

Abstract
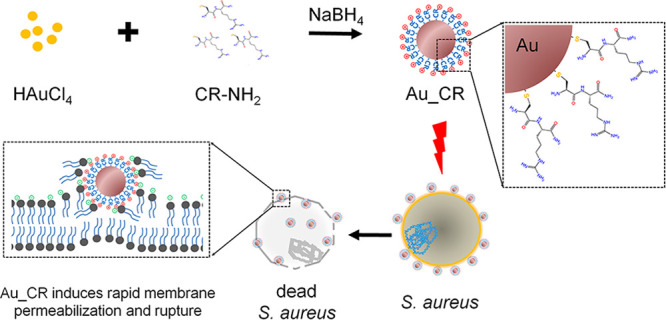
Antibiotic resistance is a global threat. Antimicrobial peptides (AMPs) are highly desirable to treat multidrug-resistant pathogen infection. However, few AMPs are clinically available, due to high cost, instability, and poor selectivity. Here, ultrashort AMPs (2–3 residues with an N-terminal cysteine) are designed and assembled as gold nanoparticles. Au–S conjugation and ultrashort size restrict nonspecific reactions and peptide orientation, thus concentrating positively charged residues on the surface. The nanostructured assemblies enormously enhance antimicrobial abilities by 1000–6000-fold and stability. One representative (Au-Cys-Arg-NH2, Au_CR) shows selective antibacterial activity against Staphylococcus aureus with 10 nM minimal inhibitory concentration. Au_CR has comparable or better in vivo antimicrobial potency than vancomycin and methicillin, with low propensity to induce resistance, little side effects, and high stability (17.5 h plasma half-life). Au_CR acts by inducing collapse of membrane potential and rupture of the bacterial membrane. The report provides insights for developing AMP–metal nanohybrids, particularly tethering nonspecific reactions and AMP orientation on the metal surface.
Keywords: dipeptide, gold nanoparticles, antibiotic-resistance, antimicrobial peptides
The continued overuse of antibiotics has been accompanied by the rapid emergence of antibiotic-resistant bacteria.1,2 According to a disturbing prediction by the World Health Organization (WHO), by the year 2050, 10 million deaths will be caused annually by drug-resistant infections.3 Many problematic multidrug-resistant (MDR) bacteria are emerging, such as MDR Pseudomonas aeruginosa; vancomycin-resistant enterococci (VRE); methicillin-resistant Staphylococcus aureus (MRSA); vancomycin-resistant MRSA; carbapenem-resistant Acinetobacter baumannii, Escherichia coli, and Klebsiella pneumoniae; and extensively drug-resistant (XDR) Mycobacterium tuberculosis.4,5 As an important opportunistic pathogen, S. aureus persistently colonizes about 20% of the human population and can cause both superficial and invasive, potentially life-threatening, infections such as sepsis, endocarditis, and pneumonia. MRSA is prevalent in hospitals, and hypervirulent MRSA strains are spreading throughout the community.6−10
Antimicrobial peptides (AMPs) show excellent potential for combating these threats due to their low tendency to select for resistance, rapid killing action, and desirable clinical efficacy against several MDR pathogens. However, several factors curtail their full utilization and approval for clinical application.11−13 One of the limiting factors is AMPs’ sensitivity to proteolytic digestion, and toxicity and efficacy of AMPs are other major drawbacks. Moreover, compared to conventional antibiotics, most druggable AMPs are expensive to produce. Therefore, current efforts are being directed at developing strategies to improve the efficacy of AMPs in vivo, design shorter peptides, enhance selectivity for microbial cells while reducing cytotoxicity, increase stability, and resist proteolytic degradation.14 Chemical modifications such as the addition of D-amino acids, cyclization, or acetylation are important strategies to solve these issues.14,15 However, additional modifications add to the production costs. Another possible strategy to improve the stability and efficacy of AMPs is the use of carrier delivery systems.16 Of particular interest is the use of nanocarriers, which are designed and conjugate AMPs to prevent self-aggregation and improve chemical stability and release profiles of AMPs to target sites.17−20 Gold nanoparticles (NPs) are comparatively safer than other metallic NPs due to the inert and nontoxic nature of gold and high biocompatibility.21,22 In this study, we aim to develop novel antibacterial agents by designing a series of ultrashort AMPs (2–3 residues) and engineering them to conjugate with gold NPs by a one-step strategy. The formulation reported here may be useful as a general platform to develop innovative antibacterial agents.
