

Abstract
The molecular mechanisms that underlie maturation and egress of Epstein-Barr virus (EBV) virions are only partially characterized. We have recently shown that the BFRF1 gene, the EBV positional homolog of herpes simplex virus type 1 and pseudorabies virus UL34, is expressed early during EBV lytic replication and that it is found predominantly on the nuclear membrane (A. Farina, R. Santarelli, R. Gonnella, R. Bei, R. Muraro, G. Cardinali, S. Uccini, G. Ragona, L. Frati, A. Faggioni, and A. Angeloni, J. Virol. 74:3235-3244, 2000). These data suggest that the BFRF1 protein might be involved in viral primary envelopment. To precisely determine the function of this protein, we have constructed an EBV mutant devoid of the BFRF1 gene (BFRF1-KO). 293 cells carrying BFRF1-KO showed no differences in comparison with wild-type EBV in terms of DNA lytic replication or expression of late viral proteins upon induction of the lytic cycle. However, binding assays and infection experiments using cell lines or human cord blood lymphocytes showed a clear reduction in the viral mutant titers. Complementation experiments with BFRF1-KO and a BFRF1 expression vector restored viral titers to levels similar to those for the wild-type control, showing that the modifications that we introduced were limited to the BFRF1 gene. Electron microscopic observations showed that the reduction in viral titers was due to sequestration of EBV nucleocapsids in the nuclei of lytically induced cells. This suggests that BFRF1 is involved in transport of the maturing virion across the nuclear membrane. This hypothesis was further supported by the observation that BFRF1 is present in maturing intracellular virions but not in their extracellular counterparts. This implies that BFRF1 is a key protein for EBV maturation.
Epstein-Barr virus (EBV) is one of the eight known human herpesviruses. This member of the gammaherpesvirus subfamily infects B lymphocytes, in which it establishes a latent infection characterized by the expression of a limited set of viral genes (25). Viral reactivation from the latent state either occurs spontaneously or is induced by a variety of different stimuli (11, 30, 32, 49, 55), leading to viral lytic replication and shedding of viral progeny. The EBV lytic program consists of the sequential activation of three distinct classes of viral genes: immediate early, early, and late. The two transactivators BZLF1 (ZEBRA) and BRLF1 (Rta) are immediate-early genes that can initiate the switch between latency and lytic replication (14, 24, 41). Early genes are frequently but not exclusively involved in viral DNA replication; these genes include, among many others, those for the viral DNA polymerase (31) and its processivity factor BMRF1 (5), the bcl-2 homolog BHRF1 (38), and the major DNA binding protein BALF2 (8). Late genes are known to encode predominantly structural proteins, such as gp350/220, the most abundant glycoprotein of the viral envelope. gp350/220 mediates the binding of the virus to its cognate receptor CR2 (50). Herpesvirus DNA replication and nucleocapsid assembly take place in the nucleus. In order to reach the extracellular environment, herpesviruses must therefore traffic through several cellular membranes. This trafficking is an active process that involves successive envelopments and de-envelopments of the viral nucleocapsid. Two herpesvirus proteins, the products of the UL34 and UL31 genes, have been shown to play an essential role during primary envelopment that is characterized by egress through the inner nuclear membrane (reviewed in reference 34). UL34 and UL31 are conserved among several human and animal herpesviruses, including herpes simplex virus type 1 (HSV-1), HSV-2, pseudorabies virus (PrV), murine cytomegalovirus, and equine herpesvirus 1 (15, 20, 26, 35, 36, 42-45, 47, 53-55).
We have recently identified and characterized the product of the BFRF1 open reading frame (ORF), which is expressed early in the viral replication process (1, 12). BFRF1 shows a degree of homology to UL34, and both proteins are located in the nuclear membrane of replicating cells, preferentially in areas where budding of the nucleocapsids underneath occurs (13, 15, 43). This suggests that BFRF1 indeed shares with UL34 the same functions during viral maturation. However, structural and positional homologies between alpha- and gammaherpesviruses are not necessarily equivalent to functional identity. To unequivocally address this issue, we have constructed a recombinant EBV in which the BFRF1 gene has been disrupted, and we report here the phenotype of this viral mutant.
MATERIALS AND METHODS
Cells.
The 293 cell line is a human embryonic epithelial kidney cell line that has been transformed by the introduction of the E1a and E1b genes from adenovirus type 5 DNA (19). Raji is an EBV-positive human B-cell line derived from a Burkitt's lymphoma that carries a defective genome unable to replicate viral DNA and to express late viral genes (40). DG75 is an EBV-negative human Burkitt's lymphoma cell line (2). 2A8 is an EBV-negative Akata cell clone, kindly provided by J. Sixbey (6). HeLa is a human cervix adenocarcinoma cell line, and HaCaT is an immortalized human keratinocyte cell line (3). All cell lines were maintained in RPMI 1640 medium supplemented with 10% fetal calf serum.
Recombinant EBV plasmid.
