

Significance
In this study, we show that KMT2D truncation mutations compromise MLL4/COMPASS (Complex of Proteins Associated with Set1) function at enhancers by altering the subcellular localization of the H3K4 histone lysine mono-methyltransferase MLL4 (Mixed Lineage Leukemia 4) from the nucleus to the cytosol. Concomitant changes in the localization of the H3K27 histone lysine demethylase UTX (KDM6A) and stability of the transcriptional coactivator NCOA6 cause a loss of transcripts from genes proximal to MLL4/COMPASS-regulated enhancers. We also identify KMT2D (MLL4) truncation mutations as potential biomarker for patient stratification in bladder cancer, demonstrating that the subcellular localization of the truncated MLL4 protein can be exploited by immunohistochemistry for potential targeted therapy in KMT2D (MLL4) truncation mutation-relevant cancers, Kabuki Syndrome and other diseases.
Keywords: chromatin modifiers, epigenetics, mutation, cancer biomarker, treatment
Abstract
Cancer genome sequencing consortiums have recently catalogued an abundance of somatic mutations, across a wide range of human cancers, in the chromatin-modifying enzymes that regulate gene expression. Defining the molecular mechanisms underlying the potentially oncogenic functions of these epigenetic mutations could serve as the basis for precision medicine approaches to cancer therapy. MLL4 encoded by the KMT2D gene highly mutated in a large number of human cancers, is a key histone lysine monomethyltransferase within the Complex of Proteins Associated with Set1 (COMPASS) family that regulates gene expression through enhancer function, potentially functioning as a tumor suppressor. We report that the KMT2D mutations which cause MLL4 protein truncation also alter MLL4’s subcellular localization, resulting in loss-of-function in the nucleus and gain-of-function in the cytoplasm. We demonstrate that isogenic correction of KMT2D truncation mutation rescues the aberrant localization phenotype and restores multiple regulatory functions of MLL4, including COMPASS integrity/stabilization, histone H3K4 mono-methylation, enhancer activation, and therefore transcriptional regulation. Moreover, isogenic correction diminishes the sensitivity of KMT2D-mutated cancer cells to targeted metabolic inhibition. Using immunohistochemistry, we identified that cytoplasmic MLL4 is unique to the tissue of bladder cancer patients with KMT2D truncation mutations. Using a preclinical carcinogen model of bladder cancer in mouse, we demonstrate that truncated cytoplasmic MLL4 predicts response to targeted metabolic inhibition therapy for bladder cancer and could be developed as a biomarker for KMT2D-mutated cancers. We also highlight the broader potential for prognosis, patient stratification and treatment decision-making based on KMT2D mutation status in MLL4 truncation-relevant diseases, including human cancers and Kabuki Syndrome.
Somatic mutations are major drivers of cancer. During the past two decades, sequencing studies led by The Cancer Genome Atlas and Catalogue of Somatic Mutations in Cancer (COSMIC) have highlighted genes (oncogenes and tumor suppressors) that are frequently mutated in a spectrum of human cancers (1). Some of these gene mutations are detected not only in tumors but also in normal tissues before cancer development, suggesting that early-stage mutations may confer competitive advantages for clonal expansion (2, 3). Among this category of early-stage somatic mutation-affected genes are the H3K4 mono-methyltransferases MLL3 (KMT2C) and MLL4 (KMT2D) and their cofactor H3K27 demethylase UTX (KDM6A), all of which are members of the Complex of Proteins Associated with Set1 (COMPASS) chromatin modifier family. MLL4 (KMT2D) is highly mutated in a variety of solid tumors and hematological malignancies including bladder cancer (4), colorectal cancer (5), lung cancer (6), esophageal cancer (7), endometrial cancer (8), diffuse large B-cell lymphoma (9, 10), and acute lymphoblastic leukemia (11, 12). Mutations of the KMT2D gene that cause truncation of its protein product (nonsense, frameshift insertion/deletion, and splice site mutations) in particular are characterized as putative cancer driver mutations which are believed to contribute to cancer initiation (13, 14). For the multisystem disorder called Kabuki syndrome, in which mutations in KMT2D and KDM6A genes cause developmental abnormalities, approximately 60% of KMT2D mutations also result in protein truncation (15–17). However, the molecular mechanisms by which these mutations alter MLL4/COMPASS function and how these alterations contribute to cancer initiation, progression, or the etiology of disease including Kabuki syndrome are all questions that remain poorly understood.
The vast majority of KMT2D mutations catalogued from genome-wide studies in COSMIC are heterozygous (~96.1%) (1). We, therefore, hypothesized that mutations affecting MLL4/COMPASS could have dosage-dependent functional effects. In this study, we sought to examine whether MLL4 protein truncation due to KMT2D mutation could also cause aberrant MLL4 protein localization. By creating isogenic cell lines with wild-type or truncation mutant allele-specific MLL4 knockout, we were able to demonstrate the nuclear-to-cytosolic translocation of the truncated MLL4 protein product of a known cancer driver mutation in KMT2D. Then, using a CRISPR knock-in (KI) approach to correct the truncation-causative mutation, we were able to demonstrate that multiple MLL4/COMPASS regulatory functions, including nuclear UTX import, NCOA6 stabilization, H3K4 mono-methylation, and regulated gene expression, are all subject to dosage-dependent effects of MLL4 truncation. Overall, our study uncovers the molecular mechanisms by which MLL4 truncation due to KMT2D mutation alters MLL4/COMPASS function. Of interest to the clinical application of this finding, our study sheds light on the potential not only for the development of MLL4 truncation as a biomarker for patient stratification but also for the development of targeted therapeutics for cancers and other diseases characterized by KMT2D mutation.
Results
KMT2D Truncation Mutations Are Prevalent in Human Cancers.
