heterogeneous populations in terms of their localization within the tumor and their functions. Extensive transcriptional analyses in mouse brain tumors indicated that microglia and TAMs share both proinflammatory M1-like and anti-inflammatory M2-like phenotypes69; in human glioblastoma, they additionally display expression profiles of non-polarized M0 macrophages70.
Detailed analyses of the immune populations in primary and metastatic human brain tumors14,15 have found that glioblastomas are populated by TRMs, microglia, and BM-derived TAMs, whereas metastatic brain tumors are populated predominantly by BM-derived TAMs, consistent with findings in a mouse model of proneural glioma71. In addition, the TME of human glioblastoma imposed a distinct pattern of gene regulation on microglia and BM-derived macrophages compared with that found in metastatic tumors14,15. These findings provide evidence for tumor-specific roles in the transcriptional imprinting of recruited monocyte-derived and TRMs in tumors and set the basis for further investigation.
Macrophage diversity in cancer
The inherited ability of macrophages to develop distinct adaptations to slight alterations of microenvironmental stimuli, including an intratumoral gradient of nutrients, metabolites, or oxygen, leads to a significant diversity of TAMs among different cancer types, but also within the same tumor72. The diversity of TAMs was previously streamlined in the simplified concept that TAMs have a polarization program resembling M2 macrophages and are skewed away from M1-polarized phenotype3. Classical M1 polarization has been defined by the expression of CD80, CD86, MHCII, iNOS, and CD68, correlated with the tumoricidal function of TAMs that could engulf cancer cells and recruit T cells. Conversely, M2 polarization has been characterized by the expression of CD206, CD204, VEGF, CD163, and Arg-1 and is associated with an immune quiescent profile3. The M1–M2 programs were thought to rely mainly on metabolism, because proinflammatory M1 macrophages are supported by glycolysis whereas anti-inflammatory M2 macrophages utilize mainly fatty acid oxidation (FAO)73. Although this concept is no longer considered appropriate, most studies continue to utilize M1–M2-associated markers for characterization of TAMs, because there is extensive experience based on the correlation between their expression and prognosis in tumor models and human cancers25,74.
It is becoming increasingly clear that macrophage metabolism is much more complex than the selective utilization of glucose or fatty acids as an energy source. It is now known that lipid utilization goes beyond fatty acid catabolism in FAO, and has an essential role in the ability of TAMs to function as potent APCs. Cancer-produced β-glucosylceramide drives reshuffling of lipid composition on the ER membrane, leading to IRE1-dependent ER stress responses75. The co-engagement of the IRE1–XBP1 and IRE1–STAT3 pathways during the ER stress response promoted pro-tumorigenic polarization and pro-survival properties of TAMs. Conversely, targeting IRE1–XBP1 and IRE1–STAT3 signaling or preserving lipid composition of the ER membrane by genetic and pharmacological approaches diminished the pro-tumorigenic ability of TAMs and inhibited tumor progression75. Thus, metabolic adaptations with significant impact on TAM function depend on lipid composition in a manner independent of their utilization as an energy source.
The significance of nutrients in TAM diversity is highlighted by the role of glutamine, which is indispensable for cellular functions, such as nucleotide and amino acid production, redox balance, and protein glycosylation. Small-molecule inhibitor blocking of glutamine metabolism reduced tumor growth and metastases in a mouse model of breast cancer by enhancing macrophage activation and inhibiting MDSC generation76. Targeting glutamine metabolism with an inhibitor of the enzyme glutamine synthetase, which generates glutamine from glutamate, converted TAMs into effector APCs, which mediated potent anti-tumor function in three highly metastatic mouse models77. An integrated high-throughput transcriptional-metabolic profiling showed that an immunosuppressive M2-like macrophage profile is supported by glutamine catabolism and is compromised by glutamine deprivation78. Consistent with these findings, production of α-ketoglutarate (αKG) via glutaminolysis is important for M2-like activation of macrophages, including engagement of FAO and epigenetic reprogramming of M2 genes79. This M2-promoting mechanism is further modulated by a high αKG/succinate ratio, whereas a low ratio strengthens the M1-like phenotype79. The immunometabolic properties of TAMs correlate not only with TAM functional diversity, but also cancer prognosis. For example, diversity of MHCIIlo and MHCIIhi TAM subsets correlate with distinct metabolic signatures and abilities to utilize lactate, a metabolite that is abundantly present in the TME. In MHCIIlo TAMs, lactate supports oxidative metabolism, increases l-arginine metabolism, and enhances their T cell suppressive capacity80. These observations underline the significance, and the complexity, of targeting the metabolic function of the diverse TAM subsets for therapeutic purposes.
