

Abstract
Objectives:
Microglia are a primary mediator of the neuroinflammatory response to neurologic injury, such as that in traumatic brain injury. Their response includes changes to their cytokine expression, metabolic profile, and immunophenotype. Dexmedetomidine (DEX) is an α2 adrenergic agonist used as a sedative in critically ill patients, such as those with traumatic brain injury. Given its pharmacologic properties, dexmedetomidine may alter the phenotype of inflammatory microglia.
Methods:
Primary microglia were isolated from Sprague-Dawley rats and cultured. Microglia were activated using multiple mediators: LPS, Poly I:C, and TBI damage-associated molecular patterns (DAMP) from a rat that sustained a prior controlled-cortical impact injury. After activation, cultures were treated with dexmedetomidine. At the 24-hour interval, cell supernatant and cells were collected for the following studies: cytokine expression (TNF-α, IL-10) via ELISA, 6-phosphofructokinase enzyme activity assay, immunophenotype profiling with flow cytometry. Cytokine expression and metabolic enzyme activity data were analyzed using two-way ANOVA. Cell surface marker expression was analyzed using FlowJo software.
Results:
In LPS-treated cultures, DEX treatment decreased the expression of TNF-alpha from microglia (mean difference = 121.5 ± 15.96 pg/mL; p<0.0001). Overall, DEX-treated cultures had a lower expression of IL-10 than non-treated (mean difference = 39.33 ± 14.50, p<0.0001). DEX decreased IL-10 expression in LPS-stimulated (mean difference = 74.93 ± 12.50, p = 0.0039) and Poly I:C-stimulated microglia (mean difference = 23.27 ± 6.405, p = 0.0221). In DAMP-stimulated microglia, DEX decreased the activity of 6-phosphofructokinase (mean difference = 18.79 ± 6.508 units/mL; p = 0.0421). Microglial immunophenotype was altered to varying degrees with different inflammatory stimuli and DEX-treatment.
Conclusions:
Dexmedetomidine may alter the neuroinflammatory response of microglia. By altering the microglial profile, dexmedetomidine may affect the progression of neurologic injury.
Keywords: dexmedetomidine, microglia, neuroinflammation, traumatic brain injury
Introduction
Microglia are the resident tissue macrophages of the central nervous system (CNS) (1). They respond to various stimuli and inflammatory insults in the CNS, such as traumatic brain injury (TBI) (2). Non-activated microglia are considered to be in a “resting” or surveillance state. In this state, their long, ramified processes are extended to survey the cerebral microenvironment for any inflammatory stimuli (3, 4). Upon encountering a marker of inflammation or neurologic injury, microglia activate and subsequently adjust their phenotype in response (4). Classically, microglia, like macrophages, were defined in a binary system of M1 pro-inflammatory versus an alternative activation M2 state, which is usually more anti-inflammatory (5). However, activated microglia more likely display features of both the pro- and anti-inflammatory states; thus, the microglial phenotype is likely better defined by a spectrum than a binary classification (1).
An overwhelmingly pro-inflammatory response can cause excessive tissue damage that propagates further neurologic injury after the initial insult. Thus, characterizing microglial inflammatory response is important to understand neuroinflammation. Part of microglial activation is the release of certain cytokines, such as IFN-γ, TNF-α and IL-10 (6–9). These cytokines induce signaling cascades which lead to secondary responses and ultimately either tissue repair or further tissue damage.
Another phenotypic change of microglia is their metabolism. By altering their metabolic balance between primarily aerobic or anaerobic metabolism, microglia regulate energy and substrate production necessary for their inflammatory phenotype (10). In immune cells, metabolic shifts can also induce changes in gene transcription and production of certain inflammatory proteins and markers (11).
Adrenergic-signaling influences metabolic shifts in cells. Microglia preferentially express beta-adrenergic receptors, but will express alpha-adrenergic receptors under settings of inflammation (12). Given the presence of sympathetic changes present in neuroinflammation and microglial expression of adrenergic receptors, it is worth considering these signaling pathways as important in the inflammatory phenotype of microglia. Adrenergic-signaling may present a specific target to modify the microglial response to neuroinflammation.
Dexmedetomidine (DEX) is an α2 adrenergic agonist administered intravenously (13). DEX is commonly used in critically ill patients as a sedative for its anesthetic, amnestic, and analgesic properties (14). DEX is known to affect sympathetic signaling; common side effects from autonomic signaling include hypotension and bradycardia (13, 14). There is also evidence in in vitro and in vivo studies that DEX may also provide a degree of neuroprotection in the setting of neurologic injury (15). In experimental TBI models, DEX provides some neuroprotection through an upregulation of anti-apoptotic proteins, decreasing oxidative stress, and limiting neuronal autophagy (15–17). Given its use in critically ill patients, adrenergic activity, we sought to determine dexmedetomidine’s effect on microglial inflammatory and metabolic profile in an in vitro setting with various inflammatory activators.
Methods
Animal Care
All animal experiments had approval by the institutional Animal Welfare Committee of the University of Texas Health Science Center at Houston, TX, USA. Adult Sprague-Dawley male rats (Envigo Labs, Indianapolis, IN; 5–7 weeks) were used as a source of microglia. Animal experiments were in compliance with our institutional and ARRIVE guidelines. Rats were housed in microisolators under 12-hour light/dark cycles. Housing temperature was controlled. Food and water were available to rats ad libitum.
Rat brain neural dissociation and microglia isolation
Rats were sacrificed, and whole brains were harvested; for the following experiments described, a total of 4 rats were sacrificed for microglia isolation. The brain tissue was taken through a previously described neural dissociation protocol (18). Rat brains were washed in Dulbecco’s PBS, and placed in a MACS C tube. Enzymatic and mechanical digestion of the tissue was performed with a MACS dissociator gentleMACS following Adult Rat Neural Dissociation protocol. The tissue digestion was then mixed with a solution of 30% Percoll with Hanks buffered salt solution (HBSS); this was centrifuged at 700g for 10 minutes with no brakes to separate the cells from the myelin. After neural dissociation, microglia were isolated from the cell pellet using CD11bc magnetic selection beads per manufacturer protocol (Miltenyi Biotec). Cells were then moved to microglia culture media (DMEM (Gibco), 10% FBS, penicillin-streptomycin, and Glutamax (Gibco)) similar to other studies (19, 20). For studying cytokine production and metabolic enzyme activity, cells were plated at a density of approximately 20,000 cells/mL.
Rat microglia activation and treatment
Our “DAMP” mediator was formulated by the following method: a controlled cortical impact injury was performed on a rat as previously described (18, 21). Briefly, rat was anesthetized with 4% isoflurane in oxygen with a maintenance rate of 1.5L/min 2–3%. The animal was secured to a stereotactic frame. The head was prepped with iodine and alcohol, and 2% lidocaine was administered as a local anesthetic. After a midline scalp incision, the calvarium was exposed by blunt dissection. A craniectomy (6–7mm in diameter) was created. The CCI-device (Impact One Stereotaxic Impactor, Leica Microsystems, Buffalo Grove, IL) was positioned over the craniectomy. Cortical injury was administered with a 4mm impactor tip with the following parameters: depth 2.7mm, impact velocity 5.6m/s, dwell time 150microseconds. After the injury, the scalp was closed with wound clips and allowed to recover in a clean microisolator cage.
At 6 hours post-injury, the rat was sacrificed, and the brain was harvested. The ipsilateral hemisphere of the rat brain was washed in PBS, and mechanically digested with a MACS C tube in a MACS dissociator. No chemical or enzymatic digestion was performed. The resulting digestion was filtered through a 70μm filter. After this, the filtered solution was centrifuged at 1000g for 15 minutes. A protein quantification was performed, and standardized aliquots of the lysate were made. This tissue extract was used to simulate TBI microenvironment and is referred to as a “DAMP.” One rat was used for the development of DAMP.
To induce an expected inflammatory response, microglia were stimulated with three different inflammatory markers: LPS (100ng/mL), poly I:C (10μg/mL), or TBI damage associated molecular patterns (1mg/mL, DAMP) similar to other studies (22–24). Naïve unstimulated microglia cultures served as controls. LPS is a TLR4 agonist, and Poly I:C is a TLR3 agonist. The DAMP stimulus was used to replicate TBI-induced neuroinflammation.
