

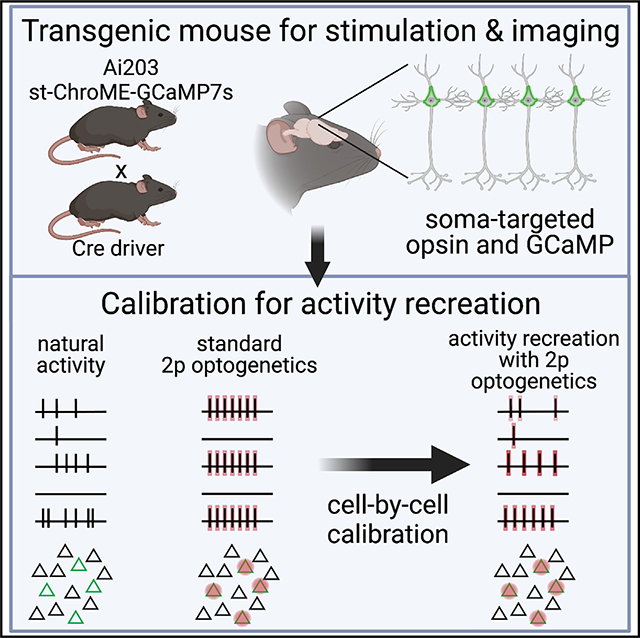
SUMMARY
Determining which features of the neural code drive behavior requires the ability to simultaneously read out and write in neural activity patterns with high precision across many neurons. All-optical systems that combine two-photon calcium imaging and targeted photostimulation enable the activation of specific, functionally defined groups of neurons. However, these techniques are unable to test how patterns of activity across a population contribute to computation because of an inability to both read and write cell-specific firing rates. To overcome this challenge, we make two advances: first, we introduce a genetic line of mice for Cre-dependent co-expression of a calcium indicator and a potent soma-targeted microbial opsin. Second, using this line, we develop a method for read-out and write-in of precise population vectors of neural activity by calibrating the photostimulation to each cell. These advances offer a powerful and convenient platform for investigating the neural codes of computation and behavior.
In brief
Bounds et al. develop and validate a TIGRE2 transgenic line that co-expresses an opsin and calcium indicator, facilitating two-photon all-optical read/write experiments. In this line, the authors calibrate holographic photostimulation to each targeted cell to accurately recreate visually evoked spike rates across a population of neurons.
Graphical abstract
INTRODUCTION
Many of the features of the neural code that underly sensation, cognition, and action remain unknown. Interventional tools that combine the simultaneous monitoring and manipulation of neural activity are proving essential to overcoming this gap. Recently, all-optical techniques using two-photon (2p) excitation have emerged as one class of tool for investigating the neural code with high spatial and temporal precision. 2p optogenetics can achieve near-single-cell resolution control over neural activity, thereby overcoming a major limitation of conventional optogenetic and electrical microstimulation,1–16 helping to reveal basic rules of circuit connectivity and behavior.17–29
2p optogenetics as a technique, however, is not yet routine, and the lack of a simple, robust, and convenient system to achieve widespread, stable co-expression of an optogenetic actuator (opsin) and an activity indicator in genetically defined neural populations has limited the use of all-optical techniques. Most 2p optogenetic studies have so far relied on viral expression,2,8,9,13,17–25,27,30 which typically requires invasive surgery, produces heterogeneous expression, can lead to toxic expression of the transgenes31,32 or may affect synaptic physiology.33 In contrast, transgenic lines promise convenient, stable, and widespread expression of transgenes,34–37 yet no transgenic line for co-expression of a calcium sensor and an optogenetic protein has been reported.
While experimenters have targeted specific, functionally defined neural ensembles, none have controlled the relative distribution of activity among the targeted neurons. Recent advances in the computational analysis of population activity have yielded new theories for how populations encode information and drive behavior. These include new insights into the geometry of neural representations and hypotheses about the importance of neural manifolds, communication subspaces, and more.38–44 Testing these new theories requires perturbations that align with the estimated coding topology or deviate from it in defined ways.44,45 Doing so necessitates a system that can both read and write specific firing rates into individual neurons across a population. Existing all-optical techniques, however, have yet to achieve this for two reasons. First, it is not straightforward to infer the spike rates underlying calcium signals. Although many models for transforming calcium signals into estimates of spike trains exist, the biological variability of the spike-to-calcium signal transformation constrains their performance.46–51 Second, it is challenging to ensure the optogenetic activation of specific numbers of spikes in targeted neurons due to a combination of the variability of opsin expression and intrinsic excitability between neurons.22,52 Both issues imply that the models for inferring spike rates from calcium signals and for driving in specific spike rates with optogenetics must be precisely tuned for each neuron to achieve the desired neural manipulations.
To overcome these obstacles, we developed a transgenic mouse line and a paradigm for all-optical reproduction of precise activity patterns. For the genetic line, we devised a single fusion protein of a high-performance calcium indicator, GCaMP7s,53 and a potent microbial opsin, ChroME,52 to ensure reliable co-expression of the two proteins. A somatic targeting sequence targets both to the soma. The sub-cellular targeting of the opsin to the somatic membrane is necessary to minimize off-target activation of unwanted neurons in targeted photostimulation.13,52,54–56 Currently there are no transgenic lines that express a soma-targeted excitatory opsin. Among high-potency opsins, ChroME has the fastest kinetics, which enables, under parallel illumination, precise temporal control at high frequencies,52 similar to its parent opsin, Chronos.57,58 Among genetically encoded calcium sensors, GCaMP7s strikes a useful balance between the sensitivity of GCaMP8s and the greater dynamic range and linearity of GCaMP6s.47,53,59 We leveraged the TIGRE2.0 system to achieve robust but non-toxic levels of expression of both transgenes34 and demonstrate that this mouse line provides high-fidelity monitoring and optogenetic control of neural activity with 2p excitation.
Next, we combined the unique features of this mouse line with 2p holographic optogenetics to develop a pipeline for precisely calibrating the transformation from 2p excitation pulses into spikes and spikes into calcium signals in individual neurons. We show that this multi-step, cell-specific calibration process enables the holographic optogenetic recreation of precise sensory-driven population activity vectors by driving in firing rates tailored to each neuron in the targeted ensemble. Taken together, this approach and mouse line will empower neuroscientists to probe the precise geometry of neural codes and their relationship to behavior.
RESULTS
Ai203 transgenic reporter line
We first generated a transgenic reporter line using the TIGRE2.0 system, which provides Cre-dependent co-expression of the potent, ultra-fast opsin, ChroME, and the sensitive calcium indicator GCaMP7s34,52,53. We used the Kv2.1 soma-targeting (st) sequence to enrich opsin expression at the soma.34,52,54–56,60 To ensure co-expression of the calcium sensor and the opsin, we fused GCaMP7s directly to the opsin (st-ChroME-GCaMP7s), producing a soma-targeted calcium indicator that will produce stochiometric expression of both proteins.61,62 The construct also includes an FLAG tag for antibody labeling in postmortem tissue when necessary. We termed this line TITL-st-ChroME-GCaMP7s-ICL-nls-mRuby3-IRES2-tTA2 (Ai203) (Figure 1A).
Figure 1. Characterization of expression, photocurrents, and GCaMP fluorescence in VGlut1-Cre;Ai203 mice.
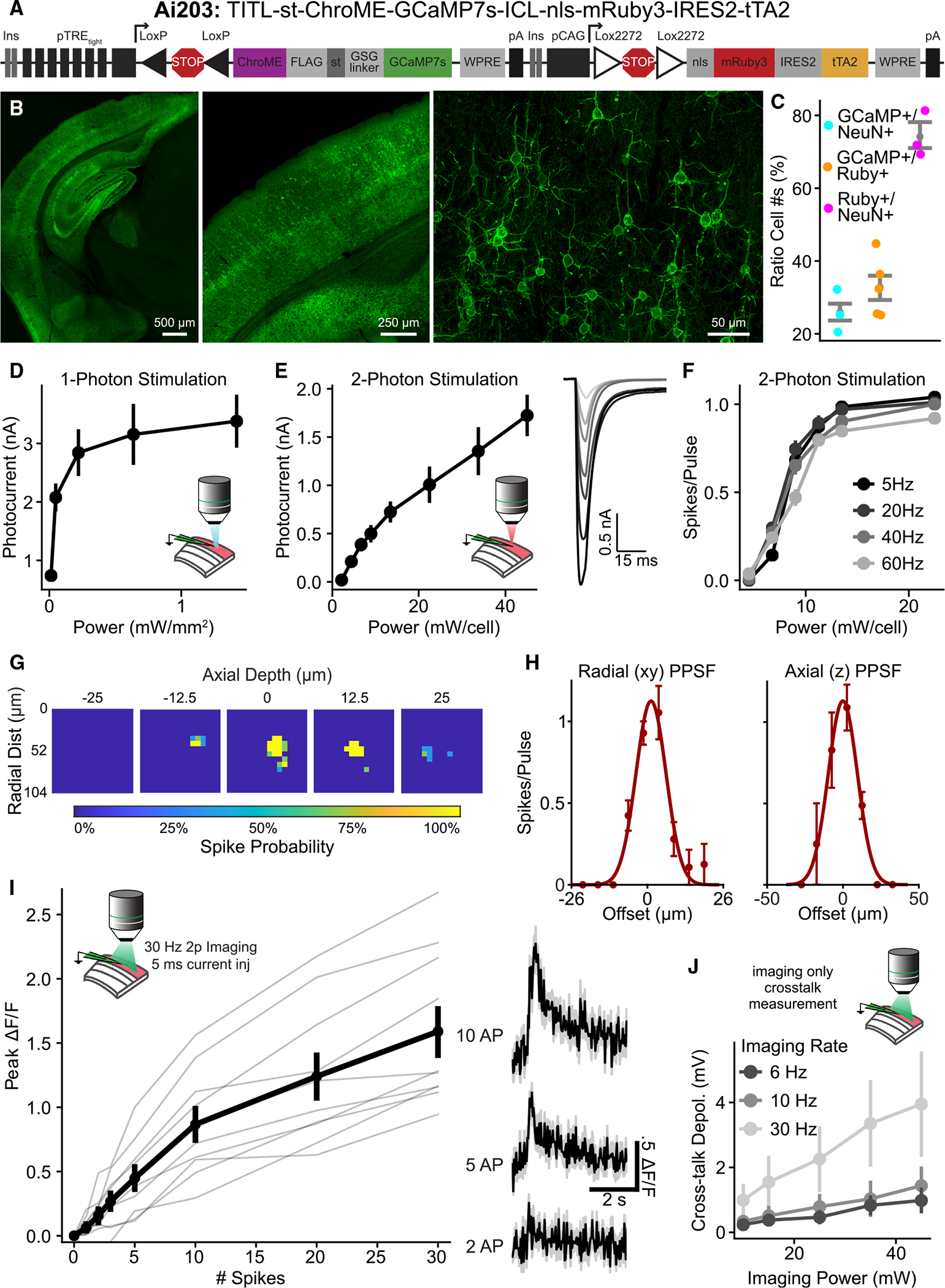
(A) The Ai203 transgene.
(B) Confocal images of a brain section from a representative VGlut1-Cre+/−;Ai203+/− mouse showing GCaMP7s (green) expression in V1 at different magnifications. Images stitched from multiple fields of view. Right, maximum z projection across several depths. See also Figures S1 and S2.
(C) Quantification of expression of GCaMP7s, anti-NeuN, and mRuby3 in V1, expressed as ratios of the number of cells for each category. n = 5 mice for mRuby3 and GCaMP7s counts, n = 3 for anti-NeuN counts.
(D) Photocurrents in opsin+ L2/3 cells evoked by 1p illumination for 5 ms at 510 nm at varying powers, measured in voltage clamp in brain slices (n = 8 cells).
(E) Left, same as (D) but for 2p illumination for 5 ms at 1,035 nm (n = 11 cells). Right, averaged photocurrent traces for an example cell; power increases from light to dark.
(F) Number of spikes per illumination pulse as a function of power, measured by loose-patch recordings during 2p stimulation with a train of 10 5-ms pulses at different frequencies (n = 11 cells).
(G and H) Assessment of physiological point-spread function (PPSF) of photostimulation-evoked spiking measured by loose-patch recordings.
(G) Heatmaps from an example cell depicting spike probability across five planes (power = 13.5 mW). Depth z = 0 corresponds to the plane of the patched cell.
(H) Across-cell mean PPSF for radial (left) and axial (right). Radial FWHM, 15 ± 2; axial FWHM, 28 ± 4 μm (n = 4 cells).
(I) Characterization of GCaMP response. Simultaneous whole-cell recording and 2p imaging were conducted and cells were electrically stimulated with trains of current injections at 30 Hz. 2p imaging was done at power 35 mW, 30 Hz, in a 680-μm FOV. Left: GCaMP fluorescence response vs. the number of induced spikes. Gray lines, individual cells (n = 10 cells). Right: calcium traces for an example cell.
(J) Mean depolarization induced by the imaging laser when imaging at different frequencies and powers with FOV size 680 × 680 μm and average dwell time 46 ns/μm (n = 4 cells). See also Figure S4.
(D–H and J) Data are presented as mean ± SEM. (C and I) Data are presented as mean ± bootstrapped 68% confidence interval.
To test the Ai203 transgenic line, we crossed it to the forebrain excitatory neuron driver, Slc17a7-IRES2-Cre (henceforth referred to as VGlut1-Cre).63 Heterozygous double-transgenic VGlut1-Cre;Ai203 mice exhibited high levels of st-ChroME-GCaMP7s expression across the entire forebrain (Figures 1B and S1). As has been noted for some other combinations of Cre driver and TIGRE2.0 reporter lines, the tetracycline response element (TRE)-dependent st-ChroME-GCaMP7s was only expressed in a subset of excitatory neurons even though VGlut1-Cre labels nearly all forebrain excitatory cells.34,63 This is thought to be due primarily to promoter silencing of the TRE. In contrast, the CAG-driven nls-mRuby3 was densely expressed, consistent with it labeling a large fraction of Cre+ glutamatergic cells (Figures 1C and S1). Quantification of expression in primary visual cortex (V1) showed that the ratio of GCaMP7s+ to mRuby3+ cells was 0.33 ± 0.4 (Figure 1C, n = 5 mice), implying that about one-third of cortical excitatory neurons expressed the st-ChroME-GCaMP7s transgene.
We next examined expression when we crossed Ai203 to other Cre lines. Cux2-Cre-ERT2;Ai203 mice displayed expression across the upper layers of cortex, as expected for this Cre driver63 (Figures S2A–S2D). GCaMP7s appeared to be largely restricted to the membrane of the soma and the proximal dendrites, matching other soma-targeted ChroME preparations52,64 (Figures S2E and S2F). To examine Ai203 expression in other neural populations, we crossed Ai203 mice with Vgat-IRES-Cre mice to label all GABAergic neurons.65 We observed expression in subcortical and cortical regions (Figures S2G–S2J). Thus, Ai203 appears to express well across many neural populations, but, like many other TIGRE2.0 lines,34 it may not express strongly in cortical inhibitory neurons.
