

Abstract
Epigenetic modification of chromosome structure has increasingly been associated with alterations in secondary metabolism and sporulation defects in filamentous fungal pathogens. Recently, the epigenetic reader protein SntB was shown to govern virulence, spore production and mycotoxin synthesis in the fruit pathogen Penicillium expansum. Through immunoprecipitation-coupled mass spectrometry, we found that SntB is a member of a protein complex with KdmB, a histone demethylase and the essential protein RpdA, a histone deacetylase. Deletion of kdmB phenocopied some but not all characteristics of the ΔsntB mutant. KdmB deletion strains exhibited reduced lesion development on Golden Delicious apples and this was accompanied by decreased production of patulin and citrinin in host tissue. In addition, ΔkdmB mutants were sensitive to several cell wall stressors which possibly contributed to the decreased virulence observed on apples. Slight differences in spore production and germination rates of ΔkdmB mutants in vitro did not impact overall diameter growth in culture.
Keywords: epigenetics, virulence, secondary metabolism, fungal biology, post-harvest disease, mycotoxin
Introduction
Plant pathogenic filamentous fungi mediate interactions with their environments utilizing different strategies. Necrotrophic pathogens strategies include tissue acidification of the host tissues, cell wall degrading enzyme secretion, effector proteins delivery and secondary metabolite production and secretion (van Kan, 2006). Secondary metabolites are small bioactive molecules that are produced by many organisms including pathogenic filamentous fungi (Keller, 2019). These molecules are diverse in structure and function and mediate varied ecological interactions. Many secondary metabolites have beneficial properties and have been utilized by humans as drugs or agricultural treatments, but others can have detrimental effects and are classified as mycotoxins.
Penicillium expansum is a post-harvest pathogen of pome fruits and the causative agent of blue mold disease. This necrotrophic pathogen produces the mycotoxins patulin and citrinin which pose a food safety threat to consumers of apples and their processed food products (Luciano-Rosario et al., 2020). Patulin is difficult to remove from contaminated food products due to its heat and acidity resistance and thus the threshold levels imposed by international agencies like the World Health Organization WHO, FDA, and EU often lead to destruction of contaminated products (Ioi et al., 2017). Other secondary metabolites produced by P. expansum include andrastins, roquefortine, chaetoglobosins, and communesins (Tannous et al., 2018), although these compounds are only occasionally reported to contaminate apple products and the potential health effects of their consumption are not characterized (Olsen et al., 2019; Andersen et al., 2004; Larsen et al., 1998; Tannous et al., 2018). The production of secondary metabolites is regulated by diverse mechanisms at a molecular level. Endogenous regulatory mechanisms include regulation of secondary metabolite gene clusters – also known as biosynthetic gene clusters (BGCs) - by cluster-specific transcription factors or by proteins that mediate chromatin accessibility of the transcriptional machinery.
In filamentous fungi, chromatin regulation couples BGC gene expression and thus secondary metabolite production with developmental processes such as virulence, sporulation, and resistance to stressors (Choi et al., 2022; Strauss and Cánovas, 2021; Bachleitner et al., 2021; Wu et al., 2021). Typically, chromatin regulation occurs through histone post-translational modifications (PTMs) to histone tails (Pfannenstiel and Keller 2019). There are many possible PTMs such as acetylation, methylation, and phosphorylation among others and these modifications are created by protein complexes that physically interact with and modify histone tails. The different classes of histone tail modification proteins include “readers” which are proteins that identify a histone tail modification and mediate the response to the modification, “writers” or proteins that add modifications to histone tails and “erasers” or proteins that remove histone tail modifications (Pfannenstiel and Keller 2019).
The first reader protein to be identified in filamentous fungi was HepA, a protein that acts on H3K9me3 which represses transcription of A. nidulans BGCs (Reyes-Domingez et al., 2010). .SntB, is a homolog of Snt2 in Saccharomyces cerevisiae which was found to interact with an eraser enzyme, the histone deacetylase (HDAC) Rpd3 (Baker et al., 2013). Through a random mutagenesis approach, SntB was identified in Aspergillus nidulans as a regulator of the mycotoxin sterigmatocystin (ST) (Pfannenstiel et al., 2017). Homologs of SntB in filamentous pathogens have been shown to mediate virulence in Fusarium oxysporum (Denisov et al., 2011) and A. flavus (Pfannenstiel et al., 2019) where, in the latter species, loss of SntB also resulted in decreased aflatoxin synthesis but increased production of normally unexpressed metabolites. Recently, we described the role of SntB in virulence, secondary metabolism and development of P. expansum where the ΔsntB mutant was altered in conidiation, germination rate, virulence and patulin production in apples (Tannous et al., 2020).
Considering reader proteins partner with ‘erasers’ or ‘writers’ in yeast, efforts also focused on determining the SntB complex in filamentous fungi. By using tandem affinity purification (TAP) and mass-spec analysis, SntB was found to interact with the HDAC RpdA (homolog of Rpd3) and the histone demethylase KdmB in A. nidulans (Bayram et al., 2012; Gacek-Matthews et al., 2016; Bauer et al., 2020; Karahoda et al., 2022). Further TAP experiments using KdmB showed it complexed with SntB and the “writer” enzyme EcoA thus establishing a multimeric KERS complex (Karahoda et al., 2022). Deletion of KdmB presented considerable similarity to the SntB deletion phenotype in A. nidulans whereas RpdA deletion yielded a more severe phenotype and EcoA is an apparent essential protein and could not be deleted.
Here we sought to determine if a similar SntB complex could operate in P. expansum and, if so, whether the loss of any complex members could impact fungal development and/or virulence of P. expansum. Through immunoprecipitation and mass spec analysis we found that SntB interacts with KdmB and RpdA, similarly as in A. nidulans. Although unable to delete rpdA, presumably due to lethality, we generated and assessed three isogenic ΔkdmB strains for fungal physiology, stress resistance, secondary metabolite production and virulence on apple. The mutant strains presented a similar phenotype as ΔsntB most notably with a decrease in virulence and mycotoxin synthesis on apple.
Materials and Methods
Background Strain and Culture Conditions
The experiments were conducted using the P. expansum Pe21 as the background strain (Hadas et al., 2007). For all the experiments, conidia were obtained from a fresh culture on Glucose Minimal Media (GMM) incubated for 5 days at 25°C. Conidia were harvested using a solution of sterile 0.01% Tween 80 and quantified using a hemacytometer.
ΔkdmB and 3XFLAG-tagged SntB knock-in Strains Construct Generation
To design and generate the transformation cassettes for the ΔkdmB strains, we used double joint PCR as described by Lim et al., 2012. Briefly, 1.5 kb upstream and downstream fragments of the kdmB open reading frame (ORF) (PEXP_096530) were amplified using Pfu DNA polymerase (Agilent, Santa Clara, CA, United States). In addition, A. fumigatus pyrG gene was amplified in the same manner. Later, the obtained PCR products were gel extracted and combined in a (1:3:1) ratio for double joint PCR. The resulting PCR product was used as template DNA to amplify the construct which was purified using a G-50 column. The primer sequences are listed on Table S1A. A 3XFLAG-tagged SntB (PEXP_027800) knock-in strain was also generated in order to determine the subcellular localization of SntB and its interacting proteins. An epitope tagging of the protein was done with a FLAG tag at its C-terminus in the P. expansum Pe-21 WT strain. The 3X-Flag tag sequence is inserted upstream of the sntB stop codon through homologous recombination. A PCR fragment carrying the 3x-Flag sequence (GACTACAAAGACCATGACGGTGATTATAAAGATCATGATATCGATTACAAGGATGACGATGACAAG) and the TrpC terminator was amplified from plasmid pSK529 (Hartmann et al., 2010; Jimenez-Ortigosa et al., 2012) using the primer pair FLAG:trpC(t)_F and FLAG:trpC(t)_R. This PCR product was fused to the hygromycin resistance gene hph under control of the gpdA promoter amplified from plasmid pSK529 (Hartmann et al. 2010; Jimenez-Ortigosa et al., 2012) using the primer pair Hyg_Flag F and Hyg_Flag R. The fragment containing the hygromycin resistance gene as a selectable marker and the 3X-flag sequence was used as the middle fragment in the double joint fusion PCR. The 5’ fragment corresponds to the last 1kb of the sntB transcript missing the stop codon. This fragment was amplified using the primer pair PesntB_flagtag_5’F/R. The last fragment corresponds to the 3’ flank of the sntB gene and was amplified using the primer pair PesntB_flagtag_3’F/R. The diagram of this construct is shown in Fig. S1C. The three fragments were fused together using a double joint PCR protocol described by Lim et al. (2012). All primers used to make the construct can be found in Table S1. In addition, we attempted to generate ΔrpdA and rpdAKD (PEXP_031050) strains but were unsuccessful leading to infer that this locus is essential for P. expansum as it is for Aspergillus nidulans (Karahoda et al., 2022)
Protoplast Isolation and PEG-mediated Transformation
To obtain transformants, we used gene replacement by homologous recombination as previously described by Greco et al., 2019 with modifications described in Luciano-Rosario et al., 2022. We used TDL 9.1, a ΔpyrG mutant, as a background strain for generating the ΔkdmB strains. The obtained transformants were screened and confirmed by PCR and Southern Blot. All the strains used in this study are listed in Table S1B.