Somatic KMT2D mutations that cause MLL4 protein truncation, including nonsense mutations, frameshift insertions, or deletions, and splice site mutations, are prevalent in human cancer samples of all types within the COSMIC database (1) (Fig. 1A). Because they eliminate the catalytic Su(var)3-9, Enhancer-of-zeste and Trithorax (SET) domain from MLL4, potentially conferring growth advantage, these truncating mutations are also considered to be primary candidate cancer driver mutations (2, 3). However, the exact molecular mechanisms underlying the prevalence of KMT2D truncation mutations are still unclear. We speculated that both the amino acid composition and functional role of MLL4 contribute to the prevalence of KMT2D truncation mutations in human cancers. First, to analyze the amino acid composition, we compared the relative abundance of individual amino acids within MLL4 to that of the total human proteome (18) (Fig. 1B). Within MLL4, both proline (P) and glutamine (Q) are apparent outliers in terms of their relative abundance compared to the human proteome (Fig. 1C). When we analyzed which KMT2D mutation types impact each amino acid in MLL4, we found that frameshift mutations tend to impact Proline (P) while nonsense mutations tend to impact glutamate (E), glutamine (Q), and arginine (R), presumably due to the different codon usage of these amino acids (Fig. 1D). The MLL4 protein sequence is also enriched for sequence stretches containing multiple consecutive Q (poly-Q). Thus, we believe that the unique properties of the MLL4 protein sequence may at least partially underlie the tendency toward the elevated mutational burden found for KMT2D in a variety of human cancers. We also analyzed the amino acid composition of the COMPASS family subunits MLL3 (KMT2C), UTX (KDM6A), and UTY(UTY), due to the high rate of KMT2C and KDM6A mutation (19) and the homology between UTX and UTY (SI Appendix, Fig. S1 A–C). MLL3 is slightly enriched for Proline (P) and Glutamine (Q) (SI Appendix, Fig. S1A), while the amino acid compositions of UTX and UTY follow the general trend of relative abundance found within the human proteome (SI Appendix, Fig. S1 B and C). When analyzing which mutation types impacted each amino acid for these proteins (SI Appendix, Fig. S1 D–F), we noticed a general tendency toward KDM6A splice site mutations, whereas UTY is rarely mutated (20). Indeed, UTY is frequently subject to deep deletion, which has occurred in more than one-third of all male cancer cell lines (SI Appendix, Fig. S1G). UTY copy number aberrations are frequently observed in lineages related to the digestive system, such as esophageal, pancreas, gastric, liver, and colorectal lineages, in which KMT2D mutations are also commonly observed (SI Appendix, Fig. S1 H and I). UTX and UTY share 85.45% sequence similarity (20). UTY was originally believed to have no observable histone demethylase activity (21). However, a later study revealed that UTY does possess some H3K27 demethylase activity in vitro, albeit significantly less than that of UTX, accounting for approximately 2.6% of UTX's activity (22). However, to date, no activity of UTY—catalytic or catalytic-independent—has been assessed within the context of MLL3/4-COMPASS in vivo. Due to the frequent loss of UTY gene copy number and the exclusion of UTY from the interdependent stability relationship between KMT2D, KDM6A, and NCOA6 (Fig. 1E), we focused further study on the impacts of genetic mutation on the interaction between MLL4 and UTX within COMPASS, excluding any compensatory effect of UTY. We also noticed that the codependency pattern is dramatically altered in cell lines with MLL4 damaging mutation, where the codependency score for KDM6A decreased from 0.55 to 0.29 (Fig. 1F) and for NCOA6 from 0.42 to 0.094 (Fig. 1G).
Fig. 1.

KMT2D mutations are prevalent in human cancers. (A) Lollilop ProteinPaint visualization highlighting KMT2D mutations in human cancers (https://pecan.stjude.cloud/). 6,210 somatic mutations found within 199 histology types from the COSMIC database were included. (B) Amino acid composition of the human proteome with the 20 amino acids in descending order of relative abundance (18). (C) Amino acid composition of the MLL4 protein with amino acids in the same order as in B. (D) Total counts of MLL4 mutations occurring at each amino acid, grouped by mutation type. Fusion and splice site mutations were included. Mutation data were retrieved from the cBio database (https://www.cbioportal.org/) for all types of human cancers (13, 14). (E) KMT2D codependent genes in 1,095 cancer cell lines. KDM6A and NCOA6 are the top codependent genes of KMT2D. (F and G) Codependency of KMT2D with KDM6A (D) and NOCA6 (E) grouped by damaging mutations and nondamaging mutations in CCLE cell lines. Pearson correlation and P-value (lingress) are calculated. Gene CRISPR effect data were downloaded from the public DepMap 23Q2 database.
KMT2D Truncation Mutations Lead to Cytosolic Accumulation of Truncated MLL4.
Among the many studies investigating the function of MLL4 during early stages of cancer development and progression, the main shared conclusion is a tumor suppressive function for MLL4. These studies have relied on the homozygous deletion of both KMT2D alleles (either complete knockout or knockout of only the catalytic domain) in various mouse models (6, 9, 23–25). However, there is increasing evidence for dosage-dependent MLL4 function (10, 26, 27), indicating that the heterozygous deletion phenotype should also be critically evaluated. When we retrieved mutation information for samples with known zygosity from COSMIC v96 (1), we noticed that 96.1% of the KMT2D mutations are indeed heterozygous (Fig. 2A), strongly suggesting that at least one wild-type copy of MLL4 protein remain in the cancer cells. To distinguish between loss- or gain-of-function due to MLL4 truncation, we took advantage of the HCT116 colorectal cancer cell line. HCT116 cells are heterozygous for KMT2D, with both wild-type and truncation mutation alleles, homozygous for wild-type UTX, and have deep deletion of UTY (50% of cells lacked the Y chromosome in G-band karyotypes) (Fig. 2B). MLL4 truncation is caused by frameshift due to a single-nucleotide insertion (p.R2443Sfs*6) in KMT2D in HCT116 cells (which also bear the missense mutation p.V160M, of unknown significance and not evaluated in our study). Using a pair of sgRNAs targeting the promoter proximal region of KMT2D, in isogenic background, we generated pseudohomozygous (MLL4WT and MLL4Mut) cell lines by allele-specific knockout and also generated a homozygous MLL4 knockout (MLL4KO) cell line (Fig. 2B). We confirmed the parental (KMT2D+/Mut), WT (KMT2D+/−), Mut (KMT2D−/Mut), and KO (KMT2D−/−) genotypes by examining the aligned reads from bam files (SI Appendix, Fig. S2A) and gene expression levels in RNA-seq (SI Appendix, Fig. S2B). The genotypes were also confirmed by western blot analysis using an antibody against amino acids 1 to 181 of the MLL4 N terminus, which can detect both full-length and truncated MLL4 proteins (Fig. 2C and SI Appendix, Fig. S2C). The MLL4/COMPASS components UTX and NCOA6 are significantly down-regulated in both MLL4Mut and MLL4KO cell lines (Fig. 2C), suggesting that the full-length MLL4 protein may be required to maintain the stability of MLL4/COMPASS components, consistent with our previously reported findings (28). We further observed rapid loss of NCOA6 protein from MLL4Mut but not WT cells upon translation inhibition with cycloheximide, indicating dramatically decreased NCOA6 protein stability in the absence of WT MLL4 (SI Appendix, Fig. S2 D and E). Additionally, bulk levels of the histone modifications H3K4me1 and H3K27Ac were decreased in MLL4Mut and to an even greater extent in MLL4KO cell lines (Fig. 2D), which confirms that these modifications are driven by the catalytic activity within the full-length MLL4 protein, while also indicating that the relationship between MLL4 and histone H3 methylation is MLL4 dosage-dependent. When we performed immunofluorescence staining for MLL4, we observed differential localization of full-length and truncated MLL4 protein to the nuclear and cytoplasmic compartments, respectively (Fig. 2E). Further cellular fractionation confirmed the differential localization of full-length and truncated MLL4 (Fig. 2F). Concomitant to loss of the full-length, nuclear MLL4 WT protein, we observed diminished levels of nuclear NCOA6 and UTX, as well as cytoplasmic UTX accumulation (Fig. 2F).
Fig. 2.

