

Significance
Antigen-specific antibody responses alone are insufficient to combat viruses that are constantly evolving. Currently, there is a great need for a broadly protective vaccine that induces robust humoral and cell-mediated immune responses. Herein, we developed two superior mRNA vaccines, TU88mCSA and ALCmCSA, using early SARS-CoV-2 spike sequence, optimized untranslated region (Ces1d and AP3B1), and advanced LNP formulations that effectively induce humoral immune response and activate stronger CD4+ and CD8+ T cell responses in spleen and lung as compared to existing SARS-CoV-2 spike mRNA construct. Notably, TU88mCSA and ALCmCSA induced robust control of BQ.1 infection in the upper respiratory airways. Our platform could be readily applied to future mRNA vaccine development for timely deployment in the face of rapid viral mutations.
Keywords: untranslated region, lipid nanoparticle, mRNA vaccine, lung tissue-resident memory T cells
Abstract
The emergence of highly transmissible severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern (VOCs) that are resistant to the current COVID-19 vaccines highlights the need for continued development of broadly protective vaccines for the future. Here, we developed two messenger RNA (mRNA)-lipid nanoparticle (LNP) vaccines, TU88mCSA and ALCmCSA, using the ancestral SARS-CoV-2 spike sequence, optimized 5′ and 3′ untranslated regions (UTRs), and LNP combinations. Our data showed that these nanocomplexes effectively activate CD4+ and CD8+ T cell responses and humoral immune response and provide complete protection against WA1/2020, Omicron BA.1 and BQ.1 infection in hamsters. Critically, in Omicron BQ.1 challenge hamster models, TU88mCSA and ALCmCSA not only induced robust control of virus load in the lungs but also enhanced protective efficacy in the upper respiratory airways. Antigen-specific immune analysis in mice revealed that the observed cross-protection is associated with superior UTRs [Carboxylesterase 1d (Ces1d)/adaptor protein-3β (AP3B1)] and LNP formulations that elicit robust lung tissue-resident memory T cells. Strong protective effects of TU88mCSA or ALCmCSA against both WA1/2020 and VOCs suggest that this mRNA-LNP combination can be a broadly protective vaccine platform in which mRNA cargo uses the ancestral antigen sequence regardless of the antigenic drift. This approach could be rapidly adapted for clinical use and timely deployment of vaccines against emerging and reemerging VOCs.
Waning vaccine-induced immunity against SARS-CoV-2 and the emergence of the highly contagious Omicron (B.1.1.529) variants prompted the development of new vaccine formulations and additional booster doses (1–4). Two years after the initial SARS-CoV-2 (WA1/2020 or Wuhan-Hu-1) first swept the globe, Omicron BA.1 became the dominant variant of concern (VOC) in November 2021. Omicron BA.1 and other subvariants (BA.2, BA.4, BA.5, BA.2.75, BA.4.6, and BQ.1) fueled record COVID-19 cases and have demonstrated substantial escape from neutralizing antibodies induced by the first-generation of SARS-CoV-2 mRNA vaccines, such as BNT162b2 (Pfizer-BioNTech) and mRNA-1273 (Moderna) (2, 5–9). To this end, several groups have developed Omicron-Spike (S) specific vaccines or evaluated third-dose booster vaccinations (10–15). In Fall 2022, updated bivalent vaccines against WA1/2020 and Omicron BA.4/BA.5 developed by Pfizer/BioNTech and Moderna were authorized by the FDA. However, these booster immunizations did not show remarkably distinct reactogenicity profile from the initial vaccines (13, 16). On the other hand, bivalent SARS-CoV-2 vaccines use both original and Omicron variant S mRNA (mS) with two prolines substitution at residues K986 and V987 (hereafter denoted as “mSpp”). These tailor-made vaccines are difficult to mass-produce in an expeditious manner and in the face of continuous viral mutations. These constrains make it difficult to develop effective vaccines against VOCs for rapid deployment. To address these concerns, a next-generation vaccine with original mS coding sequence (CDS) that can concurrently recognize both Omicron (B.1.1.529) subvariants and ancestral SARS-CoV-2 (WA1/2020) are yet to emerge (13). As Omicron BQ.1, BQ.1.1, XBB.1.1, and BA.2.86 become the next dominant VOCs (17, 18), original sequence-based approaches to mRNA vaccine are imperative to be developed. In this study, we present optimized monovalent mRNA vaccine candidates that provide substantial protection against WA1/2020, Omicron BA.1, and BQ.1, using only the mSpp CDS.
Although recent neutralizing antibody mapping and molecular modeling studies have confirmed that individuals vaccinated with two doses of mRNA-S (BNT162b2) demonstrate poor neutralizing activity against VOCs, such as Omicron BA.1 and BQ.1 (19–21), most antigen MHC class I restricted T cell epitopes from Omicron S protein were not yet affected at the amino acid sequence level (20, 22). As a second layer of defense, polyepitopic T cell responses play a vital role in recognition of the Omicron S glycoprotein. Of BNT162b2-induced CD8+ T cell response, the risk of immune evasion by Omicron BA.1 was evaluated and the Immune Epitope Database (IEDB) indicated that targets of most T cell responses remain conserved in the Omicron variant and remain capable of detecting Omicron S glycoprotein epitopes (20). Based on these properties, a SARS-CoV-2 vaccine with broad spectrum protection against both Omicron subvariants and ancestral strain can be achieved by significantly enhancing mRNA translatability, efficient in vivo delivery, and immune cell response.
To augment the translatability of antigen mRNA, structural elements, including the 5′ cap, 5′ and 3′ untranslated region (UTRs), antigen-CDS, and polyadenylated tail (Poly An) can be modified. Among these components, the 5′UTR and 3′UTR are unique regulators of mRNA expression (23–25). Multiple regulatory elements within the UTRs are critical for the stability and translation of mRNA into proteins. Moreover, the secondary structure of UTRs within mRNA profoundly influences the stability of mRNA and is associated with certain human diseases. Thus, 5′UTR and 3′UTR selection is critically important to antigen yield and vaccine efficacy. Recent progress in massive parallel oligonucleotide synthesis and sequencing provides opportunities for mRNA UTR optimization either from endogenous genes or de novo design approaches (26–29). Over thousands of human genes have been fine mapped and screened for UTR effectiveness using reporter genes (25–27). By mining these data, a range of genes with the high ribosome reads were selected to constitute a UTR library in this study.
In addition to UTR modulation, biodegradable lipid nanoparticles (LNPs) also play a vital role in mRNA vaccine performance by protecting mRNA from degradation en route to target organs in vivo. Currently, leading ionizable lipids, including ALC-0315 and SM-102, exhibit high transfection effects of long-chain mRNA in immune cells in vivo and were used in the development of COVID-19 mRNA vaccines (22, 30, 31). SM-102 has been optimized by Moderna for intramuscular (I.M.) vaccination, notably in its SARS-CoV-2 vaccine (mRNA-1273) (31–33). ALC-0315 from Acuitas has been used in Pfizer-BioNTech SARS-CoV-2 vaccine (BNT162b) (22, 32, 34). Vaccine from CureVac (CVnCoV) and University College London (LNP-nCoV saRNA-2) also selected ALC-0315 as vehicle, although the exact lipid formulation remains to be disclosed (35). Of these LNPs, Acuitas-ALC-0315 is one of the most representative LNPs and highly potent in liver and lymph node (LN) transfection, but its spleen transfection efficiency is yet to be investigated. Our group has previously developed high-performance LNP formulations for immune cell-targeting mRNA delivery, such as 113O12B, 93-O17S, and 9322-O17S, which specifically deliver mRNAs into the LN or spleen, leading to efficient gene recombination in CD8+ T lymphocytes (36, 37). In this study, we further optimized the LNP delivery system using our synthetic lipid and ALC-0315 (32) by loading an upgraded version of mRNA cargo.
Using both optimized mRNA UTRs and LNPs with high delivery efficiency, we generated two highly potent SARS-CoV-2 vaccine candidates, Acuitas-ALC-0315-Ces1d-Spp-AP3B1 mRNA vaccine (designated as ALCmCSA) and Tufts-LNP88-Ces1d-Spp-AP3B1 mRNA vaccine (designated as TU88mCSA) (SI Appendix, Fig. S1 and Table S1). ALCmCSA is comprised of Ces1d-Spp-AP3B1 mRNA (mCSA, UTR was substituted with Ces1d or AP3B1 gene) and an improved ALC-0315 formulation, while the TU88mCSA is composed of mCSA and LNP88 formulation. In mice, we demonstrated that both monovalent vaccines (ALCmCSA and TU88mCSA) using the ancestral Spp CDS (Wuhan-Hu-1 strain) elicited a more robust S-specific antibody (IgG) and T cell response in both circulating and respiratory immune compartments compared to those of Spp mRNA construct using globin as UTRs (denoted as “mrS”). A robust induction of S-specific lung-resident T cells was detected in ALCmCSA or TU88mCSA vaccinated mice. We performed golden Syrian hamster challenge studies with SARS-CoV-2 WA1/2020, Omicron BA.1 and BQ.1 to evaluate breadth of protection conferred by TU88mCSA or ALCmCSA vaccines. A two-dose of TU88mCSA or ALCmCSA vaccine provided complete or near-complete protection against WA1/2020, Omicron BA.1, and BQ.1 challenge in hamsters. Notably, the lung viral titers in Omicron BA.1 challenge model decreased to undetectable levels, and lung viral RNA copies or viral titers in BQ.1 challenge model were significantly reduced following either TU88mCSA or ALCmCSA vaccination. Taken together, our mRNA vaccine platform with optimized UTRs and LNP formulations can induce broad protection against current SARS-CoV-2 VOCs and likely future VOCs.