During the past few years, progress in genomics, single-cell RNA-sequencing, and time-of-flight (CyTOF) technologies has revealed the previously unsuspected diversity of TAMs. Macrophages can now be classified into multiple distinct clusters based on distinct combinations of genes expressed. Spatial distribution of TAMs correlates with their distinct gene profiles and specific functional properties in many cancers, including lung, renal, brain breast, and ovarian cancer, head and neck carcinoma, melanoma, and colorectal cancer14,15,72,81–84. The evolution of such technologies might guide the development of new therapies targeting unique properties of tumorigenic TAMs85.
TAms in inflammation and immunosuppression
The association between inflammation and cancer has been extensively documented and is currently considered an integral component of cancer evolution (reviewed in refs.86,87). Tumors were proposed to behave as wounds that do not heal88; indeed, several features of tissue injury and healing identified in wounds characterize the TME, including infiltration by inflammatory cells such as neutrophils, monocytes, and macrophages, tissue remodeling, and enhanced coagulation88. However, in contrast with the sequential process of tissue injury, inflammation, and healing observed in wounds, features that characterize injury and healing co-exist and persist in the TME86. Cancer-related inflammation is likely initiated by soluble factors, such as hematopoietic growth factors (for example, M-CSF, GM-CSF), cytokines (for example, IL-6, IL-1, IL-8), and chemokines (for example, CCL2, CCL5, CXCL12) produced as a consequence of the oncogenes whose expression is induced by driver mutations (Fig. 3), including KRASG12D (ref.89), p53 (ref.90), BRAFV600E (ref.91), and BRCA1 (ref.92).
Fig. 3 |. Cancer-related inflammation is initiated by hematopoietic growth factors, cytokines, and chemokines produced by cancer cells as a consequence of oncogene-mediated malignant transformation.
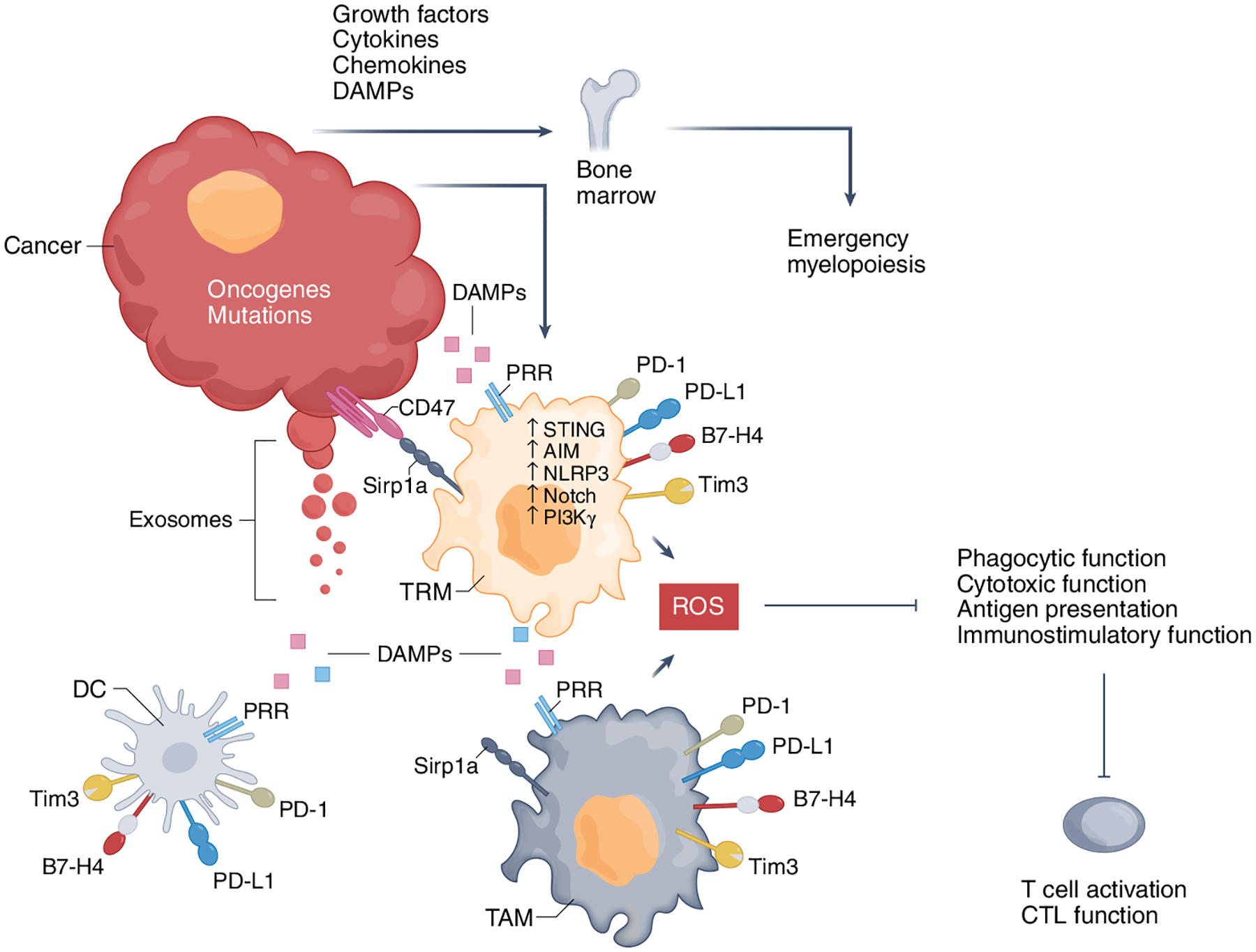
There is proinflammatory activation from DAMPs produced by cancer cells due to rapid replication and apoptosis, nutrient starvation, and hypoxia. Exosomes released from cancer cells, containing tumor DNA or RNA, contribute to the proinflammatory TME. Cytokines activate immune and cancer cells, whereas DAMPs are recognized by pattern recognition receptors (PRRs) expressed by macrophages and other cells of the innate immune system, such as dendritic cells (DCs), as well as cancer cells, to initiate proinflammatory cascades. In response to inflammatory activation, TAMs express Tim3, Tim4, PD-1, and PD-L1 checkpoint inhibitors, which can inhibit macrophage functions and synergize with the Sirp1a–CD47 pathway to inhibit phagocytosis of cancer cells. Cancer-produced cytokines and DAMPs also act on BM progenitors through cytokine and growth factor receptors and PRRs to induce myelopoiesis, which gives rise to immature immunosuppressive PMN-MDSCs and M-MDSCs, the latter being recruited into the tumor to become TAMs. CTL, cytotoxic T lymphocyte.
