

Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the cause the ongoing pandemic of Coronavirus Disease 2019 (COVID19). One key feature associated with COVID-19 is excessive proinflammatory cytokine production that leads to severe acute respiratory distress syndrome (ARDS). While the cytokine storm induces inflammatory cell death in the host, which type of programmed cell death (PCD) mechanism that occurs in various organs and cells remains elusive. Using an in vitro culture model of polarized human airway epithelium (HAE), we observed that necroptosis, but not apoptosis or pyroptosis, plays an essential role in the damage of the epithelial barrier of polarized HAE infected with SARS-CoV-2. Pharmacological inhibitors of necroptosis, nacrostatin-2 (N2) and necrosulfonamide (NSA), efficiently prevented cell death and epithelial barrier dysfunction caused by SARS-CoV-2 infection. Moreover, the silencing of genes that are involved in necroptosis, RIPK1, RIPK3, and MLKL, ameliorated airway epithelial damage of the polarized HAE infected with SARS-CoV-2. This study, for the first time, confirms that SARS-CoV-2 infection triggers necroptosis that disrupts the barrier function of human airway epithelia in vitro.
Keywords: SARS-CoV-2, human airway epithelium, necroptosis, epithelial barrier function
Introduction
Coronaviruses are a group of positive-sense, single-stranded RNA viruses. A large portion of the viral genome encodes two large open reading frames (orfs), orf1a and orf1b, which are overlapped and encode 16 nonstructural proteins. The remaining genome at the 3’ end encodes structural proteins, nucleoprotein (N), spike glycoprotein (S), envelope glycoprotein (E), and membrane glycoprotein (M). 1,2 Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) belongs to the β-coronavirus genus of the family Coronaviridae. SARS-CoV-2 infection causes Coronavirus Disease 2019 (COVID-19), which has become a worldwide pandemic. The symptoms of COVID-19 vary from mild fever, cough, and myalgia to severe acute respiratory distress syndrome (ARDS). 3–5 Even though it is rapidly transmitted among people, the mortality rate reached 2% early in the pandemic, and the most severe cases were found in immunocompromised patients and patients with underlying conditions. 6
SARS-CoV-2 infection causes diverse symptoms throughout the lung/airways, from mild upper respiratory tract symptoms 7 to severe damage in the lung, 8–11 in particular the alveoli. 12,13. Strikingly, ~80% of the cases of SARS-CoV-2 infection present mild symptoms. 14 During the early pandemic, individuals with asymptomatic or presymptomatic infections accounted for 57% of the cases in the US 15 and contributed to the spread of COVID-19. 16 In these cases, the detection of high viral loads and successful isolation of the virus from early throat swabs suggest the presence of active virus replication in the upper airways, 7 implying the importance of COVID-19 containment at the early stage of infection. While SARS-CoV-2 RNA can be detected in nasal swabs, nasopharyngeal aspirates, and bronchoalveolar lavage fluids throughout the airways, 17–21 how the virus infects epithelial cells at different levels of the respiratory tree and the underlying pathogenesis remain unclear.
Respiratory epithelium is involved in the physical segregation of hosts from a vast array of infectious pathogens. The respiratory epithelial cells also serve as a sensor of infectious agents to trigger inflammatory responses along with cell death by infection. 22 The first line of defense in the airways is predominantly based on mechanical barriers, such as the tight junction between the epithelial cells, the mucus blanket covering the airway epithelium, and the mucociliary clearance executed by the ciliated cells. These features comprise a part of innate immunity. 23 When the respiratory epithelial barrier fails to restrict microbial infections, and inflammation spreads to the interstitial part of the lung, programmed cell death (PCD) is critical for cleaning the infected cells. 22,24. The innate immune system recognizes the pathogen molecules conserved in viruses and engages a rapid response by causing inflammation and eventually PCD. For the infection of most respiratory pathogens, the PCD includes pyroptosis, apoptosis, and necroptosis. 25 Apoptosis is a caspase-3-dependent cell death pathway, and apoptotic cells are rapidly phagocytized and cleared without inflammatory responses. 26 Conversely, pyroptosis and necroptosis are two similar inflammatory PCDs that represent lytic forms of cell death pathways. Pyroptosis requires the activation of caspase-1 and the pore-forming proteins gasdermin D (GSDMD). 27 Necroptosis, which requires the activation of receptor-interacting protein kinase (RIPK) or Z-binding protein 1 (ZBP1), is executed by the mixed lineage kinase domain-like (MLKL) protein 28,29 and is an effective modality for immunogenic PCD. 30 Activated/phosphorylated MLKL undergoes oligomerization and forms pores on the cytoplasmic membrane, which results in osmotic pressure imbalance and membrane rupture that leads to cell death. 26
Studies have demonstrated that the cytokine storm is an important factor in the disease progression of COVID-19, eventually leading to multiple organ failures and death. 31 The mechanism of the severe symptom is that a variety of cytokines, including the tumor necrosis factor α (TNF-α), interferon (IFN)-γ, and interleukin (IL)-1, IL-6, IL-18, are released in a pulse pattern during the pathogen invasion. Along with TNF-α, these proinflammatory cytokines play an essential role in acute phase responses of inflammation. TNF-α and IFN-γ synergistically induce PANoptosis, which is featured by the activation of pyroptosis, apoptosis, and necroptosis, leading to tissue and organ damage in COVID-19 patients. 32,33 Virus-induced apoptosis, necroptosis, and inflammatory response were also observed in the lung of a hACE2 transgenic mouse infection model. 33. SARS-CoV-2 triggered necroptosis was also found in infected monolayer cultured cell types, such as macrophages, with activation of MLKL and RIPK3. 33,34 However, most of these studies were descriptive and carried out in monolayer cultures (dividing cells) or mouse models. How SARS-CoV-2 infection damages human airway epithelia and disrupts the epithelial barrier function is not well understood, particularly in the in vitro culture model of polarized human airway epithelium (HAE), which is composed of multiple functionally distinct cell types. 35
In this study, pseudostratified HAE cultured at an air-liquid interface (ALI) was used to model SARS-CoV-2 infection. These cultures were generated by the differentiation of primary airway epithelial cells isolated from the bronchial airways of normal lung donors without expansion, or the immortalized human airway epithelial cell line CuFi-8, 36 into mucociliary columnar airway epithelium. Our study found that necroptosis was induced, whereas apoptosis and pyroptosis were not detected, in SARS-CoV-2 infected human airway epithelia. Furthermore, the activation of both RIPK1 and RIPK3 promoted the phosphorylation of MLKL, which leads to cell necroptosis.
Results
SARS-CoV-2 infection upregulates cell death-related gene expression of polarized primary human airway epithelium.
In our previous study, we reported that SARS-CoV-2 infection of primary HAE cultured at an ALI (HAE-ALI) distorted the tight-junction structure, a protective barrier of human airway epithelia, and caused moderate to severe loss of the cilial structure. 35 In addition, immunofluorescent staining demonstrated nuclear damage of the infected cells. To determine which gene expressions were involved in the infection-caused cell death, total RNA samples were extracted for RNA-seq analysis from the cells of HAE-ALI infected with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.2 and 2, respectively, as well as from mock-infected cells as a control. The sequencing data have been deposited in the NIH-sponsored BioProject database under accession number PRJNA698337. We analyzed the differentiated gene expression profiles for the upregulated genes with a p-value of < 0.05 (Figure 1 A). We further compared the expression of genes enriched in both MOIs of 2 and 0.2 groups, and found that the upregulated genes were broadly consistent (Spearman R2=0.98) (Figure 1B). This result confirmed that SARS-CoV-2 infection of HAE-ALI changed host gene expression.
Figure 1. Cell death-related genes are upregulated in SARS-CoV-2 infected HAE-ALI.
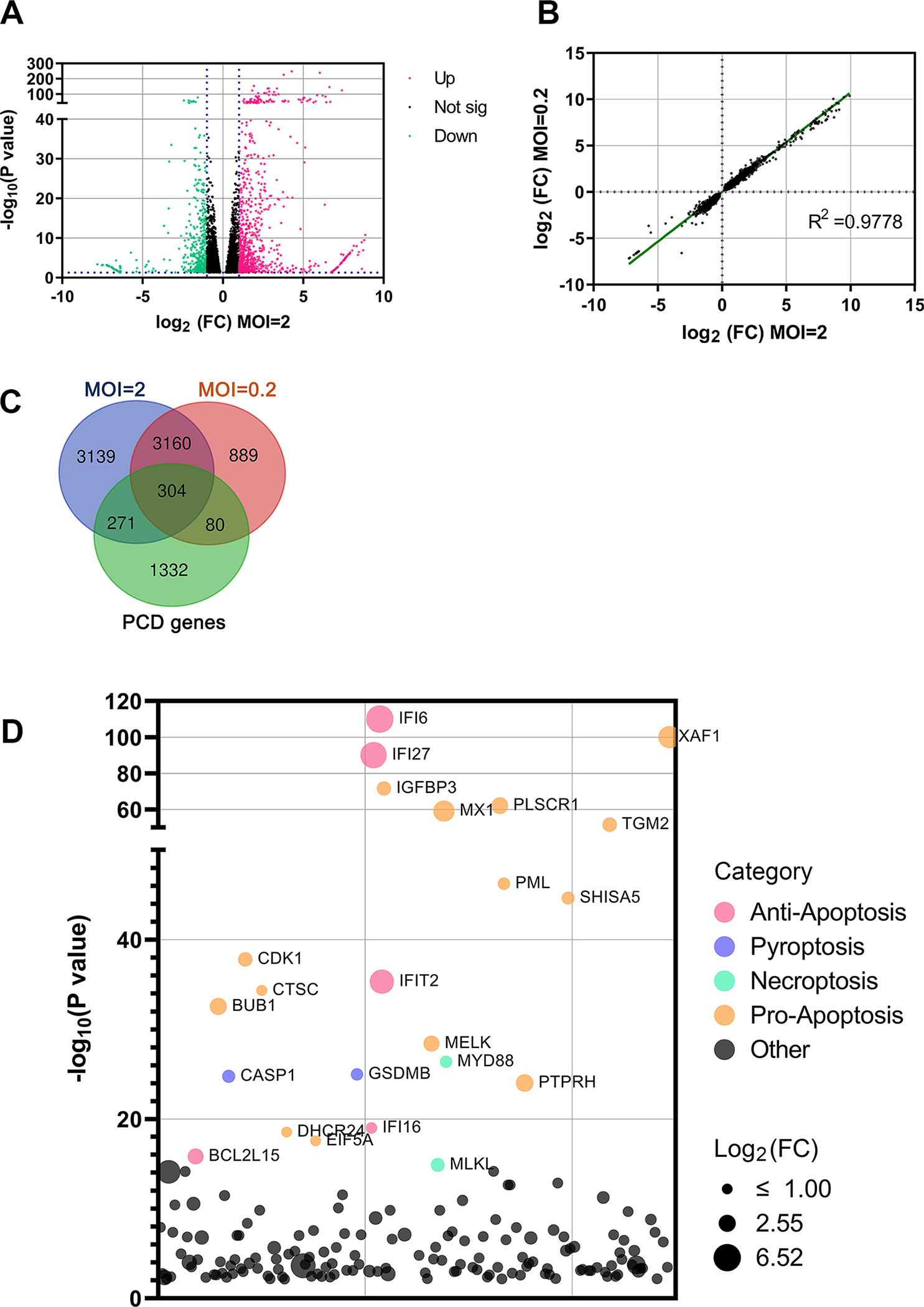
(A) Analysis of RNA-seq. The volcano plot displays statistical significance (−log10 P value) versus magnitude of change (log2 fold change) on the differentially expressed genes from RNA-seq data between HAE-ALI infected with SARS-CoV-2 (MOI=2) and mock-infected. All data selected at the criteria p < 0.05. Red dots indicate upregulated genes of >2-fold increase, and green dots indicate downregulated genes of >2-fold increase. (B) Data correlation between MOIs. Scatter plot of the fold change on all genes of HAE-ALI infected with SARS-CoV-2 at MOIs of 2 and 0.2, respectively, compared to the mock infected. The correlation R2 value is 0.9778. (C) Changes of overlapped genes. VENN plot of the genes related to PCD (Green) with the upregulated genes in the HAE-ALI infected with SARS-CoV-2 at MOIs of 2 (Blue) and 0.2 (Pink). (D) The top 100 upregulated genes related to cell death of the HAE-ALI infected with SARS-CoV-2 at MOI of 2. The y axis shows significance of hit based on the analysis. The bubble diameter indicates the fold change of individual gene enriched in SARS-CoV-2 infected HAE-ALI, compared to unselected controls. Different colors indicate the variety of the cell death pathway category.
