

Abstract
Neisseria gonorrhoeae is naturally competent for DNA transformation. In contrast to other natural prokaryotic DNA transformation systems, single-stranded donor DNA (ssDNA) has not previously been detected during transformation of N. gonorrhoeae. We have reassessed the physical nature of gonococcal transforming DNA by using a sensitive nondenaturing native blotting technique that detects ssDNA. Consistent with previous analyses, we found that the majority of donor DNA remained in the double-stranded form, and only plasmid DNAs that carried the genus-specific DNA uptake sequence were sequestered in a DNase I-resistant state. However, when the DNA was examined under native conditions, S1 nuclease-sensitive ssDNA was identified in all strains tested except for those bacteria that carried the dud-1 mutation. Surprisingly, ssDNA was also found during transformation of N. gonorrhoeae comA mutants, which suggested that ssDNA was initially formed within the periplasm.
Neisseria gonorrhoeae is naturally competent for DNA transformation (27). Previous studies have revealed several factors that appear to be essential for this process; these include the requirement for the pilus organelle on the cell surface (27); the elaboration of various pilus-associated proteins (e.g., PilC and PilT) (2, 13, 15, 24); the production of two periplasmic proteins, ComL and TPC (10); and the incorporation of ComA into the cytoplasmic membrane (9). In addition, the absence of the RecA protein precludes the successful incorporation of donor DNA into the recipient chromosome (14). However, despite the fact that all of these factors have been shown to be required for competence through the use of defined N. gonorrhoeae mutants, their specific molecular functions in the transformation process are not well understood.
DNA transformation requires the transport of donor DNA across the cell envelope as well as the incorporation of the donor DNA into the recipient chromosome. The molecular details governing the transformation process are reasonably well understood for the naturally competent gram-positive bacteria, e.g., Bacillus subtilis and Streptococcus pneumoniae (reviewed in reference 20). In these organisms, one strand of the transforming donor DNA is degraded while the complementary strand is transported across the cytoplasmic membrane (6, 17, 20, 23). Once the donor DNA has been introduced into the cytoplasm, the nascent single-stranded DNA (ssDNA) molecule then interacts with a RecA-like protein, which facilitates exchange of the donor DNA with its homolog on the recipient chromosome. In addition, gram-positive bacteria appear capable of transporting DNA in a non-species-specific manner (20). In contrast, transformation is less well defined for the naturally competent gram-negative bacteria, e.g., Haemophilus influenzae and N. gonorrhoeae. A major difference from their gram-positive counterparts is that gram-negative bacteria appear to preferentially take up only genus-specific donor DNA, due to the requirement for a specific DNA uptake sequence (DUS) to be present on the transforming DNA molecule (5, 7, 11, 20, 26). Molecular analysis of noncompetent H. influenzae mutants suggests that, following accretion of the donor DNA onto the cell surface, the transforming DNA is first sequestered from exogenous DNase I digestion. During the subsequent transit step of the donor DNA molecule into the cytoplasm, one strand of the DNA duplex is thought to be degraded, yielding a recombinogenic ssDNA molecule within the cytoplasm (20).
In contrast to other naturally transformable bacteria, previous studies have not detected ssDNA during transformation of N. gonorrhoeae (3, 8). Initial studies detected only double-stranded DNA (dsDNA) by using a combination of isopycnic gradient analysis of DNase I-resistant 32P-labeled donor DNA and a genetic assessment of DNA-transforming activity in reisolated donor DNA preparations (1, 3). Subsequent studies, which attempted to demonstrate S1 nuclease-sensitive intermediates, appeared to corroborate these earlier findings (8). However, despite this lack of evidence for ssDNA intermediates during transformation, theoretical considerations of the mechanics of homologous recombination would seem to demand that a ssDNA intermediate be formed at some point during the transformation process (16). Therefore, either its formation in the gonococcus is transient or its detection may require a more sensitive technique.