An additional mechanism responsible for cancer-related inflammation involves the generation of DAMPs that are produced by cancer cells owing to rapid replication and apoptosis, nutrient starvation, and hypoxia (Fig. 3). These DAMPs are recognized by PRRs expressed by monocytes, macrophages, other cells of the innate immune system, and cancer cells, and initiate proinflammatory cascades. Exosomes released from cancer, containing tumor DNA, recognized by STING and AIM2 (refs.93,94), or RNA, recognized by TLR3 (ref.95), induce monocyte recruitment to the primary tumor and the metastatic niche96. These pathways, along with NLRP3, which recognizes ATP97, induce production of inflammatory cytokines and chemokines, recruiting T cells. At the early stages of the anti-tumor immune response, T cells can successfully eliminate cancer by cell-mediated killing, which in turns generates a third level of inflammatory mediators in the TME. The cancer-produced cytokines and DAMPs also act on the BM progenitors to induce emergency myelopoiesis, giving rise to immunosuppressive PMN-MDSCs and M-MDSCs, the latter being recruited into the tumor to become TAMs (Fig. 3)52.
Paradoxically, the proinflammatory pathways that promote the cancer-related inflammation and generation of immunosuppressive TAMs are the same mediators of protective proinflammatory immune responses in macrophages against pathogens, such as those regulated by type I IFN induced by viral infections98. These pathways also have an instrumental role in the recruitment and retention of CD8+ T cells in tumors, an outcome that can be recapitulated by treatment with type I IFN or DNA-damaging chemo- and radiotherapies99, but can be hijacked by the tumor to initiate cancer-related inflammation and immunosuppression100. Notch signaling, which is involved in myeloid cell development and hematopoiesis and is needed for the differentiation of M1-like effector macrophages101,102, is also involved in the differentiation of TRMs into TAMs41. PI3K, a critical regulator of effector immune responses, acts as a mediator of cancer-related inflammation49.
Although the precise mechanisms of this paradox remain poorly understood, activation of proinflammatory pathways in TAMs might concomitantly lead to increased expression of inhibitory receptors and ligands, thereby favoring immunosuppression (Fig. 3). After phagocytosis of cancer cells by macrophages, AIM2, which is activated by tumor DNA, cleaves cGAS and upregulates PD-L1 and IDO, overriding the anti-tumor function of macrophages94. In response to TLR signaling or phagocytosis of cancer cells, TAMs also express Tim3, Tim4, PD-1, and PD-L1, which can inhibit macrophage functions including phagocytosis, inflammasome activation, and production of effector cytokines18–21,103, and synergize with Sirp1a, which transmits phagocytosis-inhibitory signals after engagement by CD47 (ref.104). Such mechanisms might perpetuate proinflammatory activation but impaired effector function of TAMs while inhibiting T cell activation through co-inhibitory receptors and TAM-generated reactive oxygen species (Fig. 3). Macrophages might be subjected to PD-L1-mediated inhibition as macrophage-specific blockade of PD-L1 can induce macrophage activation and proliferation105.
