

Abstract
Female APOE4 carriers are at greatest risk of Alzheimer’s disease (AD). The potent estrogen 17β-estradiol (E2) may mediate AD risk, as the onset of memory decline coincides with the menopausal transition. Whether APOE genotype mediates E2’s effects on memory and neuronal morphology is poorly understood. We used the APOE+/+/5xFAD+/− (EFAD) mouse model to examine how APOE3 homozygote (E3FAD), APOE3/4 heterozygote (E3/4FAD), and APOE4 homozygote (E4FAD) genotypes modulate effects of E2 on object and spatial memory consolidation, dendritic spine density, and dorsal hippocampal estrogen receptor expression in 6-month-old ovariectomized EFAD mice. Dorsal hippocampal E2 infusion enhanced memory consolidation and increased CA1 apical spine density in E3FAD and E3/4FAD, but not E4FAD, mice. CA1 basal mushroom spines were also increased by E2 in E3FADs. E4FAD mice exhibited reduced CA1 and mPFC basal spine density, and increased dorsal hippocampal ERα protein, independent of E2. Overall, E2 benefitted hippocampal memory and structural plasticity in females bearing one or no APOE4 allele, whereas two APOE4 alleles impeded the memory-enhancing and spinogenic effects of E2.
Keywords: Alzheimer’s disease, apolipoprotein E, APOE4, 17β-estradiol, spatial memory, object recognition, hippocampus, medial prefrontal cortex
1. Introduction
Alzheimer’s disease (AD), the most common form of dementia, poses a monumental global health crisis. Despite efforts by researchers to identify pharmacological treatments and drug targets, there are few disease-modifying therapies that mitigate the devastating impact of AD. Other than aging, risk factors for AD include APOE4 genotype and female sex. APOE4 genotype renders individuals more susceptible to AD than other APOE genotypes, increasing risk by as much as 2-fold in the presence of a single APOE4 allele (Bertram, 2009; Roses, 1996). Women, especially APOE4 carriers, are at significantly greater risk for developing AD compared to men (Altmann et al., 2014; Bretsky et al., 1999), making the examination of factors contributing to this increased risk in women critical for AD research.
One potential mediator of heightened risk for AD in women is the loss of circulating estrogens coincident with the onset of the menopausal transition. Multiple studies implicate the precipitous loss of 17β-estradiol (E2), the most prevalent circulating estrogen, at menopause as a contributing factor for age and disease-associated cognitive decline (Jacobs et al., 2016; Paganini-Hill and Henderson, 1996). In fact, female AD patients have lower levels of endogenous E2 than age and sex-matched controls (Manly et al., 2000; Tsolaki et al., 2005). However, clinical trials of estrogen therapy paint a conflicting portrait of its effects on AD risk; early studies offered support for the idea that E2 may reduce risk of AD (Baldereschi et al., 1998; Joffe et al., 2006; Kawas et al., 1997; Tang et al., 1996), whereas other studies and more recent clinical trials do not (Gleason et al., 2015; Rapp et al., 2003; Shumaker et al., 2003). These discrepant reports may be related to key factors influencing estrogen therapy efficacy including age at initiation, specific estrogen treatment regimen (e.g., estradiol vs. conjugated equine estrogens), dose, length of treatment, and physical health (Henderson, 2006; Paganini-Hill and Henderson, 1996). Another critical element of cognitive responsiveness to estrogen therapy is APOE status. Importantly, cognition in women APOE4 carriers appears to benefit less from estrogen therapy than in their APOE4-negative counterparts (Yaffe et al., 2000). This clinical work is consistent with evidence from basic research; APOE2 and APOE3, but not APOE4, act synergistically with E2 to promote neurite outgrowth in mixed cell cultures, suggesting that APOE4 expression renders cells uniquely nonresponsive to E2 (Nathan et al., 2004). Combined, the above results suggest that E2 may be beneficial for carriers of APOE3 alleles, and unhelpful or even deleterious for APOE4 carriers.
In adult female mice, treatment with exogenous E2 facilitates memory via rapid activation of cell signaling within the dorsal hippocampus (DH), which is accompanied by an increase in dendritic spine density in the DH and in the medial prefrontal cortex (Boulware et al., 2013; Fernandez et al., 2008; Fortress et al., 2013; Tuscher et al., 2016a). The spinogenic effects of E2 have been linked to improvements in learning and memory (Inagaki et al., 2012; Phan et al., 2015; Smith et al., 2009), suggesting that spinogenesis may be used as a morphological biomarker of the pro-cognitive effects of E2. Importantly, spine density loss in the hippocampus and cortex is an established correlate of cognitive decline in AD patients, and in some cases is associated more strongly with aberrant memory function than other pathological hallmarks of AD (DeKosky and Scheff, 1990; Selkoe, 2002). Unsurprisingly, APOE4 has deleterious effects on spine density relative to APOE3 in both transgenic mice and in humans, which may drive, or be a driver of, accelerated disease progression (Dumanis et al., 2009; Ji et al., 2003). Given that synaptic integrity and dendritic spine density are closely linked to learning and memory, spinogenic compounds, such as E2, may be of benefit to AD patients. However, whether the spinogenic or mnemonic benefits of E2 are modulated by APOE genotype is unknown.
The present study was designed to test whether APOE genotype interacts with E2 to modulate multiple forms of memory, dendritic spine density in the DH and medial prefrontal cortex (mPFC), and DH cell signaling and estrogen receptor expression in the well-characterized EFAD mouse model of Alzheimer’s disease (Liu et al., 2015; Tai et al., 2017; Youmans et al., 2012). Female EFAD-transgenic (Tg) mice (APOE+/+/5xFAD+/−) express 5 familial AD (FAD) mutations (5xFAD), and human APOE3 (APOE3/3/5xFAD+/−; E3FAD), APOE3/4 (APOE3/4/5xFAD+/−; E3/4FAD), or APOE4 (APOE4/4/5xFAD+/−; E4FAD). At 6 months of age, the hippocampus and frontal cortex of E4FAD mice express striking AD-like pathology, with significantly higher accumulation of total Aβ42, soluble Aβ42, soluble oligomeric Aβ42, amyloid plaques, and total APOE relative to E3FAD mice (Youmans et al., 2012). Thus, this model allows examination of the influence of APOE genotype on the cognitive and neural response to E2 against a background of AD-like pathology, which makes the EFAD model ideal for this study. First, ovariectomized female E3FAD, E3/4FAD, and E4FAD mice received bilateral dorsal hippocampal (DH) infusion of E2 immediately after training in object recognition (OR) and object placement (OP) tasks. Given the vulnerability of the DH and mPFC to excitatory synapse loss in AD, we next quantified dendritic spine density in the CA1 region of the DH and the mPFC. Because previous studies in wild-type mice showed that E2 rapidly increases activation of estrogen receptors and numerous cell-signaling kinases in the DH (e.g., Boulware et al., 2013; Kim et al., 2016, 2019), we also used western blotting to assess DH levels of kinase activity, estrogen receptor alpha (ERα), estrogen receptor beta (ERβ), and G-protein-coupled estrogen receptor (GPER). We hypothesized that E3FADs would be the most responsive to E2, such that DH E2 infusion would promote memory consolidation in both tasks and would increase dendritic spine density in both the DH and mPFC. We also hypothesized that E4FADs would be the least responsive to E2, and that E2’s efficacy in E3/4FADs would be intermediate between E3FADs and E4FADs. Based on our previous study of sex differences in E3FAD and E4FAD mice (Taxier et al., 2022a), we expected estrogen receptor alpha (ERα) levels to be highest in the DH of E4FAD mice but predicted effects on other estrogen receptors were unclear based on a lack of previous data on expression of these receptors in EFAD mice. We also anticipated that mnemonic benefits of E2 in EFADs, if present, could be linked to rapid activation of cell signaling. DH infusion of E2 facilitated memory consolidation and an increase in CA1 apical dendritic spine density in ovariectomized E3FAD and E3/4FAD, but not E4FAD, mice. Thus, in mice bearing significant AD-like pathology, E2 benefitted memory and structural plasticity in mice with zero or one copy of APOE4, whereas two copies of APOE4 blocked the memory-enhancing and spinogenic effects of E2. Understanding how APOE4 impedes the actions of E2 may provide new insights into the variable effects of E2 on AD risk in women.
2. Methods
2.1. Subjects
APOE-TR+/+/5xFAD+/− (EFAD) mice express five familial AD mutations (APP K670N/M671L, I716V, V717I, PS1 M146L, and L286V) under control of the neuron-specific mouse Thy-1 promoter, and express human APOE3 or APOE4 (Tai et al., 2017; Youmans et al., 2012). Female mice for the present experiment were homozygous for human APOE3 (E3FAD) or APOE4 (E4FAD), or had one copy each of APOE3 and APOE4 (E3/4FAD). EFAD mice were bred, weaned, and genotyped at the University of Illinois Chicago (UIC; Animal use protocol 17–066) and shipped to the University of Wisconsin-Milwaukee (UWM; Animal use protocol 19–20-03) at 2 months of age, where they were aged to 6 months before the start of behavioral testing. Prior to surgery, mice were housed in groups of up to 5 per cage and were singly housed following cannulation and ovariectomy. Mice were behaviorally tested in four separate cohorts, whereas brain analyses for all mice were conducted at the same time. The first two behavioral cohorts consisted of only E3FAD and E4FAD genotypes because E3/4FAD mice were not available at that time. When sufficient numbers of E3/4FAD mice could be generated for this project, they were added to the latter two cohorts, which included mice from all three genotypes. Thus, Ns for the E3/4FAD groups are smaller than those of the E3FAD and E4FAD groups. A subset of brains was dissected and frozen for Western blotting and the rest were Golgi impregnated for spine density analyses. Mice were maintained on a 12 h light/dark cycle with ad libitum access to food and water for the duration of the study. Protocols and procedures followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Wisconsin-Milwaukee Institutional Animal Care and Use Committee.
2.2. Surgeries
At the start of surgery, mice received a 5 mg/kg dose of subcutaneous Rimadyl for pain management. They were then anesthetized with isoflurane in 100% oxygen (5% for induction, 2% for maintenance) and placed in a stereotaxic apparatus (Kopf instruments) for ovariectomy and cannula implantation, which occurred within the same surgical session (Lewis et al., 2008; Taxier et al., 2019). Stainless steel bilateral guide cannulae (C232GC, 22 gauge; Plastics One) were implanted into the DH (−1.7 mm AP, ± 1.5 mm ML, and −2.3 mm DV) and affixed to the skull using dental cement (Darby Dental). Dummy cannulae (C232DC; Plastics One) were used to prevent clogging of the guide cannulae. Mice received MediGel carprofen (ClearH20) for postsurgical analgesia and were given one week to recover prior to the start of behavioral testing.
2.3. Drugs and Infusions
Hydroxypropyl-β-cyclodextrin (HBC)-encapsulated E2 (Sigma-Aldrich) was dissolved in 0.9% sterile saline to a concentration of 10 μg/μl and was infused at a rate of 0.5 μl/min for 1 min/hemisphere (Fortress et al., 2013; Kim et al., 2019; Taxier et al., 2019). Vehicle-treated mice received HBC (Sigma-Aldrich) dissolved in saline to the same concentration of cyclodextrin as in the E2 solution. For behavioral experiments, mice received a bilateral DH infusion of vehicle or E2 immediately post-training. Two weeks separated bouts of behavioral training to ensure that any acute effects of post-training infusions had dissipated prior to the next infusion. Two weeks following the conclusion of behavioral testing, mice were reinfused with the same treatment they received previously, and tissue was collected 5 min later for Western blotting or 2 h later for Golgi impregnation and spine counting.