After a 4-hour incubation period with their inflammatory stimulus, specified cultures were treated with DEX-containing media (10μM). Each condition had a set of untreated cultures and a set of DEX-treated cultures. Cultures were incubated for a 24-hour period. Then, cells and cell culture media were collected. To study metabolic enzyme activity, cell lysate was obtained. For cytokine analysis, cell culture media was used.
Cytokine Profile
Individual enzyme-linked immunosorbent assays (ELISA) were performed to assess the production of the cytokines TNF-α and IL-10. Cytokine production was measured using cell culture supernatant. ELISAs were performed according to manufacturer protocols (BD Biosciences).
Phosphofructokinase-1 Assay
Cell lysate was obtained using the manufacturer protocol of the PFK1 assay kit (abcam). Briefly, cells were washed with cold PBS. They were then resuspended in 100μL of cold assay buffer. Cells were homogenized using vigorous pipetting, and then incubated on ice for 10 minutes. The samples were then centrifuged at 12000g for 5 minutes at 4°C. The supernatant was then collected and then used in the PFK1 activity assay (abcam).
Briefly, the cell lysate was incubated in a reaction well for a one-hour time period. The absorbance at a certain time point reflected the concentration of NADH in the reaction well. NADH in the reaction well is produced through a reaction from the assay kit’s enzyme mix, substrate, and ADP produced by PFK1 in the cell lysate. The PFK1 in the cell lysate converts fructose 6-phosphate and ATP into fructose 1,6-diphosphate and ADP; the ADP from PFK1 reactions combines with NAD+ to form AMP and NADH. The concentration of NADH was used later for calculating the enzymatic activity.
Two time points that recorded maximum changes in NADH concentration during the assay were selected. The Vmax was calculated using the change in NADH concentration during these time points. One unit of PFK1 activity is equivalent to the amount of PFK1 that will generate 1 μmol of NADH per minute. Triplicate samples were used from each culture for cytokine and enzyme activity assays.
Flow Cytometry
A multicolor flow cytometry microglia cell panel developed in our lab was used to identify microglia and their immunophenotype (25–27). Our staining method identified the following microglial markers: CD11bc, CD45, CD32, CD163, CD200R, RT1b. Aliquots of microglia cells were moved to BD Trucount tubes (BD Biosciences) to determine absolute cell counts. Data for microglial cells was acquired by an LSR-II Flow Cytometer (Beckman Coulter). Traditional flow cytometry analysis was performed with FlowJo vr10.6.1 (FlowJo, LLC; Ashland, Oregon).
Statistical Analysis
GraphPad Prism software version 9 (GraphPad Software, Inc., San Diego, CA) was used for statistical analysis. Most data were analyzed using two-way ANOVA with Šídák’s multiple comparisons test and unpaired t-test. The data in the results section is presented as the mean difference ± standard error, unless deemed otherwise. A p-value less than 0.05 was deemed statistically significant. Prism software tests for normality. Cytokine concentrations are displayed as picograms/milliliter (pg/mL).
Results
Cytokine Profile
As commonly reported, LPS and Poly I:C stimulation increased the overall expression of TNF-α in microglia (naïve [n = 3] vs LPS [n = 3] mean difference = 615.9 pg/mL ± 15.96, p < 0.0001; naïve vs Poly I:C [n = 3] mean difference = 84.04 ± 15.96, p = 0.0005). The DAMP mediator did not induce a detectable difference in TNF-α (DAMP n = 3; mean difference versus naive = 2.555 ± 15.96, p>0.9999). DEX treatment reduced TNF-α expression in LPS-stimulated microglia (n = 3; mean difference = 121.5 ± 15.96 pg/mL; p<0.0001; Figure 1). In naïve, Poly I:C-stimulated, and DAMP-stimulated microglia, there were no differences in TNF expression after DEX treatment (naïve mean difference = 1.705 ± 15.96 pg/mL; Poly I:C = 6.000 ± 15.96, p = 0.9883; DAMP = 0.1985 ± 15.96) (Figure 1). Overall, DEX treatment significantly reduced TNF-α expression in cultures (2-way ANOVA, difference between predicted means = 28.94 ± 8.491, p<0.0001).
Figure 1.
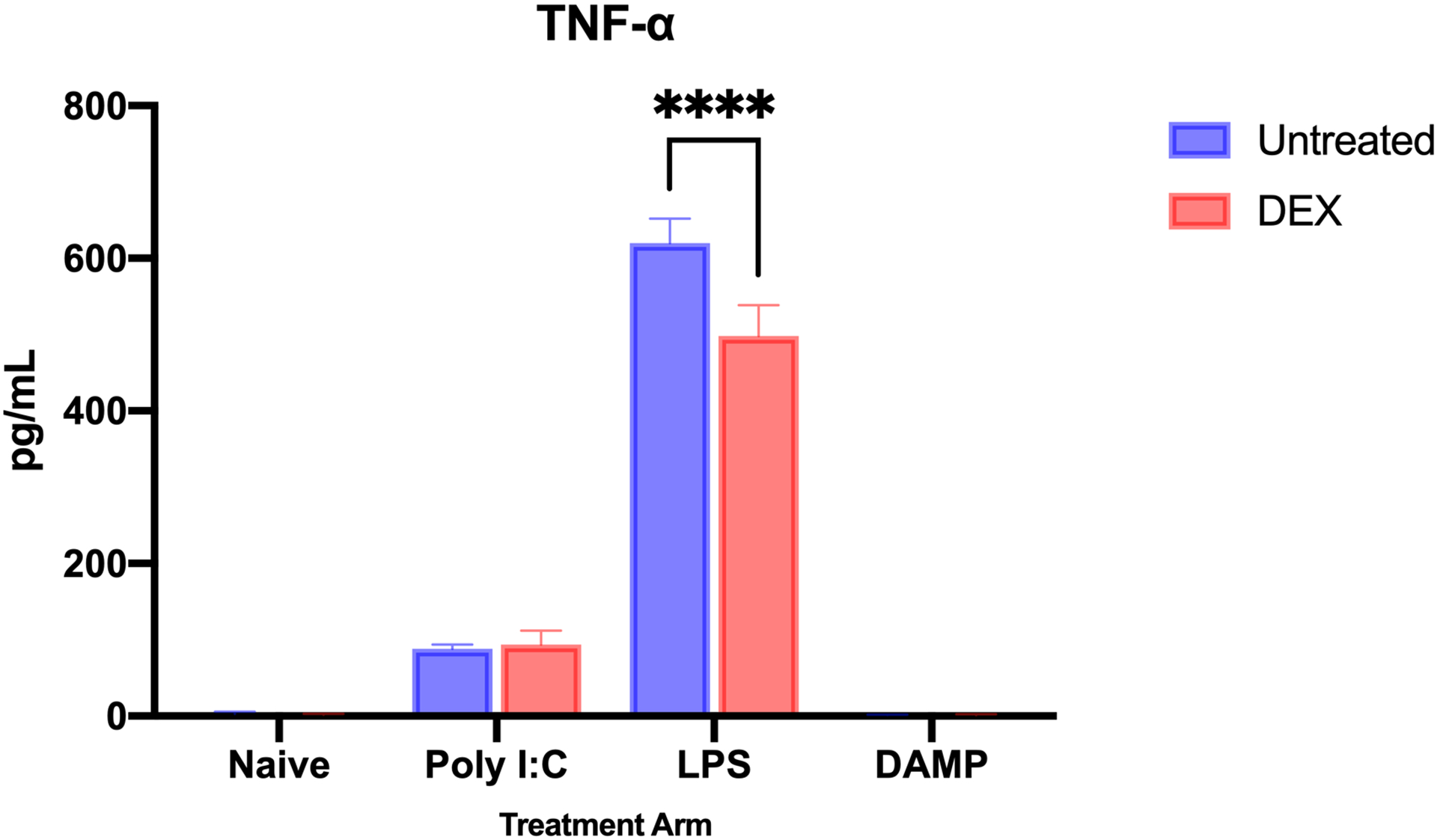
TNF-α production by microglia after inflammatory stimulation, without and with dexmedetomidine treatment. LPS and Poly I:C stimulation increased the overall expression of TNF-α in microglia (naive vs LPS mean difference = 615.9 pg/mL ± 15.96, p < 0.0001; naive vs Poly I:C mean difference = 84.04 ± 15.96, p = 0.0005). The DAMP mediator did not induce a detectable difference in TNF-α. DEX treatment reduced TNF-α expression in LPS-stimulated microglia (mean difference = 121.5 ± 15.96 pg/mL; p<0.0001). In naïve, Poly I:C-stimulated, and DAMP-stimulated microglia, there were no differences in TNF expression after DEX treatment. Overall, DEX treatment significantly reduced TNF-α expression in cultures (2-way ANOVA, difference between predicted means = 28.94 ± 8.491, p<0.0001). (n = 3 for each treatment group; **** - p<0.0001)
When compared to naïve microglia without treatment, IL-10 expression was greater overall in LPS-stimulated (n = 3; mean difference = 156.8 ± 39.00 pg/mL, p = 0.0002). DAMP and Poly I:C stimulation both decreased IL-10 production, but neither decrease was statistically significant (naïve [n = 3] vs DAMP [n = 3] mean difference = 79.27 ± 29.00, p = 0.0851; vs Poly I:C [n = 3] = 56.07 ± 29.00, p = 0.1984). When analyzing with an unpaired t-test, DEX treatment did significantly reduce the expression of IL-10 in LPS-stimulated (mean difference = 74.93 ± 12.50, p = 0.0039) and Poly I:C-stimulated microglia (mean difference = 23.27 ± 6.405, p = 0.0221). This treatment effect was not seen in naïve (51.80 ± 52.93, p = 0.3831) or DAMP-stimulated microglia (7.333 ± 19.10, p = 0.7205) (Figure 2). Overall, DEX treatment significantly reduced IL-10 expression in cultures (2-way ANOVA, difference between predicted means = 39.33 ± 14.50, p<0.0001).