In this study we have reassessed the nature of transforming DNA by using a sensitive nondenaturing native blotting technique to detect ssDNA. When this modification was used to study the molecular nature of transforming DNA, the results showed the formation of ssDNA following uptake of donor DNA into a DNase I-resistant state. Furthermore, the formation of ssDNA by comA mutants of gonococci suggests that its formation occurs primarily in the periplasm of the organism.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
Bacterial strains and plasmids used in this study are described in Table 1. The plasmid pRML115 was constructed by ligation of an oligonucleotide (SmaI-AAGTCTGCCGGACGTCCTAGGAAGTCTGCCGGAC-PstI) into the SmaI and PstI sites of pBluescript II SK (Stratagene, La Jolla, Calif.). The underlined nucleotides designate the gonococcal DUS. In addition, a 1.8-kb HindIII-SalI fragment containing the ermC gene flanked by opaC gene fragments was cloned into the SalI and HindIII sites of the plasmid. The plasmid pRML110 is identical to pRML115 except that it lacks the SmaI-PstI oligonucleotide containing the DUS. The plasmids pRML110 and pRML115 were isolated from Escherichia coli DH10B by using a Qiagen (Chatsworth, Calif.) column as described by the manufacturer.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant descriptiona | Source or reference |
|---|---|---|
| E. coli DH10B | recA1 | Life Technologies, Inc., Gaithersburg, Md. |
| N. gonorrhoeae | ||
| MS11 wt | Wild type; pilE7/30:2 | 31 |
| MS11 dud-1 | DNA uptake deficient; pilT | This study |
| P9 wt | Wild-type | 22 |
| P9 comA | comA | 9 |
| Plasmids | ||
| pRML110 | DUS−ermC | This study |
| pRML115 | DUS+ermC | This study |
Emr, erythromycin resistance.
N. gonorrhoeae was routinely grown on phosphate-buffered GC agar medium supplemented with 1% IsoVitaleX in a 5% CO2 atmosphere (30). E. coli was grown with Luria-Bertani medium (25), and when appropriate, the following concentrations of antibiotics were used: erythromycin, 200 μg/ml, and carbenicillin, 100 μg/ml.
DNA transformation.
DNA transformation of piliated N. gonorrhoeae was performed as previously described (32). The bacteria were grown on solid medium for approximately 18 h. Typically, 20 colonies were then transferred to 0.5 ml of broth medium containing 20 mM MgCl2. The plasmid pRML115 (DUS+) was added at a final concentration of 0.42 μg/ml, and the suspension was incubated at 37°C for 30 min. Following incubation, the cell suspension was diluted 10-fold in broth medium and incubated at 37°C for 5 h, after which aliquots were plated on agar plates both with and without 6.0 μg of erythromycin/ml. The frequency of transformation was determined by dividing the number of erythromycin-resistant CFU by the total CFU and is given in the text as the mean frequency ± standard error of the mean.
Isolation of DNase I-resistant DNA.
GC broth containing 20 mM MgCl2 (transformation medium) was prewarmed to 37°C and inoculated to a density of 107 to 108 CFU/ml with gonococci swabbed from GC agar plates after approximately 18 h of incubation at 37°C. Based on examination with a dissecting microscope, only piliated, nonopaque gonococcal colonies were used in this study. Plasmid DNA was added to a final concentration of 0.15 μg/ml, and the suspension was incubated at 37°C for 10 min. After incubation, 0.33 U of DNase I (U.S. Biochemicals, Cleveland, Ohio)/ml was added, and the medium was maintained at 37°C for 10 to 250 min prior to the isolation of DNase I-resistant DNA. Following DNase I treatment, the cells were pelleted by centrifugation for 10 min at 10,000 rpm in a Sorvall GSA rotor. The pellet was resuspended and washed three times in 10 ml of GC broth medium containing 0.5 M NaCl. The washed cells were resuspended in 10 ml of STE (25% sucrose–50 mM Tris–10 mM EDTA); 100 μl of proteinase K (20 mg/ml) and 1 ml of 10% sodium dodecyl sulfate were added, and the mixture was incubated for 10 min at room temperature. The lysates were then incubated for an additional 10 min at 60°C following the addition of 1.1 ml of 5 M NaCl and 1.5 ml of 10% cetyltrimethylammoniumbromide (CTAB)–0.7 M NaCl. Then, 5 ml of chloroform-isoamyl alcohol (24:1, vol/vol) was added, and the aqueous phase was separated by centrifugation at 5,000 rpm for 5 min in a Sorvall SS34 rotor. The aqueous phase was removed and extracted with 5 ml of phenol-chloroform (1:1, vol/vol). Following centrifugation, the aqueous phase was recovered and an equal volume of isopropanol was added to precipitate the DNA. Following centrifugation, the precipitate was resuspended in 1 ml of water containing 3 μg of RNase A (Boehringer Mannheim, Indianapolis, Ind.)/ml.
Blotting.