2.4. Behavioral Tasks
Object recognition (OR) and object placement (OP) were conducted as described previously (Boulware et al., 2013; Fernandez et al., 2008; Fortress et al., 2013; Kim et al., 2019; Taxier et al., 2019; Tuscher et al., 2018). Mice were handled for 30 s/day for three days prior to exposure to the behavioral arena. On the second day of handling, a Lego Duplo block (6.3 × 3.1 × 2.3 cm) was placed into each home cage to acclimate mice to objects for the remainder of handling and habituation. After handling was completed, mice were allowed to habituate to the empty behavioral arena (60 cm x 60 cm x 47 cm) for 5 min/day for two days. Light levels in the area were measured daily to ensure even illumination of 75 ± 5 lux in each corner of the arena. The following day, mice were acclimated to the empty arena for 2 min, removed to a holding cage, and immediately returned to the arena and exposed to two identical objects placed 5 cm from the upper left and right corners of the arena. Mice were then allowed to freely explore the objects until they accumulated 30 s of object exploration (or until 20 min had elapsed), defined as any time a mouse’s nose was within 2 cm of the object. The 30 s criterion was chosen to ensure that mice spent an equivalent amount of time exploring the training objects prior to drug infusion, as different amounts of time spent with these objects could alter the strength of the memory in such a way as to occlude effects of drug manipulation. Mice that did not reach the 30 s criterion within the allotted 20 min were excluded. Immediately following training, mice received bilateral DH infusion of HBC vehicle or E2 and were then returned to their home cage.
OR testing was conducted 24 h after drug infusion, at which point mice were returned to the arena in which one familiar object from the training phase was replaced by a novel object. Mice were again allowed to accumulate 30 s of object exploration. OP testing was conducted 4 h after drug infusion; here, mice were allowed to explore one familiar training object in its original location, and the second training object in a new location (bottom left or right corner of the arena). Mice that remember the identity and location of the training objects, respectively, spend more time than chance (15 s) exploring the novel or moved objects during testing (Kim et al., 2019; Taxier et al., 2019; Tuscher et al., 2016b). Time spent with the objects and time to accumulate 30 s of exploration were recorded using ANYmaze automated tracking software and scored by a researcher blinded to treatment and genotype (San Diego Instruments). The order of OR and OP testing was counterbalanced across groups, and approximately two weeks separated OR training and testing from OP training and testing to allow for acute effects of post-training E2 infusion to dissipate prior to the next training session.
Although vehicle-treated ovariectomized wild-type mice typically remember the identity and location of the training objects after a 24 h (OR) or 4 h (OP) delay, respectively (Kim et al., 2019; Taxier et al., 2019; Tuscher et al., 2016b), we have previously shown that only male E3FADs, and not female E3FADs or E4FADs of either sex, remember the training object identity or location when tested at these same delays (Taxier et al., 2022a). Thus, given that female E3FADs and E4FADs exhibit impaired memory at the 24 h (OR) and 4 h (OP) delays, mice in the present experiment were tested using these delays to assess whether DH E2 infusion could facilitate memory consolidation.
2.5. Western Blotting
Two weeks after the conclusion of behavioral testing, a subset of mice (10–12/group) were infused into the DH with vehicle or E2. Mice were cervically dislocated and decapitated 5 min later, based on previous studies in wild-type mice showing that E2 or GPER activation increases phosphorylation of numerous cell-signaling kinases, including ERK and JNK, in the DH at this time point (e.g., Boulware et al., 2013; Kim et al., 2016, 2019). Brains from each group were extracted, and the DH was bilaterally dissected immediately on ice and frozen at −80°C. Western blotting was conducted as described previously (Boulware et al., 2013; Fernandez et al., 2008; Kim et al., 2019; Koss et al., 2018; Taxier et al., 2019). Tissue samples were resuspended 1:25 weight/volume in lysis buffer containing PMSF and a protease inhibitor cocktail (Sigma-Aldrich) and homogenized using a probe sonicator (Branson Sonifier 250). Homogenates were electrophoresed on 10% TGX (Tris-Glycine eXtended) stain-free precast gels (Bio-Rad) and transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad) using the TransBlot Turbo system (Bio-Rad). Total protein transfer was then verified using a ChemiDoc MP gel imager (Bio-Rad).
Membranes were blocked in 5% milk and incubated with following primary antibodies overnight at 4°C: phosphorylated 42/44 ERK (#9101, 1:2,000, Cell Signaling Technology), total ERK (#9102, 1:20,000, Cell Signaling Technology), phosphorylated 46/54 JNK (#4668, 1:1000, Cell Signaling Technology), total JNK (#9252, 1:1000, Cell Signaling Technology), ERα (H-184, 1:1000, Santa Cruz Biotechnology), ERβ (#PA1-310B, 0.25µg/mL, Thermo Fisher), and GPER (ab39742, 1:250, Abcam). The following day, all blots were incubated at room temperature with a rabbit (#7074, 1:5000, Cell Signaling Technology) or mouse (#7076, 1:5000, Cell Signaling Technology) HRP-conjugated secondary antibody. Blots were then developed using Clarity Max chemiluminescent substrate (Bio-Rad) and protein expression was detected using a ChemiDoc MP gel imager (Bio-Rad). Densitometry was performed using Image Lab software (Bio-Rad Image Lab v 5.2). Blots were stripped and reprobed for β-Actin (#4967, 1:1000, Cell Signaling Technology) for protein normalization. Data were expressed as average volume intensity as a percentage compared to vehicle-treated E3FAD females.
2.6. Golgi Impregnation and Spine Counting
Whole brains (n=4–9/group) were collected for Golgi impregnation two weeks after the conclusion of behavioral testing. Mice were cervically dislocated and decapitated 2 h following DH infusion of vehicle or E2. The 2 h time point was chosen because previous work from our lab and others indicates that DH infusion of E2 increases DH and mPFC dendritic spine density at this delay (Murakami et al., 2006; Tuscher et al., 2016a). Golgi staining was performed as described previously (Frankfurt et al., 2011; Kim et al., 2019; Tuscher et al., 2016a) using the Rapid Golgi Stain Kit (FD Neuro Technologies). A cryostat was used to slice tissue into 100 μm sections, which were directly mounted onto gelatin-coated microscope slides. Mounted tissue was then stained according to the Rapid Golgi Stain Kit instructions, and coverslipped with Permount. Slides were coded so that the individual counting spines was blind to treatment, and slides were kept in the dark when not in use.
Secondary basal dendrites and tertiary apical dendrites were counted from pyramidal neurons in the dorsal hippocampal CA1 and layer II/III of the prelimbic/infralimbic mPFC under an Olympus BX51WI microscope (100x with oil) using NeuroLucida (v 11.08, MBF Bioscience). Accuracy of DH cannula placement was visually validated by examining sections containing DH tissue. Dendritic segments selected for spine counting were between 10–20 μm in length and 0.5–1.3 μm thick. Neurons selected for analysis were required to have well impregnated cell bodies and dendrites, and to be clearly distinguishable from adjacent cells.
Dendritic spines were identified by an experimenter scrolling through the z-plane of stained tissue in real time, and spines were reconstructed using NeuroLucida morphometric markers. Spines were classified according to three categories based on shape: mushroom, thin, or stubby (Harris et al., 1992; Kim et al., 2019). Mushroom spines had head diameters at least twice the size of their neck diameters, whereas thin spines had head diameters less than or equal to their neck diameters. Stubby spines had neck diameters relatively equal to the total length of the spine, with no discernable spine head. Two dendritic segments/neuron and 6 cells/region were included in the analysis. After segment tracing and spine marking, data were exported to NeuroLucida Explorer (MBF Bioscience), where a Branched Structure Analysis allowed for visualization of the number and type of spines on each traced dendritic segment. Spine density was calculated as the number of spines/10 μm dendrite.
2.7. Data Analysis
All statistical analyses were conducted using GraphPad Prism 9 software (La Jolla, CA). To assess within-group learning for OR and OP, one-sample t-tests were used to determine whether the time spent with each object during testing significantly differed from chance (15 s). Differences in memory between groups were assessed using two-way ANOVAs with treatment and genotype as between-subject variables. Similar two-way ANOVAs were used to analyze western blot and dendritic spine density data. Significant main effects were followed by Tukey’s post hoc tests for OR, OP, western blot, and dendritic spine data. Statistical significance was set at p < 0.05 for all statistical tests, and trends were determined by p < 0.10.
3. Results
3.1. DH infusion of E2 enhances memory in E3FAD and E3/4FAD, but not E4FAD, mice
3.1.1. Object recognition
To determine whether APOE genotype influences the ability of E2 to facilitate memory for a previously seen object or location, ovariectomized female E3FAD, E3/4FAD, and E4FAD mice were tested in the OR and OP tasks 24 h or 4 h, respectively, after receiving a post-training bilateral dorsal hippocampal infusion of vehicle (veh; E3FAD n = 21, E3/4FAD n = 12, E4FAD n = 21) or E2 (E3FAD n = 16, E3/4FAD n = 11, E4FAD n = 14). In OR, E2-treated E3FADs (Fig. 1A; t(15) = 3.406, p = 0.004) and E3/4FADs (Fig. 1A; t(10) = 2.745, p = 0.0201) spent significantly more time than chance (15 s) with the novel object during testing, indicating intact memory for the identity of the training objects. By contrast, E2-treated E4FADs, as well as all veh-treated groups, did not spend more time than chance with the novel object during testing, suggesting that two copies of the APOE4 allele prevented E2 from facilitating memory. A two-way ANOVA revealed a main effect of E2 treatment (Fig. 1A; F(1, 89) = 9.614, p = 0.003), as well as a significant genotype x treatment interaction (F(2, 89) = 4.821, p = 0.01) for time spent with the novel object during testing. These effects were driven by the selective benefits of E2 on OR memory in E3FAD and E3/4FAD females only. Tukey’s post hoc comparisons support the notion that E2-treated E3FAD and E3/4FADs drove the main effect of treatment, as there was a significant difference between vehicle- and E2-treated E3/4FADs (p < 0.031), and a trend for a difference between vehicle-and E2-treated E3FADs (p = 0.07).
Figure 1. E2 facilitates object recognition and spatial memory formation in E3FAD and E3/4FAD, but not E4FAD, females.