Figure 2.
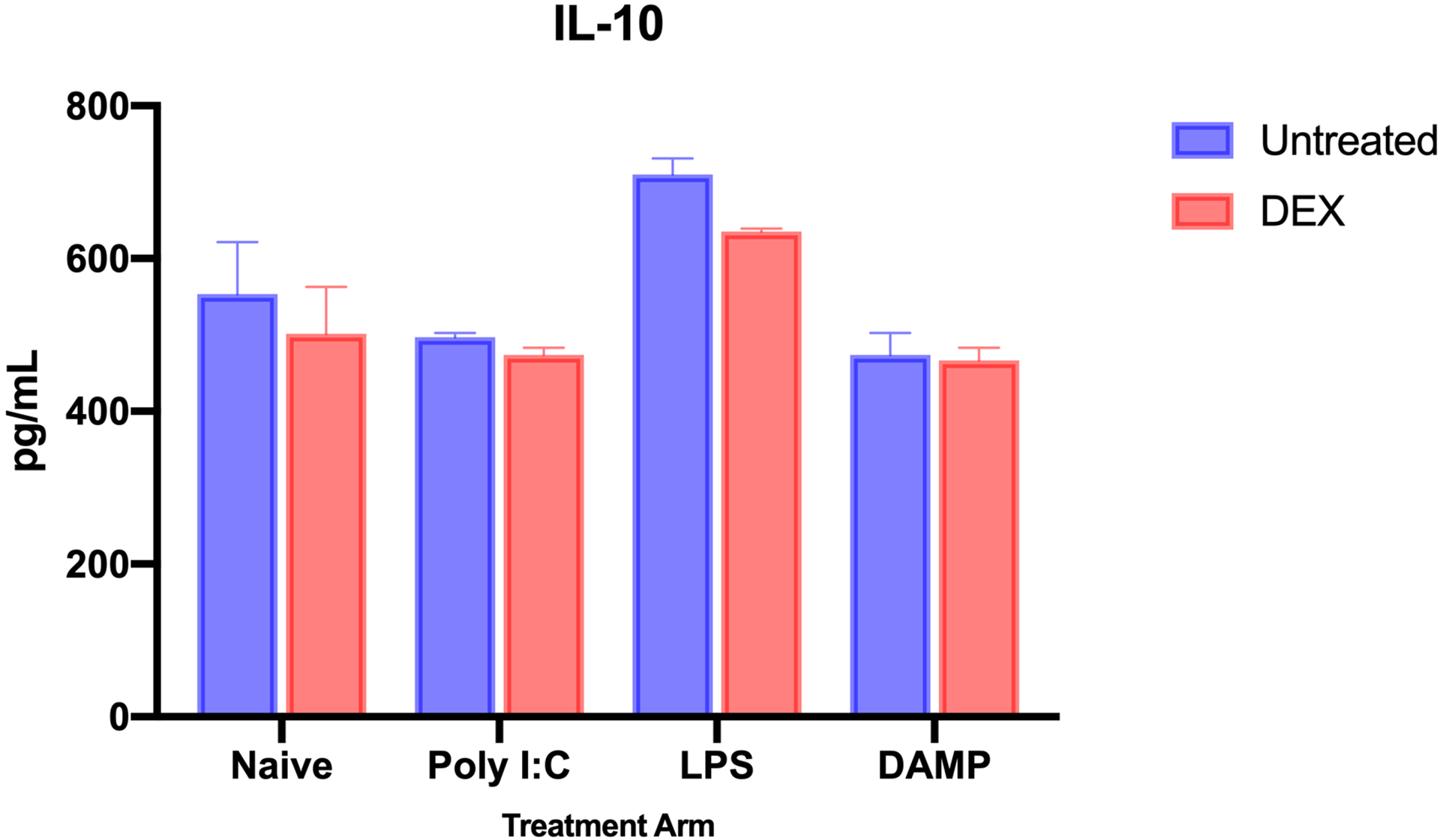
IL-10 production by microglia after inflammatory stimulation, without and with dexmedetomidine treatment. IL-10 expression increased after LPS stimulation (Naïve vs LPS mean difference = 156.8 ± 39.00 pg/mL, p = 0.0002). When using a t-test to analyze DEX treatment with certain inflammatory markers, DEX significantly decreased IL-10 expression in LPS-stimulated (mean difference = 74.93 ± 12.50, p = 0.0039) and Poly I:C-stimulated microglia (mean difference = 23.27 ± 6.405, p = 0.0221). There was no significant treatment effect from DEX observed in naïve microglia. Overall, DEX treatment significantly reduced IL-10 expression in cultures (2-way ANOVA, difference between predicted means = 39.33 ± 14.50, p<0.0001). (n = 3 for each treatment group; **** - p<0.0001)
Metabolic Profile
To study the metabolic profile of microglia, the enzymatic activity of phosphofructokinase-1 (PFK1) was measured in cell lysate. PFK1 is the second rate-limiting enzyme in glycolysis and is considered an important regulator of glycolytic activity. There was a broad treatment effect of DEX on glycolytic enzyme activity. PFK1 activity in all microglia with DEX treatment was significantly lower versus microglia without DEX treatment (mean difference = 9.126 ± 3.254 units/mL, p = 0.0127). In DAMP-stimulated microglia, DEX treatment reduced PFK1 enzyme activity (mean difference = 18.79 ± 6.508 units/mL; p = 0.0421). DEX treatment did not statistically reduce PFK1 activity in LPS-stimulated (5.701 ± 6.508, p = 0.8651) and Poly I:C-stimulated microglia (8.091 ± 6.508, p = 0.6516) (Figure 3)
Figure 3.