Peptides have multiple functional groups, including amino and thiol groups, which can immobilize peptides onto gold NPs by electrostatic interaction and Au–S coordinate covalent bonds, respectively.17,23,24 Given that covalent conjugation is reliable and conjugates show high quality and homogeneity, we designed short peptides with an N-terminal cysteine, allowing them to conjugate to gold via Au–S bond formation. To avoid nonspecific reactions with AMP side-chain amino groups that affect peptide orientation on the surface of gold to possibly decrease antimicrobial activity, we restricted the length of peptides to no more than 3 amino acid residues. In addition, a net positive charge (provided by Lys/Arg) of peptide is necessary because the cationic properties of AMPs are highly associated with their antimicrobial activity.25 To meet the requirements mentioned above, six short peptides with an amidated C terminus to improve stability and reduce negative charges, including Cys-Arg-NH2 (CR), Cys-Lys-NH2 (CK), Cys-Arg-Arg-NH2 (CRR), Cys-Lys-Lys-NH2 (CKK) (Figure 1A), Cys-Arg-Lys-NH2 (CRK), and Cys-Lys-Arg-NH2 (CKR), were designed.
Figure 1.

Characterizations of ultrashort peptide-conjugated gold nanoparticles and their antimicrobial activities. (A) Structural formulas of ultrashort peptides. TEM images of Au_CR (B), Au_CRR (C), Au_CK (D), and Au_CKK (E) (top), with diameters of 3.0 ± 0.5 nm, 3.8 ± 1.0 nm, 2.6 ± 0.7 nm, and 3.0 ± 0.7 nm, respectively (bottom). (F) Antimicrobial activities of Au_CR, Au_CRR, Au_CK, and Au_CKK and their corresponding free peptides against E. coli, B. subtilis, S. aureus, and C. albicans. MICs are shown in weight (μg/mL) and molar (μM) concentrations. MICs represent mean values of three independent experiment.
The NPs were assembled by short peptides, which were conjugated with gold via the reduction of HAuCl4 by NaBH4 in the presence of short peptides in methanol. Based on transmission electron microscopy (TEM) and statistical analysis, the diameters of the CR-gold (Au_CR), CRR-gold (Au_CRR), CK-gold (Au_CK), and CKK-gold NPs (Au_CKK) were 3.0 ± 0.5, 3.8 ± 1.0, 2.6 ± 0.7, and 3.0 ± 0.7 nm, respectively (Figure 1B–E). The corresponding ultraviolet–visible (UV–vis) absorption spectra of the short peptides and peptide-conjugated gold NPs are shown in Figure S1. A broadened peak appeared at ∼535 nm for the peptide-conjugated gold NPs, which was possibly due to their small size, and the plasmon peak was not evident on the absorbance spectra, consistent with previous findings.26 X-ray photoelectron spectroscopy (XPS) demonstrated that the molar ratios of S to Au in Au_CR, Au_CRR, Au_CK, and Au_CKK were 0.46:1, 0.27:1, 0.17:1, and 0.48:1, respectively (Figure S2 and Table S1). The numbers of CR, CRR, CK, and CKK on each gold NP were 383, 448, 91, and 406, respectively (Table S1). The zeta potentials showed that all four peptide-conjugated gold NPs were positively charged (Table S1). Although Au_CRR and Au_CKK had one more amino group than Au_CR, the zeta potentials of them were lower than Au_CR, maybe because the slight reaction between the side chain amino groups and surface of gold NPs decreased their zeta potentials. Au_CRK and Au_CKR were not focused on in this work since they showed relatively weak antimicrobial functions.
We next evaluated the antimicrobial properties of peptide-conjugated gold NPs in vitro. As illustrated in Figure 1F, the free peptides showed extremely weak antimicrobial effects (minimal inhibitory concentrations (MICs) ≥ 25 mM; i.e., ≥6 200 μg/mL). However, after conjugation with gold, they exhibited significant antimicrobial activities against S. aureus, with MICs of 1.56, 6.25, 25.00, and 6.25 μg/mL for Au_CR, Au_CRR, Au_CK, and Au_CKK, respectively (Figure 1F). They showed weak antimicrobial activities against E. coli, Bacillus subtilis, or Candida albicans (MICs > 100 μg/mL). To further assess their antimicrobial effects, we tested their antimicrobial properties against 19 clinically isolated S. aureus strains. Remarkably, Au_CR also showed the highest antibacterial activity against the clinical S. aureus strains, with MICs from 1.56 to 4.69 μg/mL (Table S2). Similarly, Au_CRR, Au_CK, and Au_CKK showed potent activities against these strains, with MICs between 6.25 and 12.50 μg/mL, 12.50 and 37.5 μg/mL, and 6.25 and 12.50 μg/mL (20–240 nM), respectively. Additionally, we also tested the antimicrobial activities of methicillin and vancomycin against these clinical strains (Table S2). Nine strains (47.4%) showed obvious methicillin resistance, with MICs of 2.34–200 μg/mL (4–339-fold increase in MICs). Vancomycin also showed antibacterial effects against most of the clinically isolated S. aureus strains (MICs of 1.17–2.34 μg/mL), with only four isolates (21.1%) exhibiting a 2-fold increase in their MICs (2.34 μg/mL).