A large body of studies have indicated that PCD caused by SARS-CoV-2 infection plays an important role in viral pathogenesis of human airways and lung disease. 32,37–42 Therefore, we compared the RNA-seq data within the genes involved in PCD (Figure 1C). We found that a large portion of the upregulated genes were PCD-related. Next, we explored which type of PCD was induced in SARS-CoV-2 infected airway epithelia. To this end, we performed a gene ontology (GO) analysis of the top 100 upregulated PCD-related genes (Figure 1D). The results revealed the upregulation of pyroptosis-associated genes, such as CASP1 (Caspase 1) and GSDMB (Gasdermin B), and the necroptosis executor gene, MLKL. While many pro-apoptosis genes were also upregulated, we noticed that the top upregulated gene was IFI6, which is a type I interferon-stimulated gene (ISG) 43 and plays an antiviral activity during virus infection. 44 The anti-apoptotic effect of IFI6 has been shown in the inhibition of cell death of dengue virus-infected endothelial cells and human bocavirus 1 (HBoV1)-infected HAE-ALI. 45,46
Collectively, our RNA-seq results of SARS-CoV-2 infected HAE-ALI suggested that SARS-CoV-2 infection activates genes of all three types of PCD in polarized human airway epithelia.
SARS-CoV-2 induced cell death is primarily mediated by necroptosis.
As the GO analysis found that both pro- and anti-apoptosis genes were upregulated in the SARS-CoV-2 infected HAE-ALI, we carried out lactate dehydrogenase (LDH) cytotoxicity assays to validate the virus infection caused cell death. The release of LDH in the culture media of the basal chamber was measured during a period of two weeks post-infection. HAE-ALI treated with nigericin at 10 μM for 3 days was used as a positive control. Nigericin is a polyether antibiotic that affects potassium ion transport and ATPase activity in mitochondria and induces cell death. 47 Compared to the mock infection, the activity of the released LDH from infected HAE-ALI was significantly increased over 14 days (Figure 2A). This result confirmed that the SARS-CoV-2 infection-induced cell death in airway epithelia.
Figure 2. SARS-CoV-2 infection of HAE-ALI induces cell death mediated by necroptosis, but not pyroptosis or apoptosis.
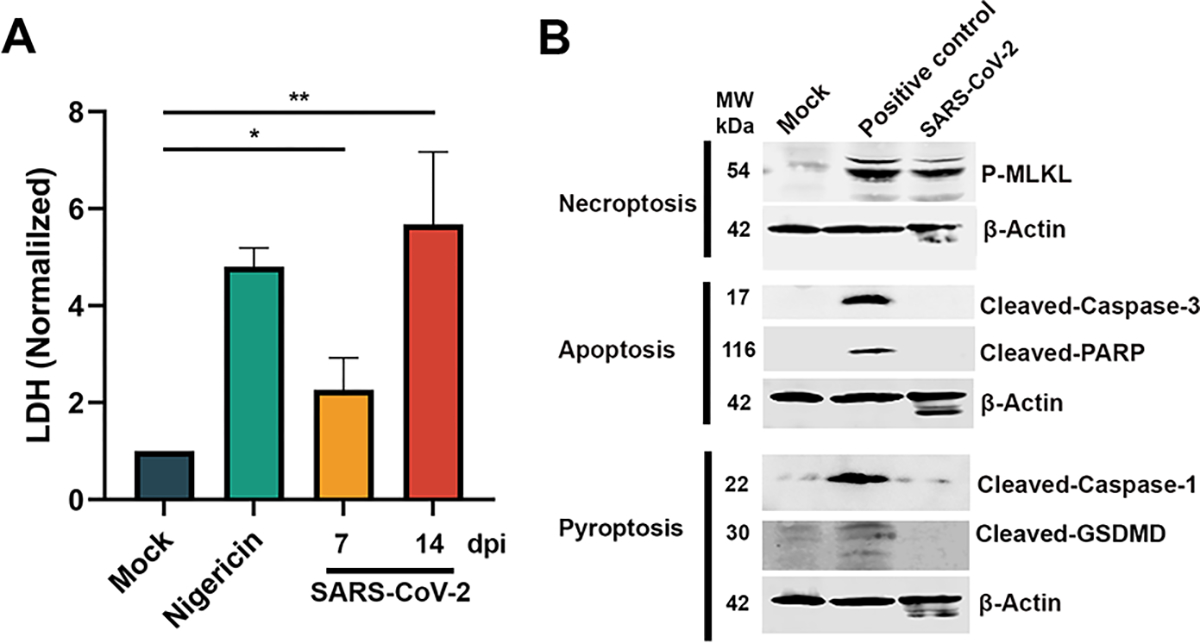
(A) SARS-CoV-2 infection of HAE-ALI induced cell death. HAE-ALI were infected with SAR-CoV-2 or mock-infected. At 7 or 14 dpi, LDH values in the basal media were measured and normalized to the mock infected cultures. Means and standard deviations (SD) were calculated from three independent infections. Mock at 14 dpi and nigericin treated HAE-ALI were used as negative and positive controls, respectively. (B) Western blotting. Cell lysates of SARS-CoV-2 infected HAE-ALI were collected at 4 dpi and were analyzed for expression of cell death hallmark proteins, as indicated. Staurosporine at 20 μM, doxorubicin at 50 μM, and hydrogen peroxide at 200 μM were used to treat HAE-ALI as controls for apoptosis, pyroptosis, and necroptosis, respectively. β-actin was detected as a loading control.
PCD caused by respiratory viruses generally includes apoptosis, necroptosis, and pyroptosis. 48–50 In our previous study on HBoV1 infection of HAE-ALI, we discovered that the infection induced pyroptosis and inhibited apoptosis. 46 To identify which PCD pathway leads to SARS-CoV-2 induced airway epithelial death, we examined the cell death protein markers in the cell lysates of the infected HAE-ALI at 3 days post-infection (dpi). In these assays, staurosporine, which induces apoptosis, 51 doxorubicin, which induces pyroptosis, 52 and hydrogen peroxide, which induces RIPK-dependent necroptosis, 53 were used to treat HAE-ALI cultures as positive controls. As indicated in Figure 2B, SARS-CoV-2 infected epithelial cells did not express cleaved caspase-3, as well as cleaved poly ADP ribose polymerase (PARP) protein, whereas staurosporine treatment resulted in cleavage of both proteins. Active caspase-3 degrades multiple cellular proteins and is responsible for morphological changes and DNA fragmentation in cells during apoptosis, and cleaved PARP indicates that cells have undergone apoptosis. 26 Thus, this result ruled out the possibility that apoptosis was induced during SARS-CoV-2 infection of HAE-ALI cultures.
We then investigated whether SARS-CoV-2 infection led to cell death via pyroptosis, which was recently reported to mediate the PCD of airway epithelial cells infected by several viruses, such as HBoV1 in polarized HAE-ALI 46 and human respiratory syncytial virus (RSV) in macrophages. 48 We detected cleaved caspase-1 and GSDMD in the HAE-ALI treated with doxorubicin (at 50 μM), but not in the SARS-CoV-2 infected HAE-ALI (Figure 2B, Pyroptosis). Since cleaved caspase-1 and GSDMD are markers of cell pyroptosis 26, this result ruled out the possibility that SARS-CoV-2 infection in HAE-ALI cultures induced pyroptosis. Lastly, we tested for necroptosis by detecting for phosphorylated (p)-MLKL, a marker of necroptosis. We found that both hydrogen peroxide treated and SARS-CoV-2 infected HAE-ALI cultures expressed p-MLKL (Figure 2B, Necroptosis).
Taken together, these results strongly indicated that SARS-CoV-2 infection of primary airway epithelia largely induced necroptosis, but not apoptosis or pyroptosis.
Necrostatin 2 and necrosulfonamide inhibit SARS-CoV-2 infection-induced necroptosis of human airway epithelia.
We next used 2 pharmacological inhibitors of necroptosis, necrostatin 2 (N2), an inhibitor of RIPK1, 54 and necrosulfonamide (NSA), an inhibitor of MLKL 55, to investigate SARS-CoV-2 infection caused necroptosis in airway epithelia. Application of NSA and N2, at 20 and 50 μM, respectively, significantly decreased the release of LDH by a half, compared with the vehicle (DMSO) control (Figure 3A), suggesting that less cell death was induced in the infected HAE-ALI by the inhibition of necroptosis. Furthermore, we detected transepithelial electrical resistance (TEER) of the infected HAE-ALI, as TEER is an indicator of the integrity of cell tight junctions. 56 Of note, we observed that the application of NSA and N2, at 20 and 50 μM, respectively, also prevented the decrease of TEER caused by viral infection, compared with the mock-infected group (Figure 3B), suggesting less epithelial damage was caused by the inhibition of necroptosis. Taken together, these results confirmed that the SARS-CoV-2 infected airway epithelia undergo necroptosis, resulting in airway epithelial barrier dysfunction, which can be prevented by necroptosis inhibitors.
Figure 3. SARS-CoV-2 infection-induced necroptosis is inhibited by pharmacological inhibitors necrostatin2 (N2) and necrosulfonamide (NSA).