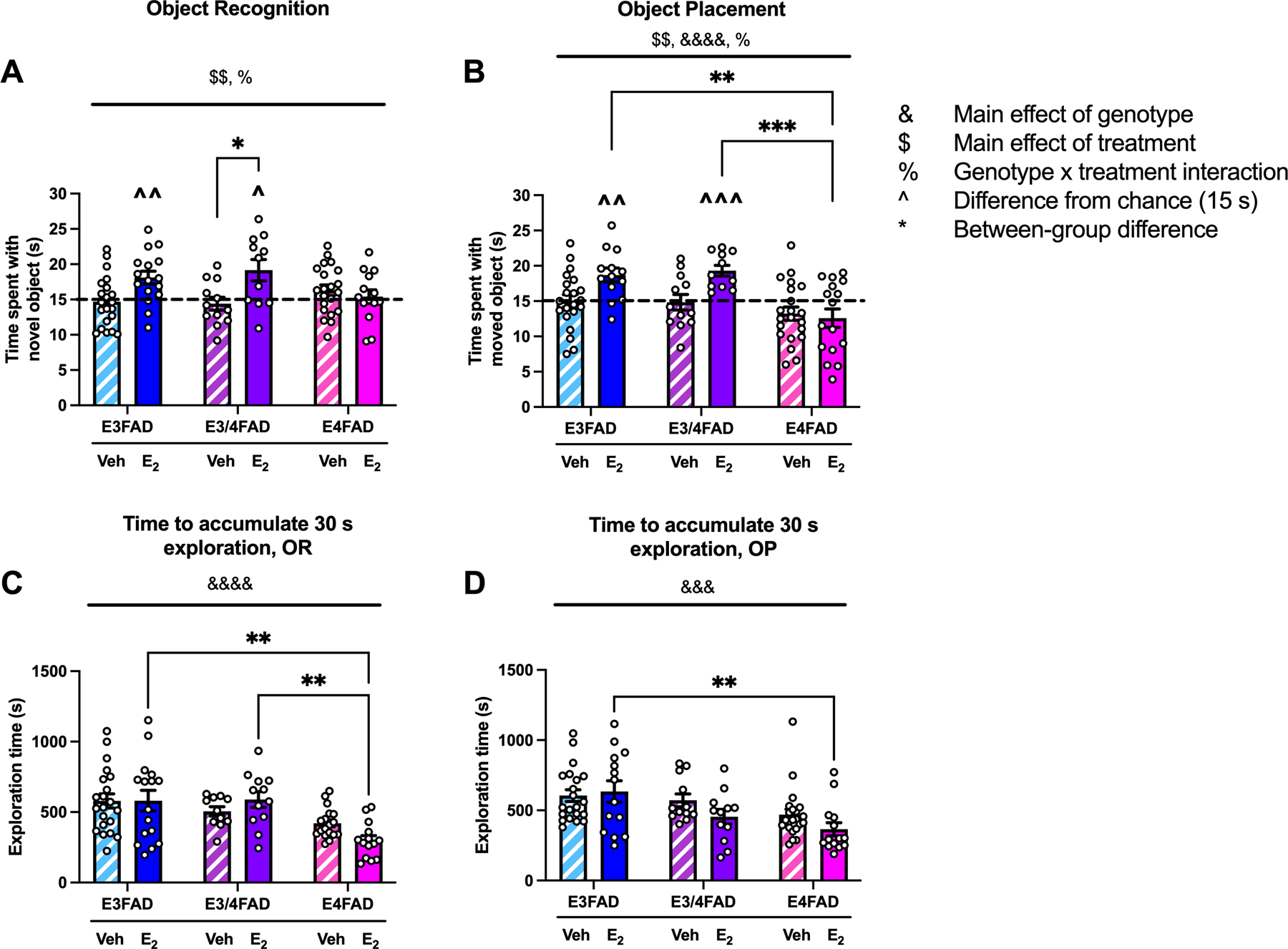
(A,B) E3FAD and E3/4FAD females treated with E2 spent significantly more time than chance (dashed line at 15 sec; ^ p < 0.05, ^^ p < 0.01, ^^^ p < 0.001) with the novel (A) and moved (B) objects during testing. In contrast, E2-treated E4FAD females did not, nor did any groups treated with vehicle (veh). The genotype-dependent effect of E2 on memory was also reflected in between-subjects analyses by a main effect of treatment ($ $p < 0.01) and treatment x genotype interaction (%p < 0.05). In OP, the main effect of genotype (&&&&p < 0.0001) indicated that E3FADs and E3/4FADs spent more time with the moved object than E4FADs. Tukey’s post hoc comparisons revealed significant differences between veh- and E2-treated E3/4FADs in OR (A; *p < 0.05), and that E2-treated E3FADs (**p < 0.01) and E3/4FADs (***p < 0.001) spent more time with the moved object in OP than E2-treated E4FADs. (C,D) The main effect of genotype was significant for both OR (C; &&&&p < 0.0001) and OP (D; &&&p < 0.001). E4FADs took less time to accumulate 30 sec of exploration in both tasks. Tukey’s post hoc comparisons indicated that E2-treated E4FADs were faster than E2-treated E3/4FADs and E3FADs in OR (C; **p < 0.01), and than E2-treated E3FADs in OP (D; **p < 0.01). Bars represent mean ± standard error of the mean (SEM).
Interestingly, time to accumulate 30 s of object exploration during testing was significantly influenced by genotype (Fig 1C; F(2, 86) = 11.93, p < 0.0001), such that E4FADs took significantly less time to reach 30 s of object exploration relative to other groups. E4FADs treated with E2 were particularly fast to complete object exploration, as demonstrated by post hoc comparisons to other E2-treated groups (Fig 1C; p = 0.002 vs E2-treated E3FADs, and p = 0.0004 vs E2-treated E3/4FADs). Given the significant genotype effect during testing, we also examined time to accumulate 30 s of object exploration during training, but found no significant effects (Veh-treated E3FADs, M = 541.35, SEM = 56.359; E2-treated E3FADs, M = 537.957, SEM = 48.977; Veh-treated E3/4FADs, M = 447.892, SEM = 65.291; E2-treated E3/4FADs, M = 538.325, SEM = 70.724; Veh-treated E4FADs, M = 557.613, SEM = 71.566; E2-treated E4FADs, M = 382.463, SEM= 57.742). This pattern of data suggests that E2-treated E4FADs were not generally hyperactive, but rather just quick to explore objects during testing.
3.1.2. Object placement
The OP task yielded similar results to OR in that E2 treatment selectively improved memory in E3FAD and E3/4FAD females. E2-treated E3FAD (Fig. 1B; t(12) = 3.741, p = 0.003) and E3/4FAD (Fig. 1B; t(10) = 5.644, p = 0.0002) females spent significantly more time than chance with the moved object during testing, indicating intact memory for the identity of the training objects. As in OR, E2-treated E4FAD and veh-treated mice of all genotypes did not spend more time than chance with the moved object during testing, again suggesting that APOE4 homozygotes were unresponsive to the mnemonic benefits of E2. As with OR, the main effect of treatment was significant (Fig. 1B; F(1, 88) = 8.183, p = 0.005), as was the genotype x treatment interaction (Fig. 1B; F(2, 88) = 3.591, p = 0.032), reflecting the selective benefit of E2 for E3FAD and E3/4FAD females. However, unlike OR, the main effect of genotype was also significant (Fig. 1B; F(2, 88) = 10.72, p < 0.0001), such that E3FAD and E3/4FADs exhibited better memory for object location than E4FADs. In support of these conclusions, Tukey’s post hoc comparisons indicated that E2-treated E4FADs spent significantly less time with the moved object than E2-treated E3FADs (Fig. 1B; p = 0.002) and E3/4FADs (Fig. 1B; p = 0.0009).
Similar to OR, time to accumulate 30 s of object exploration during testing was significantly modulated by genotype (Fig 1D; F(2, 89) = 8.765, p = 0.0003), such that E4FADs were faster to accumulate 30 s of object exploration relative to other groups. Exploration time in E2-treated E4FADs was significantly lower than that of E2-treated E3FADs (Fig 1D; p = 0.007). We again examined time to accumulate 30 s of object exploration during training, but found no significant effects (Veh-treated E3FADs, M = 495.968, SEM = 59.455; E2-treated E3FADs, M = 400.2, SEM = 56.820; Veh-treated E3/4FADs, M = 431.2, SEM = 77.794; E2-treated E3/4FADs, M = 399.033, SEM = 34.655; Veh-treated E4FADs, M = 430.119, SEM = 67.464; E2-treated E4FADs, M = 326.927, SEM = 36.685).
Combined, data from both OR and OP tasks suggest that E2 supports memory consolidation in E3FAD, and E3/4FAD, but not E4FAD, females.
3.2.1. ERα is elevated in the DH of E4FAD mice
Because we previously found that APOE4 increased ERα expression in the DH of gonadally-intact EFAD mice of both sexes (Taxier et al., 2022a), we examined effects of DH vehicle or E2 infusion on levels of this protein and two other estrogen receptors, ERβ and G-protein-coupled estrogen receptor (GPER). Two-way ANOVA revealed a significant main effect of genotype, but not treatment, nor an interaction, for levels of ERα (Fig. 2A,B; F(2,48) = 6.956, p = 0.002), driven by elevated ERα in E2-treated E4FADs relative to veh-treated E3FADs (p = 0.019 veh-treated E3FADs vs E2-treated E4FADs, p = 0.052 veh-treated E3FADs vs veh-treated E4FADs). There were no significant effects of treatment or genotype in DH protein expression of either ERβ or GPER (Fig. 2A,C,D).
Figure 2. ERα levels in the DH were increased in E4FAD females.
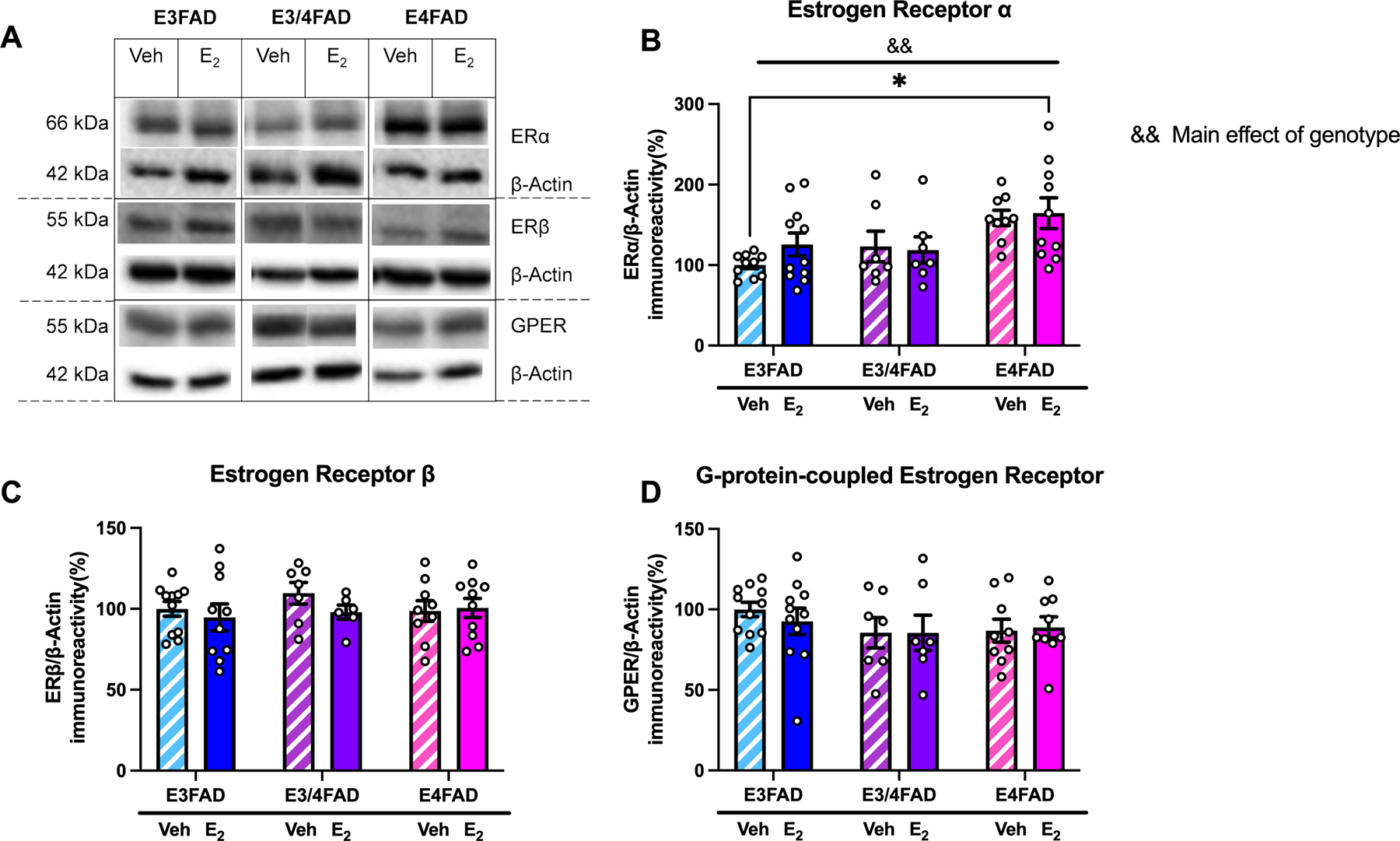
(A) Representative Western blot images. (B) ERα was affected by APOE genotype (&&p < 0.01 = main effect of genotype) such that levels were highest in E4FAD females. ERα levels were significantly higher in E2-treated E4FAD mice than in veh-treated E3FAD mice (*p < 0.05). (C,D) Levels of ERβ (C) and GPER (D) were unaffected by treatment or genotype.