Results
In Vitro Evaluation of Different 5′UTRs and 3′UTRs in mRNA Translation Efficiency.
To identify optimal 5′UTRs and 3′UTRs, we selected UTRs previously associated with high mRNA translatability for in vitro and in vivo screening (SI Appendix, Tables S2 and S3) (26, 28). We first inserted four 5′UTR fragments into a DNA plasmid (pcDNA3.0-eGFP) and compared their expression levels (SI Appendix, Fig. S2A). To rapidly assess the effect of these 5′UTRs on eGFP expression, HEK293 cells were transiently transfected with these modified pcDNA3.0-eGFP plasmids, and eGFP protein levels were monitored from 36 h to 108 h post-transfection. Flow cytometry analysis showed that 70 nt and Ces1d 5′UTRs yielded higher eGFP expression than other UTRs at 60 h and 84 h, respectively, reaching 69.2% and 69.6% eGFP+ cells (SI Appendix, Fig. S2B).
Based on the performance of Ces1d and 70 nt 5′UTRs, pcDNA3.0-70 nt-eGFP and pcDNA3.0-Ces1d-eGFP were constructed with different 3′UTR candidates. We first evaluated a series of pcDNA3.0-Ces1d-eGFP-3′UTR constructs in HEK293 cells from 48 to 120 h (SI Appendix, Fig. S2C). At 72 h post-transfection, human α-globin (hα-globin), FAM171A1 (Astroprincin), POTEE (prostate, ovary, testis, and placenta expressed ankyrin domain family member E), complement component 3 (C3) and TIAM1 (T-cell lymphoma invasion and metastasis 1) gave higher eGFP expression relative to other 3′UTRs. Among them, hα-globin and C3 displayed the highest eGFP expression, reaching 78.6% and 75.8% eGFP+ cells, respectively. Variations in the performance of these UTR combinations suggest that specific 3′UTRs modulate mRNA translation by forming favorable or unfavorable secondary structures with 5′UTRs. Similarly, WIPI2 (WD Repeat Domain, Phosphoinositide Interacting 2), S0_M_T1012, P450 2E1 (cytochrome P450 family 2 subfamily E member 1), Apo A-II (apolipoprotein), AP3B1, and YY2 TF all promoted protein expression at 96 h post-transfection, with P450 2E1 performing the best at 71.1% eGFP+ cells. Notably, AP3B1 was the only 3′UTR that continued to increase protein expression levels through 120 h post transfection, peaking at 62.6% eGFP+ cells. Together, the 3′UTRs highlighted in SI Appendix, Fig. S2C, including hα-globin, WIPI2, C3, TIAM1, P450 2E1, or AP3B1, were selected as candidates to pair with 5′UTR (Ces1d) for subsequent in vivo mRNA screening.
The 70 nt-based 3′UTRs screening was performed in HEK293 cells using the same protocol. Flow cytometry data exhibited a similar pattern to the Ces1d-based experiments, with groups of 3′UTRs peaking at different time points posttransfection (SI Appendix, Fig. S2D). 3′UTRs with peak expression levels at 72 h posttransfection included S0_M_T1012, hα-globin, Apo A-II, and FAM171A1 gene fragments, with Apo A-II performing the best among them at 89.7% eGFP+ cells. Among them, hα-globin and S0_M_T1012 modified plasmids demonstrated superior performance to the rest of the 3’UTRs, showing 85.4% and 74.7% transfection efficiency, respectively. C3, WIPI2, TIAM1, YY2 TF, and AP3B1 modified plasmids reached peak expression levels at 96 h, with AP3B1 and YY2 TF performing the best at 79.7% and 73.6% eGFP+ cells, respectively. Plasmids inserted with POTEE, OXR1, P450 2E1, and MS10433 still showed increasing protein expression levels 120 h post transfection. OXRI produced the highest level of eGFP-positive cells, peaking at 84.4%. POTEE and MS10433 were close, with 79.4% and 77.9% eGFP+ cells, respectively. Together, the 3′UTRs highlighted with red circles in SI Appendix, Fig. S2D, including S0_M_T1012, hα-globin, Apo A-II, POTEE, MS10433, YY2 TF, AP3B1 or OXR1, were chosen as candidates to pair with 5′UTR (70 nt) for in vivo mRNA screening.
In Vivo UTRs Screening Using LNP88 Formulation via Subcutaneous Route (S.C.).
All active lipids used for in vivo mRNA delivery (Lipid 88, ALC-0315, and 113O12B) were synthesized. The synthesis and characterization were shown in SI Appendix, Fig. S3. Lipid nanoparticle 88 (LNP88) has shown significantly higher luciferase expression in the liver compared to ALC-0315 for firefly Luciferase mRNA (mLuc) delivery (SI Appendix, Fig. S4). ALC-0315 and 113O12B were reported to target the LNs and/or the liver and used as vaccine or vaccine candidate when they were formulated with the excipient compounds cholesterol (Chol), distearoylphosphatidylcholine (DSPC), and 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG-PEG) (37, 38). We used lipid 88 and formulated it with same excipient compounds but with a series of weight ratios. These LNP88 formulations showed similar physical properties (diameter, polydispersity, and zeta potential) (SI Appendix, Figs. S4A and S5). We assessed their capability for mLuc delivery (SI Appendix, Fig. S4C) and found LNP88 (Lipid 88: Chol: DSPC: DMG-PEG) at a weight ratio of 16:8:4:3 is the top-performing LNP88 formulation. We chose this LNP88 for in vivo mRNA-UTRs screening.
We then evaluated the in vivo translatability of mRNA containing different pairs of 5′ and 3′ UTRs using LNP88 via S.C. administration. Fluc and eGFP gene (Luc-GFP) fusion mRNA (mLuc-GFP) was first in vitro synthesized with Ces1d or 70 nt as the 5′UTR paired with different 3′ UTRs (Fig. 1A). These 3′ UTRs include Mus musculus beta globin (Mmβ-globin), human alpha globin (hα-globin), complement component 3 (C3), TIAM1, P450 2E1, AP3B1, and WIPI2. mLuc-GFP flanked with α-globin (5′UTR) and β-globin (3′UTR) fragments from SBI was used as a standard control (SI Appendix, Tables S2 and S3). All mRNA from the same batch was confirmed using 1% agarose gel electrophoresis which showed similar quality of mRNA with different UTR combinations (SI Appendix, Fig. S6A). Fig. 1 B and C showed the luciferase expression of mLuc-GFP constructed with Ces1d as 5′ UTR paired with different 3′ UTRs. We found AP3B1 is the most efficacious 3′UTR when paired with Ces1d and exhibited the highest expression level in both livers (1.5 × 108 bioluminescence units) and LNs (1.5 × 106 bioluminescence units) among all tested UTR combinations, including the SBI control (α-globin-Luc-GFP-β-globin mRNA). Interestingly, hα-globin, C3, and TIAM1 produced low to undetectable bioluminescence level when paired with Ces1d as the 5′UTR, suggesting that mRNA secondary structures or UTR conformations may limit ribosome access and other translation factors involved in mRNA processing. We then used a similar procedure to evaluate the performance of 3′ UTRs when 70 nt is used as 5′ UTR. A series of mLuc-GFP with 70 nt 5′ UTR paired with different 3′ UTRs were constructed. The mRNA quality was checked on an agarose gel, and the results showed similar quality and negligible mRNA degradation (SI Appendix, Fig. S6B). The in vivo screening results showed that Apo A-II is the top-performing 3′UTR for the 70 nt 5′UTR with Fluc expression level around 8 × 107 bioluminescence units in livers and LNs, while other 3’UTRs were around 5 × 107 luminescence units (Fig. 1 D and E). These results suggest that Apo A-II 3′ UTR significantly enhances mRNA translatability when paired with 70 nt 5′ UTR. Based on the screening results, we therefore selected two following UTR combinations for mRNA vaccine development, including 5′UTR-Ces1d-AP3B1-3′UTR and 5′UTR-70 nt-Apo A-II-3′UTR.
Fig. 1.