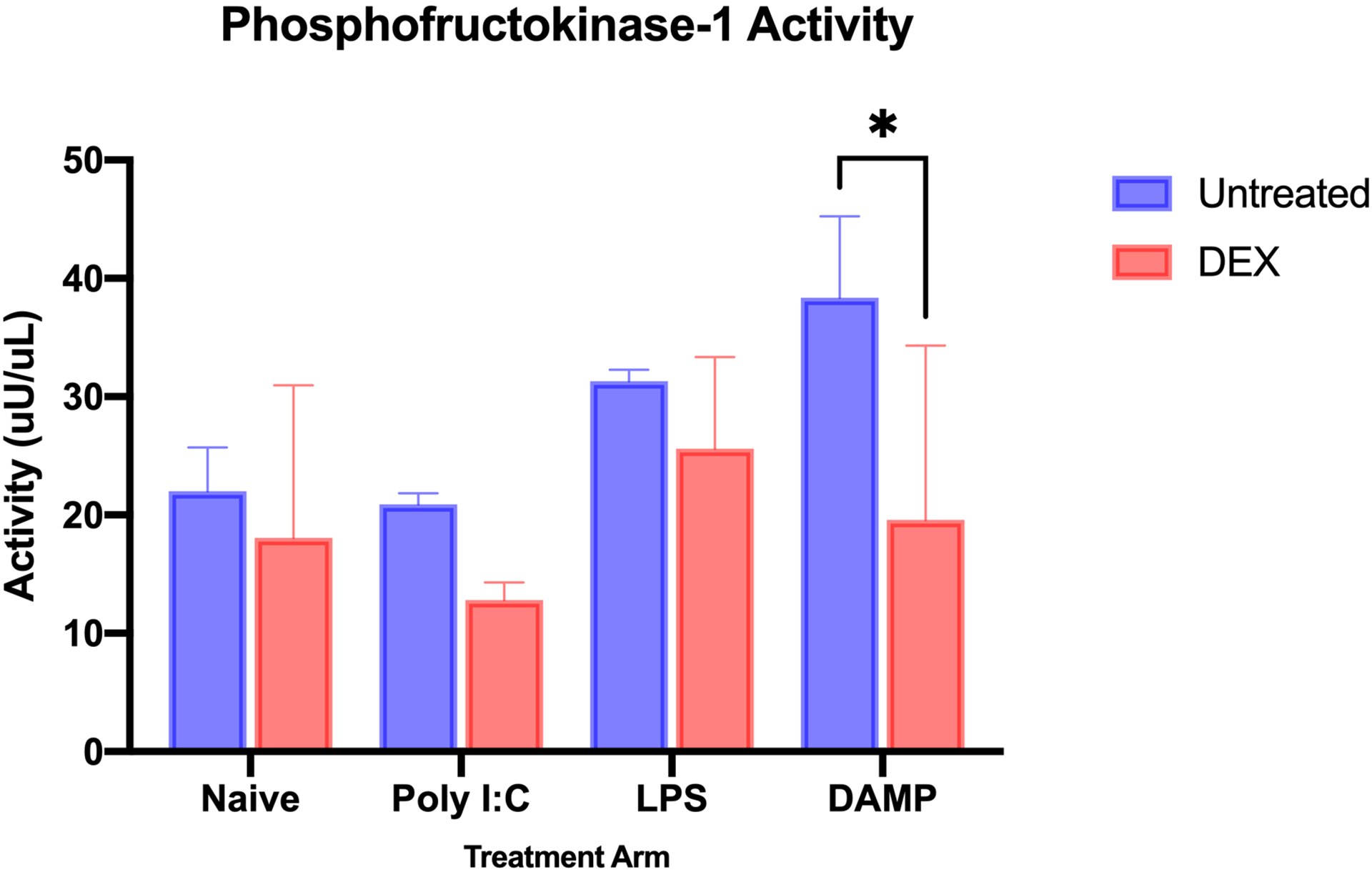
Phosphofructokinase-1 (PFK1) activity in microglia after inflammatory stimulation, without and with dexmedetomidine treatment. DEX treatment overall reduced PFK1 activity (mean difference = 9.126 ± 3.254 units/mL, p = 0.0127). Specifically in the DAMP-stimulated group, there was a significant reduction in PFK1 activity (mean difference = 18.79 ± 6.508 units/mL; p = 0.0421). (n = 3 for each treatment group; * - p<0.05)
Immunophenotype
To characterize potential effect of DEX on the immunophenotype of microglia, we used flow cytometry to measure the expression of the following microglial surface markers: CD11bc, CD32, CD45, CD163, CD200R, RT1b. As expected, all microglia expressed a high degree of CD11bc. Microglia were further defined into the following sub-populations based on their CD45 expression: CD11bc+CD45low, CD11bc+CD45mid, CD11bc+CD45high.
Most changes in CD11b+CD45low cells were in CD32, CD163, and RT1b expression (Figure 4A). LPS-stimulation decreased CD200R expression in comparison to naïve cells (MFI Naïve = 5803, LPS = 4756); DEX treatment did increase CD200R expression in this sub-population after LPS stimulation (MFI LPS = 4756, LPS + DEX treatment = 5167). LPS stimulation increased CD32 (MFI Naïve = 18646, LPS = 28104), and DEX treatment increased CD32 expression in naive and LPS-stimulated cells (Naïve + DEX treatment = 22519, LPS + DEX treatment = 31937). There was a notable reduction in CD32 expression after DAMP stimulation when compared to naive cells, without and with DEX treatment (MF DAMP = 12446, DAMP + DEX = 13205). CD163 expression increased after DEX treatment in naive and LPS-stimulated cells (MFI LPS = 465, LPS + DEX. = 525). RT1b expression decreased with either inflammatory stimulus in comparison to naïve cells (MFI Naïve = 3119, LPS = 1486, DAMP = 2155), while DEX treatment after exposure to an inflammatory stimulus increased RT1b expression (MFI LPS + DEX = 1670, DAMP + DEX = 2393). A reverse effect was seen in naïve cells treated with DEX, as RT1b expression increased (MFI Naïve = 3119, Naïve + DEX = 2101).
Figure 4.
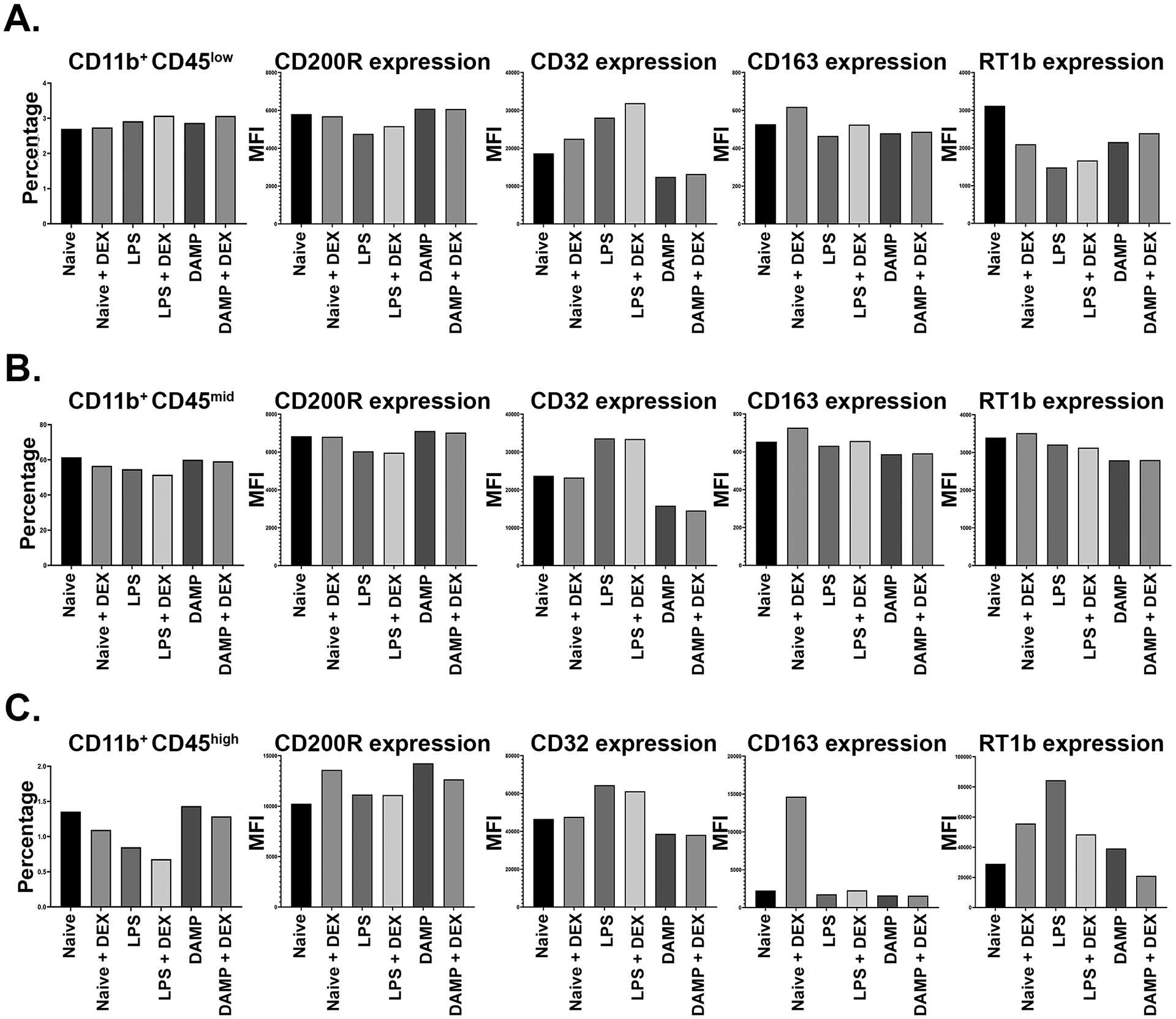
Immunophenotype of microglia in settings of inflammation, without and with dexmedetomidine treatment. Represented in each chart are the percentages of CD11+CD45low (A), CD11+CD45mid (B), and CD11+CD45high (C) cells and mean fluorescent intensity (MFI) of CD200R, CD32, CD163, and RT1b, respectively, in each treatment group.
