

Abstract
Purpose of review
The discovery of several genetic variants associated with erythroid traits and subsequent elucidation of their functional mechanisms are exemplars of the power of the new genetic and genomic technology. The present review highlights findings from recent genetic studies related to the control of erythropoiesis and dyserythropoiesis, and fetal hemoglobin, an erythroid-related trait.
Recent findings
Identification of the genetic modulators of erythropoiesis involved two approaches: genome-wide association studies (GWASs) using single nucleotide polymorphism (SNP) arrays that revealed the common genetic variants associated with erythroid phenotypes (hemoglobin, red cell count, MCV, MCH) and fetal hemoglobin; and massive parallel sequencing such as whole genome sequencing (WGS) and whole exome sequencing (WES) that led to the discovery of the rarer variants (GFI1B, SBDS, RPS19, PKLR, EPO, EPOR, KLF1, GATA1). Functional and genomic studies aided by computational approaches and gene editing technology refined the regions encompassing the putative causative SNPs and confirmed their regulatory role at different stages of erythropoiesis.
Summary
Five meta-analysis of GWASs identified 17 genetic loci associated with erythroid phenotypes, which are potential regulators of erythropoiesis. Some of these loci showed pleiotropy associated with multiple erythroid traits, suggesting undiscovered molecular mechanisms and challenges underlying erythroid biology. Other sequencing strategies (WGS and WES) further elucidated the role of rare variants in dyserythropoiesis. Integration of common and rare variant studies with functional assays involving latest genome-editing technologies will significantly improve our understanding of the genetics underlying erythropoiesis and erythroid disorders.
Keywords: erythroid traits, erythropoiesis, genetic variants
INTRODUCTION
Healthy human adults produce approximately 2.5 × 1011 new red blood cells (RBCs) per day and clear an equal number of RBCs [1]. Failure to maintain this output and clearance results in anemias of various sorts and erythrocytosis [2]. Maintenance of this rapid turnover requires efficient erythropoiesis through proliferation and differentiation of the immature erythroid progenitor population, a progeny of self-renewing stem cells [2]. Early studies routinely measured various erythroid parameters (Table 1) with the purpose of deciphering the genetic variability, as well as underlying genes and sequence variants associated with erythropoiesis, during health and disease [3]. These erythroid parameters vary significantly among individuals, with 40–90% of this phenotypic variation being heritable [4∎∎]. Genetic and environmental factors, as well as age and sex, contribute to this variability as confirmed by early twin studies [5–7]. Of the erythroid traits, fetal hemoglobin (HbF) showed the highest genetic heritability [3]. The present review provides an update on how common and rare genetic variations affect the process of erythropoiesis, and how integration of experimental (functional) and bioinformatics approaches can enhance our understanding on the genetic control of erythropoiesis.
Table 1.
Main erythrocyte phenotypes (traits) routinely measured in clinic
| Erythroid trait | Definition | Unit |
|---|---|---|
|
| ||
| Mean corpuscular hemoglobin (MCH) | Average mass of hemoglobin per red blood cell | Picogram (pg) |
| Mean corpuscular volume (MCV) | Average volume of red blood cells | Femtoliter (fL) |
| Red blood cell (RBC) count | Count of red blood cells per microliter | Million cells per microliter (×l06/μL) |
| Hemoglobin (Hb) | Hemoglobin concentration | Gram per deciliter (g/dL) |
| Hematocrit (Het) | Percentage of the blood volume occupied by erythrocytes | Percentage (%) |
| MCH concentration (MCHC) | Average concentration of hemoglobin in red blood cells (hemoglobin divided by hematocrit) | Gram per deciliter (g/dL) |
| RBC distribution width (RDW) | Distribution of red blood cell volume | Percentage (%) |
GENOME-WIDE ASSOCIATION STUDIES AND IDENTIFICATION OF COMMON VARIANTS AFFECTING ERYTHROPOIESIS
Identification of variants regulating fetal hemoglobin: an exemplar of genome-wide association studies
HbF is an erythroid parameter that has been the focus of much of the recent research efforts, as its induction ameliorates clinical symptoms associated with the β-hemoglobinopathies [8]. In normal healthy adults, HbF is present at residual levels (<0.6%) restricted to a small number of erythrocytes termed F cells. The percentage of F cells and HbF are highly correlated (R2 = 0.97). HbF and F cells are quantitative traits, with a predominantly genetically controlled heritability of 0.87 [3], compared to the other erythroid traits (Hb, Hct, RBC, MCV, MCH, MCHC) that were measured in the same twin study. HbF varies considerably (up to 20-fold) in healthy adults; the variable increases are amplified in patients with sickle cell anemia and β-thalassemia. This variable persistence of HbF in adults constitutes the historical entity of heterocellular hereditary persistence of fetal hemoglobin (HPFH); multiple genes, together with a small environmental component, determine the HbF value measured in any individual. Xmn1-HBG2 (rs7482144) on chromosome 11p, HBS1L-MYB intergenic region on chromosome 6q23, and BCL11A on chromosome 2p16 are the three major quantitative trait loci (QTL) for HbF, contributing to the complex inheritance of heterocellular HPFH. The remaining variation (‘missing heritability’) is likely to be accounted for by many loci with relatively small effects, and/or rare variants with significant quantitative effects on gamma-globin gene expression that are typically missed by GWAS population studies [8,9]. One such rare variant is KLF1 (also known as EKLF) [10∎∎].
Identification of variants regulating erythropoiesis
The HBS1L-MYB locus on chromosome 6q23 was first shown to have a pleiotropic effect on non-HbF erythroid traits in 2007 [11]. Subsequently, five independent meta-analyses [12–16] involving a range of multiethnic cohorts (Table 2), identified 17 common variants, including HBS1L-MYB locus (Table 3), associated with erythropoiesis and erythroid disorders. Of these, five were previously discovered QTLs (HFE, TFR2, TMPRSS6, HBS1L-MYB, and BCL11A), and 11 were novel, some of which were annotated for genes known to be involved in iron homeostasis (TFRC) and erythropoiesis (ABO, CCND3, CITED2, SH2B3, SPTA1). Of the 17 loci, HBS1L-MYB locus, ITFG3, TMPRSS6, and glucose-6-phosphate dehydrogenase (G6PD) showed greatest pleiotropy, achieving genome-wide significant associations with five erythroid traits (Table 3).
Table 2.
Meta-analysis studies researched for common variants associated with erythroid traits for this review
| Ancestry | Sample number (n) | Erythroid traits | Number of significantly associated loci | Meta-analysis study reference |
|---|---|---|---|---|
|
| ||||
| 6 European cohorts | 13943 | RBC, Hb, MCH, MCHC, MCV | 22 | (HaemGen Consortium) [15] |
| 6 European Cohorts | 24167 | RBC, Hb, Het, MCH, MCHC, MCV | 23 | (CHARGE consortium) [13] |
| 10 Japanese disease groups | 14700 | RBC, Hb, Het, MCH, MCHC, MCV | 49 | [14] |
| 32 European and 3 South Asian Cohorts | 71861 | RBC, Hb, Het, MCH, MCHC, MCV | 75 | [16] |
| 7 African American cohorts | 16500 | RBC, Hb, Het, MCH, MCHC, MCV, RDW | 7 | (COGENT network) [12] |
Table 3.