The concentration of DNase I-resistant DNA was determined spectrophotometrically. Typically, 50 μg of DNA from each sample was lyophilized, resuspended in 20 μl of water, and separated in a 0.7% agarose Tris-borate-EDTA gel by electrophoresis. To confirm that an equal amount of DNA was loaded on the gel for each sample, the gels were stained with ethidium bromide and the DNA was visualized with a UV transilluminator. For Southern analysis, the gel was immersed in denaturing solution (0.5 M NaOH, 1.0 M NaCl) followed by neutralization (0.5 M Tris [pH 7.4], 1.5 M NaCl) prior to transfer of the DNA to Nytran (Schleicher & Schuell, Keene, N.H.) by capillary action. A similar procedure was followed for native blotting except that the denaturation step was omitted (19). To detect pRML110 and pRML115, an ermC-specific probe was labeled with [α-32P]dATP (Dupont Corp., Boston, Mass.), using a random-primer DNA-labeling kit (Promega Corp., Madison, Wis.) as instructed by the manufacturer. Hybridization and washing were performed under stringent conditions (65°C, 0.5× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 0.1% sodium dodecyl sulfate) essentially as previously described (25). DNAs were visualized by autoradiography with X-Omat film (Eastman Kodak Co., Rochester, N.Y.).
S1 nuclease treatment.
After lyophilization, total DNA was resuspended in 20 μl of S1 nuclease buffer (33 mM sodium acetate, 50 mM NaCl, 0.03 mM ZnSO4, pH 4.5) containing 0.25 U of S1 nuclease (Boehringer Mannheim)/ml. For control reactions, the DNA was resuspended in S1 nuclease buffer without S1 nuclease. Both the control and test solutions were then incubated for 1 h at 37°C prior to electrophoresis.
RESULTS
DUS-specific uptake of plasmid DNA.
As an initial step to evaluate the molecular fate of transforming DNA, two isogenic plasmids (pRML115 and pRML110) that differed with respect to the presence of the neisserial DUS were constructed. The frequency of transformation of strains MS11 and P9 with pRML115 (DUS+) was 3.2 × 10−4 ± 1.0 × 10−4 and 2.2 × 10−4 ± 0.6 × 10−4, respectively; no transformants were obtained with pRML110 (DUS−). For comparison, the frequency of transformation of MS11 and P9 with gonococcal genomic DNA that conferred resistance to nalidixic acid was approximately 100 times greater than that obtained with pRML115 (data not shown). Piliated, nonopaque N. gonorrhoeae MS11 or DNA-uptake-deficient N. gonorrhoeae MS11 dud-1 cells were suspended in transformation medium that contained one of the two isogenic plasmids (nonopaque strains were used throughout this study to eliminate any variation in DNA uptake that could potentially result from Opa-mediated interaction with donor DNA). Donor DNA was incubated with the cells for 20 min to allow for DNA uptake. DNA uptake was then terminated by the addition of DNase I, which degraded any extracellular donor DNA (the DNase I concentrations used were subinhibitory with respect to gonococcal viability [unpublished observations]). DNase I-resistant total DNA preparations were then isolated, and the purified DNA was analyzed by Southern blotting with a radiolabeled probe that was specific for the ermC gene present on both pRML110 and pRML115. N. gonorrhoeae MS11 sequestered several electrophoretically distinct forms of pRML115 (DUS+) (Fig. 1, lane 1). Among these various DNase I-resistant DNAs, a 4.6-kb form that comigrated with a linear plasmid DNA control was observed (not shown). Analysis of the input pRML115 donor DNA preparation showed that this linear form of the plasmid was not detected in the donor DNA plasmid preparation (Fig. 1, lane 5). In addition to the linear form of the plasmid, at least four alternative forms that comigrated with the various plasmid conformational forms that were present in the input transforming donor DNA were also sequestered in a DNase I-resistant state (Fig. 1, cf. lanes 1 and 5). Collectively, these observations confirmed previous studies that demonstrated linearization of plasmid DNA following uptake into competent cells (1) as well as indicating that circular forms of plasmid donor DNAs may also enter into a DNase I-resistant state. Whether the circular forms actually entered the cytoplasm is currently unknown.
FIG. 1.