In vivo screening of UTRs via S.C. route. (A) Schematic illustration of UTR optimization in mice. In the mRNA construct, mLuc-GFP was capped with CleanCap AG or ARCA cap, tailed with 120 bases of polyadenosine (Poly A120), incorporated with N1-methylpseudouridine (m1Ψ) and fused with Ces1d or 70 nt as the 5′UTR. The 3′UTRs are substituted with UTR fragments of interest to assess their performance on mLuc-GFP translation in vivo. (B) Images of in vivo bioluminescence at 6 h and 30 h following S.C. injection of a series of mLuc-GFP constructs with Ces1d as the 5′UTR paired with different 3′UTRs in LNP88 formulation. (C) Quantification of the Fluc expression level at 6 h (a) and 30 h (b) in each organ area (Livers, LNs, and Local sites) (n = 2). (D) Images of mice bioluminescence at 6 h and 30 h following S.C. injection of a series of mLuc-GFP constructs with 70 nt as the 5′UTR paired with different 3′UTRs in LNP88 formulation. mLuc-GFP construct with SBI UTRs substitution was used as control. (E) Quantification of Fluc expression over time, 6 h (a) and 30 h (b), at the location of bioluminescence distribution in mice (Livers, LNs and Local sites) (n = 2). Unless specified otherwise, scale represents radiance (p/sec/cm2/sr) min = 4.38e4 to max = 5.58e5. Average ± SD (n = 2 mice per group), biological replicates shown.
In Vivo Optimization of LNP88 and ALC-0315 Formulations via I.M. Injection.
We investigated the in vivo delivery efficacy of ALC-0315 or LNP88 via I.M. injection of LNP-mLuc to mimic immunization in humans. Based on the data from tail-based S.C. delivery, we screened a range of weight ratios of fine-tuned ALC-0315 and LNP88 formulations (ionizable lipid: Chol: DSPC: DMG-PEG, w/w) (SI Appendix, Fig. S7A). The LNP-mRNA nanocomplex was characterized using dynamic light scattering (DLS) and transmission electron microscopy (TEM) (SI Appendix, Figs. S8 and S9). These LNP formulations were then assessed for in vivo mLuc mRNA delivery efficacy via I.M. in Balb/c mice (5 µg mLuc per mouse). Compared with 113O12B (SI Appendix, Fig. S10), a higher Fluc expression level was detected in both LNP88 and ALC-0315 treated groups as early as 6 h after I.M. injection and then decreased progressively with an average rate of one log per day from 24 h to 80 h before reaching baseline activity at 80 h post-injection (SI Appendix, Fig. S7 A and B). 113O12B-mLuc mainly accumulated at the local injection site, while both LNP88 and ALC-0315 delivered mLuc to both liver and LNs. LNP88 delivery resulted in similar biodistribution as ALC-0315 (SI Appendix, Fig. S7A), but kinetic analysis suggests that LNP88 may support more durable mRNA expression over ALC-0315 (SI Appendix, Fig. S7B). These results showed that LNP88 is as potent as ALC-0135 for in vivo mRNA delivery. We further investigated the effect of ratio between mRNA to LNP88 on mRNA delivery via I. M. injection. For this study, we kept the lipid ratio in the LNP88 constant at a weight ratio of 16:8:5:3 (Lipid88: Chol: DSPC: DMG-PEG). The weight ratio of ionizable lipid 88 to mRNA was chosen from 10/1 to 30/1. We found that 30/1 weight ratio (lipid 88: mRNA) resulted in significantly higher delivery efficiency in total flux in livers and LNs when compared to those of 10:1 and 20:1 ratios (SI Appendix, Fig. S7 C and D). Together, these results show that both LNP88 and ALC-0315 performed best when formulated with 50% ionizable lipid, 25% cholesterol, 15.6% DSPC, and 9.4% DMG-PEG weight percentage. For LNP88 formulation, a weight ratio of 30:1 (lipid 88: mRNA) showed the best luciferase expression when delivered through I. M. injection.
Tissue Distribution and Immune Cell–Targeting Efficacy of LNP88 and ALC-0315 via I.M. Injection.
We characterized the firefly luciferase expression in different tissues of mice 6 h after I.M. injection with LNP88-mLuc and ALC-0315-mLuc (SI Appendix, Fig. S11). As shown in Fig. 2A, the mRNA expression from LNP88-mLuc or ALC-0315-mLuc was mainly in the liver and LNs as expected. Notably, LNP88 also produced a significant luciferase expression in the spleen with approximately 40% of total bioluminescence intensity of all organs, while the spleen from ALC-0315-mLuc only produced 12.8% of total bioluminescence intensity (Fig. 2B). As the spleen and LNs are the primary sites of immune responses, delivering into the spleen may enhance vaccine performance. We then characterized the specific cell types transfected by LNP88 and ALC-0315 within the spleen and LNs. Cre mRNA (mCre) was used as cargo and tested in an Ai14D mouse model. Upon Cre-expression, the loxP-flanked STOP cassette is deleted, and the tdTomato expression is turned on (SI Appendix, Fig. S12).
Fig. 2.
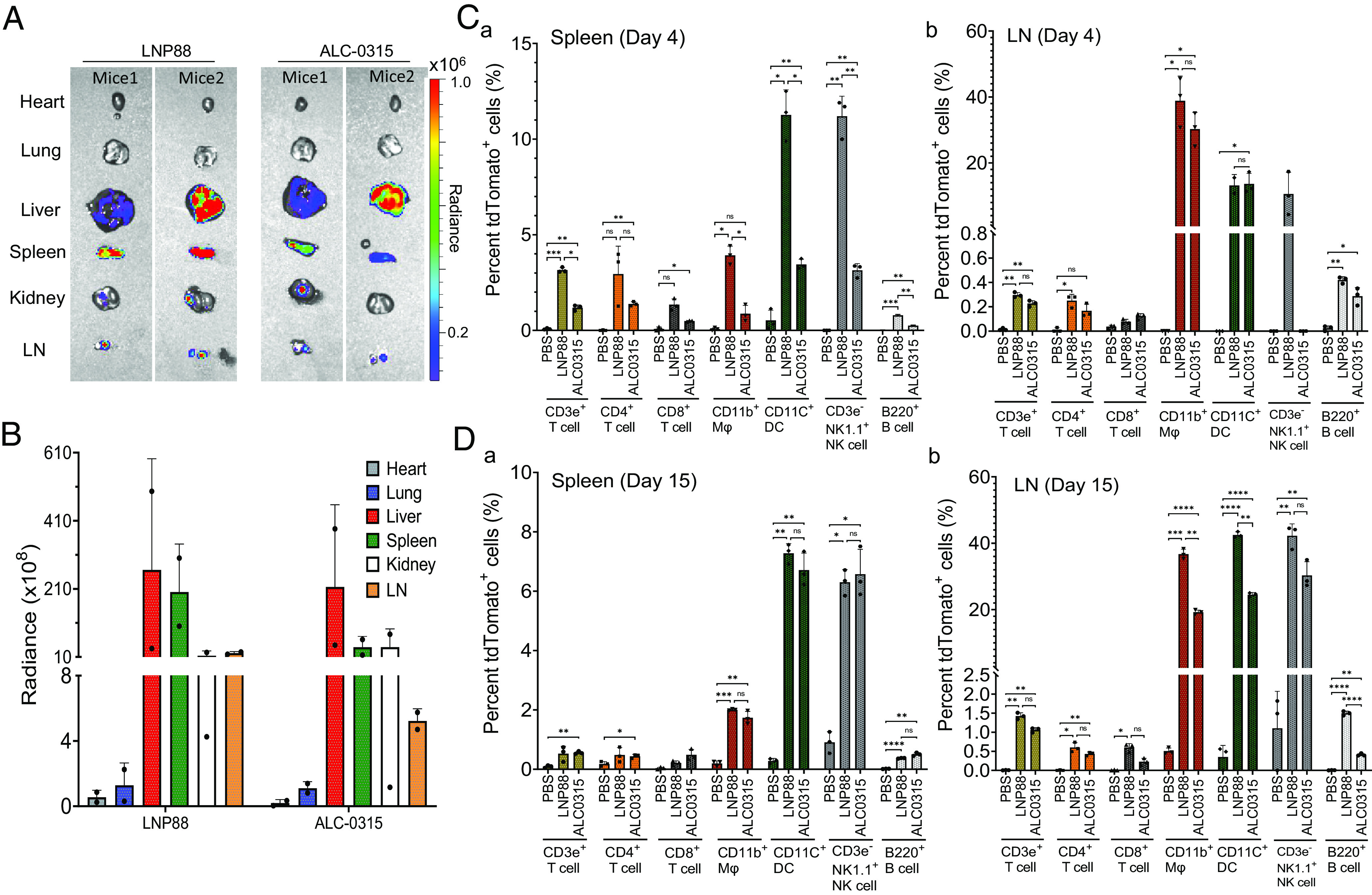
In vivo LNPs-mediated mLuc delivery in Balb/c mice and mCre delivery in Ai14D mice to explore LNPs biodistribution via I.M injection. (A) Representative ex vivo image of organs collected from LNP88-mLuc or ALC-0315-mLuc treated mice. (B) Quantification of the Fluc protein expression level in each organ (heart, lung, liver, spleen, kidney, and LN). Scale represents radiance (p/sec/cm2/sr), min = 5.0e4 to max = 1.0e6. Average ± SD (n = 2). (C) Percent tdTomato+ immune cells [T cells, macrophages (Mϕ), dendritic cells (DC), NK cells, and B cells] in the spleen (a) and LN (b) measured by flow cytometry at day 4 post LNP88-mCre or ALC-0315-mCre delivery to Ai14D mice. (n = 3 mice per group). (D) Percent tdTomato+ immune cells [T cells, macrophages (Mϕ), dendritic cells (DC), NK cells, and B cells] in the spleen (a) and LN (b) measured by flow cytometry at day 15 post LNP88-mCre or ALC-0315-mCre delivery to Ai14D mice. (n = 3 mice per group). Significance was statistically determined by the two-way ANOVA Tukey test, ns, no significance, *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001. Average ± SD, biological replicates shown.