Genomic loci associated with erythroid traits and erythropoiesis
| Gene or genomic region | Chromosome | Erythroid trait | Meta-analysis study reference |
|---|---|---|---|
|
| |||
| HBS1L-MYB | 6 | Het, MCH, MCHC, MCV, RBC | [13–16] |
| ITFG3 | 16 | Hb, MCH, MCHC, MCV, RBC | [12,13] |
| TMPRSS6 | 22 | Hb, Het, MCH, MCHC, MCV | [13–16] |
| G6PD | X | Het, Hb, MCV, RBC, RDW | [12] |
| HFE | 6 | Hb, Het, MCH, MCV | [13,15,16] |
| ABO | 9 | Hb, Het, RBC, MCHC | [14] |
| CCND3 | 6 | MCH, MCV, RBC | [13–16] |
| TFR2 | 7 | Het, MCV, RBC | [13,15,16] |
| TFRC | 3 | MCH, MCV | [13,16] |
| PDGFRA-KIT | 4 | MCV, RBC | [13,14,16] |
| CITED2 | 6 | MCH, MCV | [13,14,16] |
| HMOX2 | 16 | MCH, MCV | [12] |
| SPTA1 | 1 | MCHC | [13] |
| BCL11A | 2 | MCV | [13,16] |
| PRKCE | 2 | Het | [12] |
| CD 164 | 6 | MCH | [12] |
| SH2B3 | 12 | Hb | [16] |
HBS1L-MYB locus on chromosome 6q
High-resolution genetic mapping refined the 6q QTL to single nucleotide polymorphism (SNPs) in two clusters at −84 and −71 kb, respectively, upstream of MYB, one of the flanking genes. Functional studies in transgenic mice and primary human erythroid cells provide overwhelming evidence that the SNPs at these two regions disrupt binding of key erythroid enhancers affecting long-range interactions with MYB and MYB expression, providing a functional explanation for the genetic association of the 6q HBS1L-MYB intergenic region with levels of HbF and F cell as well as other erythroid traits. The MYB transcription factor is a key regulator of hematopoiesis and erythropoiesis, and modulates the erythroid traits via two mechanisms: indirectly through alteration of the kinetics of erythroid differentiation; low MYB levels accelerate erythroid differentiation leading to release of early erythroid progenitor cells that are larger, and still synthesizing predominantly HbF; and directly via activation of KLF1 and other repressors (e.g. nuclear receptors TR2/TR4) of gamma-globin genes [17–19]. The polymorphisms at this locus have an occupancy of erythroid TFs (TAL1/SCL, E47, GATA1, and RUNX1/AML1) critical for erythroid cell differentiation [17].
The HBS1L-MYB intergenic enhancers do not appear to affect expression of HBS1L, the other flanking gene. Further, in-vitro cellular studies also excluded HBS1L as having a role in the regulation of HbF and erythropoiesis [20]. In whole-exome sequencing of rare uncharacterized disorders, loss-of-function mutations in the HBS1L gene were identified in a female child [21]. The child had normal blood counts and normal HbF levels.
BCL11A on chromosome 2p
Functional studies in primary human erythroid progenitor cells and transgenic mice demonstrated that BCL11A acts as a repressor of gamma-globin gene expression that is effected by SNPs in intron 2 of this gene [22]. Fine-mapping demonstrated that these HbF-associated variants, in particular, rs1427407 and rs7606173, localized to an enhancer that is erythroid-specific and not functional in lymphoid cells. BCL11A interacts with several co-repressor complexes occupying discrete regions in the HBB complex leading to reconfiguration of the locus [23,24]. The composite BCL11A erythroid-specific enhancer has three DNase I hypersensitive sites (DHSs) at +55, +58, and +62 kb from the transcription start site of this gene, with DHS at +58 having the greatest effect on HBG gene expression [22,25].
Experimental studies so far have shown that BCL11A deficiency results in HbF induction with minimal effect on erythropoiesis [26∎,27∎]. Although these studies, which were performed on mice models, showed no major perturbation in erythropoiesis, there were reductions in Hct, RBC, and Hb values. However, 2 BCL11A SNPs (rs2540917 and rs243070) have also been associated with MCV (Table 3), suggesting that BCL11A is a candidate gene for erythrocyte variation resulting from the perturbation of erythropoiesis, as supported from a recent finding that BCL11A is indispensable for hematopoietic stem cell (HSC) function [28].
Variants associated with iron homeostasis and heme metabolism
Iron metabolism and erythropoiesis are closely related; iron being essential for hemoglobin synthesis during terminal erythropoiesis [29∎]. Genetic factors have a significant impact on iron homeostasis, defects in iron metabolism result in hereditary anemias and iron overload [29∎]. Early GWASs have implicated variants in the iron regulatory genes – TMPRSS6, HFE, TFR2, TFRC (Table 3) – in the variability of various erythroid traits. TMPRSS6 inhibits hepcidin, which is essential for iron absorption and effective erythropoiesis. Complete loss-of-function mutations of TMPRSS6 result in a rare disorder of iron-refractory iron deficiency anemia (IRIDA) [29∎]. Genetic variants in TMPRSS6 may lead to iron deficiency anemia in individuals with or without other predisposing factors [30]. The hereditary hemochromatosis (HFE) protein is a key component of the signaling pathway through which transferrin stimulates hepcidin synthesis [31,32], thereby modulating erythropoiesis by affecting dietary iron absorption and erythroid iron intake [33]. Transferrin receptors TFRC (also called TFR1) and TFR2 play a critical role in erythropoiesis. TFRC is one of the most abundant membrane proteins of the erythroblasts [34] and plays a dominant role in the delivery of transferrin-bound iron from the blood to developing erythroid precursors in the bone marrow, making it essential for erythropoiesis. TFRC deficiency results in defective hemoglobinization and anemia. TRF2 is a component of EPO (erythropoietin) receptor (EPOR) complex in erythroid cells that modulates EPO sensitivity and maintains the balance between the RBC production and iron availability [35]. TFR2 knockdown has shown to delay terminal differentiation, leading to inefficient erythropoiesis [36].
