

Abstract
Abdominal aortic aneurysms (AAAs), a prototypic proteolytic cardiovascular disorder, are localized expansions of the aortal wall. Chronically upregulated and overexpressed proteases irreversibly degrade and disrupt the elastic matrix, which provides stretch and recoil properties to the aortal wall. Adult vascular smooth muscle cells are inherently unable to produce sufficient elastin to form new elastic fibers to naturally repair the aortal wall and the AAA continues to grow until fatal rupture. Surgical intervention is reserved for AAAs with a high risk of rupture, but there is currently no treatment for small, still growing AAAs. We have previously developed matrix regenerative PEG-PLGA nanoparticles (NPs) with pro-elastogenic and anti-proteolytic properties that act synergistically with a released therapeutic. However, strategies are required to effectively deliver these NPs to the disease site to avail of these benefits. We have identified cathepsin K, a protease overexpressed in AAA tissue, as a potential substrate for antibody based active targeting. We sought to assess the safety and biocompatibility of NPs with anti-cathepsin K antibodies conjugated to the NP surface (cat K Ab-NPs) and then assess their biodistribution and retention in both the targeted aorta and non-target organs in a rat AAA model. In this work, we show that cat K Ab-NPs can selectively target the aneurysmal aorta in a rat AAA model. However, there is unwanted NP uptake and retention in non-target organs that can be addressed in future work. Still, cathepsin K is a viable target for active delivery of NPs in an AAA model.
Keywords: Polymer Nanoparticles, Targeting, Aneurysm
Graphical Abstract
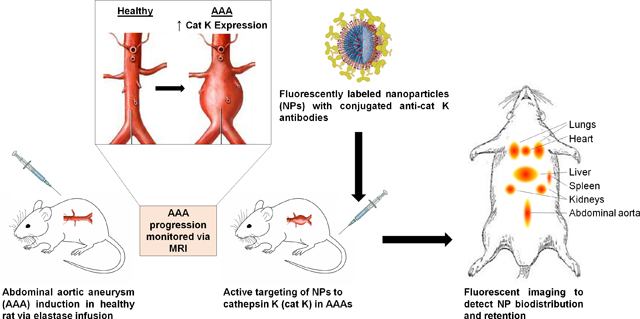
1. Introduction
Extracellular matrix (ECM)-degradative enzymes, or proteases, can be upregulated and chronically expressed in several disorders of cardiovascular tissues. This can upset the balance between the normal breakdown and remodeling of the ECM. One key component of the ECM that is particularly affected in such disorders is the elastic matrix, which provides stretch and recoil properties to blood vessels [1,2]. The elastic matrix consists primarily of elastic fibers and is especially vulnerable to proteolytic degradation by overexpressed matrix metalloproteinases (MMPs) [3,4]. The disruption and loss of the elastic matrix is generally considered naturally irreversible due to the inherent inability of adult vascular smooth muscle cells (SMCs) to produce sufficient elastin, the key protein component of elastic fibers, and their impaired ability to organize elastin precursor molecules (tropoelastin) into mature, crosslinked fibers [5,6]. This is especially evident in abdominal aortic aneurysms (AAA), a prototypic proteolytic cardiovascular disorder characterized by a localized expansion of the aortic wall which slowly grows until potential fatal rupture. Currently, there are no established pharmaceutical treatments to reverse AAA pathophysiology, including elastic matrix aberrations. Surgical intervention is only an option to treat large, rupture prone aortas, due to associated surgical risk and complication, and lack of any deterring effects on AAA growth [7]. There is a thus a pressing need to prevent the proteolytic breakdown of the elastic matrix while simultaneously stimulating new elastin production to reverse the growth of small (<50% increase in diameter), still expanding AAAs.
We have previously developed biodegradable polymer-based nanoparticles (NPs) that are designed to provide anti-proteolytic and pro-elastogenic stimuli due to both the effects of a released drug (e.g., doxycycline) and chemical moieties presented on the NP surface [8,9]. However, strategies are needed to efficiently deliver the NPs to the AAA wall. Active targeting modalities promise prospects for localized NP delivery to effectively impart their regenerative properties at the disease site and also reduce the side effects resulting from systemic drug delivery [10,11]. In active targeting, the NP surface is functionalized with a targeting moiety specific to a receptor in the disease site [11]. There are several classes of targeting moieties, each with unique advantages and disadvantages. Small molecules are low cost, stable, and easily conjugated to the NP, but typically have low specificity and affinity and there is no systematic development approach [11,12]. Aptamers are highly specific, non-immunogenic, and small in size, but they are suspect to degradation by nucleases unless they undergo complicated and expensive modification [12]. Proteins, such as antibodies, are also commonly used due to their high specificity and strong binding to substrates, however, Ab conjugation can have limitations in terms of higher cost of production and, in cases, limitations to high density presentation on the NP surface [11].
We have identified cathepsin K (cat K), a lysosomal protease overexpressed in aneurysmal tissue, as a potential substrate to target [13,14]. Previous work in our lab has shown cat K to be a viable target and antibodies against cat K can be conjugated to the surface of our NPs (cat K Ab-NPs) to act as targeting moieties [15]. In this work, we seek to study the targeting ability and efficacy of cat K Ab-NPs in a rat AAA model and study their biodistribution and retention in non-target specific organs compared to generic, non-active targeting IgG Ab conjugated NPs. Additionally, we have sought to assess the safety and biocompatibility of these cat K Ab-NPs.
2. Methods
2.1. Rat AAA Animal Model
The Institutional Animal Care and Use Committee (IACUC) at the Cleveland Clinic (ARC # 2017–1861) approved all animal procedures. The Clinic animal facility is AAALAC-approved and has animal assurance (#A3145–01). AAAs were induced by elastase infusion in adult male Sprague-Dawley rats (100–120 g, Charles River Laboratory, Wilmington, MA) and allowed to develop over a period of 3 weeks, as we have described in a prior publication [16]. Briefly, the rats were placed under general anesthesia (2% w/v isoflurane) and the infrarenal abdominal aorta surgically exposed by a laparotomy. The aorta was clamped to occlude blood flow in the infrarenal aortal region. The aorta segment between the vascular clamps was slowly infused with porcine pancreatic elastase (MP Biomedical, Solon, OH: 44 U/mL; 30 min) using a 27 ½ G needle to disrupt the elastic matrix and create a bulge in the segment. After the infusion, the clamps were removed, the intestines were replaced, the incision sites in the muscle and dermal layers were closed with 4–0 Vicyrl suture (Ethicon, Somerville, NJ) and 4–0 Monosof suture (Covidien, Dublin, Ireland), respectively. Topical tissue glue was applied along the incision. Buprenorphine was injected subcutaneously twice a day for 48 hours after the surgery for pain management. The aneurysm was allowed to develop over a period of 3 weeks. AAA induction parameters were optimized to ensure that maximal AAA diameter increases did not exceed 50%, to be consistent with the size increases seen in the small human AAAs that we ultimately seek to treat. Aneurysmal aortic SMCs (EaRASMCs) used in the in vitro safety assessments were harvested from the same model using an explant culture method and characterized as previously published [15].