Various changes in surface marker expression were seen in CD11b+CD45mid cells. CD200R decreased with LPS stimulation (MFI Naïve = 6834, LPS = 6043). When compared to naive cells, CD32 increased after LPS stimulation but decreased after DAMP stimulation (MFI Naïve = 23739, LPS = 33625, DAMP = 15815). CD163 expression decreased in LPS and DAMP stimulated microglia in comparison to naive microglia (MFI Naïve = 654, LPS = 632, DAMP = 588). DEX increased CD163 expression in naive and LPS-stimulated microglia (MFI LPS + DEX = 657). RT1b expression decreased in LPS- and DAMP-stimulated microglia in comparison to naive cells (MFI Naïve = 3395, LPS = 3128, DAMP = 2793), but minimal change was noted with DEX treatment in this sub-population for RT1b expression.
In naive CD11b+CD45high cells (Figure 4C), DEX treatment increased CD163 expression (MFI Naïve = 2250, Naïve + DEX = 14664); this effect was not seen with CD163 in cells with an inflammatory stimulus. DEX also increased CD200R in naive cells in this sub-population (MFI Naïve = 10252, Naïve + DEX = 13603). LPS-stimulation increased CD32 expression in this subpopulation (MFI Naïve = 46575, LPS = 64467), while DAMP stimulation decreased it (DAMP = 38680). RT1b increased markedly after LPS-stimulation. DEX treatment after LPS-stimulation in CD11b+CD45high cells decreased RT1b expression (MFI LPS = 84474, LPS + DEX = 48570). A similar effect on RT1b was seen in cells stimulated by DAMP, without and with DEX treatment (MFI DAMP = 39151, DAMP + DEX = 21071). Of note, the opposite was seen in naïve cultures in this sub-population, as DEX treatment increased RT1b expression (MFI Naïve = 28937, Naïve + DEX = 55747).
Discussion
In this study, we isolated primary rat microglia and examined parts of their inflammatory profile in vitro in response to LPS, Poly I:C, and DAMP: cytokine production, metabolism, immunophenotype. We also studied the effects of dexmedetomidine on microglia to determine if dexmedetomidine may present an alternative therapy to attenuate their neuroinflammatory response (Figure 6 – schematic). In this series of experiments, we found that dexmedetomidine attenuated the release of TNF-α (Figure 1) and IL-10 (Figure 2). Dexmedetomidine decreased the activity level of PFK-1, a rate-limiting enzyme in glycolysis, in microglia, particularly after stimulation with TBI DAMP (Figure 3). Dexmedetomidine had some effects on the microglial immunophenotype in different settings of inflammation (Figure 4).
In neurologic injury, microglia are a central figure in propagating the neuroinflammatory response. As the resident phagocytic cell of the CNS, they can clear damaged cells and promote tissue repair and recovery. However, an aberrant response from microglia can lead to unnecessary tissue damage. Microglia with this kind of response in TBI and CNS trauma typically have a pro-inflammatory profile that persists for an extended period of time, extending days, weeks, and years after injury (28, 29). PET imaging studies have indicated that microglial activation can persist for up to years after traumatic brain injury. This chronic microglial activation appears to localize more to the thalamus, and the degree of activation correlates with the severity of cognitive dysfunction after TBI (29). Thus, studying microglial activation remains important to understand how neuroinflammation can persist and to identify therapeutic targets.
Inflamed microglia release cytokines like TNF-α, IL-1ß, and IFN-γ to invoke a downstream response to neurologic injury from other microglia and neighboring immune cells. TNF-α induces cell death in neurons and microglia (6, 9). TNF-α induces neuronal cell death (30, 31) and perpetrates inflammation. Thus, targeting TNF-α secretion by pro-inflammatory microglia may attenuate detrimental effects. Chio et al found that treatment of rodents in an experimental TBI model with etanercept, a TNF-α inhibitor, decreased the number of TNF-α positive microglia in the area of injury. Etanercept treatment also decreased the size of the ischemic area due to neurologic injury, improved motor function testing, and decreased neurological severity scores (9).
IL-10 is a primarily anti-inflammatory cytokine seen in immune responses to neurologic injury. In a murine model of intracranial hemorrhage, IL-10 is primarily secreted by microglia; a deficiency of IL-10 results in a delayed clearance of ICH hematoma in mice (8). IL-10 from astrocytes may modulate the activation phenotype in the setting of neurologic injury (32). Interestingly, our results regarding the effect of dexmedetomidine on IL-10 secretion from microglia contrast previous studies (33, 34). In our study, dexmedetomidine treatment decreased the production of IL-10 from microglia (Figure 2). It is possible that dexmedetomidine may attenuate overall microglial activation, including the anti-inflammatory profile. As discussed below, dexmedetomidine treatment may also induce metabolic changes that cause microglia to use their substrates for other purposes in response to inflammation.
In our study, we found that overall dexmedetomidine treatment decreased TNF-α production in microglia, and more specifically in LPS-stimulated microglia (Figure 1). In comparison, Qiu et al found a similar treatment effect of dexmedetomidine in BV-2 microglial cells. Dexmedetomidine treatment of LPS-stimulated BV-2 microglia decreased their secretion of TNF-α (33).
Damage-associated molecular patterns (DAMP) are mediators released from damaged or injured tissue; examples of DAMP mediators include factors released from the nucleus like DNA and the cytosol and mitochondria like ATP. These mediators activate immune cells. In CNS injury, DAMPs stimulate microglia (35). For example, IL-33 released in TBI-related cell injury recruits microglia to the site of injury (36). ATP stimulates the construction and activation of the NLRP3 inflammasome in microglia (37). Heme is another mediator that can induce pro-inflammatory responses from microglia; heme in the extracellular environment can increase TNF-α expression in primary microglia (38). These are a few examples of the importance of considering DAMP in altering the microglia profile and response. While LPS is the classic mediator to use for in vitro studies, it may be equally important to evaluate factors that more accurately represent sterile inflammation seen in neurologic pathologies like TBI. Admittedly, one of the limitations of our method of evaluating DAMP is that the solution we developed may have a high level of heterogeneity and variability. We do feel that that is an important factor to consider in studying microglia activation, as in the clinical setting of TBI, it is more likely a multitude of factors that influence microglial activation as opposed to individual mediators.
Recently, immunometabolism has grown as a prominent field in studying immune cell response, and in the context of neuroinflammation, it offers a different perspective from which to view the microglial response. Microglia are capable of utilizing multiple different types of substrates to produce energy (39, 40). In their surveillance state, microglia utilize glucose in oxidative phosphorylation to produce ATP. After activation, particularly to a pro-inflammatory profile, microglia produce most of their energy through glycolysis (41). The activity of glycolytic enzymes will increase alongside their expression of glucose transporters on the cell surface (42, 43). Inflammatory stimulation of microglia can upregulate the PFKB3, an allosteric activator of PFK-1. PFKB3 augments aerobic glycolysis, which is a feature seen in inflammatory cells shifting their metabolic profile to rapidly increase their ATP supply from glucose. (44). This shift allows microglia to also use metabolic substrates for other purposes, such as free radical production (39, 42). Ultimately, the metabolic profile of a microglia can determine how it responds to an inflammatory stimulus, and presents a potential therapeutic target.