To generate a BFRF1-negative mutant, the recBCD Escherichia coli strain BJ5183 (22) was transformed with the recombinant EBV plasmid p2089. The prokaryotic backbone of this plasmid carries the chloramphenicol resistance gene, the gene for the green fluorescent protein (GFP), and the hygromycin resistance gene as a selectable marker in eukaryotic cells (9). We used the pGEM-BamF B95-8 EBV subclone, which contains the BamHI F fragment (EBV coordinates 54853 to 62249), to construct the BFRF1 targeting vector. The BamHI F EcoRV-PmlI fragment containing the BFRF1 gene was replaced by tetracycline resistance gene flanked by Flp recombinase binding sites from plasmid pCP16 (4) to yield pGEM-F634TET. This plasmid therefore carries a truncated version of the BFRF1 gene with a deletion of amino acids 80 to 291 of the BFRF1 protein (EBV coordinates 58875 to 59493). pGEM-F634TET was cleaved with BamHI to generate a fragment of 6,761 bp consisting of the modified BFRF1 gene flanked by EBV-specific sequences. The linearized plasmid was transformed into BJ5183 carrying p2089 to induce homologous recombination via EBV flanking regions. Bacterial clones were selected with chloramphenicol (15 μg/ml) and tetracycline (10 μg/ml). Plasmid DNAs from antibiotic-resistant clones were prepared and analyzed with different restriction enzymes to identify clones with the correct recombination pattern. The tetracycline resistance gene was removed by using the Flp recombinase cloned onto the temperature-sensitive plasmid pCP20 (4), which also carries the ampicillin resistance gene. After selection on chloramphenicol-ampicillin, the bacterial clones obtained were incubated on chloramphenicol plates at 42°C to induce the loss of the pCP20 plasmid. The resistant clones were subjected to restriction analysis to confirm the correct restriction pattern. Recombinant DNA from wild-type EBV was digested in parallel and provided an appropriate control. Plasmid DNA from a properly recombined clone was then electroporated into recA E. coli strain DH10B (Invitrogen) for further propagation of the recombinant EBV plasmid.
DNA transfection and selection of a stable transfectant.
Cells were transfected by using Lipofectamine (Invitrogen) as described elsewhere (23). Selection of stable 293 cell clones carrying the EBV recombinant plasmid was performed with hygromycin (100 μg/ml). Cell clones surviving selection were first checked for GFP fluorescence and successively expanded for further investigation. The cell lines were called 293-BFRF1-KO.
Southern blot analysis.
DNA extraction, enzymatic cleavage, and hybridization were performed as described previously (23). To analyze the BFRF1 gene, we used a specific 618-bp probe as previously described (12). The terminal repeat (TR) region was investigated by using the 3.7-kb MluI fragment of p2124 (9). The integrity of the BFRF1-contiguous ORF of BFRF2 was analyzed by Southern blotting with the PmlI/XmnI fragment of the pGEM-BamHI F plasmid (genomic coordinates 59797 to 60403) as a probe.
Plasmid rescue in E. coli.
Circular EBV plasmid DNA was extracted from 293-BFRF1-KO cells as described previously (21). Extracted DNA was electroporated (1800 V, 25 mF, 100 W) into E. coli strain DH10B, and transformed bacterial clones were selected on chloramphenicol plates.
Production of recombinant EBV particles.
293-BFRF1-KO cells carrying the BFRF1-KO EBV genome and 293-2089 cells carrying the wild-type B95-8 EBV genome were transfected with an expression plasmid carrying the BZLFI gene under the control of the cytomegalovirus promoter. To this end, cells were seeded in six-well cluster plates and transfected with 0.5 μg of BZLF1 per well when they reached 70% confluence. To complement the phenotype of the BFRF1-negative mutant, cells carrying the BFRF1-KO recombinant were cotransfected with an expression plasmid carrying the BFRF1 gene and with the BZLF1 expression plasmid (12). At 72-h posttransfection, viral supernatants were harvested and filtered through a 0.8-μm-pore-size filter. Supernatants were ultracentrifuged at 20,000 × g for 2 h, and the virus pellet was resuspended in medium to obtain a 20-fold-concentrated solution ready to be used for infection experiments.
Cell infection, B-lymphocyte immortalization, and PCR-mediated amplification.
Cell lines were infected with 1 ml of concentrated supernatant containing either BFRF1 mutant viruses, complemented BFRF1 mutant viruses, or wild-type EBV. At 3 days postinfection, the percentage of GFP-positive cells was determined by counting the fluorescent cells under an inverted UV microscope and by fluorescence-activated cell sorter analysis (FACS). Primary B lymphocytes from umbilical cord blood were incubated overnight with 1 ml of filtered supernatant, and 1.5 × 105 cells were plated on a 96-well cluster plate and fed with fresh medium once a week. Ten outgrowing clones immortalized by using wild-type EBV, 3 clones from BFRF1 mutant virus, and 10 clones from complemented mutant virus were expanded for PCR analysis after DNA extraction. PCR-mediated amplification of both the BFRF1 and EBER-1 genes was performed with the following primers: BFRF1 up (5′-CCTAGATCTCGAGAATCATG-3′), BFRF1 d (5′-CCTGGAGAATTCCCGCTCCC-3′), EBER1 up (5′-AGGACCTACGCTGCCCTAGA-3′), and EBER1 d (5′-AAAACATGCGGACCACCAGC-3′) (12, 52).