2.2. Characterization of AAAs
Two independent techniques were used to track and quantify AAA formation and growth. Magnetic Resonance Imaging (MRI) was used as a non-invasive technique to measure the percent increase in diameter and volume of the aorta. The abdominal aorta was scanned just prior to, and 3 weeks post-AAA induction. Briefly, at both time points, the rats were sedated with 2.5% w/v isoflurane and positioned in a BioSpec 70/20 USR MRI System (Bruker, Billerica, MA) with the abdominal aorta in focus. Phase contrast angiography (PCA) scans were taken to visualize moving fluid (e.g. blood) without requiring contrast agents so as to obtain images of the major blood vessels, the abdominal aorta and inferior vena cava. For each rat, baseline and aneurysmal aortic volume and diameter measurements were made using Microview Parallax software (Parallax Innovations, Ilderton, ON, Canada). A 3-D rendering was created with this software as well to isolate the aorta and vena cava from the background signal for analysis. Briefly, the aneurysmal segment of the AAA and the number of image slices (i.e. the length of the segment) were identified so that an equal segment length could be measured in the baseline pre-AAA induction surgery scan to allow for accurate percent increase in aortal size calculations. The cross sectional image slices in the transverse plane were analyzed with region of interest (ROI) tools to measure the aortic diameters in the frontal and sagittal planes/direction. Additionally, the volume of the segment was measured by using ROI tools to trace the contours of the circumference of the transverse plane cross-sections along the length of the aorta segment and generate a 3D ROI with volumetric data.
To corroborate MRI measurements, stereoscope images were taken of the exposed aorta, with an adjacent scale measure, with an Olympus SZ61 Infinity 2 camera during the AAA induction surgery and before aorta excision at the endpoints of the biodistribution study. Images of the aneurysmal aorta segment and the non-aneurysmal aorta segment (below site of elastase infusion) were analyzed and diameters measured using Image J software. The two measurements were used to calculate the percent increase in aorta diameter caused by AAA induction.
2.3. In Vivo Cat K Expression
Overexpression of cat K in aneurysmal tissue in vivo was detected non-invasively by Fluorescence Molecular Tomography (FMT) following administration of a cat K activated fluorescent probe. Briefly, 12 nmol of Cat K 680 FAST (Perkin Elmer, Waltham, MA), a quenched probe that fluoresces upon cleavage of a peptide specific to cat K, was injected via the tail vein into healthy and aneurysmal rats. After 6 hours of circulation, the rats were imaged in situ with the In Vivo Imaging System (IVIS) Spectrum CT (Perkin Elmer) at the machine defined settings specific for the probe (λ = 675 nm excitation, λ = 693 nm emission). Subsequently, the rats were sacrificed and the aortas excised for imaging. Analysis of the fluorescence was performed using the Living Image Software (Perkin Elmer) to draw regions of interest (ROIs) around the samples to measure the total radiant efficiency (TRE), which is a measure of photon flux from the fluorescent probe, normalized by exposure time, area of emission, and solid angle of the detector. This measurement allows us to accurately compare fluorescence from different samples.
2.4. NP Formulation and Ab Conjugation
The NPs used in the following studies were formulated as we have previously described [8,9]. Briefly, PEG-PLGA-COOH was synthesized using free-radical polymerization and subsequently fabricated into NPs via a double-emulsion solvent evaporation technique with 1% w/v didodecyldimethylammonium bromide (DMAB; Sigma Aldrich, St. Louis, MO) as the stabilizer [17]. A rabbit anti-rat polyclonal cat K Ab (PA5–18950; Thermo Fisher, Waltham, MA) or generic IgG Ab (31220, Invitrogen, Carlsbad, CA) were conjugated to the NPs by reacting the primary amine of the Ab with the NP terminal carboxylic acid group using EDC chemistry to form cat K Ab-NPs or IgG Ab-NPs [15]. NP diameter and zeta potential were measured with a Malvern Zetasizer Nano Series to confirm that NP size and charge meet desired specifications (NP average size of 300 ± 25 nm diameter, average surface charge of 25 ± 5 mV). Vivotag 800 (745 nm ex, 820 nm em; Perkin Elmer) encapsulated NPs were similarly created for use in in vivo biodistribution studies. For cell culture experiments, we treated cell cultures with 0.5 mg/mL of NPs since this represented the midpoint of the concentration range that assured non-cytotoxicity. For experiments to assess C3 complement activation, the NPs concentrations tested were calculated based on an infusion concentration of 10 mg of NPs/mL and circulating blood volumes of between 10 and 16 mL, typical for rats. Our NPs were formulated using sterile materials and aseptic techniques, and the NP suspension was filter-sterilized.
2.5. Assessments of In Vitro NP Safety
2.5.1. NP Effects on Cell Viability
The effects of cat K Ab-NPs on cell viability was assessed using a Live/Dead cell viability assay kit (Invitrogen). EaRASMCs were seeded at 2.5 × 104 cells/well in a 12 well plate and cultured in DMEM-F12 growth medium supplemented with 10% v/v fetal bovine serum (FBS; Invitrogen) and 1% v/v Penicillin-streptomycin (Invitrogen) over 7 days. The cells were then incubated with the cat K Ab-NPs (0.5 mg/mL in the above medium) for 24 h and then stained with the Live/Dead assay kit. Images were taken using an Olympus IX51 fluorescence microscope. Cells fluorescing green are deemed viable and those fluorescing red are deemed dead.