3.2.2. ERK and JNK are not rapidly phosphorylated by E2 in EFAD mice
In wild type C57BL/6 mice, the memory-enhancing effects of E2 are facilitated by ERα and ERβ signaling via phosphorylation of extracellular signal-regulated kinase (ERK) (Boulware et al., 2013). Although activation of GPER also facilitates memory, it does so independently from ERK and from DH E2 infusion via c-jun N-terminal kinase (JNK) activation (Kim et al., 2016). Because the cell-signaling events underlying E2-facilitated memory are uncharacterized in EFADs, we examined whether E2 facilitates memory via activation of ERK or JNK signaling in this model. Surprisingly, there were no significant effects of E2 or genotype on ERK or JNK phosphorylation in the DH (Table 2).
Table 2.
CA1 and mPFC spine density measures unaffected by E2 treatment or genotype
| Brain region | Spine type | E3FAD | E3/4FAD | E4FAD | |||
|---|---|---|---|---|---|---|---|
| Veh | E2 | Veh | E2 | Veh | E2 | ||
| CA1 | Apical thin | 3.69 ± 0.18 | 4.51 ± 0.29 | 4.06 ± 0.28 | 4.02 ± 0.11 | 4.26 ± 0.27 | 4.32 ± 0.21 |
| Apical mushroom | 3.05 ± 0.11 | 3.06 ± 0.25 | 3.30 ± 0.23 | 3.71 ± 0.30 | 3.52 ± 0.28 | 3.20 ± 0.11 | |
| mPFC | Apical total | 11.6 ± 0.88 | 10.35 ± 0.46 | 11.43 ± 0.95 | 10.03 ± 0.72 | 10.19 ± 0.85 | 9.84 ± 0.48 |
| Apical thin | 2.74 ± 0.24 | 2.27 ± 0.06 | 2.41 ± 0.26 | 2.24 ± 0.10 | 2.75 ± 0.28 | 2.51 ± 0.14 | |
| Apical stubby | 5.32 ± 0.48 | 4.77 ± 0.32 | 5.42 ± 0.43 | 4.95 ± 0.59 | 4.59 ± 0.55 | 4.46 ± 0.38 | |
| Basal stubby | 7.36 ± 0.46 | 6.63 ± 0.28 | 6.80 ± 0.26 | 6.95 ± 0.57 | 7.82 ± 0.38 | 6.79 ± 0.24 | |
| Basal mushroom | 2.84 ± 0.20 | 2.96 ± 0.23 | 2.42 ± 0.32 | 2.73 ± 0.50 | 2.32 ± 0.27 | 2.61 ± 0.29 | |
Values represent mean spines/10 µM ± SEM
3.3. DH and mPFC spine densities are affected by APOE genotype and E2 treatment
We next examined apical and basal dendritic spine density on pyramidal neurons in hippocampal area CA1 and in the prelimbic/infralimbic area of the mPFC to assess whether APOE genotype influenced the ability of E2 to increase dendritic spine density 2 h following DH infusion. Representative images of vehicle- and E2-treated spines for each genotype are shown in Figs. 3A, 4A, and 5A, whereas representative images of mushroom, stubby, and thin spines are shown in Fig. 3B. There were no effects of genotype or E2 treatment, nor an interaction, on mPFC apical spines (Table 1). Treatment and genotype effects on CA1 apical, and CA1 and mPFC basal spines, are detailed below.
Figure 3. E2 increases CA1 apical spine density in E3FAD and E3/4FAD, but not E4FAD, females.
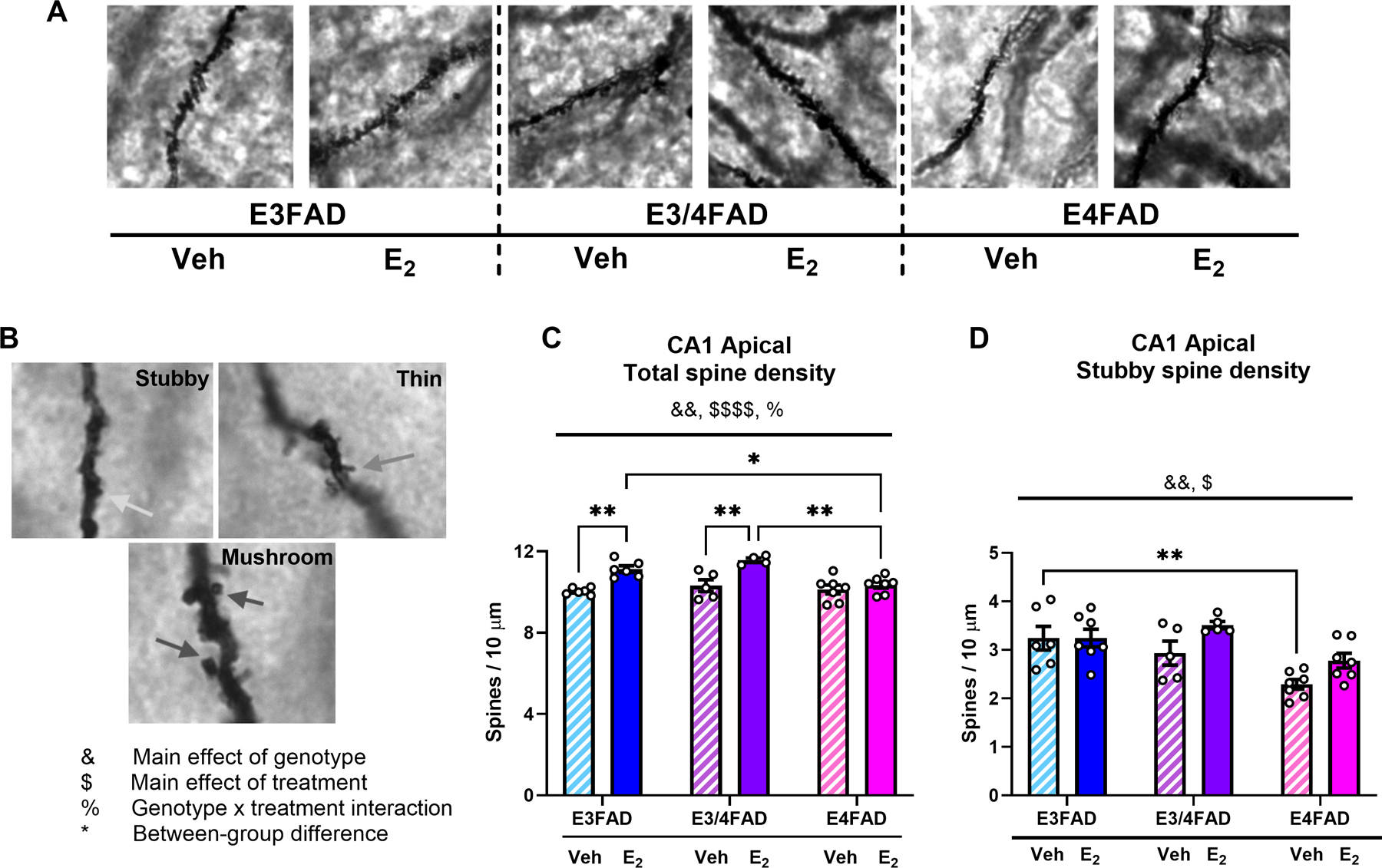
(A) Representative images of CA1 apical dendritic spine segments at 100X magnification. (B) Representative images of stubby (top left), thin (top right), and mushroom (bottom) spines. Arrows indicate individual examples of each spine class. (C) Total CA1 apical dendritic spine density was increased by DH E2 infusion ($ $ $ $p < 0.01 = main effect of treatment) and reduced by two copies of APOE4 (&&p < 0.05 = main effect of genotype; %p < 0.05 = interaction). In both E3FAD and E3/4FAD females, E2 treatment increased total apical spine density relative to veh (**p < 0.01), but no benefit of E2 was observed in E4FAD mice. Moreover, apical spine density was significantly lower in E2-treated E4FAD mice than in E2-treated E3FAD (*p < 0.05) and E3/4FAD (**p < 0.01) mice. (C) CA1 apical stubby spines was modestly increased by E2 ($p < 0.05 = main effect of treatment) and reduced by two copies of APOE4 (&&&p < 0.001 = main effect of genotype). No differences were observed between veh and E2 within any genotype, but veh-treated E3FADs had significantly more stubby spines than veh-treated E4FADs (**p < 0.01).
Figure 4. CA1 basal spine density was reduced by APOE4.
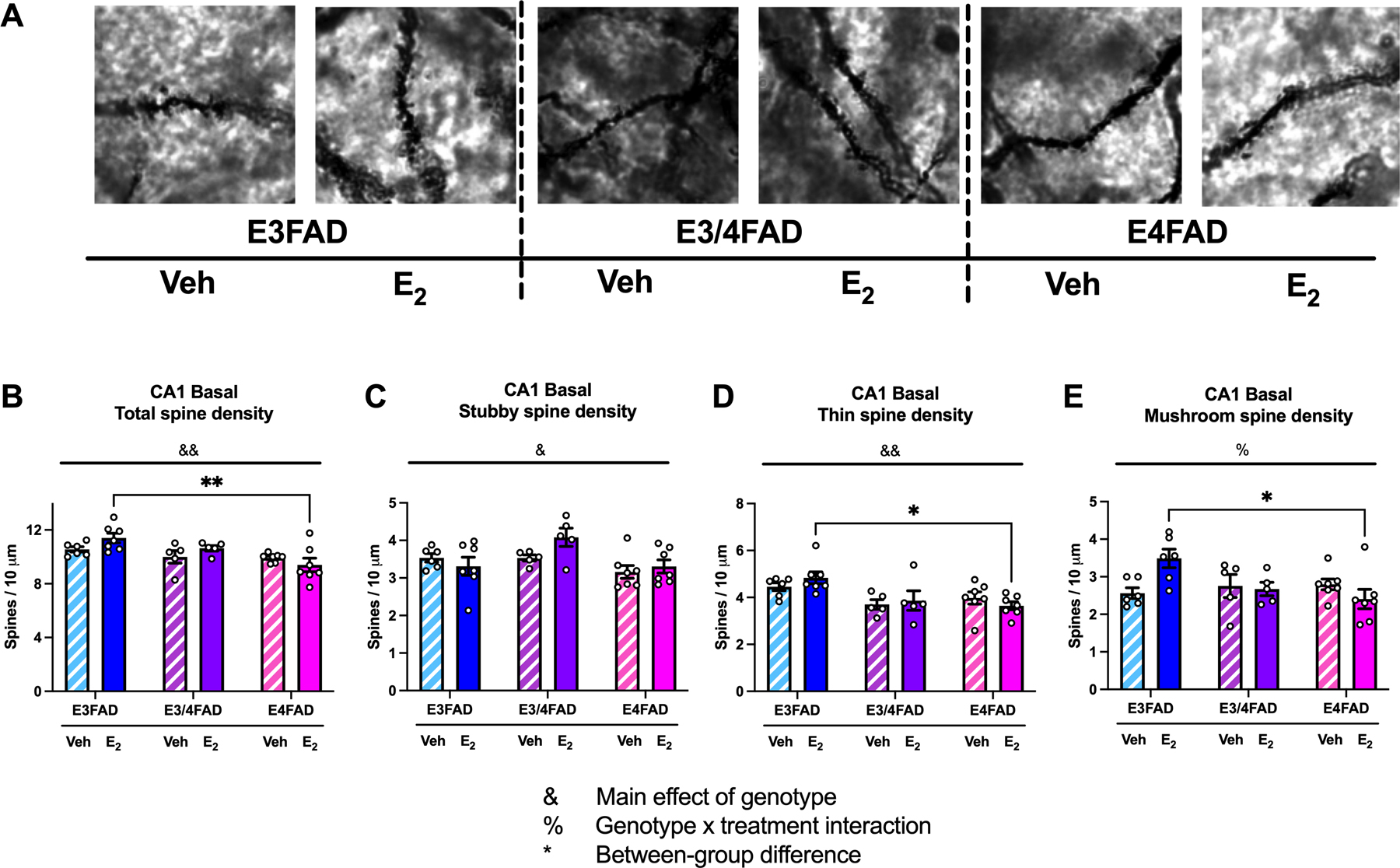
(A) Representative images of CA1 basal dendritic spine segments at 100X magnification. (B-E) Total CA1 basal spine density (B), stubby spine density (C), and thin spine density (D) was decreased in E4FADs, regardless of treatment (&p < 0.05, &&p < 0.01 = main effect of genotype). The treatment x genotype interaction was significant for mushroom spines (E; %p < 0.05). E2-treated E3FAD females had more total spines, thin spines, and mushroom spines than E2-treated E4FAD females. (*p < 0.05, **p < 0.01).