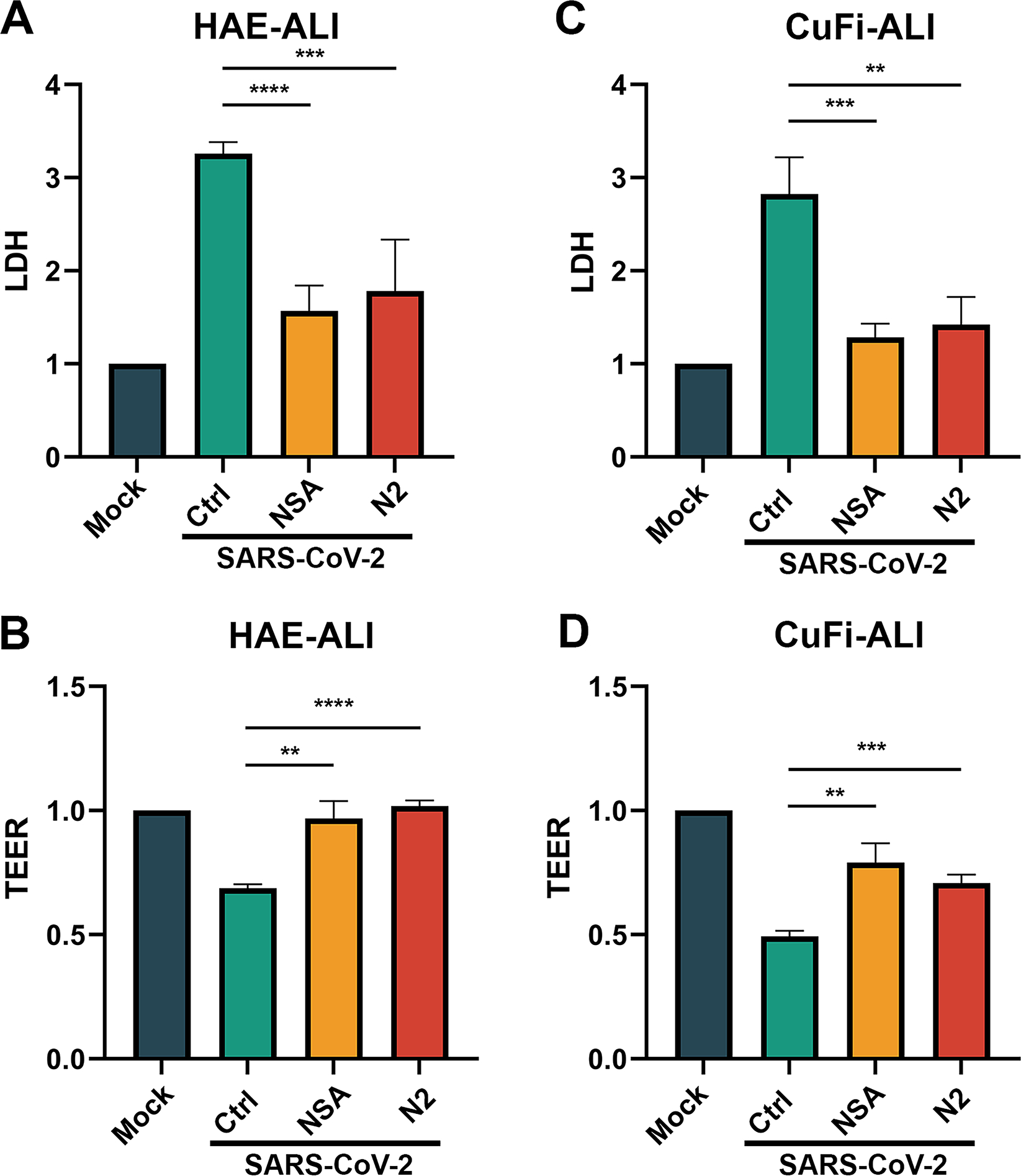
(A&C) LDH release in basal media of infected ALI cultures. NSA (at 20 μM) and N2 (at 50 μM) were added into the Transwell chambers of HAE-ALI (A) and CuFi-ALI (C), respectively, at 1 h prior to infection with SARS-CoV-2 at an MOI of 2. Dimethylsulfoxide (DMSO) was used as a vehicle Control (Ctrl). LDH releases in the basal media were measured at 4 dpi, and the levels related to the mock infected culture are shown. (B&D) TEER measurement. TEER values in HAE-ALI (B) or CuFi-ALI (D) cultures after SARS-CoV-2 infection and treatment NSA and N2 are normalized to mock-infected ALI cultures, and relative TEER values are shown. Student’s t-test was used, and data were shown as means with standard deviation (SD) of three replicates.
CuFi-8 is an immortalized human airway epithelial cell line established by exogenous expression of the telomerase reverse transcriptase and E6/E7 genes of human papillomavirus (HPV). 36 CuFi-8 cells retain the ability to differentiate into mucociliary columnar airway epithelium when cultured at an ALI. CuFi-ALI cultures have been shown to be the ideal model for studying respiratory pathogen infections, 57,58 demonstrating several advantages over the primary HAE cultures, such as no donor variations, convenience to maintain, and allowance for genetic manipulation. 59 We applied the CuFi-ALI culture model to SARS-CoV-2 infection in the conditions with or without a necroptosis inhibitor. We observed that the infection resulted in similar patterns of the release of LDH and changes of TEER (Figure 3C&D) as seen from the infections in the primary HAE-ALI cultures (Figure 3A&B). Thus, we decided to use CuFi-ALI cultures to investigate SARS-CoV-2 infection of human airway epithelia.
In our previous studies, 35 we have found that the SARS-CoV-2 infection of HAE-ALI severely damaged the tight junctions and induced the loss of cilia. Therefore, immunofluorescent assays were performed to examine the epithelial damage of the infected CuFi-ALI cultures by staining ZO-1, a marker of tight junction, 60 and tubulin, a marker of cilia. 61,62 As shown in Figure 4A, in contrast to the mock-infected cells that displayed dense tubulin expression and intact ZO-1 distribution, the infected CuFi-ALI (vehicle control) showed a large portion of tubulins were absent and a distorted structure of tight junction (Figure 4, Mock vs Control), while the treatment of NSA and N2 gave protection to a larger extent with a significantly higher expression of tubulin and a well-organized ZO-1 distribution (Figure 4A, NSA&N2 vs Control). Quantification of tubulin expression showed that the tubulin was decreased by 70% in the infected CuFi-ALI (Ctrl), compared with the mock control (Figure 4B). However, treatment with NSA or N2 prevented the damage of tubulin, which decreased tubulin expression only by ~20% and 40%, respectively (Figure 4B).
Figure 4. NSA and N2 protect CuFi-ALI cultures from disruption of epithelial structures caused by SARS-CoV-2 infection.
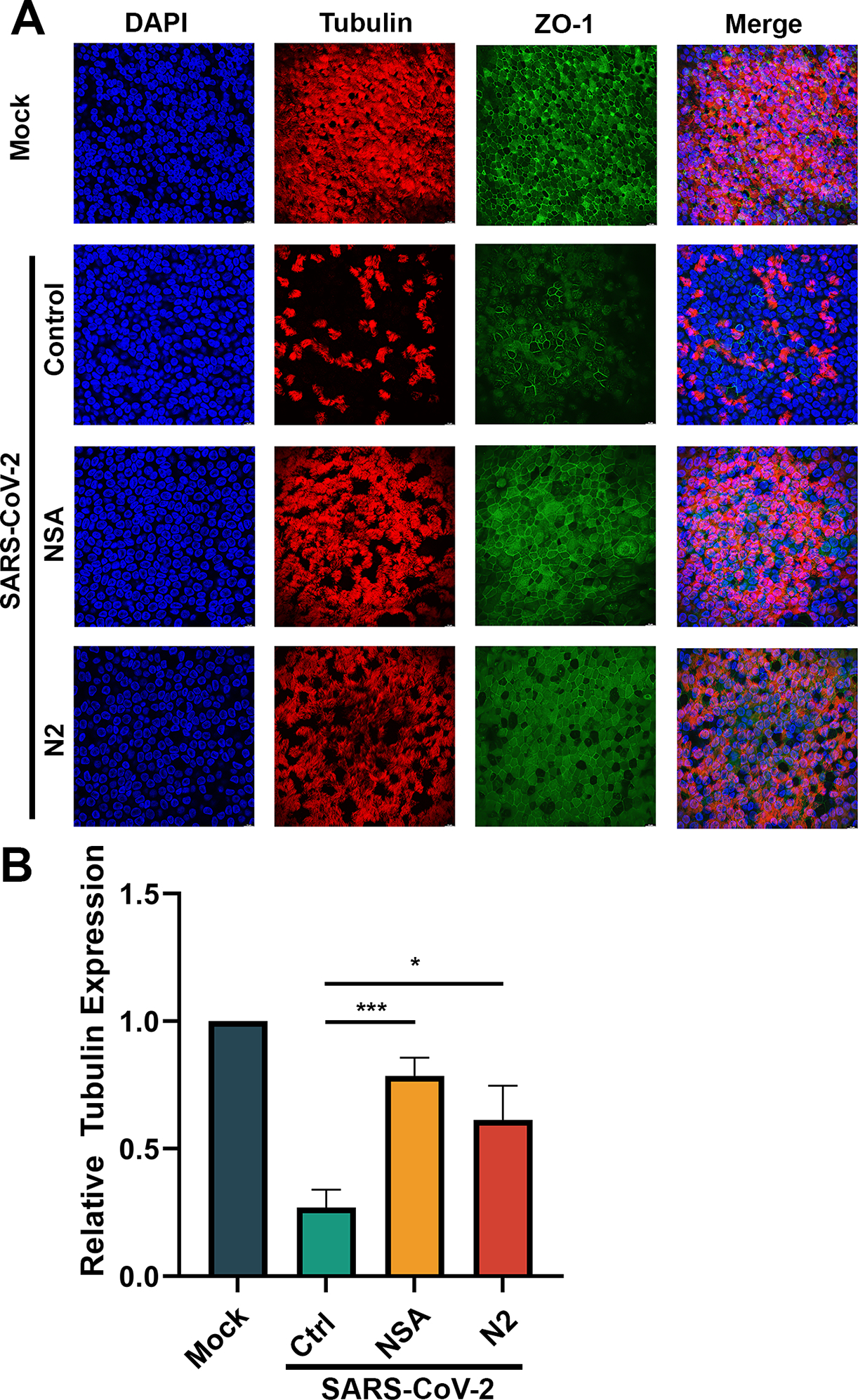
(A) Immunofluorescent assay. CuFi-ALI cultures were treated with NSA (at 20 μM) and N2 (at 50 μM) treatment, respectively, at 1 h prior to infection of SARS-CoV-2 (MOI=2). DMSO was used as a vehicle Control (Ctrl). At 4 dpi, both virus- and mock-infected CuFi-ALI were co-stained with anti-β-tubulin IV and anti-ZO-1 antibodies. Confocal images were taken at a magnification of × 40. Nuclei were stained with DAPI (blue). (B) Quantification of tubulin expression. Levels of tubulin in each infected CuFi-ALI culture were quantified using NIH Image-J software. Data were shown as means ± SD of three replicates.
Collectively, we concluded that the necroptosis inhibitors, NSA and N2, protected the physiological barrier structures of airway epithelia from the damage caused by SARS-CoV-2 infection.
SARS-CoV-2 infection activates the classic necroptosis pathway.
Similar to the observation of SARS-CoV-2 infection in HAE-ALI, Western blotting demonstrated the activation of MLKL (p-MLKL) in SARS-CoV2-infected CuFi-ALI (Figure 5A). The analyses also revealed that at 50 μM, N2 treatment of CuFi-ALI infected with SARS-CoV-2, almost completely inhibited the phosphorylation of RIPK1, and thus subsequently inhibited the downstream MLKL phosphorylation (Figure 5A). In contrast, as the inhibitor directly targeted MLKL, NSA treatment at 20 μM blocked the activation of MLKL without significant reduction in the phosphorylation levels of RIPK1 and RIPK3 (Figure 5B), which were also confirmed by immunofluorescent assays (Figure 5C&D, NSA). Our results demonstrated that the NSA and N2 are potent inhibitors of SARS-CoV-2 induced necroptosis that function in different roles in the classic necroptosis pathway. N2 inhibits the activation of RIPK1, an upstreaming signal of necroptosis, whereas NSA is a direct inhibitor of MLKL activation, which is the executor of necroptosis. While NSA did not inhibit phosphorylation of RIPK3, N2 inhibited RIPK3 (Figure 5A), suggesting that RIPK3 is downstream of RIPK1.