Expression of PD-1 is induced by TLR signaling in myeloid cells and is correlated with impaired M1-like polarization106. In the context of infection, PD-1 expression in macrophages suppresses the innate inflammatory response to sepsis107 and inhibits the phagocytosis of Mycobacterium tuberculosis108. PD-1 upregulation in TAMs during tumor progression compromises their phagocytic potency in a mouse model of colon cancer18. In peripheral blood monocytes from individuals with chronic lymphocytic leukemia (CLL), PD-L1- or antibody-mediated triggering of PD-1 hampers BTK signaling, glycolysis, and phagocytosis20. Conversely, selective PD-1 ablation19 or macrophage-specific PD-1 blockade18 in murine tumor models, or PD-1 blockade in monocytes from people with CLL20, significantly enhance anti-tumor responses. Thus, checkpoint inhibitors have an active role in the immunosuppressive properties of TAMs, but it is poorly understood under what conditions, and in which TAM subsets, these inhibitory mediators are expressed and operate. Studies in this direction might provide opportunities for tumor immunotherapy.
Resolution of inflammation
An attractive hypothesis regarding the mechanisms involved in TAM-mediated inflammation in cancer is that unidentified modifications might occur in the regulation of proinflammatory pathways during cancer evolution. According to the concept of cancer immunoediting, the immune system can control cancer development during the phase of immunosurveillance and elimination, when cancer cells are successfully recognized and cleared (Fig. 4)109. Macrophages have a central role in this process by mediating phagocytosis and clearance of cancer cells110 and the presentation of cancer neoantigens to T cells2. Under immune-mediated pressure, cancer cells undergo immunoediting, which results initially in an immune equilibrium phase and, subsequently, full bypassing of the mechanisms of antigen recognition and anti-tumor immune response, leading to tumor escape109. It is possible that, during the immune equilibrium phase, the properties of TAMs gradually change as their proinflammatory pathways are increasingly activated (Fig. 4), but lose their efficacy. For example, PRR signaling, which is controlled by posttranslational, metabolic, and epigenetic modifications111,112, might operate differently during immune equilibrium and immune escape than during elimination. In support of this hypothesis, only acute engagement of pro-immune inflammatory pathways during immune escape can override the continuing cancer-promoting inflammation and elicit anti-tumor immunity113. Under such conditions, inflammasome activation with oxidized phospholipids can induce strong and long-lasting protection against cancer and eradicate various tumors that are resistant to standard checkpoint immunotherapy113.
Fig. 4 |. Changes of TAms during cancer immunoediting.
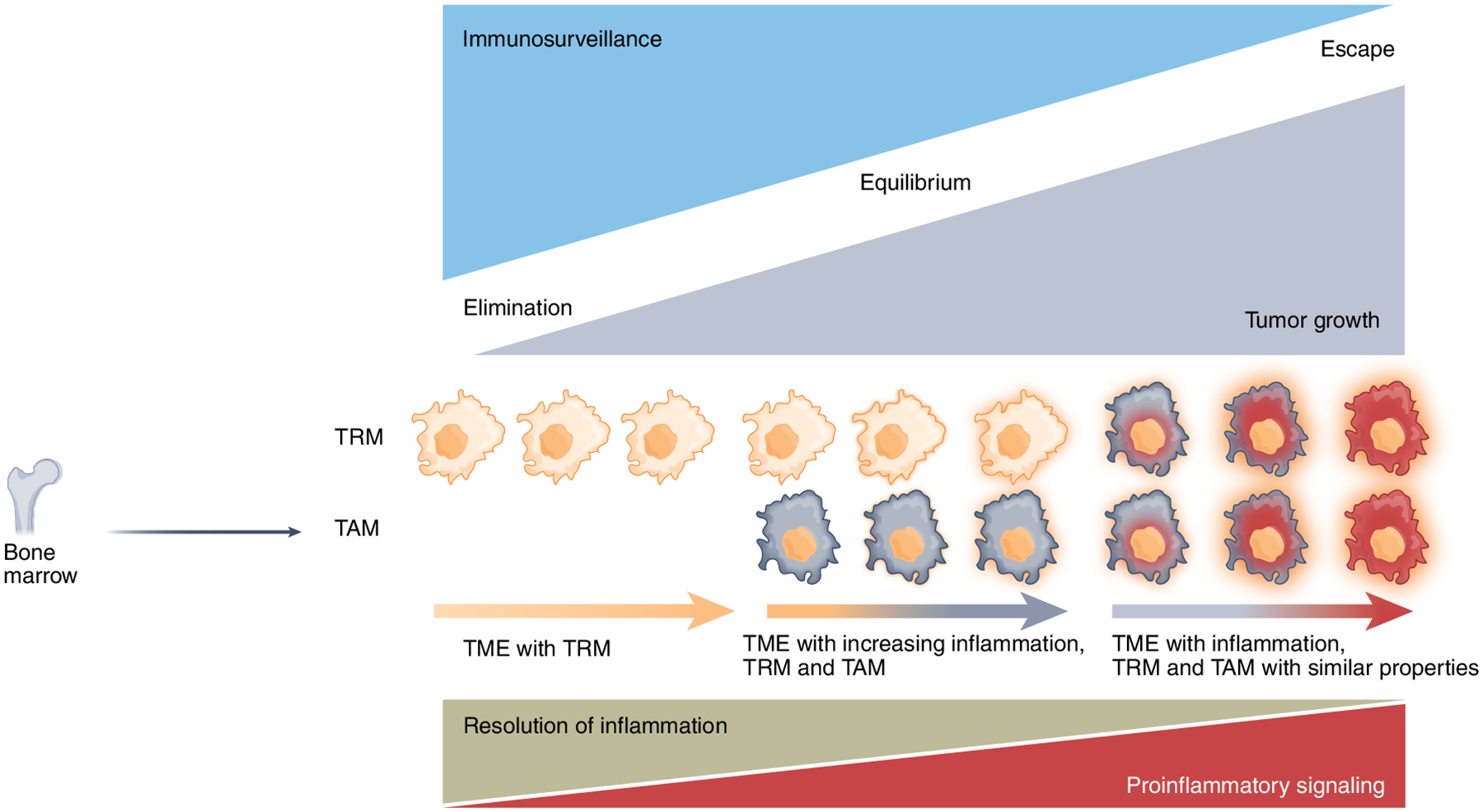