Photostimulation and calcium imaging in Ai203 mice in vitro
A key test of this line is whether st-ChroME-GCaMP7s is expressed at sufficient levels for both photostimulation and calcium imaging under a variety of conditions. Under conventional 1p illumination, we observed strong photocurrents with peak amplitudes of 3.4 ± 0.4 nA when measured in voltage clamp at −70 mV (Figure 1D, n = 8 cells). Using 2p illumination (via 3D Scanless Holographic Optogenetics with Temporal focusing [3D-SHOT]; see Figure S3 and STAR Methods) peak photocurrents were 1.7 ± 0.2 nA, comparable with that previously reported for virally expressed ChroME52 (Figure 1E; n = 11 cells). To test if these 2p-induced photocurrents were sufficient to evoke spiking, we made cell-attached recordings and illuminated the cells with trains of 10 5-ms light pulses at varying frequencies. Indeed, this reliably drove spiking at high frequencies (60 Hz) and at low powers (13.5 mW/cell) (Figure 1F; n = 11 cells). Neurons followed pulse trains with sub-millisecond jitter (0.75 ± 0.08 ms at 13.5 mW; n = 11 cells) across all stimulation frequencies tested. These data demonstrate that Ai203 mice provide robust expression of ChroME that drives temporally precise, high-fidelity spiking with 2p activation.
To test the spatial resolution of 2p holographic stimulation in Ai203 mice, we recorded light-evoked spiking from opsin-positive L2/3 pyramidal neurons in loose patch while stimulating each point in a small 3D grid surrounding the cell (Figures 1G and S3B; lateral spot size, 15.6-μm full-width half-maximum [FWHM]; axial spot size, 20.7-mm FWHM). We found an effective spatial resolution on par with previous reports using 3D-SHOT with soma-targeted opsins (Figure 1H; FWHM at 13.5 mW stimulation power, 15 ± 2 μm radial; 28 ± 4 μm axial, n = 4 cells).52,66 The actual effective resolution in an experiment will depend on many factors, including the optical resolution of the microscope, the spatial distribution of the opsin due to soma-targeting, and the power used, but these results demonstrate that the Ai203 line can provide spatial resolution comparable with other soma-targeted opsin preparations.
Next, we asked whether the opsin-fused and soma-targeted GCaMP7s sensor is capable of sensitively reporting neuronal activity. We made whole-cell current clamp recordings from stChroME-GCaMP7s+ L2/3 neurons in brain slices, and induced spiking with short (5-ms) current injections through the patch pipette while measuring GCaMP7s fluorescence using 2p calcium imaging. We observed robust fluorescence responses for even small numbers of action potentials (Figure 1I). Peak ΔF/F was 0.9 ± 0.1 for 10 action potentials (Figure 1I; n = 10 cells).
In all-optical read/write experiments, the scanning laser used for calcium imaging can cause unintended activation of the opsin, a phenomenon termed imaging “crosstalk.”9,52,57,64 To quantify crosstalk in VGlut1-Cre;Ai203 we made whole-cell current clamp recordings and recorded voltage changes induced by 2p imaging for cells in the center of the field of view (FOV). We recorded at different zooms (changing dwell times), frame rates (at constant dwell times), and powers. Under conditions commonly used for volumetric imaging in all-optical experiments (imaging rate, 6 Hz; 46-ns/μm average dwell time), mean depolarization caused by the imaging laser was 1.0 ± 0.4 mV at 45 mW of imaging power (Figures 1J, S4A, and S4B; n = 4 cells). To test crosstalk in vivo, we made loose-patch recordings in lightly anesthetized animals and recorded spike rates at baseline and under different imaging conditions (Figure S4C; powers, 40–70 mW; imaging rates, 6–10 Hz). We found that there was no change in normalized spike rate across conditions even at higher than typical powers (Figure S4C; effect of power, p = 0.88; effect of imaging rate, p = 0.24; repeated measures ANOVA; n = 4 cells). Cells are more hyperpolarized under anesthesia,67 so it is possible similar conditions could yield crosstalk in awake animals, but the difference is likely to be small. These experiments show that the st-ChroME-GCaMP7s fusion is compatible with 2p laser scanning imaging without excessive crosstalk.
Sensory-evoked calcium responses and 2p optogenetic stimulation in Ai203 mice
To determine whether the expression of st-GCaMP7s in Ai203 mice is sufficient to report sensory-evoked neural activity, we imaged calcium responses of L2/3 neurons in V1 through a cranial window (Figure 2A).68,69 We used multiplane 2p imaging with three planes imaged at ~6 Hz (see Figure S3; STAR Methods). We observed robust st-GCaMP7s responses at commonly used 2p imaging powers (Figure 2B, ~50 mW at 920 nm). Importantly, a similar proportion of L2/3 cells in Ai203 mice were visually responsive compared with another transgenic mouse line that expresses cytoplasmic GCaMP6s (using Camk2a-tTA;tetO-GCaMP6s) and that were intracranially injected with an adeno-associated virus (AAV) encoding ChroME (Figure 2C; percentage visually responsive: Ai203, 25% ± 5%, n = 11 FOVs; virus, 24% ± 4%, n = 18 FOVs, p = 0.86, t test). Furthermore, a similar fraction of cells were orientation tuned (Figure 2D; percentage orientation tuned: Ai203, 27.9% ± 1.3%, n = 5 FOVs; virus, 33.3% ± 2.1%, n = 8 FOVs, p = 0.095, t test), and their orientation selectivity was similar, albeit slightly lower (Figure 2E; orientation selectivity index [OSI]: Ai203, 0.56 ± 0.04, n = 8 FOVs; virus, 0.66 ± 0.01, n = 5 FOVs, p = 0.017, t test). Finally, we measured contrast response functions and found no difference between st-ChroME-GCaMP7s and cytoplasmic GCaMP6s (Figure 2F; p = 0.68, two-way ANOVA on contrast response curves). Cells expressing st-ChroME-GCaMP7s exhibited strong responses to high-contrast drifting gratings, only slightly lower than those of cytoplasmic GCaMP6s (Figure 2G, 75th percentile ΔF/F; Ai203, 3.9, n = 2686 cells; virus, 4.2, n = 2875 cells; p = 0.000025, Wilcoxon rank-sum test). This slightly lower response magnitude may also explain the slightly lower OSIs in Ai203. Altogether, these data demonstrate that the Ai203 line is suitable for recording sensory-evoked neural responses and that visual responses in V1 neurons are largely normal, implying transgene expression does not alter functional properties.
Figure 2. 2p imaging of in vivo visually and holographically evoked responses in VGlut1-Cre;Ai203 mice.
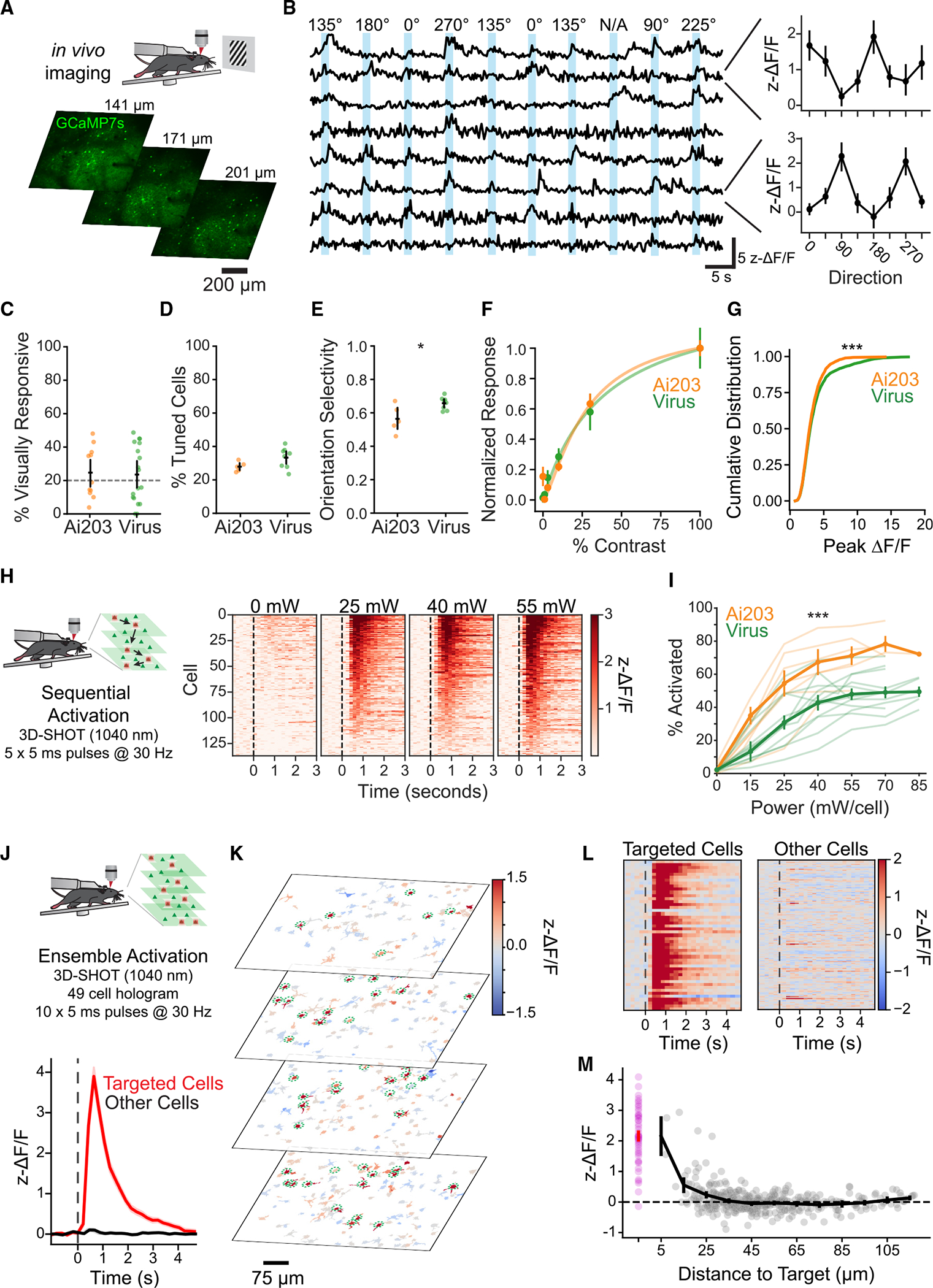
(A) Top: schematic of in vivo imaging paradigm. Bottom: example fields of view for three-plane (6.4-Hz volumetric imaging rate, FOV size ~820 μm) imaging at power 50 mW.
(B) Left: example traces of fluorescence responses to drifting gratings in V1 cells expressing st-ChroME-GCaMP7s, showing z-ΔF/F (Z-scored ΔF/F, see STAR Methods). Right: direction tuning curves for two example cells.
(C–G) Comparisons of visually evoked activity between VGlut1-Cre;Ai203 mice (Ai203, orange) and Camk2a-tTA;tetO-GCaMP6s mice expressing viral st-ChroME (virus, green).
(C) Comparison of percentage visually responsive cells between Ai203 (n = 11 FOVs) and virus (n = 18 FOVs). p = 0.85, t test. FOVs with <20% visually responsive cells were excluded from further analysis (dashed line).
(D) Comparison of percentage significantly tuned cells between Ai203 (n = 5 FOVs) and virus (n = 8 FOVs). p = 0.095, t test.
(E) Comparison of orientation-selectivity index of significantly orientation-tuned cells, mean per FOV for Ai203 (n = 5 FOVs) and virus (n = 8 FOVs). p = 0.016, t test.
(F) Contrast response functions (points) and corresponding Naka-Rushton fits (lines) to contrast modulated noise in Ai203 (n = 6 FOVs) and virus (n = 4 FOVs). p = 0.68, two-way ANOVA.
(G) Cumulative distribution of peak ΔF/F response to drifting gratings for Ai203 (n = 2,686 cells) and virus (n = 2,875 cells). p = 0.000025, rank-sum test.
(H) Left: schematic sequential holographic stimulation experiment. Targeted cells were sequentially activated, with imaging as in (A). Right: example experiment. Heatmaps of z-ΔF/F responses at different powers for 137 significantly activated cells out of 159 targeted cells (15, 110, 108, and 137 cells were activated at 0, 25, 40, and 55 mW, respectively. p < −0.05, one-tailed rank-sum test).
(I) Quantification of the fraction of neurons per FOV activated at various powers for experiments in Ai203 (n = 7 FOVs) and virus (n = 12 FOVs). Thin lines, individual sessions. p = 0.00012, repeated measures ANOVA.
(J) Top: schematic of ensemble stimulation. Neurons across four planes (4.8-Hz volumetric imaging rate, 75-mW imaging power) were targeted simultaneously in a single hologram with 45 mW/cell of power. Ten pulses of duration 5 ms were delivered at 30 Hz. Bottom: average responses from targeted (red) and non-targeted (black) cells.
(K) Images showing individual cell responses during stimulation; cell masks are colored by z-ΔF/F.
(L) Right: heatmap showing z-ΔF/F for targeted (n = 49) and non-targeted cells (n = 445); 46 out of 49 target cells were activated by stimulation (p < 0.05, one-tailed rank-sum test).
(M) Response of target cells (magenta) and non-targeted cells (gray) by distance to nearest coplanar target cell. Black, mean based on binned distance; gray, individual cells plotted by real distance. Data are presented as mean ± SEM.
A primary use for the combined expression of st-ChroME and GCaMP7s is in vivo targeted optogenetic manipulation using 2p optogenetics. To validate the utility of Ai203 in 2p optogenetics experiments in vivo, we used 3D-SHOT to holographically illuminate individual or ensembles of neurons while simultaneously imaging st-GCaMP7s responses. 2p photostimulation consistently drove large increases in st-GCaMP7s fluorescence in targeted cells (Figures 2H and 2I; Video S1). Compared with mice intracranially injected with an AAV encoding st-ChroME (same mice as used above), Ai203 mice had a higher percentage of ChroME+ cells activated at all powers tested (Figure 2I; proportion activated, p = 0.00012, two-way repeated measures ANOVA, Ai203, n = 7; virus, n = 12). This high sensitivity enables the co-activation of large ensembles of neurons with a single hologram, which we demonstrated by illuminating 49 L2/3 neurons simultaneously (Figure 2J). Even during this large ensemble stimulation, neighboring non-targeted cells showed minimal activation, demonstrating that the Ai203 reporter is suitable for large-scale, high-resolution photostimulation (Figures 2L and 2M).
Electrophysiological properties, expression stability, and visual behavior in Ai203 mice
To examine the electrophysiological properties of cortical neurons in Ai203, we used whole-cell patch-clamp recording in brain slices. We patched cortical pyramidal neurons in V1 of VGlut1-Cre;Ai203 mice blind to their GCaMP fluorescence to avoid any biases in cell selection. Cells were divided post hoc into opsin+ and opsin− by the presence of ChroME photocurrent. We measured action potential threshold via electrical stimulation and observed no differences in action potential threshold (Figure S5A; opsin+, −40 ± 1 mV, n = 16 cells; opsin−, −39 ± 0.7 mV, n = 14 cells; p = 0.38, t test), although we did find that opsin+ cells exhibited a slightly more depolarized resting membrane potential (Figure S5B; opsin+, −74 ± 2 mV, n = 16 cells; opsin−, −80 ± 1 mV, n = 14 cells; p = 0.002, t test). However, resting membrane potential was not correlated with photocurrent, implying that the expression of the opsin-GCaMP transgene per se did not explain the depolarized resting voltage (Figure S5C; slope = −0.016, r2 = 0.0142, p = 0.66, F test). To test whether expression of a GCaMP-ChroME fusion alone causes depolarized membrane potential, we conducted experiments in mice electroporated in utero with either GCaMP6s or an st-ChroME-GCaMP6s fusion and again measured resting membrane potentials. We observed no differences (Figure S5D; GCaMP6s, −70 ± 2 mV, n = 20 cells; st-ChroME-GCaMP6s, −68 ± 2 mV, n = 15 cells; p = 0.36, t test), suggesting that fusion proteins alone do not cause depolarization relative to expression of GCaMP. We further characterized synaptic properties by analyzing miniature excitatory postsynaptic currents (mEPSCs). Opsin+ cells exhibited a slightly higher rate and amplitude of mEPSCs (Figures S5E–S5G) (inter-event interval: opsin+, 0.27 ± .06 s, opsin−, 0.28 ± .04 s; p < .0001, ks test. Amplitude: opsin+, 10.1 ± .3 pA, opsin−, 9 ± 1 pA; p < .0001, ks test, n = 4 cells per group). These results could be explained if opsin+ neurons in VGlut1-Cre;Ai203 represent a specific subtype of excitatory neuron with slightly different physiological features. Alternatively, expression of the st-ChroME-GCaMP7s transgene might alter synaptic properties. Since opsin+ neurons exhibit normal contrast sensitivity and orientation tuning in vivo (Figures 2C–2F), these different properties might only occur following brain slicing or not affect the normal function of these neurons in the intact circuit.