2.5.2. NP Uptake by Macrophages
Since macrophages are present within aneurysmal wall tissue, we sought to investigate NP uptake by macrophages. Raw 264.7 macrophages (TIB-71, ATCC, Manassas, VA) were seeded at 1.5 × 104 cells/well in glass chamber slides and cultured in growth medium over 7 days. Macrophages were then polarized to the M1 phenotype via the addition of lipopolysaccharide (LPS; 100 ng/mL, Sigma Aldrich) for 24 h. The cells were serum starved for 3 h in growth medium with 1% v/v FBS and then incubated with cy3 (Lumiprobe, Hunt Valley, MD) encapsulated cat K Ab-NPs (0.5 mg/mL in growth medium) for 24 h. Cell layers were washed with PBS and then stained with CellMask Deep Red (ThermoFisher) for 15 min and fixed with 4% PFA for 15 min at room temperature. Cell nuclei were stained with and mounted with Vectashield and 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI; Vector Laboratories, Burlingame, CA) using a Leica SP8 confocal microscope.
2.5.3. NP-Induced Generation of Reactive Oxygen Species (ROS)
NP mediated Reactive Oxidative Species (ROS) and Superoxide generation was assessed using a Cellular ROS/Superoxide Detection Assay kit (Abcam, Cambridge, MA). EaRASMCs, Raw 246.7 Macrophages (polarized to the M1 phenotype, as done in section 2.5.2), and rat primary hepatocytes (Sekisui Xenotech, Kansas City, KS) were separately seeded at 104 cells/well in a black wall, clear bottom 96 well tissue culture plate, cultured in growth medium until confluent and treated with cat K Ab-NPs (0.5 mg/mL in growth medium; 200 μL) for 24 h. The assay was performed according to the kit manufacturer’s protocol with the kit. During the assay, cells deemed positive controls were incubated with the kit included ROS inducer pyocyanin and cells deemed negative controls were incubated with the kit included ROS inhibitor N-acetyl-L-cysteine.
2.5.4. Complement C3 Activation
To assess activation of complement C3 in plasma by our NPs, equivalent volumes of rat plasma (Sigma Aldrich), veronal buffer saline (Boston BioProducts, Ashland, MA), and either cat K Ab-NPs (0, 0.625, and 1 mg/mL in PBS), cobra venom factor (positive control; 50 U, Millipore Sigma) or PBS (i.e. negative control) were incubated for 30 min at 37°C [18]. Activation of Complement C3 was assessed with a commercial ELISA kit (Novus Biologicals, Centennial, CO).
2.6. NP Biodistribution
Prior to studying the regenerative effects of our NP formulation in vivo, we sought to investigate efficacy of targeting our cat K Ab-NPs to the AAA wall. Three weeks after AAA induction, rats were injected via tail vein with 500 μL suspensions of cat K Ab-NP or IgG Ab-NPs (control) in saline (10 mg of NPs/mL). The biodistribution of the cat K Ab-NPs was assessed at 1, 4, 7, or 14 days and the IgG Ab-NPs at 1 or 14 days after infusion (n = 6 rats per time point). At each designated time point, the rats were sacrificed and the organs (heart, lungs, liver, spleen, kidney, and aorta) excised and individually imaged with the IVIS Spectrum CT at the machine defined wavelengths for Vivotag 800 (745 nm ex, 820 nm em). The total radiant efficiency of each organ, as described in section 2.3, was quantified with Living Image software® (Perkin Elmer) and normalized to a negative control organ containing no NPs to eliminate background fluorescence.
2.7. Statistical Methods
In prior studies with aneurysmal SMC cultures, we showed matrix and other outcomes to exhibit near-Gaussian distribution with the distribution parameters being their mean and standard deviation [16,19,20]. In vitro cell culture experiments and in vivo animal studies were performed with 6 replicates per case, unless mentioned otherwise, as deemed appropriate to obtain necessary significant outcomes, as deemed by power analysis [21] of our published in vitro and in vivo data [16,19,20]. The Student t-test or one-way ANOVA, where appropriate, was used to analyze data since our data is discrete and sampled from a population exhibiting near-Gaussian distribution. Data is reported as mean ± standard error and differences in results were considered statistically significant if p values were ≤ 0.05 in all of the comparisons.
3. Results
3.1. AAA Characterization
Aneurysm formation was analyzed via measurements of MRI and stereoscope images for each rat (representative images in Figs. 1A and 1B). Fig. 1C shows the percent increase of aortal dimensions (volume and diameter) 3 weeks after AAA induction compared to the healthy baseline, quantified from measurements made on MRI scans. Fig. 1D shows the percent increase of aortal diameter 3 weeks after AAA induction compared to the non-aneurysmal aorta segment. The AAAs are in the range of what we consider to be small AAAs (< 50% increase in diameter).
Fig. 1.

Analysis of abdominal aortic aneurysm (AAA) induction surgeries via elastase infusion. Panel A: Representative magnetic resonance images of healthy and aneurysmal aortae. Phase contrast angiography scans provide views in the frontal and sagittal planes. The red box outlines the induced AAA 3 weeks post induction surgery. Panel B: Representative stereoscope image of the isolated AAA prior to excision at the biodistribution end point. Black arrows denote the AAA segment that was clamped and received the elastase infusion and the non-AAA segment that was inferior to the infusion site. Scale marker is 1.75 mm. Panel C: Analysis of MRI scans for percent increase in aorta dimensions 3 weeks post AAA induction surgery. Results shown are based on analysis of n = 36 aortae and represent mean ± SE. Panel D: Analysis of stereoscope images for percent increase in diameter of the aneurysmal aortal segment compared to the non-aneurysmal aortal segment. Results shown are based on analysis of n = 36 aortae and represent mean ± SE.
3.2. In Vivo Cat K Expression
The in vivo expression of cat K in a rat AAA model was evaluated using a Cat K 680 FAST probe. Results (Fig. 2) from IVIS imaging show significantly greater fluorescence and radiant efficiency values, indicative of higher cat K expression and activity, in aneurysmal rat aortae compared to healthy rat aortae.
Fig. 2.

IVIS analysis of healthy and aneurysmal aortae after injection of a Cat K 680 FAST probe to assess expression and activity of cat K in vivo. The probe is quenched until cleavage of peptides specific to cat K, so radiant efficiency is indicative of cat K expression and activity. Results shown are based on analysis of n = 3 excised aortas per group and represent mean ± SE. * denotes p = 0.04.
