

SUMMARY
Lung cancer treatment has benefited greatly through advancements in immunotherapies. However, immunotherapy often fails in patients with specific mutations like KEAP1, which are frequently found in lung adenocarcinoma. We established an antigenic lung cancer model and used it to explore how Keap1 mutations remodel the tumor immune microenvironment. Using single-cell technology and depletion studies, we demonstrate that Keap1-mutant tumors diminish dendritic cell and T cell responses driving immunotherapy resistance. This observation was corroborated in patient samples. CRISPR-Cas9-mediated gene targeting revealed that hyperactivation of the NRF2 antioxidant pathway is responsible for diminished immune responses in Keap1-mutant tumors. Importantly, we demonstrate that combining glutaminase inhibition with immune checkpoint blockade can reverse immunosuppression, making Keap1-mutant tumors susceptible to immunotherapy. Our study provides new insight into the role of KEAP1 mutations in immune evasion, paving the way for novel immune-based therapeutic strategies for KEAP1-mutant cancers.
Graphical abstract
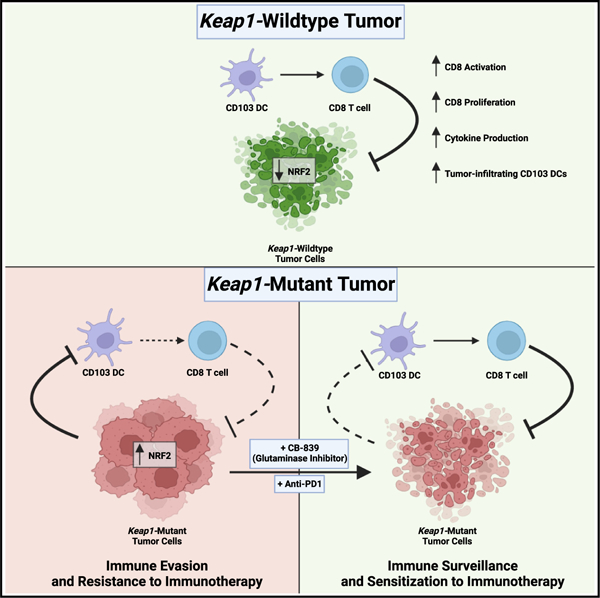
In brief
Zavitsanou et al. show that KEAP1 mutations in lung cancer promote immune evasion by suppressing dendritic and T cell responses, elucidating the mechanisms underlying immunotherapy resistance of KEAP1-mutant tumors. These findings underscore the importance of stratifying patients by KEAP1 mutation status and pave the way for novel therapies.
INTRODUCTION
Lung cancer is the leading cause of cancer-related deaths worldwide.1 Non-small cell lung cancer (NSCLC), including lung adenocarcinoma (LUAD), accounts for 85% of lung cancer cases.2,3 Despite improvements in therapy, NSCLC mortality remains high, with fewer than 20% of patients surviving after 5 years.2 Recently, immunotherapy has emerged as an important therapeutic intervention for a range of cancer types, including NSCLC.4 Immunotherapeutic approaches include the use of monoclonal antibodies against major checkpoints, such as cytotoxic T lymphocyte antigen protein 4 (CTLA4) and programmed death receptor 1 (PD1) or its ligand PDL1 (reviewed in Sharma et al.5). Despite promising results of immune checkpoint blockade (ICB) in NSCLC, either alone or with chemotherapy, improvements in survival are modest, and only a subset of patients achieve long-lasting remission.5–8 Understanding what limits the efficacy of these treatments is critical for development of future therapies.
Emerging evidence suggests that T cell presence in the tumor immune microenvironment (TIME) has an impact on immunotherapy response.9 Cytotoxic T lymphocytes (CTLs) are the main drivers of the anti-tumor immune response and are associated with improved outcomes and responses to ICB therapies.10,11 Accordingly, CTL chemoattractants such as CXCL9/CXCL10 and cytokines like type I and type II interferons have also been associated with favorable outcome, as they can promote CTL effector function.12–14 In addition, Batf3-dependent conventional type I dendritic cells (CD103 DCs) can modulate anti-tumor T cell responses via chemokine production and presentation of tumor-associated antigens, together with providing co-stimulatory or inhibitory signals.15–17 Importantly, CD103 DCs have been shown to control ICB responses in preclinical models and correlate with improved survival in humans.18–21 The full spectrum of tumor-intrinsic and host-dependent factors that determine the strength of anti-tumor immune responses remains to be fully elucidated.
One approach to address immunotherapy resistance is to define tumor-intrinsic genetic mutations that modulate the TIME and therapy response. Kelch-like ECH-associated protein 1 (KEAP1) is the third most frequently mutated gene in LUAD.22 KEAP1 is an adaptor protein for the Culin 3 (CUL3)/Ring box 1 (RBX1) E3 ubiquitin ligase complex. It targets the antioxidant transcription factor NRF2 for degradation, thereby preventing its nuclear accumulation and activation of the antioxidant program.23–25 The majority of somatic mutations in KEAP1 are missense or truncating events that can generate dominant-negative forms of KEAP1.26–29 We previously established that KEAP1 is a tumor-suppressor gene by demonstrating that Keap1 loss accelerated Kras-driven LUAD progression30 and metastasis.31 Clinical data have revealed that KEAP1-mutant lung cancers are resistant to standard-of-care radiation,32 covalent KRASG12C inhibitors,33–35 and ICB.36–40 Furthermore, KEAP1-mutant lung cancers are associated with a TIME lacking T cells,41 suggesting that mutations in KEAP1 can have an impact on immune surveillance.
In this study, we investigated whether tumor-intrinsic Keap1 mutations remodel the TIME and promote resistance to ICB. Using an antigenic mouse model of LUAD, we demonstrate that Keap1-mutant tumors suppress CD103 DC-mediated CD8 T cell immunity and drive resistance to immunotherapy. Consistent with this, our analysis of patient biospecimens showed decreased T cell and DC infiltration in KEAP1-mutant tumors. Using CRISPR-Cas9 editing, we confirmed that NRF2, one of the main KEAP1 binding partners, promotes immune evasion in Keap1-mutant tumors. Finally, we demonstrated that combining glutaminase inhibition with ICB effectively reverses this immunosuppression and sensitizes this LUAD subtype to immunotherapy. Taken together, our study reveals how tumor-intrinsic KEAP1 mutations subvert anti-tumor immune responses and opens a new therapeutic avenue of combining metabolic modulators targeting glutamine metabolism and ICB therapies for malignancies characterized by these mutations.
RESULTS
Keap1 mutation accelerates tumor growth in antigenic and autochthonous mouse models of lung adenocarcinoma
Given that previous studies have implicated tumor-intrinsic KEAP1 mutations in failure to respond to ICB,36–40 we sought to examine the impact of KEAP1 mutations on cancer growth and immune surveillance. We generated an orthotopic transplant model of KrasG12D/+; p53−/− (KP) lung cancer42 with cells expressing either a wild-type or a dominant-negative form of Keap1 (Keap1 R470C), the most frequently listed mutation of KEAP1 in the COSMIC database (Figures 1A and S1A).28,43 Because earlier work has demonstrated that Kras-driven mouse LUAD models do not elicit robust T cell responses and do not respond to immune checkpoint inhibition therapy,44–48 we elected to establish a Y chromosome-driven antigenic mouse model of LUAD. We utilized gene-targeted male tumor cells (Figure S1B) and introduced them orthotopically into immunocompetent female hosts (Figure 1A). We hypothesized that the presence of Y chromosome antigens will elicit an antigenic response in female recipients as previously shown in the context of lymphoma and liver cancer.49,50 We validated our H-Y-driven system by ensuring that male KP tumor cells trigger an immune response in females but not in males. Indeed, KP cells grew rapidly in male mice, while tumor growth was reduced in female hosts (Figure 1B). We then performed antibody-mediated depletion of CD4 or CD8 T cells in female and male hosts. Depletion of either CD4 or CD8 T cells resulted in increased tumor burden in female (Figure 1C), but not in male mice (Figure 1D).
Figure 1. Loss of Keap1 promotes immune evasion and accelerated tumor growth in a novel antigenic model of LUAD.
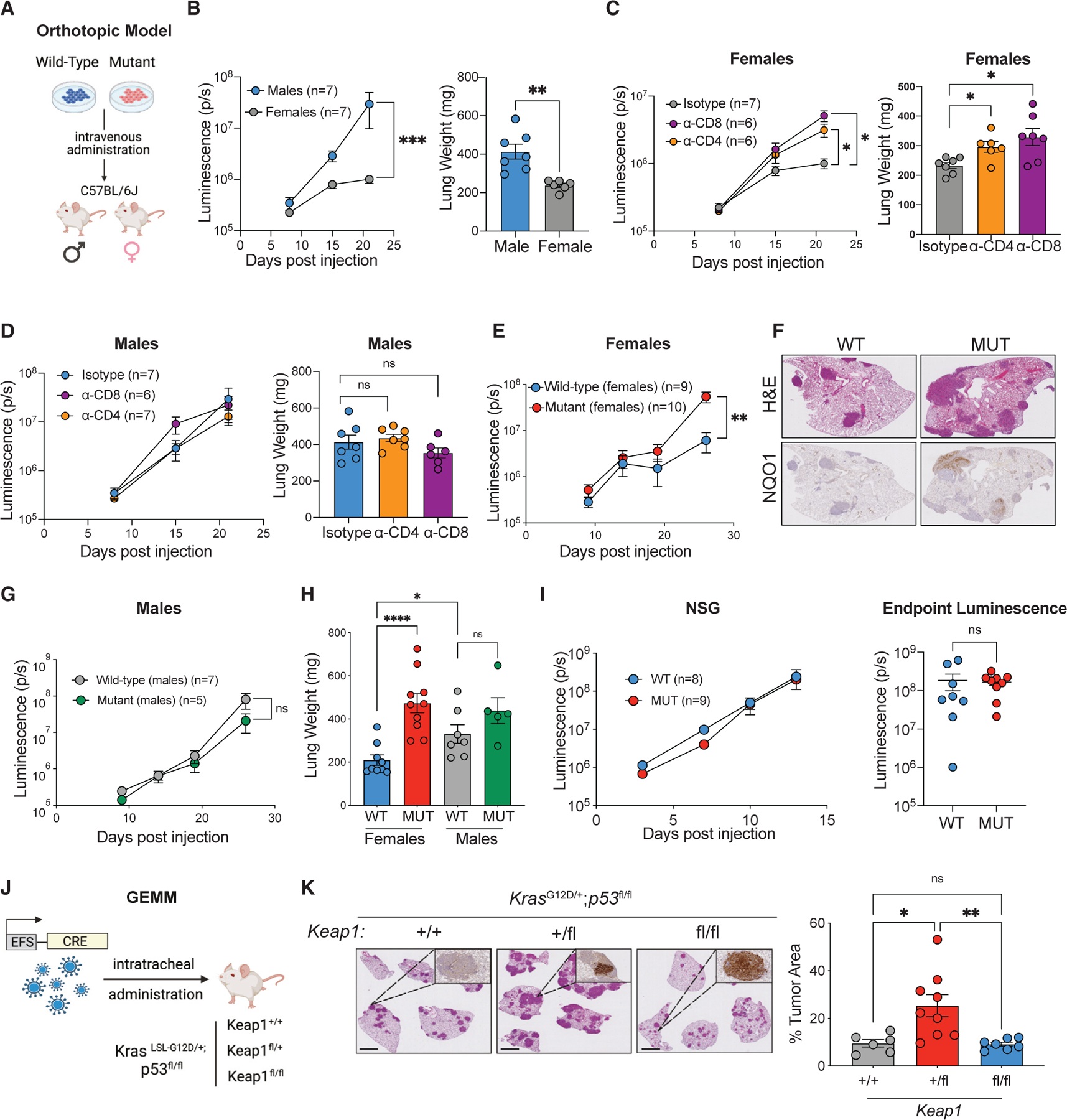
(A) Schematic of our antigenic H-Y-driven orthotopic mouse model.
(B) Growth kinetics (left) of male KP tumors established in female or male hosts with endpoint lung weights (right).
(C) Growth kinetics (left) of male KP cells grown in female hosts following antibody-mediated depletion of CD4 or CD8 T lymphocytes with endpoint lung weights (right).51
(D) Growth kinetics (left) and endpoint lung weights (right) of male KP cells injected into male hosts following antibody-mediated depletion of CD4 or CD8 T lymphocytes.
(E) Growth kinetics of Keap1 wild-type and mutant KP cells in females.
(F) Representative images of H&E and NQO1 immunohistochemical staining of Keap1 wild-type and mutant KP tumors in female hosts.
(G) Growth kinetics of Keap1 wild-type and mutant KP cells injected into male hosts.
(H) Lung weight measured on day 26 in female and male mice bearing Keap1 wild-type and mutant tumors.
(I) Growth kinetics (left) and endpoint luminescence (right) of Keap1 wild-type and mutant male cells injected into immunodeficient (NSG) female mice.
(J) Schematic of KP Keap1+/+, Keap1fl/+, or Keap1fl/fl GEMM mice infected with 20K TU Cre-expressing lentivirus.
(K) Representative H&E and NQO1 immunohistochemical staining of Keap1+/+, Keap1fl/+, and Keap1fl/fl KP tumors and tumor area quantification of mice infected with Cre-expressing lentivirus and sacrificed 3.5 months post infection. Scale bars, 2 mm. Each experimental subgroup had n ≥ 6 mice. Each symbol represents an individual mouse. *p < 0.05; **p < 0.01; ***p < 0.001; ***p < 0.0001.
To investigate the role of Keap1 mutation in modulating antitumor immune responses, we injected Keap1 wild-type and mutant male mouse lung cancer cells into immunocompetent female hosts (Figure 1E). Critically, Keap1-mutant tumors displayed accelerated growth kinetics compared with controls in female mice (Figure 1E) and increased tumor burden measured by histological analysis (Figure 1F). However, no difference in tumor growth based on Keap1 mutation was observed in male recipients (Figures 1G, 1H, and S1F). To confirm loss of KEAP1 function in our dominant-negative model, we analyzed NQO1 levels through immunohistochemistry, since Keap1 loss is known to activate the NRF2 pathway.30,52,53 Our staining confirmed that Keap1-mutant tumors expressed higher levels of NQO1, a downstream target of NRF2 (Figure 1F). In line with this, Keap1-mutant cells expressed higher levels of NRF2 target genes (Nqo1, Blvrb, and Slc7a11) in vitro compared with Keap1 wild-type cells (Figure S1C).
In vitro proliferation analysis of Keap1 wild-type and mutant cells showed no differences, suggesting that Keap1 mutation does not have an impact on the intrinsic proliferative capacity of LUAD cells (Figure S1D). To control for Keap1 overexpression, we also generated LUAD cells carrying an empty control vector. The in vitro and in vivo growth of tumor cells transduced with an empty vector was similar to that of cells transduced with Keap1 wild-type vector (Figures S1D–S1F). We hypothesized that if Keap1 mutation promotes tumor growth by dampening immune responses, then Keap1-mutant and wild-type tumors should grow at similar rates in NOD-SCID IL2Rgammanull (NSG) female mice that lack B, T, and natural killer (NK) cells. Indeed, we observed no differences, suggesting that Keap1 mutations elicit faster tumor growth by subverting the anti-tumor immune response (Figure 1I).