Transgenic lines promise to provide more stable expression of transgenes,34–37 in contrast to AAVs, which can lead to toxic expression of the transgenes.31,32 To demonstrate the stability of the st-ChroME-GCaMP7s expression in Ai203, we measured responses in one FOV for one animal at 1 month and 6 months after windowing (Figure S6A). We found similar OSI and contrast sensitivity between the two time points (Figures S6B and S6C) (OSI: 1 month, 0.61 ± 0.02; 6 months, 0.55 ± 0.03. Contrast C50: 1 month, 18% ± 2%; 6 months, 15% ± 2%). Fewer cells were activatable by the same power of stimulation (Figure S6D; percentage activatable at 70 mW: 1 month, 81%; 6 months, 54%), which may reflect expected degradation in window quality over this time.70,71 Overall, these data show consistency of visual responses over time, demonstrating stable expression of the transgenes.
Transgenic opsin and GCaMP expression is particularly advantageous in behavioral training paradigms where long-term stability is essential. However, it is important that transgene expression does not itself affect learning or behavior. Thus, we trained VGlut1-Cre;Ai203 mice on a visual contrast detection task to test both their ability to learn an operant behavior and to quantify their visual contrast sensitivity as a basic metric of vision (Figure S7A). Similar detection tasks are known to be V1 dependent.72–74 We then compared contrast sensitivity, quantified as detection threshold, of Ai203 mice with C57BL6 wild-type (WT) mice and Camk2a-tTA;tetO-GCaMP6s mice trained on the same task. VGlut1-Cre;Ai203 mice performed as well as WT and better than the Camk2a-tTA;tetO-GCaMP6s strains (Figures S7B and S7C; p = 0.0038, one-way ANOVA with post hoc Tukey test: Ai203 vs. WT, p = 0.31; Ai203 vs. Camk2a, p = 0.0046; Camk2a vs. WT, p = 0.21; n = 16 mice). Thus, VGlut1-Ai203 mice learn and perform this visual detection task comparably with WT mice, implying that transgene expression does not negatively alter learning or basic visual function.
Finally, we asked if optogenetic stimulation in VGlut1-Cre;Ai203 mice could alter behavioral performance on the visual contrast detection task. Indeed, photostimulation via a 470-nm optic fiber over V1 through a cranial window induced a dramatic increase in the false-alarm rate and the hit rate for weak stimuli (Figure S7D) (false-alarm rate: no light 23% ± 3%, light 56% ± 5%, p = 0.0016; paired t test; n = 4 mice. Hit rate: no light 62% ± 4%, light 75% ± 3%, p = 0.019). In contrast, WT animals showed no change (Figure S7F) (hit rate: no light 57% ± 1%, light 58% ± 2%, p = 0.51. False-alarm rate: no light 20% ± 1%, light 24% ± 2%, p = 0.25; paired t test; n = 3 mice). Similar results were observed from activation of excitatory neurons in V1 in other studies.72,75 Taken together with the previous findings, these data demonstrate that Ai203 mice are suitable for a diverse set of neurobiological experiments.
Programming precise population activity patterns with all-optical calibration
Neural codes depend not only on which neurons fire at a given moment but also on the distribution of firing rates across a given neural population.44,76–78 This distribution constitutes the geometry of the neural code. We sought to advance 2p optogenetics from the uniform activation of cells in previous studies to a new paradigm that enables differential distribution of precise spike counts across the population to recreate or alter natural population vectors.44,45 We reasoned that we could employ 3D-SHOT in combination with 2p imaging to achieve this goal, and that the Ai203 line would provide a particularly convenient platform for designing and validating such an approach owing to its strong, reliable expression of GCaMP7s and ChroME.
We considered two major challenges to reading and writing spike rates using all-optical techniques. First, to optogenetically write in a specific firing rate to each neuron in a targeted ensemble using holographic illumination pulses, we must ensure that each illumination pulse drives approximately one action potential. If we can achieve this, we can easily drive different spike rates into different neurons by varying the number and rate of illumination pulses delivered to each neuron. However, the heterogeneity of both the intrinsic excitability and opsin expression across neurons makes it difficult to know, a priori, how much laser power to direct to each neuron to drive one spike per illumination pulse.52 The second challenge is that reading out a specific firing rate from a calcium signal is likewise complicated by the large heterogeneity in the transfer function between action potentials and GCaMP fluorescent transients across cells, even of the same type.46,47 Therefore, we designed a two-step calibration process fitting these transformations in each neuron using rapidly acquired all-optical experimental data and validated this process by recording simultaneous electrophysiology and imaging data in vivo.
The goal of the first step of this process, which we call “power calibration,” is to accurately identify the stimulation power needed to reliably elicit one spike per illumination pulse (the “target power”) individually for each neuron in an FOV. In electrophysiological recordings from brain slices from Ai203 mice, we observed that the response to a series of brief (5-ms) illumination pulses with increasing power was a monotonic saturating spike probability function that saturated at approximately one spike per pulse (Figure 1F).52,64 Thus, there is a power above which neurons reliably exhibit single evoked spikes for each illumination pulse. If we can determine this target power for each neuron in an FOV, we could reliably drive specific spike rates into each neuron by illuminating it with a specific pattern of pulses at its target power. As it would be impractical to measure each neuron’s response to stimulation electrically, this approach must rely on the fluorescent GCaMP response.
To test this possibility, we made a series of simultaneous electrophysiological and calcium recordings of individual opsin-expressing neurons in vivo in lightly anesthetized mice. We photostimulated each neuron with a train of five 5-ms illumination pulses of increasing power to determine the target power (Figures 3A and 3B). Consistent with our slice recordings, we found that most neurons exhibited a saturating response of evoked spike probability to increasing illumination power (Figure 3B; evoked spikes/pulse at 28 mW, 1.08 ± 0.20; at 45 mW, 1.09 ± 0.03; n = 13 cells). We then estimated a target power from these electrophysiological recordings where one illumination pulse generates just about one spike/pulse by fitting the response function with a modified Hill function (STAR Methods). We found that the fitted target power reliably indicated spiking saturation (estimated spikes at GCaMP-based target power: 0.96 ± 0.02 spikes/pulses). Next, we asked whether we could accurately estimate the same target power from the calcium responses measured simultaneously in the same neurons using a similar fitting procedure (Figures 3C and 3D). Indeed, we found that the target powers estimated from electrophysiology and those from GCaMP responses were very similar (Figure 3D; error, 3.4 ± 0.9 mW, n = 13 cells). These ground-truth experiments confirm that our all-optical power calibration procedure enables us to empirically determine the illumination power needed in each neuron to drive a set number of spikes.
Figure 3. Calibration of stimulation, spiking, and GCaMP fluorescence.
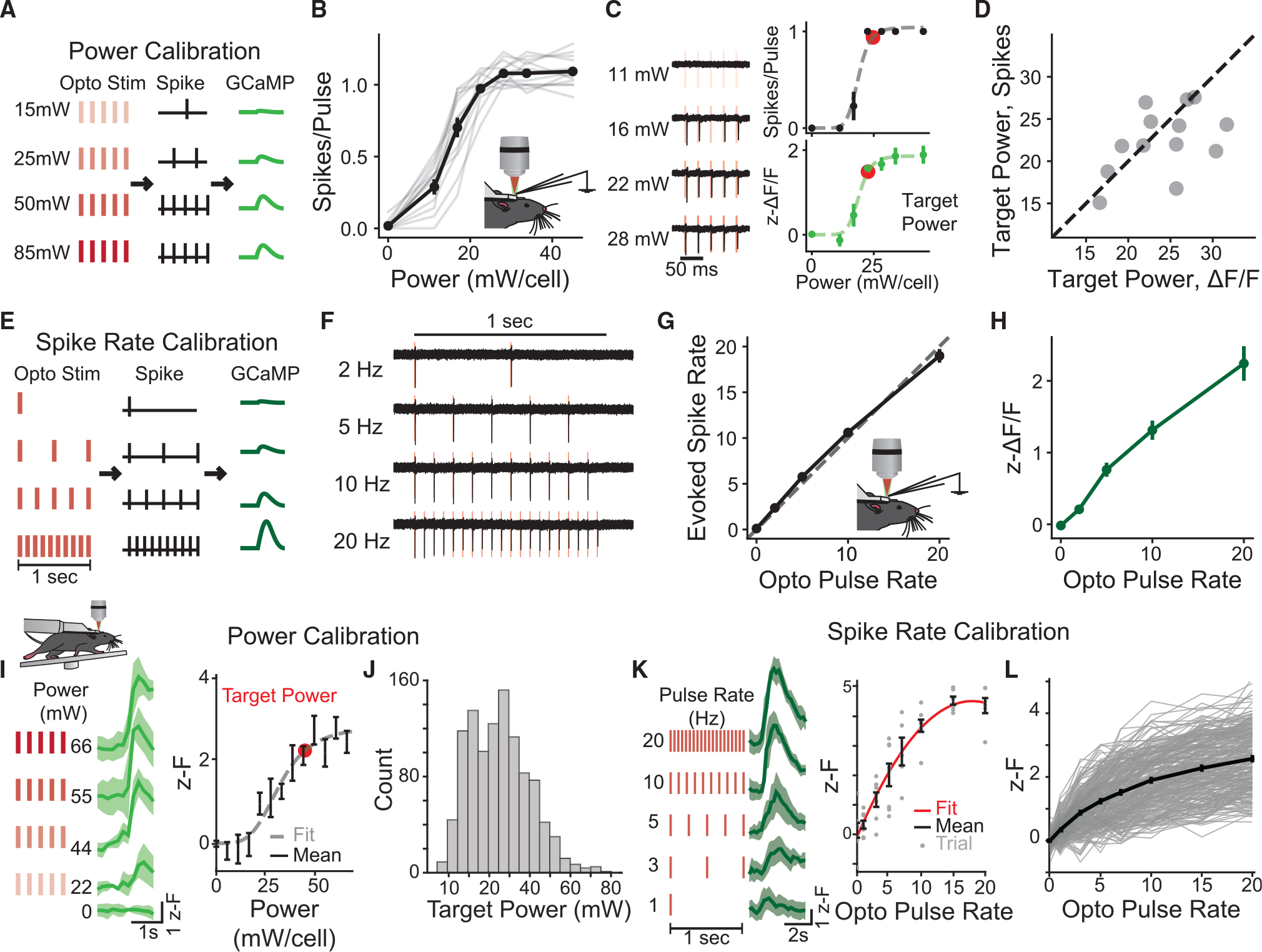
(A) Schematic of the power calibration procedure. Cells are stimulated with five 5-ms pulses at varying power to determine the power necessary to evoke a single spike per pulse.
(B) Number of spikes per pulse evoked in response to a train of five pulses at different powers recorded in vivo in lightly anesthetized mice. Gray lines, individual cells. n = 13 cells.
(C) Left: single-trial light-induced spike responses at different powers in an example cell. Right: spiking (top) and z-ΔF/F response (bottom) from the same cell. Dashed line, fit curve. Red, target power.
(D) Target power (i.e., the power needed to drive one spike per pulse) identified from the electrophysiological data (y axis) fit versus estimated target power from the fluorescence response fit (x axis). Gray dots, individual cells; dashed line indicates perfect correspondence. n = 13 cells.
(E) Schematic of the spike rate calibration procedure. Cells are stimulated at a power slightly above the target power at different spike rates for a fixed 1 s of stimulation.
(F) Example spiking responses to different photostimulation pulse rates for the same cell in (C).
(G) Evoked spike rate vs. optogenetic pulse rate during spike rate calibration. Dashed line indicates perfect correspondence. See also Figure S8.
(H) z-ΔF/F vs. optogenetic pulse rate for the cells in (G).
(I–L) In vivo awake, all-optical calibrations performed with three-plane imaging at imaging power 50 mW.
(I and J) All-optical power calibration.
(I) Calcium responses of an example cell stimulated with five optogenetic pulses at varying power. Left: calcium responses at different illumination powers, mean ± 95% confidence interval. Right: power-to-fluorescence response curve for example cell.
(J) Histogram of target powers of all fitted cells, n = 1,057 cells, eight FOVs, five mice.
(K and L) All-optical spike rate calibration.
(K) Calcium responses in an example cell photostimulated with trains at different pulse rates for 1 s (total pulses = pulse rate). Left: traces at different spike rates, mean ± 95% confidence interval. Right: response of an example cell. Black, mean; gray dots, individual trials.
(L) Fluorescence response vs. optogenetic pulse rate for all 281 fitted cells from six FOVs in four mice. Light gray, individual cells; black, mean. Data are presented as mean ± SEM unless otherwise noted.
Using this ability to write in a known number of spikes with 2p optogenetics, we performed a “spike rate calibration” where we then directly measured the spike-rate-to-calcium transformation in each cell. In prior work, this transformation has been studied either directly with simultaneous electrophysiological recordings of single neurons or indirectly, using post hoc deconvolution algorithms.46,47 Using the identified target power, we reason we should be able to all-optically directly measure this transform driving in specific spikes rates to each neuron with 2p optogenetics while reading out the response with calcium imaging (Figure 3E). We directly tested this hypothesis on a subset of the neurons from the power calibration experiments above, again performing simultaneous electrophysiological and calcium imaging measurements in vivo. For a fixed 1-s window of stimulation, we varied the rate of light pulses delivered, thus also varying the total number of pulses (2–20 Hz, 2–20 total pulses) and measured the evoked spike rate. At approximately the target power, we found that cells reliably followed pulse trains across the full range of tested rates (Figures 3F, 3G, and S7; spikes/pulse at 2 Hz, 1.17 ± 0.09; spikes/pulse at 20 Hz, 0.95 ± 0.04, n = 11 cells). The corresponding calcium responses demonstrated a similar monotonically increasing calcium response function (Figure 3H). Thus, with this procedure, we could map the relationship between evoked spike rate and GCaMP response for each cell individually and all optically without requiring electrophysiological recordings or spike inference.
Having validated the above calibrations with electrophysiology, we performed all-optical power and spike calibrations in V1 of an awake animal through a cranial window. Like our cell-attached experiments, we found that most neurons (1,057 out of 1,730 cells, eight FOVs, five mice) exhibited a monotonic and saturating calcium response to increasing photostimulation power that was well fitted by a modified Hill function (Figure 3I; mean R2 = 0.620 ± 0.006). Of the well-fitted cells, the mean target power was 34.8 ± 0.4 mW, although there was a broad range (Figure 3J; range, 7 to 89 mW, N = 1057 cells, eight FOVs, five mice), emphasizing the importance of this individual, per-cell calibration. We then used the target power for each neuron to perform the spike rate calibration. As above, by varying the pulse rate for 1 s of stimulation, we obtained a response curve relating spike rate to fluorescence changes in each imaged neuron, which we fitted with a quadratic function for each cell (Figure 3K; see STAR Methods). Although we observed high cell-to-cell variability in the gain of each transfer function, 80% (281 out of 350 cells) of targeted cells were well fitted (Figure 3L; R2 = 0.48 ± 0.01, n = 281 cells, six FOVs, four mice). These data demonstrate that we can effectively use our all-optical calibration procedures on large populations of neurons in awake mice in a single experimental session.