Virion purification.
Biochemical characterization of extracellular virions was performed by precipitating viruses from infectious supernatants by using a polyethylene glycol (PEG)-containing solution (0.5% [wt/vol] PEG 6000 in 5 M NaCl). Viruses were further collected by centrifugation at 9,000 × g for 20 min. To purify intracellular virions, lytically induced cells were extensively washed and sequentially frozen in a dry ice bath and thawed at 37°C three times. Cells were spun down at 5,000 × g for 10 min, and supernatants were filtered with a 0.8-μm-pore-size filter. Viruses present in these supernatants were further PEG precipitated as described for extracellular virions. Purified virions were suspended and analyzed by Western blotting.
Immunofluorescence and binding assays.
An indirect immunofluorescence assay was performed as described previously (12). Antibodies specific to BFRF1 (monoclonal antibody E7), gp350/220, and EA-D (ABI); Rta (Argene Biosoft); and BLRF2 (a gift of G. Miller) were used at a 1:100 dilution. The binding assay aims at detecting virions bound to the surface of Raji cells by immunofluorescence with a gp350/220-specific antibody, as previously described (7). Briefly, Raji cells were incubated with different viral supernatants for 3 h at 4°C, washed in phosphate-buffered saline, and fixed on glass slides by using acetone (15 min, 25°C). The slides were incubated with the monoclonal antibody directed against gp350/220 for 1 h at 25°C. After extensive washings, bound gp350/220 antibody was detected with a fluorescein-conjugated goat anti-mouse immunoglobulin G antibody.
Gardella gel electrophoresis.
Viral linear DNAs from different induced cell lines were detected by using the agarose gel electrophoresis system described by Gardella et al. (16). Cells (5 × 105) were directly lysed in gel slots to avoid shearing of viral DNA. Southern blot hybridization was performed with an EBV-specific probe for TRs as described before (9).
Electron microscopy.
Lytically induced cells were washed three times in phosphate-buffered saline and fixed with 2% glutaraldehyde in the same buffer at 4°C. Samples were postfixed in 1% osmium tetroxide in veronal acetate buffer (pH 7.4) for 1 h at 25°C, stained with 0,1% tannic acid in the same buffer for 30 min at 25°C and with uranyl acetate (5 mg/ml) for 1 h at 25°C, dehydrated in acetone, and embedded in Epon 812. Thin sections were examined unstained or poststained with uranyl acetate and lead hydroxide.
RESULTS
Construction of a BFRF1-negative EBV mutant strain.
The EBV mutant devoid of BFRF1 was constructed by homologous recombination between the linearized BFRF1 targeting vector and the wild-type EBV genome in the recA-positive, recBC-negative E. coli strain BJ5183. After double selection for chloramphenicol and tetracycline, single outgrowing bacterial colonies were analyzed with several restriction enzymes, and plasmids showing the expected recombination pattern were selected for further manipulations (data not shown). The tetracycline resistance gene was then excised from the recombinant EBV plasmid DNA by using the Flp recombinase (Fig. 1A). Plasmid DNA from this properly recombined viral mutant clone was then electroporated into the recA E. coli strain DH10B for further propagation. Figure 1B shows the BamHI restriction patterns of both the BFRF1-negative mutant and the wild-type EBV DNA. As expected, the BFRF1-KO restriction pattern shows a shift of the BamHI F fragment.
FIG. 1.

Mutant EBV lacking BFRF1. (A) Construction of the BFRF1-negative EBV mutant. Homologous recombination between the wild-type EBV genome and a linearized fragment carrying the BamHI F region in which 634 bp of the BFRF1 gene was replaced by the gene for the tetracycline resistance was performed in the recBCD E. coli strain BJ5183. Clones carrying the recombinant EBV were selected for chloramphenicol and tetracycline resistance. In order to excise the Tet box, selected clones were transformed with a plasmid encoding the Flp recombinase (pCP20) that recombined with the two Flp recombination target (Frt) sequences flanking the Tet box. (B) Restriction fragment analysis of EBV BFRF1-KO DNA. The restriction pattern of BFRF1-KO mutant DNA was compared to that of the wild-type (wt) EBV DNA. The modified DNA pattern is indicated by arrows. As expected from the predicted BamHI restriction pattern, the deletion of the BFRF1 fragment led to a shift of the BamHI F region with respect to that in the wild-type DNA. (C) Southern blot analysis of 293-BFRF1-KO cell clones compared to 293-2089 cells carrying the wild-type EBV genome. Genomic DNA from stably transfected clones harboring the BFRF1-KO mutant was extracted and cleaved with BamHI restriction enzyme. After agarose gel electrophoresis, Southern blot analysis was performed with a probe specific for the BFRF1 region and for the TR region. As expected, the BamHI F fragment showed a shift due to the deletion in the BFRF1 gene. In contrast, the TR fragment was unchanged, confirming that passaging in E. coli did not reduce the number of repeats within the virus.