We corroborated our findings of the impact of KEAP1 deficiency on tumor growth by evaluating additional LUAD orthotopic models. While the R470C mutation is one the most frequently occurring KEAP1 mutations in LUAD, we wanted to validate that the increased proliferation of KP tumors also occurs upon genetic loss of Keap1. Utilizing the parental male KP cell lines, we generated Keap1-deficient cells (sgKeap1) via CRISPR-Cas9 gene editing. We confirmed increased expression of NRF2 target genes in knockout cells, including Nqo1 (Figure S1G), compared with cells that received the control sgRNA (sgControl). Consistent with the Keap1-mutant R470C cell line, sgKeap1 tumor cells grew significantly faster than their controls (Figure S1H) and tumor-bearing lungs had increased weight, indicative of increased tumor burden (Figure S1I).
We next sought to investigate how Keap1 mutation affects tumor growth and immune surveillance in an autochthonous genetically engineered mouse model (GEMM) of LUAD. We generated KrasLSL-G12D/+; Trp53fl°x/fl°x mice expressing wildtype Keap1 (Keap1+/+), mice heterozygous for the floxed Keap1 conditional allele (Keap1fl/+), or animals homozygous for the conditional allele (Keap1fl/fl) (Figure S1J). Intratracheal administration of Cre-expressing lentivirus leads to the induction of LUAD tumors in these animals. Tumor burden quantification based on histological analysis revealed that partial loss of Keap1 resulted in increased tumor burden (Figure 1K). Interestingly, mice in which Cre-mediated deletion resulted in homozygous loss of Keap1 had similar tumor burden compared with animals with a wild-type Keap1 allele (Figure 1K), which is consistent with prior studies53 and data from human tumors suggesting that more aggressive tumors first acquire a missense or truncating mutation in one allele and then undergo loss of heterozygosity.26,27 Immunohistochemical staining for NQO1 displayed the expected gradient, where Keap1+/+ tumors had little to no staining, Keap1fl/+ tumors had focal staining, and Keap1fl/fl were strongly positive (Figure 1K).
To investigate the mechanisms by which Keap1-mutant lung tumors could drive immune evasion, we sorted tumor cells from the lungs of immunocompetent female mice with KP tumors carrying a wild-type or Keap1-mutant allele and performed transcriptional profiling by RNA sequencing (RNA-seq). We found that 655 genes were upregulated, while 1,108 were downregulated in mutant compared with wild-type cells (Figure S2A). Pathway enrichment analysis revealed that NRF2-regulated pathways were upregulated and interferon pathways were the top downregulated pathways in Keap1-mutant tumor cells (Figure S2B). Interferon production has been shown to be critical for activation of cross presentation of tumor antigens by DCs to CD8 T cells, as well as for T cell recruitment into the tumor microenvironment.15,54,55 Dysregulated chemokine signaling may contribute to immune evasion, as chemokines are essential for migration of both activating and immunosuppressive leukocytes in the TIME.12–14,16,56,57–60 We examined chemokine expression in wild-type and mutant tumor cells. A total of seven chemokines were differentially expressed (Cxcl9, Cxcl10, Cxcl16, Ccl20, Cxcl3, Cxcl5, and Cxcl15), with all of them being expressed at lower levels in Keap1-mutant compared with wild-type tumor cells (Figure S2C). The reduced expression of these chemokines can contribute to diminished infiltration of key anti-tumor immune cells.
To examine whether genotype-specific differences were affecting the ability of tumor cells to produce interferons (IFNs), we stimulated cells with poly(dA:dT), poly(dG:dC), or IFN-α. Poly (dA:dT) is able to stimulate both DNA and RNA sensors, while poly(dG:dC) specifically stimulates DNA sensors. IFN-α binds to IFN receptors on the surface of tumor cells and activates the JAK-STAT pathway and subsequent IFN production. We treated Keap1 wild-type, Keap1-mutant, and wild-type cells pretreated with the small molecule NRF2 activator Ki-696 with stimulators of IFN signaling and assessed the expression of type I IFN genes using quantitative real-time PCR (Figure S2D). We observed that Keap1 mutation or hyperactivation of the NRF2 pathway did not have an impact on the ability of tumor cells to produce type I IFNs and had no apparent impact on the IFN-stimulated genes. This result suggests that the diminished IFN response gene signature observed in the RNA-seq analysis of ex vivo isolated Keap1-mutant tumors could be specific to the altered TIME of Keap1-mutant tumors, rather than tumor-intrinsic upregulation of IFN response pathways. To investigate whether altered type I IFN signaling influences Keap1-mutant tumor growth in vivo specifically, we blocked IFN signaling using an anti-IFN-α/β receptor (anti-IFNAR) antibody.54 After tumor engraftment, administration of anti-IFNAR antibody drastically accelerated the growth of Keap1 wild-type tumors (Figure S2E). Remarkably, blockade of type I IFN signaling had no impact on the growth dynamics of Keap1-mutant tumors (Figure S2E). These findings suggest that dampened IFN signaling within the TIME is one of the mechanisms by which Keap1-deficient tumors evade immune surveillance.
Keap1 mutation prevents CD103 DC accumulation and activation in lung adenocarcinoma tumors
To investigate how Keap1 mutation alters the immune microenvironment, we performed single-cell RNA-seq (scRNA-seq) of wildtype and mutant lung tumors. We sorted lung-infiltrating immune cells (while excluding circulating leukocytes using intravenous injection of fluorescently tagged CD45 antibody). We identified 14 clusters in our scRNA-seq analysis representing major immune cell lineages with granulocyte, T lymphocyte, B cell, macrophage, and DC subsets being the most abundant populations (Figures S3A–S3D). Given the critical roles of DCs and T cells in anti-tumor immunity, we wanted to investigate how Keap1 mutation affects DC-mediated T cell immune surveillance. Tissue-resident DCs consist of two subsets: Batf3-dependent CD103/CD8 DCs specialized in cross presenting antigens to CD8 T cells61 and CD11b DCs, which drive CD4 helper T cell responses.62,63 CD103 DCs are essential for anti-tumor immune responses in multiple preclinical mouse models, including the KP lung cancer model.15–19,47,54,55,64 To get a more detailed look at TIME-associated DC populations, we further subclustered the DCs into six populations (Figure 2A). The CD103 DC cluster expressed the canonical genes Itgae, Xcr1, Clec9a, and Naaa, while CD11b DC characteristic transcripts included Itgam, Mgl2, and CD209a.65 In addition to bona fide CD103 and CD11b DC genes, the proliferating CD103 and CD11b DC clusters also expressed the cell-cycle and proliferation markers Birc5, Mki67, Top2a, Stmn1, Cks1b, Hist1h2ae, Hist1h1b, and Hist1h2ap. The mreg DC cluster expressed Ccr7, Fscn1, and Il4i1, as previously shown.47 pDCs expressed the canonical genes Bst2, Siglech, Ccr9, Ly6d, and Klk1 (Figure S3E).66 Our scRNA-seq analysis of Keap1-mutant tumors revealed decreased numbers of CD103 DCs, increased frequency of pDCs, and modestly increased CD11b and mreg-DC numbers (Figures 2B and 2C).
Figure 2. Absence of CD103 DC-mediated anti-tumor immune responses in Keap1-mutant tumors.
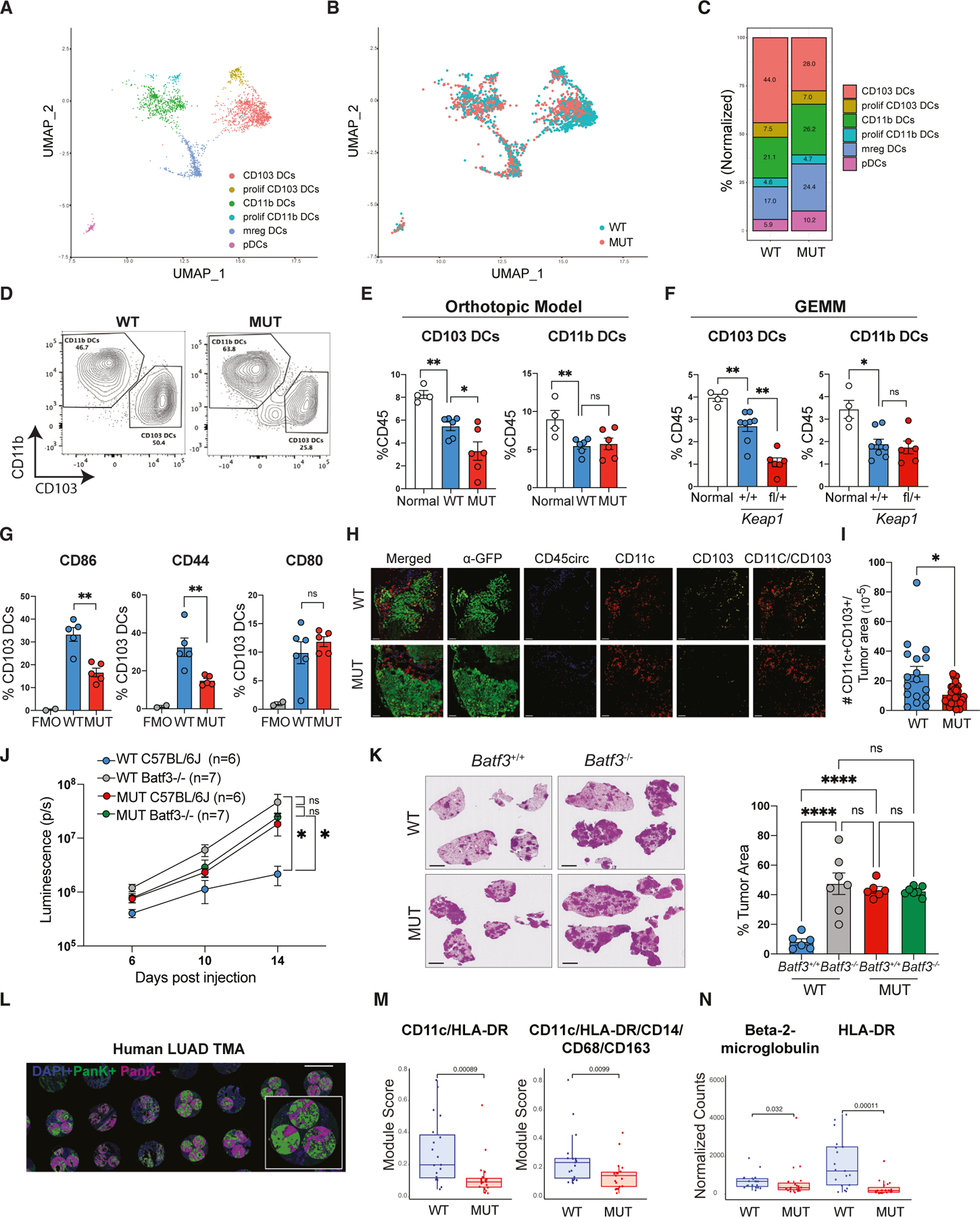
(A) UMAP of DC subclusters identified by scRNA-seq colored by cell type.
(B) UMAP of the distribution of DC subclusters in Keap1 wild-type (WT, blue) and mutant (MUT, red) lung tumors.
(C) Bar plot showing distribution of the DC clusters in Keap1 wild-type and mutant mouse lung tumors.
(D) Representative flow cytometry plots for CD103 versus CD11b within a CD11c+MHCII+ DC gate.
(E) Percentage of CD103 and CD11b DCs out of total tissue-infiltrating immune cells (CD45+CD45circ−) in normal (non-tumor) lung and lungs with orthotopic Keap1 wild-type and mutant tumors.
(F) Percentage of CD103 and CD11b DCs out of total tissue-infiltrating immune cells (CD45+CD45circ−) in normal (non-tumor) lung and lungs with autochthonous genetically engineered mouse model (GEMM) Keap1 wild-type and mutant tumors.
(G) Percentage of CD86, CD44, and CD80 among CD103 DCs in wild-type and mutant Keap1 tumors. FMO (fluorescence-minus-one) control shown for all markers.
(H) Confocal images of lung tumor sections from Keap1 wild-type and mutant KP tumors. GFP (tumors) is shown in green, circulating CD45+ cells are shown in blue, CD11c is shown in red, and CD103 is shown in yellow. Scale bars, 100 μm. Images are representative of individual tumors from ≥5 mice per genotype.
(I) Quantification of tissue-infiltrating CD103 DCs (CD11c+CD103+CD45circ−) in tumor (GFP+) areas. Each symbol represents an individual tumor.
(J) Growth kinetics of Keap1 wild-type and mutant lung orthotopic tumors in C57BL/6J and Batf3−/− female mice measured by bioluminescence.
(K) Quantification of tumor burden (tumor area/total lung area) of Keap1 wild-type and mutant tumors in C57BL/6J and Batf3−/− based on H&E staining. Scale bars, 2 mm. Each experimental subgroup had at least n = 6 mice.
(L) Representative image of the LUAD tissue microarray. Scale bars, 1 mm. Three regions of interest per tumor sample were quantified. Pancytokeratin-positive staining is shown in green, negative is shown in magenta, and DAPI is shown in blue.
(M) Boxplots showing expression of CD11c/HLA-DR and CD11C/HLA-DR/CD14/CD68/CD163 protein modules in KEAP1 wild-type (n = 19) and mutant (n = 19) patient tumor samples from tumor microarray containing LUAD patient samples. The microarray was stained and analyzed using the Nanostring GeoMx platform. Expression is shown for pancytokeratin-positive areas.
(N) Boxplots showing expression for individual proteins: β-2-microglobulin and HLA-DR. Each symbol represents an individual mouse or human tumor core. *p < 0.05; **p < 0.01; ****p < 0.0001.
Using our flow cytometry strategy, we were able to distinguish CD103 DCs and CD11b DCs (Figure S4A). We found fewer CD103 DCs in Keap1-mutant tumors compared with wild-type tumors, while CD11b DCs remained unchanged (Figures 2D, 2E, and S4B). Consistent with the immunosuppressive role of Keap1 mutation, there was no difference in CD103 DC infiltration between wild-type and mutant tumors in the less antigenic setting of male hosts (Figure S4C). When examining our other LUAD models, CD103 DC infiltration was lower in the Keap1-knockout tumors (Figure S4D) compared with controls, as well in the autochthonous model comparing Keap1fl/+ with Keap1+/+ tumors (Figure 2F). Together, these findings indicate that tumor-intrinsic loss of Keap1 leads to diminished CD103 DC infiltration. When we examined markers of DC activation, we observed that CD103 DCs from Keap1 wild-type tumors displayed higher levels of co-stimulatory molecules such as CD44 and CD86 compared with DCs from mutant tumors (Figure 2G). To examine the spatial distribution of CD103 DCs in Keap1 wildtype and mutant tumors, we conducted multi-color immunofluorescence staining. We observed that CD103 DCs were primarily found in the tumor periphery (Figure 2H). Analysis of Keap1 wildtype tumors revealed notably increased intratumoral infiltration (Figure 2H) and higher overall CD103 DC numbers (Figure 2H) compared with Keap1-mutant tumors. We reasoned that if Keap1-mutant tumors suppress CD103 DC-mediated anti-tumor immune responses, then Keap1 wild-type and mutant tumors should grow similarly in CD103 DC-deficient Batf3−/− hosts. Analysis of Batf3+/+ and Batf3−/− female C57BL/6J hosts demonstrated that, although mutant tumors displayed accelerated growth in wild-type mice, the difference in growth kinetics and tumor burden between these tumor genotypes was no longer evident in Batf3−/− hosts (Figures 2J and 2K).