Figure 5. Both NSA and N2 inhibit necroptosis via RIPK1 to MLKL pathway.
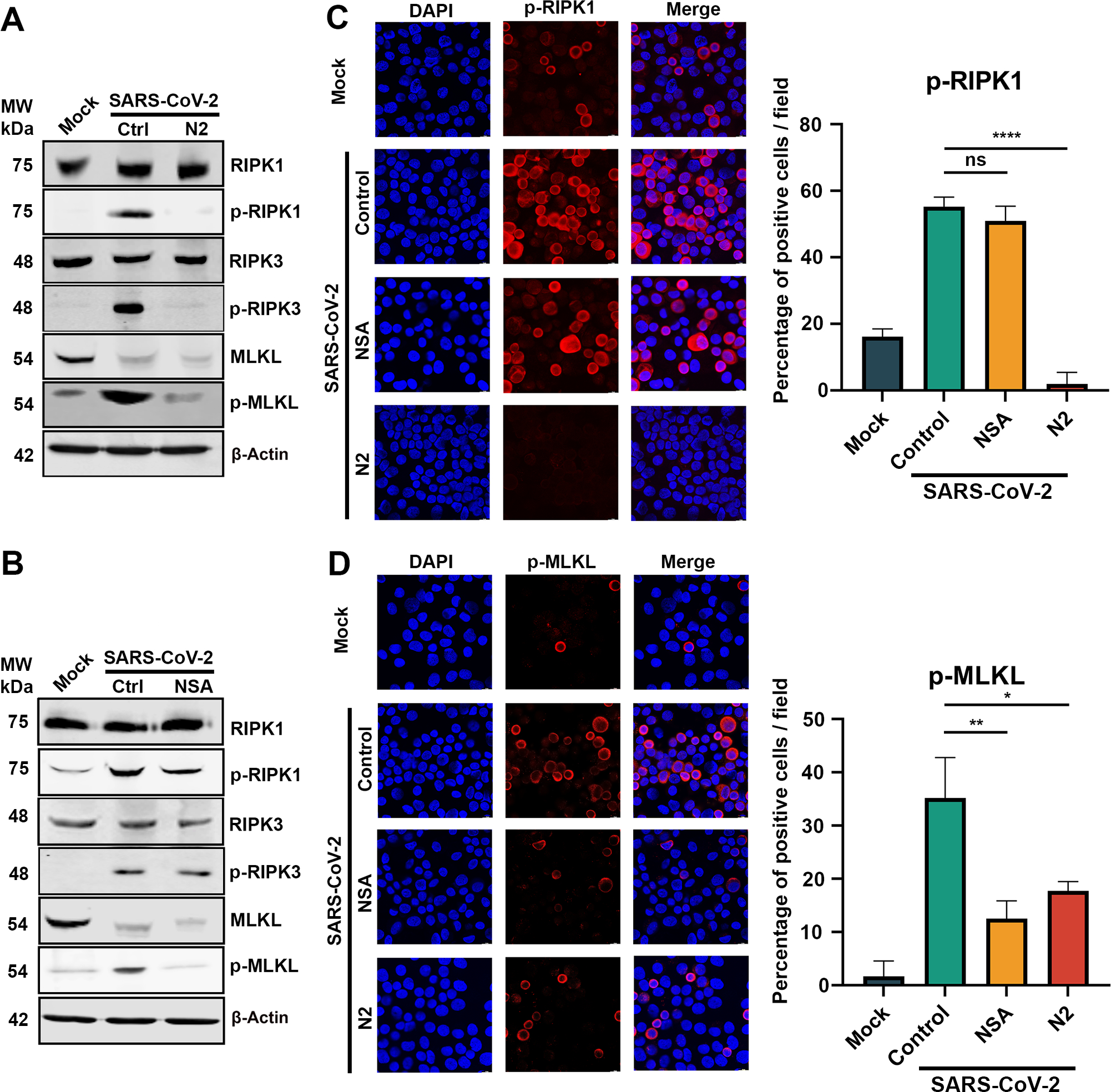
CuFi-ALI cultures were treated with NSA (A) and N2 (B) at 20 and 50 μM, respectively, followed by infection of SARS-CoV-2 at an MOI of 2. DMSO was used as a vehicle Control (Ctrl). At 3 dpi, cell lysates were collected. (A&B) Western blotting. The lysates were separated on SDS-10%PAGE and blotted for the expression of both phosphorylated (p-) and unphosphorylated RIPK1, RIPK3, and MLKL. β-actin was detected as a loading control. (C&D) Immunofluorescence analysis. The collected cells were cytospun onto slides, stained with anti-p-RIPK1 (B) and anti-p-MLKL(D), respectively. Confocal images were taken at a magnification of × 63. Nuclei were stained with DAPI (blue). Positive stained cells were counted, and percentages of the positive cells were plotted at the right side of panels C&D.
Knockdown of the necroptosis pathway genes protects epithelial damage from SARS-CoV-2 infection.
To confirm the role of necroptosis in the disruption of the airway barrier function during SARS-CoV-2 infection, we silenced RIPK1, RIPK3, and MLKL genes, respectively, in CuFi-ALI cultures. To this end, we used a lentiviral strategy to integrate specific shRNA expression into CuFi-8 cells, to selectively silence the expression of the target gene. Monolayer cultured CuFi-8 cells were infected with lentiviral vectors harboring shRNA against RIPK1, RIPK3, MLKL and scramble control, respectively (Figure 6A). At 3 days post-transduction, we confirmed the knockdown efficiency (data not shown) and seeded the transduced cell onto Transwell inserts. After 4 weeks of culturing at an ALI for differentiation, TEER values of these CuFi-ALI were greater than 1,000 Ω, indicative of well differentiated and polarized epithelia. 46 At this time point, we collected the cell lysate to confirm the gene knockdown efficiency by Western blotting (Figure 6B). Transduction with a scrambled shRNA (shScram)-expressing lentivirus served as a negative control.
Figure 6. Silencing of RIPK1, RIPK3, and MLKL decreases LDH release and retains TEER in SARS-CoV-2 infected CuFi-ALI cultures.
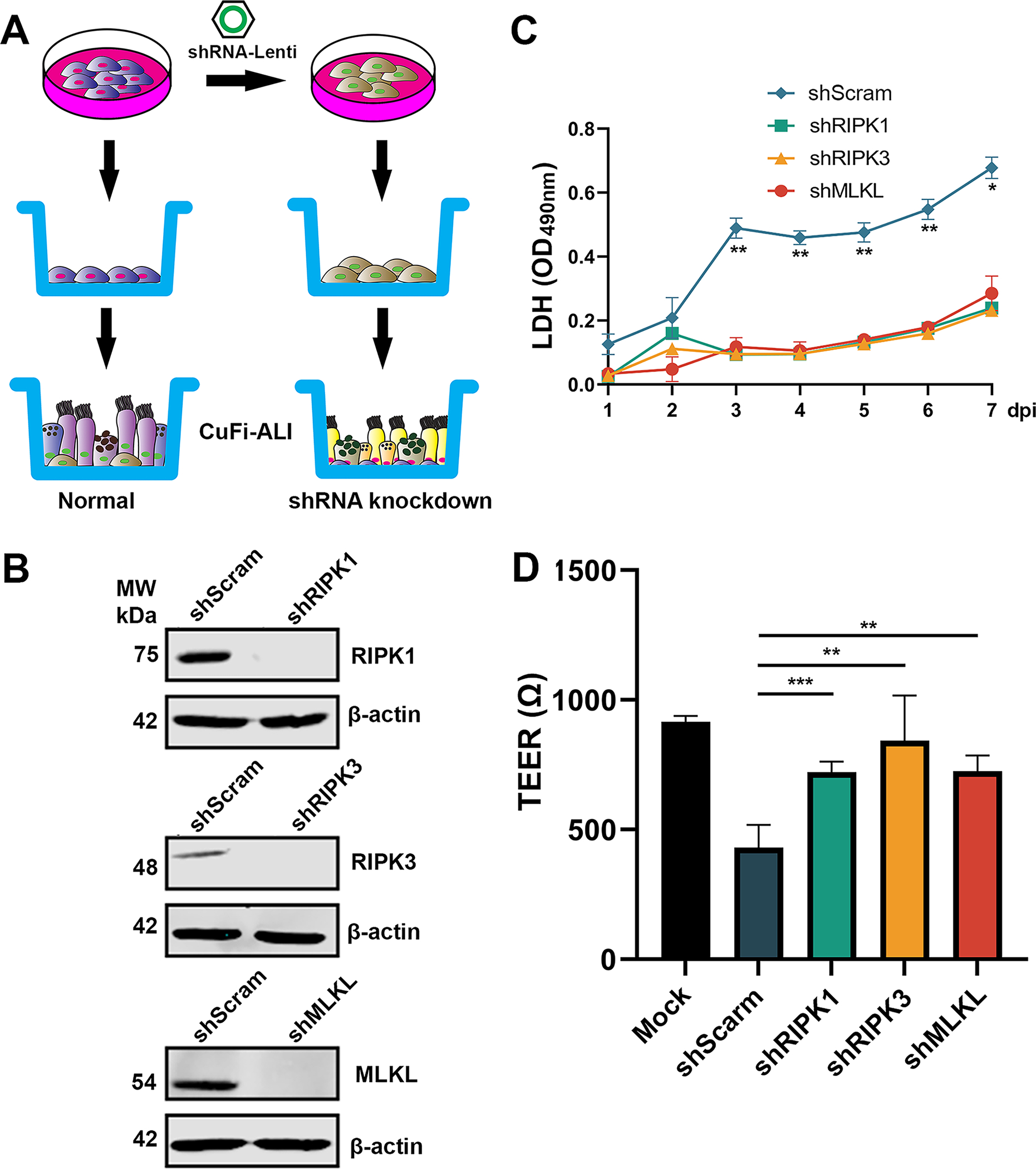
(A&B) Knockdown of RIPK1, RIPK3, and MLKL in CuFi-ALI. (A) Schematic diagram shows the development of gene knockdown of CuFi-ALI cultures. (B) Western blotting validated the knockdown of RIPK1, RIPK3, and MLKL in CuFi-ALI cultures. (C&D) SARS-CoV-2 infection. Gene silenced CuFi-ALI cultures were infected with SARS-CoV-2 at an MOI of 2. Basolateral media were daily measured for LDH release from 1 to 7 days post-infection (C). The actual values at OD490nm values are shown. At 7 dpi, the TEER of infected ALI cultures was measured (D). All data were shown as means ± SD of three replicates. Student’s t-test was used to calculate the P values. Time points that had statistical significance are marked.
Next, we infected the shRNA-expressing CuFi-ALI cultures with SARS-CoV-2. We monitored LDH release, TEER, cell morphology, and apical viral release over a period of one-week post-infection. Compared with the scramble shRNA, the RIPK1, RIPK3, and MLKL shRNA expression groups significantly decreased the SARS-CoV-2 induced LDH releases by 2-fold from 3–6 dpi and 3-fold at 7 dpi (Figure 6C). The result confirmed that silenced expression of RIPK1, RIPK3, and MLKL resulted in a significant reduction of virus-induced epithelial cell death.