3.3. In Vitro NP Safety Assessments
3.3.1. NP effects on Cell Viability
NP mediated cell death was evaluated using a LIVE/DEAD assay kit. Results (Fig. 3A) show no significant cell death upon exposure to NPs.
Fig. 3.

In vitro safety assessments of cat K Ab-NPs. Panel A: Images of EaRASMC cell layers left untreated or incubated with 0.5 mg/mL cat K Ab-NPs for 24 h before washing and staining with Live/Dead reagents. Green indicates viable cells and red indicates dead cells. Images were taken at 20× and scale bar = 100 μm. Panel B: Confocal images of macrophage cell layers left untreated or incubated with 0.5 mg/mL cy3 (green) encapsulated cat K Ab-NPs for 24 h. Cell layers in both cases were washed, fixed, and stained with CellMask Deep Red (red) and DAPI (blue). Green specks or yellow within the macrophage indicates uptake of NPs by the cells. Images were taken at 40× and scale bar = 100 μm. A digital zoom was performed after image capture. Panel C: EaRASMC cell layers incubated with 0.5 mg/mL cat K Ab-NPs for 24 h experienced similar levels of oxidative stress compared to untreated cell layers. Results shown are based on analysis of n = 3 samples per group and represent mean ± SE. * denotes p < 0.0001 and ** denotes p < 0.002. Panel D: Macrophage cell layers incubated with 0.5 mg/mL cat K Ab-NPs for 24 h experienced comparable levels of oxidative stress compared to untreated cell layers. Results shown are based on analysis of n = 6 samples per group and represent mean ± SE. * denotes p < 0.0001, ** denotes p = 0.039, # denotes p = 0.006, and ## denotes p = 0.004. Panel E: Hepatocyte cell layers incubated with 0.5 mg/mL cat K Ab-NPs for 24 h experienced significantly lower ROS and superoxide production compared to the controls. Results shown are based on analysis of n = 6 samples per group and represent mean ± SE. * denotes p = 0.003, ** denotes p = 0.004, # denotes p < 0.0001, and ## denotes p = 0.0007. Panel F: Cat K Ab-NPs at two concentrations did not induce activation of C3 complement in rat plasma. Results shown are based on n = 3 and represent mean ± SE. * denotes p < 0.0001.
3.3.2. NP Uptake by Macrophages
NP uptake by macrophages was evaluated using confocal microscopy imaging of fluorescently labeled NPs (green) and fluorescently stained macrophages and nuclei (red and blue, respectively). Results (Fig. 3B) show some uptake of NPs by macrophages, indicated by the presence of green specks and yellow hue within the red-stained macrophages.
3.3.3. NP-Induced Generation of Reactive Oxygen Species (ROS)
NP-mediated ROS and superoxide production by EaRASMCs, macrophages, and hepatocytes was evaluated using a ROS/Superoxide detection assay kit. Results of EaRASMC cultures (Fig. 3C; n = 3) show significantly lower production of ROS and superoxide in untreated and NP treated EaRASMCs compared to the positive control. While there was no significant difference in ROS production between the untreated and NP treated EaRASMCs, there was significantly less superoxide production in untreated EaRASMCs compared to NP untreated. Results (Fig. 3D) show significantly lower production of ROS and superoxide in untreated and NP treated macrophages compared to the positive control. There was significantly more ROS production in untreated EaRASMCs compared to NP treated, but there was no significant difference in superoxide production between the untreated and NP treated EaRASMCs. Results (Fig. 3E) show significantly lower ROS production in NP treated hepatocytes compared to the positive control. Additionally, there was significantly lower superoxide production between both untreated and NP treated hepatocytes compared to the positive control hepatocyte cultures.
3.3.4. Complement C3 Activation
NP induced activation of complement C3 in plasma samples was evaluated using an ELISA assay. Results (Fig. 3F) show no significant difference in active complement C3 between the NP treated samples and the negative control. However, there was significantly lower amounts of active C3 complement in the NP treated and negative control compared to the positive control containing an inducer of active C3 complement.
3.4. Cat K Ab-NP Active Targeting and Biodistribution
The effect of cat K antibodies as a targeting moiety to actively deliver NPs to the aneurysmal site for long term retention was assessed. Additionally, the biodistribution and retention of the NPs in the major organs was evaluated. Generic IgG Ab modified NPs were used as controls to assess active targeting. Results (Fig. 4A) show that after one day of NP circulation, there is significant NP uptake by non-target organs- lungs, liver, spleen, and kidneys. Antibody type has no effect on NP uptake in the non-target organs. At the 1 day time point, the presence of cat K Ab-NPs in the AAA segment was apparently higher compared to the IgG Ab-NPs, although the differences were deemed not statistically significant. Results (Fig. 5A) show that after 14 days of NP circulation, there is significant NP uptake by non-target organs- lungs, liver, spleen, and kidneys. Antibody type again has no effect on NP uptake in the non-target organs. However, there is a significant increase in the presence of cat K Ab-NPs in the aorta compared to the IgG Ab-NPs. Results (Fig. 6A) show that the presence of cat K Ab-NPs remains fairly consistent over the studied 14 days, suggesting that the NPs are taken up after 1 day of circulation and are retained up to the studied 14 days.
Fig. 4.

Comparison of the biodistribution of Vivotag800 encapsulated cat K Ab-NPs and IgG Ab-NPs after 1 day of NP circulation. Comparisons were made between antibody types and to the negative controls. Panel A: Total radiant efficiency of each test organ, indicative of NP presence, was normalized to negative control organs to control for background fluorescence. Results shown are based on analysis of n = 6 excised samples per group and represent mean ± SE. # denotes p < 0.05 compared to the Negative Control. Panel B: Representative images of each analyzed organ.
Fig. 5.

Comparison of the biodistribution of Vivotag800 encapsulated cat K Ab-NPs and IgG Ab-NPs after 14 days of NP circulation. Comparisons were made between antibody types and to the negative controls. Panel A: Total radiant efficiency of each test organ, indicative of NP presence, was normalized to negative control organs to control for background fluorescence. Results shown are based on analysis of n = 6 excised samples per group and represent mean ± SE. # denotes p < 0.05 compared to the Negative Control and * denotes p = 0.01 compared to IgG Ab-NP samples. Panel B: Representative images of each analyzed organ.
Fig. 6.