At 4 and 15 d after I.M. injection of LNP88-mCre and ALC-0315-mCre, the mice were killed, and the spleen and LNs were collected for immune cells analysis. At day 4, LNP88-mCre showed positive mRNA expression in ~3.9% macrophages (CD11b+ Mφ) and ~11.3% DCs within the spleen while ALC-0315-mCre produced Cre expression in ~0.9% Mφ and ~3.5% dendritic cells (CD11c+ DCs) (Fig. 2C and SI Appendix, Fig. S13). LNP88 successfully delivered mCre to 3.2% CD3ε+ T cells, 1.3% CD8a+ cytotoxic T cells, and 3.4% CD4+ helper T cells. Expression levels of tdTomato were onefold to twofold higher than those of ALC-0315, indicating that LNP88 exhibits higher efficiency in delivering to T cells compared with ALC-0315 formulation. At day 15, the delivery to antigen-presenting cell using LNP88 remained relatively higher compared with ALC-0315 in the spleen (Fig. 2D). Even though the percentage of tdTomato+ T cells was reduced on day 15 post LNP88 treatment, the transfection efficiency of LNP88 in T cells remained comparable to ALC-0315.
We also analyzed the immune cell types transfected by LNPs in the LNs (Fig. 2 C and D). On days 4 and 15, robust APCs were transfected by LNP88-mCre and ALC-0315-mCre. Cre expression levels peaked on day 15 following LNP88 treatment with ~36.7% Mϕ and ~42.5% DCs, both comparable to ALC-0315 treatment. LNPs delivery in T cells was lower in the LNs than in the spleen on day 4. In the 15 d after mRNA-LNP administration, tdTomato+ APCs and T cells were continuously increased in the LNs under LNP88 transfection, while the percent of tdTomato+ immune cells in both the spleen and LNs remained stable following ALC-0315 delivery, suggesting some APCs and T cells might migrate to other organs, reducing the number of APCs and T cells in the spleen. In addition, ~10.6% natural killer (NK) cells in the spleen (day 4) and ~42.2% NK cells in the LNs (day 15) were positive for tdTomato following LNP88-mediated delivery while ~6.6% NK cells in the spleen (day 15) and ~30.3% NK cells in the LNs (day 15) were positive following ALC-0315-mediated delivery. Together, both LNP88 and ALC-0315 are capable of delivering mRNA to the spleen and LNs and effectively transfect a range of immune cells, particularly APCs, T cells, and NK cells.
In Vivo Screening of Top-Performing Vaccine Candidates.
In the previous UTR optimization study (Fig. 1), we found that mRNA with two pairs of 5′ and 3′ UTR (Ces1d/AP3B1, 70 nt/ApoA2) led to high protein expression. Here, we constructed the SARS-CoV-2 spike mRNA using these two UTR pairs, referred to as mCSA and m70SA, respectively. The mRNA with α-globin/β-globin UTR combination (mrS) was constructed as control for comparison. The details regarding the construction and characterization of mRNA in addition to the verification of in vitro spike protein expression can be found in supporting information (SI Appendix, Figs. S14 and S15).
Three spike mRNAs (mrS, mCSA, and m70SA) were formulated with LNP88 to generate three mRNA vaccines (TU88mrS, TU88mCSA, and TU88m70SA). We determined the ability of each of these mRNA vaccines to produce antibodies against the spike protein in a murine model. Four groups of mice were immunized with phosphate-buffered saline (PBS; mock), TU88mrS, TU88mCSA, or TU88m70SA (1 µg per mouse) at week 0 and 3, followed by serum collection at week 4 (SI Appendix, Fig. S16A). S-specific antibody endpoint titers (EPTs) were determined by an ELISA using serum samples with 1:100 initial dilution followed by a X4 serial dilution. The data showed that the median IgG EPT of TU88mCSA vaccination (927,560) is significantly higher than those of TU88mrS (282,480) and TU88m70SA (190,736) mRNA vaccines (SI Appendix, Fig. S16B). Thus, mCSA construct was selected for further immunological study of the vaccine in mice, and mrS was used as a control for comparison. We chose both LNP88 and ALC-0315 as carriers, and generated four mRNA vaccines (TU88mCSA, TU88mrS, ALCmCSA, and ALCmrS) for following study.
Five groups of C57BL/6 mice (n = 5 per group) were immunized with PBS (mock control), TU88mCSA (1 µg), TU88mrS (1 µg), ALCmCSA (1 µg), or ALCmrS (1 µg) via I.M. administration at weeks 0 (prime) and 3 (boost) (Fig. 3A). We first examined antibodies in mouse bronchoalveolar lavage fluid (BALF) to evaluate the immune response in the respiratory tract after booster vaccination (week 5). Compared to PBS controls, immunization with TU88mCSA and ALCmCSA induced an S-specific-binding IgG response in BALF. TU88mCSA and ALCmCSA elicited a notably higher level of BALF IgG than TU88mrS and ALCmrS (Fig. 3 B, a). No significant S-specific IgA in BALF was detected in the vaccinated mice compared to the PBS control (Fig. 3 B, b).
Fig. 3.

Top-performing SARS-CoV-2 vaccine screening based on S-specific IgG titer and T Cell response in mice (A) Schematic illustration of mice experimental design and timeline. Five groups of mice (n = 5 per group) were vaccinated I.M. with mock (PBS), TU88mCSA (1 µg), TU88mrS (1 µg), ALCmCSA (1 µg), and ALCmrS (1 µg) for each at weeks 0 and 3. Two weeks after booster vaccination (week 5), mice were euthanized and subjected to analysis of immune response in the spleen, lung, and bronchus. (B) Comparison of bronchoalveolar lavage fluid (BALF) S-specific IgG or IgA optical density (OD450) values between different vaccine groups after booster (week 5) vaccination is shown. OD450 values for individual BALF samples after booster vaccination are shown. (C) Shown is the comparison of percent S-specific spleen memory CD8+ and activated CD4+ T cells in the total spleen cells between different groups. (D) Shown is the comparison of percent S-specific lung-resident memory CD8+ and activated CD4+ T cells in the total lung cells between different groups. Unless specified otherwise, significance for (B–D) was statistically determined by the one-way ANOVA Tukey test, *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001. Average ± SD (n = 5 mice per group), biological replicates shown.
Many studies have shown that T cells are involved in the protection against SARS-CoV-2 infection (39–42). Moreover, recent findings clarified that memory CD8+ T cells (CD8+ TM) with conserved specificities of SARS-CoV-2 peptides are highly abundant in patients with mild COVID-19 symptoms and exhibit strong protective efficacy (43, 44). It remains controversial whether I.M. mRNA immunization induces antigen-specific memory T cells in the respiratory system. We investigated T cell responses in both the spleen and lungs of mice following vaccination. TU88mCSA, ALCmCSA, and ALCmrS induced strong CD8+ TM and CD4+ T cell activation in the spleen compared to PBS controls based on the expression of CD44+, a surface marker representing T cell activation and memory (Fig. 3 C, a, b, e, and f). To measure vaccine-specific CD8+ TM cells, we used a SARS-CoV-2 S539–546 (VNFNFNGL)-specific major histocompatibility complex class I (MHC-I) tetramer (H-2Kb) (SI Appendix, Fig. S17A). Consistent with total number of CD44+ CD8+ T cells, TU88mCSA, ALCmCSA, and ALCmrS induced strong S-specific CD8+ TM responses in the spleen reaching 0.40%, 0.84%, and 0.50% (% of total spleen cells), respectively, which was sevenfold to 14-fold higher than the PBS controls (0.06%) (Fig. 3 C, c and d). Overall, mCSA elicited more S-specific CD8+ TM cells than mrS in the spleen (P < 0.05 between ALCmCSA and ALCmrS), which is in line with the S-specific BALF IgG response. These results suggest the potent systemic immune responses are majorly induced by TU88mCSA and ALCmCSA.