Moreover, the decreases in TEER in the shMLKL-, shRIPK3-, and shMLKL-expressing CuFi-ALI cultures were significantly less than that observed in the shScram-expressing CuFi-ALI (Figure 6D). We further carried out immunofluorescent staining for the expression of tubulin and ZO-1. The tubulin expression in the shRIPK1-, shRIPK3-, and shMLKL-expressing CuFi-ALI largely maintained >70% intact as compared with the mock-infected control; however, most of the tubulins of the scramble control were lost to <10% of the level of the mock-infected control (Fig. 7, shScram/Tubulin). On the other hand, although in the scramble control, most tight junctions as shown by ZO1 staining were disrupted or merged, the shRIPK1-, shRIPK3-, and shMLKL-expressing CuFi-ALI had a ZO-1 expression that remained intact (Fig. 7, ZO-1). This result supported that the silencing of the necroptosis genes protects the airway epithelia from the barrier dysfunction caused by SARS-CoV-2 infection.
Figure 7. Gene silencing of necroptosis genes protects CuFi-ALI cultures from epithelial damage caused by infection of SARS-CoV-2.
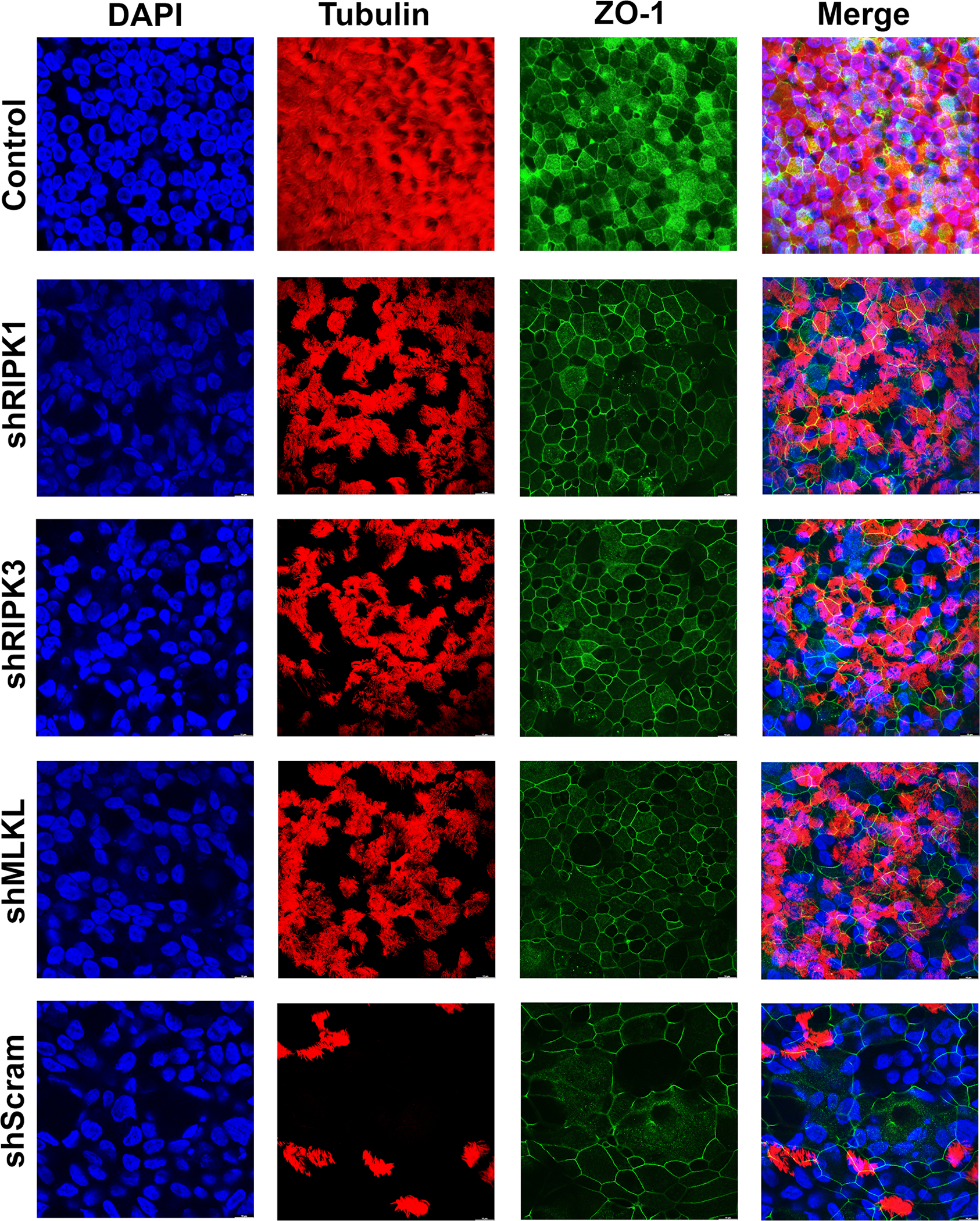
CuFi-ALI cultures that had RIPK1, RIPK3, or MLKL gene silenced or scrambled shRNA expressed were infected with SARS-CoV-2 at an MOI of 2. At 4 dpi, all CuFi-ALI cultures were fixed and co-stained with anti-β-tubulin IV and anti-ZO-1 antibodies. Mock infected CuFi-ALI was used as a control. Confocal images were taken at a magnification of × 63. Nuclei were stained with DAPI (blue).
Collectively, these results demonstrated that the silencing of necroptosis pathway genes, RIPK1, RIPK3, and MLKL, protected the epithelial damage from the SARS-CoV-2 infection. The results also suggest that silenced expression of RIPK1, RIPK3, and MLKL did not drastically increase other forms of cell death.
Inhibition of necroptosis increases virus release from the apical side of the SARS-CoV-2 infected airway epithelia.
Composed of multiple functionally distinct cell types, the in vitro human airways epithelium culture model HAE-ALI enables us to trace the fate of the infected cells and the bystander effects on the non-infected cells. Using this model, we discovered that, despite accompanying epithelial damage, SARS-CoV-2 infection in HAE-ALI can be long-lasting with several recurrent replication peaks over a period up to 51 days. 35 As the inhibition of necroptosis reduced the epithelial damage in the SARS-CoV-2 infected HAE-ALI, we anticipated that blocking the necroptosis pathway by either pharmacological inhibitors or silencing the necroptosis-related gene expression would result in more virus replication.
Thus, we first investigated the virus release kinetics of the infected CuFi-ALI cultures treated with or without the necroptosis inhibitors. The apical wash from infected CuFi-ALI cultures was collected to titrate pfu of the viruses released from the apical side. Notably, we found that at 3 dpi, there was no significant difference in apical virus release; however, at 7 dpi, both NSA- and N2-treated CuFi-ALI released more viruses than the untreated control (Figure 8A). We reasoned that the necroptosis inhibitors inhibited cell death of infected cells; thus, more viruses were released from infected ciliated cells at the apical membrane. For further confirmation, we infected CuFi-ALI with the green fluorescent protein-expressing recombinant virus, mNG-SARS-CoV-2, 63 with or without the necroptosis inhibitors. At 14 dpi, we found that the NSA-treated HAE-ALI showed the highest percentage of infected cells, compared with the N2 treated or untreated control (Figure 8B), correlating with the decreased apical virus release. These findings indicated that the use of the necroptosis inhibitors significantly increased virus replication. Our hypothesis was also supported by the results of apical virus release from the infected shRNA-expressing CuFi-ALI cultures. While it demonstrated that shRIPK1 and shRIPK3 did not significantly enhance apical virus release, compared with scramble shRNA control, shMLKL significantly increased the apical viral release by >1-log from day 4 to day 7 (Figure 8C).
Figure 8. Inhibition of necroptosis in SARS-CoV-2 infected CuFi-ALI increases apical viral releases.
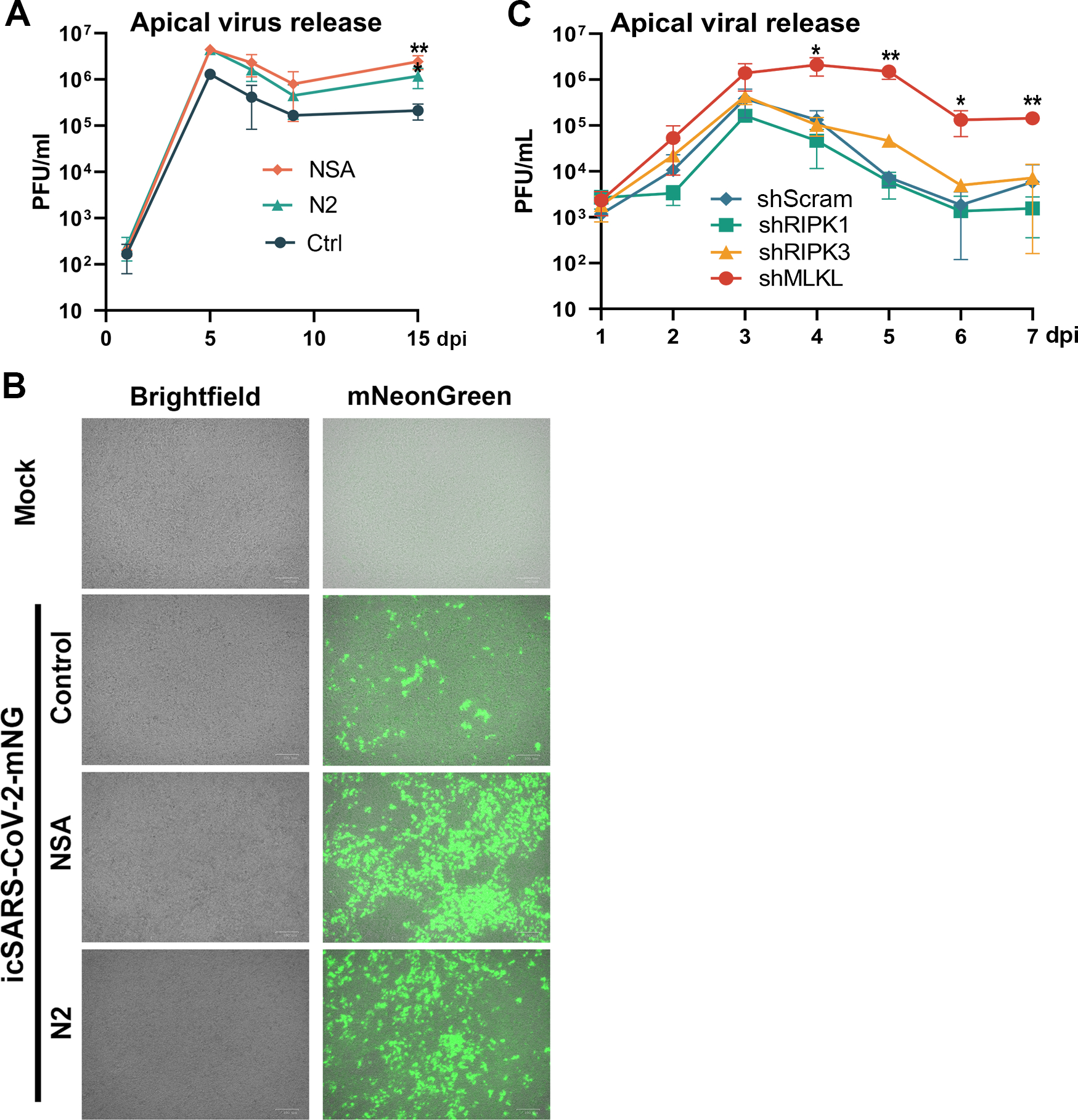
(A&B) Necroptosis inhibitors enhanced virus replication. (A) Apical virus releases. NSA and N2 treated CuFi-ALI cultures were infected with SARS-CoV-2 at an MOI of 2 from the apical side. At the indicated dpi, the apical surface was washed with 100 μl of D-PBS to collect the released virus. Virus titers in pfu were determined (y-axis) and plotted to the days of post-infection. (B) Infection of icSARS-CoV-2-mNG. NSA and N2 treated CuFi-ALI cultures were infected with icSARS-CoV-2-mNG at an MOI of 2 from the apical side. At 7 dpi, virus infected cells (green) were visualized under a Nikon inverted microscope. (C) Silencing of MLKL gene, but not RIPK1 and RIPK3, in CuFi-ALI increased apical virus release. Gene knockdown CuFi-ALI cultures were infected with SARS-CoV-2 at an MOI of 2. Apical washes were collected daily and quantified by plaque assays for virus release for 7 days post-infection. Virus titers in pfu were plotted to the days of post-infection. Values were shown as means ± SD of three replicates. Student’s t-test was used to calculate the P values. Time points that had statistical significance are marked.