Cationic peptides usually show a certain level of hemolysis and/or cytotoxicity near their therapeutic concentrations, which greatly impedes their clinical use as therapeutics.2,27 To understand the potential toxicity of peptide-conjugated gold NPs, we next evaluated their hemolytic and cytotoxic activities. Notably, at a high concentration of 100 μg/mL (64-fold higher than the Au_CR MIC against standard S. aureus), the four gold NPs exhibited negligible hemolytic activity (less than 3% hemolysis, Figure S3). We also tested their cytotoxicity against human HaCaT keratinocyte cells and human umbilical vein endothelial cells (HUVECs). As illustrated in Figure S4, Au_CR showed little effect on cell viability, and Au_CRR, Au_CK, and Au_CKK exerted a moderate degree of cytotoxicity. Thus, Au_CR showed the highest antibacterial activity and negligible toxicity, suggesting high potential as an ideal antimicrobial candidate.
We next determined the killing kinetics of Au_CR against S. aureus. As shown in Figure 2A, Au_CR showed rapid and dose-dependent killing effects, with complete S. aureus death within 60 min at 1 × MIC, 30 min at 5 × MIC, and 10 min at 10 × MIC. In contrast, vancomycin could not kill S. aureus within 3 h, even at 10 × MIC. This may be because vancomycin only inhibited bacterial cell wall synthesis without killing the bacteria within the tested time (3 h). As Au_CR killed S. aureus quickly, bacteria may have difficulty developing resistance. We therefore evaluated the selection of resistance through serial passaging (up to 60 passages) in the presence of varied concentrations of Au_CR or vancomycin. As shown in Figure 2B, S. aureus did not easily develop resistance to Au_CR, and the MIC increased only 1.5-fold after 60 passages. However, exposure to vancomycin resulted in the rapid development of resistance after the first five passages and led to a 6-fold increase in the MIC after 20 passages. Collectively, these results suggest that Au_CR exhibits rapid and potent bactericidal effects with a low propensity to induce resistance.
Figure 2.

Au_CR shows rapid killing effect, low propensity to induce resistance, and potent effects to kill biofilm and persister cells. (A) Killing kinetics of Au_CR against S. aureus. (B) Resistance development of S. aureus following long-term exposure (60 passages) to Au_CR or vancomycin. Inhibitory effects of Au_CR on biofilm formation of S. aureus ATCC2592 (C) and Z (D, clinically isolated strain). Effects of Au_CR on established biofilm eradication of S. aureus ATCC2592 (E) and Z (F). Bactericidal activities of Au_CR against persister cells of S. aureus ATCC2592 (G) and Z (H). Data represent means ± SD of three individual experiments. *p < 0.05, **p < 0.01, and ***p < 0.001; one-way ANOVA with Dunnett post hoc test compared with control (PBS).
Pathogenic biofilms are formed by a thick and dense extracellular matrix, which can prevent antimicrobial agents from contacting the cells, thus contributing to drug resistance.28,29 Thus, we next investigated the effects of Au_CR on antibiofilm formation and eradication. Notably, Au_CR inhibited the biofilm formation of the standard (Figure 2C) and clinically isolated (Figure 2D) S. aureus strains in a dose-dependent manner. Once formed, biofilms are very difficult to eradicate, resulting in chronic and persistent infections.30 Interestingly, the preformed S. aureus biofilms were eliminated by Au_CR in a dose-dependent manner (Figure 2E,F).
All bacteria can form persisters, a drug-tolerant subpopulation of dormant cells, which have been implicated in biofilms and in chronic and recurrent infections.31,32 As illustrated in Figure 2G,H, Au_CR showed potent killing effects against S. aureus persisters, even at a low concentration of 0.5 × MIC. More than half of the persister cells of the standard (Figure 2G) and clinical (Figure 2H) S. aureus strains were eliminated by 1 × MIC of Au_CR. Thus, together, the effects of Au_CR make it a promising antibiofilm and antipersister agent.