Next, we sought to recreate a natural population activity pattern in the brain with known distributions of spikes entirely with optogenetics. In a subset of the all-optical calibration experiments described above, we measured the activity of the neurons to drifting gratings of various orientations. We then selected calibrated neurons that exhibited strong, orientation-tuned visual responses. Using our cell-specific spike rate to GCaMP transfer functions, we calculated the number of illumination pulses needed to recreate each neuron’s observed mean fluorescence response to each grating (but not specific temporal dynamics). We generated a pattern of optogenetic stimulation that would drive the desired number of spikes in each neuron (Figures 4A and 4D). Consistent with our goals, we observed a marked correspondence between the visually evoked activity and the optogenetically induced activity of the targeted neurons (Figures 4B and 4C). To quantify the accuracy of this approach on a neuron-by-neuron basis, we computed the cosine similarity of the visual and the optogenetic response of each neuron, and these showed a high degree of similarity (Figure 4C) (mean similarity: recreated, 0.61 ± 0.04; shuffled, 0.18 ± 0.04; p < 3e–17, rank-sum test; n = 155 cells, five FOVs, three mice). At the population level, trajectories of the target population visualized via principal-component analysis appeared highly similar between optogenetically recreated and visually evoked patterns (Figure 4E). As with single-neuron analysis, we found that the optogenetically induced population activity vectors matched their intended visually induced target activity vectors well (cosine similarity 0.70 ± 0.03). Moreover, the induced patterns matched their target vectors better than they matched other visual induced vectors (0.32 ± 0.01, p < 5.5e–16, rank-sum test), spontaneous activity vectors (−0.03 ± 0.04, p < 5.5e–14, rank-sum test), or artificially shuffled vectors (Figures 4F and 4G; 0.46 ± 0.03, p < 1.9e–6 rank-sum test; n = 40 patterns, five FOVs, three mice). These experiments establish and validate a general paradigm for recreating precise population vectors of neural activity in the intact brains of awake animals using 2p holographic optogenetics.
Figure 4. Reproducing naturalistic responses with 2p stimulation.
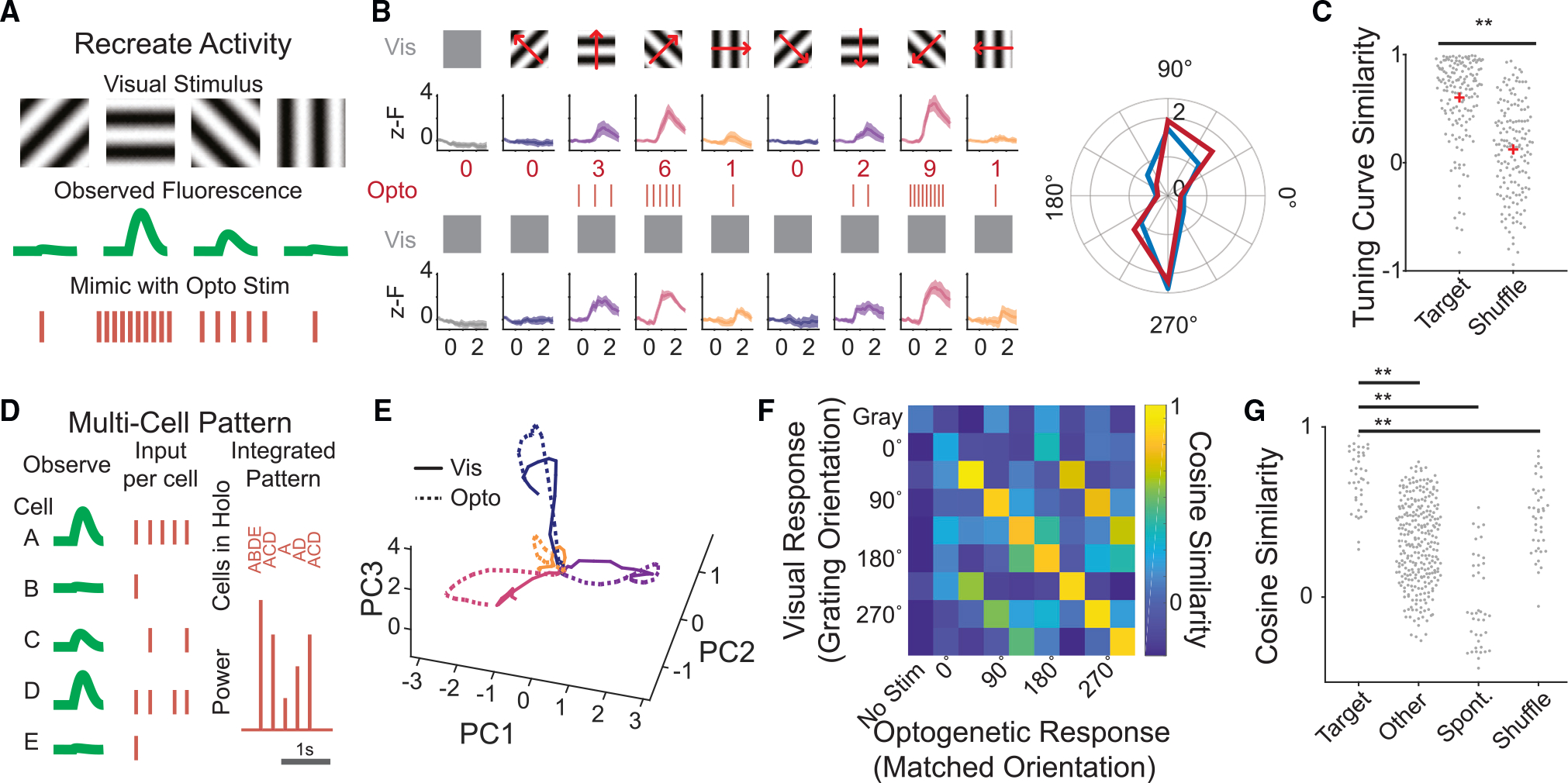
(A) Schematic of the strategy for recreating observed activity using calibrated 2p optogenetics.
(B) Top: visually evoked calcium responses to oriented gratings in an example neuron. Traces are mean ± 95% confidence interval. Middle: the calculated number of pulses needed to recreate the visual response with optogenetic stimulation alone. Bottom: calcium responses of the same cell during 2p optogenetic stimulation intended to recreate its visual response to each grating. Right: polar plot of the cell’s tuning curve, visually evoked (blue) or by optogenetics alone (red). (C) Tuning curve similarity of all visually evoked tuning curves vs. the optogenetically evoked tuning curves compared with shuffled controls. p < 3e–17 rank-sum test. n = 155 cells, five FOVs, three mice.
(D) Schematic of the multi-cell pattern writing approach.
(E) Population activity from stimulated cells in an example FOV during visual stimulus (solid lines) or optogenetic-only stimulation (dotted lines), for four drifting gratings, colors as in (B). Dimensionality is reduced via principal-component analysis.
(F) The cosine similarity between the visually evoked population activity and that evoked by optogenetic stimulation, for all tested holographic patterns from anexample FOV.
(G) Cosine similarity of an optogenetically evoked population vector to its target visually evoked pattern (“target”), a different visually evoked pattern (“other”),spontaneous activity (“spont”), or a shuffled dataset (“shuffle”). p values determined by rank-sum test. Target vs. other, p < 5.5e–16; target vs. spont, p < 5.5e–14; target vs. shuffle, p < 1.9e–6. n = 40 optogenetically evoked patterns, five FOVs, three mice. Ensembles contained between 19 and 43 cells.
DISCUSSION
Here, we report two advances: a multifunctional reporter line for all-optical circuit interrogation and an approach for recreating precise population activity patterns with 2p holographic optogenetics. The mouse line we developed provides cell-type-specific co-expression of a potent opsin, ChroME, and a high-performance calcium indicator, GCaMP7s, as a single soma-targeted fusion protein: st-ChroME-GCaMP7s. This mouse enables 2p all-optical interrogation of neural circuits with high spatial and temporal precision with strong expression of a potent opsin. Transgenic lines expressing opsins have been used for 2p holographic photostimulation without simultaneous imaging,6,9 but the Ai203 line improves on these by expressing a highly potent, soma-targeted opsin as well as GCaMP. Because the Ai203 transgenic line obviates the need for one or more viruses to co-express the opsin and the calcium indicator, it will facilitate a variety of all-optical experiments by providing more reliable, reproducible, and stable transgene expression. This facilitated our development of a paradigm that advances 2p optogenetic stimulation beyond the standard uniform activation of cells to reproducing naturalistic distributions of activity patterns in the population.
The robust, but stable, levels of ChroME expression in VGlut1-Cre;Ai203 mice allowed us to photostimulate opsin-expressing neurons with even lower laser powers than mice expressing ChroME virally (via AAVs). This strong expression is helpful for a wide variety of experiments as one can activate more total neurons with the same laser budget. Here, we demonstrated simultaneous activation of nearly 50 neurons in vivo, a number that exceeds what is sufficient to induce behavioral changes.17,19,23,25,27 The low powers required to reach reliable spiking allow flexibility in designing patterns of population stimulation, which is essential for our recreation of population activity.
Using the flexibility provided by the Ai203, we developed a paradigm to artificially recreate population vectors of neural activity. In contrast to prior approaches that activate specific cells or cell types without control over each stimulated neuron’s firing rate17–19,21–25,30,79 or match fluorescence in closed-loop,14 our approach permits the user to distribute specific numbers of spikes to each of the target neurons arbitrarily. Perhaps even more importantly, by using 2p holographic optogenetics to calibrate the transformation from spikes into calcium signals in each neuron, the user can read out and write in firing rate distributions that approximate those driven naturally by sensory stimuli or during behavior. The accuracy of this recreation will be limited by errors in the readout of activity via calcium sensors and the write-in via holographic optogenetics, but our electrophysiological recordings imply that, at least under the condition used here, these errors for write-in can be small. This paradigm could be particularly useful for questions about the geometry of neural codes. A growing array of studies argue that neural coding operates along specific manifolds, through subspaces, or is organized into distinct topologies that affect discrimination and generalization.38–43 Our approach can optogenetically drive populations to align with or deviate from coding topologies in specific directions to causally determine whether or how different dimensions of activity influence behavior.
Inferring underlying spiking from GCaMP fluorescence changes is a difficult problem due to the many sources of noise and variability within and across cells. Here, we sought to determine the spiking response of a neuron to a given stimulus so that we could reproduce an observed response. While we succeeded in matching optogenetically induced fluorescence to observed fluorescence in most cases, it is possible that our approach does not necessarily match spike counts accurately. Various types of errors could lead to such inaccuracy: for example, high-frequency spike bursts, which might generate larger calcium responses than low-frequency firing of the same total spike count,46,80,81 would lead to overestimates in the inferred spike rate, although prior work shows that most spikes in L2/3 pyramidal neurons, even in the awake state, do not occur in bursts.82
Spike inference models are another option for estimating the underlying spike rates that give rise to a calcium response. Some approaches even try to estimate the underlying times on a single-trial basis. However, even state-of-the-art methods suffer from many inaccuracies,48–51 which may stem from cell-to-cell variabilities that make estimating the calcium response to a single action potential challenging. The spike calibration approach we introduce here could be used in concert with deconvolution algorithms, especially for extending our approach to recreate precise spike timing. Currently, our system has limited ability to measure or recreate realistic spike timing since it is limited by the imaging frame rate and the kinetics of the calcium sensor. Incorporating faster imaging techniques,83,84 faster calcium sensors,59 or perhaps voltage sensors,85,86 as well as deconvolution models, can address this issue.
We note that, while this process benefits from the Ai203 transgenic, it does not require it, and it could be performed using standard viral approaches for transgene expression. However, the specific choice of opsin is crucial. Opsins with fast off-kinetics (such as ChroME) minimize the chance that single illumination pulses generate more than one spike because they allow the neuron to quickly repolarize.13,52,64 Shorter illumination pulse lengths may be able to overcome this for some slower opsins,27,87 but experimental validation of the power-to-spike transformation for any new conditions would be required.
While Ai203 is a useful line for expressing ChroME, there are several important issues to consider when employing it for experiments. First, Ai203 provides only incomplete coverage of transgene expression in Cre+ cells. Other combinations of Cre driver and TIGRE1.0 and 2.0 reporter lines have shown similar sparsity, which is thought to be due in part to promoter silencing of TRE, and new strategies to overcome this would benefit the field.34,36 While sparse labeling limits the coverage of all-optical control, it also offers advantages in many 2p optogenetic experiments due to reduced activation of neighboring cells.1
A second consideration is the impact of the transgene on the physiological properties of the expressing cells, the network, and the animal as a whole. In vivo, we found that VGlut1-Cre;Ai203 mice had visually evoked neural responses similar to those of another GCaMP-expressing transgenic line, and behavioral sensitivity to contrast similar to wild-type controls. This indicates that overall circuit function, at least in the visual system, is not compromised. However, in brain slices, we found that opsin+ cells had slightly more depolarized membrane potentials and different synaptic properties than opsin− cells. This difference could be due to opsin+ cells representing a specific subtype of L2/3 pyramidal cell that differs from the unlabeled pool. Alternatively, these differences could be due to expression of the st-ChroME-GCaMP7s itself. However, we saw no evidence for this, as opsin levels were not correlated with change in resting membrane potentials, and expression of a similar st-ChroME-GCaMP protein via in utero electroporation did not change membrane potential. Still, possible effects of the transgene on physiological cell properties should be considered when using this and any other transgenic line. In the Ai203, though, these putative differences appear to minimally affect circuit function in vivo.
The soma-targeted GCaMP construct here was produced by fusing GCaMP7s with an opsin that is soma targeted via the Kv2.1 sequence. This was done to ensure stochiometric expression of both transgenes as other strategies such as 2A peptides do not ensure stochiometric expression and, in the TIGRE locus, have been shown to cause significantly lower expression of the second protein.34 Future transgenics may develop new strategies for improved dual expression without fusion. Kv2.1 has previously been used to target GCaMP expression to the soma, but this approach has not been well characterized for functional imaging in vivo.62,88 We observed robust fluorescence changes with GCaMP7s in response to small numbers of action potentials and reliably observed sensory-evoked activity in vivo. Furthermore, we observed minimal crosstalk activation of the opsin by the 2p scanning laser under the imaging conditions used in this study.
The Ai203 reporter line and the ability to recreate population vectors both represent significant advances in all-optical experimental techniques. The more reliable and stable expression of the opsin and the calcium sensor in the Ai203 line compared with viral approaches will make all-optical experiments easier and more reproducible between animals and between studies. Recreating population vectors opens up a range of experiments testing the geometry of neural codes, not just identity codes. Whether used together or separately, these advances are sure to promote discoveries about neural coding.
Limitations of the study
Our approach to recreating visually evoked activity all-optically succeeded in accurately recreating fluorescence responses, but it does not replicate natural spike timing and may not match spike counts accurately in some cases. The same GCaMP response can possibly represent several underlying spike rates when averaged over a fixed window, such as when high-frequency spike bursts can generate larger responses than low-frequency firing of the same total spike count.46,80,81 These would lead to errors in our estimation of inferred spike rate and write-in that are not detectable with our method. We show that the Ai203 line provides robust and stable expression in excitatory pyramidal cells, but it may work less well in other cell types. Furthermore, it shows incomplete expression that may limit some experiments. We also observed small physiological differences between transgene-expressing and non-expressing cells that, while they did not appear to affect overall circuit function, should be taken into consideration.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hillel Adesnik (hadesnik@berkeley.edu).