For the 3XFLAG-tagged SntB knock-in strains, the protoplast transformation of P. expansum WT strain was conducted as described in Tannous et al. 2018. Transformants were screened on Sorbitol Minimal Media (SMM) supplemented with hygromycin at a concentration of 100 µg/mL. Ten transformants were randomly picked and subjected to southern blotting to confirm the correct tagging of sntB and the single insertion of the construct. Only one transformant (labeled TJT23.1) was chosen for further study (Fig. S1D).
Physiology: Radial Growth, Conidiation, and Germination
For radial growth measurements, point inoculations were performed using 106 spores in GMM and the colony diameter was measured for 7 days. For conidiation assay, 106 spores were overlayed in GMM plates. Then, agar plugs were collected, homogenized and the spores quantified using a hemocytometer. For evaluating germination, 1mL of 1X105 spores/mL in liquid GMM were added to wells of a 24 well-plate. A total of 50 spores were tracked for 23 hours after inoculation and the proportion of germinated spores was recorded every hour. Strains were assessed in triplicate.
Cell Wall Stressor Assay
GMM plates were prepared containing the assessed cell wall stressors: Calcofluor White (CFW), Congo Red (CR) and Sodium Dodecyl Sulfate (SDS) at the described final concentrations. Point inoculations at different spore concentrations (105, 104, and 103) were photographed 3 days post inoculation and compared to cultures in non-amended GMM plates.
Mycotoxin extraction from cultured media
A 106 fungal conidia/ml solution (100 µl) was inoculated onto 55 mm petri dishes containing 10 ml of solid YES media (20 g bacto yeast extract, 150 g sucrose and 15 g bacto agar per liter). The plates were incubated at 28°C in the dark for 3-7 days as needed for sample collection. pH was measured directly in the agar cultures with a double pore slim electrode connected to a Sartorius PB-11 Basic Meter (Sartorius, Göttingen, Germany).
Patulin was extracted from 1 g of the homogenized medium containing fungal mycelium, which was mixed with 2 mL of HPLC grade ethyl acetate (BioLab, Jerusalem, Israel) and placed in an orbital shaker at 150 rpm for 30 min at room temperature. The sample was centrifuged for 10 min at 6000 g; the supernatant was transferred to a clean glass tube and was evaporated to dryness under a stream of gaseous nitrogen at 50 °C. The residue was redissolved in 1 mL of the mobile phase (0.02 M ammonium acetate:acetonitrile 9:1 v/v) and filtered through a 0.22 μM PTFE syringe filter prior to HPLC analysis.
Citrinin was extracted from 1 g of the homogenized YES medium with fungal mycelium using 1 mL of methanol (HPLC grade; BioLab, Jerusalem, Israel). The sample was shaken in an orbital shaker at 150 rpm for 30 min at room temperature and then centrifuged for 10 min at 6000 g. The supernatant was filtered through a 0.22 μM PTFE syringe filter and kept at −20 °C prior to HPLC analysis.
RNA isolation and qRT-PCR analyses
Mycelia grown on agar plates were harvested on day 3 post-inoculation, frozen in liquid nitrogen, lyophilized for 24 hours and kept at −80 °C until use. Total RNA was extracted from 100 mg of lyophilized tissue of the selected samples using the Hybrid-R RNA isolation kit (GeneAll, Seoul, South Korea) according to the manufacturer’s protocol. The DNase and reverse-transcription reactions were performed on 1 µg of total RNA with the Maxima First-Strand cDNA Synthesis Kit (Thermo Scientific, Waltham, MA, United States) according to the manufacturer’s instructions. The cDNA samples were diluted 1:10 (v/v) with ultrapure water.
Quantitative real-time PCR was performed using Fast SYBR green Master Mix (Applied Biosystems, Waltham, MA, USA) in a StepOnePlus Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). The PCR conditions were as follows: 95 °C for 20 sec, followed by 40 cycles of 95 °C for 3 sec and 60 °C for 20 sec. The samples were normalized using β-tubulin as endogenous control and the relative expression levels were measured using the 2(−ΔΔCt) analysis method. Results were analyzed with StepOne software v2.3. Primer sequences used for qRT-PCR analysis are listed in Table S1C.
Virulence and mycotoxin assessment in apples
Golden Delicious apples were obtained from a local supermarket. Fruits were subjected to surface sterilization using 1% sodium hypochlorite solution for 1 min and immediately rinsed in sterile distilled water. A 10 μL conidial suspension containing 107 conidia/mL was injected directly into the sterilized fruits at 2 mm depth. Following inoculation, the fruits were incubated in covered plastic containers at 28 °C for 16 days for symptom monitoring and sample collection. The diameter of the rotten spots was recorded at selected time points. The pH of apple tissues was measured by inserting a double pore slim electrode directly into the tested area.
Patulin was extracted from 1 g of homogenized apple tissue samples using 2 mL ethyl acetate in a 15 mL glass tube. Each sample was vortexed for at least 5 min; then, 2 mL of 1.5% sodium bicarbonate solution was added, and the sample was vortexed vigorously again for 1 min. Next, 20 μL of acetic acid was added, and after shaking in an orbital shaker at 250 rpm for 5 min, the sample was left to stand for a few minutes at room temperature for phase separation. The upper phase was transferred to a new glass tube and evaporated to dryness under a stream of gaseous nitrogen at 50 °C. The samples were reconstituted in the mobile phase (0.02 M ammonium acetate:acetonitrile 9:1 v/v) and filtered through a 0.22 μm PTFE syringe filter into a glass injection vial to prepare for the HPLC analysis.
Citrinin was extracted from 1 g of homogenized apple tissue using 2 mL of methanol. Each sample was then vortexed for 5 min and shaken using an orbital shaker at 250 rpm for 30 min before centrifuging at 6000 g for 10 min. The aliquots were filtered through a 0.22 μm PTFE membrane syringe filter into a glass injection vial and stored at −20 °C prior to HPLC analysis.
HPLC analysis
Both patulin and citrinin were quantitatively analyzed by injection of 20 μL filtered samples into a reverse-phase UHPLC system (Waters ACQUITY Arc, FTN-R, Milford, MA, USA) using a Kinetex 3.5 μm XB-C18 (150 × 4.6 mm) column (Phenomenex, Torrance, CA, USA). The column temperature was maintained at 30 °C, and the flow rate was 0.8 and 1 ml/min, respectively. For patulin, the separation was achieved with a mobile phase composed of 0.02 M ammonium acetate:acetonitrile (9:1 v/v); citrinin mobile phase consisted of 1% acetic acid in water and acetonitrile (50:50 v/v). The patulin peak was detected with a photodiode array (PDA) detector at 276 nm; citrinin was detected with a fluorescence detector (331 nm excitation, 500 nm emission). Both mycotoxins were quantified by comparing peak areas with calibration curves of the standard mycotoxins (Fermentek, Jerusalem, Israel).