We next examined whether human tumors with KEAP1 mutation display signs of immune evasion. To assess infiltration of antigen-presenting cells (DCs) we used Nanostring GeoMx digital spatial profiling. Using a tissue microarray (TMA) containing KEAP1 wild-type and mutant LUAD human samples, we acquired GeoMx data from three different areas of each sample (Figure 2L). We developed protein modules that describe DCs (CD11c/HLA-DR) and mononuclear phagocytes (CD11c/HLA-DR/CD14/CD68/CD163) and evaluated the TMAs in which we distinguished between tumor and adjacent normal tissue based on pankeratin staining (Figures 2L and 2M). We observed a reduction in the expression of both DC modules in KEAP1-mutant tumors compared with patient samples in which the allele had no evident mutations (Figure 2M), consistent with our preclinical mouse model data. Interestingly, KEAP1 mutation status negatively correlated with levels of proteins involved in antigen presentation (β-2-microglobulin and HLA-DR)15,67 (Figure 2N), in accordance with a reduction in DC-associated signatures (Figure 2M). This suggests that genetic inactivation of KEAP1 results in decreased DC infiltration in both mice and humans.
Keap1 mutation suppresses CD103 DC-mediated CD8 T cell immunity
CD103 DCs are responsible for taking up dead tumor cells and cross priming CD8 and CD4 T lymphocytes.18,61,68,69 Furthermore, they can also promote recruitment and local expansion of CD8 T lymphocytes and support their effector function.70 We thus sought to determine whether reduced number and functionality of CD103 DCs observed in Keap1-mutant tumors was driving defective T cell responses. We identified 10 phenotypically distinct populations of T cells in our scRNA-seq dataset based on gene expression profiles (Figure 3A). Based on patterns of gene expression, we identified CD8 and CD4 naive cells (Sell+Ccr7+Dapl1+Igfbp4+) and early activated CD8 and CD4 T cells characterized by intermediate expression of Sell, Ccr7, and Lef1 and upregulation of activation markers common to both CD4 and CD8 T cells (Ccl5 and Nkg7) or unique to either CD8 (Klrk1, Xcl1, Cd7, and Ly6c2) or CD4 T cells (Cd28) (Figure S5A). We also observed effector/exhausted CD4 and CD8 T cells that robustly expressed markers of exhaustion (Pdcd1, Tigit, Lag3, and Bhlhe40) (Figures 3A and S5A). Another cluster was a CD8 population that appeared to be a cell state between early activated and exhausted cells, as it expressed markers of activation but had no notable expression of exhaustion markers. There were two additional CD4 clusters that appeared to be T regulatory cells (Il2ra, Ctla4, Klrg1, Foxp3, and Tnfrsf4) and Th1 cells (Vim, Ahnak, Id2, Crip1, Lgals1, and S100a4) based on gene expression profiles (Figures 3A and S5A).71,72
Figure 3. Keap1-mutant tumors suppress CD8 T cell responses and promote T cell exhaustion.
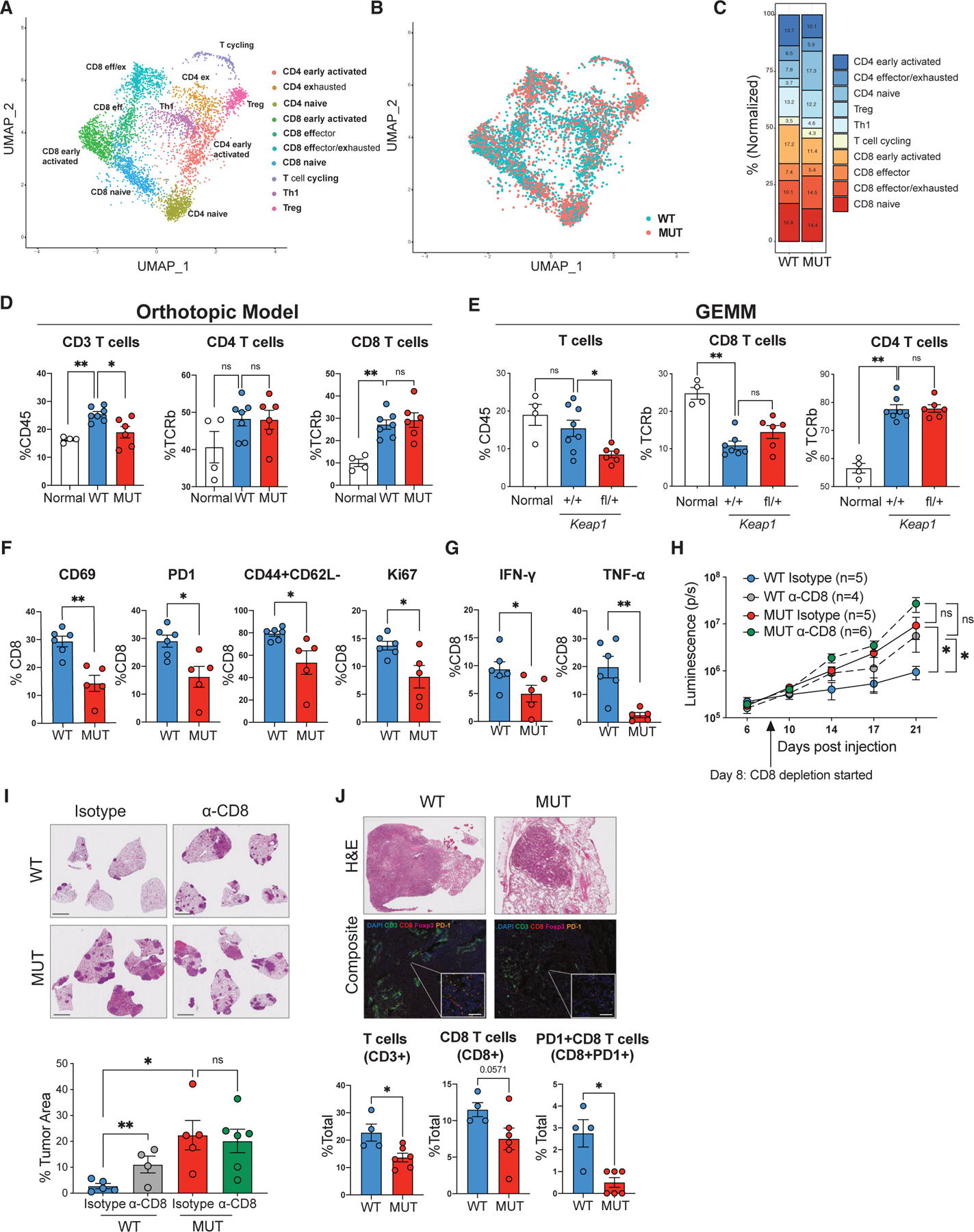
(A) UMAP of T cell subclusters identified by scRNA-seq, colored by cell type.
(B) UMAP of the distribution of T cell subclusters in Keap1 wild-type (WT, blue) and mutant (MUT, red) lung tumors.
(C) Bar plot showing distribution of T cell clusters in Keap1 wild-type and mutant mouse lung tumors.
(D) Percentage of CD3, CD4, and CD8 T cells among tissue-infiltrating immune cells (for CD3) or of tissue-infiltrating TCRβ+ T cells in non-tumor lung (normal) and lung with Keap1 wild-type or mutant orthotopic tumors. Each symbol represents an individual mouse. Each experimental subgroup had n ≥ 4 mice.
(E) Percentage of CD3, CD4, and CD8 T cells among tissue-infiltrating immune cells (for CD3) or of tissue-infiltrating TCRβ+ T cells in non-tumor lung (normal) and lung with Keap1 wild-type or mutant GEMM tumors. Each symbol represents an individual mouse. Each experimental subgroup had n ≥ 4 mice.
(F) Percentage of CD69, PD1, CD44+CD62L−, or Ki67 among CD8+ T cells in wild-type and mutant Keap1 tumors. Each symbol represents an individual mouse. Each experimental subgroup had n ≥ 5 mice.
(G) Percentage of intracellular IFN-γ- or TNF-α-positive cells among the CD8+ T lymphocytes in wild-type and mutant Keap1 tumors. Each symbol represents an individual mouse. Each experimental subgroup had n ≥ 5 mice.
(H) Growth kinetics of Keap1 wild-type and mutant tumors measured by bioluminescence in female hosts upon CD8 T cell depletion.
(I) Representative images of lung tumor burden and quantification by H&E staining. Scale bars, 2 mm.
(J) Representative H&E (top) and immunofluorescence (bottom) images of KEAP1 wild-type and mutant human tumors. DAPI is shown in blue, CD3 in green, CD8 in red, Foxp3 in magenta, and PD1 in orange. Scale bars, 20 μm. Quantification of T cells (CD3+), CD8 T cells (CD8+), and PD1+ CD8 T cells (CD8+PD1+) in KEAP1 wild-type and mutant human tumors is shown at the bottom. Tumor area was identified based on H&E staining. *p < 0.05; **p < 0.01.
Keap1 wild-type tumors had a notable enrichment of both CD4 and CD8 early activated cells (Figures 3B and 3C), consistent with CD103 DCs facilitating T cell activation.16,19,20,54,55 Keap1-mutant tumors had higher numbers of CD8 exhausted T cells. In line with a more immunosuppressive TIME of mutant tumors, wild-type tumors had more Th1 cells, while Keap1-mutant tumors were enriched in T regulatory cells (Tregs) (Figures 3B and 3C). Reconstruction of the T cell receptor (TCR) repertoire from our scRNA-seq data revealed that lungs with Keap1-mutant and wild-type tumors were infiltrated with clonally expanded T cells, suggesting antigen-specific responses (Figure S5B). Notably, while the CD8 activated/exhausted T cell cluster was characterized by clonal T cells in both tumor genotypes, Keap1-mutant tumors were characterized by the presence of clonally expanded Tregs.
Flow cytometric analysis (gating strategy in Figure S6A) revealed an overall reduction in T cell infiltration in the lungs of mice with Keap1-mutant tumors compared with lungs of animals with KP tumors (Figures 3D, 3E, and S6B). The difference in number of recruited T cells appears to be driven by a decrease in both CD4 and CD8 T cell subsets in Keap1-mutant tumors (Figures 3D and 3E). CD8 T cells infiltrating Keap1-mutant tumors expressed lower levels of activation (CD69, PD1, and CD44) and proliferation (Ki67) markers (Figure 3F). Importantly, we observed fewer CD8 T cells producing the effector cytokines IFN-γ and TNF-α critical for tumor control in mutant tumors (Figure 3G). Consistent with the Keap1 R470C mutant model, we found that Keap1-knockout tumors also had reduced activated CD8 T cells and diminished proliferation of CD8 lymphocytes (Figure S6C). Furthermore, CD8 T cells from the sgKeap1 condition also had blunted IFN-γ and TNF-α production compared with wild-type KP tumors (Figure S6D).
Examining CD4 T cells of wild-type and mutant orthotopic tumors, we found no significant differences in their activation or proliferation. However, mutant tumors had fewer cytokine-producing CD4 T cells (Figures S6E–S6G). Interestingly, CD4 T cells were skewed toward Tregs and away from a pro-inflammatory Th1 cell phenotype in Keap1-mutant tumors, consistent with the immunosuppressive role of this mutation (Figure S6H). Strikingly, there was no difference in total T cells or frequency of Tregs in the less antigenic setting of male hosts (Figure S6I).
To further investigate whether Keap1 mutation inhibits CD8 and/or CD4 anti-tumor immunity, we performed antibody-mediated depletion of CD8 and CD4 T cells to examine what role Th and cytotoxic T cells play in tumor immune surveillance. Depletion of CD8 T cells resulted in accelerated growth (Figure 3H) and increased tumor burden of Keap1 wild-type tumors without affecting the growth of Keap1-mutant tumors (Figure 3I). This suggests the presence of active CD8 T cell immune surveillance in wild-type, but not Keap1-mutant, tumors. Depletion of CD4 T cells resulted in accelerated growth of both wild-type and mutant tumors, suggesting that there is active CD4 T cell immune surveillance in both tumor genotypes (Figures S6J and S6K).
We next examined whether human tumors with KEAP1 mutation display defects in T cell immune surveillance. We previously identified KEAP1-mutant LUAD tumors using targeted exome capture and validated these results via NQO1 staining.30 Using multiplex immunofluorescence, we found that KEAP1 wild-type tumors have increased infiltration of total CD3 T cells, CD8 T cells, and PD1-expressing CD8 T cells compared with mutant tumors (Figure 3J). In line with results from our mouse models of this malignancy, we did not observe any significant differences in the CD4 T cell infiltration patient samples irrespective of their KEAP1 mutation status (Figure S6L).
In summary, we demonstrated that Keap1-mutant tumors suppress CD8 T cell immune surveillance, promoting T cell exhaustion marked by reduced activation, diminished proliferation, and decreased effector molecule production. Importantly, KEAP1-mutant human tumors also exhibit a similar deficit in CD8, but not CD4, T cell infiltration. Overall, our characterization of the immune landscape in Keap1 wild-type and mutant tumors, along with depletion studies, provides critical insight into how loss of KEAP1 function disrupts the CD103DC-CD8 anti-tumor axis and compromises immune surveillance.
Keap1 mutation impairs response to immune checkpoint blockade
Engagement of CD103 DCs and T cells is a critical determinant of success of immunotherapies in several preclinical models of cancer.18–21 Moreover, recent evidence implicates tumor intrinsic mutations in KEAP1 in immunotherapy resistance in humans.36–40 This prompted us to investigate whether suppression of the DC-CD8 T cell axis by Keap1-mutant tumors can drive resistance to ICB. Despite several efforts to establish genetically defined KRAS-driven LUAD mouse models that induce T cell responses and can respond to ICB therapy,44,45,48,73–75 to the best of our knowledge, none have been shown to respond to ICB therapy.46,48 We took advantage of our H-Y-driven orthotopic mouse model to study how Keap1-mutation subverts anti-tumor immune responses to ICB.
To investigate immune responses and tumor growth kinetics of wild-type and mutant tumors following immunotherapy, we injected wild-type and mutant male KP cell lines orthotopically into C57BL/6J female mice (Figure 4A). After engraftment, we treated mice with anti-PD1 monoclonal antibody (clone 29F.1A12) or isotype control. We selected this clone following testing of different anti-PD1 clones in an MC38 model (Figures S7A and S7B). Since higher tumor burden can promote resistance to ICB,76 we initiated anti-PD1 treatment when both genotypes had equivalent tumor burden (Figure S7C). Overall, Keap1 wild-type tumors responded better to anti-PD1 treatment than mutant tumors (Figures 4A–4C, S7D, and S7E). These results validate the clinical studies and confirm that the central role of KEAP1 in resistance to ICB.36,37 We did not observe changes in the numbers of T lymphocytes or DC populations (Figures 4D and 4E) upon anti-PD1 treatment. However, immunotherapy treatment resulted in increased CD8 T cell proliferation irrespective of Keap1 status (Figure 4F). We then investigated the production of effector molecules by T lymphocytes. We observed increased expression of IFN-γ and TNF-α in both CD4 and CD8 T cells in Keap1 wild-type but not mutant tumors (Figures 4G and S7F). These results indicate that, although antibody treatment promotes proliferation of CD8 T cells in both genotypes, anti-tumor T cell responses are blunted in Keap1-mutant tumors.
Figure 4. Keap1 mutation drives immunotherapy resistance in antigenic Kras-driven lung adenocarcinoma mouse model.