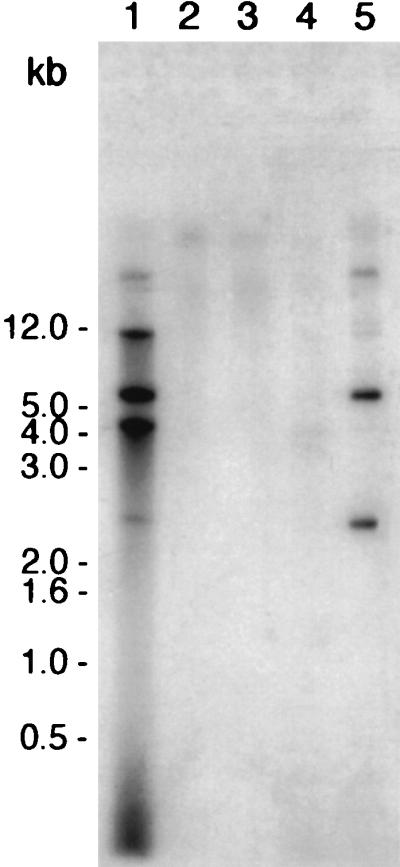
DUS-specific uptake of DNA into a DNase I-resistant state by competent gonococci. N. gonorrhoeae wild-type and N. gonorrhoeae dud-1 strains were resuspended in transformation medium that contained pRML115 (DUS+) or, as negative controls, pRML110 (DUS−) or no transforming DNA for 20 min at 37°C. Following addition of DNase I to degrade extracellular DNA, DNase I-resistant DNA was isolated and analyzed by Southern blotting with a probe specific to the ermC gene present in both pRML115 and pRML110. Lanes: 1, MS11 incubated with pRML115 (DUS+); 2, MS11 incubated with pRML110 (DUS−); 3, MS11 (no transforming DNA added); 4, MS11 dud-1 incubated with pRML115 (DUS+); 5, MS11 lysate plus pRML115 (positive control). The blot was exposed to film at −70°C for approximately 20 h.
In contrast, MS11 did not take up pRML110 (DUS−) into a DNase I-resistant state (Fig. 1, lane 2), nor did MS11 cells carrying the dud-1 mutation protect pRML115 (DUS+) from nuclease digestion (Fig. 1, lane 4). These results show that protection of transforming donor DNA from exogenous DNase I digestion in this experimental system requires both a functional pilus organelle and donor DNAs that contain the neisserial DUS.
Single-stranded DNA is formed during DNA transformation.
In order to detect ssDNA within the DNase I-resistant total DNA preparations, donor DNA isolated over various time points following the transformation of strain MS11 with pRML115 (DUS+) was examined by using a combination of Southern blotting and native blotting (Fig. 2). Piliated, nonopaque MS11 cells were suspended in transformation medium that contained pRML115 and were incubated for 10 min at 37°C. Following the DNA uptake period, DNase I was added, and large sample volumes (200 ml) were harvested after an additional 10-, 30-, and 120-min incubation period at 37°C. Consistent with the results presented in Fig. 1, Southern blotting showed the presence of several electrophoretically distinct forms of pRML115 in the DNase I-resistant samples, including (i) high-molecular-weight forms that were stable over time (120 min post-DNase I treatment) and that comigrated with the various circular forms of the plasmid (Fig. 2A) and (ii) an approximately 2.7-kb species present in the 10-min sample (Fig. 2A, lane 1) that was not detected at subsequent time points (Fig. 2A, lanes 2 and 3). When the identical samples were analyzed under native conditions, the 2.7- and 4.6-kb migrating bands were faintly visible in the 10-min sample but were not detected in subsequent samples (Fig. 2B, cf. lanes 1 to 3). In addition, a fast-migrating smear of ermC-hybridizable DNA was detected under native conditions even though the double-stranded circular forms of the plasmid, which were detected under denaturing conditions (Fig. 2A), were not detected. Consequently, these observations indicate that the various signals represent ssDNA intermediates (Fig. 2B, lanes 1 to 3). These conclusions are supported by the controls presented in Fig. 2C (where 5 ng of alkali-denatured, linear pRML115 provided a strong hybridization signal [Fig. 2C, lane 1] whereas 5 ng of double-stranded, linear pRML115 [Fig. 2C, lane 2] remained undetected) and by the fact that alkali-denatured pRML115 migrated at approximately 2.7 kb. Similar results were obtained with strain P9 and are representative of those obtained from repeated experiments under ostensibly identical experimental conditions (data not shown).
FIG. 2.

Southern blot and native blot analyses of DNase I-resistant DNA. MS11 was incubated with pRML115 (DUS+) for 10 min at 37°C. DNase I was added, samples were removed 10, 30, and 120 min after the addition of DNase I, and total nucleic acid was isolated. The DNase I-resistant DNA was subsequently analyzed by Southern blotting (A) and native blotting (B) with an ermC-specific radiolabeled probe. (C) As a control, native blotting of 5 ng of linear alkali-denatured pRML115 reacted with the ermC probe (lane 1), in contrast to 5 ng of linear double-stranded pRML115 that did not react with probe (lane 2). The arrows to the right of each panel indicate the positions of migration of both double- and single-stranded pRML115. The Southern blot and native blot were exposed to film at −70°C for approximately 18 and 36 h, respectively.