Materials availability
All animal strains used in this study are available from Jackson Laboratories. All plasmids and viral vectors generated for the study have been made available from Addgene or can be obtained from the lead contact upon request.
Data and code availability
Data for main figures and additional histology images are available at https://doi.org/10.5281/zenodo.8118399, all additional data will be shared by the lead contact upon request. Custom code for online and offline analysis has been deposited at Zenodo and is publicly available. DOIs are listed in the key resources table. Any additional information or code required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti-NeuN | Abcam | RRID:AB_2532109 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| PhP.eB-TRE-ChroME-P2A-H2B-mRuby3 | This paper | N/A |
|
| ||
| Deposited data | ||
|
| ||
| Data and analysis code | This paper | Zenodo: https://doi.org/10.5281/zenodo.8118399 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: B6; DBA-Tg(tetO-GCaMP6s)2Niell/J | Jackson Laboratory | RRID: IMSR_JAX: 024742 |
| Mouse: B6; CBA-Tg(Camk2a-tTA)1Mmay/J | Jackson Laboratory | RRID: IMSR_JAX: 003010 |
| Mouse: B6; 129S-Slc17a7tm1.1(cre)Hze/J, Slc17a7-IRES2-Cre | Jackson Laboratory | RRID:IMSR_JAX:023527 |
| Mouse: B6.Cg-Igs7<tm203(tetO-sChroME*/ GCaMP7s,CAG-mRuby3,-tTA2)Tasic>/J, Ai203 | Jackson Laboratory | JAX stock #037939 |
| Mouse: B6(Cg)-Cux2tm3.1(cre/ERT2) Mull/Mmmh, Cux2-CreERT2 | MMRRC | RRID:MMRRC_032779-MU |
| Mouse: B6J.129S6(FVB)-Slc32a1tm2(cre)Lowl/ MwarJ, Vgat-IRES-Cre | Jackson Laboratory | RRID:IMSR_JAX:028862 |
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000664 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCAG-stChroME-GCaMP6s | This paper | N/A |
| pCAG-GCaMP6s | Addgene | Addgene_171156 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Schneider et al. | https://imagej.nih.gov/ij/ |
| MATLAB | MathWorks | RRID: SCR_001622 |
| Python | Python Software Foundation | RRID: SCR_008394 |
| Suite2p | Pachitariu et al.89 | RRID: SCR_016434 |
| ScanImage | Vidrio Inc. | RRID: SCR_014307 |
| CaImAn | Giovannucci et al.90 | RRID:SCR_021152 |
| live2p | This paper | Zenodo: https://doi.org/10.5281/zenodo.8117788 |
| Code for all-optical calibration and activity recreation experiments | This paper | Zenodo: https://doi.org/10.5281/zenodo.8139926 |
| Custom code for analyses | This paper | Zenodo: https://doi.org/10.5281/zenodo.8118399 |
| Easy Electrophysiology | Easy Electrophysiology Ltd | RRID:SCR_021190 |
|
| ||
| Other | ||
|
| ||
| Coherent Monaco 2p stimulation lasers | Coherent | https://www.coherent.com/lasers/ultrashort-pulse/monaco |
| Spatial Light Modulator | Meadowlark | HSP1920-1064-HSP8-HB |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
All experiments on animals were conducted with approval of the Animal Care and Use Committee of the University of California, Berkeley. In all experiments we attempted to use male and female mice equally. Mice used for experiments in this study were wild-type C57BL/6J mice, CD-1 ICR for in utero electroporation (Charles River), Ai203 (Jax stock #037939). Cux2-CreERT2 (MMRC stock #032779-MU), PV-Cre (Jax stock #017320), SST-IRES-Cre (Jax stock #013044), Vgat-IRES-Cre (Jax stock #028862), Slc17a7-IRES2-Cre (VGlut1-Cre, JAX stock #023527), Camk2a-tTA (Jax stock# 003010) and tetO-GCaMP6s (Jax stock # 024742). All transgenic lines were maintained in a C57BL/6J background and all experimental animals were heterozygous for all transgenes. Mice were housed in cohorts of five or fewer in a reverse light:dark cycle of 12:12 h, with experiments occurring during the dark phase.
METHOD DETAILS
Mouse transgenesis
To generate the Ai203 transgenic mouse, the targeting vector was constructed using gene synthesis and standard molecular cloning techniques. It contains the following components: FRT3 – 2X HS4 chicken beta globin insulators – TREtight promoter – LoxP – ORF-3X stops – hGH polyA, PGK polyA – LoxP – ChroME-FLAG-Kv2.1-GSGlinker-GcaMP7s– WPRE-bGH polyA – 2X HS4 chicken beta globin insulators – CAG promoter – Lox2272 – ORF-3X stops – hGH polyA, TK polyA – Lox2272 – nls-mRuby3-IRES2-tTA2 – WPRE – bGH polyA – PhiC31 attB- PGK promoter – Hygro1-SD – FRT5. Targeting of the transgene cassette into the TIGRE locus was accomplished via Flp-recombinase mediated cassette exchange using circularized targeting vector and a CAG-FlpE vector (Open Biosystems Inc).34,36 Correctly targeted ES cells were identified using PCR and injected into blastocysts to obtain chimeras and subsequent germline transmission. Resulting mice were then crossed to C57BL/6J mice and maintained in C57BL/6J congenic background.
Tamoxifen induction
For Cux2-CreERT2; Ai203 mice, induction was done by intraperitoneal injection of tamoxifen (15 mg/mL in corn oil) at 100 mg/kg for five days.
Histology
Mice were anesthetized with ketamine and transcardially perfused with phosphate-buffered saline (PBS) and then 4% paraformaldehyde (PFA). Brains were left in PFA overnight, then transferred to 30% sucrose for 1–2 days, then cut into 40 μm-thick coronal sections using a freezing microtome and stored in PBS until use.
For NeuN immunostaining, sections were blocked for 1 h at 4° on a rocker in PBS containing 3% normal goat serum (NGS), 0.6% Triton X-100, 0.2% Tween 20, and 3% bovine serum albumin (BSA). For primary staining, sections were incubated overnight at 4° on a rocker in Rabbit anti-NeuN (Abcam) diluted 1:10,000 in blocking solution. The next day, sections were washed once in PBS with 0.25% Triton X-100 (PBS-T). For secondary staining, sections were incubated for 1 h on the rocker at room temperature in blocking solution with 1:1000 Goat anti-Rabbit IgG H&L Alexa Fluor 405 (Thermo Fisher Scientific). Then sections were washed once in PBS-T. Sections were mounted and coverslipped with Vectashield (Vector Laboratories Inc).
Images were collected on an Olympus FV3000 (Olympus Corporation) confocal laser scanning microscope. For cell counting, to obtain adequate z-sectioning, fields of view tiled at 212 × 212 μm (800 × 800 pixels) each were taken spanning an area of V1, stitched in ImageJ with a stitching plugin.91 Then images were rotated so that the pia was horizontal and cropped so that the counting area was a rectangular column of cortex containing all layers, to evenly count all layers. Cells were counted in ImageJ using built-in tools.
Two-photon holographic microscopy
Slice electrophysiology and in vivo experiments were performed using two setups capable of 3D scanless holographic optogenetics with temporal focusing (3D-SHOT)52,64,66 as shown in Figure S3. Briefly, both setups were custom-built on the Movable Objective Microscope (MOM; Sutter Instrument Co.) platform, with three combined optical paths: a 3D two-photon (2p) photostimulation path, a fast resonant-galvo raster scanning 2p imaging path, and a widefield one-photon (1p) epifluorescence/IR-transmitted imaging path, merged by a polarizing beamsplitter before the microscope tube lens and objective. In the 3D-SHOT photostimulation path, a custom temporally focused pattern (CTFP) was generated from the photostimulation laser with a blazed diffraction grating (33010FL01–520R Newport Corporation). The beam was relayed through a rotating diffuser to randomize the phase pattern and expand the temporally focused beam to cover the area of the high-refresh-rate spatial light modulator (SLM; HSP1920–1064-HSP8-HB, 1920 × 1152 pixels, Meadowlark Optics). Holographic phase masks were calculated using an iterative implantation of the Gerchberg-Saxton algorithm92 and displayed on the SLM placed in the Fourier plane to replicate the CTFP in 3D. The photostimulation path was then combined with the imaging path using a polarizing beamsplitter placed immediately prior to the tube lens. L1 and L2 are a beam expander, all lenses after grating are in 4F. The diffuser is on the imaging plane. Following initial alignment, L6 is adjusted to match imaging and holography planes. The photostimulation laser was synchronized with the scan phase of the resonance galvos using an Arduino Mega (Arduino), gated to be on only the edges of every bidirectional line scan, in order to reduce photostimulation artifacts contaminating the imaging data. Multiplane imaging was accomplished using an electrically-tunable lens (Optotune) placed in the light path prior to the resonance scanners.
Setup #1, used for in vitro characterization of Ai203 and in vivo loose patch (Figures 1, 3A–3H, S4, S5, and S8) used a Monaco 1035–80-60 737 (1035 nm, 2 MHz, 300 fs, Coherent Inc.) for photostimulation and a Mai Tai Ti:sapphire laser (Spectra Physics Inc.) for 2p calcium imaging. This setup contained additional lenses to expand and shrink the beam due to the greater power of laser used (Figures S3 and Table S1). The laser was tuned to 1 MHz for all slice experiments in Figures 1, S4, and S5. Setup #2, for all-optical in vivo characterization of Ai203 mice (Figures 2, 3I–3L, 4, and S6), a similar setup was used except for a Monaco 1035–80-40 (1035 nm, 2 MHz, 276 fs, Coherent Inc.) was used for photostimulation, and a Chameleon Ultra Ti:sapphire laser (Coherent Inc.) was used for 2p calcium imaging.
In vitro electrophysiology
In vitro slice recordings were performed on 300mm-thick coronal slices obtained from the V1 of both male and female mice aged P12-P30 (calcium sensor characterization) or P21-P42 (opsin characterization). The slices were prepared using a microslicer (Zero 1N; DSK). To prepare, the extracted brain was trimmed on both anterior and posterior sides of the V1, glued to a slicing chamber, and finally submerged in ice-cold (~4°C) sucrose-rich ACSF (in mM: NaCl 83, KCl 2.5, MgSO4 3.3, NaH2PO4 1, Glucose 22, Sucrose 72, and CaCl2 0.5). Per mouse, up to 8 slices containing the V1 were obtained. The slices were then recovered in the same sucrose-rich solution saturated with carbogen at 34°C for 30 min, and then continually stored at room temperature for the duration of the experiment. Whole-cell patch-clamp protocols were performed in inline heating-controlled (33°C) standard ACSF bath solution (in mM: NaCl 119, NaHCO3 26, Glucose 20, KCl 2.5, CaCl2 2.5, MgSO4 1.3, NaH2PO4 1.3). Patch pipette (4–7 MOhm) were pulled from borosilicate glass filaments (Sutter Instrument Co.) and filled with potassium (K)-gluconate solution (GCamp7s characterization; in mM: 110 K-gluconate, 10 HEPES, 1 EGTA, 20 KCl, 2 MgCl2, 2 Na-ATP, 0.25 Na-GTP, 10 Phosphocreatine, 295 mOsm, pH = 7.45) or Cesium (Cs2+)-based internal solution (opsin characterization; in mM: 135 CeMeSO4, 3 NaCl, 10 HEPES, 0.3 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 1 QX-314, 5 TEA-Cl, 295 mOsm, pH = 7.45) also containing 50 mM Alexa Fluor hydrazide 488 or 594 dye (ThermoFisher Scientific). Data was recorded at 20 kHz using 700b Multiclamp Axon Amplifier (Molecular Devices). For loose-patch recordings the pipettes were filled with standard ACSF. The headstage with the electrode holder (G23 Instruments) was controlled by Motorized Micromanipulator (MP285A; Sutter Instrument Co.). All data was acquired and analyzed with custom code written in MATLAB using the National Instruments Data Acquisition (DAQ) Toolbox (NI). Membrane (Rm) and series (Rs) resistance were monitored before and throughout experiments to ensure quality of acquired data. Cells were excluded if initial access resistance was over 30 MOhm or if access resistance changed more than 30% during the experiment, cells were excluded.
In vitro characterization of intrinsic properties Slices were taken from mice P24–29. Resting membrane potential was measured using K-gluconate internal solution and determined as the median membrane potential of 5 s of baseline measurement immediately following establishment of whole-cell configuration. Action potential threshold was analyzed using code (in MATLAB) adapted from Spike_threshold_PS function developed by the Rune Berg lab. This function determines threshold as the point of maximum positive slope in a phase plot of membrane potential and its first derivative dV/dt.93 Miniature EPSCs (mEPSC) were recorded at −70mV holding potential in bath solution containing tetrodotoxin (TTX: 0.5 μM) using Cs-based internal solution. A stable baseline was acquired for 3 min, followed by additional 5 min of recording. Postsynaptic events were detected and quantified across time bins using Easy Electrophysiology v.2.4.0 (Easy Electrophysiology Ltd, RRID:SCR_021190), then statistical analyses were done using in-house code in MATLAB.
In vitro characterization of opsin response characteristics
For 1p and 2p characterization, cells positive for st-ChroME-GCaMP7s were identified through fluorescence from widefield one-photon illumination using a Spectra X light engine (Lumencor). For measurements of 1p- and 2p-photoactivated peak currents (whole-cell voltage clamp) and spiking (loose-patch), the duration of photostimulation was 5 ms. To measure 1p evoked photocurrents, cells were illuminated with widefield light using the Spectra X Light engine at 510 nm, held in voltage clamp (−70mV) and stimulated at various powers. To measure peak 2p evoked photocurrents, a single holographic spot was aligned to a cell patched in whole-cell configuration and held in voltage clamp (−70mV) and stimulated across a series of laser powers (4.5, 7, 9, 13.5, 22.5, 45 mW) at 2 Hz. To measure spike probability, a cell in loose-patch configuration was stimulated with a single spot across laser powers (4.5, 7, 9, 11, 13.5, 22.5 mW) at different frequencies (5, 20, 40, 60 Hz). Stimulation conditions were randomized across an experiment.
The effective spatial resolution of photostimulation was measured in st-ChroME-GCaMP7s+ cells via loose-patch. We then measured spiking in response to photostimulation in the space surrounding the target cell by creating a voxelized three-dimensional volume spanning 97.5 × 97.5 × 50 μm in lateral and axial dimensions and at 6.5 × 6.5 × 12.5 μm per voxel. Spike probability was measured per stimulation of a voxel, then mapped according to the spatial location where the values were obtained. Lateral (x, y) and axial (z) physiological point-spread functions (PPSF) were quantified along grid planes where the highest spike probabilities per stimulated voxel were obtained. The spatial resolution at both lateral and axial dimensions were quantified as the full-width half-maximum (FWHM) of Gaussian profiles fitted to the PPSFs.
In vitro imaging
For GCaMP characterization (Figure 1), GCaMP7s positive cells were identified in 1P or 2P illumination and patched. After establishing stable access, cell parameters (Rm, Rs) were logged and 5 ms duration step current pulses of increasing amplitude were used to determine neuron rheobase. A train of 1, 2, 3, 5, 10, 15, 20 or 30 action potentials were triggered at 30 Hz, using 5 ms duration current pulses 25 pA higher than rheobase while simultaneously imaging calcium fluorescence at a 30 Hz frame rate. Similar imaging laser power was used (35 mW) for all cells. FOV size was 680 μm. Imaging analysis is described with in vivo analysis.