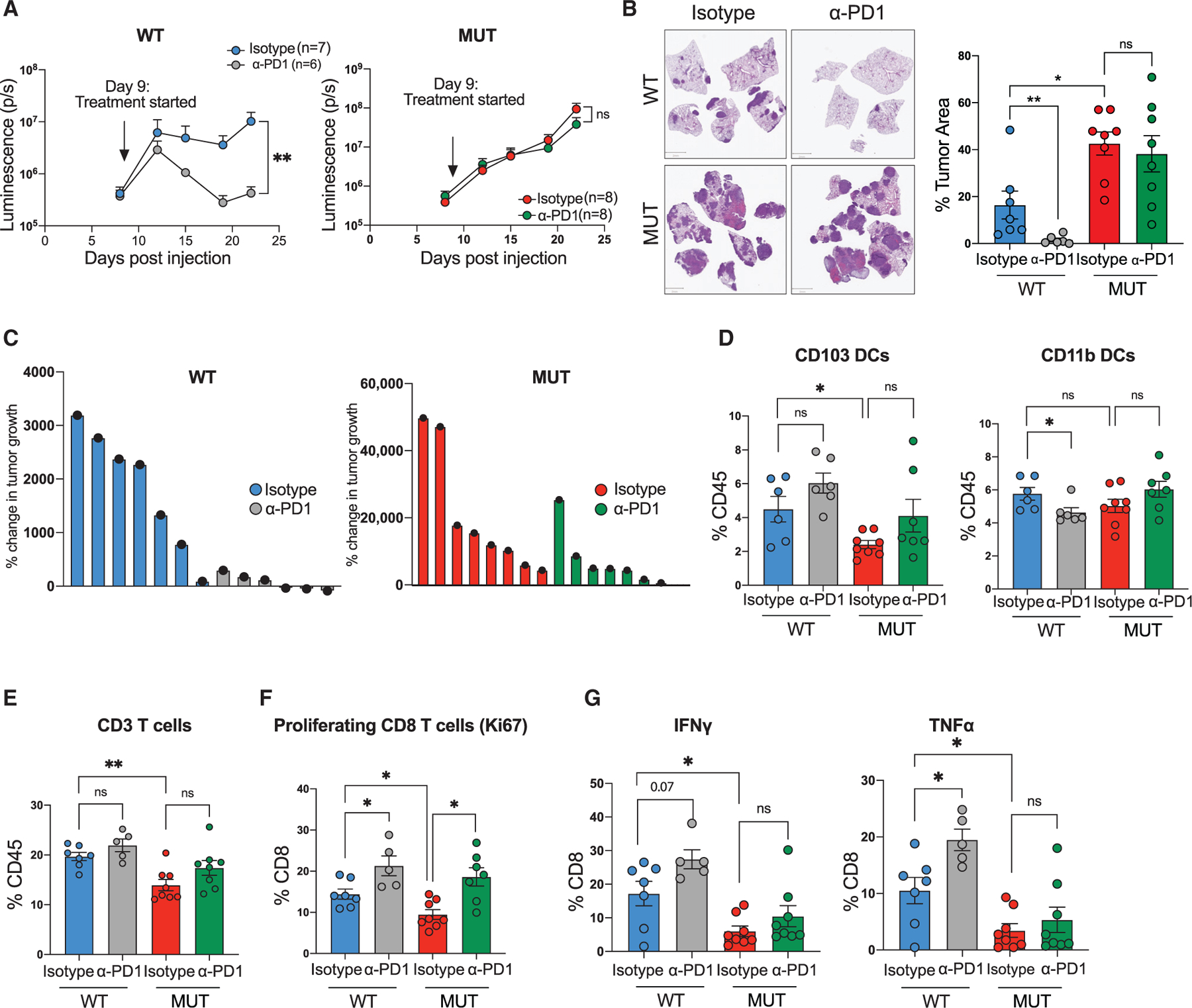
(A) Growth kinetics of Keap1 wild-type (WT, left) and mutant (MUT, right) KP cells injected intravenously in female C57BL/6J hosts upon treatment with anti-PD1 monoclonal antibody or isotype control. Each experimental subgroup had n ≥ 6 mice.
(B) Representative images of lung tumor burden and quantification (tumor area/total lung area) by H&E staining of Keap1 wild-type and mutant orthotopic tumors treated with either isotype or α-PD1. Scale bars, 2 mm. Each experimental subgroup had n ≥ 6 mice.
(C) Waterfall plots showing the percentage of tumor growth of Keap1 wild-type and mutant lung tumors treated with isotype control or anti-PD1.
(D) Percentage of CD103 and CD11b DCs among tissue-infiltrating immune cells in the lungs of animals with Keap1 wild-type or mutant tumors treated with anti-PD1 or isotype control.
(E) Percentage of CD3+ lymphocytes among tissue-infiltrating immune cells. Each experimental subgroup had n ≥ 5 mice.
(F and G) Percentages of Ki67+ cells (F) or intracellular IFN-γ- and TNF-α-positive cells within the CD8 T cell gate in Keap1 wild-type and mutant tumor-bearing mice treated with anti-PD1 or isotype control. Each experimental subgroup had at least n = 5 mice. Each symbol represents an individual mouse. *p < 0.05; **p < 0.01.
NRF2 signaling pathway promotes immune evasion in Keap1-mutant tumors
Although KEAP1 binds with many proteins, cancer genome sequencing revealed that mutations in KEAP1 and NRF2 genes are mutually exclusive. This implies that NRF2 hyperactivation is primarily responsible for tumorigenic effects of a mutated KEAP1/NRF2 pathway.77 Given that several lines of evidence point to the immunoregulatory role of the NRF2 antioxidant pathway,78–82 we performed functional studies to determine whether NRF2 is the major substrate of KEAP1 that promotes immune evasion. Using CRISPR-Cas9 editing and two different guides against Nrf2, we generated Nrf2-deficient (Nrf2−/−) Keap1-mutant KP cells. Both NRF2 and its downstream target gene Hmox1 showed decreased protein levels in Nrf2−/− Keap1-mutant cells, similar to the Keap1 wild-type cells (Figure S8A). Other NRF2 target genes (Nqo1, Blvrb, and Slc7a11) were also downregulated, confirming successful reversal of NRF2 hyperactivation in Keap1-mutant cells (Figure S6B). In vitro proliferation of Nrf2-deficient cells was unchanged compared with Nrf2 wild-type cells (Figure S8C), indicating that Nrf2 loss does not have an impact on tumor-intrinsic Keap1-mutant cell proliferation.
To investigate whether the NRF2 pathway is responsible for driving immune evasion in Keap1-mutant tumors, we injected male Keap1-mutant cells, wild-type (sgTom) cells, or cells deficient for Nrf2 into female mice (Figure 5A). We found that Keap1-mutant tumors lacking Nrf2 grew considerably slower compared with Nrf2-sufficient Keap1-mutant tumors (Figure 5A). Flow cytometric analysis revealed that downregulation of the NRF2 antioxidant pathway in Keap1-mutant tumors resulted in increased infiltration of CD103 DCs, while CD11b DCs remained unchanged (Figure 5B). Importantly, although we did not observe any changes in the number of total T cells, both CD8 and CD4 T cells infiltrating Nrf2-deficient Keap1-mutant tumors had increased cytokine production (IFN-γ and TNF-α) compared with Keap1-mutant tumors with a hyperactive NRF2 pathway (Figures 5C and S8D). These data demonstrate that tumorintrinsic hyperactivation of the NRF2 pathway impairs accumulation of CD103 DCs, thereby subverting immune surveillance by T lymphocytes.
Figure 5. NRF2 pathway drives immune evasion in Keap1-mutant tumors, and glutamine modulation synergizes with ICB to reverse immunosuppression and immunotherapy resistance.
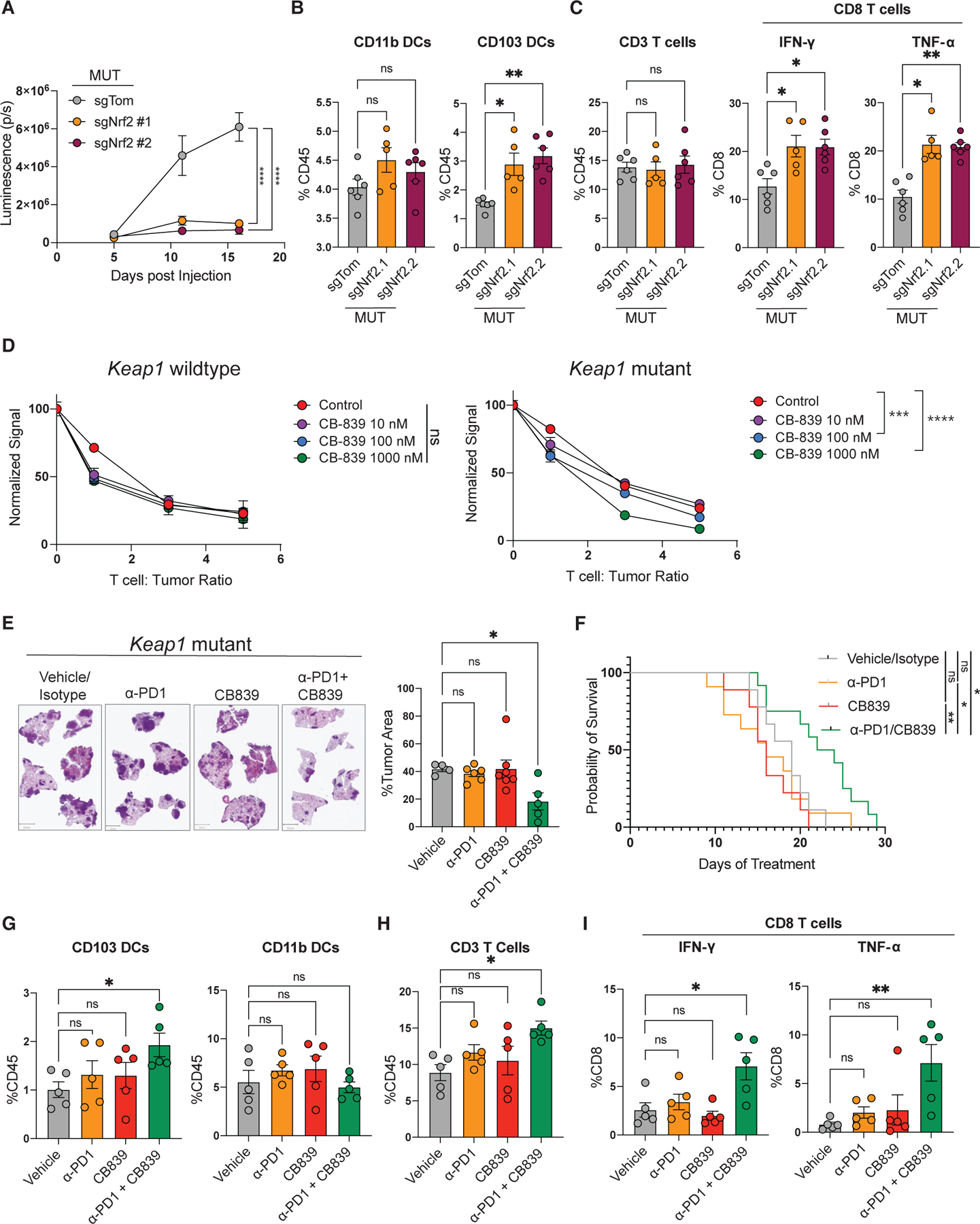
(A) Growth kinetics of Nrf2-wild-type and Nrf2-deficient Keap1-mutant KP cells injected intravenously into female C57BL/6J hosts measured by bioluminescence.
(B) Percentage of CD103 and CD11b DCs among tissue-infiltrating immune cells in the lungs of animals with wild-type or Nrf2-deficient Keap1-mutant tumors.
(C) Percentage of CD3 lymphocytes among tissue-infiltrating immune cells and percentage of intracellular IFN-γ- and TNF-α-positive cells among the CD8 T lymphocytes. Each experimental subgroup had n ≥ 5 mice.
(D) Activated OTI CD8 T cells were co-cultured with LucOS-expressing Keap1-mutant and wild-type KP cell lines in the presence of CB-839 (n = 3). After 24 h, the bioluminescent signal was measured and plotted normalized to the zero T cell condition.
(E) Representative images of lung tumor burden and quantification by H&E staining of Keap1-mutant tumors treated with vehicle/isotype control, α-PD1, CB-839, or combination of α-PD1 and CB-839. Histological samples were collected from a sample other than those used for survival studies, and all tissues came from the same time point. Scale bars, 2 mm.
(F) Survival of C57BL/6J female mice injected with Keap1-mutant KP cells and treated with vehicle/isotype control, α-PD1, CB-839, or a combination of α-PD1 and CB-839, with n ≥ 11.
(G) Percentage of CD103 and CD11b DCs among tissue-infiltrating immune cells in the lungs of animals with Keap1-mutant tumors treated with vehicle/isotype control, α-PD1, CB-839, or a combination of α-PD1 and CB-839.
(H) Percentage of CD3+ lymphocytes among tissue-infiltrating immune cells.
(I) Percentage of intracellular IFN-γ- and TNF-α-positive cells among the CD8 T lymphocytes infiltrating the lungs of animals with Keap1-mutant tumors treated with vehicle/isotype control, α-PD1, CB-839, or a combination of α-PD1 and CB-839. Each symbol in the bar plots represents an individual mouse. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Glutamine blockade synergizes with immunotherapy to reverse immunosuppression and resistance in Keap1-mutant tumors
We previously showed that a hyperactive NRF2 pathway in Keap1-mutant tumors drives significant metabolic rewiring that makes these tumors dependent on exogenous glutamine.30,83 Importantly, Keap1-mutant cells are highly sensitive to CB-839, a glutaminase inhibitor, which blocks the conversion of glutamine to glutamate.30,83–85 Since glutamine availability is critical for effector T cell function, we hypothesized that targeting the NRF2-driven metabolic rewiring of Keap1-mutant tumors using CB-839 may enhance anti-tumor T cell responses. We modeled this in vitro by examining T cell killing of tumors in the presence of CB-839. We generated Keap1 wild-type and mutant cell lines expressing LucOS, which contains luciferase fused to T cell antigens.44,73 These cells were then co-cultured with different ratios of activated OTI T cells with varying doses of CB-839, and viability was measured by bioluminescence after 24 h. Addition of CB-839 did not enhance T cell-mediated killing of Keap1 wild-type tumor cells (Figure 5D). However, in a dose-dependent manner, CB-839 significantly increased the killing of Keap1-mutant tumors by OTI T cells (Figure 5D), demonstrating that inhibiting glutaminase can augment anti-tumor T cell function.