In one reiteration of the experiment, the two ssDNA species that were detected in only the 10-min sample shown in Fig. 2B (i.e., those ssDNAs with an apparent molecular size of approximately 2.7 and 4.6 kb) remained stable throughout the entire time course of the experiment (Fig. 3A). In this single experiment, the faster-migrating smear of ermC-hybridizable DNA was again prominent only in the 10-min sample. The basis for the longevity of these large-molecular-size ssDNAs in this experiment is unknown and may represent slight differences in culture conditions. Taken together, the results presented in Fig. 2 and 3 are representative of all the observations that were obtained from repeated experiments.
FIG. 3.

S1 nuclease treatment of DNase I-resistant DNA. Following transformation of strain MS11 with pRML115, samples were removed 10, 40, 70, and 130 min after the addition of DNase I. Selected DNA samples were treated with S1 nuclease prior to electrophoresis and were compared to untreated samples under native conditions with an ermC-specific probe. (A) Untreated DNA; (B) S1 nuclease-treated DNA. The arrows to the right of each panel indicate the positions of migration of both double- and single-stranded pRML115. The blots were exposed to film overnight at −70°C.
To determine if the donor ssDNA was susceptible to S1 nuclease, total DNA preparations were treated with S1 nuclease prior to electrophoresis. No difference was apparent in the hybridization pattern of DNase I-resistant DNA when assessed under denaturing conditions (data not shown), which confirmed a previous report (8) and showed that the S1 nuclease treatment did not degrade all the DNA. In contrast, S1 nuclease did degrade the ssDNA that migrated at approximately 2.7 and 4.6 kb when analyzed under native conditions (Fig. 3B, lanes 5 to 7). Surprisingly, however, the DNA comprising the smear was still detected (Fig. 3B, lanes 5 to 7). This latter observation may indicate that the DNA comprising the smear was modified in such a way that it was not susceptible to S1 nuclease or, alternatively, that so much dsDNA carrying the ermC gene fragment remained following nuclease treatment that the specificity of the native DNA blotting technique for ssDNA was compromised (note that a faint signal is detectable in Fig. 2C [lane 2] when 5 ng of donor dsDNA was assessed under native conditions). Furthermore, the presence or absence of the smeared hybridization signal was dependent upon the time allowed for uptake of the donor DNA prior to DNase I treatment, suggesting that the material that comprised these DNA species was either being utilized or degraded during the transformation process.
Formation of transforming ssDNA in MS11 comA.
MS11 comA mutants are noncompetent, presumably due to a defect in transporting donor DNA across the cytoplasmic membrane (8). To determine if ComA is involved in the formation of ssDNA during transformation, transformation assays were performed with isogenic P9 and P9 comA strains, and their donor DNA profiles were compared. Southern analysis showed that both P9 and P9 comA sequestered linear and circular forms of pRML115 (DUS+) in a DNase I-resistant state and that S1 nuclease digestion had no discernible affect on the migration of the DNA fragments as reported previously (data not shown and reference 8). However, when these same DNA samples were examined under nondenaturing conditions, various S1 nuclease-sensitive ssDNA species were observed in both P9 and P9 comA samples (Fig. 4). The smeared hybridization patterns displayed in Fig. 4 suggest that a heterogeneous mix of ssDNA molecules are produced in both cell types. However, closer inspection of the DNA species that are obtained following transformation of the P9 comA cells seems to indicate that a greater proportion of the ssDNA comigrates with the alkali-denatured linear pRML115 (DUS+) control (Fig. 4, lane 5). Furthermore, these qualitative differences between P9- and P9 comA-derived donor DNAs with respect to migration and the relative amount of ssDNA that is detected by native blotting were consistent among repeated experiments and with results obtained with MS11 and MS11 comA strains (not shown).
FIG. 4.

ssDNA is formed by comA mutants of gonococci. Following the incubation of P9 and P9 comA with pRML115 (DUS+), DNase I-resistant DNA was analyzed by native blotting with an ermC-specific probe. Lane 1, P9; lane 2, P9 treated with S1 nuclease; lane 3, P9 comA; lane 4, P9 comA treated with S1 nuclease; lane 5, alkali-denatured pRML115 (control). The blot was exposed to film at −70°C for approximately 96 h.