Isolation of crude protein extracts for pull-down assay
The mycelia from vegetative cultures were frozen using liquid nitrogen and pulverised with a mortar and pestle. Protein crude extracts were prepared by re-suspending the pulverised mycelia in 1ml protein extraction buffer (300mM NaCl, 50mM Tris-HCl pH 7.5, 10% glycerol, 1mM EDTA, 0.1% NP-40) that had been supplemented with 1mM DTT, 1X complete EDTA-free protease inhibitors (Roche), 1mM benzamidine, 0.5mM PMSF and 1X phosphatase inhibitors (1mM NaF, 0.5mM sodium orthovanadate, 8mM β-glycerol phosphate) immediately prior to use. Samples were mixed vigorously by vortexing and centrifuged at 13,000 RPM for 10-15 minutes at 4°C. 1ml of the crude protein supernatant was transferred to a new 1.5ml microcentrifuge tube.
Immunoprecipitation of FLAG-tagged proteins
Previously published GFP-TRAP protocol for A. nidulans and A. fumigatus was modified and used for FLAG-tagged fusion proteins (Manfiolli et al., 2019; de Assis et al., 2021) For the immunoprecipitation of FLAG-tagged fusion proteins, 20μl anti-FLAG M2 magnetic beads (Sigma-M8823) were washed twice with 180μl protein extraction buffer, containing supplements. After each wash, the bead solution was placed on a magnetic rack and the protein extraction buffer was removed. The anti-FLAG M2 magnetic beads were then resuspended in 50μl protein extraction buffer and added to 1ml crude protein extract. This mixture was left to incubate on a rotator for 3 hours at 4°C. Samples were placed in a magnetic rack and the supernatant was discarded. Beads were washed twice with 1ml protein extraction buffer (without supplements) and were then washed for a third time with the same buffer containing 1mM DTT. All liquid was removed and the beads were stored at −80°C until further use.
Sample preparation for LC-MS/MS peptide identification
Isolated FLAG-tagged proteins were resuspended in 50mM ammonium bicarbonate. 1μl of 0.5M DTT was added and samples were incubated at 56°C for 20 minutes. 2.7μl of iodoacetamide (0.55M) was added and samples were incubated in the dark for 15 minutes. 1μl of 1% (w/v) ProteaseMAX (Promega) was added, followed by addition of 1μl trypsin (1μg/μl) (Promega). Samples were left to incubate overnight at 37°C. The next day, 1μl of Trifluoroacetic acid (TFA) was added to each and samples were vortexed briefly and left to incubate for 5 minutes at room temperature. Beads were collected on a magnetic rack and the supernatant was transferred to new tubes. The supernatants were centrifuged at 13,000 RCF for 10 minutes and dried in a SpeedVac for 3 hours. Samples were stored at −20°C until further use.
Peptide samples were resuspended in 20 μl resuspension buffer (0.5% TFA) and sonicated for 3 minutes, followed by a brief centrifugation. ZipTip C18 pipette tips (Millipore) were used to purify peptide samples prior to mass spectrometric analysis. To equilibrate the ZipTips, a wetting solution (0.1%, 80% acetonitrile) was aspirated 5 times, followed by aspiration of an equilibration buffer (0.1% TFA) 5 times. ZipTips were then used to pipette the peptide samples up and down 15-20 times. Then, the equilibration buffer was aspirated again 5 times, followed by elution of the peptides via aspiration of an elution buffer (0.1% TFA, 60% acetonitrile) 5 times into a new microcentrifuge tube. This solution was dried in a SpeedVac for 2 hours and peptide samples were stored at −20°C.
LC-MS/MS analysis and peptide identification
Immediately prior to loading, peptide samples were resuspended in 10μl Q-Exactive loading buffer (2% acetonitrile, 0.5% TFA) and 8μl were added to mass spectrometry vials (VWR). Digested peptides were separated using reverse phase liquid chromatography with an Ultimate 3000 NanoLC system (Dionex Corporation, Sunnyvale, CA, USA) followed by mass identification using a Q-Exactive mass spectrometer (Thermo Fisher Scientific). Samples were loaded by an autosampler onto a C18 trap column, which was switched on-line with an analytical Biobasic C18 Picofrit column (C18 PepMap, 75 μm id × 500 mm, 2 μm particle size, 100 Å pore size; Dionex). The peptides generated were eluted over 65 minutes. Full MS scans in the 300-1700 m/z range were recorded with a resolution of 140,000 (m/z 200) with a blocking mass set to 445.12003.
LC-MS identification of peptides were performed using the Proteome Discoverer Daemon 1.4 software (Thermo Fisher) and organism-specific taxon-defined protein databases. As controls, anti-FLAG magnetic beads were added to crude protein extracts from wild type strains. These samples were prepared for mass spectrometry analysis as described for the immunoprecipitated FLAG-tagged fusion proteins. Confirmation of protein interactions and unique peptides were determined by isolating only those that appear in the FLAG-tagged fusion protein purifications but do not appear in any of the wild type controls.
Results
KdmB interacts with SntB and RpdA in a trimeric complex
Fungal secondary metabolite BGCs, including the pat (patulin) and cit (citrinin) BGCs, are regulated by histone PTMs in response to various external stimuli. Following previous evidence that SntB loss in P. expansum resulted in decreased patulin and citrinin synthesis and decreased virulence in apples (Tannous et al., 2020), and that SntB was a member of the KERS complex in A. nidulans (Karahoda et al., 2022), we sought to identify SntB interacting proteins and determine whether these proteins could also impact virulence and mycotoxin synthesis in P. expansum. In order to elucidate such proteins, we created a flag-tagged version of sntB to replace the native locus. One correct transformant, TJT23.1, was used for the protein immunoprecipitation experiments which were coupled with mass spectrometry analysis (Fig. S1D).
Four separate SntB-FLAG immunoprecipitations were analyzed by LC-MS/MS analysis. In all 4 replicates, SntB recruited the same top two proteins (Fig. 1A): a homolog of A. nidulans histone demethylase KdmB (PEXP_096530) and a homolog of A. nidulans histone deacetylase RpdA (PEXP_031050). The percentage of coverage and numbers of unique peptides for these two proteins were significantly high and were consistent across all replicates (Fig. 1A, Table S2). This provides strong evidence that SntB exhibits direct interactions with both KdmB and RpdA in P. expansum, forming a trimeric complex. The P. expansum KdmB and RpdA proteins exhibit 70.91% and 70.01% sequence similarity to the A. nidulans homologs respectively, implying a high level of conservation between the two species.
Fig. 1.