MLL4 truncation due to KMT2D gene mutation leads to cytosolic accumulation of MLL4 protein. (A) MLL4 mutation zygosity information from COSMIC v96. Mutations were reported to be homozygous (n = 76), heterozygous (n = 1,850) or unknown (n = 6,300) within each sample. (B) Diagram illustrating MLL4 allele-specific CRISPR/Cas9 knockout cell line establishment. A pair of sgRNAs were employed to delete the promoter region of KMT2D. Depending on the allele(s) impacted by excision, resulting cells express only the full-length protein after excision of the truncation allele (MLL4WT), express only the truncated protein after excision of the wild-type allele (MLL4Mut) or no longer express MLL4 protein after excision of both alleles (MLL4 KO). (C) Western blot showing the protein levels of MLL4, NCOA6, UTX, PTIP, RbBP5, and WDR5 in HCT116 parental or isogenic, allele-specific MLL4 KO cells using β-tubulin as the loading control. (D) Western blot showing the levels of H3K4me1, H3K4me2, H3K4me3, and H3K27Ac in HCT116 parental or isogenic, allele-specific MLL4 KO cells using total histone H3 as the loading control. (E) Immunofluorescence showing the localization of wild type and truncated MLL4 protein in HCT116 parental or isogenic, allele-specific MLL4 KO cells. DNA was stained with DAPI and β-tubulin was stained to show the cytoplasm. (F) Western blot of nuclear and cytoplasmic cell fractions. Protein levels of MLL4, NCOA6, UTX, PTIP, ASH2L, RbBP5, and WDR5 in HCT116 parental or isogenic, allele-specific MLL4 KO cells are shown. Poly (ADP-ribose) Polymerase (PARP) is used as the nuclear loading control, while Hsp90 and β-tubulin are used as the cytoplasmic loading controls.
Correction of KMT2D Truncation Mutation in an Isogenic Background Restores Multifaceted MLL4 Functions.
Due to the mislocalization and compromised function arising from MLL4 protein truncation in HCT116 cells, we sought to correct the truncation-causing mutation using a KI strategy and determine whether this correction could rescue MLL4 function. Because the parental HCT116 cell line is heterozygous for the truncation mutant allele, KI correction is also proof-of-concept for MLL4’s dosage-dependent function in an isogenic background. We first confirmed successful correction of the mutation using Sanger sequencing of genomic DNA from parental and KI HCT116 clonal cells (Fig. 3A). As expected, parental cells exhibited double peaks due to the single cytosine nucleotide insertion in the mutant allele (Fig. 3A, ‘c’ in Top). Sequencing revealed different routes to correction in KI cells: KI#1 was corrected with the sequence from the donor template, in which two sites were mutated (without amino acid change) to exclude the CRISPR PAM sequence and for identification by PCR screen; KI#2 was corrected with the sequence from wild type allele, so that single peaks were detected by Sanger sequencing; KI#3 was corrected via the deletion of four consecutive nucleotides including the single nucleotide insertion (“cCCC”), which reestablished the normal reading frame (Fig. 3A). We further confirmed the KI mutation correction by RNA-seq, for which reads aligned to the regions surrounding the KMT2D mutation site were found in all three KI clones (SI Appendix, Fig. S3A). Then, to determine whether mutation correction could rescue MLL4 function, we first confirmed restoration of full-length MLL4 protein in the representative KI clones #1 and #2 (Fig. 3B). We also checked the total protein levels of MLL4/COMPASS subunits, finding that total protein levels of the MLL4/COMPASS-specific subunits UTX and NCOA6 increased relative to parental HCT116 in the MLL4 KI cells (Fig. 3B). This finding is consistent with the codependent relationship between KMT2D (MLL4), KDM6A (UTX), and NCOA6 (Fig. 1E) and the previous study showing the role of MLL4 in regulating UTX stability (28). Further, the restored full-length MLL4 and UTX proteins were correctly localized in the nuclear compartment (Fig. 3 C and D). Lastly, bulk levels of the MLL4/COMPASS H3K4 monomethylation mark were increased in the MLL4 KI cells, while levels of the other histone marks remained unchanged (Fig. 3B). Based on these observations, we concluded that MLL4 functions, including COMPASS stabilization, chromatin association, and enzymatic activity were fully rescued in the mutation correction KI cells.
Fig. 3.
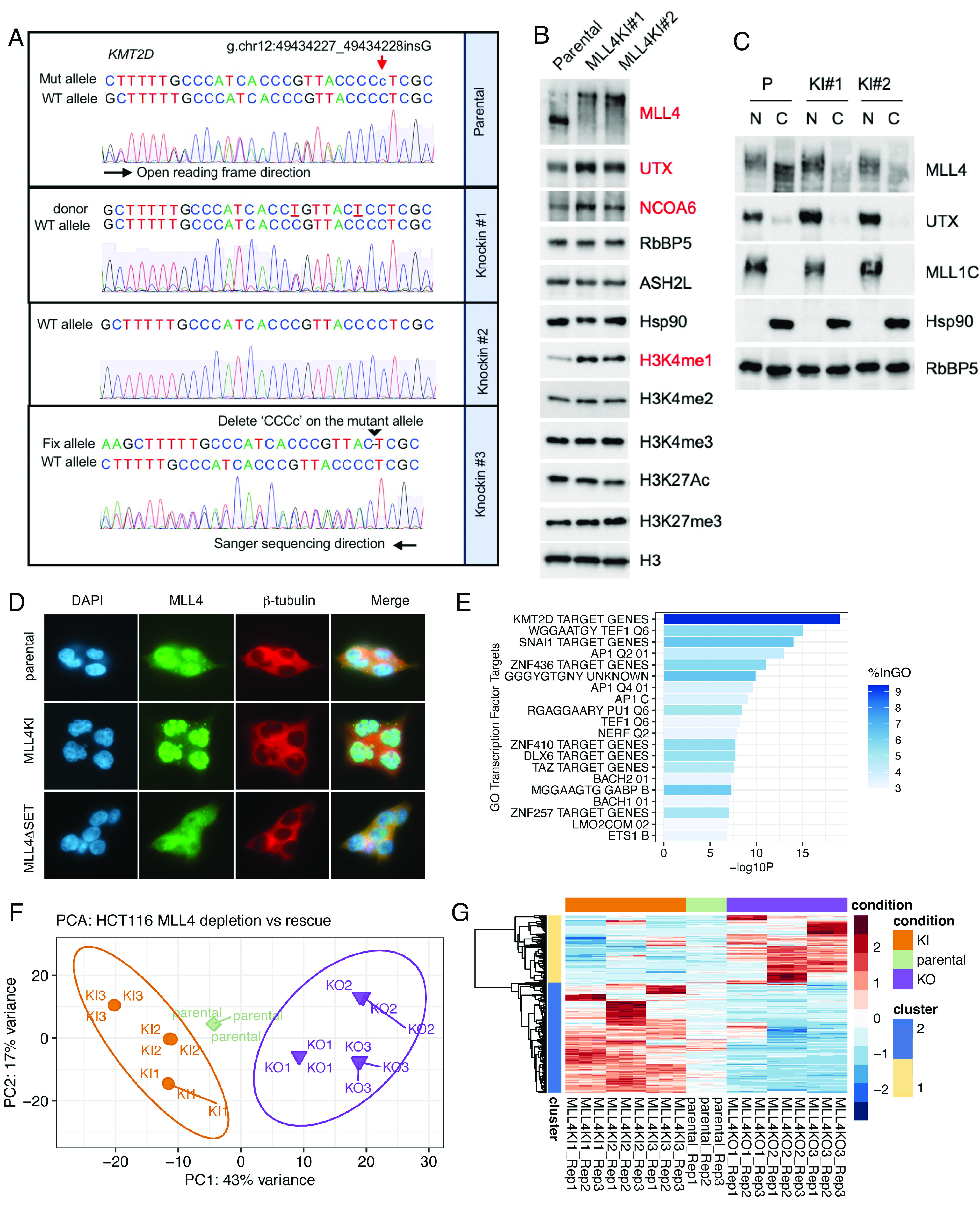
Isogenic correction of MLL4 truncation mutation restored MLL4/COMPASS protein levels and localization. (A) Sanger sequencing of genomic DNA from parental and MLL4 mutation correction KI HCT116 cells. Sequencing primer direction is 3′ to 5′ relative to the reference genome. (B) Western blot of total cell lysates from parental and MLL4 KI cells. Subunits within MLL4/COMPASS (MLL4, UTX, NCOA6, RbBP5, and ASH2L) and several key histone marks (H3K4me1/2/3, H3K27Ac, and H3K27me3) are shown. Hsp90 and total histone H3 are used as loading controls. (C) Western blot of nuclear (N) and cytoplasmic (C) fractions from parental and KI cells. Protein levels of MLL4 and UTX are shown, using MLL1 and Hsp90 as nuclear and cytoplasmic loading controls, respectively. (D) Immunofluorescence showing the subcellular localization of MLL4 in parental, MLL4KI, and MLL4ΔSET cells. DNA was stained with DAPI and β-tubulin was stained to show the cytoplasm. (E) Gene Ontology enrichment of transcription factor targets analysis for up-regulated genes in MLL4 KI clones vs. parental HCT116 cells. (F) PCA showing the clustering of KI, parental, and KO cell lines. ntop = 500 by default. (G) Top 500 genes contributing to PC1 were extracted, and the hierarchical clustering heatmap showed the gene expression changes in KI, parental, and KO cells.