We further evaluated whether our mRNA vaccine candidates induce strong T cell response in the lungs. CXCR6 has been utilized to define CD69+ tissue-resident memory T cells in mice (45–48). On the basis of CD69+ and CXCR6+, we observed that TU88mCSA and ALCmCSA induced higher frequencies of CD4+ T cells and CD8+ T cells that are also positive for CD69 and CXCR6 in the lungs than PBS controls (Fig. 3 D, a–c and g–i). Consistent with S-specific IgG response in BALF and CD8+ T cell response in the spleen, TU88mCSA and ALCmCSA both outperformed TU88mrS and ALCmrS with regards to the induction of CD4+ and CD8+ T cell activation in the lung, suggesting mCSA is a superior mRNA cargo over mrS and capable of eliciting stronger T-cell response in the respiratory tract. We also used SARS-CoV-2 S539–546 (VNFNFNGL) MHC-I tetramer to identify S-specific CD8+ T cells in the lungs (SI Appendix, Fig. S17B). ALCmCSA elicited stronger S-specific CD8+ T cells (1.22% in total lung cells) than ALCmrS and TU88mCSA (Fig. 3 D, d–f ).
These results show that I.M. immunization with mCSA elicits strong IgG responses in the serum and BALF. TU88mCSA and ALCmCSA induce stronger CD4+ and CD8+ T-cell responses in both lung and spleen when compared with TU88mrS and ALCmrS. Thus, TU88mCSA and ALCmCSA are highly immunogenic and induce systemic and respiratory immune response and were used for challenge studies against VOCs.
Evaluation of TU88mCSA Efficacy in WA1/2020 and Omicron BA.1 Challenge Models.
To evaluate protection of TU88mCSA vaccine against SARS-CoV-2, we established a small animal model for WA1/2020 and Omicron BA.1 using golden Syrian hamsters (Mesocricetus auratus) (Fig. 4A and SI Appendix, Fig. S18). In the hamster model, following prime/boost TU88mCSA vaccine, animals were infected with WA1/2020 [dose of 4 × 104 plaque-forming units (pfu) per hamster] or Omicron BA.1 (dose of 1.3 × 104 pfu per hamster) via the intranasal route (I.N.) and monitored for body weight changes. In WA1/2020 challenged groups, unvaccinated hamsters experienced gradual weight loss up to 0.7% and 1.2% from 1 to 5 days postinfection (DPI) (Fig. 4B). This significant change in body weight is likely the result of viral pathogenesis. In contrast, TU88mCSA vaccine hamsters showed a healthy weight gain after infection, up to 7.1% and 8.4%, and remained in good condition through 1 to 14 DPI, suggesting that vaccinated hamsters were protected from disease and body weight loss. In Omicron BA.1 challenged groups, the weight change in hamster groups with or without vaccination showed overall no significant variation, but weight loss was observed from 5 to 7 DPI (Fig. 4C). Unvaccinated animals exhibited a gradual weight loss at 4 to 7 DPI, while TU88mCSA vaccine group showed a mild weight loss at 6 to 7 DPI followed by a recovery that significantly increased overall weight gain (Fig. 4C). Omicron BA.1 symptoms began on day 4, later than symptoms from WA1/2020 infection. The weight loss in WA1/2020 challenged groups was also more drastic relative to Omicron BA.1, suggesting that Omicron BA.1 could be a milder SARS-CoV-2 variant with lower pathogenicity. Next, we examined the safety and efficacy of two doses of TU88mCSA vaccine in the viral challenge model. Following vaccination, no signs of lesions were observed at the injection sites and hamster body weight increased in a steady manner over 14 DPI. As shown in Fig. 4 B and C, animals in vaccinated groups gained weight and did not show any signs of morbidity, suggesting that TU88mCSA is safe at the 5 µg mRNA/150 µg LNP88 dose per hamster.
Fig. 4.

TU88mCSA protects golden Syrian hamsters from WA1/2020 and Omicron BA.1 challenge. (A) Schematic illustration of hamster experimental design. Four groups of hamsters were investigated. The first two groups were I.M. vaccinated with mock (Saline, n = 12) or TU88mCSA (5 µg, n = 12) at weeks 0 and 3, followed by SARS-CoV-2 WA1/2020 (4 × 104 pfu) challenge at day 31 and viral load analysis was performed on 4 DPI (n = 4 per group) and hamster body weights were monitored for 14 d (n = 8 per group). The other two groups of hamsters (n = 4 per group) were vaccinated mock (Saline) or TU88mCSA at weeks 0 and 3, followed by I.N. challenge with SARS-CoV-2 variant Omicron BA.1 (1.3 × 104 pfu) at day 31. On 4 DPI (n = 2), lung tissues (n = 2 per group) were harvested for analysis of viral titers. Hamster body weights were monitored for 14 days after Omicron BA.1 inoculation (n = 2 per group). See SI Appendix, Fig. S18 for hamster assignments to mock control and vaccinated groups. The challenge study was repeated to reproduce the data but was increasing hamsters to five for each group. The data from two challenge studies at SI Appendix, Fig. S21 were combined together in this figure. (B) Hamster body weight change (%) following WA1/2020 challenge (4.0 × 104 pfu/hamster, n = 8 per group). (C) Hamster body weight change (%) after Omicron BA.1 challenge (1.3 × 104 pfu/hamster, n = 2 per group). (D) SARS-CoV-2 S-specific IgG titer in hamster serum day 7 post-booster (n = 16) determined by ELISA. *P < 0.05, two-tailed t test. Average ± SD. (E) Comparison of lung viral titers in WA1/2020 or Omicron BA.1-infected hamsters with or without TU88mCSA immunization. All data from two challenge studies were combined together in the same graph. Nine hamsters per group in WA1/2020 challenge model and seven hamsters per group in Omicron BA.1 challenge model. LOD: 100 pfu/g lung. Unless specified otherwise, significance for (B) and (C) was statistically determined by two-way ANOVA Šídák's multiple comparisons test. Significance for (E) was statistically determined by the one-way ordinary ANOVA Tukey test, *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001. Average ± SD. Biological replicates shown.
We then analyzed LNP88 hepatotoxicity in mouse livers (two doses per mouse) on day 2.5 following the second immunization (SI Appendix, Fig. S19). Three groups of mice were administered with TU88mCSA, ALCmCSA, or TU113mCSA delivering a spike mRNA dose of 1 µg/mouse, respectively, via the I.M., I.V., or S.C. route. Compared with ALCmCSA and TU113mCSA, TU88mCSA induced relatively lower alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase activity although no LNPs resulted in significant hepatotoxicity during this trial. No significant changes in these markers demonstrated negligible systemic toxicity using LNP88 in mice. This conclusion was further supported by histological data in SI Appendix, Fig. S20.
The S-binding serum IgG titer induced by TU88mCSA was analyzed to confirm TU88mCSA established humoral immunity in hamsters. Hamsters vaccinated with TU88mCSA at 5-µg dose elicited a strong binding antibody response, of which IgG geometric EPT reached 3,122,226, directed against the full S protein (Fig. 4D). To evaluate the efficacy of the TU88mCSA for protection against SARS-CoV-2, hamster lungs were removed for viral load analysis on 4 DPI (Fig. 4A). WA1/2020 was detected in the lungs of all nine unvaccinated hamsters (9/9), with a median viral titer of 9.2 × 105 pfu/g of lung tissue. However, vaccination with TU88mCSA substantially controlled the infectious virus, reducing it to 219.2 pfu/g of lung tissue in all hamsters. Nevertheless, low lung viral titers remained detectable in four out of the nine vaccinated hamsters (4/9). Similarly, in the Omicron BA.1 challenge model, the lung viral titers of the mock group can be detected in all animals (7/7), and their median reached 3.7 × 104 pfu/g lung (n = 7), while TU88mCSA vaccination induced complete viral control with no detectable titers in any of the hamsters (0/7), 100 pfu/g lung (n = 7) at LOD (Fig. 4E), demonstrating that TU88mCSA vaccination confers near-complete protection of the hamsters against both ancestral SARS-CoV-2 and the Omicron BA.1 variant.
Consistent with the hamster weights monitoring, WA1/2020 infected hamster lungs displayed higher viral titers (9.2 × 105 pfu/g lung) compared to the Omicron BA.1 model (3.7 × 104 pfu/g lung), reiterating that Omicron BA.1 is a milder SARS-CoV-2 variant than WA1/2020 (Fig. 4E and SI Appendix, Fig. S21). The reproducibility of this challenge data was also fully verified (SI Appendix, Fig. S21). Together, this study provided direct evidence that TU88mCSA as a monovalent vaccine using early Spp sequence has the potential to completely control Omicron BA.1 in the lung regardless of its S antigenic drift.
Evaluation of TU88mCSA and ALCmCSA Protection Efficacy on Hamster against Omicron BQ.1 Challenge.
We also investigated the efficacy of TU88mCSA and ALCmCSA vaccination against the SARS-CoV-2 Omicron sublineage (BQ.1) (Fig. 5A). Three groups of hamsters (n = 10) were vaccinated with mock (PBS), ALCmCSA (5 µg), or TU88mCSA (5 µg) on day 0 and day 21. Two weeks after the booster dose (day 35), all hamsters were infected via the I.N. route with Omicron BQ.1 strain with doses of 2 × 104 pfu. On 2 DPI (n = 5) and 4 DPI (n = 5), vaccine protection was evaluated on the basis of body weight changes and viral loads in the lungs and nasal washes.
Fig. 5.