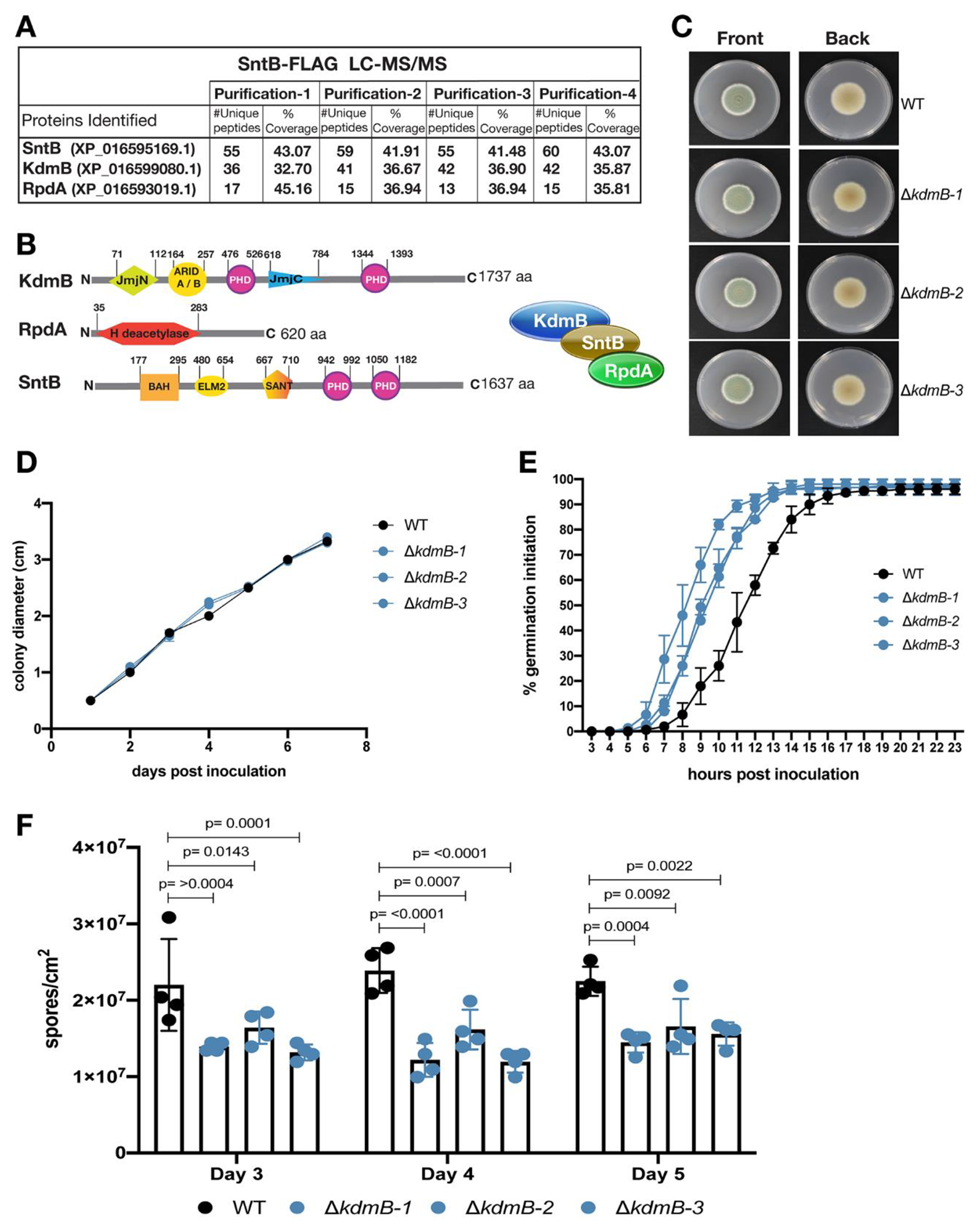
Identification of KdmB-RpdA-SntB complex in Penicillium expansum and phenotypes of ΔkdmB strains. (A) Table showing number of unique peptides and coverage of SntB interacting proteins from four different purifications of SntB-FLAG tag fusion pull downs during vegetative growth of the fungus. (B) Sizes and domains of three subunits of the KRS complex. For the description of the domains, please see the text in results section. (C) Front and back colony pictures of three independent transformants of ΔkdmB strains in comparison to WT. GMM plates were imaged 7 days post inoculation after (D) measuring their colony diameters. (E) Germination initiation was recorded for 23 hpi and (F) conidia production was quantified for 3 consecutive days upon overlay inoculation using 106 spores. Error bars denote standard deviation of the mean (SD).
The P. expansum KdmB protein is a large multi-domain protein of 1,737 amino acids (aa), similar to A. nidulans KdmB which is 1,717 aa (Fig. 1B). According to Uniprot (https://www.uniprot.org), P. expansum KdmB possesses the following domains: JmjN at aa position 71-112, ARID (A/B) at aa position 164-257, 2 PHD-type domains, one at aa position 476-526 and the other at aa position 1344-1393, a C5HC2 zinc finger domain at aa position 873-932 and lastly a JmjC domain at aa position 618-784. The P. expansum RpdA protein is 620 aa, slightly smaller than A. nidulans which is 687 aa. Both proteins contain a single histone deacetylase domain. This domain is present at aa position 35-283 in P. expansum, while it exists at aa position 26-424 in A. nidulans. The SntB protein in P. expansum is a large, multidomain protein of 1,637 aa, similar to the A. nidulans homolog which is 1,596 aa (Karahoda et al., 2022). P. expansum SntB contains the following domains: BAH at aa position 177-295, ELM2 at aa position 480-654, SANT at aa position 667-710 and two PHD-type domains at aa positions 942-992 and 1050-1182. The levels of sequence identity and domain structure for these proteins between P. expansum and A. nidulans suggest that they perform similar roles and function as a complex in both species.
Interestingly, unlike what is reported in A. nidulans (Karahoda et al., 2022) and our companion piece in A. flavus (Karahoda et al. in submission), the P. expansum EcoA homolog (PEXP_003320) was not detected in any of our pulldown data. The P. expansum EcoA protein exhibits 71.91% sequence similarity to the respective A. nidulans homolog. P. expansum EcoA is 376 aa, while the EcoA protein in A. nidulans is 401 aa (Karahoda et al., 2022). P. expansum EcoA contains a ZF domain at aa position 132-168 and an acetyltransferase domain at aa position 305-373. These two domains are present in A. nidulans EcoA at similar positions, indicating a high degree of conservation between the two species. This may imply that this protein could be performing similar roles and could be associated with the KERS complex in both species. However, lack of EcoA in any pulldown data suggests that SntB does not interact with EcoA directly in P. expansum, which differs from what was observed for the KERS complex in A. nidulans (Karahoda et al., 2022). However, it is still possible that EcoA exists as a member of this complex as it could interact with KdmB and/or RpdA and thus could be associated with SntB indirectly. The interaction of EcoA with the complex could also be weak or transient, existing only during certain stages of fungal development or under specific growth conditions or stresses.
ΔkdmB strains show enhanced germination initiation rate and decreased conidia production
To investigate the role of SntB interacting members in P. expansum, we aimed to generate gene deletion mutants for KdmB and RpdA. We successfully obtained ΔkdmB strains but could not obtain deletion nor knock-down strains for rpdA despite multiple attempts suggesting that this is an essential gene in P. expansum (Fig. S1A). We also designed a cassette for complementation of ΔkdmB strains. This construct was 14,208bp and was designed to be inserted in the ku70 locus as the background strain is Δku70. The construct included the kdmB ORF, 900bp upstream for its promoter region, and the sequence for an excisable construct of hygromycin resistance gene. The large cassette could not be generated for successful transformation. As we were unable to generate a kdmB complementation strain, we analyzed three independent deletion strains for the following experiments (Fig. 1C).
Colony diameter, germination initiation rate and conidia production for the WT and three ΔkdmB sibling strains is shown in Fig. 1. We observed no difference in colony diameter when the strains were grown in GMM media and no obvious phenotypical difference between the ΔkdmB and WT strains in this media (Fig. 1CD). This contrasted with ΔsntB that showed a decrease in diameter growth compared to WT (Tannous et al., 2020). The ΔkdmB strains showed a one hour increase in germination initiation when compared to the WT strain although all the strains achieved 96% germination by the final recorded timepoint (Fig. 1E), a similar observation as reported for ΔsntB (Tannous et al., 2020). In addition, we observed decreased conidia production per area at days 4 and 5 for two out of the three ΔkdmB strains when compared to the WT strain (Fig. 1F).
ΔkdmB strains show susceptibility to cell wall stressors
Considering that Choi et al., 2022 showed that the A. fumigatus ΔkdmB strain is more sensitive to H2O2 than the WT strain, we were curious to evaluate if P. expansum ΔkdmB strains were more sensitive to chemical stressors. We assessed the response of the WT and ΔkdmB strain to Calcofluor White (CFW) and Congo Red (CR) as cell wall stressors, and Sodium Dodecyl Sulfate (SDS) as a cell membrane stressor. All the ΔkdmB strains showed increased susceptibility to CFW at 75 μg/mL and 100 μg/mL (Fig. 2A) but minimal sensitivity to CR and SDS (Fig. 2BC). The enhanced sensitivity to CFW suggests a possible alteration in chitin synthesis in the mutants.
Fig. 2.

Wildtype (WT) and ΔkdmB strains phenotypes challenged with cell wall stressors. (A) Calcofluor White (CFW), (B) Congo Red (CR), (C) Sodium Dodecyl Sulfate (SDS).
