

Significance
Phase separation of proteins/nucleic acids is used to explain diverse biological processes. Because many phase separation studies relied on protein overexpression in cells and in vitro studies, the physiological relevance of phase separation is often unclear. PERIOD proteins, central mammalian circadian clock components, are previously shown to associate with other clock proteins. Here, we showed that PER (PEROD) exhibits phase separation behavior in cells when overexpressed. Super-resolution microscopy of the endogenous PER2 protein, however, revealed that it exists in rapidly diffusing microbodies distinct from phase-separated condensates and its interaction with other clock proteins is transient and dynamic. This study demonstrates a key feature of mammalian circadian clock mechanism and highlights the importance of examining endogenous proteins in mechanistic studies.
Keywords: circadian clock, phase separation, protein interaction, PERIOD protein
Abstract
Liquid–liquid phase separation (LLPS) underlies diverse biological processes. Because most LLPS studies were performed in vitro using recombinant proteins or in cells that overexpress protein, the physiological relevance of LLPS for endogenous protein is often unclear. PERIOD, the intrinsically disordered domain-rich proteins, are central mammalian circadian clock components and interact with other clock proteins in the core circadian negative feedback loop. Different core clock proteins were previously shown to form large complexes. Circadian clock studies often rely on experiments that overexpress clock proteins. Here, we show that when Per2 transgene was stably expressed in cells, PER2 protein formed nuclear phosphorylation-dependent slow-moving LLPS condensates that recruited other clock proteins. Super-resolution microscopy of endogenous PER2, however, revealed formation of circadian-controlled, rapidly diffusing nuclear microbodies that were resistant to protein concentration changes, hexanediol treatment, and loss of phosphorylation, indicating that they are distinct from the LLPS condensates caused by protein overexpression. Surprisingly, only a small fraction of endogenous PER2 microbodies transiently interact with endogenous BMAL1 and CRY1, a conclusion that was confirmed in cells and in mice tissues, suggesting an enzyme-like mechanism in the circadian negative feedback process. Together, these results demonstrate that the dynamic interactions of core clock proteins are a key feature of mammalian circadian clock mechanism and the importance of examining endogenous proteins in LLPS and circadian clock studies.
The core mammalian circadian oscillators consist of autoregulatory transcription- and translation-based negative feedback loops. In the core mammalian circadian negative feedback loops, the heterodimeric CLOCK/BMAL1 complex acts as the positive element that activates transcription of clock genes by binding to E-boxes on gene promoters to rhythmically activate transcription (1–3). PERIOD proteins PER1 and PER2 and CRYPTOCHROME proteins CRY1 and CRY2 have been shown to interact with each other and function as the negative elements in the negative feedback loop by interacting and inhibiting the CLOCK/BMAL1 complex, leading to the closing the circadian negative feedback loop (2, 4–10). The functional circadian negative feedback loops lead to rhythmic gene expression and robust rhythmic PER abundance (2, 10). PER proteins associate stably with casein kinase 1 (CK1), and PER proteins are progressively phosphorylated after their synthesis and become hyperphosphorylated during the late night (10–12). PER phosphorylation plays critical roles in the mammalian circadian clock function by regulation of PER stability, its repressor activity, and its subcellular localization (11, 13–17). Due to their stable association, PER acts a scaffold for CK1 to promote phosphorylation of PER and of CLOCK (18, 19). The PER-dependent CLOCK phosphorylation leads to efficient removal of CLOCK-BMAL1 complexes from E-boxes on DNA, a process that is also dependent on CRYs. On the other hand, CRYs can repress CLOCK-BMAL1 activity on DNA independently of PER (20–23). These dual feedback mechanisms ensure robust circadian rhythmicity. We showed previously that the disruption of the PER-CK1 interaction abolishes PER phosphorylation, impairs circadian negative feedback process, and results in severe loss of circadian gene expression (18).
Like the Drosophila PERIOD protein (24), most human PER2 protein regions flanking the PAS domain are predicted to be intrinsically disordered (Fig. 1A). Previous studies showed that PER, CRY, CK1δ, BMAL1, and CLOCK proteins can be immunoprecipitated with each other, and most of these proteins are present in a 1.9-MDa nuclear complex (2, 9, 10, 25). Single-particle electron microscopy revealed that the nuclear complexes are ~40 nm in diameter (25). In addition, isothermal titration calorimetry assays using recombinant proteins indicate that mCRY1-mPER2 affinity has a Kd in the lower nanomolar range in vitro and CRY and PER can form a stable complex in vitro (4, 5). On the other hand, glycerol gradient and chromatography-based approaches, however, suggest the presence of mostly discreet CLOCK-BMAL1 and PER-CRY-CK1δ complexes in mice liver extracts (26). In addition, the peaks of PER2 and CRY1 in these fractionation assays did not co-migrate in these fractionation assays (26). ChIP (Chromatin immunoprecipitation) assays revealed that PER, CRY, BMAL1, and CLOCK were found to be rhythmically enriched on chromatin E-boxes (27). Imaging studies using PER2 knock-in (KI) reporter in mice and cell lines revealed that PER proteins are highly enriched in the nucleus (27–30). By expressing fluorescent fusion proteins via lentiviral transduction in U2OS/NIH/3T3 cells, studies showed that PER2 is mostly immobile in the nucleus and PER, CRY, BMAL1, and CLOCK proteins have high affinity for each other and exhibit very low nuclear diffusion coefficients (which indicate the ability to diffuse) with that of PER2 the lowest (27, 31). Furthermore, fluorescence recovery after photobleaching results suggests that most PER2 proteins are immobile in the nucleus of SCN (suprachiasmatic nucleus) cells (28, 30). These results are consistent with the proposal that most clock components associate with each other on chromatin (27, 31). However, lentiviral transduction normally resulted in clock protein overexpression. How endogenous clock proteins interact and function remains unclear.
Fig. 1.
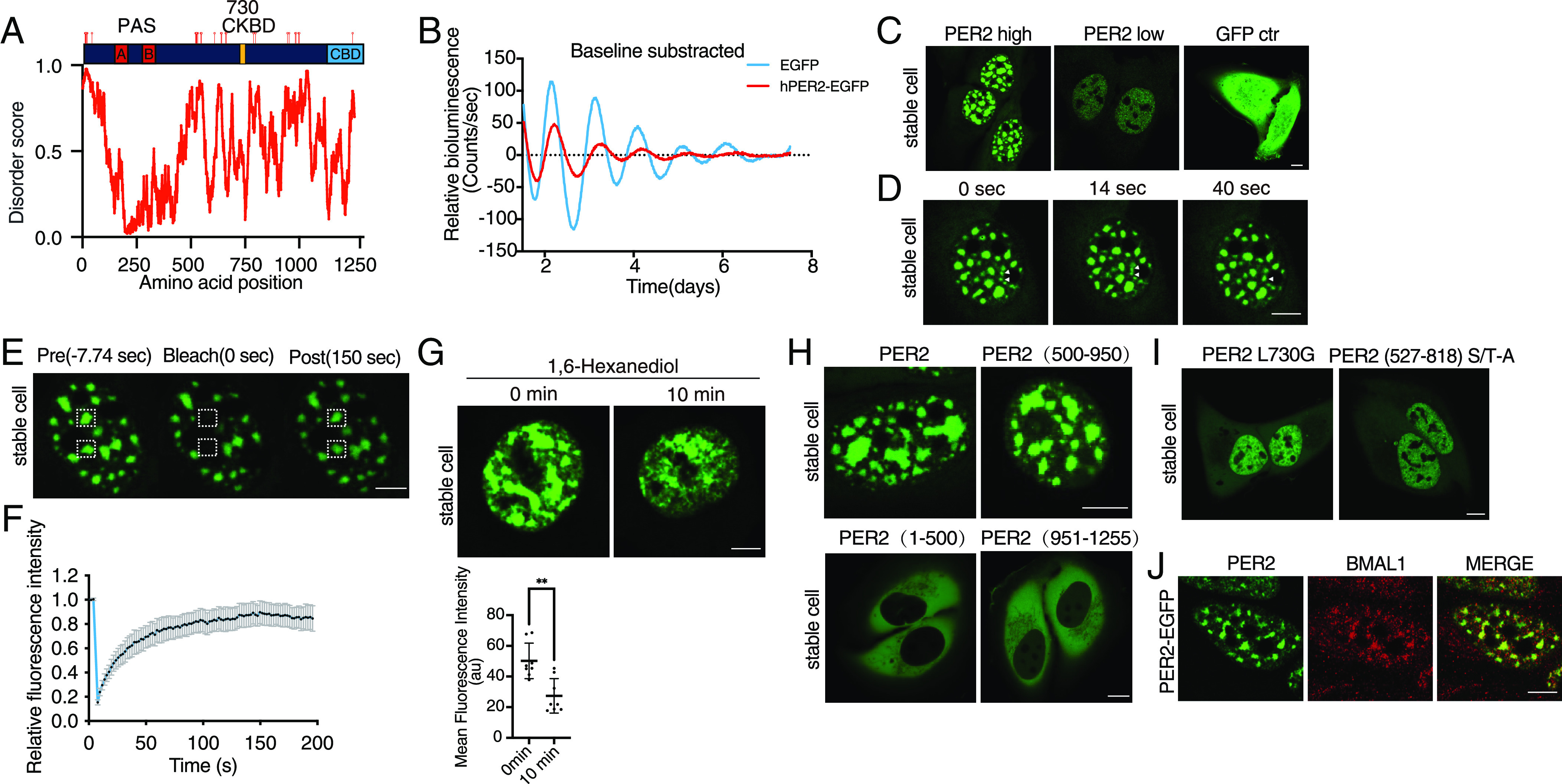
LLPS behavior of PER2-EGFP when it is constitutively overexpressed in U2OS cells. (A) Top: A diagram depicting human PER2 domains and phosphorylation sites. CKBD:Casein Kinase Binding domain. CBD: CRY binding domain. Location of the known phosphorylation sites was indicated (11). Bottom: Disorder score across the human PER2 protein as predicted by IUPred. (B) Representative bioluminescence rhythms of the U2OS cells that stably express either EGFP or PER2-EGFP as well as a Per2(E2)-Luc reporter. Rhythms were monitored by LumiCycle, and baseline-subtracted bioluminescence data are plotted (n = 3). (C) Representative confocal images of cells stably express PER2-EGFP (high and low levels) and of control cells that express EGFP. (Scale bar: 5 μm.) (D) Images of a live cell that stably expresses PER2-EGFP over time. Two condensates that fused during the time course are indicated. (E) Images of a live U2OS cell that stably expresses PER2-EGFP before and after photobleaching of regions marked by squares. (F) Relative fluorescence intensity of PER2-EGFP condensates after photobleaching in the PER2-EGFP stable cells. Data are presented as mean ± SD (n = 11). (G) Top: Images of live cells that express PER2-EGFP at 0 min and 10 min after treatment with 1,6-hexanediol (1.5%). Bottom: Quantification of mean fluorescence intensity of PER2-EGFP nuclei at 0 min and 10 min after treatment with 1,6-hexanediol. n = 8 cells. Data are presented as mean ± SD, unpaired two-tailed Student’s t test, ∗∗P < 0.01. (H) Representative confocal images of U2OS cells that overexpress the indicated PER2-EGFP proteins. (I) Confocal images cells that constitutively express PER2(L730G)-EGFP or PER2(527-818 S/T-A)-EGFP (PER2 with known and potential phosphorylation sites within aa 527-818 mutated to alanines). (J) Fluorescence immunostaining of endogenous BMAL1 in cells that stably overexpresses PER2-EGFP. Endogenous BMAL1 was stained with a BMAL1 antibody, and PER2-EGFP was monitored using the GFP channel.