ALCmCSA and TU88mCSA vaccination confer protection against challenge with the SARS-CoV-2 Omicron BQ.1 in hamsters. (A) Schematic illustration of hamster challenge study. Three groups of hamsters (n = 10 per group) were vaccinated I.M. with mock (PBS), ALCmCSA (5 µg for each) or TU88mCSA (5 µg for each) at weeks 0 and 3, followed by I.N. challenge with SARS-CoV-2 Omicron BQ.1 strain (2 × 104 pfu) at week 5. On 2 (n = 5) and 4 DPI (n = 5), lung tissues were harvested for analysis of viral titers; nasal washes were collected for analysis of viral titer; hamster body weights were also monitored. (B) A comparison of hamster body weight changes is shown between different groups from 0 to 4 DPI. Significance was statistically determined by the two-way ANOVA Tukey test, *P < 0.05. Average ± SD (n = 5 mice per group). (C) Analysis of S-specific IgG (a) and IgA (b) EPT in the hamster serum at week 5 post ALCmCSA or TU88mCSA immunization (mock, n = 4; vaccine group, n = 10). (D) Comparison of viral RNA copies in hamster lungs (log10 viral copies per milligram) between mock and vaccine groups are shown for tissues collected on 2 and 4 DPI. (E) Comparisons of viral titers in the hamster lungs (log10 FFU per gram) between mock and vaccine group are shown for tissues collected on 2 and 4 DPI. (F) A comparison of viral RNA copies in the nasal washes (log10 viral copies per milliliter) is shown between the indicated groups on 2 and 4 DPI. (G) Comparisons of viral titers in the hamster nasal washes (log10 FFU per milliliter) between the mock and vaccine groups are shown for samples collected on 2 and 4 DPI. Unless specified otherwise, significance for (C) and (D–G) was statistically determined by the one-way ordinary ANOVA Tukey test, ns, no significance, *P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001. Average ± SD. biological replicates shown.
Body weight analysis highlighted the difference between unvaccinated and vaccinated hamsters following Omicron BQ.1 infection. Infection in both mock control and ALCmCSA vaccinated hamsters resulted in a progressive weight loss of up to 3.3% and 1.5% by 3 DPI, respectively, whereas the TU88mCSA vaccine group slightly reduced by 2 DPI but overall maintained steady growth in weights through 4 DPI (Fig. 5B). Notably, the ALCmCSA vaccine group began to recover from 3 DPI onward, while mock groups were still far from recovering. These weight change data indicate that both ALCmCSA and TU88mCSA vaccination protect hamsters from Omicron BQ.1 induced morbidity. This protection efficacy is consistent with their robust serum S-specific IgG and IgA production at week 5 (ALCmCSA, IgG EPT: 194,400 and IgA EPT: 16,200; TU88mCSA, IgG EPT: 172,800 and IgA EPT: 19,800) (Fig. 5 B and C).
We assayed viral loads in lungs. At 2 DPI, compared to the mock control, ALCmCSA and TU88mCSA induced moderate control of Omicron BQ.1 in the lungs based on viral RNA copies (ALCmCSA, 17.8-fold reduction; TU88mCSA, 3.6-fold reduction) and insignificant infectious titers for all test articles. On 4 DPI, critically, ALCmCSA and TU88mCSA induced more robust control of BQ.1 based on viral RNA copies (ALCmCSA, 59.1-fold reduction; TU88mCSA, 51.0-fold reduction) and infectious viral titers (ALCmCSA, 4.1-fold reduction; TU88mCSA, 3.3-fold reduction; both under limit of detection), compared to mock control (Fig. 5 D and E). Viral titer analysis showed consistent results: At 4 DPI, no infectious virus was detectable in all five hamsters (0/5) with ALCmCSA or TU88mCSA vaccination, whereas three of five hamsters (3/5) in the mock control had detectable infectious virus (Fig. 5 D and E). Together, these data suggest that ALCmCSA and TU88mCSA effectively reduce and even eradicate infectious virus Omicron BQ.1 in the lung and protect hamsters from BQ.1 lung infection.
To further assess the effect of vaccine on BQ.1 infection in the hamster upper respiratory tract, we analyzed the viral RNA copies and infectious viral titer in the nasal washes. At 2 DPI, compared to mock control, TU88mCSA or ALCmCSA vaccination significantly reduced viral RNA copies to 17.5-fold or 4.8-fold, and infectious titers to 19.9-fold or 193.6-fold, respectively (Fig. 5 F and G). At 4 DPI, TU88mCSA or ALCmCSA induced 18.1-fold or 24.9-fold reduction in viral copies and 5.3-fold or 25.2-fold reduction in infectious titers respectively, relative to mock control. Notably, there was no detectable virus in four of five hamsters (4/5) in the ALCmCSA vaccine group and two of five hamsters (2/5) in the TU88mCSA vaccine group while all hamsters in mock control had detectable virus, suggesting ALCmCSA and TU88mCSA vaccination provides effective protection of hamster and extra control of Omicron BQ.1 in the upper respiratory airways.
Discussion
Here, we show that two potent vaccine candidates, TU88mCSA and ALCmCSA, are safe, well tolerated, and elicit a robust S-specific IgG titer and T cell response in mice. Both TU88mCSA and ALCmCSA provided effective protection against WA1/2020, Omicron BA.1, and BQ.1 challenge based on lung viral load reduction, viral clearance in the upper respiratory tract, and recovery of animal weights in hamsters. Our study demonstrates the feasibility of developing a monovalent mRNA vaccine using the early spike gene sequence (Spp) against broad VOCs, including Omicron BQ.1.
TU88mCSA and ALCmCSA vaccine utilized mCSA constructs composed of structural elements, such as the CleanCap AG, Ces1d (5′UTR), AP3B1 (3′UTR), Poly A120 tail, and m1Ψ incorporation, which have a substantial impact on translation efficiency. 5′ and 3′UTR sequence optimization represents an emerging strategy to improve mRNA vaccine performance in specific immune organs and immune cells. To evaluate the impact of UTRs on mRNA translatability, UTR fragments were introduced into plasmids for in vitro screening and introduced into mRNA constructs for in vivo screening. In comparison of UTR performance across all screening steps, notable findings include the following: i) Identical UTR variants demonstrate variable Fluc expression levels in mice and HEK293 cells, indicating that these UTRs sequences result in distinct performances in vitro and in vivo, especially in different organs and cell types. To evaluate the impact of UTRs on cell-specific mRNA translation, further investigation is necessary. ii) The performance of Ces1d (5′UTR) and AP3B1 (3UTR) combinations in mLuc translation outperformed commercial UTR combinations, including α-globin (5′UTR) and β-globin (3′UTR) segments used in SBI mRNA products and leading SARS-CoV-2 mRNA vaccines. iii) Ces1d acting as 5’UTR was not compatible with some 3′UTR fragments, including hα-globin, C3, and TIAM1, which inhibited mRNA translation in mice, but significantly enhanced the mRNA translatability by forming favorable secondary structures with AP3B1 as 3′ UTR. Alterations in the UTRs sequence may hinder ribosome access and prevent other translation factors from coordinating with ribosome to process the mRNA sequence. iv) The UTR optimization platform established here enables rapid UTR data mining and screening as a strategy for maximizing mRNA translatability in specific organs or cell types in vivo. These findings establish 5′UTR-Ces1d-AP3B1-3′UTR as a superior UTR combination for enhanced mRNA vaccine efficacy and targeted immune cell expression.
In addition to the optimized UTR construct, we designed a mRNA delivery vehicle, LNP88, and an improved ALC-0315 formulation to enhance the antigen-mRNA expression in specific immune organs and even in different immune cell types. In this study, LNP88 and ALC-0315 formulations were composed of ionizable active lipids, Chol, phospholipids, and DMG-PEG. We optimized the formulation parameters with these components for LNP88 and ALC-0315 stepwise using the S.C or I.M routes. We determined that the optimal formulation for LNP88 contained 50.0% lipid 88, 25.0% cholesterol, 15.6% DSPC, and 9.4% DMG-PEG (16:8:4:3, w/w) via the S.C. route while 51.6% lipid 88, 25.8% cholesterol, 16.1% DSPC, and 9.7% DMG-PEG (16:8:5:3, w/w) via the I.M. route with a 10:1 weight ratio of LNP88-mLuc. The weight ratio of LNP88-mLuc was further extended to 30:1, producing total flux up to 2 times over that of 10:1 in mice via I.M injection. ALC-0315 formulation was fine-tuned based on published formulation conditions (32) and ultimately the weight ratio of ALC-0315: Chol: DSPC: DMG-PEG at 16:8:5:3 performed best for mRNA delivery in total flux in vivo. These optimized LNP88 and ALC-0315 formulations enabled robust mRNA expression in the spleen and LN besides in the liver, suggesting LNP88 has the potential to transfect immune cells in the spleen and LNs as well as ALC-0315.