Figure 5. mPFC basal spine density was reduced by APOE4.
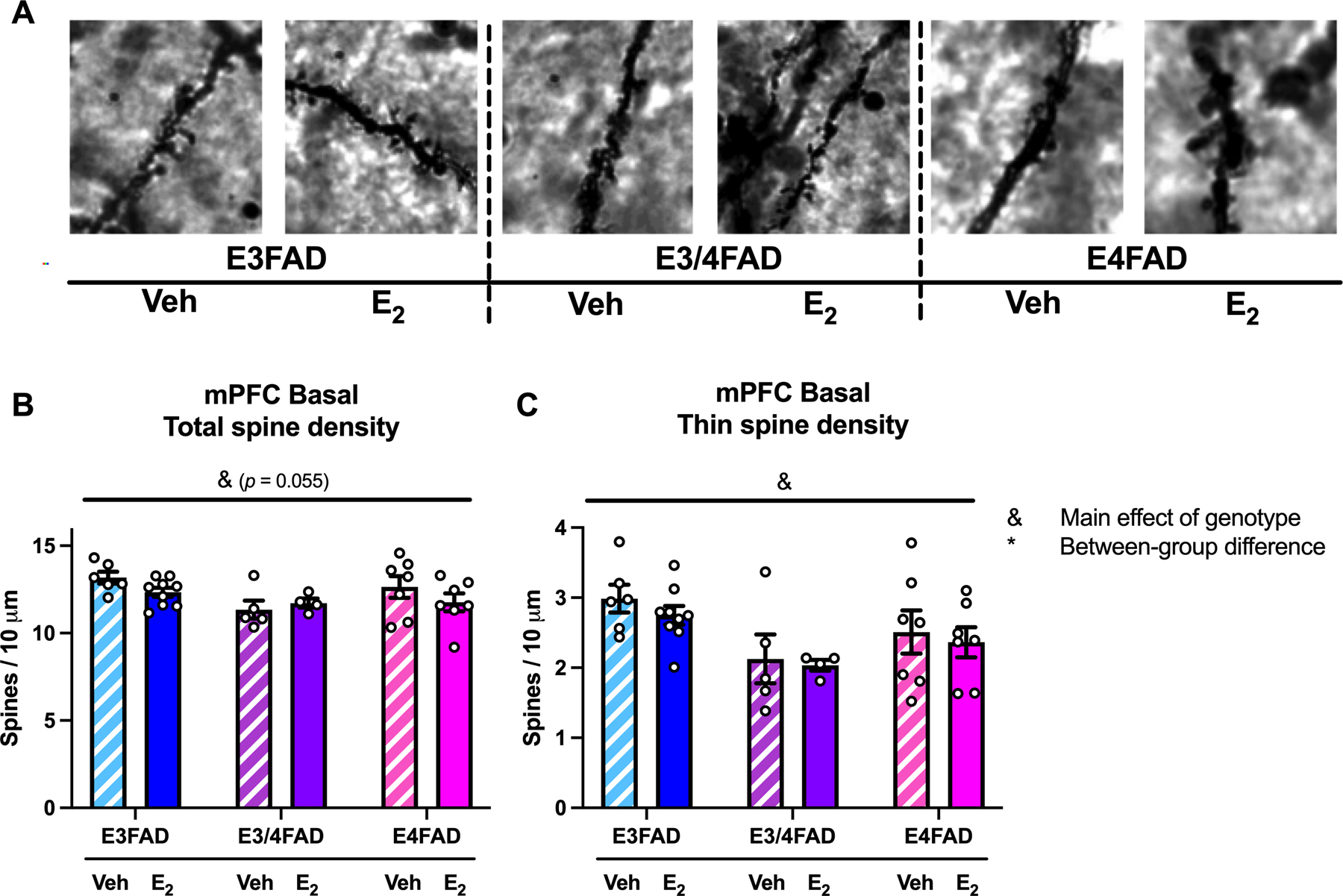
(A) Representative images of mPFC basal dendritic spine segments at 100X magnification. (A) APOE4 genotype reduced mPFC basal spines, as suggested by a modest main effect of genotype for total spines (B; &p = 0.055) and a significant main effect in thin spines (C; &p < 0.05). No between-group differences were observed.
Table 1.
Levels of phosphorylated ERK and JNK were unaffected by E2 treatment or genotype
| Protein* | E3FAD | E3/4FAD | E4FAD | |||
|---|---|---|---|---|---|---|
| Veh | E2 | Veh | E2 | Veh | E2 | |
| p42 ERK | 100.0 ± 10.56 | 93.21 ± 10.05 | 109.39 ± 20.84 | 112.56 ± 16.44 | 109.45 ± 15.76 | 110.17 ± 14.46 |
| p44 ERK | 100.0 ± 9.32 | 119.51 ± 8.41 | 97.37 ± 12.20 | 124.85 ± 9.37 | 133.63 ± 18.73 | 111.44 ± 9.96 |
| p46 JNK | 100.0 ± 2.05 | 89.52 ± 3.88 | 93.15 ± 6.28 | 94.97 ± 4.50 | 102.83 ± 5.25 | 94.02 ± 4.60 |
| p54 JNK | 100.0 ± 5.86 | 89.70 ± 7.51 | 96.39 ± 8.67 | 90.60 ± 7.57 | 97.68 ± 7.94 | 8.35 ± 8.05 |
All proteins were normalized to the vehicle-treated E3FAD group, whose values were set to 100.
Values represent mean % immunoreactivity (± SEM) for the target phosphorylated protein divided by unphosphorylated total protein.
3.3.1. CA1
APOE4 homozygosity blocked E2’s effects on CA1 total apical spine density, as suggested by the fact that E2 significantly increased CA1 total apical spine density in E3FADs and E3/4FADs, but not E4FADs. Significant main effects of genotype (Fig. 3C; F(2,29) = 6.746, p = 0.0039) and E2 treatment (Fig. 3C; F(1,29) = 30.25, p < 0.0001) were observed, as well as a genotype x treatment interaction (Fig. 3C; F(2,29) = 4.536, p = 0.0193). Tukey posthoc tests revealed that total apical spine density was significantly higher in E3FAD (p = 0.0032) and E3/4FAD (p = 0.0033) mice treated with E2 than with vehicle, whereas there were no effects of E2 found in E4FAD mice (p = 0.932). Moreover, apical spine density was significantly lower in E2-treated E4FAD mice than in E2-treated E3FAD (p = 0.0451) and E3/4FAD mice (p = 0.002). Further analysis by spine subtype revealed no significant effects of genotype or treatment, nor interactions, on CA1 apical thin or mushroom spines (Table 1). However, the main effects of genotype (Fig. 3D; F(2,31) = 4.261, p = 0.0002) and treatment (Fig. 3D; F(1,31) = 9.712, p = 0.012) were significant for CA1 apical stubby spines. Here, the only significant between-group differences were between the E3FAD and E4FAD vehicle groups, such that veh-treated E3FAD mice had more stubby apical spines than veh-treated E4FAD mice (p = 0.006).
CA1 basal spine density was influenced by genotype only. The main effect of genotype was significant (Fig. 4B; F(2,31) = 7.774, p = 0.002), but the treatment effect and interaction were not. This effect was driven largely by elevated total spine density in E2-treated E3FAD mice, who had significantly higher basal spine density than E2-treated E4FADs (Fig. 4B; p = 0.002). Further analysis by spine subtype revealed that the main effect of genotype on total CA1 basal spine density was driven by significant effects of genotype on CA1 basal stubby spines (Fig. 4C; F(2,31) = 4.49, p = 0.019) and thin spines (Fig. 4D; F(2,31) = 7.887, p = 0.002). E2-treated E3FADs exhibited elevated thin basal spines relative to E2-treated E4FADs (Fig. 4D; p = 0.013). The genotype x treatment interaction was significant for CA1 basal mushroom spines (Fig. 4E; F(2, 30) = 5.151, p = 0.012), such that mushroom spine density was elevated in E2-treated E3FADs relative to E2-treated E4FADs (Fig. 4E; p = 0.012).
3.3.2. mPFC
Neither genotype nor E2 treatment affected mPFC apical spine density in terms of total spines or spine subtypes (Table 1). However, there were modest effects of genotype on mPFC basal spines, such that E4FADs again had the lowest spine density. For total basal spines, a two-way ANOVA revealed a trend towards a significant main effect of genotype (Fig. 5B; F(2, 30) = 3.178, p = 0.055), which appeared to be driven by a significant reduction among E4FAD mice in thin spines (Fig. 5C; F(2, 30) = 5.212, p = 0.011). Although no post hoc comparisons were significant, E3FADs generally had elevated mPFC basal thin spines relative to EFADs of other genotypes, regardless of treatment. There were no significant genotype, treatment, nor genotype x treatment effects on levels of mPFC basal stubby or mushroom spines (Table 1).
4. Discussion
Because APOE4 status and female sex are unmodifiable risk factors that act synergistically to increase AD risk (Altmann et al., 2014), evaluating interventional approaches that might benefit female APOE4 carriers is a crucial step towards providing individualized treatment to AD patients. Here, we investigated whether APOE genotype interacts with the ability of E2 to promote memory consolidation, modulate estrogen receptor expression, and increase CA1 or mPFC dendritic spine density in an EFAD mouse model of AD designed to model APOE-associated disease risk against a background of AD-like pathology. We hypothesized that E2 would have the most beneficial effects on memory consolidation and spine density in ovariectomized E3FAD mice, and that E2 may exert lesser effects in E3/4FAD mice due to the presence of one APOE4 allele. Based on data from women showing no protective effect of estrogen therapy on cognitive decline in APOE4 carriers (Yaffe et al., 2000), we expected that E2 would have little to no influence on memory or spine density in E4FAD females and intermediate effects in E3/4FAD mice due to the presence of one APOE4 allele. We found that DH infusion of E2 facilitated object memory consolidation in both E3FAD and E3/4FAD mice, but not E4FAD mice, indicating that two copies of the APOE4 allele blocks the memory-enhancing effects of E2. The effects of E2 in E3FAD and E3/4FAD females were associated with an increase in total CA1 apical spine density. Independent of E2 treatment, APOE4 genotype alone reduced CA1 and mPFC spine density and increased levels of ERα in the DH.