During the elimination phase, macrophages eliminate cancer cells by phagocytosis and activation of anti-tumor T cell responses by presentation of tumor-associated antigens, whereas these immune functions are compromised during cancer progression to immune equilibrium and escape. While TRMs predominate during elimination, BM-derived TAMs increasingly enter the tumor during the immune equilibrium phase, leading to increasing local inflammation. During the immune escape phase, in response to tissue residence and cues of the TME, TRMs and BM-derived TAMs acquire similar properties, characterized by enhanced proinflammatory and diminished pro-resolving signaling.
Because macrophages have a physiological role in mediating resolution of inflammation and promoting tissue remodeling and healing2, it is possible that the cancer-promoting proinflammatory effects of TAMs are mediated by impaired mechanisms of inflammation resolution (Fig. 4). During physiologic immune responses, nuclear hormone receptors program an anti-inflammatory and pro-resolving function in macrophages114. The nuclear hormone receptor PPARγ has an important role in this process and mediates resolution of inflammation by inducing expression of the scavenger receptor CD36 and the nuclear hormone receptor LXR115,116. Notably, LXR activation can suppress cancer growth in multiple murine tumor models117. Macrophages also promote inflammation resolution by production of pro-resolving cytokines. Following tissue injury or infection, eicosanoids initiate the inflammation process. The prostaglandins PGE2 and PGI2, which are involved in vasodilation, and the leukotriene LTB4, which is involved in chemotaxis and adhesion, stimulate the recruitment of neutrophils as first responders. Subsequently, lipoxins, resolvins, protectins, and maresins, collectively called specialized pro-resolving mediators (SPMs), are produced118. Switching from eicosanoids to SPMs mediates a ‘stop signal’ to the acute inflammatory response. SPMs counteract the proinflammatory mediators and stimulate the recruitment of monocytes, which become resolving macrophages, clear apoptotic cells by efferocytosis and promote antigen presentation and engagement of adaptive immune responses119. In a mouse tumor model, debris of various cancer cell types killed by chemotherapy or targeted therapy induced production of proinflammatory cytokines by TAMs and tumor growth, whereas administration of SPMs induced inflammation resolution, T cell activation, and suppression of cancer growth120. Thus, a plausible scenario might be that, during the immune equilibrium phase, the anti-inflammatory pathways of TAMs are progressively suppressed and eventually lose efficacy during cancer immune escape (Fig. 4). A spatiotemporally aberrant and unconcerted activation of proinflammatory and pro-resolving mechanisms of TAMs might have a central role in pro-tumorigenic inflammation.