Crosstalk characterization,64 was performed in current clamp with crosstalk measured across FOV sizes (980, 680, and 527μm), imaging frequencies (6, 10, 30 Hz), and powers (10, 15, 25, 35, 45 mW). Imaging frequencies were obtained by scanning the FOV at 30Hz while using the Pockels cell (Conoptics) to gate the scanning laser every n-th frame corresponding to frequency of interest. Cells were kept at resting potential throughout the protocol. Each trial consisted of a baseline period before 2 s of a scanning period. Only the cells with stable resting potential (<4 mV change) over the protocol were accepted for data analysis. Data obtained were baseline-subtracted, then median-filtered to remove action potentials.
Viral injection and surgical procedures
For comparison with Ai203 reporter mice, double transgenic mice expressing CaMKII-tTA; tetO-GCaMP6s37 were crossed with transgenic SST-Cre94 or PV-Cre95 mice to create triple-transgenic mice expressing GCaMP6s in excitatory (tTA-expressing) neurons. Mice were intracranially injected with PhP.eB-TRE-ChroME-P2A-H2B-mRuby3.96 Notably, as the tetracycline response element (TRE) viral promoter drives expression in cells expressing tetracycline trans-activator (tTA),97 viral transduction was coincident with excitatory neurons transgenically expressing GCaMP6s, and not dependent on the particular Cre line used (PV-Cre or SST-Cre).
Mice 6 weeks or older were anesthetized with 2% isoflurane and administered 2 mg/kg of dexamethasone (to reduce cerebral edema) and 0.5 mg/kg of buprenorphine (as an analgesic). Mice were then secured in a stereotaxic frame (Kopf) and kept warm with a heating pad. A small incision was made to expose the skull and 2 burr holes were placed over left V1 (2.7 mm lateral, 1 mm anterior of lambda) approximately 1 mm apart. 300 nL of virus was slowly injected between 200 and 300 μm below the dura, at a rate of 50 nL/s using a microinjector (Micro4; World Precision Instruments) and a glass pipette (Drummond Scientific). The glass pipette was kept in place for 5–10 min after injection to ensure adequate spread and prevent backflow of virus during retraction of the pipette. Headplating and cranial windowing (below) were performed immediately after viral injection.
Mice were headplated with custom-made titanium headplates. To ensure secure placement, the skull was lightly etched with a scalpel and the headplate was initially attached over V1 with Vetbond (3M). Metabond (C&B) and OrthoJet (Lang Dental) were used to permanently attach and secure the headplate. A craniotomy was made over the injection sites and V1 using a 3.5 mm biopsy punch. Once the skull was removed, bleeding was controlled with cold PBS and Gelfoam (Pfizer Inc.). A cranial window was constructed by gluing together two 3 mm diameter circular coverslips to the bottom of a 5 mm diameter circular coverslip, then placed onto the craniotomy, and secured into place with Metabond (C&B). Mice were allowed to recover in a heated recovery cage.
For in utero electroporation, surgeries were done on pregnant CD-1 (ICR, Charles River) mice at E15. Mice were anesthetized with 2% isoflurane and kept on a heated pad to maintain body temperature. Warm, sterile PBS was intermittently perfused over the pups throughout the procedure. Recombinant DNA, pCAG-mRuby3 for screening and either pCAG-stChroME-GCaMP6s or pCAG-GCaMP6s (4:1 proportion for all), was injected into the left ventricle of each embryo using a micropipette. Successful injection was visualized using Fast green (Sigma-Aldrich). Both sides of the embryo’s head were grabbed with platinum-plated 5mm Twee-zertrodes (BTX Harvard Apparatus) connected to the negative pole, and a third electrode connected to the positive pole was placed slightly below lambda to target V1. Electroporation was conducted with 6 pulses at 30V with a 1s delay between each pulse using an Electro Square Porator (BTX Harvard Apparatus). Each mouse was then allowed to recover and come to term. Pups were screened for GCaMP6s and mRuby3 expression and allowed to develop normally.
Awake, all-optical in vivo imaging and photostimulation
For 2-photon calcium imaging in vivo in awake animals (Figures 2, 3I–3L, 4, and S6), mice were head-fixed and allowed to run on a freely moving circular treadmill under a 20× magnification (1.0 NA) water-immersion objective (Olympus Corporation) and imaged with a Sutter MOM 2p resonance scanning microscope controlled with MATLAB-based ScanImage software (Vidrio Technologies) (see also microscope design, above, Figure S3). Fast, 3 or 4 z-plane volumetric imaging (3 planes: 6.36 Hz, 4 planes: 4.77 Hz, average field-of-view: 821 ± 2.58 μm × 761 ± 1.53 μm, n = 5 defocus levels corresponding to 0, 20, 30, 40, 60 μm axially) was accomplished using an electrically-tunable lens (Optotune) placed in the light path prior to the resonance scanners. We observed only minimal changes in FOV size through the ETL at the defocus levels used within our experiments (see quantification above). A 20 ms delay was added to the Y-galvo flyback time to provide adequate settle-time for the ETL and ensure a non-distorted field-of-view (FOV). Z-planes were placed 20 or 30 μm apart for 4 or 3 plane recordings, respectively. The imaging laser (Chameleon; Coherent Inc.) was tuned to 920 nm and restricted to ≤75 mW average power to limit scanning-induced crosstalk. 3-plane imaging with 50 mW imaging power was used all in vivo experiments except Figures 2J–2L which used 4-plane imaging with 75 mW of power.
ChroME-expressing cells were identified by nuclear-localized mRuby3 fluorescence imaged at 1040 nm and using a custom 2d-peak detection algorithm in MATLAB. Detected cell locations were used as targets for holographic photostimulation. Custom written MATLAB code was used to synchronize the experiment via NI data acquisition boards.
Visual stimuli were presented on a 2048 × 1536 Retina iPad LCD display (Adafruit Industries) placed 10 cm from the mouse. The monitor backlight was synchronized with the galvos such that it came on only during the turnaround time, so that light from the monitor did not contaminate 2p imaging. Visual stimuli were created and presented with custom MATLAB code and Psychophysics ToolBox. Drifting gratings (50 visual degrees, 1 Hz, .08 cycles per degree, 100% contrast) of different orientations were randomly presented for 1 s each trial and interleaved with a grey-screen (“blank”) condition.
Power and spike calibration
All data was processed online via a real-time implementation of CaImAn OnACID90 (coined ‘live2p’ (www.github.com/willyh101/live2p). Briefly, the autodetected red nuclei (or, in some cases, manually selected green cells) used as targets for photostimulation were used as seed locations for fluorescent sources. Images were streamed from ScanImage Tiffs into multithreaded processing queues for each plane, allowing for simultaneous processing of multiple imaging z-planes in separate threads. Motion correction and signal extraction was performed during the experiment only on cells used for seeding to improve online performance.
Throughout all photostimulation experiments, all multi-target holograms are corrected for diffraction efficiency and overall efficiency, as measured by calibration of 3D-SHOT.66This ensures each target receives the intended power regardless of where its located, or what other targets are present.
A subset of detected cells were power calibrated. Groups of 5 targets per hologram were driven with five 5ms pulses at 30Hz. Targets were arranged in holograms to maximize the distance between any two simultaneously shot targets. A ramp of powers from 0 up to 85mW (4–12 steps total) was used, with 5–10 repetitions per power step. Afterward, mean denoised, neuropil-decontaminated fluorescence, as measured by CaImAn OnAcid, 1 s after stimulation was taken. The highest and lowest fluorescence trial for each power step were excluded to handle outliers. The data was fit with a ‘hill function’, below.
Where represents the fluorescence at saturation, is the power that produces 50% of the saturating response, is the optogenetic stimulation power, and can be either 4, 6, or 8 and controls the slope of the function. is determined by fitting a curve for each value and selecting the value that produces the greatest adjusted . The ‘target power’ is defined as the power needed to reach 80% of the saturating fluorescence, . Cells were considered not fit and excluded from further analysis if , or the ‘target power’ was below 10mW or above 1.5x the max power tested.
Cells that were fit by power calibration and were visually responsive were spike calibrated using a similar design. As before, cells were grouped into holograms containing 5 spread out targets, with each stimulated at 10–20% more than their fit target power. Each cell was stimulated for 1s with 1–20 pulses. The observed fluorescence was fit as a function of spike number by a quadratic function with the 0-spike intercept fixed at 0 fluorescence. Cells with an adjusted were considered not fit and excluded.
Next, we presented visual stimuli, using the protocol described above, in 8 directions and measured response to each. Stimuli were 1s long. Neural responses were measured as the mean responses in the 1 s during visual stimulation. We used the fit model from spikes to calcium to estimate the number of optogenetic pulses it would take to match the observed mean fluorescence from the visual stimulus.
To generate a multi-cell pattern, a 1 s stimulation period was divided into 50 holograms encoding 0 to 10 targets each and triggered at 50Hz. A single hologram can activate at least 50 cells simultaneously (Figures 2I and 2J), and with a 300 Hz SLM we can stimulate and switch holograms at 125Hz, allowing a wide variety of large-scale population vectors to be written.52,64 A custom built gradient descent discrete optimizer was designed to calculate the precise pattern of which cells are stimulated in which hologram. After assigning the desired number of spikes per cell, the cost function of this optimizer attempted to maximize each cell’s ISI, while keeping the number of targets per hologram relatively low. Two target holograms were prevented, as there it is difficult to deliver the correct power to both targets. One target holograms often deliver more power than multi-target holograms, and therefore were penalized but not prevented. Each hologram was triggered 5ms before a 5ms optogenetic pulse, each cell received 10–20% more power than the target power. Ensembles contained 19 to 43 cells with up to 168 spikes added in total.
For analysis of similarity between recreated, real, and shuffled datasets, shuffles were performed by randomly shuffling trial labels while keeping cell labels correct.
In vivo imaging and electrophysiology
Two-photon guided cell-attached electrophysiological recordings were performed in adult mice aged >P35. Mice were surgically prepared for recording by first anesthetizing with 1.5%–2% isoflurane. A stainless steel headplate integrated with a recording chamber was attached to the skull over the V1 with Metabond. A circular craniotomy of 2.5mm diameter was made over the V1 by slowly thinning the skull over the boundary of the circle with a 0.25 mm carbide burr. The skull fragment was removed to expose the brain, with minimal bleeding and with the dura left intact. Finally, the craniotomy was covered with 1.5% agarose and sterile HEPES-buffered ACSF.
During recording the mouse was kept under minimal anesthesia to immobilize (isoflurane; <0.7%). Glass pipettes (5-MΩ resistance) were filled with HEPES ACSF and red fluorescent dye (Alexa Fluor 594; 50 μM). Under two-photon guidance, cells were identified using GCaMP7s-mediated green-fluorescent calcium activity while imaging at 30–60 mW at 920 nm, then approached with the electrode to loosely patch (attach but not break the membrane). Cells were initially screened for spontaneous activity and then one-photon LED stimulation (Lumencor, SPECTRA X)-evoked action potential(s) to confirm co-expression of ChroME (475nm, 10hz, 5ms pulse duration). Data were acquired using a Multiclamp 700B Amplifier (Axon Instruments) and digitized at 20 kHz (National Instruments).
Imaging was performed at an FOV size of 680 μm, imaging either 1 plane (30 Hz, 6 cells) or 3 planes (6 Hz, 8 cells). Imaging power was between 60 and 83 mW, mean power was 66 mW. For all parts of the protocol, electrophysiological measurements and imaging were performed simultaneously.
First, for power calibration cells were stimulated with five 5ms pulses at 30 Hz at either six or seven different powers from 0 to 45 mW. Then for cells for which good electrophysiological connection was maintained, a suitable power for stimulation was determined by eye due to the time constraints of in vivo recording. This power was on average only 3 mW different than the fit spike target power determined after the experiment as the true target power. The power was used for the second part of the protocol, spike rate calibration, where cells were stimulated for 1 s at a rate of either 2,5,10, or 20 Hz.
Visual contrast detection task
Apparatus
The visual behavioral apparatus for the contrast detection task was controlled using an Arduino DUE synchronized with Raspberry Pi 3+/4+, which interfaced with a custom-written code in Python, Arduino and Java. Mouse licking was detected by changes in the capacitive load using a capacitive touch sensor (Adafruit, AT42QT1070) forming a circuit between the 0.05-inch diameter steel lick-port and the mouse’s tongue. Water was delivered by gravity though the lick port using a 2- way normally closed isolation valve control (Neptune Research Inc.). Visual stimuli were generated using a custom-written code in Python and presented using a gamma corrected LCD monitor (Podofo, 15.7 × 8.9 cm, 1024×600 pixels, 60 Hz refresh rate) located 12 cm from the right eye. The entire apparatus was mounted on an 8.0 × 8.0 × 0.5 inch (MB8; Thorlabs Inc.) aluminum breadboard and enclosed in a light isolation box (80/20).
Task design
The contrast detection task consisted of uncued GO or CATCH trials that occurred after random intertrial intervals (3–8 s). A no lick period was enforced in the 2 s preceding the trial, licking during this period caused a time-out punishment (5–9 s). In GO trials a visual stimulus of variable contrast was presented, if the mouse responded within the 500 ms response window, it received a reward. CATCH trials had identical timing, but no stimulus was presented and if the mouse licked within the response window it received a time out punishment. Time outs were 5–9 s. The 500 ms response window immediately followed the 600 ms stimulus presentation. Contrasts were presented in randomly shuffled blocks wherein each contrast must occur three times.
Behavioral training
Training began after headplate and cranial window implantation surgery, recovery (7 days) and water restriction (5–8 days). Prior to training, mice were habituated for two 30 min sessions to head fixation in the tube with the monitor on. The training schedule was divided into four main stages. During the first stage of behavioral training, mice were classically conditioned to lick in response to the presence of a visual stimulus consisting of a square patch of a drifting sinusoidal grating (2Hz, 0.08 cycles/degree, 600 ms, 100% contrast, 34 visual degrees, black background luminescence). On every trial, the visual stimulus was paired with a water reward delivered at the beginning of the response window. No catch trials occurred in this stage. Once mice reliably licked correctly within the response window in more than 80% of the trials, they moved onto the operant conditioning phase of training.
During the second stage of training, mice were trained to respond to presence of the visual stimulus. Catch trials were introduced as 25% of trials. The mouse must lick in response to the visual stimulus within the reward window to receive a water reward and with-hold licking otherwise to avoid time out punishments. Mice started this paradigm with the same visual stimuli features as stage 1(100% contrast, 34 VD, black background luminescence). Once mice reached performance criteria (>90% hit rate and <30% false alarm rate) for two consecutive days, mice were transitioned to stage three.
In stage three, background luminescence was ramped up until it reached mean luminance, then stimulus size was ramped down until it reached 18 VD. Ramping occurred within and across sessions as long as performance criteria were maintained. Once ramping was complete, mice had to maintain performance at criteria for one day to progress to the next stage.
In stage four, mice had to detect stimuli of varying contrasts. For strain comparisons (Figures S6B and S6C), 10 different contrasts (not including 0% catch trials) were used and catch trials were 7.5% of all trials. For optogenetic testing (Figures S6D–S6F), six different contrasts were used and catch trials were 25% of all trials.