The beneficial effects of acute DH E2 infusion on object recognition and spatial memory consolidation in 6-month-old ovariectomized E3FAD and E4FAD mice are consistent with an extensive literature from our lab and others showing similar benefits of DH E2 infusion in 2–3-month-old wild-type ovariectomized C57BL/6 mice (Fernandez et al., 2008; Fortress et al., 2013; Gross et al., 2021; Packard and Teather, 1997; Taxier et al., 2019). A primary difference is that the delays between training and testing used here (24 h for OR and 4 h for OP) were shorter than those at which E2 benefits wild-types (48 h for OR and 24 h for OP), but this difference is to be expected given the significant AD-like pathology present in the brains of EFAD mice (Tai et al., 2017; Youmans et al., 2012). Although others have shown a potential benefit of chronic E2 treatment for decreasing amyloid accumulation in ovariectomized EFAD mice (Kunzler et al., 2014) and in ovariectomized 3xTg mice (Carroll et al., 2007), our data are the first to demonstrate that a single acute dose of E2 can facilitate memory consolidation in E3FAD and E3/4FAD, but not E4FAD mice. Previous work demonstrated that chronic E2 treatment initiated in 3-month-old EFADs can significantly reduce amyloid burden (Kunzler et al., 2014). We find it particularly striking that E3FAD and E3/4FAD mice remain responsive to a single acute dose of E2 until at least 6 months of age, at which point there is already substantial AD-like pathology evident (Tai et al., 2017). Although recent clinical studies suggest that estrogen therapy is not efficacious for women with AD, these findings are clouded by differences in treatment regimen, age of experimental subjects and menopausal status, and lack of stratification by APOE status in some instances. Thus, whether APOE3+ women with AD who express 0 or 1 copies of APOE4 can benefit cognitively from E2 treatment after substantial disease progression remains a question for future study.
Although there was a significant interaction between genotype and treatment in both the OR and OP tasks, the pro-cognitive effects of E2 were more apparent for OP, suggesting that spatial memory may be more sensitive to E2 in ovariectomized E3FAD and E3/4FAD mice than recognition memory. Alternatively, the longer 24-hour delay between training and testing for OR may have been more demanding on memory function than the shorter 4-hour delay for OP. Interestingly, E4FAD mice took less time than other genotypes to accumulate 30 seconds of object exploration during testing, but not during training, which is an unusual finding in our extensive experience using these tasks to examine estrogenic regulation of memory. One possible explanation is that E4FAD mice noticed a change during testing that caused an increase in exploratory behavior, but without the ability to discriminate between old and new objects and locations, this increase did not result in a preference for novelty. Our previous work with gonadally-intact female EFAD mice indicated that during object memory habituation trials, E4FAD females explored the outer third of the open field box more than E3FAD females (Taxier et al., 2022b). Our objects are located in this peripheral zone, and so a possible training-induced increase in exploration in this zone could have caused this group to contact and explore the objects faster than other groups that were exploring the entirety of the testing arena. Future studies will need to further address this possibility.
Importantly, our results also suggest that individuals carrying a single copy of the APOE4 allele may still benefit from the cognitive benefits of E2 treatment, whereas individuals with two copies would not. This finding is somewhat consistent with data from human patients, from which the conclusion is sometimes reached that estrogens are ineffectual or in some instances harmful for cognition in APOE4 carriers (Burkhardt et al., 2004; Kang and Grodstein, 2012). Others have reported neuroprotective effects and cognitive benefits of estrogens in human APOE4 carriers, especially when treatment is initiated within a critical window (Jacobs et al., 2013; Mahoney et al., 2020). Because individuals carrying two APOE4 alleles are relatively rare in human populations (Heffernan et al., 2016), therefore making sufficiently-powered sample size a challenge, studies with human patients do not often differentiate between women carriers of one or two APOE4 alleles (but see Mahoney et al., 2020). Data from the present study suggests that this distinction is important because heterozygous E3/4FAD female mice responded to E2, whereas homozygous E4FADs did not. More work remains to determine what factors mediate the neuroprotective properties of E2 in carriers of one or both copies of an APOE4 allele.
Surprisingly, the memory-enhancing effects of E2 in the present study occurred in the absence of rapid p42 ERK phosphorylation in the DH, which has been documented many times in young adult wild type C57BL/6 mice and is necessary in these mice for E2 to enhance memory consolidation in the object recognition and object placement tasks (e.g., Boulware et al., 2013; Fernandez et al., 2008; Fortress et al., 2013). Therefore, data from the current study suggest that the benefits of E2 in ovariectomized E3FAD and E3/4FAD mice occur either via a different time course of ERK activation or an as-of-yet uncharacterized estrogen-sensitive signaling event. The absence of rapid JNK phosphorylation in the present study also suggests GPER-independent signaling, given that the effects of GPER activation on object recognition and object placement memory consolidation in young ovariectomized wild type mice are dependent upon activation of this kinase (Kim et al., 2016, 2019). It is possible that pathological changes in the DH of these mice (e.g., Youmans et al., 2012) altered the timing of kinase activation relative to wild types, so collecting tissue from E2-infused EFADs at extended time points would allow for a more complete picture of E2-related cell signaling across genotypes.
Our previous study of gonadally-intact male and female E3FAD and E4FAD mice revealed that E4FADs of both sexes had significantly higher levels of ERα in the DH (Taxier et al., 2022a). The present study is consistent with this finding and extends it to ovariectomized E4FAD mice. Our data thus far suggest that the increase in ERα among E4FAD mice is associated with impaired memory consolidation (Taxier et al., 2022a) and an inability of E2 to enhance memory consolidation (present study). Consistent with these data, others have shown that ERα is elevated in the hippocampus of AD patients, particularly APOE4 carriers (Ishunina and Swaab, 2003). It is unclear why this is so. It could be that ERα is increased in response to changes in levels of other ERs. However, levels of ERβ and GPER were unaffected by genotype or treatment, so this is unlikely. The increase in ERα could come in response to reduced levels of E2 synthesized within the hippocampus. Levels of E2 and mRNA levels of the E2-synthesizing enzyme aromatase were lower in frontal cortices and cerebella of postmortem brains from female AD patients than in non-AD controls (Yue et al., 2005), supporting the possibility that ERα levels may increase in an attempt to compensate for lower levels of brain E2. However, it is unknown if or how levels of E2 or aromatase are altered by genotype in EFAD mice. Thus, the relationship between APOE4 homozygosity and ERα levels warrants future study, especially in light of previous findings from rat hippocampus that APOE mRNA and APOE protein expression are upregulated 24 h following ERα activation, either with E2 or an ERα agonist (Wang et al., 2006). Future work should examine whether hippocampal APOE expression is altered following E2 treatment in E3FAD, E3/4FAD, or E4FADs. With respect to the lack of E2 effects on ER levels in the present study, it should be noted that 5 minutes is a very rapid timepoint at which to see effects on receptor expression of a single E2 treatment, so future studies should examine longer time points and/or longer durations of E2 treatment. Better understanding the extent to which levels of the three ERs are altered by genotype and in response to E2 treatment across genotypes could provide valuable information as to whether selective estrogen receptor modulators could be potentially useful therapeutics for preventing or treating cognitive decline in APOE4+ women.
The effects of E2 on CA1 apical dendritic spine density were remarkably consistent with its beneficial effects on memory. In line with its memory-enhancing effects in ovariectomized E3FAD and E3/4FAD females, E2 increased CA1 total apical spine density in E3FAD and E3/4FAD, but not E4FAD, females two hours after infusion. This effect is consistent with previous findings from our lab showing that CA1 total apical spine density is increased two hours after E2 infusion in wild-type ovariectomized mice (Tuscher et al., 2016). Here, we also showed that E2 modestly increased CA1 apical stubby spine density in EFADs, although levels of apical stubby spines were similar in E3FADs regardless of treatment. Although thin spines are typically considered more likely than stubby spines to transition to other spine types, previous work in another AD model suggests that stubby spines can similarly take on this dynamic role in pathological conditions (Spires-Jones et al., 2007). Therefore, an increase in apical stubby spines in CA1 may reflect ongoing spine remodeling or spinogenesis stimulated by E2. Interestingly, E2 also increases CA1 and mPFC basal spines two hours post intrahippocampal E2-infusion in wild-type ovariectomized mice (Tuscher et al., 2016). However, we did not see a corresponding increase in CA1 and mPFC basal spines following E2 infusion in E3FADs or E3/4FADs, which may be reflective of reduced interactions or connectivity between mPFC and CA1 in E2-treated EFADs relative to wild-type mice. Nevertheless, our analyses suggest that E2 may exert its memory-enhancing effects in E3FAD and E3/4FAD mice via increased CA1 apical spine density.
E2 also had a striking effect on CA1 basal mushroom spines, for which the genotype x treatment interaction was significant. This finding, in which E2-treated ovariectomized E3FADs had the highest number of CA1 basal mushroom spines, whereas E2-treated E4FADs had the lowest, represents a key measure by which E2 benefits APOE3 carriers, while harming APOE4 carriers. Because mushroom spines are thought to be mature spines (Bourne and Harris, 2007; Hayashi and Majewska, 2005), the beneficial effects of E2 on memory in E3FADs, and its inefficacy in E4FADs, may be linked to the respective E2-induced increase of CA1 mushroom spines in E3FADs, and E2-induced decrease of this same spine subtype in E4FADs. That is, in addition to effects on CA1 apical spines, E2 may facilitate memory in E3FADs by increasing the density of mature spines on CA1 basal dendrites and impair memory in E4FADs by reducing basal mature spine density. However, this possibility is limited by the fact that the same effect of E2 on CA1 basal mushroom spines was not present in E3/4FADs at the single time point we examined. Recent work suggests that dendritic spine loss does not occur at random in AD patients; rather, spines are lost in clusters depending on the presence or absence of tau pathology in examined cells (Mijalkov et al., 2021). Therefore, because cells and dendritic segments in the present study were semi-randomly chosen for analyses based on stain penetrance, our analyses are limited by the fact that we do not know for certain whether spines were counted from neurons in which AD-like pathology was present. The use of advanced methods such as immuno-EM in tissue collected from EFADs could provide ultrastructural information about the presence or absence of pathological hallmarks of AD in specific cellular compartments (Takahashi et al., 2010).