Taken together, these results suggested that blockade of the upstream kinases of necroptosis was not as efficient as the downstream executor, MLKL, in promoting apical virus release at late infection of SARS-CoV-2 in CuFi-ALI cultures.
Discussion
In this study, we demonstrated that necroptosis is mainly responsible for the SARS-CoV-2 infection caused cell death in human airway epithelia, whereas apoptosis and pyroptosis are not obviously activated. Following SARS-CoV-2 infection, RIPK1 and RIPK3 are activated, leading to the phosphorylation of MLKL and necroptosis. Moreover, pharmacological inhibitors of necroptosis, NSA and N2, blocked necroptosis in SARS-CoV2-infected human airway epithelia, and protected the airway epithelium from barrier dysfunction, including retaining cilia and tight junctions. Silencing of MLKL, RIPK1, and RIPK3 genes in human airway epithelia demonstrated the protective effects of SARS-CoV-2 infection. All together, we propose that the RIPK1/RIPK3-MLKL signaling pathway triggers necroptosis in respiratory epithelial cells during SARS-CoV-2 infection.
Besides the upregulated necroptosis genes, we noticed several anti-apoptotic genes were upregulated drastically, such as IFI6, which were observed in HBoV1 infection of airway epithelia. 46 HBoV1 infection induced pyroptosis through inhibition of apoptosis. We speculated that the upregulation of IFI6 may also execute a role in the inhibition of apoptosis in airway epithelia infected with SARS-CoV-2. Additionally, a key pyroptosis regulated gene CASP-1 was upregulated, but the level of cleaved CASP-1 was not increased in SARS-CoV-2 infected polarized HAE.
SARS-CoV-2 primarily infects the epithelial cells in human airways, including the upper and lower airways, causing lung injuries. The lower airways consist of large airways (trachea and proximal bronchi), small airways, and alveoli. 64 The majority of COVID-19 patients show flu-like symptoms; 4 however, in a specific population, such as immunosuppressed patients, the infection rapidly develops into ARDS, 65 which is a lethal respiratory failure caused by pulmonary edema. In ARDS, the alveoli are damaged, and the epithelial permeabilities decrease. As the results, the accumulation of protein-rich alveolar edema fluid eventually blocks gas exchange, leading to hypoxemia, reduced carbon dioxide excretion, and ultimately acute respiratory failure. 66 The damage to the alveoli epithelial cells is due to the dysregulation of the innate immune system, which releases many cytokines and causes a cytokine storm and consequent cell death. 66 However, how the virus infection damages the bronchial airway epithelia largely remains elusive. Airway epithelial cells are exposed to various pathogens, and typically apoptosis is a pathophysiological consequence of these external stimuli. 67 Pyroptosis and necroptosis are two PCD pathways similar in that they are lytic forms of cell death driven by activation of the pore-forming proteins GSDMD and MLKL, respectively. 28,68,69 Although most respiratory pathogens cause cell death via apoptosis, 49 there are some exceptions. In our previous study on HBoV1, virus infection kills human airway epithelial cells of HAE-ALI cultures through pyroptosis. 46 In the case of influenza A virus infection, it causes cell death by inducing multiple types of cell death, including apoptosis, necroptosis, and pyroptosis, referred to as PANoptosis. 25 In COVID-19 patients, both apoptosis and necroptosis have been observed in various infected tissues, including lung, 32,33 as well as in vitro infected cell lines. 33,34 In SARS-CoV2 infected animals, a diffuse alveolar damage with extensive apoptosis has been observed. 70 In the current study, we proved that SARS-CoV-2 infection of HAE-ALI led to necroptosis with a significant decrease in TEER. Necroptosis often causes the pathogenesis of ARDS in the lung 71 and is a type of host defense against virus infection. 72 It is noted that the in vitro polarized airway epithelia are differentiated from primary bronchial epithelial cells. The polarized ALI cultures are composed of multiple functionally distinct epithelial cell types, including ciliated, goblet, club and basal stem cells, without immune cells. 35 This indicates that during SARS-CoV-2 infection of tracheobronchial airways, and likely nasal airways, necroptosis is the key cell death pathway.
However, necroptosis acts as a double-edged sword. The inhibition of necroptosis via pharmacological inhibitors, NSA or N2, increased the virus release from the apical side of the in vitro human airway epithelial cultures. Consistent with this, MLKL knockdown significantly increased the viral release by 2-log, and NSA is an inhibitor directly targeted MLKL. However, the knockdown of RIPK1 and RIPK3 did not increase the apical viral release. In fact, it has been reported that the viral nonstructural protein NSP12 directly interacted with RIPK1, which induces expression of host factors, including ACE2 that promotes viral entry into cells. 38 Thus, the RIPK1 knockdown may balance the virus replication by the reduced ACE2 expression on airway epithelial cells, which requires further investigation. Notably, multiple RIPK1 inhibitors (e.g., Nec-1s, GSK’481/GSK’772) have been in human clinical trials to treat severe COVID-19 patients. 73,74 Considering the respective knockdown of RIPK1, RIPK3 and MLKL expression all largely alleviated the epithelial damage in the infected CuFi-ALI cultures, but only MLKL knockdown increased the apical virus release, highly specific-RIPK1 or RIPK3 but not MLKL-specific inhibitors should be considered to treat severe COVID-19 patients.
There are several pathways leading to cell necroptosis. The classic necroptosis pathway is conducted by forming a RIPK1-RIPK3 cell death platform, called the necrosome, via RIPK1/3 RHIM-RHIM domain interactions. 75,76 The phosphorylation of RIPK1 and RIPK3 are hallmarks for the activation of necroptosis 27,77 and leads to MLKL phosphorylation. The activated MLKL then damages the cell membrane, leading to cell death. 78 Although autopsy detection revealed that the expression of the downstream signaling molecules of RIPK3 was found to be very low, and phosphorylated RIPK3 and MLKL were also undetectable, there are lines of evidence of RIPK1 activation in lung biopsies of COVID-19 patients and SARS-CoV-2 infected human lung organoids and ACE2 transgenic mice. 32,79 Therefore, our current study further validated the importance of RIPK1 and RIPK3 in the induction of necroptosis during SARS-CoV-2 infection.
As ZBP-1 acts as a vital innate immune sensor by regulating necroptosis and inflammatory responses by signaling for NF-κb and potential interferon regulatory factor (IRF3/7) activation, 29,80–83 we probed both ZBP-1 and NF-κb expression in infected HAE-ALI. We found they were highly activated, and the application of NSA obviously decreased the ZBP-1 expression in a dose-dependent manner (Supplemental Figure S1). Thus, it is likely that ZBP-1 plays a role as an RNA sensor to signal the NF-κb pathway to induce necroptosis during SARS-CoV-2 infection of airway epithelia, like influenza virus A. 81,84–86 Further studies to examine the detailed mechanism underlying the necroptosis triggered during SARS-CoV-2 infection of airway epithelia are warranted.
In summary, by taking advantage of the in vitro cultures of human airway epithelia, HAE-ALI, and CuFi-ALI cultures, we confirmed that SARS-CoV-2 infected airway epithelia undergo necroptosis and verified the role of RIPK1 and RIPK3 in the induction of necroptosis. To date, there is no effective pharmacotherapy for SARS-CoV-2-caused ARDS, and the treatment remains primarily supportive with lung protective ventilation and a conservative fluid management strategy. 87 The inhibition of programmed cell death, specifically RIPK driven necroptosis, in severe COVID-19 patients could be a direction for clinical medicine development.
Materials and Methods
Ethics statement:
A biosafety protocol to work on SARS-CoV-2 in the BSL3 Lab was approved by the Institutional Biosafety Committee (IBC) of the University of Kansas Medical Center. Primary human bronchial epithelial cells used for generation of HAE-ALI cultures were isolated from the lungs of healthy human donors at the Cells and Tissue Core of the Center for Gene Therapy, University of Iowa with the approvals of the Institutional Review Board (IRB).
Cell lines and viruses.
Cell lines:
Vero-E6 cells (ATCC CRL-1586) were used for plaque assay and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (GE Healthcare Life Sciences, Logan, UT, HyClone, # SH30022.01) supplemented with 10% fetal bovine serum (FBS) (Sigma, St. Louis, MO, # F0926) at 37°C under a 5% CO2 atmosphere. CuFi-8 cells were immortalized from human primary airway cells by expressing hTERT and HPV E6/E7 genes. 36 CuFi-8 cells were cultured on collagen-coated 100-mm dishes in Ex Plus medium (StemCell, #05040).
Viruses:
SARS-CoV-2 (NR-52281), isolate USA-WA1/2020, was obtained from BEI Resources, NIAID, NIH. icSARS-CoV-2-mNG is an infectious recombinant SARS-CoV-2 obtained from UTMB’s World Reference Center for Emerging Viruses and Arboviruses. It expresses a bright monomeric yellow-green fluorescent protein (mNeonGreen) reporter, which is incorporated into the OFR7 of the viral genome. 63 These two viruses were propagated, titrated in Vero-E6 cells, and stored at −80°C. The stocks were sequenced to confirm the retention of the furin cleavage site in the viral genome.
Polarized human airway epithelial cultures at an air-liquid interface (ALI).
Polarized primary human airway epithelium at an air-liquid interface (HAE-ALI):
The primary human nasal and bronchial airway epithelial cells were seeded onto the collagen IV-coated semipermeable membrane of the Costar 6.5 mm Transwell with 0.4 μm Pore Polycarbonate Membrane Insert (#3413, Corning Inc, Corning, NY) and differentiated to pseudostratified epithelium at an ALI.
Polarized immortalized human airway epithelium of CuFi at an ALI (CuFi-ALI):
CuFi-8 cells were seeded onto a collagen-coated 100-mm dish with Ex Plus medium for 3 days until confluence, and then were digested with Accutase (Innovative Cell Technologies, Inc., San Diego, CA) and transferred onto the semipermeable polycarbonate membrane inserts (1.12 cm2 Costar Transwell, # 3460, Corning, New York), and grown at an ALI with PneumaCult-ALI medium (StemCell, # 05001) as previously described. 88 The cultures were maintained in the ALI medium. After 4 weeks of culture at an ALI, the polarized cultures were fully differentiated.
The polarity of both the HAE- and CuFi-ALI cultures were determined for a transepithelial electrical resistance (TEER) using Millicell ERS-2 volt-ohm meter (MilliporeSigma, Burlington, MA). ALI cultures with a TEER value of 1,000 Ω·cm2 or higher were selected for use in the experiments.