Given the enhancement in T cell-mediated killing by CB-839 in vitro, we then asked whether CB-839 can sensitize Keap1-mutant tumors to anti-PD1 treatment. We treated orthotopic Keap1-mutant tumors with CB-839 and monoclonal α-PD1 antibody alone or in combination. Interestingly, we observed that the combination of CB-839 with α-PD1 was more effective compared with single agents or vehicle control, resulting in decreased tumor burden and extended survival (Figures 5E and 5F). KP cells expressing a Keap1 dominant-negative mutant were not responsive to CB-839 in vivo, in contrast to prior studies using Keap1-null cells,30,83 which could be the result of CB-839 acting on various immune cell populations that have been shown to have diverse metabolic programs and differential sensitivity to glutamine metabolism modulation.86–89 Consistent with this idea, it is important to highlight that those prior in vivo CB-839 studies were conducted using non-immunogenic tumor cells or immunodeficient mouse models.30,83,84
Finally, we wanted to assess whether treatment of α-PD1 in combination with CB-839 is sufficient to reverse the immunosuppressive changes driven by Keap1-mutant tumors. Combination therapy resulted in a robust increase in CD103 DCs, while CD11b DCs remained unchanged (Figure 5G). We then examined whether augmented CD103 DC infiltration could promote a T cell-mediated anti-tumor immune response. Interestingly, we observed increased infiltration of T lymphocytes (Figure 5H) and enhanced expression of the effector molecules IFN-γ and TNF-α in CD8 and CD4 T cells (Figures 5I and S6D). On the other hand, treatment with either CB-839 or α-PD1 alone did not reverse the immunosuppressive TIME state established by Keap1-mutant tumors (Figures 5G–5I). These findings pave the way for a new therapeutic approach that combines metabolic modulators targeting glutamine metabolism with ICB therapies to treat lung cancer patients with KEAP1 mutation and a hyperactive NRF2 signaling pathway.
DISCUSSION
Cancer immune evasion is a major obstacle in designing effective anti-cancer therapies.90 Tumors can diminish their immunogenicity through multiple mechanisms: impaired antigen presentation, compromised recruitment and activation of T cells, or an increased presence of suppressive cells and modulators. Emerging clinical data highlight that alterations in KEAP1 are associated with poor prognosis, resistance to multiple therapies including immunotherapy,32,36,37 and reduced T cell infiltration.41 Understanding the mechanisms of KEAP1-dependent immune evasion can pave the way for new immune-based therapeutic interventions. Here we demonstrated that Keap1 loss of function dampens type I IFN signaling and CD103 DC-orchestrated CD8 T cell immunity, thereby driving immunotherapy resistance. Furthermore, analysis of human samples demonstrated that tumor-intrinsic KEAP1 mutation similarly promotes immune evasion in cancer patients. These findings indicate that KEAP1 inactivation in LUAD could function as a key immune evasion mechanism.
Our ex vivo analysis indicates that Keap1 inactivation is associated with reduced IFN and IFN-stimulated chemokine signatures (Figures S2A–S2C), pathways that are essential for anti-tumor immunity.12–14 However, in vitro analysis of tumor lines showed equivalent IFN production in Keap1 wild-type and mutant tumor cells in response to various stimuli (Figure S2D). This result suggests that our in vitro methods may not adequately capture the complexity of the tumor microenvironment (TME). Further in vivo experiments revealed that blocking type I IFN had no impact on Keap1-mutant tumors but did accelerate the growth of Keap1 wild-type tumors (Figure S2E). This points to a suppression of type I IFN signaling, although the precise mechanism remains elusive. Additional studies are needed to fully delineate the role of IFN signaling pathways in KEAP1-mediated immunosuppression and the dynamic interaction of DCs and T cells in the TIME.
T cell infiltration in the TME has been correlated with better prognosis and responses to ICB.9,91 Although DCs were primarily studied for their ability to prime T cells in the lymph nodes, they are now also recognized as key players at the primary tumor site.13,19,70 Here we demonstrated that Keap1-mutant lung tumors are characterized by decreased CD103 DC infiltration and activation (Figures 2D–2I, S4B, and S4C), resulting in diminished CD8 effector T cell responses (Figures 3H and 3I). Our analysis reveals that CD8 T cells from Keap1-mutant tumors appear more exhausted and have lower levels of activation markers, decreased proliferative capacity, and reduced production of effector molecules (Figures 3A–3G).
Since Keap1-mutant tumors drastically modulate the TIME via suppression of CD103 DCs and CD8 T cells, we wanted to investigate whether Keap1 mutation could be driving resistance to ICB. Most immunogenic cancer models rely on the expression of model antigens, such as ovalbumin and LCMV-derived peptides,44,45,73,74 or mismatch repair genetic variants.48,75 Importantly, none of these lung cancer models have been shown to respond to ICB therapy,46,48 hindering studies aimed at investigating immunotherapy responses. We hypothesized that the lack of responses might be driven by the absence of a diverse antigenic landscape. We confirmed that our H-Y-driven antigenic model can drive anti-tumor T cell responses (Figures 1B–1D). Moreover, our examination of the T cell repertoire showed evidence of clonal immune responses within the tumors (Figure S5B). Future studies will be required to define the specific antigens present in these models and to establish the tumor reactivity of the clonal T cells.
Our in vitro assays demonstrated that inhibition of glutaminase with CB-839 can promote the CD8-dependent killing of KEAP1-mutant cancer cells (Figure 5D). These data are supported by prior studies in T cells that demonstrate that inhibition of glutamine metabolism results in enhanced tumor killing.86,92 In the immunogenic LUAD mouse model, glutaminase inhibition can sensitize Keap1-mutant tumors to immunotherapy (Figures 5E and 5F). Given the synergistic benefit we observed between glutaminase inhibition and ICB therapy, an exciting future research direction would be to examine how the rewiring of glutamine metabolism in Keap1-mutant tumors affects anti-tumor immune responses. Our group, along with others, has shown that Keap1-mutant tumors have elevated antioxidant production83,85 and a heightened dependency on exogenous non-essential amino acids such as glutamine and serine.30,83,85 Glutamine88,93,94 and serine95 are essential for T cell activation. Examining the TIME metabolic landscape will give additional insight into how Keap1-mutant tumors are driving immunosuppression.
NRF2, the most well characterized target of KEAP1, has been suggested to transcriptionally regulate immunomodulatory pathways such as chemokine production and the cGAS/STING-induced type I IFN pathway, albeit primarily in an in vitro or non-tumor setting.79,81,82 Given that point mutations within KEAP1 can impair targeting and degradation of multiple ETGE motif-containing substrates (PALB2, MCM3, IKKB, and DPP3),96–99 we sought to investigate whether NRF2 is the KEAP1 primary binding partner promoting immune evasion. Interestingly, we showed that loss of Nrf2 in Keap1-mutant tumors results in delayed tumor kinetics in our immunogenic model and reverses the majority of immunosuppressive phenotypes in the TME. Given the pleiotropic role of the KEAP1/NRF2 pathway in tumorigenesis, it remains to be determined which of the downstream targets of NRF2 is underlying the immune evasion phenotype of KEAP1-mutant tumors.
Finally, our work has important clinical implications. We have demonstrated that KEAP1-mutant LUAD patients display an immunosuppressive phenotype (Figures 2, 3, and 5) and are resistant to ICB (Figure 4). Our group has previously shown that Keap1-mutant tumors are highly sensitive to the glutaminase inhibitor CB-839,30,83,85 which blocks the conversion of glutamine to glutamate. We combined CB-839 glutaminase inhibition with α-PD1 treatment, effectively sensitizing Keap1-mutant tumors to immunotherapy. This combination was more effective in suppressing Keap1-mutant tumor growth than either agent alone. Importantly, the combination therapy reversed the immunosuppressive phenotypes associated with Keap1-mutant tumors. We hope our studies will encourage development of new therapeutic strategies combining ICB with glutamine metabolism inhibitors for patients with KEAP1-mutant tumors that are refractory to the standard-of-care combination of chemotherapy and immunotherapy.
Limitations of the study
Our study defines the impact of Keap1-mutant lung tumors in the suppression of anti-tumor T cell responses. A possible mechanism of immunosuppression is due to depletion of metabolites, such as glutamine, from the TME, which in turn impairs T cell function. One way to elucidate the metabolites involved would be to perform metabolic rescue experiments in the context of in vitro T cell killing assays. We also found that Keap1-mutant tumors grow independent of DC function using Batf3−/− mice, yet it remains unknown if changes in tumor metabolism affect antigen cross presentation. In addition, our study highlights the synergy of CB-839 and anti-PD1 in enhancing anti-tumor T cell responses. Yet, a recent phase 2 trial found no clinical benefit of CB-839 (telaglenastat) combined with standard-of-care immunotherapy in KEAP1-mutant patients. Further studies will assess the efficacy of combining checkpoint inhibitors with newer, broader-acting, glutamine antagonists. Furthermore, while our TCR analysis revealed clonal responses within the tumors, antigen-specific T cell responses to the tumor itself remain unproven. Functional studies of the clonal TCRs identified would be essential to determine the specificity of T cells from the TME toward tumor antigens. Finally, we demonstrated that Keap1-mutant tumors suppress type I IFN signaling using an anti-IFNAR antibody. However, it is unclear whether this suppression occurs in tumor cells or in surrounding immune cells. To understand if tumor-intrinsic IFN signaling is affected, further studies employing IFNAR-knockout mice or tumor-specific knockout of IFNAR are necessary.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Sergei Koralov (sergei.koralov@nyulangone.org).
Materials availability
Plasmids generated are available upon request.
Data and code availability
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD3 | eBioscience | 500A2 |
| anti-mouse CD4 | eBioscience | RM4–5 |
| anti-mouse CD8 | BD bioscience | 53–6.7 |
| anti-mouse CD45 | BD bioscience | 30-F11 |
| anti-mouse CD4 | BD Bioscience | GK1.5 |
| anti-mouse CD44 | eBioscience | IM7 |
| anti-mouse CD62L | eBioscience | MEL-14 |
| anti-mouse CD69 | eBioscience | H1.2F3 |
| anti-mouse GR1 | eBioscience | RB6–8C5 |
| anti-mouse Rorgt | BD Bioscience | Q31–378 |
| anti-mouse Ki-67 | BD Bioscience | B56 |
| anti-mouse IFN gamma | eBioscience | XMG1.2 |
| anti-mouse TNF alpha | eBioscience | MP6-XT22 |
| anti-mouse FoxP3 | eBioscience | FJK-16s |
| anti-mouse Tbet | eBioscience | 4B10 |
| ant-mouse GATA3 | eBioscience | TWAJ |
| anti-mouse TCRb | eBioscience | H57–597 |
| anti-mouse PD1 | eBioscience | J43 |
| anti-mouse CD86 | BD Bioscience | GL1 |
| anti-mouse CD11b | Biolegend | M1–70 |
| anti-mouse CD11c | Biolegend | N418 |
| anti-mouse CD103 | Biolegend | 2E7 |
| anti-mouse CD64 | Biolegend | X54–5/7.1 |
| anti-mouse CD80 | Biolegend | 16–10A1 |
| anti-mouse IA/IE | eBioscience | M5/114.15.2 |
| anti-mouse SiglecF | BD Bioscience | E50–2440 |
| anti-human CD3 | Biocare Medical | LN10 |
| anti-human CD8 | CST | C8/144 |
| anti-human PD1 | CST | D4W2J |
| anti-human FOXP3 | Thermo Scientific | 236A/E7 |
| anti-mouse CD11c | Biolegend | N418 |
| anti-mouse CD4 | Biolegend | GK1.5 |
| anti-mouse CD103 | Biolegend | M290 |
| anti-mouse NQO1 | Sigma Aldrich | HPA007308 |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| IFN-alpha | R&D | 12100–1 |
|
Deposited Data | ||
| scRNA-seq and RNA-seq data | GEO | GSE241482 |
|
Experimental Models: Cell Lines | ||
| KrasLSL-G12D/+; p53fl/fl lung adenocarcinoma cells (1233, 1234) | Tyler Jacks Lab | N/A |
|
Experimental Models: Organisms/Strains | ||
| B6.129S(C)-Batf3tm1Kmm/J | Jackson Laboratories | 013755 |
| Keap1 fl/fl | Taconic | 8799 |
| KrasLSL-G12D/+; p53fl/fl | Jackson Laboratories | 032435 |
|
Oligonucleotides | ||
| UTY | UTY-F: 5’-GAGGTTTTGTGGCATGGGAG -3’; UTY-R: 5’-TGCAGAAGATAACGAAGGAGCTA-3’ |
|
| Blvrb | Blvrb-F: 5’-CCGATGTGGACAAGACTGTG-3’; Blvrb-R: 5’-TCGGACATTACTGTAGTGGGACT-3’ |
|
| Nqo1 | Nqo1-F: 5’-AGCGTTCGGTATTACGATCC-3’ Nqo1-R: 5’-AGTACAATCAGGGCTCTTCTCG-3’; |
|
| Slc7a11 | Slc7a11-F: 5’-GATTCATGTCCACAAGCACAC-3’; Slc7a11-R: 5’-GAGCATCACCATCGTCAGAG-3’; |
|
| ISG15 | ISG15-F 5’- GGAACGAAAGGGGCCACAGCA-3’; ISG15-R 5’- CCTCCATGGGCCTTCCCTCGA-3’ |
|
| IFN beta | IFNβ -F 5’ – GCTCCTGGAGCAGCTGAATG-3’; IFNβ – R 5’ – CGTCATCTCCATAGGGATCTTGA-3’; |
|
| CXCL10 | CXCL10 – F 5’ – TGATTTCAAGCTTCCCTATGGC – 3’; CXCL10 – R 5’ – ATTTTCTGCCTCATCCTGCT – 3’; |
|
| IRF7 | IRF7 – F 5’ – GAAGAGGCTGGAAGACCAACTTC-3’; IRF7 – R 5’- GAATTGTCTTGGCGCAAGATAA; |
|
|
Software and Algorithms | ||
| GraphPad Prism v9 | N/A | |
| FlowJo | N/A | |
METHOD DETAILS
Mice
All animal procedures were approved by the NYU Langone Medical Center Institutional Animal Care and Use Committee (IACUC). Animals were housed according to IACUC guidelines in ventilated caging in a specific pathogen free (SPF) animal facility. C57BL/6J mice were bred in house or purchased from Jackson Laboratories. OT-I mice were obtained from the Schwab laboratory at NYU. B6.129S(C)-Batf3tm1Kmm/J were purchased from Jackson Laboratories. Animals acquired from Jackson laboratories were housed for at least one week in our SPF facility prior to experiment initiation. KrasLSL-G12D/+; p53fl/fl were crossed to Keap1fl/fl (Taconic, 8799). For tumor induction, mice 6–10 weeks of age were infected intratracheally with Cre-expressing lentivirus as previously described.100 Males or female mice were used as specified in the figure legends.
Tumor cell line generation
KrasG12D/+; p53−/− cells were isolated from a KrasLSL-G12D/+; p53fl/fl C57BL/6J male mouse that was infected with adenovirus expressing constitutive Cre recombinase. No clonal selection was performed. We generated Keap1 overexpressing cells by cloning the mouse Keap1 cDNA into Gibson compatible lentiviral backbone with hygromycin resistance cassette. Keap1R470C point mutation was generated by QuickChange II Site-directed Mutagenesis (Agilent) per manufacturer’s protocol. Cells infected were selected with 600μg/ml hygromycin. All cell lines tested negative for mycoplasma (PlasmoTest, InvivoGen). Cells were maintained in DMEM (Cellgro, Corning) supplemented with 10% fetal bovine serum (FBS) (Sigma Aldrich) and gentamycin (Invitrogen). For Nrf2 and Keap1 knockout, cells were generated by transducing cells with the plasmid lenticrispr V2 puro (Addgene #98290) cloned with a specific guide. Two days after transduction cells were selected with 8 ug/mL puromycin for 5 days. Sequence of CRISPR guides: Nrf2.1:
5’-CACCGTGGAGGCAAGACATAGATCT-3’;
5’-AAACAGATCTATGTCTTGCCTCCAC-3’;
Nrf2.2: 5’-CACCGAGGTTGGGGCAGCACCTGCT-3’;
5’-AAACAGCAGGTGCTGCCCCAACCTC-3’;
Keap1: 5’-CACCGCCGAAGCCCGTTGGTGAACA-3’;
5’- AAACTGTTCACCAACGGGCTTCGGC-3’.