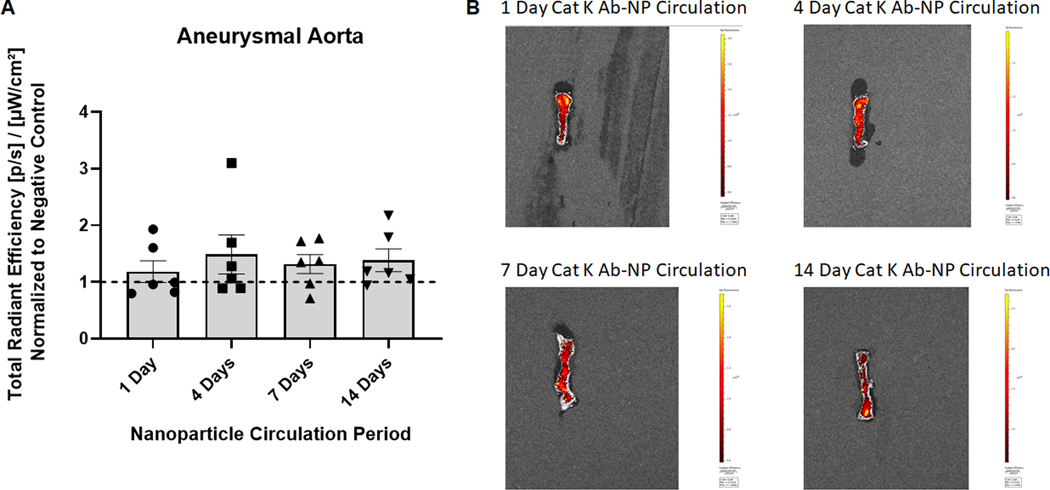
Cat K Ab-NP retention in target organ over time. Panel A: Total radiant efficiency of each aorta at their respective time point was normalized to negative control organs to eliminate background fluorescence. Cat K Ab-NPs were injected via tail vein and circulated for 1, 4, 7, and 14 days to assess NP targeting and retention over time. Results shown are based on analysis of n = 6 aortae per group and represent mean ± SE.
4. Discussion
The chronic overexpression of matrix degrading proteases in the aorta wall is a key determinant of AAA pathophysiology [1,4]. In particular, the significant overexpression of cathepsin K that has been reported in the aneurysmal wall [14,15,22], but not significant in healthy aortae or in circulation [23], presents a unique opportunity to actively target our matrix regenerative NPs to the AAA wall to locally impart regenerative benefits while reducing systemic effects. Furthermore, cat K is expressed early in AAA pathophysiology, making it an attractive target to deliver NPs to small, still growing AAAs. Additionally, targeting cat K also inhibits its activity, thus aiding in decreasing proteolytic activity in the AAA microenvironment separate from the effects of the NP. In a prior publication, we determined significant overexpression of cat K in an aneurysmal smooth muscle cell (SMC) cultures compared to healthy cells, suggesting its utility as a substrate for targeted binding of NPs, and showed the cat K to be expressed on the surface of the cells [15]. We also showed that conjugating anti-cat K antibodies to the NP surface was useful to significantly enhance NP binding and retention on aneurysmal SMCs to effect long term functional (pro-elastogenic and anti-proteolytic) benefits in in vitro cultures. In this work, we sought to assess the safety and biocompatibility of the cat K Ab-NPs and then assess their targeting efficacy in vivo in a rat model of AAAs.
While there are several known animal models of AAAs [24], none exhibit all known characteristics of human AAAs. The rat elastase injury model was chosen due to its similarity to human AAAs [25] and because the sub-primary SMCs generated from primary aneurysmal SMC isolates maintain their diseased phenotype in culture in a manner similar to human AAA SMCs [16,20]. Key pathophysiologic aspects of clinically manifested small infrarenal aortic aneurysms, including disrupted medial elastic lamellae, vessel expansion, inflammatory and immune cell infiltration, and chronic overexpression and activity of elastolytic proteases, are also present in the elastase injury rat model. This has been described both in literature [25] and confirmed by our own prior studies [16,20]. MRI and stereoscope imaging (Figs. 1A and 1B) were utilized to confirm the formation of AAAs and to quantify the increase in aortal dimensions compared to the healthy baseline before the induction surgery. Analysis of MRI scans (Fig. 1C) of healthy, pre-elastase injured aorta and the aorta 3 weeks post injury allows for the quantification of increases in the diameter of the aorta in two different anatomical planes as well as volumetric increases. Vessel wall expansions in aneurysms are not always regular in shape, so the ability to also analyze increases in volume is advantageous. In this study, the percent increase of the aortal diameter after AAA induction was fairly consistent in the frontal and sagittal aortal plane and was in the range of size increases typical of small human aneurysms (< 50% increase in diameter). We specifically seek to target small AAAs for treatment because a) endovascular interventions are not effective in arresting or regressing their growth, b) risk of rupture is too low to justify open surgery which has high procedural risk, and c) no other drug treatments are available that can be administered to slow or arrest their growth during their slow growth to rupture. Stereoscope images of the aorta were taken at the time of excision for analysis (Fig. 1D) of the increase in diameter of aneurysmal segment compared to the non-aneurysmal segment. By comparing different segments of the aorta at the same time point, we can control for any additional growth in rats with the longer biodistribution end points. Each condition saw the required ~50% increase in aortal diameter and thus suits our needs for this animal model. Both methods of imaging confirm the induction of a small AAA.
Studies in both humans [13,14,22,26–28] and in animal models of AAAs [15,29], have provided evidence that cathepsin K is specifically overexpressed with no significant increases in plasma levels. This provides an opportunity for targeting our NPs to the AAA wall with cat K antibodies. While our lab has previously shown the overexpression of cat K in cell cultures derived from aneurysmal rats [15], we sought to confirm in vivo cat K overexpression in our rat model using a Cat K 680 FAST probe. However, IVIS imaging of whole rats proved difficult since they tended to grow to a large size in the post-AAA induction and post-intervention periods and had substantial auto-fluorescence of many peripheral organs. We thus had to excise and image the aortae to isolate the fluorescence from the probe and found cat K was significantly overexpressed in the aorta of rats with induced AAAs compared to healthy controls (Fig. 2). This work and previous literature supports the use of cat K as a substrate to actively target with our matrix regenerative NPs.