Liquid–liquid phase separation (LLPS) results in formation of membraneless, spherical condensates of proteins or nucleic acids (32–34). Numerous studies have demonstrated that protein-containing LLPS condensates are involved in diverse biological processes (32, 33, 35–38). The LLPS behavior of biomolecular molecules is concentration-dependent and driven by multivalent macromolecular interactions. Intrinsically disorder regions (IDRs), also called low complexity domains (LCDs), have often been shown to drive LLPS behavior due to their ability to mediate multivalent interactions (33, 39, 40). Characteristics of LLPS condensates include fusion of small condensates to become larger condensates and fluorescence recovery after photobleaching (32, 41, 42). Sensitivity of condensates to treatment of compounds such as 1,6-hexanediol, which interfere with weak multivalent hydrophobic interactions, is commonly used as an indicator of LLPS behavior (41–43). In addition, protein condensates are often regulated by post-translational modifications, and mutations that alter both LLPS and biological functions are also presumed to reflect physiological functions of LLPS (40, 44).
Although LLPS offers an attractive mechanism to explain many biological processes, most prior LLPS studies were based on protein overexpression in cells or in vitro biochemical experiments using high concentration of recombinant proteins (42). As a result, concerns have been raised regarding the physiological relevance of LLPS for endogenous proteins (41, 42). Nonetheless, the LLPS mechanism is often taken as physiologically relevant despite the lack of rigorous evidence on endogenous proteins. Studies of RNA polymerase II and transcription factors suggest that endogenous proteins may form “hubs” rather than LLPS condensates (42, 45, 46). However, the same multivalent LCD-LCD interactions appear to mediate the formation of both protein hub and LLPS condensates.
Here, we studied the dynamics and interactions of the endogenous core human circadian clock proteins. We observed that stably expressed PER2 from transgene forms nuclear condensates with typical LLPS-like behavior. Super-resolution fluorescence microscopy of the endogenous PER2, however, revealed that PER2 is present in rapidly diffusing microbodies that are distinct from LLPS. Surprisingly, most PER2, BMAL1, and CRY1 exist in microbodies that do not colocalize in cells and in mice tissues. Together, these results demonstrate the dynamic nature of clock protein interactions and the importance of examining endogenous proteins in studies of the biological functions.
Results
LLPS Behaviors of PER2-GFP When Constitutively Expressed in U2OS Cells.
To determine the cellular distribution of IDR-rich PER2 in human cells with a functional circadian clock, U2OS cells were transduced with a lentiviral vector for constitutive expression of a PER2-EGFP (Enhanced green fluorescent protein) fusion protein under the control of the EF-1a promoter (SI Appendix, Fig. S1A). Control cells were transduced with the vector for expression of EGFP only. We did not use commonly used transient transfection to express the PER2-EGFP fusion protein because it resulted in approximately 100x higher levels of PER2-EGFP than detected in stably transduced cells (SI Appendix, Fig. S1B). We then transfected a Per2(E2)-Luc reporter plasmid into the U2OS cells that stably express PER2-EGFP and into control U2OS cells (47). The constitutive expression of PER-EGFP caused severe dampening of the circadian bioluminescence rhythm compared to control cells (Fig. 1B). Constitutive overexpression of PER2 mediated by adenoviral infection was previously shown to abolish circadian rhythm in MEF (mouse embryonic fibroblasts) cells (48). The dampened rhythm observed in our case is likely due to a reduced PER2-GFP overexpression level. Analysis using conventional confocal microscopy revealed that PER2-EGFP fluorescence was highly enriched in the nucleus (Fig. 1C), which is similar to previous observations in the knock-in mouse cells and fibroblasts (28, 29). In contrast, upon transient transfection of a vector for expression of PER2-EGFP into U2OS cells, PER2-EGFP was mostly cytoplasmic during the first 2 to 3 d after transfection (SI Appendix, Fig. S1C), which is similar to previously reported results (49–51). This difference is likely caused by the drastic overexpression of PER2-EGFP by transient transfection (SI Appendix, Fig. S1B). However, it is also important to note that the viral transduction resulted PER2-EGFP expression in stable cells still led to PER2 overexpression that is ~20-fold higher than that of the endogenous PER2 level (see below).
After transduction, fluorescence signal was detected in about two dozen discrete puncta in the nucleus in most of the cells that express PER2-EGFP. These puncta were mainly spherical when small but large irregularly shaped condensates were also observed (Fig. 1C). The small condensates were able to fuse, and this resulted in the larger one (Fig. 1D and SI Appendix, Supplemental Data File 1). In cells with low levels of PER2-EGFP fluorescence, however, the PER2-EGFP condensates were not observed and signals were evenly distributed in the nucleus (SI Appendix, Fig. S1C), indicating that the PER2-EGFP condensate formation is concentration-dependent. In contrast, in control cells with high EGFP expression level, fluorescence signal was evenly distributed in both cytoplasm and nucleus (Fig. 1C), indicating that the condensate behavior of PER2-EGFP is not caused by EGFP. Although EGFP is normally monomeric, it can dimerize at high concentrations (52). To exclude the effect of dimerization on our findings, we expressed the PER2 fused to EGFP(A206K); this mutation prevents dimerization (52). Condensate formation was similar whether PER2 was fused to EGFP or the mutant (SI Appendix, Fig. S1D). This result indicates that the condensation of PER2-EGFP is not caused by EGFP dimerization.
The PER2-EGFP condensates are somewhat stationary in the nuclei of live cells, which allowed us to perform the fluorescence recovery after photobleaching assay. EGFP fluorescence in the condensates showed a rapid recovery after photobleaching with a t1/2 of ~23 s (Fig. 1 E and F). The alcohol 1,6-hexanediol interferes with weak hydrophobic interactions, and LLPS condensates dissolve upon treatment with 1,6-hexanediol (41, 42). When U2OS stable cells that express PER2-EGFP were treated with 1.5% 1,6-hexanediol, there was marked decrease of the fluorescence signal of the PER2-EGFP condensates after 10 min (Fig. 1G) due to that monomeric PER2-eGFP protein is below the detection limit of the microscope. Together, these results demonstrate that constitutively expressed PER2 exhibits LLPS behavior in the nucleus.
LLPS behavior of many proteins is regulated by post-translational modifications (33, 41). PER phosphorylation has been shown to play important roles in circadian clock function (11–13). Thus, a role for PER phosphorylation in regulating its LLPS behavior would suggest that PER LLPS has a biological role. To identify the PER2 domain involved in LLPS, we created U2OS cell lines that stably express the N-terminal (amino acids 1 to 500), middle (amino acids 501 to 950), or C-terminal (amino acids 951 to 1,255) regions of the protein fused to EGFP. We found that the expression of the middle region, but not the N- or C-terminal domains, resulted in nuclear PER2-EGFP condensates (Fig. 1H). The PER2(1-500)-EGFP and PER2(951-1255)-EGFP proteins were found to be evenly distributed in the cytoplasm. The middle region of PER2 contains the previously identified PER-CK1 interaction domain and IDRs with many phosphorylation sites (Fig. 1A) (11, 15, 18, 53). To determine the role of PER2 phosphorylation in condensate formation, we stably expressed PER2-EGFP with the L730G mutation in the PER-CK1 interaction domain, which disrupts the PER-CK1 interaction and abolishes PER2 phosphorylation in cells and in mice (18). Fluorescence in cells that expressed the mutant was distributed throughout the nucleus, and no nuclear condensates were observed, even though PER2(L730G)-EGFP was expressed at a much higher level than the PER2-EGFP (Fig. 1I and SI Appendix, Fig. S1 E and F). The PER2-EGFP condensate formation was also abolished when 34 known and potential PER2 phosphorylation sites between aa 527 to 818 were mutated to alanine (Fig. 1I and SI Appendix, Fig. S1F). Thus, the formation of the PER2-EGFP condensates is dependent on PER phosphorylation.