Another component of iron-regulatory system is HMOX2, which has an important role in erythroid differentiation and erythropoiesis [37]. The lead SNP rs7192051, discovered in the meta-analysis involving African-American ancestry, is located in the second intron of HMOX2, and is associated with lower MCH and MCV [12] (Table 3). However, validation of this association failed in two independent population-based African-American and European (CHARGE consortium) samples. Interestingly, another HMOX2 SNP, rs4786504, is associated with an adaptive trait among Tibetans who live in high-altitude hypoxic conditions, where the variant is associated with increase in HMOX2 expression, facilitating the breakdown of heme and helping to maintain low hemoglobin levels at high altitude [38].
ABO blood group polymorphisms
The ABO blood group antigen system is complex; several weak phenotypes or subgroups are caused by SNPs mostly attributed to the coding variants [39–41]. However, there are ABO subgroups that are not related to variants in coding regions or splicing sites. For example, a positive regulatory element in intron 1 appears to enhance the activity of the ABO promoter in an erythroid cell–specific manner, through binding of erythroid-specific TF GATA binding protein 1 (GATA1) [42]. The ABO antigen is initially expressed on cells derived from erythroid blast colony forming units and colony forming units, with a gradual increase in expression during erythroid maturation [43,44]. Recent GWASs have identified the ABO gene locus as being significantly associated with erythroid traits (Table 3) [14,45,46]. Of the two SNPs (rs495828 and rs8176746) discovered in the Japanese GWAS study, rs8176746 which is nonsynonymous, serves as one of the deterministic variants of the B-antigen, and is associated with increase in erythroid parameters [14,46]. This SNP has also been mapped to the erythroid enhancer regions marked by p300 and colocalized erythroid transcription factors (TFs) KLF1 and TAL1 [47], suggesting a possible regulation of this gene during erythroid cell differentiation. Thus, it seems that the erythroid cell-specific regulatory activity of ABO expression is dependent on the binding of GATA1 and its co-factors (KLF1 and TAL1) during erythropoiesis [42]. In this context, the ABO SNPs discovered from GWAS, and their association with erythroid traits (Hb, Hct, and RBC) may further explain the molecular basis of the subphenotyes associated with this blood group during erythropoiesis.
CCND3 and CITED2
Cyclin D3 (CCND3) plays a key role in HSC expansion; its absence in mice results in ineffective erythropoiesis and anemia [48]. CCND3 variants (rs3218097, rs9349205, rs11970772, rs9349204) have also been implicated in their association with erythroid traits (MCV, MCH, RBC) by four different meta-analysis studies (Table 3). The region surrounding SNP rs9349205 was determined to be the region of erythroid enhancer element of CCND3, with TAL1, GATA1, and KLF1 chromatin occupancies [49]. The follow-up functional studies involving CCDN3 knockout animal models and knockdowns involving humans ex vivo models confirmed that CCND3 regulates the number of cell divisions during terminal erythropoiesis and that reduced levels of CCND3 correlate with fewer terminal erythroid cell divisions, resulting in fewer but larger terminally differentiated erythrocytes [49].
GWAS meta-analyses also revealed significant association of Cbp/P300 Interacting Transactivator With Glu/Asp Rich Carboxy-Terminal Domain 2 (CITED2) variants (rs628751, rs632057, rs643381, rs590856, rs632057) with erythroid traits MCH and MCV (Table 3). CITED2 is a master regulator of stem cell fate with a key role in the adult HSC maintenance [50]. A proper coordination among growth factors EPO, SCF, Forkhead box O3 (FOX3A), and STAT5 is essential for the induction of CITED2 expression and regulation of gene expression program in erythroblasts [51]. Also, CITED2 regulates iron homeostasis and erythropoiesis via hypoxiainducible factor 1-alpha (HIF1A) and GATA1 [52,53].
SH2B Adaptor Protein 3 polymorphism
A nonsynonymous SNP in SH2B3 gene (rs318504), which results in R262W substitution, is significantly associated with high hemoglobin levels (Table 3) [16]. SH2B3 negatively regulates hematopoietic cytokine signaling [54]. Targeted suppression of SH2B3 expression in mice primary hematopoietic stem and progenitor cells (HSPCs) improves erythroid expansion and differentiation, and increases hemoglobin [55]. In humans, targeted SH2B3 suppression and inactivation facilitated erythroid expansion and maturation by augmenting both the EPO and KIT signaling pathways [56∎∎]. Thus, SH2B3 deficiency can enhance erythropoiesis in vitro and production of RBCs for transfusion purposes. Genetic variants of SH2B3 at the population-level have been associated with increased RBC levels, thus recapitulating the results of the in-vitro functional studies.
Other genome-wide association studies identified common variants and their role in erythropoiesis
Three SNPs (rs218237, rs172629, rs218238) in the PDGFRA-KIT intergenic region were found to be associated with erythroid traits MCV and RBC (Table 3). These three SNPs are located in the intergenic region downstream of the platelet-derived growth factor receptor α polypeptide gene (PDGFRA) and upstream of the human homolog of the proto-oncogene c-kit gene (KIT). Until recently, the function of PDGFRA on erythropoiesis was unknown. A recent study [57] has identified PDGFRA as a negative regulator in erythroid differentiation, and is part of the miR-146b, PDGFRA, and GATA1 regulatory circuit. KIT has a primary role in erythropoiesis. Considering their close proximity, we speculate that the three SNPs influence MCV and RBC through their regulation of PDGFRA and KIT.
Rare intronic and low frequency nonsynonymous coding variants in ITFG3 (also known as FAM234A) are African-American-specific and were found to be associated with various erythroid traits (Table 3: Hb, MCH, MCHC, MCV, RBC). Although the role of ITFG3 in erythropoiesis is unknown, the variants of this gene encompass the α-globin (HBA2-HBA1) locus and can be disrupting long-range enhancers of alpha-globin [58∎∎].
Two SNPs, rs762516 and rs1050828, in G6PD on the X chromosome have been associated with multiple erythroid traits (Table 3: Hct, Hb, MCV, RBC, RDW); rs1050828 is associated with increased risk of anemia in African-American women [12]. G6PD is indispensible for erythropoiesis and its deletion or deficiency results in accelerated erythropoiesis with increased red cell deformability in fetal erythrocytes and abrogation of the embryonic-adult hemoglobin developmental switch [59,60].
Similarly, variants in spectrin (SPTA1), protein kinase C epsilon (PRKCE) and endolyn (CD164) are associated with erythroid traits through their effect on erythropoiesis. An intronic (rs857721) [13] as well as a nonsynonymous variant (rs857725) in SPTA1 [61] are associated with MCHC. Mutations in SPTA1 are implicated in erythroid differentiation, regulation of cell cycle, and ineffective erythropoiesis [62,63]. PRKCE variant rs10495928 is associated with Hb and Hb-related traits in African Americans, Europeans, and Japanese [12]. Similarly, CD164 variant (rs9386791) is associated with lower MCH in African Americans, whereas other variants are associated with RBC, MCH, and MCV in Japanese (rs11966072) and with MCV in Europeans (rs9374080). This suggests ethnic-specific allelic heterogeneity for erythroid traits for these two genes.