Tumor cell injections
Tumor cells were harvested by trypsinization and washed three times with PBS. For lung orthotopic experiments, 7–10-week-old mice were injected intravenously with 2x105 KP cells expressing GFP and luciferase in 200ul endotoxin-free PBS. For MC38 subcutaneous injections, cells were injected into both right and left flack of each recipient mouse at 5x105 cells in 100ul of endotoxin free PBS containing 2.5mM EDTA.
Virus preparation
Lentivirus was produced by co-transfection of HEK293 cells with viral vector and packaging plasmids (psPAX2, pMD2G) using PEI Pro (PolyPlus). Virus containing media was collected 48h and 72h post transfection and filtered through 0.45uM PVDF filter. For cell line generation, harvested virus was added onto target cells in the presence of 8μg/ml polybrene (Millipore). For in vivo experiments, harvested virus was concentrated by ultracentrifugation at 25000rpm for 2h at 4°C, dissolved in PBS and stored at −80°C. Virus titration was performed using the Green-Go reporter cell line as previously described.101
Proliferation assay
For cell proliferation assays, cells cultured in DMEM/10% FBS were trypsinized and plated into 6 well plates at 2*10^5 cells and 2ml DMEM/10%FBS media per well. Cells were counted at days 1, 3 and 6 post plating.
qPCR
mRNA was harvested from tumor cells using RNeasy mini kit (Qiagen) according to manufacturer’s protocol. cDNA was synthesized from mRNA using SuperScript VILO (Invitrogen) according to manufacturer’s protocol. Real-time PCR was performed with StepOne Plus PCR system using SybrGreen master mix (Applied Biosystems). Primer sequences used were as follows:
UTY-F: 5’-GAGGTTTTGTGGCATGGGAG -3’;
UTY-R: 5’-TGCAGAAGATAACGAAGGAGCTA-3’;
Blvrb-F: 5’-CCGATGTGGACAAGACTGTG-3’;
Blvrb-R: 5’-TCGGACATTACTGTAGTGGGACT-3’;
Nqo1-F: 5’-AGCGTTCGGTATTACGATCC-3’
Nqo1-R: 5’-AGTACAATCAGGGCTCTTCTCG-3’;
Slc7a11-F: 5’-GATTCATGTCCACAAGCACAC-3’;
Slc7a11-R: 5’-GAGCATCACCATCGTCAGAG-3’;
ISG15-F 5’- GGAACGAAAGGGGCCACAGCA-3’;
ISG15-R 5’- CCTCCATGGGCCTTCCCTCGA-3’;
IFNβ -F 5’ – GCTCCTGGAGCAGCTGAATG-3’;
IFNβ – R 5’ – CGTCATCTCCATAGGGATCTTGA-3’;
CXCL10 – F 5’ – TGATTTCAAGCTTCCCTATGGC – 3’;
CXCL10 – R 5’ – ATTTTCTGCCTCATCCTGCT – 3’;
IRF7 – F 5’ – GAAGAGGCTGGAAGACCAACTTC-3’;
IRF7 – R 5’- GAATTGTCTTGGCGCAAGATAA;
Stimulation assays
Tumor cells were plated in DMEM/10% FBS overnight. Next day, they were treated with 1ug/ml polydA:dT (Inviogen), 1ug/ml polydG:dC (Invivogen), and 10 ng/ml IFN-α (R&D, 12100–1). They were collected at the timepoints indicated in the figure. Media were collected and then tumor cells were directly lysed on the plates according to RNeasy mini kit (Qiagen) protocol.
Antibody depletion experiments
For depletion of CD8 and CD4 T lymphocytes, anti-CD8a antibodies (clone: 2.43, Bioxcell) and anti-CD4 (GK1.5, Bioxcell) were used. Antibodies were diluted in PBS and injected intraperitoneally at 150μg/mouse twice a week (Monday, Friday), 7 days after tumor cell implantation until the end point of the experiment.
Labeling of circulating cells
Circulating immune cells were labeled using intravenously injected APC-conjugated CD45 antibody (30-F11, Biolegend) 3 minutes prior to sacrifice as previously described.45,102
Histology analysis
Mice were euthanized by lethal doses of ketamine and xylazine. Lungs were inflated and incubated overnight at room temperature (RT) with 10% formalin. They were subsequently transferred to 70% ethanol, and subsequently embedded in paraffin. 5uM sections were stained with H&E or subjected to other immunohistochemical staining. For immunohistochemistry (IHC) we used antibody against NQO1 (1:100, HPA007308, Sigma-Aldrich). Immunohistochemistry was performed on a Leica Bond RX and slides were imaged on a Leica SCN400F whole slide scanner. For NQO1, antigen retrieval was performed using antigen retrieval buffer pH=6 (Leica) for 20min. For detection, Leica Bond Polymer Refine Detection secondary antibody was used according to manufacturer’s protocol. Total tumor lung area was quantified via H&E-stained slides using QuPath software.103 Tumor burden and IHC analyses were done in a blinded fashion.
Immune cell isolation from lung tumors
Lungs were harvested at timepoints indicated in figure legends. Lungs were minced on glass slides followed by digestion using a cocktail of collagenase IV (125U/ml, Sigma), DNASE I (40U/ml, LifeTechnologies), 1X HEPES (Cellgro) in RPMI. Samples were incubated at 37°C for 35min and mixed by inverting every 5–8min. Digestion was quenched by adding RPMI containing 1mM EDTA final concentration. Digested samples were strained through 70μm cell strainers followed by red blood cell lysis (BD PharmLyse, BD Biosciences) for 10 min at 4°C.
Immune checkpoint blockade treatments
For immune checkpoint blockade treatment optimization on mc38 tumors anti-PD1 (RMP1–14, Bioxcell), anti-PD1 (mPD1–4H2-mIgG1-D265A, clone 6A1_RAS_Ab, Bristol Myers Squibb), anti-PD1 (29F.1A12, Bioxcell), anti-PD-L1 (10F.9G2, Bioxcell) was used. For isotype controls, IgG2b (LTF-2, Bioxcell), IgG1 (Bristol Myers Squibb), IgG2a (2A3, Bioxcell) were used. Tumor volume was measured by caliper, with volume calculated based on the following formula: (Length x Widtĥ2 x (3.14/6)). After tumor engraftment (~40–50mm3 tumor size) and before tumors reach 100mm3, animals were randomized and assigned to treatment group. For treatments of KP lung orthotopic tumors, anti-PD1 (29F.1A12, Bioxcell) was used. For anti-PD1 treatments, antibodies were diluted in PBS and injected intraperitoneally at 200ug/mouse three times a week until the end point of the experiment. For anti-PD-L1 treatment, mice were given a total of three doses intraperitoneally at 200μg/mouse every other day.
Anti-IFNAR treatment
Anti-IFNAR (MAR1–5A3) and isotype controls (IgG1, MOPC-21) were obtained from Bioxcell. Antibody was diluted in PBS. On day 0 of treatment mice were administered intraperitoneally 500 μg of either anti-IFNAR or isotype control then 250 ug every other day.
OTI/tumor in vitro killing assay
Keap1 wildtype or Keap1 mutant (R470C) KP cell lines were transduced with a lentivirus expressing LucOS and hCD2. Cells were then sorted for hCD2. OTI CD8 T cells were isolated from a spleen of an OTI mouse via negative magnetic selection. T Cells were activated in RPMI using plate bound anti-CD3 (Invitrogen 145–2C11) and anti-CD28 (Ebioscience 16–0281-82) in a 96 well flat bottom plate coated with goat anti-hamster IgG (MP Biomedical 856984). After 72 hours, T cells were expanded in a 75 mL flask with RPMI supplemented daily with recombinant mouse IL-2 (Peprotech, 25 IU/mL). On day 4 LuCOS expressing tumor cells were plated in a 96 well plate (10000 cells per well). Next day, OTI T cells were co-cultured with tumors cells (1,3, and 5:1 T cell:tumor ratio) and CB-839 (10 – 1000 nM) or vehicle (DMSO). 24 hours later cells bioluminescence was measured in a plate reader.
CB-839 treatment
Animals were treated with 200 mg/kg CB-839 or vehicle (Calithera, San Francisco, California) twice daily (morning and evening) administered through oral gavage as before.104 The vehicle contained 25% (w/v) hydroxypropyl-β-cyclodextrin in 10 mmol/L citrate (pH 2.0), and CB-839 was formulated at 20 mg/mL for a final dosing volume of 10 mL/kg.
Mouse tissue immunofluorescence
Tissues were processed as previously described.105 Harvested tissues were fixed in PLP buffer106 overnight at 4°C. Tissues were then dehydrated in 30% sucrose for 24 hours at 4°C and subsequently embedded in OCT media and stored at −80°C. 20mm sections were loaded on to racks (Shandon Sequenza) and washed with PBS. Sections were blocked with anti-CD16/CD32 (BioLegend, clone 93, 1/300) diluted in 1x PBS, 2% FBS, and 2% goat serum for 1h at RT. Staining antibodies were diluted in 1x PBS with 2% FBS, and 2% goat serum. Following blocking, sections were then incubated with fluorescently conjugated antibodies for CD11c (N418, Biolegend, 1/100), CD4, (Gk1.5, Biolegend,1/100), CD103 (M290, Biolegend, 1/100) diluted in PBS with 2%FBS and 2% goat serum for 1h at room temperature. Zeiss LSM 880 confocal microscope with the Zeiss Zen Black software was used for imaging. Confocal microscopy images were analyzed using the Bitplane Imaris x64 version 9.0.2 software. Images were filtered with the 3x3x1 median filter function to reduce background autofluorescence. To quantify different cell types within tumor lesions, “spots” were made from either the positive or negative signal combinations of CD45circ-CD11c+ CD103+ (CD103 DCs). Tumors were manually constructed into “2D surfaces” based on positive GFP signal. The “find spots close to surfaces” function was then performed on generated spots and surfaces with a threshold of 0.5 to quantify generated spots located within the surfaces. These quantifications are reported as the number of spots present within constructed tumor surfaces per surface area to control for differences in tumor area between images.
Human tissue immunofluorescence
5μm paraffin sections were stained using Akoya Biosciences® Opal™ multiplex automation kit reagents unless stated otherwise. Automated staining was performed on Leica BondRX® autostainer. The protocol was performed according to manufacturers’ instructions with the following antibodies: anti-CD3 (LN10, Biocare Medical, 1/75, Opal570), anti-CD8 (C8/144, Cell Signaling Technology, 1/500, Opal620), anti-PD-1 (D4W2J, Cell Signaling Technology, 1/200, Opal520), anti-Foxp3 (236A/E7, Thermo Fisher Scientific, 1/100, Opal780). All slides underwent sequential epitope retrieval with Leica Biosystems epitope retrieval 2 solution (ER2, EDTA based, pH9, Cat. AR9640), primary and secondary antibody incubation and tyramide signal amplification (TSA) with Opal® fluorophores. Primary and secondary antibodies were removed during epitope retrieval steps while fluorophores remain covalently attached to the epitope. Semi-automated image acquisition was performed on a Vectra® Polaris multispectral imaging system. After whole slide scanning at 20X the tissue was manually outlined to select fields for spectral unmixing and analysis using InForm® version 2.4.7 software from Akoya Biosciences. For each field of view, a tissue segmentation algorithm (tissue/no tissue) was run prior to cell segmentation. Cells were segmented based on nuclear signal (DAPI). Cells were phenotyped after segmentation using inForm’s trainable algorithm based on glmnet1 package in R. Phenotypes were reviewed for different samples during training iterations.
Flow cytometry and FACS
For surface staining, single cell suspensions were incubated for 10min with Fc receptor block (2.4G2, Bioxcell) at 4°C, followed by antibody staining for either 15min (for adaptive immune markers) or 30min (for innate immune markers) at 4°C. Staining was performed in FACS buffer (PBS, 2%BSA, 1mM EDTA). For cytokine staining, single cell suspensions were treated with a cell stimulation and protein transport inhibition cocktail, containing Golgi Plug (55029, 1/1000, BD Biosciences), Golgi Stop (555029, 1/1000, BD Biosciences), PMA (1/10000), Ionomycin (1/1000) for 3.5h. at 37°C in RPMI supplemented with 10%FBS. Cells were then surface stained as above, fixed in 2%PFA and permeabilized with 0.5% saponin. For transcription factors, staining was performed with the Foxp3 Staining Buffer kit (00552300, eBioscience). Fluorochrome conjugated antibodies with the following specificities were used: CD3 (ebio500A2, ebioscience), CD4 (RM4–5, ebioscience), CD4 (GK1.5, BD bioscience), CD45 (30-F11, BD bioscience), CD8 (53–6.7, BD bioscience), CD44 (IM7, ebioscience), CD62L (MEL-14, ebioscience), CD69 (H1.2F3, ebioscience), Gr-1 (RB6–8C5, ebioscience), Rorgt (Q31–378, BD Bioscience), Ki67 (B56, BD Bioscience), IFN-γ (XMG1.2, ebioscience), TNF-α (MP6-XT22, ebioscience), Foxp3 (FJK-16s, ebioscience), Tbet (4B10, ebioscience), GATA3 (TWAJ, ebioscience), TCRb (H57–597, ebioscience), PD-1 (J43, ebioscience), CD86 (GL1, BD Bioscience), CD11b (M1–70, Biolegend), CD11c (N418, Biolegend), CD103 (2E7, Biolegend), CD64 (X54–5/7.1, Biolegend), CD80 (16–10A1, Biolegend), IA/IE (M5/114.15.2, eBioscience), Siglec-F (E50–2440, BD Bioscience). For viability dye (1/1000, Biolegend), cells were stained in 100ul PBS for 10min at RT. Samples were acquired using BD LSR Fortessa Cell Analyzer. For sorting, single cell suspensions were resuspended in FACS buffer (PBS, 2%FBS, 0.01%Tween) and stained for viability dye (1/1000, Biolegend) in 100ul PBS for 10min at RT followed by surface staining with antibody specific for CD45 (30-F11, PercPcy55, 1/300, Biolegend). Tumor cells (for RNA-seq) were sorted for singlets, live cells, negative for CD45 and GFP positive. Immune cells (for single cell RNA seq) were sorted for singlets, live cells, negative for CD45 circulating antibody, and CD45 positive. Cells were sorted into pre-chilled tube containing DMEM supplemented with 20% FBS. Data were analyzed with FlowJo software (Tree Star Inc).