Next, we investigated how gene expression regulation is rescued by restoration of full-length, nuclear MLL4 protein and whether MLL4 has a dosage-dependent effect on gene expression. We performed a hierarchical clustering analysis of genes differentially expressed in MLL4 KI cells compared with parental HCT116 cells (SI Appendix, Fig. S3B). Consistent with the known role of MLL4 in enhancer regulation and transcriptional activation, a large number of genes were up-regulated (n = 735) while only 467 genes were down-regulated when full-length, nuclear MLL4 protein was restored (SI Appendix, Fig. S3 B and C). Pathway enrichment analysis indicated that the up-regulated genes were involved in neutrophil degranulation, actin cytoskeleton organization, signaling by receptor tyrosine kinases, and membrane organization (SI Appendix, Fig. S3D). Further, Gene Ontology transcription factor target enrichment analysis of the up-regulated genes yielded MLL4 (KMT2D) target genes as the top hit, confirming that the restoration of full-length, nuclear MLL4 restored its gene expression regulatory function (Fig. 3E). For the down-regulated genes in MLL4 KI vs. parental cells, pathway enrichment analysis indicated multiple cell cycle-related processes (SI Appendix, Fig. S3E). Finally, we examined differential gene expression between HCT116 parental, MLL4KI, and MLL4KO clones (Fig. 3 F and G). Despite variance along PC2, which reflects clonal effects, PC1 explained the majority of variance in gene expression among the distinct genotypes (Fig. 3F). The top 500 genes contributing to PC1 were disrupted in MLL4KO cells and rescued in MLL4KI cells (Fig. 3G).
Correction of MLL4 Truncation Mutation Leads to Enhancer Function Elevation.
The increase in gene expression observed in the MLL4KI cell lines coincides with increased H3K4me1 levels at gene loci such as MGLL (SI Appendix, Fig. S3G), suggesting that enhancer function is restored by MLL4 mutation correction. To further investigate enhancer function, we used H3K4me1 and H3K27Ac ChIP-seq peaks observed in parental cells to identify enhancer regions and measured the level of enhancer RNA (eRNA) using PRO-seq (precision nuclear run-on sequencing) with spike-in normalization. The PRO-seq data revealed that eRNA transcription is significantly increased at a subset of enhancers (cluster 1), in MLL4 KI cells (Fig. 4 A, B, and D). This increase in PRO-seq signal was highly reproducible, as it was observed in both KI1 and KI2 cells with three biological replicates (SI Appendix, Fig. S4A). Consistent with the increased PRO-seq signal, we observed an increase in H3K27Ac, which is associated with active transcription, at cluster 1 enhancers (Fig. 4 A and D and SI Appendix, Fig. S4B). H3K4me1 was also dramatically increased at these enhancers (Fig. 4 A and D and SI Appendix, Fig. S4B). Notably, comparison to RNA-seq data demonstrated that the majority of genes nearest to cluster 1 enhancers are up-regulated in MLL4 KI relative to parental cells (Fig. 4A). Together, these results strongly suggest that the correction of MLL4 truncation mutation restores both deposition of H3K4me1 and eRNA transcription at a specific group of enhancers, leading restored expression of nearby genes controlled by these enhancers. At the cluster 2 enhancers, mutation correction partially restored H3K4me1 but not eRNA transcription or H3K27Ac (Fig. 4 A and C and SI Appendix, Fig. S4A), suggesting that the function of MLL4 in enhancer activation is restricted to specific genomic regions, as observed previously (6).
Fig. 4.

Isogenic correction of MLL4 truncation mutation restored enhancer function regulated by MLL4. (A) Clustering of enhancers using PRO-seq data. ChIP-seq signal for H3K4me1 and H3K27Ac and the changes in PRO-seq signal are centered on enhancers and sorted by H3K4me1 occupancy in parental cells within each cluster. The changes in RNA-seq signal at the nearest genes are indicated at the Right. Log2FC, log2-fold change. RRPM, reference-adjusted reads per million. N = 4,414 (Cluster 1), 9,073 (Cluster 2). (B and C) Average PRO-seq signal in parental and MLL4KI1 cells, centered on cluster 1 (B) or cluster 2 (C) enhancers. (D) Representative track example showing PRO-seq and ChIP-seq (H3K4me1 and H3K27Ac) signal in parental, MLL4KI1, and MLL4KI2 cells at the cluster 1 enhancer.
Nuclear to Cytoplasmic Translocation of MLL4 due to KMT2D Truncation Mutations in Cancer.
After observing that the KMT2D truncation mutation in HCT116 cells causes nuclear to cytoplasmic MLL4 translocation, we examined whether this translocation was specific to HCT116 or caused by MLL4 truncation mutations found in cancer. To do so, we selected several colorectal cancer cell lines with different KMT2D mutation status: Caco2 and HT55 (KMT2D WT) and HCT116, RKO, and LoVo (KMT2D truncation mutated) (Fig. 5A). No KDM6A (UTX) mutations were detected in any of these cell lines. Upon cellular fractionation, we observed a primarily nuclear localization of the WT MLL4 protein in Caco2 and HT55 cells, while truncated MLL4 protein found in cancer was detected in the cytosolic fractions of HCT116, RKO, and LoVo cells (Fig. 5B). The ratio of nuclear to cytoplasmic UTX was strongly decreased for all of the truncated MLL4 cell lines relative to cell lines with WT MLL4 (Fig. 5 B and C).
Fig. 5.