Apart from the meta-analysis studies reported in this review, a separate GWAS study [61] identified genetic loci in genes THRB, PTPLAD1, CDT1, EPO, ALDH8A1, FBXO7, associated with erythroid traits that are known to play role in erythroid differentiation and cell-cycle regulation.
RARE VARIANTS: IDENTIFICATION AND STRATEGIES
The role of common genetic variation in hematological traits has been well characterized with the expansion of GWAS-based consortia, but rare genetic variants with minor allele frequency less than 1%, although with large effect size were typically not discovered in GWASs [64]. Recently, DNA sequencing-based approaches and custom arrays have been deployed on a large scale to comprehensively evaluate the contribution of rare genetic variants to complex traits and diseases [65].
Kruppel-like factor 1 variants
Kruppel-like factor 1 (KLF1) is a master regulator of erythropoiesis, regulating approximately 700 erythroid genes involved in wide array of molecular mechanisms [10∎∎,66,67]. Red cell disorders have rarely been attributed to KLF1 variants, until high-throughput DNA sequencing identified numerous sporadic cases, prompting population surveys. Since 2008, a range of hematologic phenotypes associated with KLF1 variants have been identified including inconsequential In(Lu) type of Lu(a-b-) blood group [68], increases in HbF as a primary phenotype or secondary to other red cell disorders [67,69], severe dyserythropoietic anemia [70], and an extreme case of hydrops fetalis [71]. Since 2010, more than 65 different KLF1 variants have been identified; these variants have varied effects on the severity of ineffective erythropoiesis and their clinical significance [10∎∎]. Although KLF1 variants appeared to be a ‘common’ variant and associated with milder thalassemia in southern China (where β-thalassemia is prevalent) compared with a northern Chinese population [72], several GWASs of HbF, including ones in sickle cell anemia patients of African descent, have failed to identify common variants in KLF1 [73,74].
GATA binding protein 1 variants
GATA1 encodes a transcription factor required for erythroid differentiation [75]. Exome sequencing in two male siblings with Diamond-Blackfan anemia (DBA) has identified mutations (a G → C transversion) in the exon 2 at a splice site that impaired production of the full-length form of the protein [76]. Interestingly, a missense mutation consisting an identical G → C transversion in the zinc fingers of GATA1 resulted in dyserythropoietic anemias and thrombocytopenias [77–79]. Although most studies attribute DBA pathogenesis to a defective ribosomal biogenesis [80], the discovery of GATA1 mutations reveal the potential for other plausible underlying mechanisms and basis for this erythroid disorder. A methodical sequencing approach of other DBA cases will further reveal the scope and extent to which GATA1 mutations contribute to this disease.
Targeted next-generation sequencing
Targeted next-generation sequencing is popular due to its cost-efficiency, and provides rapid and accurate mutation analysis. This approach has been utilized in the detection of novel mutations associated with rare congenital anemias. For example, targeted sequencing of genes from the Oxford Red Cell Panel (ORCP) [81∎] on a patient previously diagnosed with DBA, resulted in the discovery of hypomorphic mutation in the Shwachman Bodian Diamond syndrome (SBDS) gene and a revised diagnosis of Shwachman–Diamond syndrome (SDS). Similar corrections and revisions in diagnosis include a revision of Congenital Dyserythropoietic Anemia type I (CDA-1) to DBA with the discovery of a mutation in RPS19, previously shown to have a mechanistic role in erythropoiesis. Targeted resequencing using ORCP on a patient with an initial diagnosis of CDA revealed a mutation in Pyruvate Kinase, Liver And RBC (PKLR) gene and revised the diagnosis to Pyruvate Kinase deficiency. Mutations of the EPOR have been documented in families with isolated familial erythrocytosis [82]. Using targeted re-sequencing on a family of 33 individuals, the first EPOR mutation was confirmed [83]. This discovery propelled further research that led to the discovery of ten different truncating mutations as well as several point mutations [84]. Thus, targeted resequencing of a carefully curated panel of genes not only serves as an essential diagnostic tool for clinical purposes, but also provides new insights on the role of unsuspected genetic variants in the regulation of erythropoiesis.
Whole-exome sequencing
Whole-exome and targeted re-sequencing approaches have been used to identify rare, loss (or gain)-of-function coding variants segregating within families with hematologic traits at the extremes of the phenotypic distribution. Whole exome sequencing (WES) approach, involving large population-based cohorts phenotyped for hematological traits, is in developmental stages. One notable example is a study by Polfus et al. [85∎∎] where WES association analyses of hematologic quantitative traits in 15,459 individuals from European and African-American ancestry have discovered rare synonymous variant in GFI1B (rs150813342), with the follow-up knockdown experiments in primary human HSPCs revealing an alternative splicing mechanism wherein rs150813342 variant suppresses the long isoform of GFI1B that is indispensable for megakaryopoiesis and not the short isoform, which is indispensable for erythropoiesis [85∎∎]. A recent study examining WES data from a cohort of more than 450 patients with a clinical diagnosis of DBA, led to the discovery of a homozygous recessive mutation in EPO which resulted in an R150Q substitution in the mature EPO protein, affecting the erythroid differentiation and proliferation [86].
Exome arrays
Large-scale studies, such as the 1000 Genomes Project and Exome Sequence Project (ESP), have catalogued coding DNA sequence variants, facilitating the study of these rare variants using standard genotyping arrays. Exome-wide genotyping arrays (exome chips) are now commercially available, and although computationally less challenging to analyze, they are not as comprehensive as the NGS technologies which may result in missing a large amount of very rare genetic variation. Furthermore, the exome arrays are based mostly on sequence data from the European population, hence rare variants in other populations may be missed. Despite these limitations, exome chips have already been used successfully to identify rare coding variants associated with erythroid traits. A recent exome array study has identified a rare low-frequency missense variant in the erythropoietin gene EPO (rs62483572) in the high-affinity receptor binding site and associated with lower Hct and Hb values [64]. Another study, consisting of meta-analyses of seven RBC phenotypes in multiethnic individuals from studies genotyped on an exome array, have discovered rare variants associated with erythroid traits, such as MAP1A (for Hb), HNF4A (for Hct and Hb), CD36 (for RBC), and ALAS2 (for MCV) [4∎∎].
CONCLUSION
GWASs have been extremely successful in uncovering thousands of associations between common variants and complex traits as well as diseases, but much of the heritability of these traits remains unexplained and unexplored. Within the Mendelian erythroid diseases, such as SCD and β-thalassemia, most of the discoveries from GWASs were focused on the erythroid trait HbF [9,87,88,89∎]. Although this is a significant achievement when compared to GWAS-identified loci for all other traits, there is a scope for further identification and characterization of common as well as rare variants modulating HbF levels. Meta-analysis of GWASs, as well as recent rare variant studies, revealed novel loci annotated for genes known to be involved in erythropoiesis (Fig. 1). GWAS-identified trait-associated noncoding variants have small effect size, and thus the impact on the biological processes is often unknown. One way to address this challenge is to develop assays for high-throughput functional screening of GWAS loci, and complement the results with genome-editing in gene modulation assays. Another approach is to comprehensively evaluate the contribution of rare genetic variants by DNA sequencing, followed by functional characterization. Ultimately, integration of data from both common and rare variant studies, and follow-up gene functional assays will provide further insights on how the genetic variation in erythroid traits affects erythropoiesis (Fig. 2).