To examine whether the PER2 condensates colocalize with the endogenous clock protein BMAL1, we performed immunofluorescence staining of the cells using a BMAL1-specific antibody. As shown in Fig. 1J, most of the endogenous nuclear BMAl1 was found to localize in the PER2-GFP condensates, suggesting that the PER2 condensates recruit the BMAL1-CLOCK complex, which should sequester CLOCK-BMAL1 complex and inhibit its function in circadian transcriptional activation. Together, these results appear to suggest that the phosphorylation-regulated LLPS properties of PER proteins are important for circadian clock function.
Behavior of the Endogenous PER2 Tagged with EGFP by CRISPR/Cas9-Mediated Knock-In.
To determine whether the endogenous PER2 exhibits LLPS behavior, we modified a previously developed CRISPR/Cas9-based method (29) to tag the C-terminal end of the endogenous PER2 with EGFP reporter in U2OS cells (Fig. 2A). Due to the low expression levels of the endogenous PER2, direct selection of correct homologous integration clones by EGFP fluorescence signal resulted in mostly false negative clones. In the donor vector, an mCherry reporter was added upstream of the Per2 left homologous recombination arm, and the sequence encoding the ECFP reporter was added between the EGFP reporter and the right homologous recombination arm (Fig. 2A). Thus, fluorescence-activated cell sorting (FACS) of ECFP-positive but mCherry-negative cells from the blasticidin-resistant cells could be used to greatly enrich for cells in which the donor vector had been integrated at the endogenous Per2 genomic locus. Subsequently, the sequence encoding the ECFP reporter, which is flanked by LoxP sites, was removed by transfecting selected cells with a CRE expression plasmid; this restored the 3′UTR of the Per2 locus.
Fig. 2.

CRISPR/Cas9-mediated knock-in to tag the endogenous PER2 with EGFP. (A) Schematic of PER2-EGFP knock-in donor plasmid and genome editing strategy. (B) Live cell confocal imaging of PER2-EGFP KI U2OS cells and U2OS cells without KI. (Scale bar: 5 μm.) (C) Lambda scan of PER2-EGFP KI cells and control cells. The GFP wavelength was extracted, and all other wavelengths were merged as background. (D) Representative bioluminescence rhythms of the Per2(E2)-Luc reporter in the PER2-EGFP KI and control U2OS cells. Baseline-subtracted bioluminescence data are plotted (n = 3). (E) Western blot showing the PER2-EGFP levels in the PER2-EGFP KI cells and cells that stably overexpress PER2-EGFP. The protein sample from the cells that stably overexpress PER2-EGFP was diluted 20-fold.
Single clones of the resulting ECFP-negative cells were obtained by FACS sorting and confirmed by PCR and sequencing. Examination by live cell fluorescence microscopy revealed that >40% of clones had highly enriched nuclear EGFP signal (Fig. 2B), as previously reported for PER2-tagged reporter cells (28, 29). In contrast, there was no nuclear fluorescence signal in the control U2OS cells without the KI.
Fluorescence was also detected in the perinuclear regions in the cytoplasm of both knock-in and control U2OS cells (Fig. 2B). Lambda scan of the fluorescent images was used to extract this background noise spectra and the EGFP signal. Most of the perinuclear signals were indeed background noise in the KI cells (Fig. 2C). Due to the cytoplasmic background autofluorescence signals, we focused on the nuclear fluorescence signals. A Per2(E2)-Luc reporter plasmid was transfected into the PER2-EGFP KI and control U2OS cells (47). Our results revealed that both KI and control cells had similarly robust circadian bioluminescence rhythms with the same periodicity (Fig. 2D). This indicated that the EGFP KI did not affect endogenous PER2 function, which is similar to previous reporter knock-in at the PER2 C-terminus (30, 54).
Although cells that stably express PER2-EGFP also exhibited nuclei-enriched EGFP signals, comparison of PER2-EGFP protein levels by western blot analysis showed that the PER2-EGFP levels in cells that stably express this construct are more than 20-fold higher than the peak endogenous PER2-GFP levels (measured 10 h after dexamethasone synchronization) in the KI cells (Fig. 2E). This result indicated that although the viral transduction-mediated PER2 expression level in stable cells is much lower than that by transient transfection of cells, it is still much higher than those of the endogenous GFP-tagged PER2.
Consistent with previous analyses of the PER2 reporter KI cells (29), confocal fluorescence imaging of the live PER2-EGFP KI cells revealed a robust circadian rhythm of nuclear PER2-EGFP signal after the cells were synchronized by dexamethasone (DXMS) treatment. At its peak time (10 h after dexamethasone synchronization), the PER2-EGFP signal was found to be distributed throughout the nucleus; however, condensate-like structures were not observed in the KI cells (Fig. 2 B and C).
Super-Resolution Imaging Reveals Circadian Rhythm of the Endogenous PER Bodies.
To determine the high-resolution spatial distribution of the endogenous PER2, we performed Airyscan super-resolution imaging of fixed PER2-EGFP KI cells at 10 h after DXMS synchronization (55, 56). Super-resolution imaging revealed hundreds of small PER2-EGFP foci in the nucleus (Fig. 3A). To confirm that the nuclear EGFP foci contained PER2-EGFP, we used PER2-EGFP KI cells after treatment with Per2-specific siRNA and Per2KO cells (Per2 gene was disrupted by CRISPR/Cas9 using a Per2 specific sgRNA) as the negative controls (Fig. 3B and SI Appendix, Fig. S2A). In live cells (scan time ~ 1 s), although most nuclear foci were blurred which were due to their movements during the time needed for image collection, clear foci were still observed (Fig. 3A and other live cell experiments below). In addition, the distribution of PER2-EGFP signals in live cells is very similar to previously shown for endogenous PER2 fusion with other reporters in cells and mice (28–30). These results indicated that the PER2-EGFP foci are the endogenous foci and not artifacts caused by cell fixation. These nuclear foci of PER2 were referred as PER bodies because their fluorescence intensities indicate that they consist of multiple PER2 molecules in each. Although some PER bodies were also detected in the cytoplasm, they were far fewer than in the nucleus and could be masked by autofluorescence signals. Thus, we focused our analyses on the nuclear PER2 bodies.
Fig. 3.
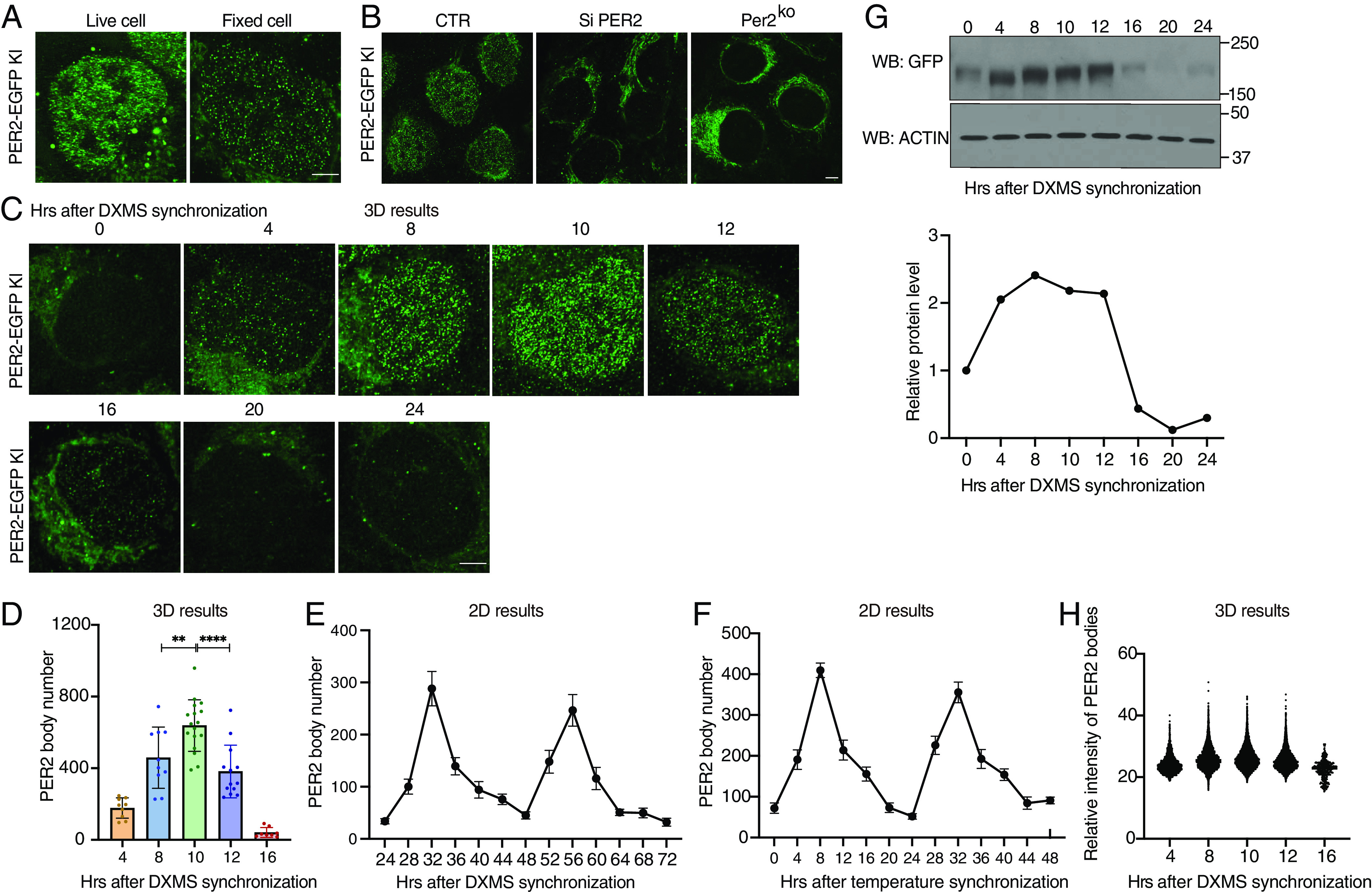
Super-resolution imaging of PER2-EGFP KI cells showing PER2 is concentrated in nuclear microbodies. (A) Airyscan confocal imaging of live (Left) and fixed (Right) PER2-EGFP KI cells in the nucleus. (Scale bar: 5 μm.) (B) Airyscan imaging of fixed PER2-EGFP KI cells (Left), KI cells treated with Per2-specific siRNA (si-PER2), and Per2KO cells. (C) Time course 3D imaging results of nuclear GFP signals in PER2-EGFP KI cells after dexamethasone synchronization. Representative 2D images from the 3D results are shown. (D) Numbers of nuclear PER bodies at different time points after dexamethasone synchronization (n = 8 to 17 cells), unpaired two-tailed Student’s t test. (E and F) Quantification of the PER2-EGFP microbodies of time course 2D imaging results of the PER2-EGFP KI cells after dexamethasone synchronization from h 24 to 72 (E) or after temperature cycle synchronization from h 0 to 48 (F). (G) Top: Western blot of EGFP in PER2-EGFP KI cells after dexamethasone synchronization. Equal amount of protein was loaded in each lane. Bottom: Quantification of PER2-EGFP levels. (H) Nuclear PER body mean fluorescence intensities after dexamethasone synchronization (n = 8 to 17 cells).