To elucidate a potential mechanism of action of Au_CR, effects of Au_CR on bacterial membrane permeabilization were studied by a fluorescence assay and flow cytometry. Compared to free S. aureus cells, 5 × MIC of Au_CR treatment for 30 min resulted in a significant increase in the number of fluorescent deposits (red), i.e., propidium iodide (PI) uptake (Figure 3A), indicating that Au_CR caused rapid membrane permeabilization. Furthermore, flow cytometry also detected immediate and increasing PI influx after Au_CR was added to the S. aureus cells (Figure 3B), further suggesting that Au_CR permeabilized the bacterial membrane quickly and in a time-dependent manner. Membrane permeabilization or collapse of membrane integrity may result from loss of membrane potential (or membrane electric potential, ΔΨ).33 To test this hypothesis, we used the fluorescent dye 3,3-dipropylthiadicarbocyanine iodide (DiSC3(5)) to evaluate whether Au_CR dissipated the membrane potential. As shown in Figure 3C, Au_CR gradually increased the fluorescence intensity of DiSC3(5) in a dose-dependent manner, indicating a gradual collapse of the membrane potential after Au_CR treatment. The pore-forming lantibiotic Nisin A also caused a similar DiSC3(5) signal profile as Au_CR, but the effect was a little faster. The potassium ionophore valinomycin was also added as a control, which instantly dissipated the membrane potential (Figure 3C).
Figure 3.

Au_CR induces rapid membrane permeabilization and rupture. (A) Fluorescence assay of S. aureus treated with Au_CR (5 × MIC) by PI staining. Image of corresponding fluorescence (right) in phase contrast mode is shown (left). (B) Kinetics of PI influx in S. aureus. S. aureus cells were first incubated with PI for 15 min, then measured by flow cytometry in the presence or absence of Au_CR (5 × MIC) for 6 min. Images are representative of at least three independent experiments. (C) Membrane potential levels of S. aureus upon addition of Au_CR (1–8 × MIC), Nisin A (4 μM), and Valinomycin (10 μM). Saline was used as control. TEM (D) and SEM (E) images showing morphological changes in S. aureus treated with Au_CR (5 × MIC, 7.8 μg/mL) or without Au_CR (Control). (F) TEM image of unstained super thin slices of S. aureus treated with Au_CR (5 × MIC) showing the internalized NPs. The graph depicts a representative measurement of three independent replicates. In panels D and F, the arrows indicate the NPs.
Membrane permeabilization may lead to disruption of the bacterial membrane structure. TEM images showed that the plasma membrane of S. aureus ruptured after Au_CR treatment, thus allowing entry of the NPs (Figure 3D). In contrast, the membranes of the untreated cells remained intact. The scanning electron microscopy (SEM) images also showed membrane rupture and pore formation in Au_CR-treated S. aureus cells (Figure 3E). TEM images of unstained slices further proved that Au_CR killed S. aureus by disrupting the bacterial membrane, with the NPs clearly internalized in the cytoplasm of the damaged S. aureus cells (Figure 3F). Together, these data demonstrate that Au_CR induces collapse of membrane potential and rupture of the bacterial membrane.
To further assess the possible antimicrobial mechanism of Au_CR, we performed all-atom (AA) and coarse-grained (CG) molecular dynamics (MD) simulations. After the construction of the AA model, the 3 nm gold core was coated with monolayer CR peptides by Au–S bonds. CR peptides were concentrated and evenly distributed on the gold surface. Guanidyl groups of arginine contributed positive charges orderly extended toward the outside (Figure S5A,B). The model of 1,2-dioleoyl-sn-glycerol-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (DOPG) with a ratio of 7:3 was used to simulate negatively charged S. aureus membranes.34,35 The root-mean-square deviation (RMSD) of Au_CR reached a steady state, ensuring structural stability in the later stage of MD simulations (Figure S5C). As shown in Figure 4A, once the simulation started, the Au_CR particles began to move toward the membrane quickly. In the production process, the lipid membrane became rough and formed a hump in another direction to accommodate the penetration of the particle. The phospholipids near the Au_CR particle became disordered, especially the DOPG molecules. The coordinates of PO4 beads in one side within 0.5 nm of the particle were extracted to depict the interface of the model membrane with the influence of the Au_CR particle (Figure 4B). With the penetration of the Au_CR particle, the membrane became bumpier. The contact curve demonstrated effective interactions between the Au_CR particle and membranes (Figure 4C). The deuterium order parameters were defined in a previous study to characterize the lipid order during simulation,36 which are calculated for the angles between the bond vectors of two consecutive tail beads and the bilayer normal for the lipid acyl chains. The first and the last 1 μs production trajectory was used for deuterium order parameter analysis, and two lipid acyl chains (sn-1 and sn-2) were analyzed. Comparing the results before and after the simulations, the lipids near the particle become more disorderly as the simulation goes on (Figure 4D). Some lipids bent out the bilayer, and their deuterium order parameters decreased, further indicating the disturbance of the membrane induced by Au_CR.