Before introducing the NPs into the animal disease model, the safety and biocompatibility of the NPs was analyzed with several in vitro assessments [30]. Fluorescent staining of cell layers was used to complement quantitative assays to provide a more comprehensive analysis of NP- induced cytotoxicity. Per the Live/Dead staining (Fig. 3A), cat K Ab-NPs did not induce cell death after incubation with EaRASMCs, the target cells the NPs are expected to interact with in the AAA wall in our rat model. This result was consistent with the limited production and generation of cytotoxic/apoptosis-inducing ROS and superoxides in EaRASMCs after incubation with NPs (Figs. 3C and 3D). EaRASMCs have been thoroughly characterized in our lab [8,16] and have been shown to be stable with consistent growth rates, therefore triplicate cultures are sufficient to show statistical significance. Macrophages are recruited to and are resident in the AAA wall and can clear NPs by phagocytosis to significantly reduce their therapeutic benefit and such phagocytosis can also trigger ROS or superoxide generation to cause cell cytotoxicity. We showed that while there is some limited uptake of NPs by macrophages (Fig. 3B), it was not sufficient to generate ROS or superoxides. The average size of our NPs (~300 nm in diameter) aids in limiting uptake by macrophages since particles typically larger than 500 nm are phagocytosed [31]. To ensure statistical significance, macrophages were cultured as n = 6 replicates to account for any variability from LPS polarization to the M1 phenotype. Although NPs accumulated in the liver, we found that the NPs did not induce ROS or superoxide production in hepatocytes, which were also cultured as n = 6 replicates to account for any variability. However, efforts still need to be made to reduce the uptake of NPs in non-target organs to increase targeting efficacy and efficiency. Since our route of administration is i.v., we also assessed the effects of cat K Ab-NPs on the activation of C3 complement, and thus the immune response, within plasma [18]. Two doses of NPs were assessed and chosen based on the concentration of circulating NPs in the rat model (0.625 mg/mL blood) and the maximum concentration recommended (1 mg/mL). Neither dose induced significant activation of C3 complement compared to the basal levels of C3 complement in the PBS vehicle/negative control so we can infer that the NPs are not inducing an immune response (Fig. 3E). The results of the safety assessments demonstrate the suitability of our NP formulation in an in vivo application.
Previous works have sought to deliver nanoparticles to AAAs to modulate the disease process, but our approach in this study differs in a) the AAA model used, b) the stage of AAA progression in which intervention is planned, c) type of NP and targeting moiety and substrate, and d) the pathophysiologic pathway that is targeted by nanotherapy. Our model of luminal elastase infusion invokes disease progression starting from the lumen and progressing out to the adventitia, which mimics clinical AAAs, as opposed to models with periadventitial application of calcium chloride. We seek to target small still growing AAAs, whose pathophysiology includes denuding of the endothelial layer and limited, but ongoing, degradation of the elastic matrix. We also seek to not only stop the growth of AAAs, but also regenerate the vessel wall by stimulating elastogenesis. Accordingly, our targeting strategy is uniquely designed to account for these conditions and hence differs from prior approaches that sought to deliver NPs appropriately surface-functionalized for active targeting to substrates such as a) αvβ3 expressed by activated luminal endothelial cells that remain intact when the AAA is induced by periadventitial injury as published in [32] or the exposed elastin protein core of highly disrupted elastic fibers in advanced AAAs wherein elastic matrix breakdown is significant [33], unlike the small AAAs we are seeking to treat wherein elastic matrix breakdown is still ongoing with regions of intact elastic fibers and lamellae. Active targeting of NPs can be achieved with a variety of ligands, but antibodies have well-documented success as a targeting moiety [11,34,35] and have been chosen as a means to validate the use of cat K as a targeting substrate for in vivo NP delivery. IgG Ab-NPs were formulated to serve as a control to determine the ability of cat K Ab-NPs to specifically target the aneurysmal aorta and to elucidate the effects of antibodies on the non-target organ uptake of NPs.
After 1 day of circulation (Fig. 4A), there was significant uptake of both cat K Ab-NPs and IgG Ab-NPs in non-target organs (heart, lungs, liver, spleen, and kidneys) but this was to be expected due to their role in blood filtration. The PEG groups on the NP surface serve to impart stealth properties on the NP and to inhibit protein adhesion in efforts to improve circulation time [36]. Extended time points were studied to determine if the NPs would be able to effectively clear these organs and continue to circulate. While not completely efficient in terms of avoiding uptake into other organs, our results do confirm that cat K Ab conjugation improves AAA wall uptake of the NPs relative to generic IgG-conjugated NPs. However, NP uptake in non-target organs was not significantly affected by the antibody type, which indicates that cat K antibodies are not contributing to active NP targeting to non-aorta organs, and by inference that cat K is not upregulated in these other organs. After 14 days of circulation (Fig. 5A), both cat K Ab-NPs and IgG Ab-NPs were present in non-target organs, but only cat K Ab-NPs reached the aorta. In the aorta, the cat K Ab-NPs appear to enter the AAA wall within 24 h of infusion, and were completely retained on site for up to 14 days (Fig. 6), suggesting their ultimate utility to effect long-term release of drug after a single NP dosing event. That said, although the radiant efficiency of Cat K Ab-NP appears higher, there is no significant difference to the negative control (NP-untreated AAAs). We believe this is related to the dose of NPs administered, and that statistically significant differences between the Cat K Ab NPs and negative controls will be obtained if the infused NP dose were to be significantly increased. The single, initial dose we tested was selected based on related literature in the field [37] and previous work in our lab [15]. Our planned follow up study will 1) study biodistribution and targeting as a function of dose and the efficacy of NPs delivering a drug, and 2) utilize a greater number of animals and higher doses to tighten the data points. This is an area to improve upon with future work to either reduce the uptake of NPs by non-target organs or to improve their clearance from these organs back into blood circulation. Despite this promise, the efficacy of NP targeting to the AAA wall is limited by a) significant uptake and retention in non-target organs, b) limited NP uptake into the AAA wall on an absolute basis. To address these issues, we propose several possible strategies which we will investigate sequentially in follow up studies. These include a) optimizing PEG surface density to reduce opsonization and improve circulation time while also providing additional binding sites to increase the presentation of targeting moieties [38,39], b) utilizing a different targeting moiety to avoid the disadvantages of antibody based targeting- namely the complex pharmacokinetics, prohibitive cost of scale up, and regulatory concerns, and c) investigating synthetic peptides based on the active binding site of the cat K propeptide, which is known to bind and inactivate mature cat K [40,41] as an alternative targeting moiety.