Concluding remarks
Macrophages have an essential and indispensable role in homeostasis and immunity, but lose their protective functions and become TAMs in the context of cancer. TAMs arise from TRMs localized at the tumor site and BM-derived monocytes that are recruited to tumors. TAMs provide a protective niche for cancer growth and invasion at primary and metastatic sites. TAMs are highly heterogeneous, and the significance and therapeutic potential of their diversity are evolving. Although TAMs might have either a supportive or a suppressive role in anti-tumor immunity, they most frequently enhance tumor growth by promoting angiogenesis and immunosuppression through hijacking proinflammatory pathways naturally programmed to provide protective immune responses. Altered signaling and metabolic properties support the immunosuppressive functions of TAMs. The immunological, biochemical, and metabolic aberrations can serve as targets of novel precision therapies for reprogramming and switching macrophage function from pro-tumorigenic TAMs to anti-tumorigenic and protective APCs. On this road, the great challenge remains of spatially guiding such interventions to achieve tumor-specific outcomes without compromising responses of healthy innate and adaptive immune cells.
Acknowledgements
This work was supported by NIH grants R01CA238263 (V.A.B.) and R01CA229784 (A. Charest and V.A.B.).
Footnotes
Competing interests
V.A.B. has patents on the PD-1 pathway licensed by Bristol-Myers Squibb, Roche, Merck, EMD-Serono, Boehringer Ingelheim, AstraZeneca, Novartis, and Dako. The authors declare no other competing interests.
References
- 1.Watanabe S, Alexander M, Misharin AV & Budinger GRS The role of macrophages in the resolution of inflammation. J. Clin. Investig 129, 2619–2628 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirayama D, Iida T & Nakase H The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int. J. Mol. Sci 19, 92 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinez FO, Sica A, Mantovani A & Locati M Macrophage activation and polarization. Front. Biosci 13, 453–461 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Wynn TA, Chawla A & Pollard JW Macrophage biology in development, homeostasis and disease. Nature 496, 445–455 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ginhoux F & Guilliams M Tissue-resident macrophage ontogeny and homeostasis. Immunity 44, 439–449 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Carlin LM et al. Nr4a1-dependent Ly6Clow monocytes monitor endothelial cells and orchestrate their disposal. Cell 153, 362–375 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Auffray C et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317, 666–670 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Geissmann F et al. Development of monocytes, macrophages, and dendritic cells. Science 327, 656–661 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol 13, 753–760 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei S et al. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J. Leukoc. Biol 88, 495–505 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ginhoux F et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ginhoux F & Merad M Ontogeny and homeostasis of Langerhans cells. Immunol. Cell Biol 88, 387–392 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Schulz C et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Klemm F et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 181, 1643–1660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friebel E et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 181, 1626–1642 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Robinson A, Han CZ, Glass CK & Pollard JW Monocyte regulation in homeostasis and malignancy. Trends Immunol 42, 104–119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mantovani A, Marchesi F, Jaillon S, Garlanda C & Allavena P Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell Mol. Immunol 18, 566–578 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon SR et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumor immunity. Nature 545, 495–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strauss L et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol 5, eaay1863 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qorraj M et al. The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia 31, 470–478 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Dixon KO et al. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature 595, 101–106 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo WI et al. Expression of VISTA on tumor-infiltrating immune cells correlated with short intravesical recurrence in non-muscle-invasive bladder cancer. Cancer Immunol. Immunother 70, 3113–3122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin H et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Investig 128, 805–815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dangaj D et al. Novel recombinant human b7-h4 antibodies overcome tumoral immune escape to potentiate T-cell antitumor responses. Cancer Res 73, 4820–4829 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sica A et al. Macrophage polarization in tumour progression. Semin. Cancer Biol 18, 349–355 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Casazza A et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24, 695–709 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Wenes M et al. Macrophage metabolism controls tumor blood vessel morphogenesis and metastasis. Cell Metab 24, 701–715 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Mantovani A, Marchesi F, Malesci A, Laghi L & Allavena P Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol 14, 399–416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manz MG & Boettcher S Emergency granulopoiesis. Nat. Rev 14, 302–314 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Veglia F, Sanseviero E & Gabrilovich DI Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev 21, 485–498 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mantovani A et al. Chemokines in the recruitment and shaping of the leukocyte infiltrate of tumors. Semin. Cancer Biol 14, 155–160 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Kitamura T et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med 212, 1043–1059 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma RY et al. Monocyte-derived macrophages promote breast cancer bone metastasis outgrowth. J. Exp. Med 217, e20191820 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qian BZ et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin EY, Nguyen AV, Russell RG & Pollard JW Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med 193, 727–740 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin EY et al. Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol. Oncol 1, 288–302 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ban Y et al. Targeting autocrine CCL5–CCR5 axis reprograms immunosuppressive myeloid cells and reinvigorates antitumor immunity. Cancer Res 77, 2857–2868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De la Fuente Lopez M et al. The relationship between chemokines CCL2, CCL3, and CCL4 with the tumor microenvironment and tumor-associated macrophage markers in colorectal cancer. Tumour Biol 40, 1010428318810059 (2018). [DOI] [PubMed] [Google Scholar]