These results unequivocally demonstrate the formation of ssDNA in comA mutants and suggest that the competence defect associated with the comA mutation may be an inability of the ssDNA to enter the cytoplasm. (It should be noted that these cells remain recombinogenic in that they are still able to undergo pilin-antigenic variation [9, 11a].)
DISCUSSION
Previous studies of DNA transformation in the gonococcus clearly indicated that donor DNA taken up by competent cells into a DNase I-resistant state remained predominantly double stranded (1, 3, 8). These biochemical observations, in conjunction with an apparent lack of an eclipse period following DNA uptake, led to the development of a novel model for gonococcal transformation, where donor dsDNA is taken up into the cytoplasm. Nonetheless, as previously reported (3, 8), at least a portion of the incoming donor DNA was likely to be converted into a single-stranded form (if only transiently) at some point in order for recombination with the host chromosome to proceed. Despite these expectations, no evidence for ssDNA intermediates was forthcoming with the available techniques. Therefore, the purpose of this study was to reexamine the nature of donor DNA during transformation of N. gonorrhoeae by using a sensitive blotting technique that specifically detects ssDNA.
Native blotting was originally developed to detect regions of nonhomology in bacteriophage λ chromosomes following crosses where one of the participating λ chromosomes contained a sizable deletion (18, 19). Subsequently, this technique has been adapted to detect ssDNA that is formed during the transformation of Acinetobacter calcoaceticus (21) as well as in detecting ssDNA intermediates in the λ Red recombination pathway following the in vivo induction of homologous recombination (12). Through the use of native blotting combined with S1 nuclease, donor ssDNA was identified during DNA transformation of N. gonorrhoeae. Control experiments where ssDNA and dsDNA were compared by native blotting showed the specificity for detecting ssDNA in this type of analysis.
Overall, our results are consistent with previous studies that indicated that the majority of donor DNA remained in the double-stranded form (1, 3, 8). Therefore, it is not surprising that reisolated DNase I-resistant donor DNA is capable of transforming competent organisms (3). Likewise, attempts to identify ssDNAs through S1 nuclease treatment and Southern analysis (8) probably failed due to the relatively low level of ssDNA that is formed within the cell.
The lack of ssDNA when the transforming DNA did not possess a DUS, or when N. gonorrhoeae dud-1 mutants were used as recipients, indicated that the single-stranded segments were formed by gonococcal enzymes and not during the preparation of the donor DNA from E. coli. At least two predominant forms of ssDNA could be detected on the basis of differences in electrophoretic mobility, in addition to a faster-migrating smear of ermC-hybridizable material. Unfortunately, the exact molecular nature of each of these forms is unclear. The ssDNA that migrated at approximately 4.6 kb is likely to represent partially single-stranded linearized donor DNA or complementary ssDNA that partially reannealed. Heat-denatured pRML115 typically migrated at approximately 4.6 kb and is therefore consistent with this interpretation. Alkali-denatured donor DNA (Fig. 2C, lane 1) migrated at approximately 2.7 kb, and thus it seems likely that the 2.7-kb donor ssDNA detected during transformation represents fully denatured donor DNA. The smear of ermC-hybridizable material may also represent various forms of ssDNA species that migrate aberrantly due to differences in the extent of the single-stranded region that is present within each molecule. However, it remains unclear if these ssDNAs are formed via a resection process using an endogenous gonococcal exonuclease, reminiscent of the formation of ssDNA intermediates in the λ Red recombination pathway (12), or are formed through the action of a DNA helicase that unwinds the linearized duplex. The diffuse nature of the hybridization pattern argues for the former explanation, in that the smeared hybridization pattern probably represents an assortment of partial duplex and/or ssDNA plasmid molecules, each containing single-stranded regions of differing lengths. The observation that the smear is transient and disappears with time may indicate that this is the DNA that is being transported to the cytoplasm; however, the smear is also resolved with time in comA mutants, suggesting that the DNA is being degraded periplasmically in a comA-independent fashion.
Based on the results of these experiments, we cannot conclude that the ssDNA is the recombinogenically active form of transforming DNA, nor can be dismiss this possibility. Previous studies indicated that plasmid DNA is subject to restriction when introduced into a noncompatible host by DNA transformation of gonococci, whereas restriction is averted when DNA is conjugally transferred (29). These previously published results appear to support a model of gonococcal DNA transformation in which dsDNA is transported to the cytoplasm, where it would be susceptible to restriction enzymes acting specifically on dsDNA. In contrast, the apparent lack of restriction following conjugal transfer was thought to correlate with the transfer of ssDNA. However the observation that ssDNA can mediate gonococcal transformation at frequencies similar to those obtained with dsDNA (28) supports the idea that gonococci can transport ssDNA to the cytoplasm, similar to the mechanism utilized by other naturally competent bacteria. In this regard, Butler and Gotschlich demonstrated that high-frequency mobilization of broad-host-range plasmids into the gonococcus by conjugation required methylation of the donor DNA, suggesting that restriction barriers were also present during the transport of ssDNA via the conjugation route (4). Thus, the presence of restriction barriers in the transfer of DNA between gonococcal strains by transformation does not necessarily preclude a mechanism of transformation whereby ssDNA is transported to the cytoplasm; restriction could result following the formation of duplex DNA from complementary single strands in the cytoplasm.