Figure 4.

Au_CR interacts with and disrupts mimetic membrane. (A) The process of Au_CR particle penetrating into the model membrane in MD simulations. The Cys residues in Au_CR are shown as green spheres, and Arg residues are shown as blue spheres. The phospholipids are represented by PO4 beads for convenience. The yellow spheres represent the DOPC molecules, and the orange spheres represent DOPG molecules. (B) The simulation snapshots with an interval of 10 μs of the model membrane interface with the influence of Au_CR nanoparticle. (C) The number of contacts between Au_CR and membrane during MD simulations. (D) Deuterium order parameter results for lipid chains within 0.5 nm near the particle during first and last 1 μs simulation. The first 1 μs is colored in blue and last 1 μs in red. (E) Fluorescence assay of liposomes labeled with DiI (red) and encapsulated Fluo-4 (green) in a solution containing 10 mM Ca2+ and 5 × MIC Au_CR. The images were captured after 0, 10, and 30 min of incubation with Au_CR (left). Percentage of leaked vesicles was also analyzed (right). Scale bar: 50 μm. ***p < 0.01, one-way ANOVA with Dunnett post hoc test compared with 0 min group. Images are representative of at least three independent experiments.
We further investigated the effects of Au_CR on biomembrane-mimicking unilamellar vesicles, which were constructed using DOPC/DOPG lipids at a 7:3 ratio, as in the MD simulations. The vesicles were labeled with DiI (red) and preloaded with Fluo-4 (green), a Ca2+-sensitive fluorescent dye, the fluorescence signal of which will be enhanced in the presence of Ca2+. As illustrated in Figure 4E, 10 and 30 min after addition of Au_CR in situ, Fluo-4 fluorescent spots were significantly increased in a time-dependent manner, indicating that Au_CR induces liposome leakage and transmembrane influx of Ca2+. Together, these data suggest Au_CR interacts with and disrupts the bacterial membrane.
Before we started to evaluate the therapeutic potential of Au_CR in vivo, metabolic stability and acute toxicity of it were first determined. As illustrated in Table S3, the incubation of Au_CR with plasma at 37 °C for 24 h had no apparent effect on its antibacterial activity against S. aureus, indicating its high plasma stability. Furthermore, pharmacokinetic analysis of Au_CR demonstrated a half-life of ∼17.5 h after intravenous (iv) injection into mice (Figure 5A and Figure S6). The injected Au_CR was predominantly localized in the kidneys, spleen, liver, and lungs with little found in the heart (Figure 5B). The high concentration of Au_CR in the kidneys indicated that it may be partially excreted via renal clearance, which is likely due to their small hydrodynamic size as smaller NPs show quicker renal clearance.37 Importantly, after administration of Au_CR at a dose of 20 or 30 mg/kg, mice showed no signs of toxicity (Figure S7), suggesting the high biocompatibility of Au_CR in vivo.
Figure 5.

Au_CR increases survival and inhibits S. aureus lung infection and inflammation. (A) Pharmacokinetic profile of Au_CR via intravenous injection at Au dose of 10 mg/kg. (B) Biodistribution of Au_CR in main organs 48 h after injection. (C) Survival curves for mice (n = 10) after iv infection with S. aureus Z. Treatment with Au_CR (0.25, 0.5, and 1 mg/kg) and vancomycin (1 mg/kg) was performed (via iv injection) at 1 and 6 h after inoculation. (D) Survival curves for mice (n = 10) after ip infection with S. aureus Z and ip administration of Au_CR (1, 2, and 4 mg/kg) and vancomycin (4 mg/kg) at 1 and 6 h after infection. (E) Mice (n = 6) were first intranasally administered with S. aureus Z (clinically isolated strain), followed by iv treatment with Au_CR (1, 2, 4 mg/kg) or vancomycin (2 and 4 mg/kg) to examine their therapeutic effects, with bacterial load in lung homogenate shown. (F) Representative histopathological images of processed lung section with H&E staining. Scale bar represents 50 μm. Mouse model experiments were repeated twice. *p < 0.05 and **p < 0.01, two-way ANOVA with Dunnett post hoc test compared with vehicle group (sterile saline). Data in panels A, B, and E represent mean ± SD values of three independent experiments.