Impaired virulence of ΔkdmB strains in Golden Delicious apples
To assess if kdmB is involved in virulence and blue mold disease, we performed an apple inoculation experiment and measured the lesion diameter following the colonization by 7 days (Fig. 3AB). All ΔkdmB strains showed reduced lesion diameter over time when compared to the WT strain suggesting that kdmB plays a role in virulence. These results are consistent with those described for the SntB deleted strain, where a marked reduction in decay development in infected apples was also observed (Tannous et al., 2020). Since acidification of host tissues is an essential virulence strategy that necrotrophic pathogens employ (Prusky et al., 2004), and the ΔkdmB strains showed reduced lesion diameters on apples, we measured the resulting pH for these strains in the rotten apple tissue 16 days post inoculation. We found that the ΔkdmB strains are impaired in tissue acidification when compared to the WT strain (Fig. S2A). Interestingly, when measuring the pH in YES media, the same trends hold but to a decreased extent (Fig. S2B). In addition, as some secondary metabolites including patulin act as virulence factors for some pathogens and patulin production is modulated by pH (Snini et al., 2016; Kumar et al., 2018), we quantified patulin and citrinin in infected apples. The ΔkdmB strains showed decreased amounts of patulin and citrinin in apples (Fig. 3CD).
Fig. 3.
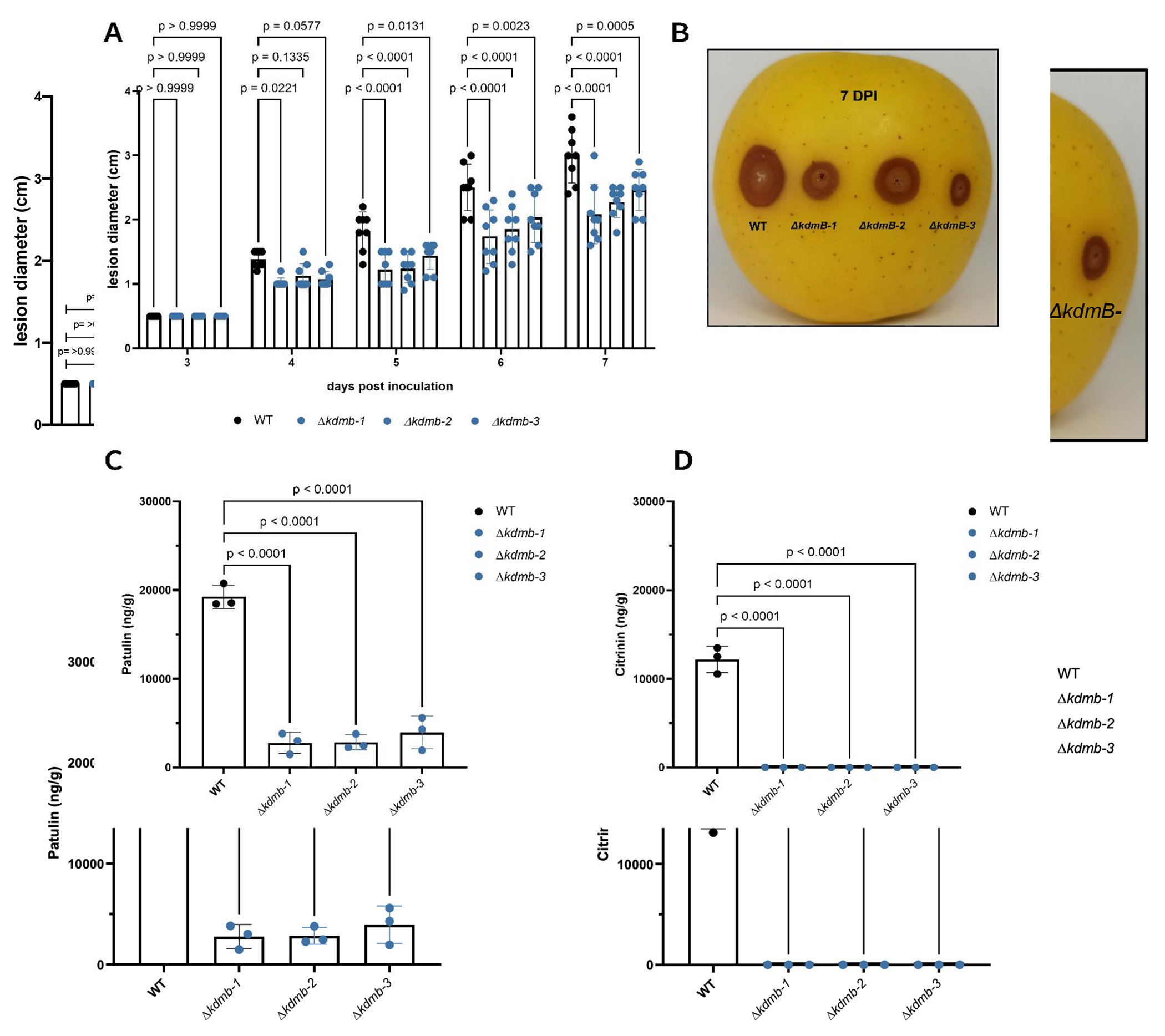
Apple lesion development and mycotoxin detection in vivo. (A) Lesion diameters were measured for 7 days upon inoculation with WT or ΔkdmB strains. (B) representative photo of an inoculated apple with the assessed strains. (C) patulin and (D) citrinin were detected from 1g of homogenized infected tissue at 16 dpi. Error bars denote standard deviation of the mean.
Mycotoxin production in host tissue is not always reflected in growth media. To determine if KdmB also impacted patulin and citrinin synthesis in laboratory conditions, we grew WT and the ΔkdmB strains in the mycotoxin producing media YES. Figure 4A and B show that patulin but not citrinin synthesis was reduced in this media. Because epigenetic mutations often result in transcriptional changes of BGC genes we then evaluated the expression of select patulin and citrinin BGC genes to see if there was any correlation with expression and mycotoxin production. Expression of the backbone polyketide synthase gene patK required for patulin synthesis was decreased in the ΔkdmB compared to the WT strain when grown in YES media (Fig. 4C), possibly contributing to the decreased patulin amounts quantified in YES media (Fig. 4A). The citrinin polyketide synthase gene citS was increased in relative gene expression in the ΔkdmB strains when compared to the WT whereas two other citrinin genes were not differentially expressed largely matching with YES metabolite data (Fig 4D).
Fig. 4.