Given that both LNP88 and ALC-0315 target secondary lymphoid organs, their delivery efficacy to a range of immune cells was assessed in the spleen and LNs of Ai14D mice. A large proportion of LNP88 and ALC-0315 targeted APCs, including macrophages and DC cells. To date, LNPs used in leading vaccines, including ALC-0315 and SM-102, have not demonstrated clear evidence of efficient mRNA delivery to both spleen and LN (22, 31, 38). Our results clearly show that LNP88 and ALC-0315 vehicle transports mRNA to both the spleen and LNs and transfects immune cells (APCs, T cells, B cells, and NK cells) via I.M administration. These results suggest LNP88 and the improved ALC-0315 as potent mRNA delivery vehicles for inducing robust immune responses.
Along with the optimization of UTRs and LNP formulations in vitro and in vivo, we have here established a platform to identify the potent mRNA-LNP for inducing robust immune responses. We evaluated the immunogenicity of mCSA-LNP88 (TU88mCSA) and mCSA-ALC-0315 (ALCmCSA) platforms in mice. The mCSA vaccine candidate elicited significantly higher S-specific serum antibody responses than mrS and m70SA. Even though mCSA employs the early Spp sequence, it protects against omicron sublineages. As local mucosal immunity in the lungs is critical for protection against virus, we evaluated TU88mCSA and ALCmCSA vaccines to induce S-specific antibodies in BALF and S-specific TM response in the lungs in addition to systemic immunity. In BALF, TU88mCSA and ALCmCSA induced higher S-specific IgG antibody compared to TU88mrS and ALCmrS, although we did not detect significant S-specific IgA production in TU88mCSA or ALCmCSA vaccine groups. On the basis of CD69+ and CXCR6+, TU88mCSA and ALCmCSA consistently induced relatively higher CD4+ TM and CD8+ TM in the lungs compared to TU88mrS and ALCmrS. Moreover, ALCmCSA induced robust S-specific CD8+ TM over other vaccine groups, especially in lungs, indicating that mCSA is an effective platform for future SARS-COV-2 mRNA vaccine development.
To develop effective vaccines against VOCs, the bivalent vaccines acquired the VOC-specific S sequences as a booster vaccine, which has emerged as a validated strategy in human subjects. However, Omicron S-specific mRNA vaccination offered no additional protection against an Omicron challenge compared to an ancestral Spp mRNA vaccine booster, indicating that a VOC S-targeted strategy may not provide broad protection against SARS-CoV-2 VOCs (49, 50). Furthermore, VOC-specific vaccine development is too slow to meet the timely deployment against new VOCs. In this study, our S mRNA sequence was designed from the ancestral or early SARS-CoV-2 sequence (Wuhan-Hu-1) but modified with optimized UTRs (Ces1d/AP3B1). Despite use of the original sequence, our data reveal that Spp with different UTR substitution enabled distinct performance in eliciting TM cell immune response and BALF IgG titer in mice. In Omicron BA.1 challenge study, there was no detectable infectious virus in all hamsters in the TU88mCSA group (0/7), whereas all hamsters in the mock group had detectable virus (7/7), confirming that TU88mCSA induced robust control of Omicron BA.1 and eradicated virus from the lungs in 4 DPI. We evaluated the protective efficacy of TU88mCSA and ALCmCSA in hamsters against Omicron variant, BQ.1, which carries multiple S mutations. Significant reductions in BQ.1 viral RNA copies and lung viral titer indicated that TU88mCSA and ALCmCSA almost completely inhibited viral replication in the lungs at 4 DPI. In lung viral titer analysis, TU88mCSA and ALCmCSA induced complete viral control with no detectable BQ.1 viral titer in all hamsters (0/5), showing that TU88mCSA and ALCmCSA provide strong protection to hamsters in the lower respiratory tract. In our BQ.1 challenge models, TU88mCSA and ALCmCSA induced robust viral control of Omicron BQ.1 in nasal wash either at 2 DPI or 4 DPI, demonstrating that TU88mCSA and ALCmCSA enable the immediate clearance of infectious Omicron BQ.1 in both lower and upper respiratory tracts. To confirm this protection efficacy was induced by neutralizing antibodies or T cell immunity, we have examined their neutralizing activity toward the challenge virus BQ.1. Our data showed that sera of hamsters vaccinated with both vaccines (ALCmCSA and TU88mCSA) were negative for neutralization (FRNT50 < 20). The data are consistent with prior research that vaccines based on the early ancestral spike sequence have much reduced neutralization activity against SARS-CoV-2 variants, especially the emerging Omicron sub-variant, such as BQ1. The data support that the protection by ALCmCSA and TU88mCSA is likely mediated by cellular immunity other than neutralizing antibodies (SI Appendix, Fig. S22). Our study provides comprehensive evidence that optimized LNP88 and ALC-0315 mRNA vaccine formulations provide enhanced protective efficacy against WA1/2020, Omicron BA.1, and BQ.1 in hamsters due to the activation of a series of immune responses in both circulating system and respiratory airway. Notably, all mRNA vaccines used in this study utilize the ancestral spike gene sequence (Spp) without any VOC mutations. By altering the CDS of antigen mRNA constructs, our platform can be readily used for future mRNA vaccine development for timely deployment in the face of constant viral mutations.
Our study has several limitations. First, the UTR screening profile is far from enough for us to do accurate prediction and selection. Recent computational advances in mRNA construct optimization may help to identify additional UTRs for enhanced expression (51). Immune cell–specific UTRs screening in vivo warrants further investigation for robust antigen expression in next-generation mRNA vaccine development. In particular, the performance of UTRs in the APCs, CD4+ T cells, CD8+ T cells, B220+ B cells, and TM requires further evaluation using a series of UTRs fused mCre in Ai14D mice. Second, in our current study, fluorescent CD45 staining to distinguish tissue-resident cells from potential circulating T cells was not conducted, and we acknowledge that this is a limitation. Nevertheless, our data have shown that while substantial tetramer+ Spike-specific CD8+ T cells were observed in the lungs following vaccination (ranging 20-40% of total CD8+ T cells), only 3 to 8% of tetramer+ Spike-specific CD8+ T cells (in total CD8+ T cells) was observed in the spleen following vaccination (Fig. 3D). These data indicate that the high levels of S-specific CD8+ T cells detected in the lungs may not be simply due to the contamination of the circulating S-specific CD8+ T cells. In addition, our data showed significant levels of CXCR6+CD69+ T cells, indicating the lung tissue residency potential of these T cells. Nevertheless, more accurate detection of TM and more comprehensive T-cell analysis need to be performed in future studies to more thoroughly understand systemic and respiratory immunity elicited by these mRNA vaccines. Third, TU88mCSA and ALCmCSA vaccine efficacy was only evaluated 1 or 2 wk after booster vaccination. Without long-term monitoring, the durability of immune response and protection efficacy is not clear. Finally, in Omicron BQ.1 challenge studies, even though the TU88mCSA and ALCmCSA induced complete control of BQ.1 replication in both the lungs and upper respiratory tract of hamsters, the precise mechanisms of upper airway protection remain ambiguous. We have detected lung CD4+ TM, CD8+ TM, and S-specific CD8+ TM immune response induced by TU88mCSA and ALCmCSA, but these data may not fully address the mechanism of complete protective efficacy of vaccination. Additional analysis of immune mechanisms should be performed. Nevertheless, we have developed a UTR-based mRNA vaccine approach targeting highly variable S and demonstrated robust and broad protection against WA1/2020, Omicron BA.1, and BQ.1 infection using the original Spp sequence alone. In the future, the TU88mCSA and ALCmCSA platform can be applied in new mRNA vaccines to induce broadly protective immune response against new emerging VOCs.
Materials and Methods
More methods and materials are detailed in SI Appendix, Methods and Materials.
mRNA Synthesis.
The pMRNA- Luc-GFP or pMRNA-S variants plasmid was used as templates for gene polyadenylation using the Tail PCR Primer A93/A95, of which reverse primer contains 120 oligodT. Generated Tail PCR product was used as template in an in vitro transcription reaction (10×T7 reaction buffer (1×), CleanCap AG or ARCA cap (10 mM), ATP (10 mM), CTP (10 mM), GTP(3.75 mM), N1-methylpseudouridine (N1mψ, 3.75 mM), each of mRNA templates (25 ng/µL), and T7 RNA polymerase mix) and treated with Turbo DNase (ThermoFisher) and Antarctic Phosphatase (New England Biolabs) and purified using a MegaClear Kit (Life Technologies). The modified nucleotides used in this study were N1mψ (TriLink), which were incorporated to completely substitute their natural counterparts UTR in mRNA synthesis.
Lipid Synthesis.
Lipid 88, ALC-0315, and 113O12B were synthesized and characterized using NMR and MS, as shown in SI Appendix, Fig. S3. All 1H NMR spectra were recorded on a Bruker AVIII 500 MHz NMR spectrometer operated in the Fourier transform mode.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Stephen Kwok at Tufts University School of Medicine for assistance in preparing the Flow Cytometry Core. We also thank Caining Jin at Hopewell for assistance in ELISA experiments. We finally thank Shinji Makino, Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston for providing SARS-CoV N protein. Part of the work was performed and supported through World Reference Center for Emerging Viruses and Arboviruses (WRCEVA) at UTMB (R24AI120942). We acknowledge the funding support from Massachusetts Consortium on Pathogen Readiness (Q.X.) and Hopewell Therapeutics Inc. (Q.X.).