Genotype modulated spine density in a manner largely consistent with our hypotheses, in that genotype effects on CA1 apical, CA1 basal, and mPFC basal spines were seemingly driven by increased dendritic spine density in E3FADs, or decreased dendritic spine density in E4FADs, relative to other groups, which may have contributed to their impaired memory consolidation. The main effect of genotype on basilar thin spine density in CA1 is of particular interest because post hoc comparisons indicated that E2-treated E3FADs had the highest density of this subtype. Our data are consistent with previous work showing that treatment with E2 facilitated cognitive function in aged rhesus monkeys, an effect dependent upon increased thin spine density (Hao et al., 2006). Furthermore, our data fit with the notion that thin spines are thought to be “learning spines,” or transient protrusions that often evolve into more mature, mushroom “memory” spines (Bourne and Harris, 2007). CA1 basal and proximal apical dendrites receive projections from CA3 to support memory processes (Li et al., 1994; Spruston, 2008; Whitlock et al., 2006), and E2 treatment evokes an increase in synaptic transmission at CA3-CA1 synapses (Kumar et al., 2015; Smejkalova and Woolley, 2010; Smith et al., 2009). Therefore, higher density of CA1 total apical spines in E2-treated E3FADs and E3/4FADs, as well as higher density of CA1 basilar thin spines in E2-treated E3FADs, could be linked to improved memory processes. The lack of genotype or treatment effects on mPFC apical spine density (Table 1), and modest genotype effects on mPFC basal spine density indicate that the memory-enhancing effects of E2 in ovariectomized E3FADs and E3/4FADs are not mediated through changes in mPFC spine density. However, the genotype effects on mPFC total and thin basal spines suggests that E3FAD females have somewhat higher basal spine density relative to E4FAD females, which is consistent with our previous findings in gonadally-intact E3FAD and E4FAD mice (Taxier et al., 2022a).
In sum, intrahippocampal E2 infusion facilitated memory consolidation and increased CA1 apical spine density in ovariectomized E3FAD and E3/4FAD mice, suggesting that mice with AD-like pathology bearing zero or one copy of APOE4 remain responsive to acute E2 treatment. Notably, two copies of APOE4 had a deleterious effect on the ability of E2 to enhance memory and increase dendritic spine density, and also increased levels of ERα in a manner possibly linked to impaired memory consolidation. In addition, the fact that E2 increased CA1 basal mushroom spine density in E3FADs and reduced these spines in E4FADs may account for the respective ability and inability of E2 to enhance memory in E3FADs and E3/4FADs versus E4FADs. These data support the notion that E2 treatment can potentially benefit individuals who carry zero or one copy of the APOE4 allele, but not those with two copies. Future experiments should examine the kinetics of both protein expression and dendritic spine density following E2 treatment at multiple time points to fully elucidate the molecular correlates of the mnemonic benefits of E2 in EFAD mice. Much more work remains to fully characterize the interactive effects between E2 and APOE4 genotype and to determine how APOE4 impedes the actions of E2. This information may provide new important new insights about the impact of E2 loss on AD risk in women and better define subpopulations of women who could benefit cognitively from estrogen therapy.
Supplementary Material
Acknowledgements
We gratefully acknowledge the primary support of an inaugural Sex and Gender in Alzheimer’s grant (SAGA-17- 419092) from the Alzheimer’s Association for this work. The Frick lab was also supported by R01MH107886 to KMF, 2R15GM118304-02 to KMF, and 1F31MH118822-01A1 to LRT, the UWM College of Letters & Science, and the UWM Office of Undergraduate Research. Additional funding of the LaDu lab was provided by R01AG056472, R01AG057008, UH2/3NS100127, R56AG058655, philanthropic support from Lou and Christine Friedrich, and UIC institutional funds. We also thank Dr. James R. Moyer, Jr. for use of his Olympus BX51WI microscope and NeuroLucida software.
Abbreviations
- 5xFAD
5 familial AD mutations
- AD
Alzheimer’s disease
- ANOVA
analysis of variance
- DH
dorsal hippocampus
- E2
17β-estradiol
- EFAD
APOE+/+/5xFAD+/−
- E3FAD
APOE3/3/5xFAD+/−
- E3/4FAD
APOE3/4/5xFAD+/−
- E4FAD
APOE4/4/5xFAD+/−
- ERα
estrogen receptor alpha
- ERβ
estrogen receptor beta
- GPER
G-protein-coupled estrogen receptor
- mPFC
medial prefrontal cortex
- OP
object placement
- OR
object recognition
- Tg
transgenic
- s
second
- min
minute
- h
hour
Footnotes
Declarations of Interest
K.M.F. is a co-founder and the Chief Scientific Officer of Estrigenix Therapeutics, Inc. The other authors have no actual or potential conflicts of interest to declare.
References
- Altmann A, Tian L, Henderson VW, Greicius MD, 2014. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann. Neurol 75, 563–573. 10.1002/ana.24135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldereschi M, Di Carlo A, Lepore V, Bracco L, Maggi S, Grigoletto F, Scarlato G, Amaducci L, 1998. Estrogen-replacement therapy and Alzheimer’s disease in the Italian Longitudinal Study on Aging. Neurology 50, 996–1002. 10.1212/wnl.50.4.996 [DOI] [PubMed] [Google Scholar]
- Bertram L, 2009. Alzheimer’s disease genetics current status and future perspectives. Int. Rev. Neurobiol 84, 167–184. 10.1016/S0074-7742(09)00409-7 [DOI] [PubMed] [Google Scholar]
- Boulware MI, Heisler JD, Frick KM, 2013. The memory-enhancing effects of hippocampal estrogen receptor activation involve metabotropic glutamate receptor signaling. J. Neurosci 33, 15184–15194. 10.1523/JNEUROSCI.1716-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne J, Harris KM, 2007. Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol 17, 381–386. 10.1016/j.conb.2007.04.009 [DOI] [PubMed] [Google Scholar]
- Bretsky PM, Buckwalter JG, Seeman TE, Miller CA, Poirier J, Schellenberg GD, Finch CE, Henderson VW, 1999. Evidence for an interaction between apolipoprotein E genotype, gender, and Alzheimer disease. Alzheimer Dis. Assoc. Disord 13, 216–221. 10.1097/00002093-199910000-00007 [DOI] [PubMed] [Google Scholar]
- Burkhardt MS, Foster JK, Laws SM, Baker LD, Craft S, Gandy SE, Stuckey BGA, Clarnette R, Nolan D, Hewson-Bower B, Martins RN, 2004. Oestrogen replacement therapy may improve memory functioning in the absence of APOE ε4. J. Alzheimers Dis 6, 221–228. 10.3233/JAD-2004-6302 [DOI] [PubMed] [Google Scholar]
- Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, LaFerla FM, Pike CJ, 2007. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. Off. J. Soc. Neurosci 27, 13357–13365. 10.1523/JNEUROSCI.2718-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW, 1990. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann. Neurol 27, 457–464. 10.1002/ana.410270502 [DOI] [PubMed] [Google Scholar]
- Dumanis SB, Tesoriero JA, Babus LW, Nguyen MT, Trotter JH, Ladu MJ, Weeber EJ, Turner RS, Xu B, Rebeck GW, Hoe H-S, 2009. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci 29, 15317–15322. 10.1523/JNEUROSCI.4026-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez SM, Lewis MC, Pechenino AS, Harburger LL, Orr PT, Gresack JE, Schafe GE, Frick KM, 2008. Estradiol-induced enhancement of object memory consolidation involves hippocampal extracellular signal-regulated kinase activation and membrane-bound estrogen receptors. J. Neurosci 28, 8660–8667. 10.1523/JNEUROSCI.1968-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortress AM, Fan L, Orr PT, Zhao Z, Frick KM, 2013. Estradiol-induced object recognition memory consolidation is dependent on activation of mTOR signaling in the dorsal hippocampus. Learn. Mem. Cold Spring Harb. N 20, 147–155. 10.1101/lm.026732.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankfurt M, Salas‐Ramirez K, Friedman E, Luine V, 2011. Cocaine alters dendritic spine density in cortical and subcortical brain regions of the postpartum and virgin female rat. Synapse 65, 955–961. 10.1002/syn.20918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason CE, Dowling NM, Wharton W, Manson JE, Miller VM, Atwood CS, Brinton EA, Cedars MI, Lobo RA, Merriam GR, Neal-Perry G, Santoro NF, Taylor HS, Black DM, Budoff MJ, Hodis HN, Naftolin F, Harman SM, Asthana S, 2015. Effects of hormone therapy on cognition and mood in recently postmenopausal women: findings from the randomized, controlled KEEPS-cognitive and affective study. PLoS Med 12, e1001833; discussion e1001833. 10.1371/journal.pmed.1001833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross KS, Alf RL, Polzin TR, Frick KM, 2021. 17β-estradiol activation of dorsal hippocampal TrkB is independent of increased mature BDNF expression and is required for enhanced memory consolidation in female mice. Psychoneuroendocrinology 125, 105110. 10.1016/j.psyneuen.2020.105110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Rapp PR, Leffler AE, Leffler SR, Janssen WGM, Lou W, McKay H, Roberts JA, Wearne SL, Hof PR, Morrison JH, 2006. Estrogen alters spine number and morphology in prefrontal cortex of aged female rhesus monkeys. J. Neurosci 26, 2571–2578. 10.1523/JNEUROSCI.3440-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B, 1992. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci 12, 2685–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Majewska AK, 2005. Dendritic spine geometry: functional implication and regulation. Neuron 46, 529–532. 10.1016/j.neuron.2005.05.006 [DOI] [PubMed] [Google Scholar]
- Heffernan AL, Chidgey C, Peng P, Masters CL, Roberts BR, 2016. The neurobiology and age-related prevalence of the ε4 allele of apolipoprotein E in Alzheimer’s disease cohorts. J. Mol. Neurosci. MN 60, 316–324. 10.1007/s12031-016-0804-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson VW, 2006. Estrogen-containing hormone therapy and Alzheimer’s disease risk: understanding discrepant inferences from observational and experimental research. Neuroscience 138, 1031–1039. 10.1016/j.neuroscience.2005.06.017 [DOI] [PubMed] [Google Scholar]
- Inagaki T, Frankfurt M, Luine V, 2012. Estrogen-induced memory enhancements are blocked by acute bisphenol A in adult female rats: role of dendritic spines. Endocrinology 153, 3357–3367. 10.1210/en.2012-1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishunina TA, Swaab DF, 2003. Increased neuronal metabolic activity and estrogen receptors in the vertical limb of the diagonal band of Broca in Alzheimer’s disease: relation to sex and aging. Exp. Neurol 183, 159–172. 10.1016/s0014-4886(03)00138-9 [DOI] [PubMed] [Google Scholar]
- Jacobs EG, Kroenke C, Lin J, Epel ES, Kenna HA, Blackburn EH, Rasgon NL, 2013. Accelerated cell aging in female APOE-ε4 carriers: Implications for hormone therapy use. PLoS ONE 8, e54713. 10.1371/journal.pone.0054713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs EG, Weiss BK, Makris N, Whitfield-Gabrieli S, Buka SL, Klibanski A, Goldstein JM, 2016. Impact of sex and menopausal status on episodic memory circuitry in early midlife. J. Neurosci 36, 10163–10173. 10.1523/JNEUROSCI.0951-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Gong Y, Gan W, Beach T, Holtzman DM, Wisniewski T, 2003. Apolipoprotein E isoform-specific regulation of dendritic spine morphology in apolipoprotein E transgenic mice and Alzheimer’s disease patients. Neuroscience 122, 305–315. 10.1016/j.neuroscience.2003.08.007 [DOI] [PubMed] [Google Scholar]