We performed 3D Airyscan super-resolution microscopy of fixed PER2-EGFP KI cells harvested at different time points. The number of PER bodies exhibited a robust rhythm, with a peak at 10 h after synchronization at approximately 650 PER bodies per nucleus and a trough at 20 to 22 h after synchronization with approximately dozens of bodies per cell (Fig. 3 C and D and SI Appendix, Supplemental Data File 2). In addition, robust rhythms of PER bodies with a similar phase and period as also seen 24 to 72 h after cell synchronization by DXMS and 0 to 48 h after temperature cycle entrainment of cells (Fig. 3 E and F and SI Appendix, Fig. S2 B and C), indicating that the rhythms of PER bodies in the first 24 h are genuine circadian rhythms. In addition to the PER body rhythm, the PER2-EGFP protein levels also exhibited a robust rhythm with a peak at approximately 8 h and a trough at approximately 20 h after synchronization (Fig. 3G). Comparison of these two rhythms revealed that although PER2-EGFP levels at 4 h were comparable to those at 10 h, the number of PER bodies was more than 3 times higher at hour 10 h than that at hour 4. In addition, the peak of PER bodies occurred at ~10 h, whereas the PER2-EGFP levels peaked at 8 h. Nuclear and cytoplasmic fractions of the cell extracts showed that the nuclear PER2-EGFP level is only modestly higher at hour 10 than that at hour 4 (SI Appendix, Fig. S2D). Thus, PER2 protein level alone does not determine the formation of PER2 bodies and at hour 4, there are relatively more PER molecules are not in PER bodies and cannot be detected.
Although PER levels at 16 h were about 80% lower than those from at 8 to 12 h and PER body numbers were also much lower, fluorescence intensities of the PER bodies, which should reflect their sizes, were not markedly different (Fig. 3H). This result contrasts with the concentration-dependent LLPS behavior exhibited by the PER2-EGFP condensates in cells that stably overexpress the fusion protein (Fig. 1C), indicating that the formation of endogenous PER2-EGFP bodies is less sensitive to protein concentration changes than the LLPS behavior of the overexpressed PER2-EGFP. The robust rhythms of PER2-EGFP protein and fluorescence are similar to the published rhythms of PER2 (10, 57).
We used two different methods to estimate the number of PER2 molecules in each PER body. Mass spectrometry–based analyses previously determined that the PER2 levels peaked at about 13,000 copies per cell (58), which led us to estimate that each PER body might have ~20 PER2 molecules. We also used known amount of recombinant EGFP protein to estimate the absolute abundance of PER2-EGFP molecules per PER2-EGFP KI cell at 10 h after synchronization (SI Appendix, Fig. S2E). Using this method, we estimate that there are on average ~23 PER2-EGFP molecules in each PER body. It should be noted that both methods likely overestimate the number of PER2 molecules in a PER body because not all PER2 molecules are in nuclear PER2 bodies. The PER bodies should also expect to include PER1, PER2, and PER3 proteins given previously established interactions of these proteins (18, 25).
Most PER Bodies Freely Diffuse in the Nucleus, but Some Are Immobile for Less than 1 s.
PER, CRY, BMAL1, and CLOCK proteins were thought to exist primarily in a 1.9-MDa nuclear repressor protein complex (25). Fluorescence correlation spectroscopy of overexpressed PER2, BMAL1, CLOCK, and CRY1 proteins in cells have very low nuclear diffusion coefficients; that of PER2 is the lowest (<0.2 µm2/s), raising the possibility that these overexpressed proteins are mostly immobile likely due to their association with chromatin (27, 59). To examine spatial dynamics of the endogenous nuclear PER bodies, we performed live cell imaging using an Airyscan super-resolution microscope in 0.35-1 s sampling intervals (SI Appendix, Supplemental Data File 3). Surprisingly, no stationary PER bodies (i.e., a body immobile in at least three consecutive frames) were detected, and it was not possible to track the movement of individual PER body due to its rapid movements. This result indicates that the endogenous PER bodies are distinct from the stationary condensates seen in cells that stably overexpress PER2-EGFP.
We then examined live KI cells using a high-resolution laser-scanning oblique plane microscope (light sheet microscopy) (60, 61), which allows for high-spatiotemporal imaging of fluorescent molecules under gentle, non-phototoxic illumination. Even here, with an exposure time of 30 ms per plane, PER body dynamics were too fast to image in a volumetric format. Thus, we settled on imaging a single oblique cross-section of the nucleus at 30-ms intervals (Fig. 4A and SI Appendix, Supplemental Data File 4). Tracking the trajectories of individual fluorescent PER bodies confirmed that the vast majority were not stationary (Fig. 4B, representative tracks 1, 2, 3, and 4), indicating that most PER bodies are not associated with chromatin. Analysis of mean square displacement (MSD) of the PER bodies as a function of time showed that they have a mean diffusion coefficient of 12.12 ± 0.5117 µm2/s (Fig. 4 C and D), which is similar to free diffusion behavior previously shown for transcription factors (62). However, 0.6% trackable traces of PER bodies revealed that some PER bodies were immobile for longer than 90 ms (Fig. 4B). In one example (track 5), the PER body was immobile during the entire trackable duration, and in another (track 6), the body first moved and then became immobile. The average time of immobility of the immobile PER2 bodies was ~0.4 s (Fig. 4E). Our estimate of the percentage of PER2 bodies that are immobile is certainly an underestimate since we could not detect PER bodies that were immobile for less than 90 ms and one PER body can be responsible for multiple tracks due to the imaging limitation of single nuclear cross-section. Nonetheless, these results demonstrate that only a very small fraction of PER bodies are on chromatin at any given time and that PER bodies can only remain on chromatin for less than 1 s.
Fig. 4.

Most PER2 bodies diffuse freely in the nucleus and are formed by a mechanism distinct from overexpression-caused LLPS condensates. (A) Representative image of nuclear PER bodies in PER2-EGFP KI cell under light-sheet fluorescence microscopy at hour 10 after synchronization. (Scale bar: 5 μm.) (B) Representative movement tracks of PER bodies. 1 to 4: freely diffusing tracks. 5: immobile tracks. 6: a diffusing PER body became immobile. (C) The averaged MSD of PER bodies computed from data collected from three cells. Data are presented as means ± SEM. (D) Mean speeds of different tracks (n = 3 cells). (E) Duration of immobility of PER2 bodies of the identified immobile PER2 bodies (data collected from three cells). (F) Representative image of nuclear PER bodies at 10 min after treatment with DMSO or 1,6-hexanediol in KI cells at hour 10 after synchronization. (G) Comparison of nuclear PER2-EGFP fluorescence intensities of KI cells at 10 min after treatment with DMSO or 1,6-hexanediol at hour 10 after synchronization. Data are presented as median fluorescence intensity in each cell. n = 14 to 18 cells. (H) Western blot of PER2-EGFP in the indicated cells at hour 10 after synchronization. The arrow indicates the phosphorylated PER2-EGFP species. * a background band. (I) Representative Airyscan images of PER bodies in the indicated cells at hour 10 after synchronization. (J) Nuclear PER2 body numbers in the indicated cells at 10 h and 16 h after dexamethasone synchronization.