In our study, dexmedetomidine treatment downregulated the activity of rate-limiting enzyme of glycolysis, 6-phosphofructokinase overall. More specifically, dexmedetomidine downregulated PFK-1 in microglia stimulated by TBI DAMP (Figure 3). PFK1 is an important rate-limiting enzyme in glycolysis, thus it is an important regulator of energy use. As one of the irreversible reactions of glycolysis, PFK1’s activity dictates how an energy substrate is utilized. One of the limitations of our study is that our evaluation of microglial metabolism was limited to only this enzyme; however, this does provide some potential directions going forward with further microglial metabolic studies. It would be worth considering an evaluation of lactate production by microglia. Lactate is an important end-product of glycolysis and anaerobic metabolism. Inflamed microglia relying primarily on glycolysis may produce an excess of lactate, which could lead to further disruption in the cell microenvironment of neurologic injury.
Microglia express a variety of surface markers that can be measured by flow cytometry. These markers give microglia an immunophenotype that changes in response to certain stimuli, such as markers of inflammation or neurologic injury. Certain surface markers are reported to be reflective of pro-inflammatory or anti-inflammatory phenotypes. We have previously established that the microglial immunophenotype after a controlled cortical impact injury in rats can be studied using flow cytometry (25–27).
Classically, inflamed microglia have been thought to exist in binary phenotypes: pro-inflammatory or anti-inflammatory. Given the heterogeneity of pathologies like traumatic brain injury and the complexity of the neuroinflammation microenvironment, the microglial immunophenotype more likely exists on a spectrum. We have previously characterized surface marker expression of microglia (26). By existing on a phenotypic spectrum, activated microglia may have variations in their response that encompass elements that are both pro- and anti-inflammatory. By doing this, it allows microglia to simultaneously perform pro-inflammatory tasks, like recruit more immune cells with certain cytokine factors and clear dead cell debris with phagocytosis, and anti-inflammatory tasks, like promote tissue repair and regeneration. Too much of one activity may lead to excessive tissue damage or delay neurologic repair and recovery.
Markers like CD200R and CD163 are important to consider in microglia, as they can decrease tissue damage in the setting of inflammation and modulate cytokine release. RT1b is important for antigen presenting activity. In the setting of neurologic injury, changes to the microglia immunophenotype reflect changes in their activity (26). As a sedative administered in the ICU and for TBI patients, some of our data suggests that there may be some immunophenotype changes to certain sub-populations of microglia with dexmedetomidine treatment.
Clinically, dexmedetomidine is a sedative given intravenously predominantly in intensive care units. DEX sedates patients without affecting respiratory rate, oxygen saturation, or arterial carbon dioxide (45). Common side effects include bradycardia and hypotension. In the bloodstream, DEX is mostly protein-bound, and it has the ability to cross the blood-brain barrier. It has an elimination half-life of 2–3 hours (13). DEX acts on presynaptic receptors in the sympathetic nervous system to inhibit norepinephrine release; it also hyperpolarizes central postsynaptic receptors to create a negative feedback loop which further diminishes sympathetic outflow (46). For patients in a neurological ICU, sedation via DEX is appealing. Compared to other sedatives that may act on GABA-signaling pathways, DEX allows patients to be more easily aroused and cooperative with a neurologic exam (46–48). It may even reduce opioid requirements in the critically ill patient (48). Dexmedetomidine’s action on the sympathetic nervous system has clinical benefit, as it can act as a treatment for paroxysmal autonomic instability. Paroxysmal autonomic instability is associated with traumatic brain injury, and can present as the sudden onset of fever, tachycardia, tachypnea, and diaphoresis (49).
Our study does have some limitations. We only included rat microglia in this work, so there may be species-specific differences. Our study also only used male rats as a source of primary microglia. It is plausible that there are some sex-based differences in the microglia response to different inflammatory mediators and dexmedetomidine in vitro. However, we conducted a recent study of sex-based differences in our experimental model of TBI in rodents; one feature of neuroinflammation we studied was the microglia profile with flow cytometry. There were few differences in the microglia profile after TBI between sexes (18). We also conducted an extensive literature review as an adjunct to this study; sex-based differences in TBI are variable and may depend on the study population in clinical studies or the TBI model in basic science studies. Our studies were performed in vitro, and it is worth considering that microglia in vivo may behave differently as a part of a more complicated response to tissue or organ injury. As stated previously, our metabolic studies were limited to a single glycolytic enzyme; similarly, our profile of cytokine secretion could be expanded as well.
Conclusions
In this study, we demonstrate that dexmedetomidine may modify the microglial response by attenuating inflammatory cytokine expression, shifting their metabolic profile, or inducing further changes to their immunophenotype. Based on our results and prior studies, dexmedetomidine potentially attenuates the progression of neuroinflammation through its effect on microglia (Figure 5).
Figure 5.
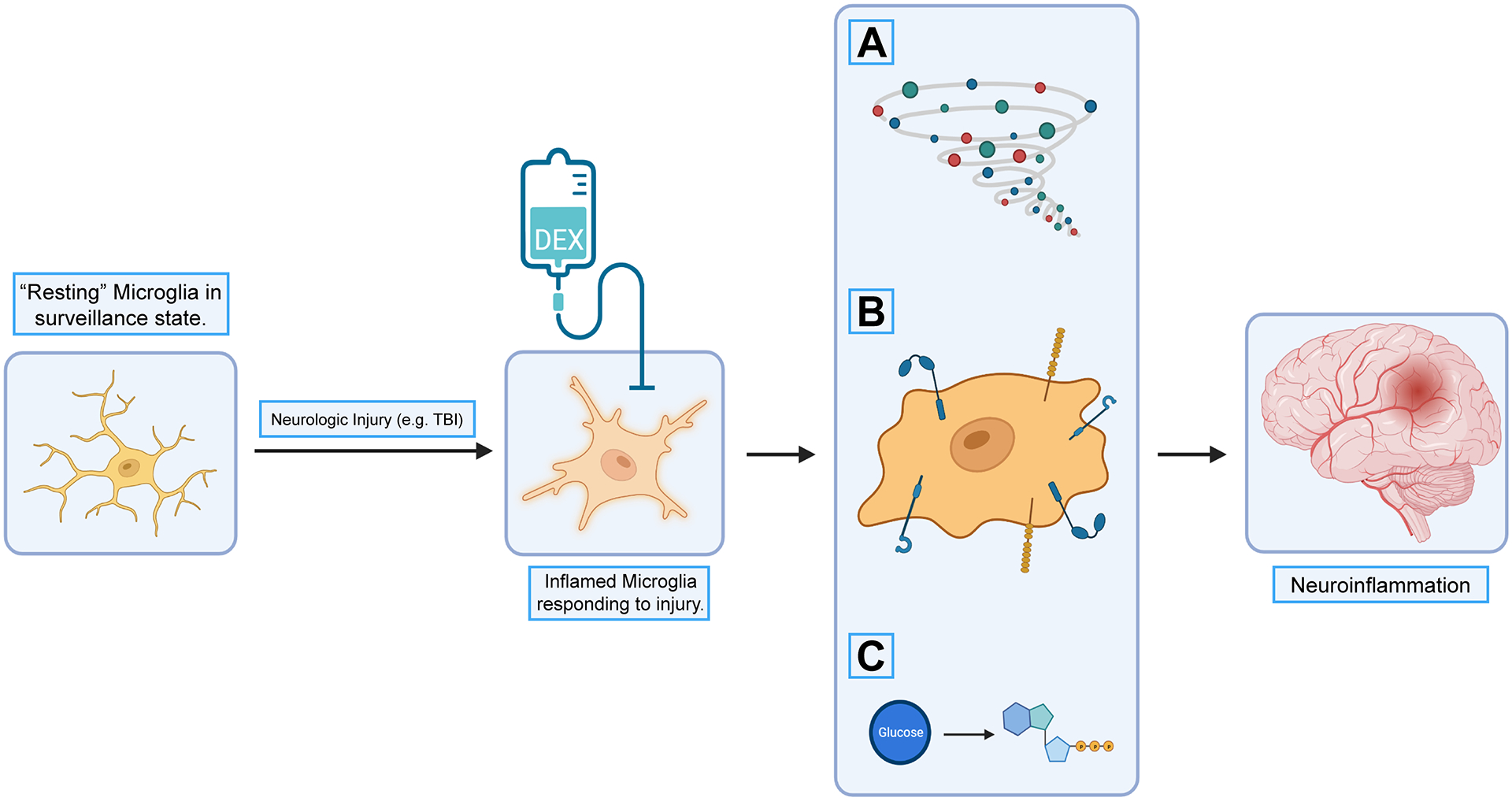
Potential downstream effects by targeting microglia with dexmedetomidine and other pharmacologic agents. “Resting” microglia in a surveillance state are stimulated to an inflammatory profile by markers of neurologic injury, like traumatic brain injury. Their response includes release of inflammatory cytokines (A), changes in their immunophenotype (B), and metabolic shifts that alter their consumption of glucose and other energy substrates (C). All of these responses affect the ensuing neuroinflammation after neurologic injury. Based on our data and previous studies, dexmedetomidine treatment in the setting of neurologic injury may alter the inflammatory profile of microglia, which could attenuate neuroinflammation.