Establishment of a BFRF1-KO producer cell line.
BFRF1 mutant EBV plasmid DNA was then transfected into 293 cells, which are permissive for EBV replication, and subjected to hygromycin selection. Hygromycin-resistant GFP-positive clones, carrying the recombinant EBV genome, were further expanded. Genomic DNA from selected hygromycin-resistant clones was extracted, and the integrity of the BFRF1 locus was analyzed by a Southern blot assay with a 618-bp probe spanning the BFRF1 ORF (Fig. 1C). Both the recombined BamHI F fragment of the recombinant virus DNA and the wild-type BamHI F were found to have the expected size. A nonspecific band of approximately 5.5 kb is visible and probably represents a degradation product. In addition, we wanted to ensure that the recombinant BFRF1-KO carried an appropriate number of TRs. This was confirmed by Southern blot analysis with a TR-specific probe (Fig. 1C).
To further confirm the structural integrity of the BFRF1-KO present in the selected 293 clones, plasmid DNA was extracted and used to transform DH10B cells (21). Restriction enzyme analysis confirmed that the 293 clones carried the intact BFRF1-KO recombinant genome (data not shown).
Deletion of the BFRF1 gene does not interfere with expression of other viral genes.
In order to test the ability of the 293-BFRF1-KO clones to support the lytic cycle of EBV, cells were transiently transfected with a plasmid to obtain the heterologous expression of ZEBRA alone or in combination with BFRF1. At 3 days posttransfection, the expression of some important viral antigens was assessed by immunostaining. As expected for the BFRF1-KO mutant, immunostaining performed with anti-BFRF1 monoclonal antibody E7 (12) on induced 293-BFRF1-KO cells did not yield any signal (Fig. 2b), whereas BFRF1 expression was restored when BFRF1 was complemented in trans (Fig. 2c). Transfection of BZLF1 alone led to a clear expression of the immediate-early antigen Rta (Fig. 2d to f) as well as of the late proteins gp350/220 (BLLF1) (Fig. 2g to i) and BLRF2 (Fig. 2j to l) in the induced 293-BFRF1-KO cells. In these experiments, induced wild-type EBV (293-2089 cells) or complemented 293-BFRF1-KO cells provided the appropriate positive controls. Similar results were obtained in Western blot analysis (data not shown) performed on transiently transfected 293-BFRF1-KO cells. In addition, a Gardella assay performed to investigate whether the deletion of BFRF1 might affect viral replication in the recombinant EBV excluded the possibility that the lytic program was inhibited in 293-BFRF1-KO cells (Fig. 3). These experiments provide evidence that BFRF1 is not essential for the expression of early and late viral genes and does not interfere with the lytic replication program of viral DNA.
FIG. 2.

Expression of early and late proteins in lytically induced 293-BFRF1-KO cell lines. The results of an immunofluorescence assay performed on lytically induced 293-2089, 293-BFRF1-KO, and BFRF1-complemented 293-BFRF1-KO cell lines are shown. Lytically induced 293-BFRF1-KO cells do not show any expression of BFRF1 (b) compared to the wild-type 293-2089 cells or to the BFRF1-complemented 293BFRF1-KO cells (a and c, respectively), while the expression of the immediate-early antigen Rta (e), as well as of the late antigens BLLF1 (h) and BLRF2 (k), is still maintained in the 293-BFRF1-KO cell line at a level comparable to that in the wild-type or the BFRF1 complemented 293-BFRF1-KO cells.
FIG. 3.
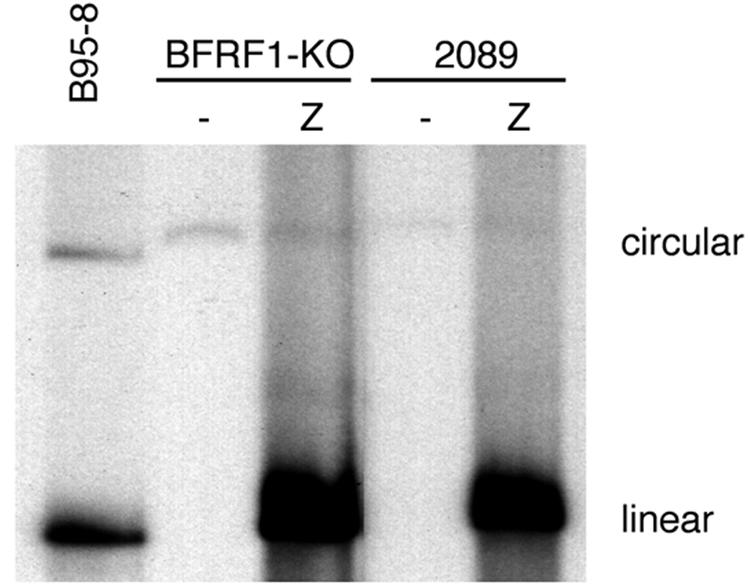
Gardella gel analysis of unit-length EBV DNA in 293-BFRF1-KO cells. Linear forms of the virus are detected in 293-BFRF1-KO cells upon induction of the lytic cycle as well as in 293-2089 producing cells that were used as a positive control. This experiment shows that BFRF1 deletion does not affect lytic viral DNA replication.