FIGURE 1.
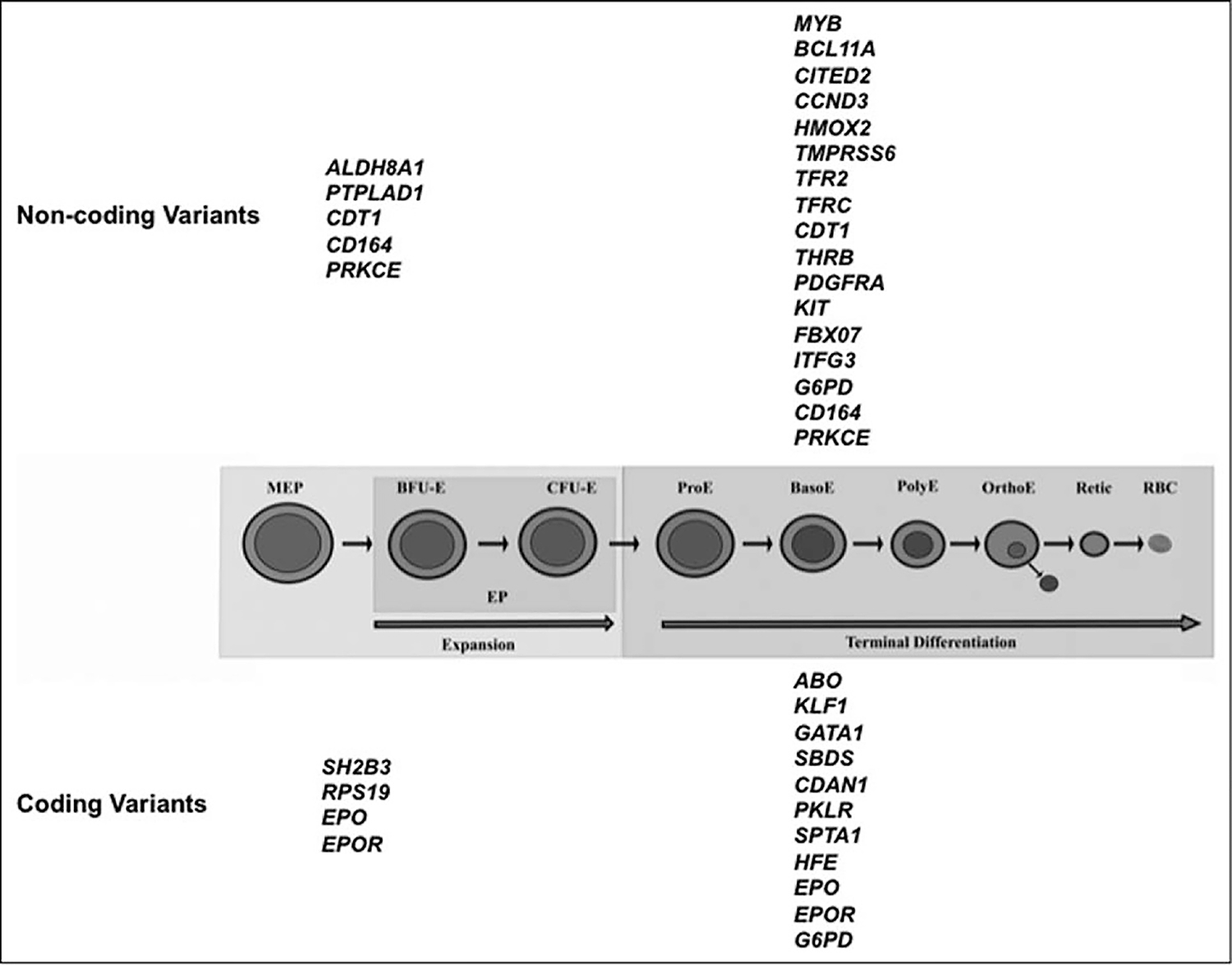
Genes implicated in erythropoiesis are associated with erythroid traits. Different stages of erythroid differentiation, from the megakaryocyte erythroid progenitor (MEP) to the mature red blood cell (RBC), are shown here: Megakaryocyte erythroid progenitor; BFU-E, blast colony forming unit-erythroid; CFU-E, colony forming unit-erythroid; ProE, proerythroblast; BasoE, basophilic erythroblast; PolyE, polychromatic erythroblast; OrthoE, orthochromatic erythroblast; Retic, reticulocyte. Genes associated with noncoding variants are shown at the top, and genes associated with coding variants are shown at the bottom.
FIGURE 2.