We first investigated whether it would rescue C57BL/6 mice infected with a lethal dose of S. aureus. As shown in Figure 5C, Au_CR treatment via iv administration significantly increased the survival of mice after iv infection with a clinically isolated S. aureus strain (Z). Survival of Au_CR-treated mice after peritoneal infection with S. aureus was also significantly higher than that of vehicle (saline)-treated mice, and treatment with Au_CR had a dose-dependent impact on the survival rate (Figure 5D).
We next used MDR S. aureus-induced lung infection and bacteremia models to evaluate the therapeutic potential of Au_CR. In the first set of experiments, C57BL/6 mice were administered intranasally with a suspension of a clinically isolated S. aureus strain (Z) to induce lung infection, followed by iv treatment with Au_CR and control samples (vehicle and vancomycin). Notably, treatment with 1, 2, and 4 mg/kg Au_CR inhibited the bacterial load in the lung by 62.7%, 77.5%, and 83.0%, while 2 and 4 mg/kg vancomycin inhibited the bacterial load by 55.1% and 74.0%, respectively (Figure 5E). Au_CR demonstrated better therapeutic potential than vancomycin. Furthermore, histopathological examination of fixed lung sections revealed that Au_CR administration significantly alleviated lung inflammation and inflammatory cell infiltration (Figure 5F), indicating a potential anti-inflammatory effect in vivo.
In the second set of experiments, mice were inoculated with S. aureus Z via intraperitoneal (ip) injection, with Au_CR and controls also administered via ip injection to assess their effects on dissemination of bacteria from the peritoneal cavity to the blood, lung, liver, and spleen. As illustrated in Figure S8A,D, compared to the vehicle treatment, Au_CR treatment significantly suppressed (2–5-fold) bacterial dissemination to the blood and organs. Similar results were observed after treatment with vancomycin. In addition, S. aureus infection led to elevated cytokine levels of interleukin (IL)-1β, IL-6, IL-10, tumor necrosis factor (TNF)-α, monocyte chemotactic protein (MCP)-1, and interferon (IFN)-γ (Figure S8E–J). However, Au_CR treatment significantly suppressed these cytokine releases, further suggesting an anti-inflammatory potential of Au_CR. Moreover, histopathological analysis of the lungs, liver, and spleen also highlighted the potent therapeutic effects of Au_CR against S. aureus dissemination in vivo (Figure S8K–M). Collectively, these results indicate that Au_CR can inhibit S. aureus infection and dissemination to target organs.
Collectively, our results indicated that the dipeptide (CR)-gold NPs exhibited highly potent bactericidal activity and specificity against S. aureus with a favorable safety profile and showed impressive therapeutic efficacy. The formulation reported here may be useful as a general platform to develop innovative antibacterial agents.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32070443 and 21761142002), Chinese Academy of Sciences (XDB31000000, SAJC201606, KFJ-BRP-008, and KGFZD-135-17-011), and Yunnan Province Grant (202302AA310015).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.nanolett.3c03909.
Materials, synthesis and characterization of nanoparticles, antimicrobial properties in vitro and in vivo, simulation setup and experimental procedures for pharmacokinetics and biodistribution study, Figures S1–S8, Tables S1–S3, and supplementary references (PDF)
Author Contributions
¶ Z.Z., Y.C., J.G., and M.Y. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Adedeji W. A. The Treasure Called Antibiotics. Annals of Ibadan Postgraduate Medicine 2016, 14 (2), 56–57. [PMC free article] [PubMed] [Google Scholar]
- Costa F.; Teixeira C.; Gomes P.; Martins M. C. L. Clinical Application of AMPs. Advances in experimental medicine and biology 2019, 1117, 281–298. 10.1007/978-981-13-3588-4_15. [DOI] [PubMed] [Google Scholar]