Supernatant from 293-BFRF1-KO cells has drastically decreased infection efficiency.
We then investigated the ability of the BFRF1-KO virus to infect a panel of cell lines, including B cells such as Raji and 2A8; epithelial cells such as HeLa, HaCaT, and 293; and primary human cord blood B lymphocytes. At 3 days postinfection, GFP-positive Raji cells could be observed after incubation with wild-type virus (Fig. 4a) or with supernatant of BFRF1-complemented induced 293-BFRF1-KO cells (Fig. 4c), but the infection efficiency was drastically reduced when Raji cells were incubated with the recombinant virus (Fig. 4b). GFP-positive 293 or 2A8 cells were observed exclusively when wild-type virus stocks or BFRF1-complemented recombinant EBV was used (Fig. 4d, f, g, and i), whereas no positive cells could be detected after incubation with supernatant of induced 293-BFRF1-KO cells (Fig. 4e and h). The percentage of positive cells resulting from these infection experiments was further evaluated by FACS analysis, and the results are given in Table 1.
FIG. 4.

Infection of different cell types with supernatants from induced 293 cells containing wild-type EBV (2089+Z), BFRF1-KO (KO+Z), or complemented BFRF1-KO (KO+Z+F), as detected by GFP expression with an inverted epifluorescence microscope.
TABLE 1.
Infection of different cell lines with wild-type EBV, BFRF1-negative EBV, and BFRF1-complemented EBV
| Cell line | Mean % (range) positive fora:
|
||
|---|---|---|---|
| Wild-type EBV | BFRF1-negative EBV | BFRF1-complemented EBV | |
| Raji | 27.9 (22.4-33.4) | 3.7 (3.3-4.1) | 31.18 (27.5-34.86) |
| 293 | 17.96 (16.4-19.52) | 0 | 20.79 (18.62-22.96) |
| HaCat | 0 | 0 | 0 |
| HeLa | 0 | 0 | 0 |
| 2A8 | 14.36 (12.51-16.21) | 0 | 10.9 (9.8-12) |
Percentages of GFP-positive cells observed after incubation with different viral supernatants and FACS analysis. The values represent means from three independent infection experiments.
The assumption that supernatants from 293-BFRF1-KO cells had reduced virus titers was confirmed in transformation assays with human cord blood B lymphocytes. As shown in Table 2, the immortalization efficiency was 100% with wild-type EBV, dropped drastically to 4.6% with the supernatant containing BFRF1-negative EBV, and was restored to 93.7% with BFRF1-complemented EBV. DNA from immortalized B-cell clones that were established after infection with wild-type EBV (Fig. 5, lanes 1, 2, and 3), with complemented BFRF1-KO EBV (lanes 4, 5, and 6), or with BFRF1-KO EBV (lanes 7, 8, and 9) and control DNA from B95-8 cells (EBV positive) or from DG75 cells (EBV negative) were amplified by PCR with BFRF1-specific sequences. The BFRF1 primers are located outside the deletion introduced in the mutant, and as a result, wild-type virus and BFRF1-KO mutants generate BFRF1 amplification products of different sizes. PCR amplification of the EBER-1 gene was performed as a control for the presence of EBV in the immortalized clones (Fig. 5, lower panel). As expected, amplification of DNAs obtained from all immortalized clones with both set of primers showed the presence of EBV-specific sequences. PCR amplification with DNA from B-cell lines established with virus stock containing wild-type EBV (Fig. 5, upper panel, lanes 1 to 3) showed the predicted 1,063-bp BFRF1 amplification product. In contrast, clones generated with the BFRF1-KO EBV showed a truncated BFRF1-specific amplification product, confirming that they indeed carry this mutant virus (Fig. 5, upper panel, lanes 4 to 9).
TABLE 2.
Transformation frequencies of umbilical cord blood lymphocytes after infection with EBV supernatants
| EBV supernatant | No. of positive wells/no. of wells plated | Immortalization efficiency (%) |
|---|---|---|
| Wild type | 64/64 | 100 |
| BFRF1 negative | 3/64 | 4.68 |
| BFRF1 complemented | 60/64 | 93.7 |
FIG. 5.

(Upper panel) PCR analysis for the BFRF1 gene of DNA from immortalized B-cell clones that were established after infection with wild-type EBV (lanes 1 to 3), with complemented BFRF1-KO EBV (lanes 4 to 6), or with BFRF1-KO EBV (lanes 7 to 9) and of control DNA from B95-8 cell lines (+) or from DG75 cell lines (−). (Lower panel) Control PCR analysis of the EBER-1 region.