Funding
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number 2T32GM008792. MCS was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number 2T32GM008792.
Disclosure of Potential Conflict of Interests
CSC has received research support from Athersys, CBR Systems, Hope Bio, Biostage, and is on the Scientific Advisory Board of Cellvation and CBR. SDO has received research support from Athersys, CBR Systems, Hope Bio, Biostage, Cellvation, and Generate Life Sciences. MCS received support from the American College of Surgeons for travel to present part of this work at the Committee on Trauma Annual Meeting 2022.
Footnotes
The following manuscript complies with all instructions provided to the authors. Authorship requirements have been met. All authors approved the final manuscript. This manuscript has not been published in another journal and is not under consideration by another journal. This study conforms to the COPE and UT Health IRB guidelines. Care for animals was in accordance with ARRIVE guidelines.
References
- 1.Scott MC, Bedi SS, Olson SD, Sears CM, Cox CS. Microglia as therapeutic targets after neurological injury: strategy for cell therapy. Expert Opin Ther Targets. 2021;25(5):365–80. Epub 2021/05/25. doi: 10.1080/14728222.2021.1934447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox CS Jr., Cellular therapy for traumatic neurological injury. Pediatr Res. 2018;83(1–2):325–32. Epub 2017/10/07. doi: 10.1038/pr.2017.253. [DOI] [PubMed] [Google Scholar]
- 3.Dubbelaar ML, Kracht L, Eggen BJL, Boddeke E. The Kaleidoscope of Microglial Phenotypes. Front Immunol. 2018;9:1753. Epub 2018/08/16. doi: 10.3389/fimmu.2018.01753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. Epub 2011/04/30. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 5.Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflammation. 2014;11:98. Epub 2014/06/04. doi: 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87(1):10–20. Epub 2011/10/26. doi: 10.1016/j.brainresbull.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lobo-Silva D, Carriche GM, Castro AG, Roque S, Saraiva M. Interferon-beta regulates the production of IL-10 by toll-like receptor-activated microglia. Glia. 2017;65(9):1439–51. Epub 2017/06/16. doi: 10.1002/glia.23172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Q, Lan X, Han X, Durham F, Wan J, Weiland A, Koehler RC, Wang J. Microglia-derived interleukin-10 accelerates post-intracerebral hemorrhage hematoma clearance by regulating CD36. Brain Behav Immun. 2021;94:437–57. Epub 2021/02/16. doi: 10.1016/j.bbi.2021.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chio CC, Chang CH, Wang CC, Cheong CU, Chao CM, Cheng BC, Yang CZ, Chang CP. Etanercept attenuates traumatic brain injury in rats by reducing early microglial expression of tumor necrosis factor-alpha. BMC Neurosci. 2013;14:33. Epub 2013/03/19. doi: 10.1186/1471-2202-14-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernier LP, York EM, MacVicar BA. Immunometabolism in the Brain: How Metabolism Shapes Microglial Function. Trends Neurosci. 2020;43(11):854–69. Epub 2020/09/23. doi: 10.1016/j.tins.2020.08.008. [DOI] [PubMed] [Google Scholar]
- 11.O’Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16(9):553–65. Epub 2016/07/12. doi: 10.1038/nri.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gyoneva S, Traynelis SF. Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem. 2013;288(21):15291–302. Epub 2013/04/04. doi: 10.1074/jbc.M113.458901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weerink MAS, Struys M, Hannivoort LN, Barends CRM, Absalom AR, Colin P. Clinical Pharmacokinetics and Pharmacodynamics of Dexmedetomidine. Clin Pharmacokinet. 2017;56(8):893–913. Epub 2017/01/21. doi: 10.1007/s40262-017-0507-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Afonso J, Reis F. Dexmedetomidine: current role in anesthesia and intensive care. Rev Bras Anestesiol. 2012;62(1):118–33. Epub 2012/01/18. doi: 10.1016/S0034-7094(12)70110-1. [DOI] [PubMed] [Google Scholar]
- 15.Unchiti K, Leurcharusmee P, Samerchua A, Pipanmekaporn T, Chattipakorn N, Chattipakorn SC. The potential role of dexmedetomidine on neuroprotection and its possible mechanisms: Evidence from in vitro and in vivo studies. Eur J Neurosci. 2021;54(9):7006–47. Epub 2021/09/26. doi: 10.1111/ejn.15474. [DOI] [PubMed] [Google Scholar]
- 16.Li F, Wang X, Zhang Z, Zhang X, Gao P. Dexmedetomidine Attenuates Neuroinflammatory-Induced Apoptosis after Traumatic Brain Injury via Nrf2 signaling pathway. Ann Clin Transl Neurol. 2019;6(9):1825–35. Epub 2019/09/04. doi: 10.1002/acn3.50878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng X, Ma W, Zhu J, Jiao W, Wang Y. Dexmedetomidine alleviates early brain injury following traumatic brain injury by inhibiting autophagy and neuroinflammation through the ROS/Nrf2 signaling pathway. Mol Med Rep. 2021;24(3). Epub 2021/07/20. doi: 10.3892/mmr.2021.12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott MC, Prabhakara KS, Walters AJ, Olson SD, Cox CS Jr., Determining Sex-Based Differences in Inflammatory Response in an Experimental Traumatic Brain Injury Model. Front Immunol. 2022;13:753570. Epub 2022/03/01. doi: 10.3389/fimmu.2022.753570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tamashiro TT, Dalgard CL, Byrnes KR. Primary microglia isolation from mixed glial cell cultures of neonatal rat brain tissue. J Vis Exp. 2012(66):e3814. Epub 2012/08/30. doi: 10.3791/3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bohlen CJ, Bennett FC, Bennett ML. Isolation and Culture of Microglia. Curr Protoc Immunol. 2019;125(1):e70. Epub 2018/11/11. doi: 10.1002/cpim.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prabhakara KS, Kota DJ, Jones GH, Srivastava AK, Cox CS Jr., Olson SD. Teriflunomide Modulates Vascular Permeability and Microglial Activation after Experimental Traumatic Brain Injury. Mol Ther. 2018;26(9):2152–62. Epub 2018/07/25. doi: 10.1016/j.ymthe.2018.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giridharan VV, Scaini G, Colpo GD, Doifode T, Pinjari OF, Teixeira AL, Petronilho F, Macedo D, Quevedo J, Barichello T. Clozapine Prevents Poly (I:C) Induced Inflammation by Modulating NLRP3 Pathway in Microglial Cells. Cells. 2020;9(3). Epub 2020/03/04. doi: 10.3390/cells9030577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh M, Xu Y, Pearse DD. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J Neuroinflammation. 2016;13:9. Epub 2016/01/14. doi: 10.1186/s12974-015-0463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao R, Ying M, Gu S, Yin W, Li Y, Yuan H, Fang S, Li M. Cysteinyl Leukotriene Receptor 2 is Involved in Inflammation and Neuronal Damage by Mediating Microglia M1/M2 Polarization through NF-kappaB Pathway. Neuroscience. 2019;422:99–118. Epub 2019/11/15. doi: 10.1016/j.neuroscience.2019.10.048. [DOI] [PubMed] [Google Scholar]