RNA-seq
Tumor cells were sorted as described above. RNA was isolated using Purelink RNA mini kit (Invitrogen) per manufacturer’s protocol. cDNA was synthesized from RNA using SMARTer PCR cDNA synthesis kit (Clontech) per manufacturer’s protocol. Sequencing libraries were prepared using Nextera XT DNA library preparation kit (Illumina) per manufacturer’s protocol. Samples were pooled at equimolar ratios. Libraries were loaded on an SP11 cycle flow cells and sequenced on Illumina NovaSeq6000. Read qualities were evaluated using FASTQC (Babraham Institute) and mapping to GRCm38 (GENCODE M25) reference genome using STAR programs107 and RSEM108 and with default parameters. Read counts, TPM and FPKM were calculated using RSEM. Identification of differentially expressed genes between wild-type and mutant tumor cells was performed using DESeq2 in R/Bioconductor. Genes without valid adjusted-p values (p-adj ==NA) were removed. All plots were generated using customized R scripts. Pathway enrichment analysis was performed using GSEA program109 based on log2FC values. Gene sets were downloaded from MsigDB.110
Single cell RNA seq
Samples were multiplexed using cell hashing antibodies. These were covalently and irreversibly conjugated to barcode oligos by iEDDA-click chemistry (“home conjugated”) as previously described.111,112 Cells were incubated for 10min with Fc receptor block (2.4G2, Bioxcell) and subsequently with hashing antibodies for 30min at 4°C. Cells were washed three times in PBS containing 2% bovine serum albumin (BSA) and 0.01% Tween followed by centrifugation (300g) for 5min at 4°C and supernatant aspiration. After final wash, cells were resuspended in PBS and filtered through 40μm cell strained. Cells from each sample were pooled and loaded into 10X Chromium. Gene expression together with Hashtag oligo (HTO) libraries were processed using Cell Ranger (v5.0.0) in count mode. UMI count matrices from each modality were imported into the same Seurat object as separate assays. Cell-containing droplets were selected using the default filtering from Cell Ranger count “filtered_feature_bc_matrix”. Viable cells were filtered based on having more than 250 genes detected and less than 55% of total UMIs stemming from mitochondrial transcripts. HTO counts were normalized using centered log ratio transformation before hashed samples were demultiplexing using the Seurat::HTODemux function (positive.quantile set to 0.93). Cells from two separate droplet emulsion reactions were combined using the standard SCTrans-form integration workflow in the Seurat (v4) R package.113 Multimodal integration was performed using the weighted-nearest neighbor (WNN) method available in Seurat. Briefly, a WNN network was constructed based on modality weights estimated for each cell using FindMultiModalNeighbors function. Cell clusters were identified using FindClusters function based on the weighted SNN (WSNN) graph using leiden algorithm at various resolutions, ranging from 1 to 1.6. Cell types were annotated based on canonical cell type markers as well as differential expressed genes of each cluster identified using FindAllMarkers function in Seurat. Clusters expressing markers of the same cell type were merged into a single cluster. Single cell TCR sequence data was processed using Cell Ranger vdj pipeline to identify TCR genes and CDR3 sequences. For each sample, full length, productive TCR beta and alpha chains were retained for downstream analysis. Clonal expansions were determined using nucleotide sequences of CDR3 sequence of both chains and TRR alpha and beta genes that appeared in at least 5 cells based on all samples. TCR information was added as metadata into the scRNASeq Seurat object based on cell barcodes and sample information.
Nanostring GeoMx digital spatial profiling
Slide preparation was performed following the GeoMx DSP instructions. Briefly, 5μm TMA sections were baked for 2 hrs at 60 degrees Celsius before loading onto an automated slide strainer (LeicaBiosystems Bond RX). Slides underwent Baking and Dewaxing steps followed by blocking for 60 minutes with buffer ‘W’ (part of the slide prep kit for GeoMx). GeoMx antibody incubation was performed overnight at four degrees Celsius with the following combination of antibody panels: Human Immune Cell Profiling Protein Core, Human Immune Activation Status Protein, Human Immune Cell Typing Protein, Human IO Drug Target Protein, Human Pan-Tumor Protein (v1.0 for all panels and core). The Human Solid Tumor TME (v1.0) morphology marker kit was used to visualize pan-cytokeratin and CD45 antigens on the slide scan. Digital counts from barcodes corresponding to protein probes were analyzed as follows: raw counts were first normalized with internal spike-in controls (ERCC) to account for system variation. Data was normalized using the geometric mean of housekeeping antibody counts for Histone H3 and S6. The module score was calculated by first rescaling each gene across measurements by dividing by the maximum value across measurements, then for each measurement, taking the mean of genes included in each module. Normalized values were visualized with boxplots. The wilcoxon test was used to calculate statistics between conditions. One outlier sample was removed. Outlier samples were determined as having values above the 98th percentile in more than 20 proteins. Ggplot and ggpubr R packages were used to generate plots and statistics.
Statistics
Values are represented as mean ± SEM. Statistical analyses were performed using Prism 9 (GraphPad Software) and a p value<0.05 was considered significant. All animal experiments contained at least n>3 mice. Mann-Whitney test for flow cytometry and lung weight experiments with less than 2 experimental groups. One-way analysis of variance (ANOVA) and Tukey’s test for lung weights and histological analysis with more than two experimental groups. Two-way ANOVA for growth in vivo growth kinetics and OTI tumor killing experiments. Survival analysis was performed using the Log-Rank (Mantel Cox) test. *P<0.05; **P<0.01; ***P<0.001; ****P < 0.0001.
Supplementary Material
Highlights.
KEAP1 mutations in lung cancer suppress anti-tumor CD103 DC and CD8 T cell responses
Hyperactivation of NRF2 is responsible for immune evasion in KEAP1-mutant tumors
Glutaminase inhibition can sensitize KEAP1-mutant tumors to immunotherapy
ACKNOWLEDGMENTS
Work in the T.P. laboratory was supported by NIH grants (R37CA222504 and R01CA227649), an American Cancer Society Research Scholar grant (RSG-17-200-01-TBE), and the Emerald Foundation Young Investigator Award. Work in the S.B.K. laboratory was supported by NIH grants (R01HL125816 and R01CA271245), a LEO Foundation grant (LF-OC-20-000351), an NYU Cancer Center Pilot grant (P30CA016087), and a Judith and Stewart Colton Center for Autoimmunity Pilot grant. W.L.W. is supported by NIH training grants (T32GM007308 and 1F30CA247020). R.P. is supported by the William Rom Fellowship, the Stony Wold-Herbert Fund, and NIH T32 training grants (T32CA009161 and T32AI100853). E.B. is supported by an NIH training grant (T32AI100853-10). Work in the K.M.K. laboratory is supported by an NIH grant (R01AI143861-01). A.T. is supported by NCI/NIH P01CA229086 and NCI/NIH R01CA252239. T.B.B. was supported by the Danish Cancer Society (Kræftens Bekæmpelse). We would like to thank Drs. M. Okuniewska and S. Schwab (NYU) for providing OT-I mice and Dr. Thomas (NIH) for providing Ki696 and anti-NRF2 antibody. CB-839 was provided by Calithera. We thank the NYU Experimental Pathology Laboratory, Flow Cytometry Core, and Animal Resources Facility staff for their support and guidance. We thank the Genome Technology Center for expert library preparation and sequencing, and the Applied Bioinformatics Laboratories for providing bioinformatics support and helping with the analysis and interpretation of the data. These cores are partially supported by NYU Cancer Center grant P30CA016087. This work has used computing resources at the NYU School of Medicine High Performance Computing Facility. We are grateful to Drs. Frey and Reizis for their feedback on the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113295.
DECLARATION OF INTERESTS
T.P. has received research support from Agios Pharmaceuticals, and T.P. and S.B.K. have received funding from Dracen Pharmaceuticals, Kymera Therapeutics, and Bristol Myers Squibb. T.P. has received honoraria from Calithera Biosciences and Vividion Therapeutics. T.P. and S.B.K. are authors on US provisional patent application 16/483,835: “Methods for treating cancers having a deregulated NRF2/KEAP1 pathway.”
REFERENCES
- 1.Siegel RL, Miller KD, and Jemal A (2019). Cancer statistics, 2019. CA. Cancer J. Clin 69, 7–34. 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, Henley SJ, Anderson RN, Firth AU, Ma J, et al. (2018). Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 124, 2785–2800. 10.1002/cncr.31551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herbst RS, Morgensztern D, and Boshoff C (2018). The biology and management of non-small cell lung cancer. Nature 553, 446–454. 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 4.Cable J, Greenbaum B, Pe’er D, Bollard CM, Bruni S, Griffin ME, Allison JP, Wu CJ, Subudhi SK, Mardis ER, et al. (2021). Frontiers in cancer immunotherapy-a symposium report. Ann. N. Y. Acad. Sci 1489, 30–47. 10.1111/nyas.14526. [DOI] [PubMed] [Google Scholar]
- 5.Sharma P, Hu-Lieskovan S, Wargo JA, and Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. 10.1016/j.cell.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howlader N, Forjaz G, Mooradian MJ, Meza R, Kong CY, Cronin KA, Mariotto AB, Lowy DR, and Feuer EJ (2020). The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med 383, 640–649. 10.1056/NEJMoa1916623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dal Bello MG, Alama A, Coco S, Vanni I, and Grossi F (2017). Understanding the checkpoint blockade in lung cancer immunotherapy. Drug Discov. Today 22, 1266–1273. 10.1016/j.drudis.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 8.Brahmer JR, Govindan R, Anders RA, Antonia SJ, Sagorsky S, Davies MJ, Dubinett SM, Ferris A, Gandhi L, Garon EB, et al. (2018). The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of non-small cell lung cancer (NSCLC). J. Immunother. Cancer 6, 75. 10.1186/s40425-018-0382-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen DS, and Mellman I (2017). Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330. 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 10.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. (2017). IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest 127, 2930–2940. 10.1172/jci91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, et al. (2012). An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother 61, 1019–1031. 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, Lanitis E, Duraiswamy J, Tanyi JL, Benencia F, et al. (2019). Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell 35, 885–900.e10. 10.1016/j.ccell.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spranger S, Bao R, and Gajewski TF (2015). Melanoma-intrinsic b-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235. 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 14.Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, and Luster AD (2019). Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 50, 1498–1512.e5. 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spranger S, Dai D, Horton B, and Gajewski TF (2017). Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 31, 711–723.e4. 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayoux M, Roller A, Pulko V, Sammicheli S, Chen S, Sum E, Jost C, Fransen MF, Buser RB, Kowanetz M, et al. (2020). Dendritic cells dictate responses to PD-L1 blockade cancer immunotherapy. Sci. Transl. Med 12, eaav7431. 10.1126/scitranslmed.aav7431. [DOI] [PubMed] [Google Scholar]
- 18.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. (2016). Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 44, 924–938. 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, et al. (2014). Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26, 638–652. 10.1016/j.ccell.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 49, 1148–1161.e7. 10.1016/j.immuni.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sánchez-Paulete AR, Cueto FJ, Martínez-López M, Labiano S, Morales-Kastresana A, Rodríguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, et al. (2016). Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti-PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov 6, 71–79. 10.1158/2159-8290.cd-15-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research Network (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550. 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, and Yamamoto M (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell Biol 24, 7130–7139. 10.1128/mcb.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harris IS, and DeNicola GM (2020). The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol 30, 440–451. 10.1016/j.tcb.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 25.Wu WL, and Papagiannakopoulos T (2020). The Pleiotropic Role of the KEAP1/NRF2 Pathway in Cancer. Annu. Rev. Cancer Biol 4, 413–435. [Google Scholar]
- 26.Berger AH, Brooks AN, Wu X, Shrestha Y, Chouinard C, Piccioni F, Bagul M, Kamburov A, Imielinski M, Hogstrom L, et al. (2016). High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell 30, 214–228. 10.1016/j.ccell.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E, et al. (2006). Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med 3, e420. 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson CJ, Chang M, Karttunen M, and Choy WY (2021). KEAP1 Cancer Mutants: A Large-Scale Molecular Dynamics Study of Protein Stability. Int. J. Mol. Sci 22, 5408. 10.3390/ijms22105408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hast BE, Cloer EW, Goldfarb D, Li H, Siesser PF, Yan F, Walter V, Zheng N, Hayes DN, and Major MB (2014). Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res 74, 808–817. 10.1158/0008-5472.can-13-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeB-oeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sánchez-Rivera FJ, et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med 23, 1362–1368. 10.1038/nm.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, Pass HI, Bhutkar AJ, Tsirigos A, Ueberheide B, et al. (2019). Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 178, 316–329.e18. 10.1016/j.cell.2019.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Binkley MS, Jeon Y-J, Nesselbush M, Moding EJ, Nabet BY, Almanza D, Kunder C, Stehr H, Yoo CH, Rhee S, et al. (2020). KEAP1/NFE2L2 Mutations Predict Lung Cancer Radiation Resistance That Can Be Targeted by Glutaminase Inhibition. Cancer Discov 10, 1826–1841. 10.1158/2159-8290.Cd-20-0282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. (2020). The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov 10, 54–71. 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F, et al. (2021). Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med 384, 2371–2381. 10.1056/NEJMoa2103695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Negrao MV, Araujo HA, Lamberti G, Cooper AJ, Akhave NS, Zhou T, Delasos L, Hicks JK, Aldea M, Minuti G, et al. (2023). Co-mutations and KRAS G12C inhibitor efficacy in advanced NSCLC. Cancer Discov, 1420. 10.1158/2159-8290.Cd-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, Lito P, Berger M, Solit DB, Hellmann M, et al. (2018). Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res 24, 334–340. 10.1158/1078-0432.Ccr-17-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marinelli D, Mazzotta M, Scalera S, Terrenato I, Sperati F, D’Ambrosio L, Pallocca M, Corleone G, Krasniqi E, Pizzuti L, et al. (2020). KEAP1-driven co-mutations in lung adenocarcinoma unresponsive to immunotherapy despite high tumor mutational burden. Ann. Oncol 31, 1746–1754. 10.1016/j.annonc.2020.08.2105. [DOI] [PubMed] [Google Scholar]