Virus infection, sample collection and titration.
Plaque assays:
Vero-E6 cells were seeded in 24-well plate at a density of 50,000 cells and grown to confluence on the second day. Apical washes were serially diluted 10-fold in D-PBS. 200 μl of the diluent were added to each well and incubated for 1 h on a rocking rotator at 37°C. The virus diluent was then removed, and 0.5 ml of overlay medium (1% methylcellulose and 5% FBS in DMEM) was added to each well. The plates were incubated at 37°C under 5% CO2. At 4 days post-infection, the methylcellulose overlays were removed, and the cells were fixed using the 10% formaldehyde solution and stained with 1% crystal violet solution, followed by extensive washing. Plaques were counted and multiplied by the dilution factor for the virus titer at the unit of plaque-forming units (pfu) per ml.
Virus infection:
For apical infection, the apical chamber of the inserts was inoculated with 100 μl of SARS-CoV-2 at a multiplicity of infection (MOI) of 2 pfu/cell. The infected ALI cultures were incubated at 37°C and 5% CO2 for 1 h, followed by aspiration of the virus and washing three times with Dulbecco’s Phosphate Buffered Saline (D-PBS; Corning #21-031-CV. The ALI cultures were further cultured at 37°C and 5% CO2.
Virus sample collection:
Viral samples were collected from the apical wash of the epithelial surface at multiple time points. In brief, 300 μl of D-PBS was added to the apical chamber for 30 min at 37°C and 5% CO2. Thereafter, D-PBS (apical wash) was aspirated carefully from the apical chamber.
Lentiviruses and transduction.
Lentiviruses: shRNA expressing lentiviruses, shMLKL (#TRCN0000003227 and #TRCN0000003226), shRIPK1 (#TRCN0000196958 and #TRCN0000200006), shRIPK3 (#TRCN0000002257 and #TRCN0000002261), and shScram (#SHC016V) were purchased from MilliporeSigma.
Lentivirus transduction: CuFi-8 cells cultured as monolayer at 50% confluence were transduced with two lentiviral vectors with shRNA targeted the selected gene or one lentiviral vector expressing shScram at an MOI of ~5. At 2 days post-transduction, the cells were cultured in the medium with puromycin at a final concentration of 2 μg/ml to select the transduced cells. At 3 days post-treatment, the transduced CuFi cells were transferred onto the semipermeable polycarbonate membrane inserts for generation of ALI culture as described above.
Western blot analysis.
Airway epithelial cells were dissociated from the inserts of ALI cultures and solubilized with the Lysis buffer [150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl, pH 7.4]. The cell lysates were mixed with 5 × protein loading buffer and boiled for 10 min. Then the samples were subjected to SDS-(10%) polyacrylamide gel electrophoresis (PAGE), followed by transferring the separated proteins onto a nitrocellulose membrane 89. After blocking in non-fat milk for 1 h, the membrane was incubated with the first antibody, followed by incubation with an infrared dye conjugated IgG (H+L) second antibody, and imaged on a Li-Cor Odyssey imager (LI-COR Biosciences, Lincoln, NB).
LDH cytotoxicity assay.
The cytotoxicity assay kit (Promega, #J2380) was used according to the manufacturer’s instructions. Briefly, the culture media were collected from ALI cultures on the indicated days and loaded in 96-wellplate wells. Then, the Cytotoxicity assay reagent was added. After 30 min incubation, the stop solution was added, and the absorbance (OD) at 490 nm in the wells was measured using a Synergy H1 microplate reader (BioTek, Winooski, VT).
Immunofluorescent confocal microscopy.
Immunofluorescence assay:
The membrane support was cut from the insert of the ALI cultures and fixed in 4% paraformaldehyde PBS at 4°C overnight. The fixed membrane was washed in PBS for 3 times and then split into 8 pieces for whole-mount immunostaining. The fixed membrane pieces were permeabilized with 0.5% Triton X-100 for 15 min at room temperature, and mounted on a slide. The slide was incubated with the primary antibody in PBS containing 2% FBS for 1 h at 37°C, followed by washing 3 times, and was incubated with fluorescein isothiocyanate- and rhodamine-conjugated secondary antibodies, followed by staining of the nuclei with DAPI (4’,6-diamidino-2-phenylindole).
Confocal microscopy:
Slides were visualized under a Leica TCS SPE confocal microscope at the Confocal Core Facility of the University of Kansas Medical Center. Images were processed with the Leica Application Suite X software.
Small molecule compounds and treatment.
Necrosulfonamide (NSA), necrostatin 2 (N2), nigericin, staurosporine, and doxorubicin were purchased from APEXBio (Houston, TX). Hydrogen peroxide was purchased from Sigma (St. Louis. MO).
NSA and N2 were added to the media in the basolateral chamber of the ALI cultures at 20 and 50 μM, respectively, at 1 h prior to infection. Nigericin (at 10 μM), staurosporine (at 20 μM), doxorubicin (at 50 μM), and hydrogen peroxide (at 200 μM) were added to the media of the ALI cultures for 3 days.
Antibodies.
Primary antibodies used were rabbit monoclonal anti-SARS-CoV-2 nucleocapsid (NP) (clone 001, #. 40143-R001; SinoBiological US, Wayne, PA) at a dilution of 1:25, mouse monoclonal anti-β-tubulin IV antibody (clone ONS.1A6, # T7941; MilliporeSigma, St. Louis, MO) at 1:100, mouse anti-ZO-1 (# 610966; BD Bioscience, San Jose, CA) at 1:100, anti-p-MLKL (Cell signaling, Necroptosis Antibody Sampler Kit #98110), anti-cleaved-caspase-3 (Cell signaling, #9662), anti-cleaved-PARP (Cell signaling, #9532), anti-ZBP-1 (Proteintech #13285-1-AP) anti-gasdermin-D (Cell signaling #43811), anti-NF-ĸB (cell signaling #8242), anti-cleaved-caspase-1(Santa Cruz, #sc-392736), and anti-β-actin (Abclonal, #AC004). Primary antibodies used were infrared conjugated anti-mouse IgG (H+L) (DyLight™ 800 4X PEG Conjugate; Cell Signaling #5257) and anti-mouse IgG (H+L) (DyLight™ 680 Conjugate; Cell Signaling #5470). Fluorescein isothiocyanate- and rhodamine-conjugated secondary antibodies were purchased from Jackson Immunoreserch Laboratories lnc.
Statistical analysis.
Virus released kinetics were determined with the means and standard deviations (SD) obtained from at least three ALI cultures by using GraphPad Prism version 8.0. Error bars represent means and SD. Statistical significance (P value) was determined by using unpaired (Student) t-test for comparison of two groups. ****, P<0.0001; ***, P<0.001; **, P<0.01; and *, P<0.05 were regarded as statistically significant and n.s. represents statistically no significance.
Supplementary Material
Acknowledgments
We thank the Cells and Tissue Core of Center for Gene Therapy, the University of Iowa, which is partially funded by NIH grant DK054759, for providing the primary HAE-ALI cultures. This study was supported by PHS grants AI150877 and AI151542 from the National Institute of Allergy and Infectious Diseases. This study was also supported by grant YAN19XX0 from the Cystic Fibrosis Foundation. The following reagent was deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281. Recombinant icSARS-CoV-2-mNG was obtained from UTMB’s World Reference Center for Emerging Viruses and Arboviruses. We are grateful to the Confocal Microscopy Core Laboratory of The University of Kansas Medical Center that was supported by NIH S10 OD 023625 for the Leica SP8 STED confocal microscope.
Footnotes
Conflict of Interest statement
The authors declare no competing financial interests.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Sequencing data have been deposited in the NIH-sponsored BioProject database under accession number PRJNA698337.
References
- 1.Kim D, Lee JY, Yang JS et al. The Architecture of SARS-CoV-2 Transcriptome. Cell. 2020; 181(4):914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Methods Mol.Biol. 2015; 12821–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chow EJ, Schwartz NG, Tobolowsky FA et al. Symptom Screening at Illness Onset of Health Care Personnel With SARS-CoV-2 Infection in King County, Washington. JAMA. 2020; 323(20):2087–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhama K, Khan S, Tiwari R et al. Coronavirus Disease 2019-COVID-19. Clin.Microbiol.Rev. 2020; 33(4):e00028–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramadori GP. SARS-CoV-2-Infection (COVID-19): Clinical Course, Viral Acute Respiratory Distress Syndrome (ARDS) and Cause(s) of Death. Med.Sci.(Basel). 2022; 10(4):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Omame A, Abbas M, Abdel-Aty AH. Assessing the impact of SARS-CoV-2 infection on the dynamics of dengue and HIV via fractional derivatives. Chaos.Solitons.Fractals. 2022; 162:112427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolfel R, Corman VM, Guggemos W et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020; 581(7809):465–469. [DOI] [PubMed] [Google Scholar]
- 8.Huang C, Wang Y, Li X et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020; 395(10223):497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen N, Zhou M, Dong X et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020; 395(10223):507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang D, Hu B, Hu C et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA. 2020; 323(11):1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu K, Fang YY, Deng Y et al. Clinical characteristics of novel coronavirus cases in tertiary hospitals in Hubei Province. Chin Med J (Engl.). 2020; 133(9):1025–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Z, Shi L, Wang Y et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir.Med. 2020; 8(4):420–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang H, Zhou P, Wei Y et al. Histopathologic Changes and SARS-CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann.Intern.Med. 2020; 179(9):629–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Verity R, Okell LC, Dorigatti I et al. Estimates of the severity of coronavirus disease 2019: a model-based analysis. Lancet Infect.Dis. 2020; 20(6):669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimball A, Hatfield KM, Arons M et al. Asymptomatic and Presymptomatic SARS-CoV-2 Infections in Residents of a Long-Term Care Skilled Nursing Facility - King County, Washington, March 2020. MMWR Morb.Mortal.Wkly.Rep. 2020; 69(13):377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He X, Lau EHY, Wu P et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat.Med. 2020; 26(5):672–675. [DOI] [PubMed] [Google Scholar]
- 17.Zhou P, Yang XL, Wang XG et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579(7798):270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu F, Zhao S, Yu B et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020; 579(7798):265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu N, Zhang D, Wang W et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N.Engl.J Med. 2020; 382(8):727–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu R, Zhao X, Li J et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020; 395(10224):565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothe C, Schunk M, Sothmann P et al. Transmission of 2019-nCoV Infection from an Asymptomatic Contact in Germany. N.Engl.J Med. 2020; 382(10):970–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vareille M, Kieninger E, Edwards MR, Regamey N. The airway epithelium: soldier in the fight against respiratory viruses. Clin.Microbiol.Rev. 2011; 24(1):210–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bustamante-Marin XM, Ostrowski LE. Cilia and Mucociliary Clearance. Cold Spring Harb.Perspect.Biol. 2017; 9(4):a028241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tesfaigzi Y Roles of apoptosis in airway epithelia. Am.J.Respir.Cell Mol.Biol. 2006; 34(5):537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Place DE, Lee S, Kanneganti TD. PANoptosis in microbial infection. Curr.Opin.Microbiol. 2021; 5942–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Zhang W, Yi W et al. Pathway of Cell Death and Its Role in Virus Infection. Viral Immunol. 2022;doi: 10.1089/vim.2022.0010. [DOI] [PubMed] [Google Scholar]
- 27.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat.Rev.Mol.Cell Biol. 2014; 15(2):135–147. [DOI] [PubMed] [Google Scholar]