In conclusion, we have demonstrated the formation of donor ssDNA during transformation of competent gonococci; however, it is unclear whether this DNA mediates transformation or represents the degradation of donor DNA following uptake. Additional information regarding the specificity of ComA for DNA transport and the use of additional defined gonococcal mutants is likely to be necessary to determine definitively the fate of donor DNA during DNA transformation of gonococci.
ACKNOWLEDGMENTS
We thank Thomas Meyer for the generous use of his competence-deficient strains, Kit Tilly and Jos van Putten for critical review of the manuscript, and Gary Hettrick and Bob Evans for help in making the figures.
REFERENCES
- 1.Biswas G D, Burnstein K L, Sparling P F. Linearization of donor DNA during plasmid transformation in Neisseria gonorrhoeae. J Bacteriol. 1986;168:756–761. doi: 10.1128/jb.168.2.756-761.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Biswas G D, Sox T, Blackman E, Sparling P F. Factors affecting genetic transformation of Neisseria gonorrhoeae. J Bacteriol. 1977;129:983–992. doi: 10.1128/jb.129.2.983-992.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biswas G D, Sparling P F. Entry of double-stranded deoxyribonucleic acid during transformation of Neisseria gonorrhoeae. J Bacteriol. 1981;145:638–640. doi: 10.1128/jb.145.1.638-640.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butler C A, Gotschlich E C. High-frequency mobilization of broad-host-range plasmids into Neisseria gonorrhoeae requires methylation in the donor. J Bacteriol. 1991;173:5793–5799. doi: 10.1128/jb.173.18.5793-5799.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danner D B, Deich R A, Sisco K L, Smith H O. An eleven-base-pair sequence determines the specificity of DNA uptake in Haemophilus transformation. Gene. 1980;11:311–318. doi: 10.1016/0378-1119(80)90071-2. [DOI] [PubMed] [Google Scholar]
- 6.de Vos W M, Venema G, Canosi U, Trautner T A. Plasmid transformation in Bacillus subtilis: fate of plasmid DNA. Mol Gen Genet. 1981;181:424–433. doi: 10.1007/BF00428731. [DOI] [PubMed] [Google Scholar]
- 7.Elkins C, Thomas C E, Seifert H S, Sparling P F. Species-specific uptake of DNA by gonococci is mediated by a 10-base-pair sequence. J Bacteriol. 1991;173:3911–3913. doi: 10.1128/jb.173.12.3911-3913.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Facius D, Fussenegger M, Meyer T F. Sequential action of factors involved in natural competence for transformation of Neisseria gonorrhoeae. FEMS Microbiol Lett. 1996;137:159–164. doi: 10.1111/j.1574-6968.1996.tb08099.x. [DOI] [PubMed] [Google Scholar]
- 9.Facius D, Meyer T F. A novel determinant (comA) essential for natural transformation competence in Neisseria gonorrhoeae and the effect of a comA defect on pilin variation. Mol Microbiol. 1993;10:699–712. doi: 10.1111/j.1365-2958.1993.tb00942.x. [DOI] [PubMed] [Google Scholar]
- 10.Fussenegger M, Facius D, Meier J, Meyer T F. A novel peptidoglycan-linked lipoprotein (ComL) that functions in natural transformation competence of Neisseria gonorrhoeae. Mol Microbiol. 1996;19:1095–1105. doi: 10.1046/j.1365-2958.1996.457984.x. [DOI] [PubMed] [Google Scholar]
- 11.Goodman S D, Scocca J J. Identification and arrangement of the DNA sequence recognized in specific transformation of Neisseria gonorrhoeae. Proc Natl Acad Sci USA. 1988;85:6982–6986. doi: 10.1073/pnas.85.18.6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11a.Hill, S. Unpublished observation.