- 39.Hanna RN et al. Patrolling monocytes control tumor metastasis to the lung. Science 350, 985–990 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kubo H, Mensurado S, Goncalves-Sousa N, Serre K & Silva-Santos B Primary tumors limit metastasis formation through induction of IL15-mediated cross-talk between patrolling monocytes and NK cells. Cancer Immunol. Res 5, 812–820 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Franklin RA et al. The cellular and molecular origin of tumor-associated macrophages. Science 344, 921–925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu Y et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 47, 323–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loyher PL et al. Macrophages of distinct origins contribute to tumor development in the lung. J. Exp. Med 215, 2536–2553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmid MC et al. Combined blockade of integrin-α4β1 plus cytokines SDF-1α or IL-1β potently inhibits tumor inflammation and growth. Cancer Res 71, 6965–6975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmid MC et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell 19, 715–727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lafuente EM et al. RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Dev. Cell 7, 585–595 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Cho EA et al. Phosphorylation of RIAM by src promotes integrin activation by unmasking the PH domain of RIAM. Structure 29, 320–329 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patsoukis N et al. The adaptor molecule RIAM integrates signaling events critical for integrin-mediated control of immune function and cancer progression. Sci. Signal 10, eaam8298 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Kaneda MM et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 539, 437–442 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmid MC et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat. Commun 9, 5379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cassetta L et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 35, 588–602 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar V et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 44, 303–315 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hughes R et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res 75, 3479–3491 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen L et al. Tie2 expression on macrophages is required for blood vessel reconstruction and tumor relapse after chemotherapy. Cancer Res 76, 6828–6838 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Twum DY et al. IFN regulatory factor-8 expression in macrophages governs an antimetastatic program. JCI Insight 4, e124267 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gui P et al. The protease-dependent mesenchymal migration of tumor-associated macrophages as a target in cancer immunotherapy. Cancer Immunol. Res 6, 1337–1351 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Sharma SK et al. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J. Immunol 194, 5529–5538 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Wu J et al. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res 72, 3977–3986 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li X et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 66, 157–167 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Matsumura H et al. Kupffer cells decrease metastasis of colon cancer cells to the liver in the early stage. Int. J. Oncol 45, 2303–2310 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Goldmann T et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol 17, 797–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoeffel G & Ginhoux F Fetal monocytes and the origins of tissue-resident macrophages. Cell. Immunol 330, 5–15 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Nimmerjahn A, Kirchhoff F & Helmchen F Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Butovsky O et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci 17, 131–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hickman SE et al. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Masuda T et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566, 388–392 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Louis DN, Wiestler OD & Cavenee WK World Health Organization Classification of Tumours of the Central Nervous System 5th ed. (International Agency for Research on Cancer, 2021). [Google Scholar]
- 68.Boussiotis VA & Charest A Immunotherapies for malignant glioma. Oncogene 37, 1121–1141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szulzewsky F et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 10, e0116644 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gabrusiewicz K et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 1, e85841 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pyonteck SM et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med 19, 1264–1272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Donadon M et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J. Exp. Med 217, e20191847 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.O’Neill LA & Pearce EJ Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med 213, 15–23 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jayasingam SD et al. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: technicalities and challenges in routine clinical practice. Front. Oncol 9, 1512 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Conza G et al. Tumor-induced reshuffling of lipid composition on the endoplasmic reticulum membrane sustains macrophage survival and pro-tumorigenic activity. Nat. Immunol 22, 1403–1415 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oh MH et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig 130, 3865–3884 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Menga A et al. Glufosinate constrains synchronous and metachronous metastasis by promoting anti-tumor macrophages. EMBO Mol. Med 12, e11210 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jha AK et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Liu PS et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol 18, 985–994 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Geeraerts X et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep 37, 110171 (2021). [DOI] [PubMed] [Google Scholar]