- 28.Malireddi RKS, Gurung P, Kesavardhana S et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J.Exp.Med. 2020; 217(3):20191644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Udawatte DJ, Rothman AL. Viral Suppression of RIPK1-Mediated Signaling. MBio. 2021; 12(4):e0172321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barbosa LA, Fiuza PP, Borges LJ et al. RIPK1-RIPK3-MLKL-Associated Necroptosis Drives Leishmania infantum Killing in Neutrophils. Front Immunol. 2018; 91818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rabiu AA, Ahmad R, Rowaiye AB et al. Targeting Specific Checkpoints in the Management of SARS-CoV-2 Induced Cytokine Storm. Life (Basel). 2022; 12(4):478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karki R, Sharma BR, Tuladhar S et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell. 2021; 184(1):149–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Zhang Y, Guan Z et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal.Transduct.Target Ther. 2020; 5(1):235–00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karki R, Lee S, Mall R et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci.Immunol. 2022; 7(74):eabo6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hao S, Ning K, Kuz CA et al. Long-Term Modeling of SARS-CoV-2 Infection of In Vitro Cultured Polarized Human Airway Epithelium. MBio. 2020; 11(6):e02852–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zabner J, Karp P, Seiler M et al. Development of cystic fibrosis and noncystic fibrosis airway cell lines. Am.J.Physiol Lung Cell Mol.Physiol. 2003; 284(5):L844–L854. [DOI] [PubMed] [Google Scholar]
- 37.Vanderheiden A, Ralfs P, Chirkova T et al. Type I and Type III Interferons Restrict SARS-CoV-2 Infection of Human Airway Epithelial Cultures. J Virol. 2020; 94(19):e00985–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu G, Li Y, Zhang S et al. SARS-CoV-2 promotes RIPK1 activation to facilitate viral propagation. Cell Res. 2021; 31(12):1230–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferreira AC, Soares VC, de Azevedo-Quintanilha IG et al. SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death.Discov. 2021; 7(1):43–00428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivanisenko NV, Seyrek K, Kolchanov NA, Ivanisenko VA, Lavrik IN. The role of death domain proteins in host response upon SARS-CoV-2 infection: modulation of programmed cell death and translational applications. Cell Death.Discov. 2020; 6(1):101–00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inde Z, Croker BA, Yapp C et al. Age-dependent regulation of SARS-CoV-2 cell entry genes and cell death programs correlates with COVID-19 severity. Sci.Adv. 2021; 7(34):eabf8609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bader SM, Cooney JP, Pellegrini M, Doerflinger M. Programmed cell death: the pathways to severe COVID-19? Biochem.J. 2022; 479(5):609–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoggins JW, Wilson SJ, Panis M et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011; 472(7344):481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyer K, Kwon YC, Liu S et al. Interferon-alpha inducible protein 6 impairs EGFR activation by CD81 and inhibits hepatitis C virus infection. Sci.Rep. 2015; 59012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang J, Li Y, Qi Y et al. Coordinated regulation of autophagy and apoptosis determines endothelial cell fate during Dengue virus type 2 infection. Mol.Cell Biochem. 2014; 397(1–2):157–165. [DOI] [PubMed] [Google Scholar]
- 46.Deng X, Zou W, Xiong M et al. Human Parvovirus Infection of Human Airway Epithelia Induces Pyroptotic Cell Death via Inhibiting Apoptosis. J.Virol. 2017; 91(24):e01533–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qin N, Xu G, Wang Y et al. Bavachin enhances NLRP3 inflammasome activation induced by ATP or nigericin and causes idiosyncratic hepatotoxicity. Front Med. 2021; 15(4):594–607. [DOI] [PubMed] [Google Scholar]
- 48.Bedient L, Pokharel SM, Chiok KR et al. Lytic Cell Death Mechanisms in Human Respiratory Syncytial Virus-Infected Macrophages: Roles of Pyroptosis and Necroptosis. Viruses. 2020; 12(9):932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Troy NM, Bosco A. Respiratory viral infections and host responses; insights from genomics. Respir.Res. 2016; 17(1):156–0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galani V, Tatsaki E, Bai M et al. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): an up-to-date cell-specific review. Pathol.Res.Pract 2010; 206(3):145–150. [DOI] [PubMed] [Google Scholar]
- 51.Zhou N, Wang R, Zhang Y et al. Staurosporine Induced Apoptosis May Activate Cancer Stem-Like Cells (CD44(+)/CD24(−)) in MCF-7 by Upregulating Mucin1 and EpCAM. J.Cancer. 2015; 6(10):1049–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Lian Z, Ge Y et al. TRIM25 Rescues Against Doxorubicin-Induced Pyroptosis Through Promoting NLRP1 Ubiquitination. Cardiovasc.Toxicol. 2021; 21(10):859–868. [DOI] [PubMed] [Google Scholar]
- 53.Zhao L, Lin H, Chen S et al. Hydrogen peroxide induces programmed necrosis in rat nucleus pulposus cells through the RIP1/RIP3-PARP-AIF pathway. J.Orthop.Res. 2018; 36(4):1269–1282. [DOI] [PubMed] [Google Scholar]
- 54.Zhou L, Yao Q, Ma L, Li H, Chen J. TAF1 inhibitor Bay-299 induces cell death in acute myeloid leukemia. Transl.Cancer Res. 2021; 10(12):5307–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang QX, Guo D, Wang FC, Ding WY. Necrosulfonamide (NSA) protects intervertebral disc degeneration via necroptosis and apoptosis inhibition. Eur.Rev.Med.Pharmacol.Sci. 2020; 24(5):2683–2691. [DOI] [PubMed] [Google Scholar]
- 56.Nicolas A, Schavemaker F, Kosim K et al. High throughput transepithelial electrical resistance (TEER) measurements on perfused membrane-free epithelia. Lab Chip. 2021; 21(9):1676–1685. [DOI] [PubMed] [Google Scholar]
- 57.Huang Q, Deng X, Yan Z et al. Establishment of a reverse genetics system for studying human bocavirus in human airway epithelia. PLoS.Pathog. 2012; 8(8):e1002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schildgen V, Mai S, Khalfaoui S et al. Pneumocystis jirovecii can be productively cultured in differentiated CuFi-8 airway cells. MBio. 2014; 5(3):e01186–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheikh Z, Bradbury P, Pozzoli M et al. An in vitro model for assessing drug transport in cystic fibrosis treatment: Characterisation of the CuFi-1 cell line. Eur.J.Pharm.Biopharm. 2020; 156121–130. [DOI] [PubMed] [Google Scholar]
- 60.Gonzalez-Mariscal L, Betanzos A, Nava P, Jaramillo BE. Tight junction proteins. Prog.Biophys.Mol.Biol. 2003; 81(1):1–44. [DOI] [PubMed] [Google Scholar]
- 61.Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc.Natl.Acad.Sci.U.S.A. 2004; 101(13):4620–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Villenave R, Thavagnanam S, Sarlang S et al. In vitro modeling of respiratory syncytial virus infection of pediatric bronchial epithelium, the primary target of infection in vivo. Proc.Natl.Acad.Sci.U.S.A. 2012; 109(13):5040–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xie X, Muruato A, Lokugamage KG et al. An Infectious cDNA Clone of SARS-CoV-2. Cell Host.Microbe. 2020; 27(5):841–848.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME. Airway epithelial cells: current concepts and challenges. Proc.Am.Thorac.Soc. 2008; 5(7):772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Batah SS, Fabro AT. Pulmonary pathology of ARDS in COVID-19: A pathological review for clinicians. Respir.Med. 2021; 176106239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huppert LA, Matthay MA, Ware LB. Pathogenesis of Acute Respiratory Distress Syndrome. Semin.Respir.Crit Care Med. 2019; 40(1):31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chambers E, Rounds S, Lu Q. Pulmonary Endothelial Cell Apoptosis in Emphysema and Acute Lung Injury. Adv.Anat.Embryol.Cell Biol. 2018; 22863–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nailwal H, Chan FK. Necroptosis in anti-viral inflammation. Cell Death.Differ. 2019; 26(1):4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sauler M, Bazan IS, Lee PJ. Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu.Rev.Physiol. 2019; 81375–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chan JF, Zhang AJ, Yuan S et al. Simulation of the clinical and pathological manifestations of Coronavirus Disease 2019 (COVID-19) in golden Syrian hamster model: implications for disease pathogenesis and transmissibility. Clin.Infect.Dis. 2020;5811871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Faust H, Mangalmurti NS. Collateral damage: necroptosis in the development of lung injury. Am.J Physiol Lung Cell Mol.Physiol. 2020; 318(2):L215–L225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Orzalli MH, Kagan JC. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol. 2017; 27(11):800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mifflin L, Ofengeim D, Yuan J. Receptor-interacting protein kinase 1 (RIPK1) as a therapeutic target. Nat Rev.Drug Discov. 2020; 19(8):553–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu L, Lalaoui N. 25 years of research put RIPK1 in the clinic. Semin.Cell Dev.Biol. 2021; 10986–95. [DOI] [PubMed] [Google Scholar]
- 75.Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: Mechanisms and Relevance to Disease. Annu.Rev.Pathol. 2017; 12103–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death.Differ. 2019; 26(1):99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Christofferson DE, Yuan J. Cyclophilin A release as a biomarker of necrotic cell death. Cell Death.Differ. 2010; 17(12):1942–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amin P, Florez M, Najafov A et al. Regulation of a distinct activated RIPK1 intermediate bridging complex I and complex II in TNFα-mediated apoptosis. Proc.Natl.Acad.Sci.U.S.A. 2018; 115(26):E5944–E5953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Riebeling T, Jamal K, Wilson R et al. Primidone blocks RIPK1-driven cell death and inflammation. Cell Death.Differ. 2021; 28(5):1610–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaiser WJ, Upton JW, Mocarski ES. Receptor-interacting protein homotypic interaction motif-dependent control of NF-kappa B activation via the DNA-dependent activator of IFN regulatory factors. J Immunol. 2008; 181(9):6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takaoka A, Wang Z, Choi MK et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007; 448(7152):501–505. [DOI] [PubMed] [Google Scholar]
- 82.Rebsamen M, Heinz LX, Meylan E et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-kappaB. EMBO Rep. 2009; 10(8):916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kuriakose T, Kanneganti TD. ZBP1: Innate Sensor Regulating Cell Death and Inflammation. Trends Immunol. 2018; 39(2):123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host.Microbe. 2012; 11(3):290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thapa RJ, Ingram JP, Ragan KB et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host.Microbe. 2016; 20(5):674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maelfait J, Liverpool L, Bridgeman A et al. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017; 36(17):2529–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Borba MGS, Val FFA, Sampaio VS et al. Effect of High vs Low Doses of Chloroquine Diphosphate as Adjunctive Therapy for Patients Hospitalized With Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection: A Randomized Clinical Trial. JAMA Netw.Open. 2020; 3(4):e208857. [DOI] [PubMed] [Google Scholar]
- 88.Yan Z, Deng X, Qiu J. Human Bocavirus 1 Infection of Well-Differentiated Human Airway Epithelium. Curr.Protoc.Microbiol. 2020; 58(1):e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheng F, Chen AY, Best SM et al. The capsid proteins of Aleutian mink disease virus (AMDV) activate caspases and are specifically cleaved during infection. J.Virol. 2009; 84(6):2687–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Sequencing data have been deposited in the NIH-sponsored BioProject database under accession number PRJNA698337.