PER Bodies and LLPS Condensates Are Formed by Distinct Mechanisms.
It was previously proposed that the multivalent protein–protein interactions mediated by IDRs/LCDs of some transcription factors can mediate the formation of protein hubs at physiological concentrations (42, 45, 63). When these proteins are overexpressed, the same LCD-LCD multivalent interactions can result in formation of LLPS condensates (45). To compare the endogenous PER bodies with the PER2 LLPS condensates, we treated with PER2-EGFP knock-in cells with 1,6- hexanediol (1.5%), which rapidly abolished the condensates in cells that stably overexpressed PER2-EGFP (Fig. 1G). In contrast, the hexanediol treatment did not affect number or fluorescence intensities of the PER bodies in the KI cells (Fig. 4 F and G and SI Appendix, Fig. S3A). This result indicates that the endogenous PER bodies and the LLPS condensates are formed by distinct mechanisms: LLPS condensates are mediated by multivalent weak interactions, whereas the endogenous PER bodies are formed by stronger protein–protein interactions that are not resistant to hexanediol treatment.
We then examined whether the endogenous PER2 bodies are sensitive to PER2 phosphorylation. We used the CRISPR/Cas9 method to mutate the PER2-CK1 interaction domain of the Per2 gene in the KI cells. We obtained a single clone-derived cell line in which Per2 has an in-frame deletion (Δ727-731) in the PER2-CK1 interaction domain (18). We further disrupted the endogenous Per1 gene by CRSPR/Cas9 in these cells. As expected, PER2-EGFP in the PER2(Δ727-731)-EGFP; Per1KO cells became severely hypophosphorylated compared to the parental cells (Fig. 4H). However, the numbers and intensities of the PER bodies were high in the PER2(Δ727-731)-EGFP KI; Per1KO cells (Fig. 4 I and J and SI Appendix, Fig. S3 B and C). Whereas the number of PER bodies decreased dramatically at 16 h after synchronization in both PER2-EGFP KI and PER2-EGFP KI; Per1KO cells, their numbers and their intensities remained high in the PER2(Δ727-731)-EGFP KI; Per1KO cells. This result is consistent with our previous study showing the importance of the PER-CK1 interaction for clock function at the molecular level (18). These experiments demonstrated that formation of endogenous PER2 bodies is not dependent on PER2 phosphorylation and PER-CK1 interaction, further confirming that PER body formation and LLPS of PER2 are mediated by distinct mechanisms. Due to heterogeneity of PER2 microbodies, we cannot exclude the possibility that a few of small LLPS-dependent structures can still exist in cells. Thus, the PER2 LLPS condensates are mostly, if not all, caused by protein overexpression and are not normally present in cells.
Interactions among PER2-, BMAL1-, and CRY1-Containing Microbodies Are Dynamic.
In cells that stably overexpress PER2-EGFP, most BMAL1 was recruited to the PER2-EGFP condensates (Fig. 1J). Similarly, the overexpression of EGFP-PER2 was previously shown to dramatically reduce the mobility of CRY1 (27). These protein overexpression-based results are consistent with the proposal that most core clock proteins, including PER1/2, are in large nuclear complexes on the chromatin (25). To examine the spatial distributions of other endogenous core clock proteins and their interactions, we inserted an in-frame mScarlet-I tag at the C-terminal end of the open reading frame of the endogenous Bmal1 and Cry1 loci in the PER2-EGFP KI cells using the CRSPR/Cas9 approach (Fig. 5A and SI Appendix, Fig. S4A). After synchronization of cells by DXMS, PER2-EGFP fluorescence exhibited a robust rhythm that peaked at 8 to 10 h after synchronization (Fig. 5 B and C). In contrast, the rhythm of BMAL1-mScarlet-I fluorescence was ~ 8 h phase advanced from the PER2 rhythm: BMAL1-mScarlet-I fluorescence peaked at 4 h and troughed at 10 to 12 h (Fig. 5B), consistent with the advanced BMAL1 protein phase in mice (18, 64). On the other hand, the CRY1-mScarlet-I fluorescence in the CRY1-mScarlet-I fluorescence did not exhibit a robust rhythm in the double KI cells (Fig. 5C). Like PER bodies, hundreds of BMAL1-mScarlet-I and CRY1-mScarlet-I foci were also detected in each cell after fixation in the 2D imaging results (Fig. 5 B and C), suggesting that BMAL1 and CRY1 also form multimeric complexes. Importantly, the merged images showed that most of the PER bodies did not colocalize with BMAL1-mScarlet-I/CRY1-mScarlet-I foci (SI Appendix, Fig. S4 B and C). Similar results were also seen for cells at hour 32 after synchronization (S4D). Live cell imaging of these double KI cells further confirmed that the distribution patterns of the PER, BMAL1, and CRY1 bodies are very similar to those in the fixed cells (Fig. 5 D and E), further indicating that the results of the fixed cells are not artifacts caused by cell fixation. We in live cells.
Fig. 5.

Airyscan imaging analyses of dual fluorescence reporter U2OS cells in which the endogenous PER2 and BMAL/CRY1 are tagged by EGFP and mScarlet-I, respectively. (A) Diagrams showing the design of the knock-in donor plasmids. (B) Time course Airyscan fluorescence imaging of PER2 and BMAL1 in fixed indicated KI cells after dexamethasone synchronization. (Scale bar: 5 μm.) (C) Time course Airyscan imaging of PER2 and CRY1 in fixed double KI cells after dexamethasone synchronization. (D and E) Airyscan fluorescence imaging of PER2 and BMAL1/CRY1 in live double KI cells.
To estimate the numbers of BMAL1/CRY1 microbodies and identify the colocalization foci with PER2 bodies, we performed 3D Airyscan fluorescence microscopy analyses (Fig. 6 A and B and SI Appendix, Supplemental Data Files 5 and 6), which revealed that the numbers of BMAL1 bodies were rhythmic but with a much lower amplitude than that of PER body rhythm (Fig. 6C). There were about 800 BMAL1 bodies at its peaks (hour 4), and at troughs, there were about 5 to 600 BMAL1 foci. On the other hand, the number of CRY1 bodies stayed ~600 at different time points after hour 0. Based on the previous estimates of ~20,000 BMAL1 and CRY1 molecules per cell (58), there are likely ~25 to 30 BMAL1 and CRY1 molecules in their respective bodies if all BMAL1 and CRY1 reside in their respective microbodies.
Fig. 6.

Airyscan imaging analyses of double KI cells showing that most PER2 bodies do not associate with BMAL1/CRY1 and the DNA residence time of BMAL1. (A and B) 3D Airyscan imaging result of PER2-EGFP and BMAL1/CRY1-mScarlet-I double KI cells at hour 10 after synchronization. Representative converted 2D nuclear images were shown. The 3D Airyscan imaging results for other time points are presented in SI Appendix, Supplemental Data Files. (Scale bar: 5 μm.) (C) Numbers of BMAL1 bodies and CRY1 bodies after dexamethasone synchronization in the double KI cells. Data are means ± SEM (n = 8 cells). (D) Numbers of PER2 and BMAL/CRY1 foci that colocalize after dexamethasone synchronization. Threshold distance for colocalization was set <100 nm. n = 8 cells. (E and F) Percentages of BMAL1/CRY1 and PER2 foci that colocalize after synchronization in the indicated PER2 and BMAL1/CRY1 double KI cells. n = 8 cells. (G) Time series of live cell Airyscan imaging of nuclear BMAL1 foci in the BMAL1-mScarlet-I knock-in cells. The arrow indicates immobile BMAL1 foci. Frame rate: 1.02 s/frame. (Scale bar: 5 μm.) (H) Summary of duration of immobility of identified immobile BMAL1 foci in the BMAL1-mScarlet-I KI cells. n = 10 cells.
We analyzed the 3D fluorescence images of the double-labeled cells and identified EGFP foci (PER2) and mScarlet-I (BMAL1/CRY1) foci that were less than 100 nm apart based on their centers using Imaris spots colocalization analysis (SI Appendix, Fig. S5 A and B), which were regarded as interacting with each other if the sizes of PER2/BMAL1/CRY1 bodies are 100 nm or larger. Most EGFP (PER2) and mScarlet-I (BMAL1 and CRY1) foci did not colocalize (Fig. 6 A and B and SI Appendix, Fig. S5 A and B). There were rhythms of the number of colocalized foci peaking at 8 to 10 h post synchronization for both PER2 with BMAL1 and PER2 with CRY1 (Fig. 6D), corresponding to the time of peak PER body number. In addition, the percentages of PER2, BMAL1, and CRY1 bodies that colocalize with each other were also rhythmic (Fig. 6 E and F). At the peak of the PER2 level after synchronization (hour 8 to 10), there were less than 10% of PER2 were considered to interact with BMAL1 and CRY1, respectively. For BMAL1 and CRY1, peak percentages associated with PER2 peaked at about 13% and 10%, respectively. The highest PER2 colocalization with BMAL1/CRY1occurred when PER2 level peaked, which is consistent with previously immunoprecipitation results (10). Single-particle electron microscopy previously estimated the super nuclear PER-containing complexes from mouse liver to be ~40-nm structures (25). When colocalization distance of 40 nm was used in our analysis, only about 2% of PER bodies were found to be colocalized with BMAL1 or CRY1 at hour 10 (SI Appendix, Fig. S6 A–C). Therefore, the vast majority of PER2 molecules are not in complex with BMAL1 or CRY1 in the nucleus at any given time, indicating that their interactions are transient.