Viral maturation is blocked in the absence of BFRF1.
The markedly reduced capacity of supernatants from 293-BFRF1-KO cells to infect various cell types could be due either to a reduction in virus titers or to production of virus with limited infectious potential. To test these hypotheses, we performed a binding assay on Raji cells to monitor the amount of virus bound from the different supernatants. The results of viral binding are shown in Table 3, and immunofluorescence images showing fluorescence dots possibly corresponding to virions, or clusters on virions (7), bound on the plasma membranes of Raji cells are shown in Fig. 6. In comparison to the case for wild-type virus, both the percentage of cells with virus bound on the cell surface and the number of fluorescent dots per cell were markedly reduced after incubation with supernatants from the 293-BFRF1-KO cells. Supernatants from complemented 293-BFRF1-KO cells had binding levels similar to those for wild-type supernatant. These results suggest reduced virus production in 293-BFRF1-KO cells.
TABLE 3.
Binding of EBV supernatants on Raji cells
| Virus supernatant | % of cells with bound virus | No. of fluorescent dots/cell (mean ± SD)a |
|---|---|---|
| B95-8 | 48.5 | 10.1 ± 2.9 |
| p2089+Z | 23 | 8.9 ± 2.7 |
| BFRF1-negative EBV | 4 | 2.6 ± 1.8* |
| BFRF1-complemented EBV | 25 | 9.2 ± 2.9** |
*versus **; P < 0.001 (Student's t test).
FIG. 6.

Virus binding on the surface of Raji cells as detected by indirect immunofluorescence staining of the major envelope glycoprotein gp350/220. (a) EBV from supernatant of B95-8 cells; (b) supernatant of induced 293-2089 cells; (c) supernatant of 293-BFRF1-KO cells; (d) supernatant of BFRF1-complemented 293-BFRF1-KO cells. Bar, 10 μm.
To further assess the validity of this hypothesis, we directly visualized viral replication in 293 cells carrying BFRF1-KO virus or BFRF1-complemented BFRF1-KO virus by using electron microscopy. Lytically induced 293-BFRF1-KO cells contained numerous fully assembled, DNA-containing EBV nucleocapsids in the nucleus (Fig. 7, left and center micrographs) and virtually no virions in the cytoplasm. In contrast, BFRF1-complemented BFRF1-KO virions were readily visible in the cytoplasm and in the extracellular environment (Fig. 7, right micrograph). In addition, only a few complemented nucleocapsids were present in the nucleus. We then further searched for the presence of nuclear membrane duplications, as they represent a typical if not specific ultrastructural feature of active EBV lytic replication (10, 51). Whereas nuclear membrane duplications were common in replicating complemented 293-BFRF1-KO cells, duplications were very rarely or never observed in cells carrying the mutant virus (Fig. 7). The results of these ultrastructural observations in comparison to those of 293 cells carrying wild-type p2089 virus are summarized in Table 4. Taken together, these observations are consistent with a block in primary egress with subsequent sequestration of immature viruses in the nucleus.
FIG. 7.

Ultrastructural observations of induced 293-BFRF1-KO cells and 293-BFRF1-KO cells complemented with BFRF1. Left and center micrographs: numerous nucleocapsids are present in the nuclei of induced BFRF1-KO cells and are distributed mainly in the proximity of the nuclear membrane (arrows). Insets show an higher magnification of fully assembled intranuclear nucleocapsids containing electron-dense material corresponding to viral DNA. Right micrograph: few nucleocapsids (arrows) are visible in BFRF1-complemented cells. A typical multilayered reduplication of the nuclear membrane (NM) is shown. An extracellular mature virion is shown at higher magnification in the inset. PM, plasma membrane; Nu, nucleus. Bars, 1 μm (0.1 μm in the insets).
TABLE 4.
Quantitation of ultrastructural modifications in 293 cells carrying wild-type virus (p2089), in 293-BFRF1 KO cells, and in BFRF1-complemented cellsa
| Cells | No. of nucleocapsids/10μm2 (nuclear area) | No. of virions/10μm2 (cytoplasmic area) | % Length of duplicated nuclear membrane/length of nuclear membrane |
|---|---|---|---|
| BFRF1-KO+Z | 3.72 ± 1.74* | 0.03 ± 0.03† | 3.14 ± 2.02‡ |
| BFRF1-KO+Z+F | 0.21 ± 0.20** | 0.23 ± 0.15†† | 19.4 ± 4.38‡‡ |
| 2089+Z | 0.29 ± 0.21*** | 0.16 ± 0.07††† | 6.53 ± 1.88‡‡‡ |
For each transfectant, 25 cell sections of cells positive for the presence of nucleocapsids in the nucleus were recorded with a charge-coupled device camera. A total of 1,947.76 μm2 of nuclear area, 2,051.1 μm2 of cytoplasmic area, and 1,281.62 μm of nuclear membrane length were measured and analyzed. Results are means ± standard deviations. * versus **, P < 0.001; * versus ***, P < 0.001; ** versus ***, P not significant; † versus ††, P < 0.05; † versus †††, P < 0.05; †† versus †††, not significant; ‡ versus ‡‡, P < 0.0001; ‡ versus ‡‡‡, P < 0.001; ‡‡ versus ‡‡‡, not significant.