Nuclear to cytoplasmic translocation of MLL4 upon truncation mutation in cancer. (A) Colorectal cancer cell lines and their MLL4 mutation status. (B) Western blot of nuclear (N) and cytoplasmic (C) fractions from colorectal cancer cell lines. Protein levels of MLL4 and UTX are shown. PARP and β-Tubulin were used as the nuclear and cytoplasmic loading controls, respectively. (C) Bar plot showing the nuclear to cytoplasmic ratio of UTX protein levels in colorectal cancer cell lines. (D and E) IHC staining of patient bladder tumor samples. MLL4 mutation status was determined by whole-exome sequencing. Immunohistochemistry staining was performed with anti-MLL4 N-terminus (823) antibody in MLL4 WT (D) and MLL4 truncation mutation (E) tumor samples.
To further examine whether CRISPR/Cas9-mediated MLL4 truncation could cause a similar protein translocation (SI Appendix, Fig. S5A), we designed a series of 18 sgRNAs to target different sequences across the KMT2D gene. After sgRNA validation, we successfully truncated MLL4 using six different sgRNA in Caco2 cells, which express WT MLL4 (SI Appendix, Fig. S5B). Truncated proteins were detected in the cytoplasmic fractions, and nuclear MLL4 expression was decreased (SI Appendix, Fig. S5C). However, unlike the decreased nuclear to cytoplasmic ratio of UTX observed in MLL4 truncated relative to WT cell lines (Fig. 5B) and the reduction of cytoplasmic UTX in MLL4 KI relative to parental HCT116 cells (Fig. 3C), CRISPR/Cas9-mediated truncation of MLL4 did not perturb the expression or localization of UTX (SI Appendix, Fig. S4C). This difference may be due to cell line-specific regulation of UTX stability or localization or a longer-term selection of the genotypes may be required to see the effects of MLL4 truncation on UTX in MLL4 WT cell lines like Caco2.
To examine whether the cytoplasmic shift due to MLL4 truncation was recapitulated in active human cancer, we performed immunohistochemistry (IHC) staining of patient bladder tumor samples with and without KMT2D mutations. The individual KMT2D and KDM6A mutation status for each sample was determined by whole-exome sequencing (patient #1 to 3, KMT2D wild type; patient #4 to 6, KMT2D truncation mutation). Using our validated antibody against the N-terminus of MLL4, we observed cytoplasmic MLL4 localization in bladder tumor tissues with KMT2D truncation mutation (Fig. 5 D and E). This finding indicates an opportunity to develop cytoplasmic MLL4 IHC as a biomarker to enable early diagnosis, patient stratification, and treatment decisions for cancers with deficient MLL4/UTX-COMPASS function.
MLL4 Truncation as a Biomarker for Predicting Sensitivity to Therapeutic Metabolic Inhibition.
Because we previously demonstrated heightened sensitivity of mESCs and human cancer cell lines with MLL3/MLL4/UTX-COMPASS mutations to the de novo purine synthesis inhibitor lometrexol (5), we sought to examine the effects of KI mutation correction on lometrexol sensitivity, finding that sensitivity to lometrexol in HCT116 cells is partially relieved by KI correction of the MLL4 truncation mutation (Fig. 6A). Next, we performed RNA-seq to explore differential gene expression in response to lometrexol in MLL4 KI vs parental HCT116 cells. Consistent with the effects of lometrexol on cell growth, altered gene expression in response to lometrexol was abrogated by KI mutation correction (Fig. 6B and SI Appendix, Fig. S6), consistent with our results in xenograft mouse models of MLL4 wild type and mutant colorectal cancer (5). Overall, our findings suggest that lometrexol could be an effective treatment for tumors with MLL3/MLL4/UTX-COMPASS mutations (5).
Fig. 6.

MLL4 mutation as a biomarker for predicting sensitivity to metabolic inhibition. (A) Percentage of cell growth inhibition in HCT116 parental and MLL4 mutation correction KI cells treated with 0, 80, or 160 nM lometrexol for 72 h, as measured by CellTiter-Glo® luminescent cell viability assay. (B) PC analysis of differentially expressed genes identified by RNA-seq in HCT116 parental and MLL4 mutation correction KI cells treated with 0, 50, or 100 nM lometrexol for 72 h. (C) Flowchart illustrating sequence of lometrexol treatment, tumor evaluation, and RNA-seq in the BBN induced bladder mouse model. (D) Tumor grade classification in dimethyl sulfoxide (DMSO) and LTX treatment groups. (E) Tumor grade classification in DMSO and LTX treatment groups stratified based on MLL4 mutation status.
After observing nuclear to cytoplasmic translocation of MLL4 in patient bladder tumor samples with MLL4 truncation, we sought a bladder cancer model to explore the effects of MLL4 genotype on lometrexol sensitivity. In the BBN mouse model of bladder cancer, administration of the carcinogen N-butyl-N-(4-hydroxybutyl)- nitrosamine (BBN) (which mimics the effects of chronic tobacco use) to C57/B6 male mice in drinking water for at least 20 wk causes muscle-invasive bladder tumors (29). The BBN mouse model also displays a high rate (~70% of tumors) of Kmt2d (Mll4) mutations, similar to rates seen in bladder cancer (29, 30). Using this model, we hypothesized that BBN-induced tumors with Kmt2d truncation mutations would be more responsive to treatment with lometrexol than Kmt2d WT tumors found in this animal model of bladder cancer. To address this hypothesis, we first administered water-containing 0.1% BBN to thirty male C57Bl/J6 mice (7 wk old) for 6 wk. The mice were then randomized into DMSO (n = 15) or lometrexol (n = 15, 15 mg/kg) treatment groups and were treated via intraperitoneal injection twice a week for 12 wk. At the end of the treatment period, bladders were removed for histology to assign tumor staging and for mRNA isolation to analyze Kmt2d mutations by RNA-seq. Finally, we assessed the effectiveness of lometrexol treatment against tumors with Kmt2d WT or mutant genotypes by comparing the relative impact of treatment on tumor stage (Fig. 6C). Lometrexol treatment significantly reduced tumor stage in mice with Kmt2d mutant but not WT tumors (Fisher’s exact test, P = 0.037) (Fig. 6 D and E). The results of this bucket trial of lometrexol effectiveness in the BBN bladder cancer model suggest that KMT2D truncation mutations could be used to stratify patients based on expected sensitivity to targeted therapies like lometrexol.
Discussion
Recurrent mutations in genes encoding the enzymes MLL3 (KMT2C) and MLL4 (KMT2D) subunits of the COMPASS family occur frequently in a large number of cancers, affecting both catalytic-dependent and -independent activities (19, 30–32). Our analysis indicates that in addition to the unique amino acid sequence properties of MLL4, its multifaceted biological functions also contribute to the high KMT2D mutation rate observed in many cancers, as well as in Kabuki Syndrome. The majority of KMT2D mutations introduce early stop codons into the gene and have therefore been assumed to generate loss-of-function alleles. However, in this study we show that these mutant transcripts evade nonsense mRNA decay to produce robust expression of truncated proteins that aberrantly localize to the cytosol. We previously demonstrated that tumors with KMT2D truncation mutations are targetably sensitive to de novo purine nucleotide synthesis inhibition (5). Our current study demonstrates that tumors with KMT2D truncation mutations have stable and identifiable truncated MLL4 protein that aberrantly localizes to the cytosol. This finding provides us with a potential biomarker for prognosis and patient stratification based on expected sensitivity to targeted therapies such as lometrexol (5). The molecular mechanisms by which KMT2D mutations cause or contribute to cancer progression are still unclear. Cells containing heterozygous KMT2D truncation mutations exhibit redistribution of UTX to the cytosol, destabilized NCOA6, decreased histone H3K4 mono-methylation, and dysregulated gene expression that cannot be entirely explained by loss-of-function of the remaining WT allele, indicating potential gain-of-function for the mutant allele. Correction of the KMT2D truncation mutation in HCT116 cells rescued MLL4/COMPASS function, suggesting that restoration of normal MLL4 function at the protein level could potentially be used as a strategy to inhibit KMT2D mutation-induced tumorigenesis or developmental disorders. Previous studies ignored the KMT2D truncation mutation when using the HCT116 cell line to study MLL4 function (28, 33, 34). Our study demonstrated that there is a KMT2D truncation mutation allele in the HCT116 colorectal cancer cell line from which truncated MLL4 protein is stably expressed and aberrantly localizes to the cytosol. Our work establishes a framework to further investigate MLL4 truncation mutations and other similarly mutated epigenetic factors in various types of cancers. We note that researchers should be careful to consider potential gain-of-function when using cell lines with epigenetic factor mutations in future studies.