One-photon optogenetic stimulation
One-photon (1p) photostimulation during contrast detection task was delivered using an optical fiber (400μm diameter, 0.39 nA; Thorlabs Inc.) coupled to a 470-nm LED (M470F3, Thorlabs) driven by an LED driver (LEDD1B; Thorlabs Inc.) and positioned using a micromanipulator over the location of V1 at the posterior of the window. Light power was calibrated at the end of the fiber using a power meter (PM160T, Thorlabs). Stimulation power was calibrated to each mouse and ranged from 0.05 to 0.4 mW out of the fiber. For control wildtype mice, 0.5 mW was used for all mice. To prevent photostimulation from interfering with behavioral performance, we attached a custom designed light-blocking cone to the mouse head and illuminated blue LED masking lights during the entire session. To further reduce any extraneous cues, we covered the left eye with a plastic eye patch. Light was delivered from the onset of the stimulus to the end of the response window on 33% of trials.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed in MATLAB or python. Information on statistical tests, n, and center and dispersion measurements is specified in results and figure legends wherever relevant. Sample sizes were not predetermined using power analysis.
Calcium imaging post-processing and analysis
For Figures 1, 2, and 3A–3I, calcium imaging data was extracted from raw TIFF files, motion corrected, and source-extracted using Suite2p.89 Putative sources were manually curated based on morphology and fluorescence statistics. Neuropil traces for each source were generated by Suite2p, multiplied by a correction coefficient (0.7), and subtracted from source fluorescence traces. Calcium traces were minimum subtracted, and was calculated as:
Where is a cell’s fluorescence response and is baseline fluorescence. For in vitro characterization in Figure 1, was set to be the mean fluorescence 1 s pre-stimulus. For in vivo characterization in Figure 2 and the electrophysiology and imaging experiments in Figures 3A–3H, was calculated by a 1-min rolling 20% quantile. Individual trialwise responses were baseline subtracted using the mean pre-stimulus (holographic or visual) activity. For Figures 2 and 3, were z-scored by subtracting the mean and dividing by the standard deviation.
Holographic targets were matched to sources identified by Suite2p by the source satisfying the argmin Euclidean distance for each target. Targets lacking a source within 15 μm were excluded from further analysis. In the rare cases where multiple targets matched to the same source, only the closest target was used.
For in vitro characterization in Figure 1, zero stimulation trials were simulated by taking fluorescence from the inter-trial interval of one spike trials, 6 s after the last spike stimulation. Mean calcium traces were calculated by taking the mean calcium trace across all trials for a given spike number for a given cell. Peak was the peak of the mean calcium trace, with the peak for 0 spike trials subtracted to account for noise.
For visual experiment analysis, neurons with significantly different responses to visual stimuli (, 1-way ANOVA) were considered as visually responsive. FOVs with fewer than 20% visually responsive cells were considered unlikely to be in V1 and excluded from further analysis. Preferred orientation was calculated as the max response from trial-averaged orientation tuning curves; orthogonal orientation was determined as the response 90° from preferred orientation. Orientation selectivity index (OSI) was then calculated as:
Where is the mean response to preferred orientation and is the mean response to orthogonal orientation. Orientation tuning curves were min subtracted prior to calculation to ensure values ranged from 0 to 1.
To construct contrast response curves, full-screen Gaussian contrast modulated noise stimuli98 were presented using custom written MATLAB scripts and Psychophysics Toolbox. Contrast response functions were fit with a modified Naka-Rushton function,99 defined as:
Where is the maximum firing rate of the neuron, is the baseline firing rate, is the contrast shown, and is the contrast corresponding to 50% maximum response, and is an exponent controlling the slope. Contrast response curves were fit by least-squares regression using SciPy’s curve_fit function in Python. B was set constant to 0, corresponding to the low baseline firing rates of pyramidal cells in vivo. n was constrained between 0 and 4.
To determine GCaMP signal-to-noise in vivo, we used the peak signal for each cell’s mean response to drifting gratings at that cell’s preferred orientation.
Cells were considered to be “stimmable” if their mean response to holographic stimulation was significantly different (, one-tailed rank-sum) from baseline.
For the all-optical data in Figures 3I–3L and Figure 4, because suite2p uses a different method than CaImAn for cell identification and analysis, for consistency with online measurements taken during calibration, to analyze the power and spike calibration data, and the writing in of population vectors after power and spike calibration, we used live2p (an implementation of CaImAn OnACID) to run offline on the entire imaging session at once, seeded to our targets. This motion corrected and source-extracted the data, and used the z-scored neuropil-corrected and denoised calcium signal for analysis. We also analyzed the data using suite2p, as and obtained similar results. In fact, we found that with suite2p, a higher % of matched targets were fit in the power balancing step, 80.5% (803/997 detected cells) were fit resulting in a similar mean target power (66.3 ± 0.9 mW). Thus, we believe that most of the cells that were not fit represent low signal to noise cells or ROIs that were falsely identified as cells.
Analysis of in vivo simultaneous electrophysiology and imaging data
Calcium data was preprocessed as described above. For analysis of evoked spiking, spikes were considered evoked if they occurred either after the start of an optogenetic pulse and either before the next start of optogenetic pulse or within 55 ms of the end of a pulse, whichever was shorter. For power calibration, for zero power stimulation, pulse times for analysis were set to start and end at the same as all other powers. For spike calibration, for zero pulse rate trials, pulse times for analysis were set to be the same as for 2 Hz stimulation.
Cells were excluded from analysis if their mean z-ΔF/F for 45 mW of stimulation (for power calibration) or 10 Hz stimulation (spike rate calibration) was less than the mean +1.5 times the standard deviation of the response to 0 power or 0 Hz stimulation, respectively. One cell was excluded from both steps of by this metric, one cell was excluded from only the first step but due to recentering the imaging between steps was included for analysis of the spike curve calibration.
For calcium data, power curves were fit with a hill function as described above to mean ΔF/F taken over 1.5 s after stimulus onset. Target power estimation from spiking data was done by the same procedure, but using evoked spikes, and target power was set to be the power at which the spike response reached 90% of saturation rather than 80%.
Supplementary Material
Highlights.
Ai203 line expresses soma-targeted ChroME opsin andGCaMP7s for all-optical studies
All-optical calibration of stimulation power per cell enables control of spike rates
Population activity vectors can be recreated on demand using this procedure
ACKNOWLEDGMENTS
We thank the members of the Adesnik lab for comments, Savitha Sridharan for advice on construct development, Janine Beyer for technical support, and Nikhil Bhatla for assistance with histology. We thank Viviana Gradinaru and Ken Chan for the initial PhP.eB ChroME virus. H.A. was a New York Stem Cell Robertson Investigator. This work was supported by the New York Stem Cell Foundation, National Institutes of Health (NIH) grant UF1- NS107574 (H.A.), NIH grant U19- NS107613 (H.A.), NIH grant RF1- RF1MH120680 (H.A.), NIH grant RO1- EY023756 (H.A.), National Science Foundation Graduate Research Fellowship DGE-1752814 (H.A.B.), NIH grant F31-EY031977 (W.D.H.), NIH grant K99-EY029758 (I.A.O.), and NIH grant U19-MH114830 (H.Z.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the National Science Foundation.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. One or more of the authors of this paper self-identifies as living with a disability. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research.
Footnotes
DECLARATION OF INTERESTS
H.A. has a patent related to this work: 3D Sparse Holographic Temporal focusing, 2016, L. Waller, N. Pegard, and H. Adesnik, Provisional Patent Application #62–429,017.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112909.
REFERENCES
- 1.Adesnik H, and Abdeladim L (2021). Probing neural codes with two-photon holographic optogenetics. Nat. Neurosci. 24, 1356–1366. 10.1038/s41593-021-00902-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrasfalvy BK, Zemelman BV, Tang J, and Vaziri A (2010). Two-photon single-cell optogenetic control of neuronal activity by sculpted light. Proc. Natl. Acad. Sci. USA 107, 11981–11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bégue A, Papagiakoumou E, Leshem B, Conti R, Enke L, Oron D, and Emiliani V (2013). Two-photon excitation in scattering media by spatiotemporally shaped beams and their application in optogenetic stimulation. Biomed. Opt Express 4, 2869–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pegard N, et al. (2017). Holographic Temporal Focusing for 3D Photoactivation With Single Neuron Resolution 3–5. [Google Scholar]
- 5.Ronzitti E, Emiliani V, and Papagiakoumou E (2018). Methods for Three-Dimensional All-Optical Manipulation of Neural Circuits. Front. Cell. Neurosci. 12, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papagiakoumou E, Anselmi F, Bégue A, de Sars V, Glückstad J, Isacoff EY, and Emiliani V (2010). Scanless two-photon excitation of channelrhodopsin-2. Nat. Methods 7, 848–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Papagiakoumou E, Ronzitti E, and Emiliani V (2020). Scanless two-photon excitation with temporal focusing. Nat. Methods 17, 571–581. [DOI] [PubMed] [Google Scholar]
- 8.Yang W, Carrillo-Reid L, Bando Y, Peterka DS, and Yuste R (2018). Simultaneous two-photon imaging and two-photon optogenetics of cortical circuits in three dimensions. Elife 7, e32671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen IW, Ronzitti E, Lee BR, Daigle TL, Dalkara D, Zeng H, Emiliani V, and Papagiakoumou E (2019). In vivo submillisecond two-photon optogenetics with temporally focused patterned light. J. Neurosci. 39, 3484–3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prakash R, Yizhar O, Grewe B, Ramakrishnan C, Wang N, Goshen I, Packer AM, Peterka DS, Yuste R, Schnitzer MJ, and Deisseroth K (2012). Two-photon optogenetic toolbox for fast inhibition, excitation and bistable modulation. Nat. Methods 9, 1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Packer AM, Peterka DS, Hirtz JJ, Prakash R, Deisseroth K, and Yuste R (2012). Two-photon optogenetics of dendritic spines and neural circuits. Nat. Methods 9, 1202–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rickgauer JP, and Tank DW (2009). Two-photon excitation of channelrhodopsin-2 at saturation. Proc. Natl. Acad. Sci. USA 106, 15025–15030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shemesh OA, Tanese D, Zampini V, Linghu C, Piatkevich K, Ronzitti E, Papagiakoumou E, Boyden ES, and Emiliani V (2017). Temporally precise single-cell-resolution optogenetics. Nat. Neurosci. 20, 1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z, Russell LE, Packer AM, Gauld OM, and Häusser M (2018). Closed-loop all-optical interrogation of neural circuits in vivo. Nat. Methods 15, 1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Russell LE, Dalgleish HWP, Nutbrown R, Gauld OM, Herrmann D, Fişek M, Packer AM, and Häusser M (2022). All-optical interrogation of neural circuits in behaving mice. Nat. Protoc. 17, 1579–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaziri A, and Emiliani V (2012). Reshaping the optical dimension in optogenetics. Curr. Opin. Neurobiol. 22, 128–137. [DOI] [PubMed] [Google Scholar]
- 17.Carrillo-Reid L, Han S, Yang W, Akrouh A, and Yuste R (2019). Controlling Visually Guided Behavior by Holographic Recalling of Cortical Ensembles. Cell. 10.1016/j.cell.2019.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrillo-Reid L, Yang W, Bando Y, Peterka DS, and Yuste R (2016). Imprinting and recalling cortical ensembles. Science 353, 691–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gill JV, Lerman GM, Zhao H, Stetler BJ, Rinberg D, and Shoham S (2020). Precise Holographic Manipulation of Olfactory Circuits Reveals Coding Features Determining Perceptual Detection. Neuron 108, 382–393.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Naka A, et al. (2019). Complementary networks of cortical somatostatin interneurons enforce layer specific control. Elife. 10.7554/eLife.43696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jennings JH, Kim CK, Marshel JH, Raffiee M, Ye L, Quirin S, Pak S, Ramakrishnan C, and Deisseroth K (2019). Interacting neural ensembles in orbitofrontal cortex for social and feeding behaviour. Nature 565, 645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chettih SN, and Harvey CD (2019). Single-neuron perturbations reveal feature-specific competition in V1. Nature 567, 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dalgleish HW, Russell LE, Packer AM, Roth A, Gauld OM, Green-street F, Thompson EJ, and Häusser M (2020). How many neurons are sufficient for perception of cortical activity? Elife 9, e58889–e58899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rickgauer JP, Deisseroth K, and Tank DW (2014). Simultaneous cellular-resolution optical perturbation and imaging of place cell firing fields. Nat. Neurosci. 17, 1816–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daie K, Svoboda K, and Druckmann S (2021). Targeted photostimulation uncovers circuit motifs supporting short-term memory. Nat. Neurosci. 24, 259–265. [DOI] [PubMed] [Google Scholar]
- 26.Hage TA, Bosma-Moody A, Baker CA, Kratz MB, Campagnola L,Jarsky T, Zeng H, and Murphy GJ (2019). Distribution and strength of interlaminar synaptic connectivity in mouse primary visual cortex revealed by two-photon optogenetic stimulation. Preprint at bioRxiv. 10.1101/2019.12.13.876128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marshel JH, Kim YS, Machado TA, Quirin S, Benson B, Kadmon J, Raja C, Chibukhchyan A, Ramakrishnan C, Inoue M, et al. (2019). Cortical layer-specific critical dynamics triggering perception. Science 365, eaaw5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oldenburg IA, et al. (2022). The logic of recurrent circuits in the primary visual cortex. Preprint at bioRxiv. 10.1101/2022.09.20.508739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buetfering C, Zhang Z, Pitsiani M, Smallridge J, Boven E, McElligott S, and Häusser M (2022). Behaviorally relevant decision coding in primary somatosensory cortex neurons. Nat. Neurosci. 25, 1225–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lerman GM, Gill JV, Rinberg D, and Shoham S (2019). Precise optical probing of perceptual detection. In Optics InfoBase Conference Papers, p. F169. [Google Scholar]
- 31.Haery L, Deverman BE, Matho KS, Cetin A, Woodard K, Cepko C, Guerin KI, Rego MA, Ersing I, Bachle SM, et al. (2019). Adeno-Associated Virus Technologies and Methods for Targeted Neuronal Manipulation. Front. Neuroanat. 13, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyashita T, Shao YR, Chung J, Pourzia O, and Feldman DE (2013). Long-term channelrhodopsin-2 (ChR2) expression can induce abnormal axonal morphology and targeting in cerebral cortex. Front. Neural Circ. 7, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackman SL, Beneduce BM, Drew IR, and Regehr WG (2014). Achieving high-frequency optical control of synaptic transmission. J. Neurosci. 34, 7704–7714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daigle TL, Madisen L, Hage TA, Valley MT, Knoblich U, Larsen RS, Takeno MM, Huang L, Gu H, Larsen R, et al. (2018). A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell 174, 465–480.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madisen L, Mao T, Koch H, Zhuo J.m., Berenyi A, Fujisawa S, Hsu YWA, Garcia AJ 3rd, Gu X, Zanella S, et al. (2012). A toolbox of Cre-dependent optogenetic transgenic mice for light-induced activation and silencing. Nat. Neurosci. 15, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wekselblatt JB, Flister ED, Piscopo DM, and Niell CM (2016). Large-scale imaging of cortical dynamics during sensory perception and behavior. J. Neurophysiol. 115, 2852–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gallego JA, Perich MG, Naufel SN, Ethier C, Solla SA, and Miller LE (2018). Cortical population activity within a preserved neural manifold underlies multiple motor behaviors. Nat. Commun. 91, 4233–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobak D, Pardo-Vazquez JL, Valente M, Machens CK, and Renart A (2019). State-dependent geometry of population activity in rat auditory cortex. Elife 8, e44526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stringer C, Pachitariu M, Steinmetz N, Carandini M, and Harris KD (2019). High-dimensional geometry of population responses in visual cortex. Nature 571, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jazayeri M, and Afraz A (2017). Navigating the Neural Space in Search of the Neural Code. Neuron 93, 1003–1014. [DOI] [PubMed] [Google Scholar]