Nuclear Movements of BMAL1 and CRY1 Bodies.
We next examined nuclear dynamics of BMAL1-mScarlet-I and CRY1-mScarlet-I in the KI cells by performing live cell Airyscan super-resolution imaging at 1-s intervals (SI Appendix, Supplemental Data Files 7 and 8). The low signal levels of the mScarlet-I-labeled proteins in the knock-in cells prevented us from examining the live cell imaging of BMAL1 and CRY1 bodies in shorter time intervals. BMAL1 and CRY1 bodies that stayed in the same location for three consecutive frames (~3 s) were considered as immobile. We were not able to identify any CRY1 bodies that were immobile using this method, suggesting that CRY1 most freely diffuse and can only stay on chromatin for less than 3 s. For BMAL1, however, some immobile bodies were identified with many immobile for 3 to 5 s or as long as 16 s (Fig. 6 G and H), consistent with the residence time of known transcription factors on chromatin (62). Fig. 6G shows that a BMAL1 body was immobile for 8 s before disappearing afterward. These results indicate that BMAL1/CLOCK are on the chromatin considerably longer than PER and CRY proteins. The short duration of PER and CRY association with BMAL1/CLOCK on chromatin suggests that the PER and CRY bodies are enzyme-like and can act on multiple BMAL1/CLOCK complex.
Spatial Distributions of PER2, BMAL1, and CRY1 and Their Associations Revealed by STED (Stimulated Emission Depletion) Microscopy and Immunodepletion Assays.
STED fluorescence microscopy can circumvent the optical diffraction limit and can achieve lateral spatial resolution of ~40 nm in cells (65, 66). We screened commercially available GFP, BMAL1, and CRY1 antibodies that can specifically detect PER2-EGFP, BMAL1, and CRY1 in the PER2-EGFP KI cells but not in control U2OS cells, MEF cells lacking BMAL1 or U2OS depleted for CRY1, respectively (57). As shown in SI Appendix, Fig. S7 A–C, anti-GFP, anti-BMAL1, and anti-CRY1 antibodies detected nuclear-specific PER2-EGFP, BMAL1, and CRY1 foci in the PER2-EGFP KI cells, but their signals were either absent or dramatically reduced in the control cells, indicating the specificity of these antibodies. 2D STED microscopy using two different antibodies (anti-GFP and anti-BMAL1 or anti-GFP and anti-CRY1) performed on the PER2-EGFP KI cells at 10 h after synchronization revealed hundreds of nuclear PER2-EGFP, BMAL1, and CRY1 foci per cell, consist with our Airyscan fluorescence data (Fig. 7 A and B). Merged images revealed that the vast majority of PER2-EGFP and BMAL1foci and of PER2-EGFP and CRY1 foci were not colocalized. Analysis of these 2D STED images revealed that 15 to 20% of the PER2-EGFP were less than 100 nm from centers of BMAL1 or CRY1 foci (Fig. 7 C and D). However, it should be noted that compared to 3D results, 2D analyses typically result in overestimation of protein colocalization by several folds.
Fig. 7.

PER, BMAL1, and CRY1 microbodies and their interactions revealed by STED microscopy. (A and B) STED microscopy images of PER2 and BMAL1/CRY1 puncta in the PER2-EGFP KI cells at 10 h after synchronization. PER2, BMAL1, and CRY1 were stained by anti-GFP (mouse), anti-BMAL1 antibody (rabbit), and anti-CRY1 antibody, respectively. (Scale bar: 5 μm.) (C and D) Percentages of PER2 and BMAL1/CRY1 bodies that colocalize at 10 h after synchronization. Distance threshold was set <100 nm. Data are presented as mean ± SEM (n = 8 cells). (E) Comparison of PER body sizes estimated by conventional anti-GFP antibody and anti-GFP nanobody. n = 5 cells. (F and G) Immunodepletion assay of CRY1 or BMAL1 in extracts prepared from the indicated double KI U2OS cells 10 h after synchronization (F) or from mice liver tissue (ZT18) (G). After immunodepletion, western blot analyses were performed using the indicated antibodies. (H) Representative immunofluorescence imaging results in the nucleus using mice SCN tissues harvested at the indicated ZT time points using a rabbit polyclonal PER2 and a mouse CRY1 antibody, respectively. Cells from the core SCN region were selected. (I) Quantification of the nuclear PER2/CRY1 foci numbers in the SCN cells at the indicated time points.
The increased resolution of the STED microscopy did, however, allowed us the estimate the sizes of the PER/BMAL1/CRY1 bodies to be ~100 nm (Fig. 7E and SI Appendix, Fig. S7D). Because the use of primary and secondary antibodies (size ~20 nm) for indirect immunolabeling in STED should increase the apparent size of PER bodies, we performed STED microscopy using an anti-GFP nanobody conjugated with STAR 635P (size < 4 nm). As expected, the sizes of PER bodies identified by the GFP nanobody were estimated at 70 nm. The actual sizes of the PER bodies are likely smaller than 70 nm due to the presence of multiple PER2 molecules in unknown orientations in each PER body. This result might be consistent with the 40-nm estimate from the previous single-particle electron microscopy analysis (25). When 40 nm was used as the colocalization distance, the percentage of PER2-EGFP foci that colocalize with BMAL1 foci and CRY1 foci dropped to 8% and 3%, respectively, in these 2D image analyses (SI Appendix, Fig. S7 E and F).
To further confirm our conclusion, we performed immunodepletion assays using protein extracts prepared from the double KI U2OS cells and the lowest concentration of CRY1 or BMAL1 antibodies that could deplete the protein of interest. As shown in Fig. 7F, the depletion of CRY1 or BMAL1 did not markedly affect the PER2 levels in the depleted cell extracts. Our conclusion was further confirmed by mPER2 immunodepletion assays by using C57BL/6 mice liver extracts (ZT18, ~peak of mPER2) (Fig. 7G), indicating that our conclusion in U2OS cells can also be applied in mice.
Most Nuclear PER2 and CRY1 Do Not Colocalize in Mice SCN Cells.
Finally, to confirm whether our conclusion can also be applied in mice SCN cells, we performed Airyscan immunofluorescence imaging experiments to examine the colocalization of PER2 and CRY1 using mice SCN tissues. PER2-specific and two different CRY1 antibodies (one mouse antibody and one guinea pig antibody) were used. As expected, mPER2 and mCRY1 foci similar to those in U2OS KI cells were found to be enriched in the nuclei of the SCN cells (Fig. 7H and SI Appendix, Fig. S7G). As expected, despite some colocalization, most mPER2 and mCRY1 foci did not colocalize. In addition, the number of mPER2 foci was significantly higher at ZT12 and CT12 than those at ZT4 and CT4, respectively, consistent with a mPER2 rhythm in SCN cells (Fig. 7I). As shown in SI Appendix, Fig. S7H, the PER2 and CRY1 antibodies had low background nuclear signals in the SCN cells prepared from the Per1−/−Per2−/−Per3−/− triple KO and Cry1−/−Cry2−/− double KO mice, respectively (67, 68), indicating the specificity of the antibodies. Together, our results demonstrated that endogenous PER2 only interacts with endogenous BMAL1 and CRY1 transiently and most PER2 bodies are not in complex with BMAL1 or CRY1 at any given time.
Discussion
The challenges of examining LLPS behavior of endogenous proteins have often meant that the conclusion of many studies were based on protein overexpression in cells or on in vitro biochemical studies using high concentrations of recombinant proteins (42). Our analysis presents an important cautionary example for LLPS and circadian studies and demonstrates the importance of examining endogenous proteins in our understanding of biological mechanisms.
PER proteins are core components of the animal circadian clocks(1–3). When PER2 was stably overexpressed in U2OS cells at a much lower level than commonly used transient transfection, PER2 LLPS condensates are formed in the nucleus. These condensates satisfies standard LLPS criteria: They are sensitive to concentration changes, can fuse with each other, rapidly recover after photobleaching, and are highly sensitive to 1,6-hexanediol treatment. Moreover, the LLPS behavior also appeared to correlate with PER2 biological function as condensate formation depends on PER phosphorylation and the condensates can recruit other clock proteins. Had we relied only on these results, we would have proposed that PER LLPS was important for clock function. The overexpressed Drosophila PER and the mammalian REV-ERBα were recently shown to exhibit LLPS behavior (69, 70). How the endogenous dPER and REV-ERBα proteins behave is still unclear.
Super-resolution imaging of the endogenous PER2 protein, however, revealed that the LLPS behavior exhibited by the overexpressed protein was not physiologically relevant. Although the endogenous PER2 formed highly concentrated protein microbodies, they were insensitive to concentration changes, resistant to 1,6-hexanediol treatment, and insensitive to the loss of PER2 phosphorylation. These results indicate that the LLPS condensates and the endogenous PER bodies are formed by distinct mechanisms. The former is likely mediated by multivalent weak and nonspecific protein interactions involving IDRs when PER2 is over expressed, but the latter is likely formed by strong specific protein–protein interactions insensitive to 1,6-hexanediol treatment and protein phosphorylation. This is different from the protein hub formation of some transcription factors, which was previously shown to be mediated by the multivalent IDR-IDR interactions (42, 45, 46). A crystal structure of a PER2 PAS domains revealed a homodimer stabilized by antiparallel packing of the PAS-B β-sheets (71), suggesting that highly specific protein–protein interactions mediated by structured protein regions (72). Based on our results, we propose that for LLPS studies, endogenous proteins should be examined whenever possible. In addition, the level of the endogenous protein should be compared to that used for LLPS studies.