- 12.Hill S A, Stahl M M, Stahl F W. Single-strand DNA intermediates in phage lambda’s Red recombination pathway. Proc Natl Acad Sci USA. 1997;94:2951–2956. doi: 10.1073/pnas.94.7.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jephcott A E, Reyn A, Birch-Andersen A. Neisseria gonorrhoeae. III. Demonstration of presumed appendages to cells from different colony types. Acta Pathol Microbiol Scand Sect B. 1971;79:437–439. doi: 10.1111/j.1699-0463.1971.tb00086.x. [DOI] [PubMed] [Google Scholar]
- 14.Koomey J M, Falkow S. Cloning of the recA gene of Neisseria gonorrhoeae and construction of gonococcal recA mutants. J Bacteriol. 1987;169:790–795. doi: 10.1128/jb.169.2.790-795.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koomey M, Fox R, Brossay L, Hebert J. Proceedings of the 9th International Pathogenesis Neisseria Conference. 1994. The gonococcal PilT protein plays an essential role in pilus-associated phenotypes of twitching motility and natural competence for transformation; p. 64. [Google Scholar]
- 16.Kowalczykowski S C, Dixon D A, Eggleston A K, Lauder S D, Rehrauer W M. Biochemistry of homologous recombination in Escherichia coli. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lacks S, Greenberg B, Carlson K. Fate of donor DNA in pneumococcal transformation. J Mol Biol. 1967;29:327–347. [Google Scholar]
- 18.Lichten M, Fox M S. Evidence for inclusion of regions of nonhomology in heteroduplex products of bacteriophage λ recombination. Proc Natl Acad Sci USA. 1984;81:7180–7184. doi: 10.1073/pnas.81.22.7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lichten M J, Fox M S. Detection of non-homology-containing heteroduplex molecules. Nucleic Acids Res. 1983;11:3959–3971. doi: 10.1093/nar/11.12.3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorenz M G, Wackernagel W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol Rev. 1994;58:563–602. doi: 10.1128/mr.58.3.563-602.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palmen R, Vosman B, Buijsman P, Breek C K D, Hellingwerf K J. Physiological characterization of natural transformation in Acinetobacter calcoaceticus. J Gen Microbiol. 1993;139:295–305. doi: 10.1099/00221287-139-2-295. [DOI] [PubMed] [Google Scholar]
- 22.Perry A C, Nicolson I J, Saunders J R. Structural analysis of the pilE region of Neisseria gonorrhoeae. Gene. 1987;60:85–92. doi: 10.1016/0378-1119(87)90216-2. [DOI] [PubMed] [Google Scholar]
- 23.Piechowska M, Fox M S. Fate of transforming deoxyribonucleate in Bacillus subtilis. J Bacteriol. 1971;108:680–689. doi: 10.1128/jb.108.2.680-689.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudel T, Facius D, Barten R, Scheuerpflug I, Nonnenmacher E, Meyer T F. Role of pili and the phase-variable PilC protein in natural competence for transformation of Neisseria gonorrhoeae. Proc Natl Acad Sci USA. 1995;92:7986–7990. doi: 10.1073/pnas.92.17.7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 26.Sisco K L, Smith H O. Sequence-specific DNA uptake in Haemophilus transformation. Proc Natl Acad Sci USA. 1979;76:972–976. doi: 10.1073/pnas.76.2.972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sparling P F. Genetic transformation of Neisseria gonorrhoeae to streptomycin resistance. J Bacteriol. 1966;92:1364–1371. doi: 10.1128/jb.92.5.1364-1371.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein D C. Transformation of Neisseria gonorrhoeae: physical requirements of the transforming DNA. Can J Microbiol. 1991;37:345–349. doi: 10.1139/m91-056. [DOI] [PubMed] [Google Scholar]
- 29.Stein D C, Gregoire S, Piekarowicz A. Restriction of plasmid DNA during transformation but not conjugation in Neisseria gonorrhoeae. J Bacteriol. 1988;56:112–116. doi: 10.1128/iai.56.1.112-116.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swanson J. Studies on gonococcus infection. XII. Colony color and opacity variants of gonococci. Infect Immun. 1978;19:320–331. doi: 10.1128/iai.19.1.320-331.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swanson J, Robbins K, Barrera O, Corwin D, Boslego J, Ciak J, Blake M, Koomey J M. Gonococcal pilin variants in experimental gonorrhea. J Exp Med. 1987;165:1344–1357. doi: 10.1084/jem.165.5.1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swanson J, Morrison S, Barrera O, Hill S. Piliation changes in transformation-defective gonococci. J Exp Med. 1990;171:2131–2139. doi: 10.1084/jem.171.6.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]