5. Conclusions
In this work, we have developed an MRI protocol to non-invasively quantify AAAs in a rat elastase infusion model that corroborates previously used stereoscope measurements while also providing a new means to characterize AAAs with volumetric analysis. We also assessed the safety and biocompatibility of an active targeting NP formulation and supported the selection of cathepsin K as a delivery target. This work provides evidence that cat K antibodies can successfully target and deliver matrix regenerative nanoparticles to the aneurysmal site. However, future work can improve upon these results. The NP surface density of PEG can be increased to take advantage of its pro-circulation properties while also providing additional binding sites for targeting moieties. Additionally, alternative targeting moieties, such as synthetic peptides, can be pursued to improve targeting efficiency while reducing costs and simplifying regulatory hurdles. However, cathepsin K remains a viable substrate for the active targeting of our matrix regenerative NPs in proteolytic cardiovascular disorders.
Statement of Significance.
We have previously developed elastic matrix regenerative polymer nanoparticles (NPs), but require strategies to efficiently target the disease site. Antibodies against cathepsin K, an overexpressed protease in abdominal aortic aneurysms, have been conjugated to the NP surface to act as a targeting moiety. In this work, we assessed NP safety and in vivo biodistribution in an aneurysmal rat model and demonstrated positive targeting and retention for up to 2 weeks within the aortal wall.
Acknowledgements
This work was supported by funding obtained through NIH grant HL139662, NSF grant 1926939 awarded to A.R., and grants from the American Heart Association 19TPA34890029 (A.R.) and 20PRE35120023 (S.C). Dr. Charlie Androjna from Cleveland Clinic’s Biomedical Engineering Department provided exceptional support and training on the use of MRI equipment and image analysis. This work utilized the Leica SP8 confocal microscope, purchased with funding from NIH SIG grant 1S10OD019972-01.
Footnotes
Author Disclosure Statement
No competing financial interests exist
References
- [1].Brauer PR, MMPs--role in cardiovascular development and disease, Front. Biosci. J. Virtual Libr 11 (2006) 447–478. [DOI] [PubMed] [Google Scholar]
- [2].Korpos E, Wu C, Sorokin L, Multiple roles of the extracellular matrix in inflammation, Curr. Pharm. Des 15 (2009) 1349–1357. [DOI] [PubMed] [Google Scholar]
- [3].Ahmed SH, Clark LL, Pennington WR, Webb CS, Bonnema DD, Leonardi AH, McClure CD, Spinale FG, Zile MR, Matrix metalloproteinases/tissue inhibitors of metalloproteinases: relationship between changes in proteolytic determinants of matrix composition and structural, functional, and clinical manifestations of hypertensive heart disease, Circulation. 113 (2006) 2089–2096. [DOI] [PubMed] [Google Scholar]
- [4].Raffetto JD, Khalil RA, Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease, Biochem. Pharmacol 75 (2008) 346–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sivaraman B, Bashur CA, Ramamurthi A, Advances in biomimetic regeneration of elastic matrix structures, Drug Deliv. Transl. Res 2 (2012) 323–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Johnson DJ, Robson P, Hew Y, Keeley FW, Decreased Elastin Synthesis in Normal Development and in Long-term Aortic Organ and Cell Cultures Is Related to Rapid and Selective Destabilization of mRNA for Elastin, Circ. Res 77 (1995) 1107–1113. [DOI] [PubMed] [Google Scholar]
- [7].Aggarwal S, Qamar A, Sharma V, Sharma A, Abdominal aortic aneurysm: A comprehensive review, Exp. Clin. Cardiol 16 (2011) 11. [PMC free article] [PubMed] [Google Scholar]
- [8].Sivaraman B, Ramamurthi A, Multifunctional nanoparticles for doxycycline delivery towards localized elastic matrix stabilization and regenerative repair, Acta Biomater. 9 (2013) 6511–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Camardo A, Seshadri D, Broekelmann T, Mecham R, Ramamurthi A, Multifunctional JNK -inhibiting nanotherapeutics for augmented elastic matrix regenerative repair in aortic aneurysms, Drug Deliv. Transl. Res 8 (2018) 964–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Couvreur P, Nanoparticles in drug delivery: Past, present and future, Adv. Drug Deliv. Rev 65 (2013) 21–23. [DOI] [PubMed] [Google Scholar]
- [11].Friedman AD, Claypool SE, Liu R, The Smart Targeting of Nanoparticles, Curr. Pharm. Des 19 (2013) 6315–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yoo J, Park C, Yi G, Lee D, Koo H, Active Targeting Strategies Using Biological Ligands for Nanoparticle Drug Delivery Systems, Cancers. 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liu C-L, Guo J, Zhang X, Sukhova GK, Libby P, Shi G-P, Cysteine protease cathepsins in cardiovascular disease: from basic research to clinical trials, Nat. Rev. Cardiol 15 (2018) 351. [DOI] [PubMed] [Google Scholar]
- [14].Sukhova GK, Shi G-P, Do Cathepsins Play a Role in Abdominal Aortic Aneurysm Pathogenesis?, Ann. N. Y. Acad. Sci 1085 (2006) 161–169. [DOI] [PubMed] [Google Scholar]
- [15].Jennewine B, Fox J, Ramamurthi A, Cathepsin K-targeted sub-micron particles for regenerative repair of vascular elastic matrix, Acta Biomater. 52 (2017) 60–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gacchina CE, Deb P, Barth JL, Ramamurthi A, Elastogenic Inductability of Smooth Muscle Cells from a Rat Model of Late Stage Abdominal Aortic Aneurysms, Tissue Eng. Part A 17 (2011) 1699–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Seshadri DR, Immuno-nanotherapeutics to Inhibit Macrophage Polarization for Non-Small-Cell Lung Cancers, Case Western Reserve University, 2017. https://etd.ohiolink.edu/pg_10?::NO:10:P10_ETD_SUBID:156749#abstract-files (accessed December 6, 2019). [Google Scholar]