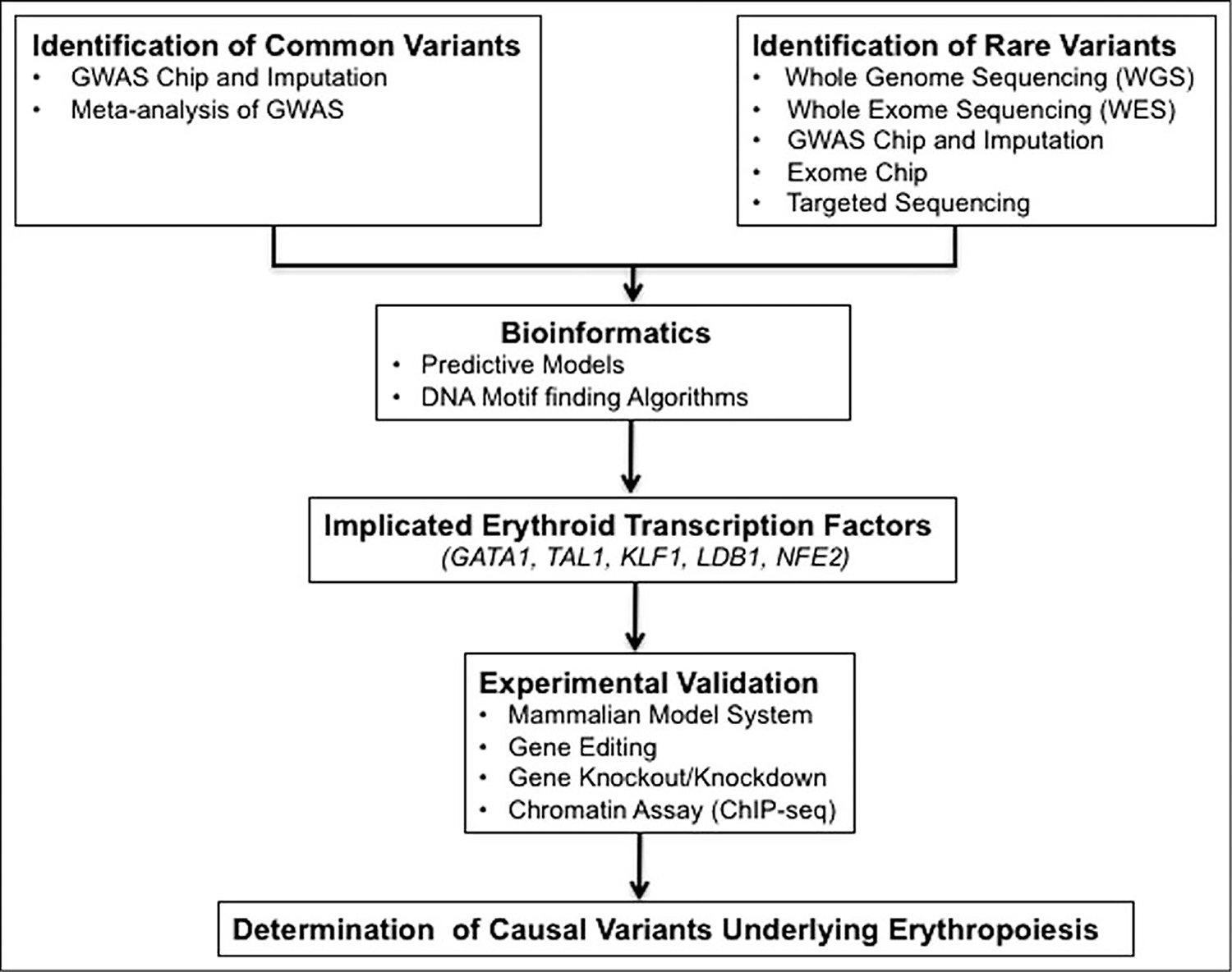
Model of the integrated approach for the genetic determinants of erythropoiesis.
KEY POINTS.
Erythroid traits are heritable and clinically important biomarkers but much of the heritability of these traits remains unaccounted for.
Understanding how genetic variants modulate erythroid traits in health (and disease) can provide us with new insights into the mechanistic underpinnings of erythropoiesis.
GWASs and meta-analyses of GWAS data have identified robust associations between many common variants and erythroid traits in healthy and disease states, but these variants have small effect size, and their impacts on biological process such as erythropoiesis remain uncertain.
Meta-analyses of GWAS data, as well as recent rare variant association studies have also identified novel genetic loci that showed pleotropic association with erythroid traits and were annotated for genes (ABO, CCND3, CITED2, SH2B3, SPTA1, GFI1B, SBDS, RPS19, PKLR, EPO, EPOR, KLF1, GATA1) involved in erythropoiesis.
Integration of common and rare variant studies with functional assays involving latest genome-editing technologies will significantly improve our understanding of the genetics underlying erythropoiesis and erythroid disorders.
Acknowledgements
We would like to thank Rusinel Amarante for her help in preparation of the manuscript.
Financial support and sponsorship
This work was supported by the Intramural Research Program of the National Heart, Lungs, and Blood Institute, NIH.
Footnotes
Conflicts of interest
There are no conflicts of interest.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
∎ of special interest
∎∎ of outstanding interest
- 1.Higgins JM, Mahadevan L. Physiological and pathological population dynamics of circulating human red blood cells. Proc Natl Acad Sci USA 2010; 107:20587–20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell 2008; 132:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garner C, Tatu T, Reittie JE, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood 2000; 95:342–346. [PubMed] [Google Scholar]
- 4.∎∎. Chami N, Chen MH, Slater AJ, et al. Exome genotyping identifies pleiotropic variants associated with red blood cell traits. Am J Hum Genet 2016; 99:8–21. This study presents the multiethnic meta-analyses of seven erythroid traits, expanding the list of rare and low-frequency coding or splice site variants associated with erythroid traits, and demonstrating the utility of ExomeChip for the genetic discovery, which can complement the GWAS approach to fine map erythroid causal genes.
- 5.Dal Colletto GM, Fulker DW, Barretto OC, Kolya M. Genetic and environmental effects on blood cells. Acta Genet Med Gemellologiae 1993; 42:245–252. [DOI] [PubMed] [Google Scholar]
- 6.Evans DM, Frazer IH, Martin NG. Genetic and environmental causes of variation in basal levels of blood cells. Twin Res 1999; 2:250–257. [DOI] [PubMed] [Google Scholar]
- 7.Whitfield JB, Martin NG. Genetic and environmental influences on the size and number of cells in the blood. Genet Epidemiol 1985; 2:133–144. [DOI] [PubMed] [Google Scholar]
- 8.Thein SL, Menzel S, Lathrop M, Garner C. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet 2009; 18:R216–R223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet 2007; 39:1197–1199. [DOI] [PubMed] [Google Scholar]
- 10.∎∎. Perkins A, Xu X, Higgs DR, et al. Kruppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood 2016; 127:1856–1862. Comprehensive review sheds light on to the broad range of previously unrelated human erythroid disorders caused by variants in KLF1, which were hitherto considered to be extremely rare causes of human genetic disease.
- 11.Menzel S, Jiang J, Silver N, et al. The HBS1L-MYB intergenic region on chromosome 6q23.3 influences erythrocyte, platelet, and monocyte counts in humans. Blood 2007; 110:3624–3626. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Tang H, Qayyum R, et al. Genome-wide association analysis of red blood cell traits in African Americans: the COGENT Network. Hum Mol Genet 2013; 22:2529–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganesh SK, Zakai NA, van Rooij FJ, et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet 2009; 41:1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kamatani Y, Matsuda K, Okada Y, et al. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet 2010; 42:210–215. [DOI] [PubMed] [Google Scholar]
- 15.Soranzo N, Spector TD, Mangino M, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat Genet 2009; 41:1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Harst P, Zhang W, Mateo Leach I, et al. Seventy-five genetic loci influencing the human red blood cell. Nature 2012; 492:369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stadhouders R, Aktuna S, Thongjuea S, et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J Clin Invest 2014; 124:1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchi E, Zini R, Salati S, et al. c-Myb supports erythropoiesis through the transactivation of KLF1 and LMO2 expression. Blood 2010; 116:e99–e110. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki M, Yamamoto M, Engel JD. Fetal globin gene repressors as drug targets for molecular therapies to treat the beta-globinopathies. Mol Cell Biol 2014; 34:3560–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang J, Best S, Menzel S, et al. cMYB is involved in the regulation of fetal hemoglobin production in adults. Blood 2006; 108:1077–1083. [DOI] [PubMed] [Google Scholar]
- 21.Sankaran VG, Joshi M, Agrawal A, et al. Rare complete loss of function provides insight into a pleiotropic genome-wide association study locus. Blood 2013; 122:3845–3847. [DOI] [PubMed] [Google Scholar]
- 22.Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science (New York, NY) 2013; 342:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sankaran VG, Xu J, Ragoczy T, et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature 2009; 460:1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jawaid K, Wahlberg K, Thein SL, Best S. Binding patterns of BCL11A in the globin and GATA1 loci and characterization of the BCL11A fetal hemoglobin locus. Blood Cells Mol Dis 2010; 45:140–146. [DOI] [PubMed] [Google Scholar]
- 25.Canver MC, Smith EC, Sher F, et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015; 527:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.∎. Brendel C, Guda S, Renella R, et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J Clin Invest 2016; 126:3868–3878. This study presents preclinical data using an innovative gene therapy approach, utilizing lineage-specific and miRNA-embedded expression of BCL11A-targeting shRNAs, for the treatment of SCD.