PER proteins promote the CRY-dependent removal of BMAL1-CLOCK complex from E-boxes on chromatin by recruiting CK1 to phosphorylate CLOCK and modulate CRY-mediated inhibition of BMAL1-CLOCK activity on chromatin (18–22). PER was thought to mediate these roles through formation of large and stable nuclear complexes with CRY1, BMAL1, and CLOCK (25, 27). By directly visualizing the nuclear distribution and dynamic movements of endogenous proteins, our results showed that PER2, BMAL1, and CRY1 all form hundreds of nuclear microbodies that are under circadian control with PER2 have the most robust rhythm. The number of PER bodies is not solely determined by PER2 levels since at 4 and 10 h after cell synchronization, levels of PER2 were about the same, but there were drastically different PER body numbers. The progressive phosphorylation of PER2 during this time frame may modulate PER body formation.
PER proteins have been previously shown to co-immunoprecipitated with BMAL1 and CRY proteins and have high affinity binding with each other in vitro (2, 4–10), suggesting that they can form large stable complex together (25). In contrast, multiple lines of results presented here demonstrate that the vast majority of PER microbodies are not in association with BMAL1 or CRY1 bodies at any time, indicating that the interaction between endogenous PER and other clock proteins is transient. These results suggest that the interaction between PER2 and CRY1/BMAL1 in vivo is regulated.
Live cell imaging of BMAL1-mScarlet-I knock-in cells showed that only a small percentage of BMAL1 bodies are immobile for up to 16 s, which is similar to observations of other transcription factors (62). Our light-sheet microscopy analyses demonstrated that most PER bodies freely diffuse in the nucleus. Those that are immobile remain so for less than 1 s (average of 0.4 s), suggesting that the association of PER bodies with BMAL1 on DNA is very transient. Thus, PER bodies transiently interact with BMAL1-CLOCK complexes to promote CLOCK phosphorylation by CK1 resulting in dissociation of BMAL1-CLOCK from DNA (18, 19). In live cell imaging of CRY1-mScarlet-I knock-in cells at 1-s intervals, we did not detect immobile CRY1 bodies, indicating that the association time of CRY1 with chromatin is also much shorter than that of BMAL1. Thus, both PER and CRY1 mediate their roles in the circadian negative feedback process by transient association of with BMAL1 on chromatin which make PER and CRY1 to be enzyme-like and act on multiple BMAL1-CLOCK complexes, achieving high efficiency of repression.
The lack of co-localization between most PER2 bodies with CRY1 bodies was surprising mCRY1 and mPER2 were previously shown to have a high affinity for each other and can be fractionated together (4, 26). Our results here, which are confirmed by fluorescence and immunofluorescence imaging in U2OS cells and SCN cells and immunodepletion assays in cells and liver tissue, indicate that the PER2 and CRY1 interaction is transient in cells. Phosphorylation of either PER or CRY is one likely mechanism that regulates their interaction in vivo. CRY can repress BMAL1-CLOCK activity on DNA independently of PER proteins (20, 22, 73). We previously showed that PER and CRY can sustain a functional (but very low amplitude) circadian negative feedback loop by repressing BMAL1-CLOCK activity independently of the PER-CK1 interaction-mediated negative feedback mechanism (18). The transient PER-CRY1 association may modulate the inhibitory function of CRY1/2 to result in rhythmic BMAL1-CLOCK activity. Clock protein overexpression is commonly used in ccircadian studies. As we demonstrated in this study that the transient transfection- and lentiviral transduction-mediated PER2 expression resulted in over 2,000x and 20x overexpression, respectively. Given the discrepancies between results obtained based on experiments relied on protein overexpression and our findings, our study highlights the importance of studying endogenous proteins to understand clock mechanisms.
Unlike our results here and previously published imaging results of PER2, BMAL1, and CRY1 (28–30, 74), the Drosophila PER and CLOCK proteins were recently found to be in a few large discrete foci at the nuclear envelope during the circadian repression phase (75). Such a difference may be caused by differences in circadian clock mechanisms between insects and mammals.
Materials and Methods
U2OS, HEK293T, and MEF cells were used in this study. HEK293T and MEF cells were cultured in Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum (FBS). U2OS cells were cultured in McCoy's 5A medium with 10% FBS and penicillin–streptomycin in 35-mm or 10-cm plates. All live cell imaging studies were performed at 37 °C with CO2. All cell lines generated in this study were derived from U2OS cells, which are originally derived from human bone osteosarcoma epithelial cells of a female patient. Detailed materials and methods are described in SI Appendix.
Supplementary Material
Appendix 01 (PDF)
A movie related to the result in Figure 1D showing the fusion of two small condensates into one.
A movie showing the 3D images related to Figure 3D showing the nuclear PER2 bodies in the PER2-EGFP KI cells.
A movie showing the live cell fluorescence imaging of the PER2-EGFP KI cells using an Airyscan super-resolution microscope in 0.35 s sampling intervals.
A movie related to the result in Figure 4A showing the nuclear movements of PER2 bodies captured by light sheet microscopy in 30ms interval.
A movie showing the 3D fluorescence images related to Figure 6A which PER2 (green) and BMAL1 (red) bodies in the PER2-EGFP and BMAL1-mScarlet-I double KI cells at different time points after synchronization.
A movie showing the 3D fluorescence images related to Figure 6B which PER2 (green) and CRY1 (red) bodies in the PER2-EGFP and CRY1-mScarlet-I double KI cells at different time points after synchronization.
A movie showing live cell imaging of the BMAL1-mScarlet-I KI cells using an Airyscan super-resolution microscope in ~1s sampling intervals showing the nuclear movements of BMAL1 bodies.
A movie showing live cell imaging of the CRY1-mScarlet-I KI cells using an Airyscan super-resolution microscope in ~1s sampling intervals showing the nuclear movements of CRY1 bodies.
Acknowledgments
We thank Drs. Katherine Phelps and Marcel Mettlen and the University of Texas Southwestern Quantitative Light Microscopy Core for assistances in imaging analyses and Drs. Michael Rosen, Steven McKnight, Carla Green, Joachim Seeman, Matthew Parker, and Xuewu Zhang for discussions and suggestions. We would like to thank the NIH (R35GM118118 to Y.L.; U54CA268072, P30CA142543, and RM1GM145399 to K.M.D.; R01NS114527 to S.Y.), the Welch Foundation (I-1560 to Y.L.), and the NSF (IOS-1931115 to S.Y.) for their generous funding. STED microscopy facility is supported by the Peter O’Donnell Jr. Brain Institute.