The seemingly conflicting results of previous studies have raised some controversy regarding the role of the MLL4 SET domain and its catalytic activity in regulating MLL4 protein stability. While the MLL4 protein is destabilized in cells with mutations rendering the SET domain catalytically dead (35), the protein is stably expressed in cells with deletion of the full SET domain (36, 37). There are two potential caveats of these previous studies: 1) they used nuclear lysates to analyze total protein levels, excluding the possibility of detecting stable but translocated MLL4 in cytoplasmic fractions, and 2) they used an antibody against the MLL4 C-terminus of MLL4, excluding the possibility of detecting truncated MLL4 protein (38). Another possible explanation for the apparent effect of the catalytic-dead mutation may be due to the specific mutational location, as point mutations within certain regions may cause destabilization of the whole protein that larger deletions or insertions would not. In this study we focused on truncation mutation and observed the stable cytoplasmic expression of truncated MLL4 proteins which lack the SET domain, consistent with the previous detection of cytoplasmic MLL4 apparently due to mutations (both truncation and missense) in the BBN carcinogen-induced mouse model (30). It will be crucial to further examine whether truncation mutations universally alter MLL4 protein localization, and which missense mutations in the SET domain or other regions may also relocalize MLL4 to the cytosol. Based on our results regarding the effects of MLL4 truncation on stability and localization of other MLL4/COMPASS components, complete information regarding the impact of MLL4 truncation and other potentially re-localizing mutations will not only contribute to biomarker development for MLL4 mutated tumors but for tumors with mutations in other epigenetic factors such as UTX.
In currently established models to study KMT2D/MLL4 loss-of-function, both KMT2D alleles are deleted in the MLL4 KO cell lines or mice for comparison to their parental counterparts. However, because approximately 96.1% of KMT2D mutations in the COSMIC database are heterozygous, residual wild-type MLL4 function may be retained in somatically mutated patient tumors. The results of our truncation mutation correction experiment highlight the possibility of elevating the degree of wild-type MLL4 function to prevent tumor progression or alleviate the impact of developmental syndromes. Restoring MLL4 function in cancers or Kabuki Syndrome is a challenging yet promising direction for future therapeutic development.
Materials and Methods
Cell Culture, shRNA Knockdown, and CRISPR/Cas9-Guided Gene Editing.
HCT116, Caco2, HT55, RKO, and LoVo cell lines were purchased from ATCC. HCT116 and RKO cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS); Caco2 and HT55 cells were grown in DMEM with 15% FBS; LoVo cells were cultured in Ham’s F12 with 10% FBS. The sequences of sgRNA, screen primers, KI donor sequences, and qRT-PCR primers are listed in SI Appendix, Table S1. Lentiviruses were packaged with psPAX2 and CMV-VSVG in 293T cells. Culture media was collected at 24 h and 48 h post transfection, passed through 0.45 µm filters, concentrated with lenti-X concentrator (Takara Bio), and resuspended in DMEM with 10% FBS. Cells were infected with lentiviruses and selected with puromycin (2 μg/mL) for 5 d before low-density seeding for picking single clones.
Antibodies and Western Blot.
The following primary antibodies were used in this study: anti-H3K4me1 (Cell Signaling Technology, #5326), anti-H3K4me2 (generated in-house), anti-H3K4me3 (Cell Signaling Technology, #9751), anti-H3K27Ac (Cell Signaling Technology, #8173), anti-H3K27me3 (Cell Signaling Technology, #9733), total H3 (Cell Signaling Technology, #4499), anti-MLL4 (N-terminus, #823, generated in-house), anti-NCOA6 (Bethyl Laboratories, A300-410A), anti-UTX (Cell Signaling Technology, #33510), anti-PTIP (Bethyl Laboratories, A300-370A), anti-RbBP5 (Bethyl Laboratories, A300-109A), anti-WDR5 (Cell Signaling Technology, #13105), anti-ASH2L (Cell Signaling Technology, #5019), anti-MLL1C (Cell Signaling Technology, #14197), PARP (Cell Signaling Technology, #9542), p53 (Santa Cruz Biotechnology, sc-126), p21 (Cell Signaling Technology, #2947), anti-Hsp90 (Santa Cruz Biotechnology, sc-13119), and anti-β-tubulin (Proteintech, 66240-1-Ig).
Cellular Fractionation.
Freshly harvested cell pellets were resuspended in buffer A (10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT) and protease inhibitor) on ice for 5 min, and 10% NP-40 was added to a final concentration of 1%. After vortexing, lysates were kept on ice for 20 min prior to centrifugation at 400 g for 5 min. Supernatants in buffer A were reserved as cytoplasmic fractions. Pellets were washed once with buffer A and centrifuged at 400 g for 5 min. Supernatants were discarded and the pellets were dissolved in buffer B (20 mM HEPES pH7.9, 1.5 mM MgCl2, 420 mM NaCl, 25% (v/v) Glycerol, 0.2 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM DTT, protease inhibitor, and benzonase). After 30 min of incubation on ice, samples were centrifuged at 16,000 g for 15 min at 4 °C. Supernatants in buffer B were reserved as nuclear fractions. To make sodium dodecyl sulfate (SDS) samples, salt concentration and final volume were adjusted equally for cytoplasmic and nuclear fractions. Western blot analysis was performed as previously described (39).
Immunofluorescence.
Cells were seeded at ~50 to 80% confluency in 6-well plates with coverslips. Twenty-four hours after cell seeding, cell culture media was removed and cells were gently washed once with phosphate-buffered saline (PBS). The cells were fixed with 4% formaldehyde solution diluted in PBS (16% stock, methanol free, Life technologies, 28908) and incubated at room temperature for 20 min under gentle shaking. After another PBS wash, cells were permeabilized with 0.2% triton X-100 in PBS for 10 min. Then, 1% BSA (w/v) in PBS is used for blocking for 30 min. Primary antibodies were diluted in 1% BSA in PBS and incubated at 4 °C overnight. The next day, coverslips were washed with PBST three times for 5 min each under shaking. Then, coverslips were incubated with fluorophore-conjugated secondary antibody (Alexa Fluor™ 488 Goat anti-Rabbit IgG (H+L), A11008; Alexa Fluor™ 568 Goat anti-Mouse IgG (H+L), A11004) in the dark for 1 h at room temperature. After three washes with PBST, the coverslips were mounted in DAPI mounting media (Vector Laboratories, H-1200).