- de Kraker M. E. A.; Stewardson A. J.; Harbarth S. Will 10 Million People Die a Year due to Antimicrobial Resistance by 2050?. PLOS Medicine 2016, 13 (11), e1002184 10.1371/journal.pmed.1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. G.; Perdreau-Remington F.; Rieg G.; Mehdi S.; Perlroth J.; Bayer A. S.; Tang A. W.; Phung T. O.; Spellberg B. Necrotizing Fasciitis Caused by Community-Associated Methicillin-Resistant Staphylococcus aureus in Los Angeles. New England Journal of Medicine 2005, 352 (14), 1445–1453. 10.1056/NEJMoa042683. [DOI] [PubMed] [Google Scholar]
- Levin A. S.; Barone A. A.; Penço J.; Santos M. V.; Marinho I. S.; Arruda E. A. G.; Manrique E. I.; Costa S. F. Intravenous Colistin as Therapy for Nosocomial Infections Caused by Multidrug-Resistant Pseudomonas aeruginosa and Acinetobacter baumannii. Clinical Infectious Diseases 1999, 28 (5), 1008–1011. 10.1086/514732. [DOI] [PubMed] [Google Scholar]
- Foster T. J.; Geoghegan J. A.; Ganesh V. K.; Hook M. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nature reviews. Microbiology 2014, 12 (1), 49–62. 10.1038/nrmicro3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo F. R.; Otto M.; Kreiswirth B. N.; Chambers H. F. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 2010, 375 (9725), 1557–68. 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Annu. Rev. Microbiol. 2010, 64, 143–62. 10.1146/annurev.micro.112408.134309. [DOI] [PubMed] [Google Scholar]
- Chambers H. F.; Deleo F. R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nature reviews. Microbiology 2009, 7 (9), 629–41. 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeo F. R.; Chambers H. F. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin. Invest. 2009, 119 (9), 2464–74. 10.1172/JCI38226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Li X.; Wang Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44 (D1), D1087–D1093. 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falanga A.; Lombardi L.; Franci G.; Vitiello M.; Iovene M. R.; Morelli G.; Galdiero M.; Galdiero S. Marine Antimicrobial Peptides: Nature Provides Templates for the Design of Novel Compounds against Pathogenic Bacteria. Int. J. Mol. Sci. 2016, 17 (5), 785. 10.3390/ijms17050785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi L.; Falanga A.; Del Genio V.; Galdiero S. A New Hope: Self-Assembling Peptides with Antimicrobial Activity. Pharmaceutics 2019, 11 (4), 166. 10.3390/pharmaceutics11040166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; Zhang M.; Lai R.; Zhang Z. Chemical modifications to increase the therapeutic potential of antimicrobial peptides. Peptides 2021, 146, 170666. 10.1016/j.peptides.2021.170666. [DOI] [PubMed] [Google Scholar]
- Mwangi J.; Hao X.; Lai R.; Zhang Z. Y. Antimicrobial peptides: new hope in the war against multidrug resistance. Zoological research 2019, 40 (6), 488–505. 10.24272/j.issn.2095-8137.2019.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstrom R.; Malmsten M. Delivery systems for antimicrobial peptides. Adv. Colloid Interface Sci. 2017, 242, 17–34. 10.1016/j.cis.2017.01.005. [DOI] [PubMed] [Google Scholar]
- Rai A.; Pinto S.; Velho T. R.; Ferreira A. F.; Moita C.; Trivedi U.; Evangelista M.; Comune M.; Rumbaugh K. P.; Simões P. N.; Moita L.; Ferreira L. One-step synthesis of high-density peptide-conjugated gold nanoparticles with antimicrobial efficacy in a systemic infection model. Biomaterials 2016, 85, 99–110. 10.1016/j.biomaterials.2016.01.051. [DOI] [PubMed] [Google Scholar]
- Wadhwani P.; Heidenreich N.; Podeyn B.; Burck J.; Ulrich A. S. Antibiotic gold: tethering of antimicrobial peptides to gold nanoparticles maintains conformational flexibility of peptides and improves trypsin susceptibility. Biomaterials science 2017, 5 (4), 817–827. 10.1039/C7BM00069C. [DOI] [PubMed] [Google Scholar]
- Bi L.; Yang L.; Narsimhan G.; Bhunia A. K.; Yao Y. Designing carbohydrate nanoparticles for prolonged efficacy of antimicrobial peptide. Journal of controlled release: official journal of the Controlled Release Society 2011, 150 (2), 150–6. 10.1016/j.jconrel.2010.11.024. [DOI] [PubMed] [Google Scholar]
- Brandelli A. Nanostructures as promising tools for delivery of antimicrobial peptides. Mini reviews in medicinal chemistry 2012, 12 (8), 731–41. 10.2174/138955712801264774. [DOI] [PubMed] [Google Scholar]