- [18].Neun BW, Ilinskaya AN, Dobrovolskaia MA, Analysis of Complement Activation by Nanoparticles, in: McNeil SE (Ed.), Charact. Nanoparticles Intend Drug Deliv., Springer; New York, New York, NY, 2018: pp. 149–160. [DOI] [PubMed] [Google Scholar]
- [19].Gacchina C, Brothers T, Ramamurthi A, Evaluating smooth muscle cells from CaCl2-induced rat aortal expansions as a surrogate culture model for study of elastogenic induction of human aneurysmal cells, Tissue Eng. Part A 17 (2011) 1945–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Deb PP, Ramamurthi A, Spatio-Temporal Mapping of Matrix Remodeling and Evidence of in-situ Elastogenesis in Experimental Abdominal Aortic Aneurysms, J. Tissue Eng. Regen. Med 11 (2017) 231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Thompson S, Basic and clinical biostatistics. Beth Dawson-Saunders and Robert G. Trapp, Appleton and Lange, Connecticut, 1990. ISBN: C-8385–4541-4, Stat. Med. 10 (1991) 1636–1637. [Google Scholar]
- [22].Lohoefer F, Reeps C, Lipp C, Rudelius M, Haertl F, Matevossian E, Zernecke A, Eckstein H-H, Pelisek J, Quantitative expression and localization of cysteine and aspartic proteases in human abdominal aortic aneurysms, Exp. Mol. Med 46 (2014) e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bromme AO,D, Role of Cathepsin K, L and S in Blood Vessel Remodeling, in: Bush R. (Ed.), Aneurysmal Dis. Thorac. Abdom. Aorta, InTech, 2011. [Google Scholar]
- [24].Anidjar S, Salzmann JL, Gentric D, Lagneau P, Camilleri JP, Michel JB, Elastase-induced experimental aneurysms in rats., Circulation. 82 (1990) 973–981. [DOI] [PubMed] [Google Scholar]
- [25].Halpern VJ, Nackman GB, Gandhi RH, Irizarry E, Scholes JV, Ramey WG, Tilson MD, The elastase infusion model of experimental aortic aneurysms: synchrony of induction of endogenous proteinases with matrix destruction and inflammatory cell response, J. Vasc. Surg 20 (1994) 51–60. [DOI] [PubMed] [Google Scholar]
- [26].Lohoefer F, Reeps C, Lipp C, Rudelius M, Zimmermann A, Ockert S, Eckstein H-H, Pelisek J, Histopathological analysis of cellular localization of cathepsins in abdominal aortic aneurysm wall, Int. J. Exp. Pathol 93 (2012) 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sukhova GK, Shi GP, Simon DI, Chapman HA, Libby P, Expression of the elastolytic cathepsins S and K in human atheroma and regulation of their production in smooth muscle cells, J. Clin. Invest 102 (1998) 576–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Reeps C, Lohöfer F, Eckstein HH, Rudelius M, Pelisek J, Cellular expression of cathepsin proteases in symptomatic and asymptomatic AAA, in: Schumpelick V, Bruch H-P, Schackert HK (Eds.), Chir. Forum DGAV Forum 2009, Springer, Berlin, Heidelberg, 2009: pp. 313–315. [Google Scholar]
- [29].Sun J, Sukhova GK, Zhang J, Chen H, Sjöberg S, Libby P, Xia M, Xiong N, Gelb BD, Shi G-P, Cathepsin K deficiency reduces elastase perfusion-induced abdominal aortic aneurysms in mice, Arterioscler. Thromb. Vasc. Biol 32 (2012) 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Savage DT, Hilt JZ, Dziubla TD, In Vitro Methods for Assessing Nanoparticle Toxicity, in: Zhang Q. (Ed.), Nanotoxicity Methods Protoc., Springer; New York, New York, NY, 2019: pp. 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mitragotri S, Lahann J, Physical approaches to biomaterial design, Nat. Mater 8 (2009) 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cheng J, Zhang R, Li C, Tao H, Dou Y, Wang Y, Hu H, Zhang J, A Targeting Nanotherapy for Abdominal Aortic Aneurysms, J. Am. Coll. Cardiol 72 (2018) 2591–2605. [DOI] [PubMed] [Google Scholar]
- [33].Nosoudi N, Nahar-Gohad P, Sinha A, Chowdhury A, Gerard P, Carsten CG, Gray BH, Vyavahare NR, Prevention of Abdominal Aortic Aneurysm Progression by Targeted Inhibition of Matrix Metalloproteinase Activity with Batimastat-Loaded Nanoparticles, Circ. Res 117 (2015) e80–e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dawidczyk CM, Kim C, Park JH, Russell LM, Lee KH, Pomper MG, Searson PC, State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines, J. Controlled Release 187 (2014) 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Byrne JD, Betancourt T, Brannon-Peppas L, Active targeting schemes for nanoparticle systems in cancer therapeutics, Adv. Drug Deliv. Rev 60 (2008) 1615–1626. [DOI] [PubMed] [Google Scholar]
- [36].Blanco E, Shen H, Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol 33 (2015) 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sinha A, Shaporev A, Nosoudi N, Lei Y, Vertegel A, Lessner S, Vyavahare N, Nanoparticle targeting to diseased vasculature for imaging and therapy, Nanomedicine Nanotechnol. Biol. Med 10 (2014) e1003–e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vasconcelos A, Vega E, Pérez Y, Gómara MJ, García ML, Haro I, Conjugation of cell-penetrating peptides with poly(lactic-co-glycolic acid)-polyethylene glycol nanoparticles improves ocular drug delivery, Int. J. Nanomedicine 10 (2015) 609–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hak S, Helgesen E, Hektoen HH, Huuse EM, Jarzyna PA, Mulder WJM, Haraldseth O, de L C. Davies, The Effect of Nanoparticle Polyethylene Glycol Surface Density on Ligand-directed Tumor Targeting Studied in vivo by Dual Modality Imaging, ACS Nano. 6 (2012) 5648–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Billington CJ, Mason P, Magny M-C, Mort JS, The Slow-Binding Inhibition of Cathepsin K by Its Propeptide, Biochem. Biophys. Res. Commun 276 (2000) 924–929. [DOI] [PubMed] [Google Scholar]
- [41].Guay J, Falgueyret J-P, Ducret A, Percival MD, Mancini JA, Potency and selectivity of inhibition of cathepsin K, L and S by their respective propeptides: Propeptide inhibition of cathepsins K, L and S, Eur. J. Biochem 267 (2000) 6311–6318. [DOI] [PubMed] [Google Scholar]