IHC Staining.
IRB approval was obtained to evaluate a retrospective cohort of patients with high-risk bladder cancer at Northwestern Memorial Hospital; a waiver of informed consent was approved by Northwestern Institutional Review Board (STU00204352) (40). Paraffin-embedded patient tissue slides were deparaffinized and the antigen was retrieved using citrate buffer pH 6.0 for 15 min, followed by hydrogen peroxide block for 10 min, 1X Tris-Buffered Saline, 0.1% Tween® 20 Detergent (TBST) wash, and protein block for 30 min (DAKO X0909). Slides were incubated with primary anti-MLL4 (homemade against N-terminus of MLL4) antibody at 1:2,000 dilution overnight at 4 °C. Slides were washed 3 times with TBST, then incubated with secondary antibody (anti-Rabbit, DAKO K4003) for 30 min at room temperature followed by 3 washes with TBST. After DAB detection, a counter stain was performed with Hematoxylin-Sigma Meyer’s for 15 min and Blue reaction with 0.5% lithium carbonate for 2.5 min. Finally, the slides underwent dehydration and mounting.
Patient Sample Library Preparation and Sequencing.
Genomic DNA was extracted from macro-dissected formalin-fixed paraffin-embedded (FFPE) sections and quantified using the Qubit 2.0 DNA HS Assay (ThermoFisher, Massachusetts, USA). A prehybridization library was produced using SureSelect XT HS and XT low input library preparation kit (Agilent Technologies) and barcoding was performed using Illumina 8-nt dual-indices.
Hybridization was performed using a customized integrated DNA technologies (IDT) discovery pool based on the manufacturer’s protocol. The enriched libraries were quantified using the Qubit 2.0 DNA HS Assay and library quality was evaluated by TapeStation HSD1000 ScreenTape (Agilent Technologies). Enriched libraries were pooled prior to sequencing on an Illumina NovaSeq S4 sequencer for 150 bp read length in paired-end mode, with an output of 27 million total reads (13.5 M reads each side) per sample.
Patient Sample Mutation Data Analysis.
Adapter sequences and low-quality bases were trimmed from Fastq files using trim_galore and FastQC was performed on the trimmed reads. Trimmed reads were mapped to human reference genome version hg38 using bwa-mem. Next, duplicate reads were marked using the markduplicate utility from Picard (GATK4), for which duplicate reads are defined as originating from a single fragment of DNA. The coverage profiles were generated using mosdepth. Finally, analysis-ready BAM files were generated by excluding reads with MAPQ < 30 using samtools. Variant detection was performed using the Pisces variant caller. Resulting variants were annotated using SnpEff and SnpSift. Finally, annotated variants were filtered out to retain high-quality somatic variants.
BBN Bladder Cancer Mouse Model.
Bladder cancer was induced in 30 seven-week-old C57BL/6J male mice by administering 0.1% BBN in drinking water for 6 wk. The mice were then randomized into DMSO (n = 15) or lometrexol treatment (n = 15, 15 mg/kg) groups and were given intraperitoneal injections of the respective treatments twice a week for 12 wk. At the end of the treatment period, the bladder tissues of the mice were harvested for hematoxylin and eosin stain (H&E) staining and RNA isolation. Each mouse's tumor grade was assessed as we have previously described (41), in alignment with the American Joint Committee on Cancer (AJCC) pathologic staging guidelines for bladder cancer, and mRNA samples were analyzed for mutations in the Kmt2d/Mll4 gene through RNA-seq.
NGS Data Processing.
RNA-seq and ChIP-seq samples were sequenced with Illumina NextSeq technology, and output data were processed with bcl2fastq. Sequence quality was assessed using FastQC v 0.11.2 (42), and quality trimming was done using Trimmomatic (43). RNA-seq reads were aligned to the hg19 genome using STAR version 2.5.2 (44), and only uniquely mapped reads with a two-mismatch threshold were considered for downstream analysis. Gene annotations from Ensembl 75 were used. Output bam files were converted into bigwig track files to display coverage throughout the genome (in RPM) using the GenomicRanges package (45) as well as other standard Bioconductor R packages. Next-generation sequencing data are deposited at Gene Expression Omnibus with accession number GSE246058.
RNA-seq Analysis.
Gene count tables were constructed using HTseq (46) with Ensembl gene annotations and used as input for edgeR 3.0.8 (47). Genes with Benjamini–Hochburg adjusted P-values less than 0.01 were considered to be differentially expressed. Pathway analysis was performed with Metascape (48).
ChIP-seq Analysis.
We performed de novo identification of enhancers in HCT116 cells by taking the genomic regions occupied by both H3K4me1 and H3K27Ac modifications. ChIP-seq peaks were called using MACS2 (49). H3K27Ac peaks that overlap H3K4me1 peaks were retained and merged with the nearest peaks if the distance between two peaks is ≤2 kb using bedtools 2.30.0 (50). The peaks were then discarded if the promoter regions are overlapped. The promoter regions were defined as TSS (transcription start site) ± 2 kb based on hg19 RefSeq Curated annotation in UCSC genome browser. The resulting peaks (N = 13,487) were defined as enhancers and used for downstream analysis. Heatmaps and metaplots were generated using deepTools 3.5.1 (51).
PRO-seq Analysis.
PRO-seq samples were prepared as described previously (52) with minor modifications. Briefly, 1 million human HCT116 nuclei mixed with 2% spike-in Drosophila S2 nuclei were used for the nuclear run-on assay and the subsequent library preparation as described in qPRO-seq method (53). Libraries were sequenced on NovaSeq 6000 (Illumina). Following the removal of low-quality bases, adapters and ribosomal RNA signal, the paired-end reads with length of 15 to 51 nt were aligned to a concatenated genome comprised of human hg19 and fly dm3 assemblies using bowtie 2.4.5 (54) with very sensitive option. To obtain PRO-seq signal in bigwig format, 5′ ends of R1 reads with MAPQ ≥ 20 were assigned to the opposite strand. Read counts were normalized to total reads aligned to the spike-in genome. Heatmaps and metaplots were generated using deepTools 3.5.1 (51).
Variant Calling for RNR-seq Data.
The GATK pipeline was used to identify variants from RNA-seq data. RNAseq fastq files (50 bp, paired end) were trimmed using Trimmomatic v0.39. Trimmed reads were aligned using STAR 2.7.9a to Ensembl GRCm39 mouse genome assembly with Ensembl v108 (GRCm39) gene annotations. Duplicate alignments were marked using GATK v4.1.0 MarkDuplicates tool, followed by alignment reformatting and base quality score correction using SplitNCigarReads, BaseRecalibrator, and ApplyBQSR. Next, HaplotypeCaller was used to call variants. The resulting gVCF file was converted to a VCF file using GenotypeGCVF and was further annotated using SnpEff v4. Information about variants commonly found in the C57BL_10J mouse (downloaded as a vcf file from the Mouse genome project) were added using dbSNP. Finally, these commonly found variants and variants with both a quality score less than 100 and Fisher strand score greater than 30 were filtered out from the annotated vcf using SnpSift.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We would like to thank Dr. Lu Wang for MLL4 mutation correction KI design, Benjamin C. Howard, Madhurima Das, and Dr. Marta Iwanaszko for NGS library preparation, Brianna M. Monroe for graphical illustration, and Dr. Elizabeth Bartom for NGS analysis discussion. Studies in regard to the MLL4 function and regulation are supported in part by the generous support of the National Cancer Institute through the Outstanding Investigator Award R35-CA197569 mechanism to A.S.