- Yang X.; Yang M.; Pang B.; Vara M.; Xia Y. Gold Nanomaterials at Work in Biomedicine. Chem. Rev. 2015, 115 (19), 10410–88. 10.1021/acs.chemrev.5b00193. [DOI] [PubMed] [Google Scholar]
- Ghosh P.; Han G.; De M.; Kim C. K.; Rotello V. M. Gold nanoparticles in delivery applications. Advanced drug delivery reviews 2008, 60 (11), 1307–15. 10.1016/j.addr.2008.03.016. [DOI] [PubMed] [Google Scholar]
- Ben Haddada M.; Blanchard J.; Casale S.; Krafft J. M.; Vallee A.; Methivier C.; Boujday S. Optimizing the immobilization of gold nanoparticles on functionalized silicon surfaces: amine- vs thiol-terminated silane. Gold Bull. 2013, 46 (4), 335–341. 10.1007/s13404-013-0120-y. [DOI] [Google Scholar]
- Kumar A.; Mandal S.; Selvakannan P. R.; Pasricha R.; Mandale A. B.; Sastry M. Investigation into the Interaction between Surface-Bound Alkylamines and Gold Nanoparticles. Langmuir: the ACS journal of surfaces and colloids 2003, 19 (15), 6277–6282. 10.1021/la034209c. [DOI] [PubMed] [Google Scholar]
- Hancock R. E.; Sahl H. G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nature biotechnology 2006, 24 (12), 1551–7. 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Beattie D. A.; Ralston J.; Sedev R. Colloid stability of thymine-functionalized gold nanoparticles. Langmuir: the ACS journal of surfaces and colloids 2007, 23 (24), 12096–103. 10.1021/la7019878. [DOI] [PubMed] [Google Scholar]
- Bobone S.; Stella L. Selectivity of Antimicrobial Peptides: A Complex Interplay of Multiple Equilibria. Advances in experimental medicine and biology 2019, 1117, 175–214. 10.1007/978-981-13-3588-4_11. [DOI] [PubMed] [Google Scholar]
- Flemming H. C.; Wingender J. The biofilm matrix. Nature reviews. Microbiology 2010, 8 (9), 623–33. 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- Stewart P. S.; Costerton J. W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358 (9276), 135–138. 10.1016/S0140-6736(01)05321-1. [DOI] [PubMed] [Google Scholar]
- Bjarnsholt T. The role of bacterial biofilms in chronic infections. APMIS. Supplementum 2013, 121, 1–58. 10.1111/apm.12099. [DOI] [PubMed] [Google Scholar]
- Allison K. R.; Brynildsen M. P.; Collins J. J. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 2011, 473 (7346), 216–20. 10.1038/nature10069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonneuve E.; Gerdes K. Molecular mechanisms underlying bacterial persisters. Cell 2014, 157 (3), 539–48. 10.1016/j.cell.2014.02.050. [DOI] [PubMed] [Google Scholar]
- Chapple D. S.; Mason D. J.; Joannou C. L.; Odell E. W.; Gant V.; Evans R. W. Structure-function relationship of antibacterial synthetic peptides homologous to a helical surface region on human lactoferrin against Escherichia coli serotype O111. Infection and immunity 1998, 66 (6), 2434–40. 10.1128/IAI.66.6.2434-2440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganewatta M. S.; Chen Y. P.; Wang J.; Zhou J.; Ebalunode J.; Nagarkatti M.; Decho A. W.; Tang C. Bio-inspired resin acid-derived materials as anti-bacterial resistance agents with unexpected activities. Chemical Science 2014, 5 (5), 2011–2016. 10.1039/c4sc00034j. [DOI] [Google Scholar]
- Kim W.; Zou G.; Hari T. P. A.; Wilt I. K.; Zhu W.; Galle N.; Faizi H. A.; Hendricks G. L.; Tori K.; Pan W.; Huang X.; Steele A. D.; Csatary E. E.; Dekarske M. M.; Rosen J. L.; Ribeiro N. d. Q.; Lee K.; Port J.; Fuchs B. B.; Vlahovska P. M.; Wuest W. M.; Gao H.; Ausubel F. M.; Mylonakis E. A selective membrane-targeting repurposed antibiotic with activity against persistent methicillin-resistant Staphylococcus aureus. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (33), 16529–16534. 10.1073/pnas.1904700116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig J.; Seelig A. Lipid conformation in model membranes and biological membranes. Q. Rev. Biophys. 1980, 13 (1), 19–61. 10.1017/S0033583500000305. [DOI] [PubMed] [Google Scholar]
- Soo Choi H.; Liu W.; Misra P.; Tanaka E.; Zimmer J. P.; Itty Ipe B.; Bawendi M. G.; Frangioni J. V. Renal clearance of quantum dots. Nature biotechnology 2007, 25 (10), 1165–1170. 10.1038/nbt1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.