Author contributions
Z.Y., S.R.B., Jianzhu Chen, H.H., and Q.X. designed research; Z.Y., S.R.B., L.G.A.M., J.A.P., J.W., Y.Z., C.X., L.L., J.H., S.G., Z.Z., K.S.P., A.G., H.H., and Q.X. performed research; Z.Y., S.R.B., C.H., Jinjin Chen, Y.L., D.S., H.H., and Q.X. contributed new reagents/analytic tools; Z.Y., S.R.B., L.G.A.M., H.H., and Q.X. analyzed data; and Z.Y. wrote the paper.
Competing interests
Q.X. and Z.Y. are inventors on a provisional patent from Tufts University. Q.X. is a founder and consultant (with Chief Technology Officer title) of Hopewell Therapeutics Inc.
Footnotes
This article is a PNAS Direct Submission. B.Z. is a guest editor invited by the Editorial Board.
Contributor Information
Haitao Hu, Email: haihu@utmb.edu.
Qiaobing Xu, Email: Qiaobing.Xu@tufts.edu.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Collie S., Champion J., Moultrie H., Bekker L. G., Gray G., Effectiveness of BNT162b2 vaccine against omicron variant in South Africa. N. Engl. J. Med. 386, 494–496 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferdinands J. M., et al. , Waning 2-dose and 3-dose effectiveness of mRNA vaccines against COVID-19-associated emergency department and urgent care encounters and hospitalizations among adults during periods of delta and omicron variant predominance—vision network, 10 States, August 2021-January 2022. MMWR Morb. Mortal Wkly. Rep. 71, 255–263 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anonymous, Enhancing response to Omicron SARS-CoV-2 variant: Technical brief and priority actions for Member States. World Health Organization (2022), pp. 1–28.
- 4.Yu J., et al. , Neutralization of the SARS-CoV-2 Omicron BA.1 and BA.2 variants. N Engl. J. Med. 386, 1579–1580 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nanduri S., et al. , Effectiveness of Pfizer-BioNTech and moderna vaccines in preventing SARS-CoV-2 infection among nursing home residents before and during widespread circulation of the SARS-CoV-2 B.1.617.2 (Delta) variant—National Healthcare Safety Network, March 1-August 1, 2021. MMWR Morb. Mortal Wkly. Rep. 70, 1163–1166 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Viana R., et al. , Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 603, 679–686 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu J., et al. , Neutralization of the SARS-CoV-2 Omicron BA.1 and BA.2 variants. N. Engl. J. Med. 386, 1579–1580 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou J., et al. , Neutralization of BA.4–BA.5, BA.4.6, BA.2.75.2, BQ.1.1, and XBB.1 with bivalent vaccine. N. Engl. J. Med. 388, 854–857 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kurhade C., et al. , Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 29, 344–347 (2023). [DOI] [PubMed] [Google Scholar]
- 10.Fang Z., et al. , Omicron-specific mRNA vaccination alone and as a heterologous booster against SARS-CoV-2. Nat. Commun. 13, 3250 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arunachalam P. S., et al. , Durable protection against the SARS-CoV-2 Omicron variant is induced by an adjuvanted subunit vaccine. Sci. Transl. Med. 14, eabq4130 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langel S. N., et al. , Adenovirus type 5 SARS-CoV-2 vaccines delivered orally or intranasally reduced disease severity and transmission in a hamster model. Sci. Transl. Med. 14, eabn6868 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chalkias S., et al. , A bivalent omicron-containing booster vaccine against covid-19. N Engl. J. Med. 387, 1279–1291 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dangi T., et al. , Pre-existing immunity modulates responses to mRNA boosters. Cell Rep. 42, 112167 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ying B., et al. , Boosting with variant-matched or historical mRNA vaccines protects against Omicron infection in mice. Cell 185, 1572–1587.e1511 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheaffer S. M., et al. , Bivalent SARS-CoV-2 mRNA vaccines increase breadth of neutralization and protect against the BA.5 Omicron variant in mice. Nat. Med. 29, 247–257 (2022). [DOI] [PubMed] [Google Scholar]
- 17.Arora P., et al. , Omicron sublineage BQ.1.1 resistance to monoclonal antibodies. Lancet Infect Dis. 23, 22–23 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.WHO Tracking SARS-CoV-2 variants (2023) (https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/).
- 19.Cele S., et al. , Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 602, 654–656 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muik A., et al. , Neutralization of SARS-CoV-2 Omicron by BNT162b2 mRNA vaccine-elicited human sera. Science 375, 678–680 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller J., et al. , Substantial neutralization escape by SARS-CoV-2 Omicron variants BQ.1.1 and XBB.1. N. Engl. J. Med . 388, 662–664 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sahin U., et al. , BNT162b2 vaccine induces neutralizing antibodies and poly-specific T cells in humans. Nature 595, 572–577 (2021). [DOI] [PubMed] [Google Scholar]
- 23.Sample P. J., et al. , Human 5’ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 37, 803–809 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao J., et al. , High-throughput 5’ UTR engineering for enhanced protein production in non-viral gene therapies. Nat. Commun. 12, 4138 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao W., et al. , Massively parallel functional annotation of 3′ untranslated regions. Nat. Biotechnol. 32, 387–391 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng C., et al. , Leveraging mRNA sequences and nanoparticles to deliver SARS-CoV-2 antigens in vivo. Adv Mater 32, e2004452 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jia L., et al. , Decoding mRNA translatability and stability from the 5’ UTR. Nat. Struct. Mol. Biol. 27, 814–821 (2020). [DOI] [PubMed] [Google Scholar]
- 28.Sultana N., et al. , Optimization of 5’ untranslated region of modified mRNA for use in cardiac or hepatic ischemic injury. Mol. Ther. Methods Clin. Dev. 17, 622–633 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwanhausser B., et al. , Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Hou X., Zaks T., Langer R., Dong Y., Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 6, 1078–1094 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corbett K. S., et al. , SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 586, 567–571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schoenmaker L., et al. , mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 601, 120586 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.ModernaTX I., A Phase 3, randomized, stratified, observer-blind, placebo-controlled study to evaluate the efficacy, safety, and immunogenicity of mRNA-1273 SARS-CoV-2 vaccine in adults aged 18 years and older. COVID-19 Clin. Res. Coalition (2020). [Google Scholar]
- 34.Hinton R. D. M., Pfizer-BioNTech COVID-19 vaccine EUA letter of authorization. Food and Drug Administration (2021). [Google Scholar]
- 35.Buschmann M. D., et al. , Nanomaterial delivery systems for mRNA vaccines. Vaccines (Basel) 9, 65 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao X., et al. , Imidazole-based synthetic lipidoids for in vivo mRNA delivery into primary T lymphocytes. Angew. Chem. Int. Ed Engl. 59, 20083–20089 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Chen J., et al. , Lipid nanoparticle-mediated lymph node-targeting delivery of mRNA cancer vaccine elicits robust CD8(+) T cell response. Proc. Natl. Acad. Sci. U.S.A. 119, e2207841119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vogel A. B., et al. , BNT162b vaccines protect rhesus macaques from SARS-CoV-2. Nature 592, 283–289 (2021). [DOI] [PubMed] [Google Scholar]
- 39.Mateus J., et al. , Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 370, 89–94 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Bert N., et al. , SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 584, 457–462 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Peng Y., et al. , Broad and strong memory CD4(+) and CD8(+) T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 21, 1336–1345 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferretti A. P., et al. , Unbiased screens show CD8(+) T cells of COVID-19 patients recognize shared epitopes in SARS-CoV-2 that largely reside outside the spike protein. Immunity 53, 1095–1107 e1093 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adamo S., et al. , Signature of long-lived memory CD8(+) T cells in acute SARS-CoV-2 infection. Nature 602, 148–155 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mallajosyula V., et al. , CD8(+) T cells specific for conserved coronavirus epitopes correlate with milder disease in COVID-19 patients. Sci. Immunol. 6, eabg5669 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar B. V., et al. , Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 20, 2921–2934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wein A. N., et al. , CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 216, 2748–2762 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheon I. S., Son Y. M., Sun J., Tissue-resident memory T cells and lung immunopathology. Immunol. Rev. 316, 63–83 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mabrouk N., et al. , CXCR6 expressing T cells: Functions and role in the control of tumors. Front. Immunol. 13, 1022136 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gagne M., et al. mRNA-1273 or mRNA-Omicron boost in vaccinated macaques elicits similar B cell expansion, neutralizing responses, and protection from Omicron. Cell 185, 1556–1571.e1518 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hajnik R. L., et al. , Dual spike and nucleocapsid mRNA vaccination confer protection against SARS-CoV-2 Omicron and Delta variants in preclinical models. Sci. Transl. Med. 14, eabq1945 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ye Z., et al. , The mRNA vaccine revolution: COVID-19 has launched the future of vaccinology. ACS Nano 17, 15231–15253 (2023). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.