- 25.Toledano Furman NE, Prabhakara KS, Bedi S, Cox CS Jr., Olson SD OMIP-041: Optimized multicolor immunofluorescence panel rat microglial staining protocol. Cytometry A. 2018;93(2):182–5. Epub 2017/10/17. doi: 10.1002/cyto.a.23267. [DOI] [PubMed] [Google Scholar]
- 26.Toledano Furman N, Gottlieb A, Prabhakara KS, Bedi S, Caplan HW, Ruppert KA, Srivastava AK, Olson SD, Cox CS Jr., High-resolution and differential analysis of rat microglial markers in traumatic brain injury: conventional flow cytometric and bioinformatics analysis. Sci Rep. 2020;10(1):11991. Epub 2020/07/21. doi: 10.1038/s41598-020-68770-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gottlieb A, Toledano-Furman N, Prabhakara KS, Kumar A, Caplan HW, Bedi S, Cox CS Jr., Olson SD. Time dependent analysis of rat microglial surface markers in traumatic brain injury reveals dynamics of distinct cell subpopulations. Sci Rep. 2022;12(1):6289. Epub 2022/04/17. doi: 10.1038/s41598-022-10419-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caplan HW, Cardenas F, Gudenkauf F, Zelnick P, Xue H, Cox CS, Bedi SS. Spatiotemporal Distribution of Microglia After Traumatic Brain Injury in Male Mice. ASN Neuro. 2020;12:1759091420911770. Epub 2020/03/10. doi: 10.1177/1759091420911770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70(3):374–83. Epub 2011/06/29. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 30.Feng W, Wang Y, Liu ZQ, Zhang X, Han R, Miao YZ, Qin ZH. Microglia activation contributes to quinolinic acid-induced neuronal excitotoxicity through TNF-alpha. Apoptosis. 2017;22(5):696–709. Epub 2017/03/21. doi: 10.1007/s10495-017-1363-5. [DOI] [PubMed] [Google Scholar]
- 31.Bras JP, Bravo J, Freitas J, Barbosa MA, Santos SG, Summavielle T, Almeida MI. TNF-alpha-induced microglia activation requires miR-342: impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 2020;11(6):415. Epub 2020/06/04. doi: 10.1038/s41419-020-2626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Recasens M, Shrivastava K, Almolda B, Gonzalez B, Castellano B. Astrocyte-targeted IL-10 production decreases proliferation and induces a downregulation of activated microglia/macrophages after PPT. Glia. 2019;67(4):741–58. Epub 2018/12/15. doi: 10.1002/glia.23573. [DOI] [PubMed] [Google Scholar]
- 33.Qiu Z, Lu P, Wang K, Zhao X, Li Q, Wen J, Zhang H, Li R, Wei H, Lv Y, Zhang S, Zhang P. Dexmedetomidine Inhibits Neuroinflammation by Altering Microglial M1/M2 Polarization Through MAPK/ERK Pathway. Neurochem Res. 2020;45(2):345–53. Epub 2019/12/12. doi: 10.1007/s11064-019-02922-1. [DOI] [PubMed] [Google Scholar]
- 34.Bao Y, Zhu Y, He G, Ni H, Liu C, Ma L, Zhang L, Shi D. Dexmedetomidine Attenuates Neuroinflammation In LPS-Stimulated BV2 Microglia Cells Through Upregulation Of miR-340. Drug Des Devel Ther. 2019;13:3465–75. Epub 2019/10/22. doi: 10.2147/DDDT.S210511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klegeris A Regulation of neuroimmune processes by damage- and resolution-associated molecular patterns. Neural Regen Res. 2021;16(3):423–9. Epub 2020/09/29. doi: 10.4103/1673-5374.293134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wicher G, Wallenquist U, Lei Y, Enoksson M, Li X, Fuchs B, Abu Hamdeh S, Marklund N, Hillered L, Nilsson G, Forsberg-Nilsson K. Interleukin-33 Promotes Recruitment of Microglia/Macrophages in Response to Traumatic Brain Injury. J Neurotrauma. 2017;34(22):3173–82. Epub 2017/05/12. doi: 10.1089/neu.2016.4900. [DOI] [PubMed] [Google Scholar]
- 37.Gaikwad S, Patel D, Agrawal-Rajput R. CD40 Negatively Regulates ATP-TLR4-Activated Inflammasome in Microglia. Cell Mol Neurobiol. 2017;37(2):351–9. Epub 2016/03/11. doi: 10.1007/s10571-016-0358-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshizaki S, Kijima K, Hara M, Saito T, Tamaru T, Tanaka M, Konno DJ, Nakashima Y, Okada S. Tranexamic acid reduces heme cytotoxicity via the TLR4/TNF axis and ameliorates functional recovery after spinal cord injury. J Neuroinflammation. 2019;16(1):160. Epub 2019/07/31. doi: 10.1186/s12974-019-1536-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Devanney NA, Stewart AN, Gensel JC. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp Neurol. 2020;329:113310. Epub 2020/04/15. doi: 10.1016/j.expneurol.2020.113310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagy AM, Fekete R, Horvath G, Koncsos G, Kriston C, Sebestyen A, Giricz Z, Kornyei Z, Madarasz E, Tretter L. Versatility of microglial bioenergetic machinery under starving conditions. Biochim Biophys Acta Bioenerg. 2018;1859(3):201–14. Epub 2017/12/24. doi: 10.1016/j.bbabio.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 41.Ghosh S, Castillo E, Frias ES, Swanson RA. Bioenergetic regulation of microglia. Glia. 2018;66(6):1200–12. Epub 2017/12/09. doi: 10.1002/glia.23271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gimeno-Bayon J, Lopez-Lopez A, Rodriguez MJ, Mahy N. Glucose pathways adaptation supports acquisition of activated microglia phenotype. J Neurosci Res. 2014;92(6):723–31. Epub 2014/02/11. doi: 10.1002/jnr.23356. [DOI] [PubMed] [Google Scholar]
- 43.Wang L, Pavlou S, Du X, Bhuckory M, Xu H, Chen M. Glucose transporter 1 critically controls microglial activation through facilitating glycolysis. Mol Neurodegener. 2019;14(1):2. Epub 2019/01/13. doi: 10.1186/s13024-019-0305-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holland R, McIntosh AL, Finucane OM, Mela V, Rubio-Araiz A, Timmons G, McCarthy SA, Gun’ko YK, Lynch MA. Inflammatory microglia are glycolytic and iron retentive and typify the microglia in APP/PS1 mice. Brain Behav Immun. 2018;68:183–96. Epub 2017/10/25. doi: 10.1016/j.bbi.2017.10.017. [DOI] [PubMed] [Google Scholar]
- 45.Venn RM, Hell J, Grounds RM. Respiratory effects of dexmedetomidine in the surgical patient requiring intensive care. Crit Care. 2000;4(5):302–8. Epub 2000/11/01. doi: 10.1186/cc712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nelson LE, Lu J, Guo T, Saper CB, Franks NP, Maze M. The alpha2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology. 2003;98(2):428–36. Epub 2003/01/29. doi: 10.1097/00000542-200302000-00024. [DOI] [PubMed] [Google Scholar]
- 47.Shelly MP. Dexmedetomidine: a real innovation or more of the same? Br J Anaesth. 2001;87(5):677–8. Epub 2002/03/07. doi: 10.1093/bja/87.5.677. [DOI] [PubMed] [Google Scholar]
- 48.Venn RM, Grounds RM. Comparison between dexmedetomidine and propofol for sedation in the intensive care unit: patient and clinician perceptions. Br J Anaesth. 2001;87(5):684–90. Epub 2002/03/07. doi: 10.1093/bja/87.5.684. [DOI] [PubMed] [Google Scholar]
- 49.Goddeau RP Jr., Silverman SB, Sims JR. Dexmedetomidine for the treatment of paroxysmal autonomic instability with dystonia. Neurocrit Care. 2007;7(3):217–20. Epub 2007/07/03. doi: 10.1007/s12028-007-0066-0. [DOI] [PubMed] [Google Scholar]