The presence of BFRF1 in virions correlates with the viral maturation stages.
The results presented so far suggest a role for BFRF1 in primary envelopment at the nuclear membrane. We next analyzed, by Western blotting, at which stages of viral maturation BFRF1 could be detected. To this end, cell-free B95-8 viruses from infectious supernatants or intracellular B95-8 viruses obtained from the producing cell line were purified, and protein extracts were analyzed by Western blotting. Figure 8 shows that, as expected, BFRF1 is present in intracellular virions and that no reactivity for BFRF1 was detectable in extracellular virions with a monoclonal antibody specific for BFRF1 (E7), whereas a strong positive signal was visible after immunoblotting with a monoclonal antibody directed against the known virion component BRLF2, confirming the presence of virions in the concentrated supernatant. This finding confirms the results of previous immunogold staining with an antibody specific to BFRF1 combined with electron microscopy, which showed that BFRF1 is present in intracellular virions but is not a component of the mature virion (13). These observations suggest that the de-envelopment and reenvelopment model of herpesviral maturation might apply also to EBV, as initially proposed by Gong and Kieff (17).
FIG. 8.

Western immunoblot analysis performed on purified virions. Fractions containing extracellular or intracellular purified virions were probed with antibodies directed against BFRF1 and BLRF2 gene products. An immunoblot of uninduced (−) and TPA-induced (+) B95-8 cell extracts is shown as a control.
DISCUSSION
In the present study, we have generated a recombinant EBV strain devoid of the BFRF1 gene in order to understand the function of the protein encoded by this gene in EBV biology. We have analyzed the phenotype of this mutant in terms of lytic replication, infection, and immortalization. The results of this analysis provide strong evidence for a role of BFRF1 in primary egress when the maturing virus particle first becomes enveloped by crossing the nuclear membrane. This assertion is based on the observed reduction in virus titers as assessed by binding assays and on the observation that mutant virions accumulate in the nucleus. In contrast, BFRF1 does not seem to affect lytic DNA replication or nucleocapsid assembly. Similarly, BFRF1 had no influence on the expression of a few late structural proteins. However, since the full cascade of lytic viral genes is far from being completely known, we cannot formally exclude the possibility that the lack of BFRF1 may affect, directly or indirectly, the expression of other viral genes. It is important to note, however, that virus production was not entirely inhibited, an observation that suggests that even if BFRF1 greatly facilitates transport across the nuclear membrane, it is not absolutely essential for this process. This observation may suggest that other viral proteins may partially share the ability to get virions through the nuclear membrane with BFRF1 (27, 29, 33, 37, 39). Alternatively, it is also possible that immature virions could reach the cell cytoplasm by trafficking through defects of the nuclear membrane. Once the viral particles reach the cytoplasm, the events that lead to viral maturation could take place, with subsequent release of the virus into the extracellular environment. Our Western blotting analysis of the expression of BFRF1 in virions at different maturation steps shows that the protein is present in intracellular virions, suggesting that BFRF1 is involved in the nuclear membrane enveloping process, whereas it is absent from the mature extracellular virion at a step when BFRF1 is apparently no longer required. These observations are in line with our previous work in which we could show that BFRF1 protein localizes preferentially on the nuclear membrane (13) and that it is an integral protein that is anchored in membranes through a transmembrane domain. The apparent discrepancy with our initial observation that BFRF1 is a virion component (12) could be explained by the fact that in that study we used virions prepared from repeatedly frozen and thawed cells, and we cannot exclude cellular contamination. The present results, based on purified virus concentrated from infected-cell supernatants, unequivocally demonstrate that BFRF1 is absent from extracellular virions.
BFRF1 appears to be conserved among the herpesvirus family, as BFRF1 shows significant DNA sequence homology with UL34 from HSV-1 and PrV. BFRF1 is also the positional homolog of UL34 in the EBV genome. The data gathered in this study about the role of the BFRF1 protein during EBV virus production indicate that this homology extends to the functional level. UL34 from both HSV-1 and PrV has been shown to be essential for primary virus egress from the nucleus, in which the capsids acquire a first envelope which is subsequently lost in the cytoplasm (15, 20, 26, 34, 35, 42-44, 47). A tegument is then organized around the nucleocapsids, and finally a second envelope is acquired during the steps that lead to the release of extracellular mature virions (reviewed in reference 34). It therefore appears that the molecular mechanisms that underlie virus maturation are conserved among several members of different herpesvirus subfamilies. A recent report described the effect of HSV-1 UL34 and UL31 gene products on the nuclear lamina and on the host cell chromatin (48). Similarly to HSV-1 UL34, BFRF1 interacts with nuclear lamina components, as reported in the accompanying paper (18). However, our ultrastructural analysis did not reveal differences in chromatin distribution in the presence or absence of BFRF1, suggesting that, in contrast to UL34, BFRF1 does not play an essential role in chromatin exclusion from envelopment sites.