- Joffe H, Hall JE, Gruber S, Sarmiento IA, Cohen LS, Yurgelun-Todd D, Martin KA, 2006. Estrogen therapy selectively enhances prefrontal cognitive processes: a randomized, double-blind, placebo-controlled study with functional magnetic resonance imaging in perimenopausal and recently postmenopausal women. Menopause 13, 411–422. 10.1097/01.gme.0000189618.48774.7b [DOI] [PubMed] [Google Scholar]
- Kang JH, Grodstein F, 2012. Postmenopausal Hormone Therapy, Timing of Initiation, APOE and Cognitive Decline. Neurobiol. Aging 33, 1129–1137. 10.1016/j.neurobiolaging.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E, 1997. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer’s disease: The Baltimore Longitudinal Study of Aging. Neurology 48, 1517–1521. 10.1212/WNL.48.6.1517 [DOI] [PubMed] [Google Scholar]
- Kim J, Szinte JS, Boulware MI, and Frick KM (2016). 17β-estradiol and agonism of G-protein coupled estrogen receptor (GPER) enhance hippocampal memory consolidation via different cell-signaling mechanisms. J. Neurosci 36, 3309–3321. 10.1523/JNEUROSCI.0257-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Schalk JC, Koss WA, Gremminger RL, Taxier LR, Gross KS, Frick KM, 2019. Dorsal hippocampal actin polymerization is necessary for activation of G-protein-coupled estrogen receptor (GPER) to increase CA1 dendritic spine density and enhance memory consolidation. J. Neurosci 39, 9598–9610. 10.1523/JNEUROSCI.2687-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss WA, Haertel JM, Philippi SM, Frick KM, 2018. Sex differences in the rapid cell signaling mechanisms underlying the memory-enhancing effects of 17β-estradiol. eNeuro 5. 10.1523/ENEURO.0267-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Bean LA, Rani A, Jackson T, Foster TC, 2015. Contribution of estrogen receptor subtypes, ERα, ERβ, and GPER1 in rapid estradiol-mediated enhancement of hippocampal synaptic transmission in mice. Hippocampus 25, 1556–1566. 10.1002/hipo.22475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzler J, Youmans KL, Yu C, LaDu MJ, Tai LM, 2014. APOE modulates the effect of estrogen therapy on Aβ accumulation EFAD-Tg mice. Neurosci. Lett 560, 131–136. 10.1016/j.neulet.2013.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis MC, Kerr KM, Orr PT, Frick KM, 2008. Estradiol-induced enhancement of object memory consolidation involves NMDA receptors and protein kinase A in the dorsal hippocampus of female C57BL/6 mice. Behav. Neurosci 122, 716–721. 10.1037/0735-7044.122.3.716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-G, Somogyi P, Ylinen A, Buzsáki G, 1994. The hippocampal CA3 network: An in vivo intracellular labeling study. J. Comp. Neurol 339, 181–208. 10.1002/cne.903390204 [DOI] [PubMed] [Google Scholar]
- Liu D, Pan X, Zhang J, Shen H, Collins NC, Cole AM, Koster KP, Ben Aissa M, Dai X, Zhou M, Tai LM, Zhu Y, LaDu MJ, Chen X, 2015. APOE4 enhances age-dependent decline in cognitive function by down-regulating an NMDA receptor pathway in EFAD-Tg mice. Mol. Neurodegener 10, 7. 10.1186/s13024-015-0002-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney ER, Dumitrescu L, Maki PM, Rapp SR, Keene CD, Corrada MMM, Hayden KM, Baker LD, Espeland MA, Resnick SM, Hohman TJ, 2020. Menopausal hormone therapy has beneficial effects on cognitive trajectories among homozygous carriers of the APOE-ε4 allele. Alzheimers Dement 16, e041482. 10.1002/alz.041482 [DOI] [Google Scholar]
- Manly JJ, Merchant CA, Jacobs DM, Small SA, Bell K, Ferin M, Mayeux R, 2000. Endogenous estrogen levels and Alzheimer’s disease among postmenopausal women. Neurology 54, 833–837. 10.1212/wnl.54.4.833 [DOI] [PubMed] [Google Scholar]
- Mijalkov M, Volpe G, Fernaud-Espinosa I, DeFelipe J, Pereira JB, Merino-Serrais P, 2021. Dendritic spines are lost in clusters in Alzheimer’s disease. Sci. Rep 11, 12350. 10.1038/s41598-021-91726-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami G, Tsurugizawa T, Hatanaka Y, Komatsuzaki Y, Tanabe N, Mukai H, Hojo Y, Kominami S, Yamazaki T, Kimoto T, Kawato S, 2006. Comparison between basal and apical dendritic spines in estrogen-induced rapid spinogenesis of CA1 principal neurons in the adult hippocampus. Biochem. Biophys. Res. Commun 351, 553–558. 10.1016/j.bbrc.2006.10.066 [DOI] [PubMed] [Google Scholar]
- Nathan BP, Barsukova AG, Shen F, McAsey M, Struble RG, 2004. Estrogen facilitates neurite extension via Apolipoprotein E in cultured adult mouse cortical neurons. Endocrinology 145, 3065–3073. 10.1210/en.2003-1707 [DOI] [PubMed] [Google Scholar]
- Packard MG, Teather LA, 1997. Intra-hippocampal estradiol infusion enhances memory in ovariectomized rats. NeuroReport 8, 3009–3013. [DOI] [PubMed] [Google Scholar]
- Paganini-Hill A, Henderson VW, 1996. Estrogen replacement therapy and risk of Alzheimer disease. Arch. Intern. Med 156, 2213–2217. [PubMed] [Google Scholar]
- Phan A, Suschkov S, Molinaro L, Reynolds K, Lymer JM, Bailey CDC, Kow L-M, MacLusky NJ, Pfaff DW, Choleris E, 2015. Rapid increases in immature synapses parallel estrogen-induced hippocampal learning enhancements. Proc. Natl. Acad. Sci 112, 16018–16023. 10.1073/pnas.1522150112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapp SR, Espeland MA, Shumaker SA, Henderson VW, Brunner RL, Manson JE, Gass MLS, Stefanick ML, Lane DS, Hays J, Johnson KC, Coker LH, Dailey M, Bowen D, WHIMS Investigators, 2003. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 289, 2663–2672. 10.1001/jama.289.20.2663 [DOI] [PubMed] [Google Scholar]
- Roses AD, 1996. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med 47, 387–400. 10.1146/annurev.med.47.1.387 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, 2002. Alzheimer’s disease is a synaptic failure. Science 298, 789–791. 10.1126/science.1074069 [DOI] [PubMed] [Google Scholar]
- Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J, WHIMS Investigators, 2003. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 289, 2651–2662. 10.1001/jama.289.20.2651 [DOI] [PubMed] [Google Scholar]
- Smejkalova T, Woolley CS, 2010. Estradiol acutely potentiates hippocampal excitatory synaptic transmission through a presynaptic mechanism. J. Neurosci 30, 16137–16148. 10.1523/JNEUROSCI.4161-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Vedder LC, McMahon LL, 2009. Estradiol and the relationship between dendritic spines, NR2B containing NMDA receptors, and the magnitude of long-term potentiation at hippocampal CA3-CA1 synapses. Psychoneuroendocrinology 34 Suppl 1, S130–142. 10.1016/j.psyneuen.2009.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, Meyer-Luehmann M, Osetek JD, Jones PB, Stern EA, Bacskai BJ, Hyman BT, 2007. Impaired spine stability underlies plaque-related spine loss in an Alzheimer’s disease mouse model. Am. J. Pathol 171, 1304–1311. 10.2353/ajpath.2007.070055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, 2008. Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci 9, 206–221. 10.1038/nrn2286 [DOI] [PubMed] [Google Scholar]
- Tai LM, Balu D, Avila-Munoz E, Abdullah L, Thomas R, Collins N, Valencia-Olvera AC, LaDu MJ, 2017. EFAD transgenic mice as a human APOE relevant preclinical model of Alzheimer’s disease. J. Lipid Res 58, 1733–1755. 10.1194/jlr.R076315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK, 2010. Co-occurrence of Alzheimer’s disease β-amyloid and tau pathologies at synapses. Neurobiol. Aging 31, 1145–1152. 10.1016/j.neurobiolaging.2008.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M-X, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R, 1996. Effect of oestrogen during menopause on risk and age at onset of Alzheimer’s disease. The Lancet 348, 429–432. 10.1016/S0140-6736(96)03356-9 [DOI] [PubMed] [Google Scholar]
- Taxier LR, Philippi SM, Fortress AM, Frick KM, 2019. Dickkopf-1 blocks 17β-estradiol-enhanced object memory consolidation in ovariectomized female mice. Horm. Behav 114, 104545. 10.1016/j.yhbeh.2019.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxier LR, Philippi SM, York JM, LaDu MJ, Frick KM, 2022a. The detrimental effects of APOE4 on risk for Alzheimer’s disease may result from altered dendritic spine density, synaptic proteins, and estrogen receptor alpha. Neurobiol. Aging 112, 74–86. 10.1016/j.neurobiolaging.2021.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxier LR, Philippi SM, York JM, LaDu MJ, Frick KM, 2022b. APOE4 genotype or ovarian hormone loss influence open field exploration in an EFAD mouse model of Alzheimer’s disease. Horm. Behav 140, 105124. 10.1016/j.yhbeh.2022.105124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsolaki M, Grammaticos P, Karanasou C, Balaris V, Kapoukranidou D, Kalpidis I, Petsanis K, Dedousi E, 2005. Serum estradiol, progesterone, testosterone, FSH and LH levels in postmenopausal women with Alzheimer’s dementia. Hell. J. Nucl. Med 8, 39–42. [PubMed] [Google Scholar]
- Tuscher JJ, Luine V, Frankfurt M, Frick KM, 2016a. Estradiol-mediated spine changes in the dorsal hippocampus and medial prefrontal cortex of ovariectomized female mice depend on ERK and mTOR activation in the dorsal hippocampus. J. Neurosci 36, 1483–1489. 10.1523/JNEUROSCI.3135-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuscher JJ, Szinte JS, Starrett JR, Krentzel AA, Fortress AM, Remage-Healey L, Frick KM, 2016b. Inhibition of local estrogen synthesis in the hippocampus impairs hippocampal memory consolidation in ovariectomized female mice. Horm. Behav 83, 60–67. 10.1016/j.yhbeh.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuscher JJ, Taxier LR, Fortress AM, Frick KM, 2018. Chemogenetic inactivation of the dorsal hippocampus and medial prefrontal cortex, individually and concurrently, impairs object recognition and spatial memory consolidation in female mice. Neurobiol. Learn. Mem 156, 103–116. 10.1016/j.nlm.2018.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JM, Irwin RW, Brinton RD, 2006. Activation of estrogen receptor alpha increases and estrogen receptor beta decreases apolipoprotein E expression in hippocampus in vitro and in vivo. Proc. Natl. Acad. Sci. U. S. A 103, 16983–16988. 10.1073/pnas.0608128103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF, 2006. Learning induces long-term potentiation in the hippocampus. Science 313, 1093–1097. 10.1126/science.1128134 [DOI] [PubMed] [Google Scholar]
- Yaffe K, Haan M, Byers A, Tangen C, Kuller L, 2000. Estrogen use, APOE, and cognitive decline: evidence of gene-environment interaction. Neurology 54, 1949–1954. 10.1212/wnl.54.10.1949 [DOI] [PubMed] [Google Scholar]
- Youmans KL, Tai LM, Nwabuisi-Heath E, Jungbauer L, Kanekiyo T, Gan M, Kim J, Eimer WA, Estus S, Rebeck GW, Weeber EJ, Bu G, Yu C, LaDu MJ, 2012. APOE4-specific changes in Aβ accumulation in a new transgenic mouse model of Alzheimer disease. J. Biol. Chem 287, 41774–41786. 10.1074/jbc.M112.407957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X, Lu M, Lancaster T, Cao P, Honda S-I, Staufenbiel M, Harada N, Zhong Z, Shen Y, Li R, 2005. Brain estrogen deficiency accelerates Aβ plaque formation in an Alzheimer’s disease animal model. Proc. Natl. Acad. Sci 102, 19198–19203. 10.1073/pnas.0505203102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.