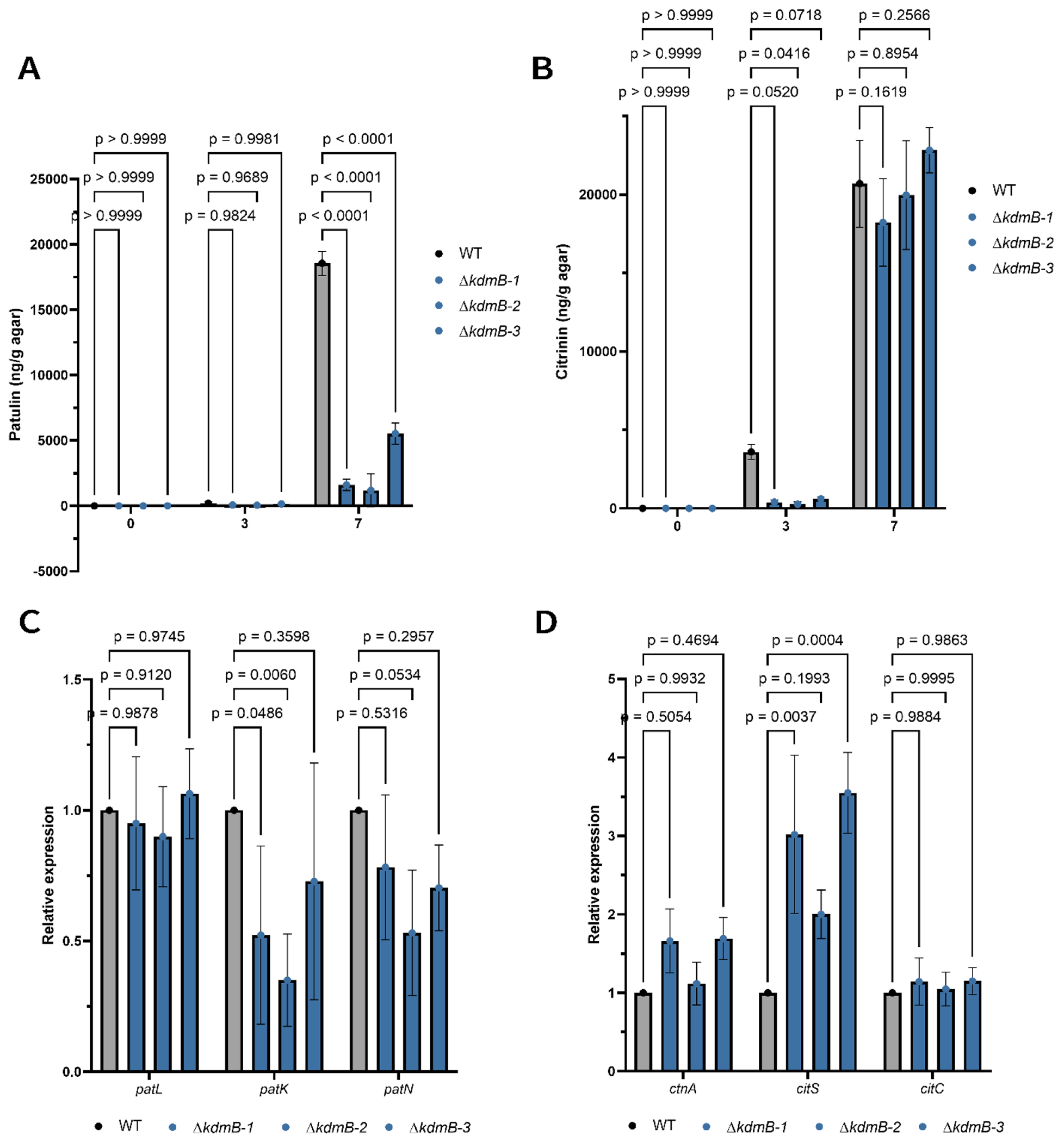
Patulin and citrinin quantification and gene expression using qPCR for WT and ΔkdmB strains in YES media. (A) Patulin and (B) Citrinin were extracted and quantified using an HPLC from YES media plates at 3 and 7 dpi. (C) Gene expression of the patulin backbone genes patL (transcription factor), patK (polyketide synthase, and patN (isoepoxydron dehydrogenase). (D) gene expression of the citrinin backbone genes ctnA (transcription factor), citS (polyketide synthase), citC (oxidoreductase). Error bars denote standard deviation of the mean.
Discussion
Fungal responses to the environment are governed by vast changes in transcriptionally active regions of the genome. Other than S. cerevisiae and Schizosaccharomyces pombe, the identity and function of chromatin modifying complexes is still in its infancy in fungi. However, recent advances such as the Hst1-Rfm1-Sum1 complex controlling virulence related genes in C. glabrata (Vázquez-Franco et al., 2022) and the discovery of the KERS complex in A. nidulans governing fungal development and mycotoxin synthesis are providing new frameworks for continuous progress in this field. KERS is a heteromeric complex containing a ‘reader’ (SntB), two ‘eraser’ (RpdA and KdmB) and one ‘writer’ (EcoA) protein. A previous study showed SntB to be a key regulator of virulence, mycotoxin production and fungal development in the blue mold pathogen P. expansum (Tannous et al., 2020). Here, as found in A. nidulans, we report that SntB recruits the Jarid1-type Jumonji domain demethylase KdmB and the class I HDAC RpdA (Fig. 1). In A. nidulans, KdmB and SntB form a bridging complex with KdmB interacting with EcoA and SntB interacting with RpdA. Similarly, in an associated paper (Karahoda et al. in submission), tagging of KdmB with HA and GFP epitope tag in the plant pathogenic fungus Aspergillus flavus led to identification of EcoA, RpdA and SntB. However, in our four purifications we could not identify EcoA as an interaction partner of KdmB-RpdA-SntB complex. We speculate that if EcoA exists as a member of the KERS complex in P. expansum, it could be interacting with proteins other than SntB, most likely KdmB based on results in Aspergillus spp. Thus, further pulldowns of KdmB, RpdA and EcoA could help clarify the direct and indirect interactions of EcoA within this complex. These pulldown data could also provide information regarding the interaction partners of the complex, as well as the independent interactions of each individual tagged protein. This could provide information regarding possible roles of each protein that are independent of the KERS complex. Efforts to delete kdmB were successful but deletions were not possible for rpdA and ecoA following that reported for A. nidulans and A. flavus. Our efforts therefore were to characterize kdmB deletion on the pathogenicity and mycotoxin synthesis of P. expansum.
KdmB has been studied in diverse filamentous fungi (Gacek-Matthews et al., 2016; Hou et al., 2020; Janevska et al., 2018; Lukito et al., 2019; Bachleitner et al., 2019; Choi et al., 2022). In these studies, in addition to establishing that KdmB demethylates H3K4me3, the data show that kdmB also plays a role in virulence, secondary metabolism, stress resistance and conidiation in a variety of fungi. In A. nidulans, kdmB was identified to regulate secondary metabolism but through an alternate mechanism that its catalytic role as a demethylase (Gacek-Matthews et al., 2016). For example, in A. fumigatus, an increased in survival rate and decreased fungal burden was reported for mice when inoculated with a ΔkdmB strain as compared to WT (Choi et al., 2022). In addition, the secondary metabolites gliotoxin and sterigmatocystin were reduced in ΔkdmB strains of A. fumigatus and A. nidulans respectively (Choi et al., 2022; Gacek-Matthews et al., 2016). Similarly, in the associated manuscript, kdmB and rpdA mutants cannot produce the mycotoxin aflatoxin in A. flavus (Karahoda et al. in submission). Botrytis cinerea ΔBcjar1 mutants showed reduced conidiation and tolerance to SDS and H2O2 (Hou et al., 2020). While reduced conidiation was observed in B. cinerea, increased conidiation was reported for F. fujikuroi and A. fumigatus (Janevska et al., 2018; Choi et al., 2022).
We also found some alterations in physiological traits of ΔkdmB mutants. These mutants showed many similarities to P. expansum ΔsntB strains such as an increased germination initiation rate and decreased conidiation (Fig. 2 and Tannous et al., 2020). These findings contrasted with ΔkdmB strains in Fusarium fujikuroi and Aspergillus fumigatus which presented increased conidia production (Janevska et al., 2018; Choi et al., 2022). While in the later species kdmB may act as a negative regulator of conidiation, the data suggests that in P. expansum it has an opposite role although we acknowledge that the cultural conditions may have a confounding impact on any conclusions on gene loss and physiology.
Since PTMs have been described to impact secondary metabolism in filamentous fungi, we hypothesized that kdmB would have a role in mediating regulation of secondary metabolism. There are many instances in which ΔkdmB strains have been shown to impact secondary metabolite production. For example, in F. fujikuroi, the ΔkdmB strain showed reduced bikaverin, fusarubins, fusarins and GA3 production when compared to the WT strain (Janevska et al., 2018). Similarly, paxilline and lolitrem B were reduced in Epichloë festucae ΔkdmB strains (Lukito et al., 2019). A similar trend was described for Fusarium graminearum, A. nidulans and A. fumigatus in which ΔkdmB strains produced less deoxynivalenol (DON), fusarin C, zearalenone, fusarielin (Bachleitner et al., 2019); sterigmatocystin (ST), emericellamide C, emericellamide D, orsellinic acid (Gacek-Matthews et al., 2016); and gliotoxin respectively (Choi et al., 2022). In concordance with these studies, we observed that P. expansum mutants showed reduced patulin and citrinin production in ΔkdmB strains in apples. We also found reduced patulin and decreased expression of its associated polyketide synthase gene in YES media which suggest that the decreased patulin but not citrinin possibly occurs through transcriptional regulation.