- 42.Churchland MM, Cunningham JP, Kaufman MT, Foster JD, Nuyujukian P, Ryu SI, and Shenoy KV (2012). Neural population dynamics during reaching. Nature 487, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Semedo JD, Zandvakili A, Machens CK, Yu BM, and Kohn A (2019). Cortical Areas Interact through a Communication Subspace. Neuron 102, 249–259.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vyas S, Golub MD, Sussillo D, and Shenoy KV (2020). Computation Through Neural Population Dynamics. Annu. Rev. Neurosci. 43, 249–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shenoy KV, and Kao JC (2021). manipulation and modeling of brain-wide neural population dynamics. Nat. Commun. 12, 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang L, Ledochowitsch P, Knoblich U, Lecoq J, Murphy GJ, Reid RC, de Vries SE, Koch C, Zeng H, Buice MA, et al. (2021). Relationship between simultaneously recorded spiking activity and fluorescence signal in gcamp6 transgenic mice. Elife 10, e51675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. (2013). Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rupprecht P, Carta S, Hoffmann A, Echizen M, Blot A, Kwan AC, Dan Y, Hofer SB, Kitamura K, Helmchen F, and Friedrich RW (2021). A database and deep learning toolbox for noise-optimized, generalized spike inference from calcium imaging. Nat. Neurosci. 249, 1324–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berens P, Freeman J, Deneux T, Chenkov N, McColgan T, Speiser A, Macke JH, Turaga SC, Mineault P, Rupprecht P, et al. (2018). Community-based benchmarking improves spike rate inference from two-photon calcium imaging data. PLoS Comput. Biol. 14, e1006157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Theis L, Berens P, Froudarakis E, Reimer J, Román Rosón M, Baden T, Euler T, Tolias AS, and Bethge M (2016). Benchmarking Spike Rate Inference in Population Calcium Imaging. Neuron 90, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friedrich J, Zhou P, and Paninski L (2017). Fast online deconvolution of calcium imaging data. PLoS Comput. Biol. 13, e1005423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mardinly AR, Oldenburg IA, Pégard NC, Sridharan S, Lyall EH, Chesnov K, Brohawn SG, Waller L, and Adesnik H (2018). Precise multimodal optical control of neural ensemble activity. Nat. Neurosci. 21, 881–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dana H, Sun Y, Mohar B, Hulse BK, Kerlin AM, Hasseman JP, Tsegaye G, Tsang A, Wong A, Patel R, et al. (2019). High-performance calcium sensors for imaging activity in neuronal populations and micro-compartments. Nat. Methods 16, 649–657. [DOI] [PubMed] [Google Scholar]
- 54.Baker CA, Elyada YM, Parra A, and Bolton MML (2016). Cellular-resolution circuit mapping with temporal-focused excitation of soma-targeted channelrhodopsin. Elife. 10.7554/eLife.14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Forli A, Vecchia D, Binini N, Succol F, Bovetti S, Moretti C, Nespoli F, Mahn M, Baker CA, Bolton MM, et al. (2018). Two-Photon Bidirectional Control and Imaging of Neuronal Excitability with High Spatial Resolution In vivo. Cell Rep. 22, 3087–3098. 10.1016/j.celrep.2018.02.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu C, Ivanova E, Zhang Y, and Pan ZH (2013). rAAV-Mediated Subcellular Targeting of Optogenetic Tools in Retinal Ganglion Cells In vivo. PLoS One 8, e66332. 10.1371/journal.pone.0066332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ronzitti E, Conti R, Zampini V, Tanese D, Foust AJ, Klapoetke N, Boyden ES, Papagiakoumou E, and Emiliani V (2017). Submillisecond Optogenetic Control of Neuronal Firing with Two-Photon Holographic Photoactivation of Chronos. J. Neurosci. 37, 10679–10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, Morimoto TK, Chuong AS, Carpenter EJ, Tian Z, et al. (2014). Independent Optical Excitation of Distinct Neural Populations. Nat. Methods 11, 338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, et al. (2021). Fast and sensitive GCaMP calcium indicators for imaging neural populations. Preprint at bioRxiv. 10.1101/2021.11.08.467793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lim ST, Antonucci DE, Scannevin RH, and Trimmer JS (2000). A Novel Targeting Signal for Proximal Clustering of the Kv2.1 K+ Channel in Hippocampal Neurons. Neuron 25, 385–397. [DOI] [PubMed] [Google Scholar]
- 61.Chen Y, Jang H, Spratt PWE, Kosar S, Taylor DE, Essner RA, Bai L, Leib DE, Kuo TW, Lin YC, et al. (2020). Soma-Targeted Imaging of Neural Circuits by Ribosome Tethering. Neuron 107, 454–469.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shemesh OA, Linghu C, Piatkevich KD, Goodwin D, Celiker OT, Gritton HJ, Romano MF, Gao R, Yu CCJ, Tseng HA, et al. (2020). Precision Calcium Imaging of Dense Neural Populations via a Cell-Body-Targeted Calcium Indicator. Neuron 107, 470–486.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harris JA, Hirokawa KE, Sorensen SA, Gu H, Mills M, Ng LL, Bohn P, Mortrud M, Ouellette B, Kidney J, et al. (2014). Anatomical characterization of Cre driver mice for neural circuit mapping and manipulation. Front. Neural Circ. 8, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sridharan S, Gajowa MA, Ogando MB, Jagadisan UK, Abdeladim L, Sadahiro M, Bounds HA, Hendricks WD, Turney TS, Tayler I, et al. (2022). High-performance microbial opsins for spatially and temporally precise perturbations of large neuronal networks. Neuron 110, 1139–1155.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vong L, Ye C, Yang Z, Choi B, Chua S Jr., and Lowell BB (2011). Leptin Action on GABAergic Neurons Prevents Obesity and Reduces Inhibitory Tone to POMC Neurons. Neuron 71, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pégard NC, Mardinly AR, Oldenburg IA, Sridharan S, Waller L, and Adesnik H (2017). Three-dimensional scanless holographic optogenetics with temporal focusing (3D-SHOT). Nat. Commun. 8, 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Berg-Johnsen J, and Langmoen IA (1987). Isoflurane hyperpolarizes neurones in rat and human cerebral cortex. Acta Physiol. Scand. 130, 679–685. [DOI] [PubMed] [Google Scholar]
- 68.Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, and Andermann ML (2014). Removable cranial windows for long-term imaging in awake mice. Nat. Protoc. 9, 2515–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andermann ML, Gilfoy NB, Goldey GJ, Sachdev RNS, Wölfel M, McCormick DA, Reid RC, and Levene MJ. (2013). Chronic Cellular Imaging of Entire Cortical Columns in Awake Mice Using Microprisms. Neuron 80, 900–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cramer SW, Carter RE, Aronson JD, Kodandaramaiah SB, Ebner TJ, and Chen CC (2021). Through the looking glass: A review of cranial window technology for optical access to the brain. J. Neurosci. Methods 354, 109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WCA, et al. (2009). Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat. Protoc. 4, 1128–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cone JJ, Bade ML, Masse NY, Page EA, Freedman DJ, and Maunsell JHR (2020). Mice Preferentially Use Increases in Cerebral Cortex Spiking to Detect Changes in Visual Stimuli. J. Neurosci. 40, 7902–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Glickfeld LL, Histed MH, and Maunsell JHR (2013). Mouse Primary Visual Cortex Is Used to Detect Both Orientation and Contrast Changes. J. Neurosci. 33, 19416–19422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruediger S, and Scanziani M (2020). Learning speed and detection sensitivity controlled by distinct cortico-fugal neurons in visual cortex. Elife 9, e59247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jin M, and Glickfeld LL (2019). Contribution of Sensory Encoding to Measured Bias. J. Neurosci. 39, 5115–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pouget A, Dayan P, and Zemel R (2000). Information processing with population codes. Nat. Rev. Neurosci. 1, 125–132. [DOI] [PubMed] [Google Scholar]
- 77.Kriegeskorte N, and Wei X-X (2021). Neural tuning and representational geometry. Nat. Rev. Neurosci. 22, 703–718. [DOI] [PubMed] [Google Scholar]
- 78.Chung S, and Abbott LF (2021). Neural population geometry: An approach for understanding biological and artificial neural networks. Curr. Opin. Neurobiol. 70, 137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Siegle JH, Ledochowitsch P, Jia X, Millman DJ, Ocker GK, Caldejon S, Casal L, Cho A, Denman DJ, Durand S, et al. (2021). Reconciling functional differences in populations of neurons recorded with two-photon imaging and electrophysiology. Elife 10, e69068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nauhaus I, Nielsen KJ, and Callaway EM (2012). Nonlinearity of two-photon Ca2+ imaging yields distorted measurements of tuning for V1 neuronal populations. J. Neurophysiol. 107, 923–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Kock CPJ, Bruno RM, Spors H, and Sakmann B (2007). Layer- and cell-type-specific suprathreshold stimulus representation in rat primary somatosensory cortex. J. Physiol. 581, 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu J, Liang Y, Chen S, Hsu CL, Chavarha M, Evans SW, Shi D, Lin MZ, Tsia KK, and Ji N (2020). Kilohertz two-photon fluorescence microscopy imaging of neural activity in vivo. Nat. Methods 173, 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Voleti V, Patel KB, Li W, Perez Campos C, Bharadwaj S, Yu H, Ford C, Casper MJ, Yan RW, Liang W, et al. (2019). Real-time volumetric microscopy of in vivo dynamics and large-scale samples with SCAPE 2.0. Nat. Methods 1610, 1054–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Villette V, Chavarha M, Dimov IK, Bradley J, Pradhan L, Mathieu B, Evans SW, Chamberland S, Shi D, Yang R, et al. (2019). Ultrafast Two-Photon Imaging of a High-Gain Voltage Indicator in Awake Behaving Mice. Cell 179, 1590–1608.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kulkarni RU, Vandenberghe M, Thunemann M, James F, Andreassen OA, Djurovic S, Devor A, and Miller EW (2018). In vivo Two-Photon Voltage Imaging with Sulfonated Rhodamine Dyes. ACS Cent. Sci. 4, 1371–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mattis J, Tye KM, Ferenczi EA, Ramakrishnan C, O’Shea DJ, Prakash R, Gunaydin LA, Hyun M, Fenno LE, Gradinaru V, et al. (2011). Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nat. Methods 9, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vierra NC, Kirmiz M, van der List D, Santana LF, and Trimmer JS (2019). 1 mediates spatial and functional coupling of l-type calcium channels and ryanodine receptors in mammalian neurons. Elife 8, e49953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pachitariu M, et al. (2017). Suite2p: beyond 10,000 neurons with standard two-photon microscopy. Preprint at bioRxiv. 10.1101/061507. [DOI] [Google Scholar]
- 90.Giovannucci A, Friedrich J, Gunn P, Kalfon J, Brown BL, Koay SA, Taxidis J, Najafi F, Gauthier JL, Zhou P, et al. (2019). Caiman an open source tool for scalable calcium imaging data analysis. Elife 8, e38173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Preibisch S, Saalfeld S, and Tomancak P (2009). Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 25, 1463–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gerchberg RW, and Saxton WO (1972). A Practical Algorithm for the Determination of Phase from Image and Diffraction Plane Pictures. Opt. Stuttg. 35, 237–250. [Google Scholar]
- 93.Sekerli M, Del Negro CA, Lee RH, and Butera RJ (2004). Estimating action potential thresholds from neuronal time-series: New metrics and evaluation of methodologies. IEEE Trans. Biomed. Eng. 51, 1665–1672. [DOI] [PubMed] [Google Scholar]
- 94.Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, et al. (2011). A Resource of Cre Driver Lines for Genetic Targeting of GABAergic Neurons in Cerebral Cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hippenmeyer S, Vrieseling E, Sigrist M, Portmann T, Laengle C, Ladle DR, and Arber S (2005). A Developmental Switch in the Response of DRG Neurons to ETS Transcription Factor Signaling. PLoS Biol. 3, e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chan KY, Jang MJ, Yoo BB, Greenbaum A, Ravi N, Wu WL, Sánchez-Guardado L, Lois C, Mazmanian SK, Deverman BE, and Gradinaru V (2017). Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 20, 1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gossen M, Freundlieb S, Bender G, Müller G, Hillen W, and Bujard H (1995). Transcriptional activation by tetracyclines in mammalian cells. Science 268, 1766–1769. [DOI] [PubMed] [Google Scholar]
- 98.Lee AM, Hoy JL, Bonci A, Wilbrecht L, Stryker MP, and Niell CM (2014). Identification of a Brainstem Circuit Regulating Visual Cortical State in Parallel with Locomotion. Neuron 83, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Naka KI, and Rushton WA (1966). S-potentials from luminosity units in the retina of fish (Cyprinidae). J. Physiol. 185, 587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for main figures and additional histology images are available at https://doi.org/10.5281/zenodo.8118399, all additional data will be shared by the lead contact upon request. Custom code for online and offline analysis has been deposited at Zenodo and is publicly available. DOIs are listed in the key resources table. Any additional information or code required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit anti-NeuN | Abcam | RRID:AB_2532109 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| PhP.eB-TRE-ChroME-P2A-H2B-mRuby3 | This paper | N/A |
|
| ||
| Deposited data | ||
|
| ||
| Data and analysis code | This paper | Zenodo: https://doi.org/10.5281/zenodo.8118399 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: B6; DBA-Tg(tetO-GCaMP6s)2Niell/J | Jackson Laboratory | RRID: IMSR_JAX: 024742 |
| Mouse: B6; CBA-Tg(Camk2a-tTA)1Mmay/J | Jackson Laboratory | RRID: IMSR_JAX: 003010 |
| Mouse: B6; 129S-Slc17a7tm1.1(cre)Hze/J, Slc17a7-IRES2-Cre | Jackson Laboratory | RRID:IMSR_JAX:023527 |
| Mouse: B6.Cg-Igs7<tm203(tetO-sChroME*/ GCaMP7s,CAG-mRuby3,-tTA2)Tasic>/J, Ai203 | Jackson Laboratory | JAX stock #037939 |
| Mouse: B6(Cg)-Cux2tm3.1(cre/ERT2) Mull/Mmmh, Cux2-CreERT2 | MMRRC | RRID:MMRRC_032779-MU |
| Mouse: B6J.129S6(FVB)-Slc32a1tm2(cre)Lowl/ MwarJ, Vgat-IRES-Cre | Jackson Laboratory | RRID:IMSR_JAX:028862 |
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000664 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pCAG-stChroME-GCaMP6s | This paper | N/A |
| pCAG-GCaMP6s | Addgene | Addgene_171156 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Schneider et al. | https://imagej.nih.gov/ij/ |
| MATLAB | MathWorks | RRID: SCR_001622 |
| Python | Python Software Foundation | RRID: SCR_008394 |
| Suite2p | Pachitariu et al.89 | RRID: SCR_016434 |
| ScanImage | Vidrio Inc. | RRID: SCR_014307 |
| CaImAn | Giovannucci et al.90 | RRID:SCR_021152 |
| live2p | This paper | Zenodo: https://doi.org/10.5281/zenodo.8117788 |
| Code for all-optical calibration and activity recreation experiments | This paper | Zenodo: https://doi.org/10.5281/zenodo.8139926 |
| Custom code for analyses | This paper | Zenodo: https://doi.org/10.5281/zenodo.8118399 |
| Easy Electrophysiology | Easy Electrophysiology Ltd | RRID:SCR_021190 |
|
| ||
| Other | ||
|
| ||
| Coherent Monaco 2p stimulation lasers | Coherent | https://www.coherent.com/lasers/ultrashort-pulse/monaco |
| Spatial Light Modulator | Meadowlark | HSP1920-1064-HSP8-HB |