- 27.∎. Smith EC, Luc S, Croney DM, et al. Strict in vivo specificity of the Bcl11a erythroid enhancer. Blood 2016; 128:2338–2342. This study presents the data that establishes the exquisite erythroid in-vivo lineage specificity of the Bcl11a intronic enhancer near the vicinity of the GWAS-identified SNPs associated with HbF levels.
- 28.Luc S, Huang J, McEldoon JL, et al. Bcl11a deficiency leads to hematopoietic stem cell defects with an aging-like phenotype. Cell Repo 2016; 16:3181–3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.∎. Camaschella C, Pagani A, Nai A, Silvestri L. The mutual control of iron and erythropoiesis. Int J Lab Hematol 2016; 38 (Suppl 1):20–26. This review updates the latest findings relating to the role of iron regulatory elements in iron homeostasis, erythropoiesis-mediated regulation of hepcidin, and that can be used for targeted therapies for anemias.
- 30.An P, Wu Q, Wang H, et al. TMPRSS6, but not TF, TFR2 or BMP2 variants are associated with increased risk of iron-deficiency anemia. Hum Mol Genet 2012; 21:2124–2131. [DOI] [PubMed] [Google Scholar]
- 31.Iron Camaschella C. and hepcidin: a story of recycling and balance. Hematology Am Soc Hematol Educ Program 2013; 2013:1–8. [DOI] [PubMed] [Google Scholar]
- 32.Gao J, Chen J, Kramer M, et al. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab 2009; 9:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramos P, Guy E, Chen N, et al. Enhanced erythropoiesis in Hfe-KO mice indicates a role for Hfe in the modulation of erythroid iron homeostasis. Blood 2011; 117:1379–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moura IC, Hermine O, Lacombe C, Mayeux P. Erythropoiesis and transferrin receptors. Curr Opin Hematol 2015; 22:193–198. [DOI] [PubMed] [Google Scholar]
- 35.Nai A, Lidonnici MR, Rausa M, et al. The second transferrin receptor regulates red blood cell production in mice. Blood 2015; 125:1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forejtnikova H, Vieillevoye M, Zermati Y, et al. Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood 2010; 116:5357–5367. [DOI] [PubMed] [Google Scholar]
- 37.Alves LR, Costa ES, Sorgine MH, et al. Heme-oxygenases during erythropoiesis in K562 and human bone marrow cells. PLoS One 2011; 6:e21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang D, Peng Y, Ouzhuluobu, et al. HMOX2 functions as a modifier gene for high-altitude adaptation in Tibetans. Hum Mutat 2016; 37:216–223. [DOI] [PubMed] [Google Scholar]
- 39.Matzhold EM, Wagner A, Drexler C, Wagner T. Novel ABO gene variants caused by missense mutations in Exon 7 leading to discrepant ABO blood typing results. Transfusion 2015; 55:1589–1590. [DOI] [PubMed] [Google Scholar]
- 40.Matzhold EM, Drexler C, Wagner T. A novel ABO O allele caused by a large deletion covering two exons of the ABO gene identified in a Caucasian family showing discrepant ABO blood typing results. Transfusion 2016; 56:2739–2743. [DOI] [PubMed] [Google Scholar]
- 41.Ogasawara K, Yabe R, Uchikawa M, Saitou N, et al. Molecular genetic analysis of variant phenotypes of the ABO blood group system. Blood 1996; 88:2732–2737. [PubMed] [Google Scholar]
- 42.Sano R, Nakajima T, Takahashi K, Kubo R, et al. Expression of ABO blood-group genes is dependent upon an erythroid cell-specific regulatory element that is deleted in persons with the B(m) phenotype. Blood 2012; 119:5301–5310. [DOI] [PubMed] [Google Scholar]
- 43.Bony V, Gane P, Bailly P, Cartron JP. Time-course expression of polypeptides carrying blood group antigens during human erythroid differentiation. Br J Haematol 1999; 107:263–274. [DOI] [PubMed] [Google Scholar]
- 44.Sieff C, Bicknell D, Caine G, Robinson J, et al. Changes in cell surface antigen expression during hemopoietic differentiation. Blood 1982; 60:703–713. [PubMed] [Google Scholar]
- 45.Lo KS, Wilson JG, Lange LA, Folsom AR, et al. Genetic association analysis highlights new loci that modulate hematological trait variation in Caucasians and African Americans. Hum Genet 2011; 129:307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yip SP. Sequence variation at the human ABO locus. Ann Hum Genet 2002; 66:1–27. [DOI] [PubMed] [Google Scholar]
- 47.Su MY, Steiner LA, Bogardus H, et al. Identification of biologically relevant enhancers in human erythroid cells. J Biol Chem 2013; 288:8433–8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kozar K, Ciemerych MA, Rebel VI, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell 2004; 118:477–491. [DOI] [PubMed] [Google Scholar]
- 49.Sankaran VG, Ludwig LS, Sicinska E, et al. Cyclin D3 coordinates the cell cycle during differentiation to regulate erythrocyte size and number. Genes Dev 2012; 26:2075–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kranc KR, Schepers H, Rodrigues NP, Bamforth S, et al. Cited2 is an essential regulator of adult hematopoietic stem cells. Cell Stem Cell 2009; 5:659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bakker WJ, van Dijk TB, Parren-van Amelsvoort M, et al. Differential regulation of Foxo3a target genes in erythropoiesis. Mol Cell Biol 2007; 27:3839–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McLachlan S, Giambartolomei C, White J, et al. Replication and characterization of association between ABO SNPs and red blood cell traits by meta-analysis in Europeans. PLoS One 2016; 11:e0156914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang FL, Shen GM, Liu XL, et al. Hypoxia-inducible factor 1-mediated human GATA1 induction promotes erythroid differentiation under hypoxic conditions. J Cell Mol Med 2012; 16:1889–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tong W, Zhang J, Lodish HF. Lnk inhibits erythropoiesis and Epo-dependent JAK2 activation and downstream signaling pathways. Blood 2005; 105:4604–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bersenev A, Wu C, Balcerek J, Tong W. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest 2008; 118:2832–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.∎∎. Giani FC, Fiorini C, Wakabayashi A, et al. Targeted application of human genetic variation can improve red blood cell production from stem cells. Cell Stem Cell 2016; 18:73–78. This study, through population analysis, discovered the association of SH2B3 rare mutation with increased hemoglobin levels, with follow-up functional studies confirming its role in the regulation of erythropoiesis.