In addition, the ΔkdmB strains showed a reduced lesion diameter in apple inoculation assays when compared to the WT strain suggesting that kdmB plays a role in virulence as well. This result is very similar to the one reported for the ΔsntB strain. Along with the reduced production of patulin in vivo, which acts as a virulence factor for some apple cultivars, an additive effect may be possible due to the observed increased susceptibility to cell wall stressors for the ΔkdmB strains as the apple environment has a plethora of stressors due to activation of PAMP-triggered immunity (PTI) such as reactive oxygen species (ROS) and cell wall reinforcement from the apple immune response (Wang et al., 2019). Also, degradative enzyme production has been linked to deletion of chromatin modifying proteins (Wu et al., 2021). We speculate that there could be some changes in enzyme activity in these mutants as P. expansum mutants decreased secretion of plant cell degradative enzymes are less virulent on apple (Jurick et al., 2020). Acidification of apple tissue by P. expansum is also an important virulence trait (Prusky et al., 2004) and possibly the reduced acidification ability of the ΔkdmB strains could also contribute to impaired colonization and decreased lesion size. While this study presents distinct data that shows P. expansum’s kdmB role in diverse aspects of fungal biology, we are still interested and curious on what the histone PTM modification pattern of the ΔkdmB strains. We hypothesize that KdmB in P. expansum has demethylase activity acting in a similar manner to other filamentous fungi that contain the KERS complex but further experimentation to assess histone methylation marks levels remains to be performed.
The present data supports an important role for KdmB in virulence of P. expansum and, via coupling with previous data of a sntB mutant (Tannous et al., 2020), we hypothesize that much of the defects in virulence observed in both mutants are mediated by a KERS-like complex in the blue mold fungus. Taken together, this work suggests this complex could be a potential target for disease control, possibly broadly applicable across filamentous ascomycetes.
Supplementary Material
Acknowledgements.
This work was supported by Research Grant Award No. IS-5323-20C from BARD, the United States – Israel Binational Agricultural Research and Development Fund, to NPK and ES, by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number T32AI055397 and the University of Wisconsin-Madison Advanced Opportunity Fellowship, Science, and Medicine Graduate Research Scholars program to DL. ÖB was funded by Science Foundation Ireland, grant 21 FFP-P 10146. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or BARD. DF was funded by an IRC postgraduate scholarship (GOIPG/2018/35).
References
- Andersen B, Smedsgaard J, Frisvad JC, 2004. Penicillium expansum: consistent production of patulin, chaetoglobosins, and other secondary metabolites in culture and their natural occurrence in fruit products. J Agric Food Chem 52, 2421–2428. 10.1021/jf035406k [DOI] [PubMed] [Google Scholar]
- Bachleitner S, Sørensen JL, Gacek-Matthews A, Sulyok M, Studt L, Strauss J, 2019. Evidence of a Demethylase-Independent Role for the H3K4-Specific Histone Demethylases in Aspergillus nidulans and Fusarium graminearum Secondary Metabolism. Front Microbiol 10, 1759. 10.3389/fmicb.2019.01759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachleitner S, Sulyok M, Sørensen JL, Strauss J, Studt L, 2021. The H4K20 methyltransferase Kmt5 is involved in secondary metabolism and stress response in phytopathogenic Fusarium species. Fungal Genet Biol 155, 103602. 10.1016/j.fgb.2021.103602 [DOI] [PubMed] [Google Scholar]
- Baker LA, Ueberheide BM, Dewell S, Chait BT, Zheng D, Allis CD, 2013. The yeast Snt2 protein coordinates the transcriptional response to hydrogen peroxide-mediated oxidative stress. Mol Cell Biol 33, 3735–3748. 10.1128/MCB.00025-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer I, Gross S, Merschak P, Kremser L, Karahoda B, Bayram ÖS, Abt B, Binder U, Gsaller F, Lindner H, Bayram Ö, Brosch G, Graessle S, 2020. RcLS2F - A Novel Fungal Class 1 KDAC Co-repressor Complex in Aspergillus nidulans. Front Microbiol 11, 43. 10.3389/fmicb.2020.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayram O, Bayram OS, Valerius O, Jöhnk B, Braus GH, 2012. Identification of protein complexes from filamentous fungi with tandem affinity purification. Methods Mol Biol 944, 191–205. 10.1007/978-1-62703-122-6_14 [DOI] [PubMed] [Google Scholar]
- Chen Y, Li B, Xu X, Zhang Z, Tian S, 2018. The pH-responsive PacC transcription factor plays pivotal roles in virulence and patulin biosynthesis in Penicillium expansum. Environ Microbiol 20, 4063–4078. 10.1111/1462-2920.14453 [DOI] [PubMed] [Google Scholar]
- Choi Y-H, Lee M-W, Shin K-S, 2022. The Lysine Demethylases KdmA and KdmB Differently Regulate Asexual Development, Stress Response, and Virulence in Aspergillus fumigatus. J Fungi (Basel) 8, 590. 10.3390/jof8060590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Assis LJ, Silva LP, Bayram O, Dowling P, Kniemeyer O, Krüger T, Brakhage AA, Chen Y, Dong L, Tan K, Wong KH, Ries LNA, Goldman GH, 2021. Carbon Catabolite Repression in Filamentous Fungi Is Regulated by Phosphorylation of the Transcription Factor CreA. mBio 12, e03146–20. 10.1128/mBio.03146-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov Y, Freeman S, Yarden O, 2011. Inactivation of Snt2, a BAH/PHD-containing transcription factor, impairs pathogenicity and increases autophagosome abundance in Fusarium oxysporum. Mol Plant Pathol 12, 449–461. 10.1111/j.1364-3703.2010.00683.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacek-Matthews A, Berger H, Sasaki T, Wittstein K, Gruber C, Lewis ZA, Strauss J, 2016. KdmB, a Jumonji Histone H3 Demethylase, Regulates Genome-Wide H3K4 Trimethylation and Is Required for Normal Induction of Secondary Metabolism in Aspergillus nidulans. PLoS Genet 12, e1006222. 10.1371/journal.pgen.1006222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco C, Pfannenstiel BT, Liu JC, Keller NP, 2019. Depsipeptide Aspergillicins Revealed by Chromatin Reader Protein Deletion. ACS Chem. Biol 14, 1121–1128. 10.1021/acschembio.9b00161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadas Y, Goldberg I, Pines O, Prusky D, 2007. Involvement of Gluconic Acid and Glucose Oxidase in the Pathogenicity of Penicillium expansum in Apples. Phytopathology 97, 384–390. 10.1094/PHYTO-97-3-0384 [DOI] [PubMed] [Google Scholar]
- Hartmann T, Dümig M, Jaber BM, Szewczyk E, Olbermann P, Morschhäuser J, Krappmann S, 2010. Validation of a Self-Excising Marker in the Human Pathogen Aspergillus fumigatus by Employing the β-Rec/six Site-Specific Recombination System. Appl Environ Microbiol 76, 6313–6317. 10.1128/AEM.00882-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Feng H-Q, Chang H-W, Liu Y, Li G-H, Yang S, Sun C-H, Zhang M-Z, Yuan Y, Sun J, Zhu-Salzman K, Zhang H, Qin Q-M, 2020. The H3K4 demethylase Jar1 orchestrates ROS production and expression of pathogenesis-related genes to facilitate Botrytis cinerea virulence. New Phytol 225, 930–947. 10.1111/nph.16200 [DOI] [PubMed] [Google Scholar]