- 81.Zilionis R et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 50, 1317–1334 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Azizi E et al. Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell 174, 1293–1308 e1236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chevrier S et al. An immune atlas of clear cell renal cell carcinoma. Cell 169, 736–749 e718 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Artyomov MN & Van den Bossche J Immunometabolism in the single-cell era. Cell Metab 32, 710–725 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mantovani A, Allavena P, Sica A & Balkwill F Cancer-related inflammation. Nature 454, 436–444 (2008). [DOI] [PubMed] [Google Scholar]
- 87.Nakamura K & Smyth MJ Targeting cancer-related inflammation in the era of immunotherapy. Immunol. Cell Biol 95, 325–332 (2017). [DOI] [PubMed] [Google Scholar]
- 88.Dvorak HF Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med 315, 1650–1659 (1986). [DOI] [PubMed] [Google Scholar]
- 89.Bayne LJ et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ubertini V et al. Mutant p53 gains new function in promoting inflammatory signals by repression of the secreted interleukin-1 receptor antagonist. Oncogene 34, 2493–2504 (2015). [DOI] [PubMed] [Google Scholar]
- 91.Sumimoto H, Imabayashi F, Iwata T & Kawakami Y The BRAF–MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med 203, 1651–1656 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mehta AK et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Cancer 2, 66–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dou Z et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Su S et al. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell 175, 442–457 e423 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Liu Y et al. Tumor exosomal RNAs promote lung pre-metastatic niche formation by activating alveolar epithelial TLR3 to recruit neutrophils. Cancer Cell 30, 243–256 (2016). [DOI] [PubMed] [Google Scholar]
- 96.Keklikoglou I et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol 21, 190–202 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Di Virgilio F, Sarti AC, Falzoni S, De Marchi E & Adinolfi E Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 18, 601–618 (2018). [DOI] [PubMed] [Google Scholar]
- 98.Hubel P et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat. Immunol 20, 493–502 (2019). [DOI] [PubMed] [Google Scholar]
- 99.Mowat C, Mosley SR, Namdar A, Schiller D & Baker K Anti-tumor immunity in mismatch repair-deficient colorectal cancers requires type I IFN-driven CCL5 and CXCL10. J. Exp. Med 218, e20210108 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.West AJ et al. Inflammasome-associated gastric tumorigenesis is independent of the NLRP3 pattern recognition receptor. Front Oncol 12, 830350 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Radtke F, Fasnacht N & Macdonald HR Notch signaling in the immune system. Immunity 32, 14–27 (2010). [DOI] [PubMed] [Google Scholar]
- 102.Wang YC et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res 70, 4840–4849 (2010). [DOI] [PubMed] [Google Scholar]
- 103.Chiba S et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol 13, 832–842 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Matlung HL, Szilagyi K, Barclay NA & van den Berg TK The CD47–SIRPα signaling axis as an innate immune checkpoint in cancer. Immunological Rev 276, 145–164 (2017). [DOI] [PubMed] [Google Scholar]
- 105.Hartley GP, Chow L, Ammons DT, Wheat WH & Dow SW Programmed cell death ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol. Res 6, 1260–1273 (2018). [DOI] [PubMed] [Google Scholar]
- 106.Chen W, Wang J, Jia L, Liu J & Tian Y Attenuation of the programmed cell death-1 pathway increases the M1 polarization of macrophages induced by zymosan. Cell Death Dis 7, e2115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang X et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl Acad. Sci. USA 106, 6303–6308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shen L et al. PD-1/PD-L pathway inhibits M.tb-specific CD4+ T-cell functions and phagocytosis of macrophages in active tuberculosis. Sci. Rep 6, 38362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schreiber RD, Old LJ & Smyth MJ Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331, 1565–1570 (2011). [DOI] [PubMed] [Google Scholar]
- 110.Lecoultre M, Dutoit V & Walker PR Phagocytic function of tumor-associated macrophages as a key determinant of tumor progression control: a review. J. Immunother. Cancer 8, e001408 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu J, Qian C & Cao X Post-translational modification control of innate immunity. Immunity 45, 15–30 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Bene K, Halasz L & Nagy L Transcriptional repression shapes the identity and function of tissue macrophages. FEBS Open Bio 11, 3218–3229 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhivaki D et al. Inflammasomes within hyperactive murine dendritic cells stimulate long-lived T cell-mediated anti-tumor immunity. Cell Rep 33, 108381 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Christofides A, Konstantinidou E, Jani C & Boussiotis VA The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 114, 154338 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chawla A et al. A PPARγ–LXR–ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol. Cell 7, 161–171 (2001). [DOI] [PubMed] [Google Scholar]
- 116.Moore KJ et al. The role of PPARγ in macrophage differentiation and cholesterol uptake. Nat. Med 7, 41–47 (2001). [DOI] [PubMed] [Google Scholar]
- 117.Tavazoie MF et al. LXR/ApoE activation restricts innate immune suppression in cancer. Cell 172, 825–840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Serhan CN Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Libreros S et al. A new E-series resolvin: RvE4 stereochemistry and function in efferocytosis of inflammation-resolution. Front. Immunol 11, 631319 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sulciner ML et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med 215, 115–140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]