- 57.Zhai PF, Wang F, Su R, et al. The regulatory roles of microRNA-146b-5p and its target platelet-derived growth factor receptor alpha (PDGFRA) in erythropoiesis and megakaryocytopoiesis. J Biol Chem 2014; 289:22600–22613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.∎∎. Astle WJ, Elding H, Jiang T, Allen D, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell 2016; 167:1415–1429.e19. This study expands the range of genes and regulatory mechanisms governing hematopoietic development in humans and opens the possibilities for targeting key pathways involved in abnormal or dysregulated hematopoiesis.
- 59.Johnson RM, Panchoosingh H, Goyette G Jr, Ravindranath Y. Increased erythrocyte deformability in fetal erythropoiesis and in erythrocytes deficient in glucose-6-phosphate dehydrogenase and other glycolytic enzymes. Pediatric Res 1999; 45:106–113. [DOI] [PubMed] [Google Scholar]
- 60.Paglialunga F, Fico A, Iaccarino I, Notaro R, et al. G6PD is indispensable for erythropoiesis after the embryonic-adult hemoglobin switch. Blood 2004; 104:3148–3152. [DOI] [PubMed] [Google Scholar]
- 61.Ding K, Shameer K, Jouni H, Masys DR, et al. Genetic Loci implicated in erythroid differentiation and cell cycle regulation are associated with red blood cell traits. Mayo Clinic Proc 2012; 87:461–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marchesi SL, Letsinger JT, Speicher DW, Marchesi VT, et al. Mutant forms of spectrin alpha-subunits in hereditary elliptocytosis. J Clin Invest 1987; 80:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vatopoulou T, Clarke B, de la Fuente J, et al. A novel alpha specrtin mutation causing severe innefective erythropoiesis. Blood 2014; 124:4002–4002. [Google Scholar]
- 64.Auer PL, Lettre G. Rare variant association studies: considerations, challenges and opportunities. Genome Med 2015; 7:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Abiola O, Angel JM, Avner P, et al. The nature and identification of quantitative trait loci: a community’s view. Nat Rev Genet 2003; 4:911–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Siatecka M, Bieker JJ. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 2011; 118:2044–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Borg J, Patrinos GP, Felice AE, Philipsen S. Erythroid phenotypes associated with KLF1 mutations. Haematologica 2011; 96:635–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Singleton BK, Burton NM, Green C, et al. Mutations in EKLF/KLF1 form the molecular basis of the rare blood group In(Lu) phenotype. Blood 2008; 112:2081–2088. [DOI] [PubMed] [Google Scholar]
- 69.Borg J, Papadopoulos P, Georgitsi M, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet 2010; 42:801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arnaud L, Saison C, Helias V, et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am J Hum Genet 2010; 87:721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Magor GW, Tallack MR, Gillinder KR, Bell CC, et al. KLF1 null neonates display hydrops fetalis and a deranged erythroid transcriptome. Blood 2015; 125:2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu D, Zhang X, Yu L, et al. KLF1 mutations are relatively more common in a thalassemia endemic region and ameliorate the severity of beta-thalassemia. Blood 2014; 124:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mtatiro SN, Mgaya J, Singh T, et al. Genetic association of fetal-hemoglobin levels in individuals with sickle cell disease in Tanzania maps to conserved regulatory elements within the MYB core enhancer. BMC Med Genet 2015; 16:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhatnagar P, Purvis S, Barron-Casella E, et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. J Hum Genet 2011; 56:316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsai S-F, Martin DIK, Zon LI, et al. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature 1989; 339:446–451. [DOI] [PubMed] [Google Scholar]
- 76.Sankaran VG, Ghazvinian R, Do R, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest 2012; 122:2439–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chou ST, Kacena MA, Weiss MJ, et al. GATA1-Related X-Linked Cytopenia. 2006. Nov 22 [Updated 2014 Apr 17]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2017. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1364/. [PubMed] [Google Scholar]
- 78.Hollanda LM, Lima CS, Cunha AF, et al. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet 2006; 38:807–812. [DOI] [PubMed] [Google Scholar]
- 79.Nichols KE, Crispino JD, Poncz M, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet 2000; 24:266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boria I, Garelli E, Gazda HT, et al. The ribosomal basis of Diamond-Blackfan Anemia: mutation and database update. Hum Mutat 2010; 31:1269–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.∎. Roy NB, Wilson EA, Henderson S, et al. A novel 33-Gene targeted resequencing panel provides accurate, clinical-grade diagnosis and improves patient management for rare inherited anaemias. Br J Haematol 2016; 175:318–330. This study employed a novel analytical and validation strategies for the development of a targetted NGS panel to be used for clinical diagnosis of previously unexplained congenital anemias.
- 82.Kralovics R, Prchal JT. Genetic heterogeneity of primary familial and congenital polycythemia. Am J Hematol 2001; 68:115–121. [DOI] [PubMed] [Google Scholar]
- 83.de la Chapelle A, Traskelin AL, Juvonen E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci USA 1993; 90:4495–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Percy MJ. Genetically heterogeneous origins of idiopathic erythrocytosis. Hematology (Amsterdam, Netherlands) 2007; 12:131–139. [DOI] [PubMed] [Google Scholar]
- 85.∎∎. Polfus LM, Khajuria RK, Schick UM, Pankratz N, et al. Whole-exome sequencing identifies loci associated with blood cell traits and reveals a role for alternative GFI1B splice variants in human hematopoiesis. Am J Hum Genet 2016; 99:481–488. This study used the whole exome sequence association analysis in identifying previously undiscovered rare variants, and successfully performed the follow-up functional studies confirming role of rare variants in the regulation of human hematopoiesis.
- 86.Kim AR, Ulirsch JC, Wilmes S, et al. Discovery of the first pathogenic human EPO mutation provides mechanistic insight into cytokine signaling. Blood 2016; 128:331–331.27252232 [Google Scholar]
- 87.Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA 2008; 105:1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA 2008; 105:11869–11874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.∎. Smith EC, Orkin SH. Hemoglobin genetics: recent contributions of GWAS and gene editing. Hum Mol Genet 2016; 25:R99–r105. This study reviews the recent work on the control of fetal hemoglobin production, and implications for human genetics and therapeutic applications for hemoglobinopathies.