Author contributions
P.X., X.X., K.M.D., S.Y., Y.X., and Y.L. designed research; P.X., X.X., C.Y., K.M.D., I.L., S.K.T.T., and S.Y. performed research; P.X., I.L., S.K.T.T., J.T., and Y.X. contributed new reagents/analytic tools; P.X., X.X., C.Y., K.M.D., S.Y., Y.X., and Y.L. analyzed data; and P.X., K.M.D., S.Y., and Y.L. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Takahashi J. S., Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 18, 164–179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Partch C. L., Green C. B., Takahashi J. S., Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 24, 90–99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hastings M. H., Reddy A. B., Maywood E. S., A clockwork web: Circadian timing in brain and periphery, in health and disease. Nat. Rev. Neurosci. 4, 649–661 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Schmalen I., et al. , Interaction of circadian clock proteins CRY1 and PER2 is modulated by zinc binding and disulfide bond formation. Cell 157, 1203–1215 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Nangle S. N., et al. , Molecular assembly of the period-cryptochrome circadian transcriptional repressor complex. Elife 3, e03674 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parico G. C. G., et al. , The human CRY1 tail controls circadian timing by regulating its association with CLOCK:BMAL1. Proc. Natl. Acad. Sci. U.S.A. 117, 27971–27979 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu H., et al. , Cryptochrome 1 regulates the circadian clock through dynamic interactions with the BMAL1 C terminus. Nat. Struct. Mol. Biol. 22, 476–484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fribourgh J. L., et al. , Dynamics at the serine loop underlie differential affinity of cryptochromes for CLOCK:BMAL1 to control circadian timing. Elife 9, e55275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langmesser S., Tallone T., Bordon A., Rusconi S., Albrecht U., Interaction of circadian clock proteins PER2 and CRY with BMAL1 and CLOCK. BMC Mol. Biol. 9, 41 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee C., Etchegaray J. P., Cagampang F. R., Loudon A. S., Reppert S. M., Posttranslational mechanisms regulate the mammalian circadian clock. Cell 107, 855–867 (2001). [DOI] [PubMed] [Google Scholar]
- 11.Vanselow K., et al. , Differential effects of PER2 phosphorylation: Molecular basis for the human familial advanced sleep phase syndrome (FASPS). Genes Dev. 20, 2660–2672 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y., et al. , Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature 434, 640–644 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Toh K. L., et al. , An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science 291, 1040–1043 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Xu Y., et al. , Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell 128, 59–70 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou M., Kim J. K., Eng G. W., Forger D. B., Virshup D. M., A period2 phosphoswitch regulates and temperature compensates circadian period. Mol. Cell 60, 77–88 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Narasimamurthy R., et al. , CK1delta/epsilon protein kinase primes the PER2 circadian phosphoswitch. Proc. Natl. Acad. Sci. U.S.A. 115, 5986–5991 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masuda S., et al. , Mutation of a PER2 phosphodegron perturbs the circadian phosphoswitch. Proc. Natl. Acad. Sci. U.S.A. 117, 10888–10896 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.An Y., et al. , Decoupling PER phosphorylation, stability and rhythmic expression from circadian clock function by abolishing PER-CK1 interaction. Nat. Commun. 13, 3991 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao X., Yang Y., Selby C. P., Liu Z., Sancar A., Molecular mechanism of the repressive phase of the mammalian circadian clock. Proc. Natl. Acad. Sci. U.S.A. 118, e2021174118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kume K., et al. , mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98, 193–205 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Griffin E. A. Jr., Staknis D., Weitz C. J., Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science 286, 768–771 (1999). [DOI] [PubMed] [Google Scholar]
- 22.Ye R., et al. , Dual modes of CLOCK:BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes Dev. 28, 1989–1998 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McManus D., et al. , Cryptochrome 1 as a state variable of the circadian clockwork of the suprachiasmatic nucleus: Evidence from translational switching. Proc. Natl. Acad. Sci. U.S.A. 119, e2203563119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fu J., et al. , Codon usage affects the structure and function of the Drosophila circadian clock protein PERIOD. Genes Dev. 30, 1761–1775 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aryal R. P., et al. , Macromolecular assemblies of the mammalian circadian clock. Mol. Cell 67, 770–782.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao X., Wang L., Selby C. P., Lindsey-Boltz L. A., Sancar A., Analysis of mammalian circadian clock protein complexes over a circadian cycle. J. Biol. Chem. 299, 102929 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koch A. A., et al. , Quantification of protein abundance and interaction defines a mechanism for operation of the circadian clock. Elife 11, e73976 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smyllie N. J., et al. , Visualizing and quantifying intracellular behavior and abundance of the core circadian clock protein PERIOD2. Curr. Biol. 26, 1880–1886 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gabriel C. H., et al. , Live-cell imaging of circadian clock protein dynamics in CRISPR-generated knock-in cells. Nat. Commun. 12, 3796 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smyllie N. J., et al. , Cryptochrome proteins regulate the circadian intracellular behavior and localization of PER2 in mouse suprachiasmatic nucleus neurons. Proc. Natl. Acad. Sci. U.S.A. 119, e2113845119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ollinger R., et al. , Dynamics of the circadian clock protein PERIOD2 in living cells. J. Cell Sci. 127, 4322–4328 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Hyman A. A., Weber C. A., Julicher F., Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 30, 39–58 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Banani S. F., Lee H. O., Hyman A. A., Rosen M. K., Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 18, 285–298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brangwynne C. P., Phase transitions and size scaling of membrane-less organelles. J. Cell Biol. 203, 875–881 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hnisz D., Shrinivas K., Young R. A., Chakraborty A. K., Sharp P. A., A phase separation model for transcriptional control. Cell 169, 13–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strom A. R., et al. , Phase separation drives heterochromatin domain formation. Nature 547, 241–245 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du M., Chen Z. J., DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li P., et al. , Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato M., et al. , Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwon I., et al. , Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell 155, 1049–1060 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alberti S., Gladfelter A., Mittag T., Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates. Cell 176, 419–434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McSwiggen D. T., Mir M., Darzacq X., Tjian R., Evaluating phase separation in live cells: Diagnosis, caveats, and functional consequences. Genes Dev. 33, 1619–1634 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kroschwald S., Maharana S., Simon A., Hexanediol: A chemical probe to investigate the material properties of membrane-less compartments. Matters 3, e201702000010 (2017). [Google Scholar]
- 44.Monahan Z., et al. , Phosphorylation of the FUS low-complexity domain disrupts phase separation, aggregation, and toxicity. EMBO J. 36, 2951–2967 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chong S., et al. , Tuning levels of low-complexity domain interactions to modulate endogenous oncogenic transcription. Mol. Cell 82, 2084–2097.e5 (2022). [DOI] [PubMed] [Google Scholar]
- 46.Chong S., et al. , Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, eaar2555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoo S. H., et al. , A noncanonical E-box enhancer drives mouse Period2 circadian oscillations in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 2608–2613 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen R., et al. , Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Mol. Cell 36, 417–430 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miyazaki K., Mesaki M., Ishida N., Nuclear entry mechanism of rat PER2 (rPER2): Role of rPER2 in nuclear localization of CRY protein. Mol. Cell Biol. 21, 6651–6659 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yagita K., et al. , Nucleocytoplasmic shuttling and mCRY-dependent inhibition of ubiquitylation of the mPER2 clock protein. EMBO J. 21, 1301–1314 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yagita K., et al. , Dimerization and nuclear entry of mPER proteins in mammalian cells. Genes Dev. 14, 1353–1363 (2000). [PMC free article] [PubMed] [Google Scholar]
- 52.von Stetten D., Noirclerc-Savoye M., Goedhart J., Gadella T. W. Jr., Royant A., Structure of a fluorescent protein from Aequorea victoria bearing the obligate-monomer mutation A206K. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 68, 878–882 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee C., Weaver D. R., Reppert S. M., Direct association between mouse PERIOD and CKIepsilon is critical for a functioning circadian clock. Mol. Cell. Biol. 24, 584–594 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoo S. H., et al. , PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc. Natl. Acad. Sci. U.S.A. 101, 5339–5346 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu X., Hammer J. A., ZEISS Airyscan: Optimizing usage for fast, gentle, super-resolution imaging. Methods Mol. Biol. 2304, 111–130 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huff J., The Airyscan detector from ZEISS: Confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods 12, i–ii (2015). [Google Scholar]
- 57.Bunger M. K., et al. , Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103, 1009–1017 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Narumi R., et al. , Mass spectrometry-based absolute quantification reveals rhythmic variation of mouse circadian clock proteins. Proc. Natl. Acad. Sci. U.S.A. 113, E3461–E3467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koike N., et al. , Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sapoznik E., et al. , A versatile oblique plane microscope for large-scale and high-resolution imaging of subcellular dynamics. Elife 9, e57681 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang B. J., et al. , Real-time multi-angle projection imaging of biological dynamics. Nat. Methods 18, 829–834 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Izeddin I., et al. , Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. Elife 3, e02230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McSwiggen D. T., et al. , Evidence for DNA-mediated nuclear compartmentalization distinct from phase separation. Elife 8, e47098 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Preitner N., et al. , The orphan nuclear receptor REV-ERBalpha controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110, 251–260 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Vicidomini G., Bianchini P., Diaspro A., STED super-resolved microscopy. Nat. Methods 15, 173–182 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Berning S., Willig K. I., Steffens H., Dibaj P., Hell S. W., Nanoscopy in a living mouse brain. Science 335, 551 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Pendergast J. S., Oda G. A., Niswender K. D., Yamazaki S., Period determination in the food-entrainable and methamphetamine-sensitive circadian oscillator(s). Proc. Natl. Acad. Sci. U.S.A. 109, 14218–14223 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vitaterna M. H., et al. , Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 96, 12114–12119 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li M., Li S., Zhang L., Phosphorylation promotes the accumulation of PERIOD protein foci. Research (Wash. D.C.) 6, 0139 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhu K., et al. , An intrinsically disordered region controlling condensation of a circadian clock component and rhythmic transcription in the liver. Mol. Cell 83, 3457–3469.e7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hennig S., et al. , Structural and functional analyses of PAS domain interactions of the clock proteins Drosophila PERIOD and mouse PERIOD2. PLoS Biol. 7, e94 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Musacchio A., On the role of phase separation in the biogenesis of membraneless compartments. EMBO J. 41, e109952 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chiou Y. Y., et al. , Mammalian Period represses and de-represses transcription by displacing CLOCK-BMAL1 from promoters in a Cryptochrome-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 113, E6072–E6079 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang N., et al. , Quantitative live imaging of Venus::BMAL1 in a mouse model reveals complex dynamics of the master circadian clock regulator. PLoS Genet. 16, e1008729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xiao Y., Yuan Y., Jimenez M., Soni N., Yadlapalli S., Clock proteins regulate spatiotemporal organization of clock genes to control circadian rhythms. Proc. Natl. Acad. Sci. U.S.A. 118, e2019756118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
A movie related to the result in Figure 1D showing the fusion of two small condensates into one.
A movie showing the 3D images related to Figure 3D showing the nuclear PER2 bodies in the PER2-EGFP KI cells.
A movie showing the live cell fluorescence imaging of the PER2-EGFP KI cells using an Airyscan super-resolution microscope in 0.35 s sampling intervals.
A movie related to the result in Figure 4A showing the nuclear movements of PER2 bodies captured by light sheet microscopy in 30ms interval.
A movie showing the 3D fluorescence images related to Figure 6A which PER2 (green) and BMAL1 (red) bodies in the PER2-EGFP and BMAL1-mScarlet-I double KI cells at different time points after synchronization.
A movie showing the 3D fluorescence images related to Figure 6B which PER2 (green) and CRY1 (red) bodies in the PER2-EGFP and CRY1-mScarlet-I double KI cells at different time points after synchronization.
A movie showing live cell imaging of the BMAL1-mScarlet-I KI cells using an Airyscan super-resolution microscope in ~1s sampling intervals showing the nuclear movements of BMAL1 bodies.
A movie showing live cell imaging of the CRY1-mScarlet-I KI cells using an Airyscan super-resolution microscope in ~1s sampling intervals showing the nuclear movements of CRY1 bodies.
Data Availability Statement
All study data are included in the article and/or supporting information.