Author contributions
Z.Z., J.J.M., and A.S. designed research; Z.Z., Y.A., C.N.P., Y.Y., L.S.J., J.Q., and J.M.Z. performed research; Z.Z., Y.A., and K.A.M. analyzed data; and Z.Z., Y.A., S.R.G., J.J.M., and A.S. wrote the paper.
Competing interests
J.J.M.: Consultant: Merck, AstraZeneca, Incyte, Janssen, BMS, UroGen, Prokarium, Imvax, Pfizer, Seagen/Astellas, Compensation for talks/educational courses: AUA, OncLive, Olympus, UroToday, Clinical Trials: SWOG, Genentech, Merck, AstraZeneca, Incyte.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Next generation sequencing data have been deposited in GEO (GSE246058) (55). All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Tate J. G., et al. , COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li R., et al. , Macroscopic somatic clonal expansion in morphologically normal human urothelium. Science 370, 82–89 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Lawson A. R. J., et al. , Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 370, 75–82 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Gui Y., et al. , Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 43, 875–878 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Z., et al. , Therapeutic targeting of metabolic vulnerabilities in cancers with MLL3/4-COMPASS epigenetic regulator mutations. J. Clin. Invest. 133, e169993 (2023), 10.1172/JCI169993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alam H., et al. , KMT2D deficiency impairs super-enhancers to confer a glycolytic vulnerability in lung cancer. Cancer Cell 37, 599–617.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.N. Cancer Genome Atlas Research et al. , Integrated genomic characterization of oesophageal carcinoma. Nature 541, 169–175 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.N. Cancer Genome Atlas Research et al. , Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ortega-Molina A., et al. , The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 21, 1199–1208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang J., et al. , Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat. Med. 21, 1190–1198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brady S. W., et al. , The genomic landscape of pediatric acute lymphoblastic leukemia. Nat. Genet. 54, 1376–1389 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindqvist C. M., et al. , The mutational landscape in pediatric acute lymphoblastic leukemia deciphered by whole genome sequencing. Hum. Mutat. 36, 118–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cerami E., et al. , The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao J., et al. , Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng S. B., et al. , Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 42, 30–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheon C. K., et al. , Identification of KMT2D and KDM6A mutations by exome sequencing in Korean patients with Kabuki syndrome. J. Hum. Genet. 59, 321–325 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Cocciadiferro D., et al. , Dissecting KMT2D missense mutations in Kabuki syndrome patients. Hum Mol. Genet. 27, 3651–3668 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brune D., Andrade-Navarro M. A., Mier P., Proteome-wide comparison between the amino acid composition of domains and linkers. BMC Res. Notes 11, 117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L., et al. , Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat. Med. 24, 758–769 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L., Shilatifard A., UTX mutations in human cancer. Cancer Cell 35, 168–176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong S., et al. , Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. U.S.A. 104, 18439–18444 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walport L. J., et al. , Human UTY(KDM6C) is a male-specific Nϵ-methyl lysyl demethylase. J. Biol. Chem. 289, 18302–18313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egolf S., et al. , MLL4 mediates differentiation and tumor suppression through ferroptosis. Sci. Adv. 7, eabj9141 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang G., et al. , CRISPR-GEMM pooled mutagenic screening identifies KMT2D as a major modulator of immune checkpoint blockade. Cancer Discov. 10, 1912–1933 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ning H., et al. , Enhancer decommissioning by MLL4 ablation elicits dsRNA-interferon signaling and GSDMD-mediated pyroptosis to potentiate anti-tumor immunity. Nat. Commun. 13, 6578 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ashokkumar D., et al. , MLL4 is required after implantation, whereas MLL3 becomes essential during late gestation. Development 147, dev186999 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Maitituoheti M., et al. , Enhancer reprogramming confers dependence on glycolysis and IGF signaling in KMT2D mutant melanoma. Cell Rep. 33, 108293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rickels R., et al. , A small UTX stabilization domain of Trr is conserved within mammalian MLL3-4/COMPASS and is sufficient to rescue loss of viability in null animals. Genes. Dev. 34, 1493–1502 (2020), 10.1101/gad.339762.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fantini D., Meeks J. J., The BBN model: A mouse bladder cancer model featuring basal-subtype gene expression and MLL3/MLL4 genetic disruption. Oncoscience 5, 172–173 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fantini D., et al. , A Carcinogen-induced mouse model recapitulates the molecular alterations of human muscle invasive bladder cancer. Oncogene 37, 1911–1925 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sze C. C., Shilatifard A., MLL3/MLL4/COMPASS family on epigenetic regulation of enhancer function and cancer. Cold Spring Harb. Perspect. Med. 6, a026427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morgan M. A., Shilatifard A., Chromatin signatures of cancer. Gene. Dev. 29, 238–249 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu D. Q., et al. , The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol. Cell Biol. 33, 4745–4754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kantidakis T., et al. , Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes. Dev. 30, 408–420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jang Y., Wang C., Zhuang L., Liu C., Ge K., H3K4 methyltransferase activity is required for MLL4 protein stability. J. Mol. Biol. 429, 2046–2054 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dorighi K. M., et al. , Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol. Cell 66, 568–576.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rickels R., et al. , Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet. 49, 1647–1653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hillman R. T., et al. , KMT2D/MLL2 inactivation is associated with recurrence in adult-type granulosa cell tumors of the ovary. Nat. Commun. 9, 2496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Z., et al. , Systematic analyses of the cytotoxic effects of compound 11a, a putative synthetic agonist of photoreceptor-specific nuclear receptor (PNR), in cancer cell lines. PLoS One 8, e75198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robertson A. G., et al. , Identification of differential tumor subtypes of T1 bladder cancer. Eur. Urol. 78, 533–537 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Piunti A., et al. , Immune activation is essential for the antitumor activity of EZH2 inhibition in urothelial carcinoma. Sci. Adv. 8, eabo8043 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andrews S., FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. (2010). Accessed 26 April 2010.
- 43.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobin A., et al. , STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawrence M., et al. , Software for computing and annotating genomic ranges. PLoS Comput. Biol. 9, e1003118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anders S., Pyl P. T., Huber W., HTSeq–A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Y., et al. , Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y., et al. , Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramirez F., et al. , deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aoi Y., et al. , SPT6 functions in transcriptional pause/release via PAF1C recruitment. Mol. Cell 82, 3412–3423.e5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Julius Judd L. A. W., et al. , A rapid, sensitive, scalable method for Precision Run-On sequencing (PRO-seq). bioRxiv [Preprint] (2020). 10.1101/2020.05.18.102277 (Accessed 19 May 2020). [DOI]
- 54.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Z. Zhao et al. , Somatic mutations of MLL4/COMPASS induce cytoplasmic localization providing insight into cancer prognosis and treatment. Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE246058. Deposited 23 October 2023. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Next generation sequencing data have been deposited in GEO (GSE246058) (55). All study data are included in the article and/or SI Appendix.