- Ioi JD, Zhou T, Tsao R, F Marcone M, 2017. Mitigation of Patulin in Fresh and Processed Foods and Beverages. Toxins (Basel) 9, E157. 10.3390/toxins9050157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janevska S, Güldener U, Sulyok M, Tudzynski B, Studt L, 2018. Set1 and Kdm5 are antagonists for H3K4 methylation and regulators of the major conidiation‐specific transcription factor gene ABA1 in Fusarium fujikuroi. Environ Microbiol 20, 3343–3362. 10.1111/1462-2920.14339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Ortigosa C, Aimanianda V, Muszkieta L, Mouyna I, Alsteens D, Pire S, Beau R, Krappmann S, Beauvais A, Dufrêne YF, Roncero C, Latgé J-P, 2012. Chitin Synthases with a Myosin Motor-Like Domain Control the Resistance of Aspergillus fumigatus to Echinocandins. Antimicrob Agents Chemother 56, 6121–6131. 10.1128/AAC.00752-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karahoda B, Pardeshi L, Ulas M, Dong Z, Shirgaonkar N, Guo S, Wang F, Tan K, Sarikaya-Bayram Ö, Bauer I, Dowling P, Fleming AB, Pfannenstiel BT, Luciano-Rosario D, Berger H, Graessle S, Alhussain MM, Strauss J, Keller NP, Wong KH, Bayram Ö, 2022. The KdmB-EcoA-RpdA-SntB chromatin complex binds regulatory genes and coordinates fungal development with mycotoxin synthesis. Nucleic Acids Research 50, 9797–9813. 10.1093/nar/gkac744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karahoda B, Pfannenstiel BT, Sarikaya-Bayram Ö, Wong KO, Fleming AB, Keller NP, Bayram Ö, in submission. The KdmB-EcoA-RpdA-SntB chromatin regulatory complex controls development, secondary metabolism and pathogenicity in Aspergillus flavus. Fungal Genetics and Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller NP, 2019. Fungal secondary metabolism: regulation, function and drug discovery. Nat Rev Microbiol 17, 167–180. 10.1038/s41579-018-0121-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D, Tannous J, Sionov E, Keller N, Prusky D, 2018. Apple Intrinsic Factors Modulating the Global Regulator, LaeA, the Patulin Gene Cluster and Patulin Accumulation During Fruit Colonization by Penicillium expansum. Front Plant Sci 9, 1094. 10.3389/fpls.2018.01094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen TO, Frisvad JC, Ravn G, Skaaning T, 1998. Mycotoxin production by Penicillium expansum on blackcurrant and cherry juice. Food Addit Contam 15, 671–675. 10.1080/02652039809374696 [DOI] [PubMed] [Google Scholar]
- Lim FY, Sanchez JF, Wang CCC, Keller NP, 2012. Toward awakening cryptic secondary metabolite gene clusters in filamentous fungi. Methods Enzymol 517, 303–324. 10.1016/B978-0-12-404634-4.00015-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano‐Rosario D, Keller NP, Jurick WM, 2020. Penicillium expansum: biology, omics, and management tools for a global postharvest pathogen causing blue mould of pome fruit. Mol Plant Pathol 21, 1391–1404. 10.1111/mpp.12990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luciano-Rosario D, Eagan JL, Aryal N, Dominguez EG, Hull CM, Keller NP, 2022. The Hydrophobin Gene Family Confers a Fitness Trade-off between Spore Dispersal and Host Colonization in Penicillium expansum. mBio 13, e0275422. 10.1128/mbio.02754-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukito Y, Chujo T, Hale TK, Mace W, Johnson LJ, Scott B, 2019. Regulation of subtelomeric fungal secondary metabolite genes by H3K4me3 regulators CclA and KdmB. Mol Microbiol 112, 837–853. 10.1111/mmi.14320 [DOI] [PubMed] [Google Scholar]
- Manfiolli AO, Mattos EC, de Assis LJ, Silva LP, Ulaş M, Brown NA, Silva-Rocha R, Bayram Ö, Goldman GH, 2019. Aspergillus fumigatus High Osmolarity Glycerol Mitogen Activated Protein Kinases SakA and MpkC Physically Interact During Osmotic and Cell Wall Stresses. Front Microbiol 10, 918. 10.3389/fmicb.2019.00918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen M, Lindqvist R, Bakeeva A, Leong S-LL, Sulyok M, 2019. Distribution of mycotoxins produced by Penicillium spp. inoculated in apple jam and crème fraiche during chilled storage. Int J Food Microbiol 292, 13–20. 10.1016/j.ijfoodmicro.2018.12.003 [DOI] [PubMed] [Google Scholar]
- Pfannenstiel BT, Greco C, Sukowaty AT, Keller NP, 2018. The epigenetic reader SntB regulates secondary metabolism, development and global histone modifications in Aspergillus flavus. Fungal Genet Biol 120, 9–18. 10.1016/j.fgb.2018.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfannenstiel BT, Keller NP, 2019. On top of biosynthetic gene clusters: How epigenetic machinery influences secondary metabolism in fungi. Biotechnol Adv 37, 107345. 10.1016/j.biotechadv.2019.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfannenstiel BT, Zhao X, Wortman J, Wiemann P, Throckmorton K, Spraker JE, Soukup AA, Luo X, Lindner DL, Lim FY, Knox BP, Haas B, Fischer GJ, Choera T, Butchko RAE, Bok J-W, Affeldt KJ, Keller NP, Palmer JM, 2017. Revitalization of a Forward Genetic Screen Identifies Three New Regulators of Fungal Secondary Metabolism in the Genus Aspergillus. mBio 8, e01246–17. 10.1128/mBio.01246-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusky D, McEvoy JL, Saftner R, Conway WS, Jones R, 2004. Relationship Between Host Acidification and Virulence of Penicillium spp. on Apple and Citrus Fruit. Phytopathology 94, 44–51. 10.1094/PHYTO.2004.94.1.44 [DOI] [PubMed] [Google Scholar]
- Snini SP, Tannous J, Heuillard P, Bailly S, Lippi Y, Zehraoui E, Barreau C, Oswald IP, Puel O, 2016. Patulin is a cultivar-dependent aggressiveness factor favouring the colonization of apples by Penicillium expansum. Mol Plant Pathol 17, 920–930. 10.1111/mpp.12338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss J, Cánovas D, 2021. Editorial SI FGB “Chromatin regulation and epigenetics.” Fungal Genet Biol 153, 103569. 10.1016/j.fgb.2021.103569 [DOI] [PubMed] [Google Scholar]
- Tannous J, Barda O, Luciano-Rosario D, Prusky DB, Sionov E, Keller NP, 2020. New Insight Into Pathogenicity and Secondary Metabolism of the Plant Pathogen Penicillium expansum Through Deletion of the Epigenetic Reader SntB. Front Microbiol 11, 610. 10.3389/fmicb.2020.00610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannous J, Keller NP, Atoui A, El Khoury A, Lteif R, Oswald IP, Puel O, 2018a. Secondary metabolism in Penicillium expansum: Emphasis on recent advances in patulin research. Crit Rev Food Sci Nutr 58, 2082–2098. 10.1080/10408398.2017.1305945 [DOI] [PubMed] [Google Scholar]
- Tannous J, Kumar D, Sela N, Sionov E, Prusky D, Keller NP, 2018b. Fungal attack and host defence pathways unveiled in near-avirulent interactions of Penicillium expansum creA mutants on apples. Mol Plant Pathol 19, 2635–2650. 10.1111/mpp.12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kan JAL, 2006. Licensed to kill: the lifestyle of a necrotrophic plant pathogen. Trends in Plant Science 11, 247–253. 10.1016/j.tplants.2006.03.005 [DOI] [PubMed] [Google Scholar]
- Vázquez-Franco N, Gutiérrez-Escobedo G, Juárez-Reyes A, Orta-Zavalza E, Castaño I, De Las Peñas A, 2022. Candida glabrata Hst1-Rfm1-Sum1 complex evolved to control virulence-related genes. Fungal Genet Biol 159, 103656. 10.1016/j.fgb.2021.103656 [DOI] [PubMed] [Google Scholar]
- Wang K, Zheng X, Zhang X, Zhao L, Yang Q, Boateng NAS, Ahima J, Liu J, Zhang H, 2019. Comparative Transcriptomic Analysis of the Interaction between Penicillium expansum and Apple Fruit (Malus pumila Mill.) during Early Stages of Infection. Microorganisms 7, 495. 10.3390/microorganisms7110495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Nakazawa T, Morimoto R, Shivani, null, Sakamoto M, Honda Y, 2021. Targeted disruption of hir1 alters the transcriptional expression pattern of putative lignocellulolytic genes in the white-rot fungus Pleurotus ostreatus. Fungal Genet Biol 147, 103507. 10.1016/j.fgb.2020.103507 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.