- 38.Aggarwal C, Thompson JC, Chien AL, Quinn KJ, Hwang WT, Black TA, Yee SS, Christensen TE, LaRiviere MJ, Silva BA, et al. (2020). Baseline Plasma Tumor Mutation Burden Predicts Response to Pembrolizumab-based Therapy in Patients with Metastatic Non-Small Cell Lung Cancer. Clin. Cancer Res 26, 2354–2361. 10.1158/1078-0432.Ccr-19-3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papillon-Cavanagh S, Doshi P, Dobrin R, Szustakowski J, and Walsh AM (2020). STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open 5, e000706. 10.1136/esmoopen-2020-000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kadara H, Choi M, Zhang J, Parra ER, Rodriguez-Canales J, Gaffney SG, Zhao Z, Behrens C, Fujimoto J, Chow C, et al. (2017). Whole-exome sequencing and immune profiling of early-stage lung adenocarcinoma with fully annotated clinical follow-up. Ann. Oncol 28, 75–82. 10.1093/annonc/mdw436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, Sher X, Liu XQ, Lu H, Nebozhyn M, et al. (2018). Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 362, eaar3593. 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dimitrova N, Gocheva V, Bhutkar A, Resnick R, Jong RM, Miller KM, Bendor J, and Jacks T (2016). Stromal Expression of miR-143/145 Promotes Neoangiogenesis in Lung Cancer Development. Cancer Discov 6, 188–201. 10.1158/2159-8290.Cd-15-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. (2019). COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–D947. 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, Crowley D, Chen J, and Jacks T (2011). Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell 19, 72–85. 10.1016/j.ccr.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, DuPage M, Tammela T, Kerper NR, Farago AF, et al. (2015). Regulatory T Cells in Tumor-Associated Tertiary Lymphoid Structures Suppress Anti-tumor T Cell Responses. Immunity 43, 579–590. 10.1016/j.immuni.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. (2016). Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 44, 343–354. 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, Chudnovskiy A, Maskey S, Walker L, Finnigan JP, et al. (2020). A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 580, 257–262. 10.1038/s41586-020-2134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martinez-Usatorre A, Kadioglu E, Boivin G, Cianciaruso C, Guichard A, Torchia B, Zangger N, Nassiri S, Keklikoglou I, Schmittnaegel M, et al. (2021). Overcoming microenvironmental resistance to PD-1 blockade in genetically engineered lung cancer models. Sci. Transl. Med 13, eabd1616. 10.1126/scitranslmed.abd1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milo I, Bedora-Faure M, Garcia Z, Thibaut R, Périé L, Shakhar G, Deriano L, and Bousso P (2018). The immune system profoundly restricts intratumor genetic heterogeneity. Sci. Immunol 3, eaat1435. 10.1126/sciimmunol.aat1435. [DOI] [PubMed] [Google Scholar]
- 50.Poillet-Perez L, Sharp DW, Yang Y, Laddha SV, Ibrahim M, Bommareddy PK, Hu ZS, Vieth J, Haas M, Bosenberg MW, et al. (2020). Autophagy promotes growth of tumors with high mutational burden by inhibiting a T-cell immune response. Nat. Cancer 1, 923–934. 10.1038/s43018-020-00110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, Ryan RJH, Iwamoto Y, Marinelli B, Gorbatov R, et al. (2012). Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. USA 109, 2491–2496. 10.1073/pnas.1113744109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, Imakado S, Kotsuji T, Otsuka F, Roop DR, et al. (2003). Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat. Genet 35, 238–245. 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 53.Kang YP, Torrente L, Falzone A, Elkins CM, Liu M, Asara JM, Dibble CC, and DeNicola GM (2019). Cysteine dioxygenase 1 is a metabolic liability for non-small cell lung cancer. Elife 8, e45572. 10.7554/eLife.45572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. (2011). Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med 208, 1989–2003. 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, and Gajewski TF (2011). Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med 208, 2005–2016. 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koyama S, Akbay EA, Li YY, Aref AR, Skoulidis F, Herter-Sprie GS, Buczkowski KA, Liu Y, Awad MM, Denning WL, et al. (2016). STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res 76, 999–1008. 10.1158/0008-5472.CAN-15-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, and Reis e Sousa C (2018). NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 172, 1022–1037.e1014. 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, and Pollard JW (2011). CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225. 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chao T, Furth EE, and Vonderheide RH (2016). CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res 4, 968–982. 10.1158/2326-6066.cir-16-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, and Lowe SW (2007). Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660. 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. (2008). Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322, 1097–1100. 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, et al. (2007). Differential antigen processing by dendritic cell subsets in vivo. Science (New York, N.Y.) 315, 107–111. 10.1126/sci-ence.1136080. [DOI] [PubMed] [Google Scholar]
- 63.Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, Tsui J, Ruhland MK, Kersten K, Abushawish MA, et al. (2019). Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell 177, 556–571.e16. 10.1016/j.cell.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, He Y, Wang L, Zhang Q, Kim A, et al. (2020). Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181, 442–459.e29. 10.1016/j.cell.2020.03.048. [DOI] [PubMed] [Google Scholar]
- 65.Brown CC, Gudjonson H, Pritykin Y, Deep D, Lavallée VP, Mendoza A, Fromme R, Mazutis L, Ariyan C, Leslie C, et al. (2019). Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 179, 846–863.e24. 10.1016/j.cell.2019.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Abbas A, Vu Manh TP, Valente M, Collinet N, Attaf N, Dong C, Naciri K, Chelbi R, Brelurut G, Cervera-Marzal I, et al. (2020). The activation trajectory of plasmacytoid dendritic cells in vivo during a viral infection. Nat. Immunol 21, 983–997. 10.1038/s41590-020-0731-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, Wurtz A, Dong W, Cai G, Melnick MA, et al. (2017). Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov 7, 1420–1435. 10.1158/2159-8290.cd-17-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 30, 324–336. 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, Davidson JT 4th, Bagadia P, Liu T, Briseño CG, et al. (2020). cDC1 prime and are licensed by CD4(+) T cells to induce anti-tumour immunity. Nature 584, 624–629. 10.1038/s41586-020-2611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, Daniel D, Hwang ES, Rugo HS, and Coussens LM (2014). Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637. 10.1016/j.ccell.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tibbitt CA, Stark JM, Martens L, Ma J, Mold JE, Deswarte K, Oliynyk G, Feng X, Lambrecht BN, De Bleser P, et al. (2019). Single-Cell RNA Sequencing of the T Helper Cell Response to House Dust Mites Defines a Distinct Gene Expression Signature in Airway Th2 Cells. Immunity 51, 169–184.e5. 10.1016/j.immuni.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 72.Connolly KA, Kuchroo M, Venkat A, Khatun A, Wang J, William I, Hornick N, Fitzgerald B, Dam M, Kasmani MY, et al. (2021). A reservoir of stem-like CD8 T cells in the tumor-draining lymph node maintains the ongoing anti-tumor immune response. Preprint at biorxiv 10.1101/2021.01.27.428467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, and Jacks T (2012). Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409. 10.1038/nature10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Damo M, Fitzgerald B, Lu Y, Nader M, William I, Cheung JF, Connolly KA, Foster GG, Akama-Garren E, Lee DY, et al. (2021). Inducible de novo expression of neoantigens in tumor cells and mice. Nat. Biotechnol 39, 64–73. 10.1038/s41587-020-0613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Downey CM, and Jirik FR (2015). DNA mismatch repair deficiency accelerates lung neoplasm development in K-ras(LA1/+) mice: a brief report. Cancer Med 4, 897–902. 10.1002/cam4.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA, et al. (2015). Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 162, 1257–1270. 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, Asamura H, Yamamoto M, and Hirohashi S (2008). Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc. Natl. Acad. Sci. USA 105, 13568–13573. 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ma Q, Battelli L, and Hubbs AF (2006). Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol 168, 1960–1974. 10.2353/ajpath.2006.051113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kitamura H, Onodera Y, Murakami S, Suzuki T, and Motohashi H (2017). IL-11 contribution to tumorigenesis in an NRF2 addiction cancer model. Oncogene 36, 6315–6324. 10.1038/onc.2017.236. [DOI] [PubMed] [Google Scholar]
- 80.Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, and Yamamoto M (2016). Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun 7, 11624. 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Olagnier D, Brandtoft AM, Gunderstofte C, Villadsen NL, Krapp C, Thielke AL, Laustsen A, Peri S, Hansen AL, Bonefeld L, et al. (2018). Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat. Commun 9, 3506. 10.1038/s41467-018-05861-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nishina T, Deguchi Y, Miura R, Yamazaki S, Shinkai Y, Kojima Y, Okumura K, Kumagai Y, and Nakano H (2017). Critical Contribution of Nuclear Factor Erythroid 2-related Factor 2 (NRF2) to Electrophile-induced Interleukin-11 Production. J. Biol. Chem 292, 205–216. 10.1074/jbc.M116.744755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sayin VI, LeBoeuf SE, Singh SX, Davidson SM, Biancur D, Guzelhan BS, Alvarez SW, Wu WL, Karakousi TR, Zavitsanou AM, et al. (2017). Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife 6, e28083. 10.7554/eLife.28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, Chen PH, Boroughs LK, Rodriguez MLM, Zhang W, et al. (2019). LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res 79, 3251–3267. 10.1158/0008-5472.Can-18-3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.LeBoeuf SE, Wu WL, Karakousi TR, Karadal B, Jackson SR, Davidson SM, Wong KK, Koralov SB, Sayin VI, and Papagiannakopoulos T (2020). Activation of Oxidative Stress Response in Cancer Generates a Druggable Dependency on Exogenous Non-essential Amino Acids. Cell Metab 31, 339–350.e4. 10.1016/j.cmet.2019.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton RJ, et al. (2018). Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 175, 1780–1795.e19. 10.1016/j.cell.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, et al. (2015). Glutamine-dependent a-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal 8, ra97. 10.1126/scisignal.aab2610. [DOI] [PubMed] [Google Scholar]
- 88.Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, Blonska M, Lin X, and Sun SC (2014). Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705. 10.1016/j.immuni.2014.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, Li K, Ma T, Wang H, Ni L, et al. (2017). Metabolic control of T(H)17 and induced T(reg) cell balance by an epigenetic mechanism. Nature 548, 228–233. 10.1038/nature23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara HMCS, et al. (2015). Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol 35, S185–S198. 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 91.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, et al. (2019). Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366, 1013–1021. 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, Turay AM, and Frauwirth KA (2010). Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol 185, 1037–1044. 10.4049/jimmu-nol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chang WK, Yang KD, Chuang H, Jan JT, and Shaio MF (2002). Glutamine protects activated human T cells from apoptosis by up-regulating glutathione and Bcl-2 levels. Clin. Immunol 104, 151–160. 10.1006/clim.2002.5257. [DOI] [PubMed] [Google Scholar]
- 95.Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, Mainolfi N, Suri V, Guak H, Balmer ML, et al. (2017). Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab 25, 482. 10.1016/j.cmet.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 96.Hast BE, Goldfarb D, Mulvaney KM, Hast MA, Siesser PF, Yan F, Hayes DN, and Major MB (2013). Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res 73, 2199–2210. 10.1158/0008-5472.can-12-4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee DF, Kuo HP, Liu M, Chou CK, Xia W, Du Y, Shen J, Chen CT, Huo L, Hsu MC, et al. (2009). KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol. Cell 36, 131–140. 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tamberg N, Tahk S, Koit S, Kristjuhan K, Kasvandik S, Kristjuhan A, and Ilves I (2018). Keap1-MCM3 interaction is a potential coordinator of molecular machineries of antioxidant response and genomic DNA replication in metazoa. Sci. Rep 8, 12136. 10.1038/s41598-018-30562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, Mehta M, Cheung KL, Ganesan S, Kong ANT, et al. (2012). PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell Biol 32, 1506–1517. 10.1128/mcb.06271-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc 4, 1064–1072. 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sánchez-Rivera FJ, Papagiannakopoulos T, Romero R, Tammela T, Bauer MR, Bhutkar A, Joshi NS, Subbaraj L, Bronson RT, Xue W, and Jacks T (2014). Rapid modelling of cooperating genetic events in cancer through somatic genome editing. Nature 516, 428–431. 10.1038/nature13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, and Masopust D (2014). Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 9, 209–222. 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bankhead P, Loughrey MB, Fernández JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: Open source software for digital pathology image analysis. Sci. Rep 7, 16878. 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23, 517–528. 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Perez OA, Yeung ST, Vera-Licona P, Romagnoli PA, Samji T, Ural BB, Maher L, Tanaka M, and Khanna KM (2017). CD169(+) macrophages orchestrate innate immune responses by regulating bacterial localization in the spleen. Sci. Immunol 2, eaah5520. 10.1126/sciimmunol.aah5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McLean IW, and Nakane PK (1974). Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J. Histochem. Cytochem 22, 1077–1083. 10.1177/22.12.1077. [DOI] [PubMed] [Google Scholar]
- 107.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf 12, 323. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, Smibert P, and Satija R (2018). Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224. 10.1186/s13059-018-1603-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.van Buggenum JAGL, Gerlach JP, Eising S, Schoonen L, van Eijl RAPM, Tanis SEJ, Hogeweg M, Hubner NC, van Hest JC, Bonger KM, and Mulder KW (2016). A covalent and cleavable antibody-DNA conjugation strategy for sensitive protein detection via immuno-PCR. Sci. Rep 6, 22675. 10.1038/srep22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-mouse CD3 | eBioscience | 500A2 |
| anti-mouse CD4 | eBioscience | RM4–5 |
| anti-mouse CD8 | BD bioscience | 53–6.7 |
| anti-mouse CD45 | BD bioscience | 30-F11 |
| anti-mouse CD4 | BD Bioscience | GK1.5 |
| anti-mouse CD44 | eBioscience | IM7 |
| anti-mouse CD62L | eBioscience | MEL-14 |
| anti-mouse CD69 | eBioscience | H1.2F3 |
| anti-mouse GR1 | eBioscience | RB6–8C5 |
| anti-mouse Rorgt | BD Bioscience | Q31–378 |
| anti-mouse Ki-67 | BD Bioscience | B56 |
| anti-mouse IFN gamma | eBioscience | XMG1.2 |
| anti-mouse TNF alpha | eBioscience | MP6-XT22 |
| anti-mouse FoxP3 | eBioscience | FJK-16s |
| anti-mouse Tbet | eBioscience | 4B10 |
| ant-mouse GATA3 | eBioscience | TWAJ |
| anti-mouse TCRb | eBioscience | H57–597 |
| anti-mouse PD1 | eBioscience | J43 |
| anti-mouse CD86 | BD Bioscience | GL1 |
| anti-mouse CD11b | Biolegend | M1–70 |
| anti-mouse CD11c | Biolegend | N418 |
| anti-mouse CD103 | Biolegend | 2E7 |
| anti-mouse CD64 | Biolegend | X54–5/7.1 |
| anti-mouse CD80 | Biolegend | 16–10A1 |
| anti-mouse IA/IE | eBioscience | M5/114.15.2 |
| anti-mouse SiglecF | BD Bioscience | E50–2440 |
| anti-human CD3 | Biocare Medical | LN10 |
| anti-human CD8 | CST | C8/144 |
| anti-human PD1 | CST | D4W2J |
| anti-human FOXP3 | Thermo Scientific | 236A/E7 |
| anti-mouse CD11c | Biolegend | N418 |
| anti-mouse CD4 | Biolegend | GK1.5 |
| anti-mouse CD103 | Biolegend | M290 |
| anti-mouse NQO1 | Sigma Aldrich | HPA007308 |
|
Chemicals, Peptides, and Recombinant Proteins | ||
| IFN-alpha | R&D | 12100–1 |
|
Deposited Data | ||
| scRNA-seq and RNA-seq data | GEO | GSE241482 |
|
Experimental Models: Cell Lines | ||
| KrasLSL-G12D/+; p53fl/fl lung adenocarcinoma cells (1233, 1234) | Tyler Jacks Lab | N/A |
|
Experimental Models: Organisms/Strains | ||
| B6.129S(C)-Batf3tm1Kmm/J | Jackson Laboratories | 013755 |
| Keap1 fl/fl | Taconic | 8799 |
| KrasLSL-G12D/+; p53fl/fl | Jackson Laboratories | 032435 |
|
Oligonucleotides | ||
| UTY | UTY-F: 5’-GAGGTTTTGTGGCATGGGAG -3’; UTY-R: 5’-TGCAGAAGATAACGAAGGAGCTA-3’ |
|
| Blvrb | Blvrb-F: 5’-CCGATGTGGACAAGACTGTG-3’; Blvrb-R: 5’-TCGGACATTACTGTAGTGGGACT-3’ |
|
| Nqo1 | Nqo1-F: 5’-AGCGTTCGGTATTACGATCC-3’ Nqo1-R: 5’-AGTACAATCAGGGCTCTTCTCG-3’; |
|
| Slc7a11 | Slc7a11-F: 5’-GATTCATGTCCACAAGCACAC-3’; Slc7a11-R: 5’-GAGCATCACCATCGTCAGAG-3’; |
|
| ISG15 | ISG15-F 5’- GGAACGAAAGGGGCCACAGCA-3’; ISG15-R 5’- CCTCCATGGGCCTTCCCTCGA-3’ |
|
| IFN beta | IFNβ -F 5’ – GCTCCTGGAGCAGCTGAATG-3’; IFNβ – R 5’ – CGTCATCTCCATAGGGATCTTGA-3’; |
|
| CXCL10 | CXCL10 – F 5’ – TGATTTCAAGCTTCCCTATGGC – 3’; CXCL10 – R 5’ – ATTTTCTGCCTCATCCTGCT – 3’; |
|
| IRF7 | IRF7 – F 5’ – GAAGAGGCTGGAAGACCAACTTC-3’; IRF7 – R 5’- GAATTGTCTTGGCGCAAGATAA; |
|
|
Software and Algorithms | ||
| GraphPad Prism v9 | N/A | |
| FlowJo | N/A | |