It has been suggested that UL34, in cooperation with UL31 and possibly other proteins, permits the anchorage of the viral capsids to the inner nuclear membrane. This somehow facilitates the envelopment at the nuclear membrane and allows the egress of the viral particles, which otherwise are too large to freely leave the nucleus through the nuclear pori (43, 54). The identification of viral proteins interacting with UL34, as well as the study of their possible cellular partners in the nuclear membrane components, would further help our understanding of this complex process (35, 46). The search for additional viral genes involved in virus maturation will be facilitated by the observation that these genes appear to cluster together in a particular segment of all herpesvirus genomes (15, 26, 34, 42-44, 47, 54). EBV BFLF2 is the positional homolog of HSV-1 UL31 and is therefore likely to interact with BFRF1 during viral envelopment (28). Our results on characterization of BFLF2 and on its interactions with BFRF1 and with the nuclear lamina in infected cells are presented in the accompanying paper (18).
Acknowledgments
This work was partially supported by grants from the MIUR, Ministero della Sanità, Progetto AIDS, Associazione Italiana di ricerca sul Cancro (AIRC), and Istituto-Cenci-Bolognetti Foundation.
Giuseppe Lucania is gratefully acknowledged for photographic work. We thank J. Sixbey for the gift of 2A8 cells and G. Miller for the anti-BLRF2 antibody.
REFERENCES
- 1.Angeloni, A., A. Farina, G. Gentile, A. Capobianchi, P. Martino, V. Visco, R. Muraro, L. Frati, and A. Faggioni. 2001. Epstein-Barr virus and breast cancer: search for antibodies to the novel BFRF1 protein in sera of breast cancer patients. J. Natl. Cancer Inst. 93:560-561. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Bassat, H., N. Goldblum, S. Mitrani, T. Goldblum, J. M. Yoffey, M. M. Cohen, Z. Bentwich, B. Ramot, E. Klein, and G. Klein. 1977. Establishment in continuous culture of a new type of lymphocyte from a “Burkitt like” malignant lymphoma (line D.G.-75). Int. J. Cancer 19:27-33. [DOI] [PubMed] [Google Scholar]
- 3.Boukamp, P., R. T. Petrussevska, D. Breitkreutz, J. Hornung, A. Markham, and N. E. Fusenig. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 106:761-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cherepanov, P. P., and W. Wackernagel. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9-14. [DOI] [PubMed] [Google Scholar]
- 5.Cho, M. S., G. Milman, and S. D. Hayward. 1985. A second Epstein-Barr virus early antigen gene in BamHI fragment M encodes a 48- to 50-kilodalton nuclear protein. J. Virol. 56:860-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chodosh, J., V. P. Holder, Y. J. Gan, A. Belgaumi, J. Sample, and J. W. Sixbey. 1998. Eradication of latent Epstein-Barr virus by hydroxyurea alters the growth-transformed cell phenotype. J. Infect. Dis. 177:1194-1201. [DOI] [PubMed] [Google Scholar]
- 7.Cirone, M., A. Angeloni, G. Barile, C. Zompetta, M. Venanzoni, M. R. Torrisi, L. Frati, and A. Faggioni. 1990. Epstein-Barr virus internalization and infectivity are blocked by selective protein kinase C inhibitors. Int. J. Cancer 45:490-493. [DOI] [PubMed] [Google Scholar]
- 8.Decaussin, G., V. Leclerc, and T. Ooka. 1995. The lytic cycle of Epstein-Barr virus in the nonproducer Raji line can be rescued by the expression of a 135-kilodalton protein encoded by the BALF2 open reading frame. J. Virol. 69:7309-7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delecluse, H. J., T. Hilsendegen, D. Pich, R. Zeidler, and W. Hammerschmidt. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 95:8245-8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Epstein, M. A., and B. C. Achong. 1979. Morphology of the virus and virus-induced cytopathologic changes, p. 23-27. In M. A. Epstein and B. G. Achong (ed.), The Epstein-Barr virus. Springer Verlag, New York, N.Y.
- 11.Faggioni, A., C. Zompetta, S. Grimaldi, G. Barile, L. Frati, and J. Lazdins. 1986. Calcium modulation activates Epstein-Barr virus genome in latently infected cells. Science 232:1554-1556. [DOI] [PubMed] [Google Scholar]
- 12.Farina, A., R. Santarelli, R. Gonnella, R. Bei, R. Muraro, G. Cardinali, S. Uccini, G. Ragona, L. Frati, A. Faggioni, and A. Angeloni. 2000. The BFRF1 gene of Epstein-Barr virus encodes a novel protein. J. Virol. 74:3235-3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farina, A., G. Cardinali, R. Santarelli, R. Gonnella, J. Webster-Cyriaque, R. Bei, R. Muraro, L. Frati, A. Angeloni, M. R. Torrisi, and A. Faggioni. 2004. Intracellular localization of the Epstein-Barr virus BFRF1 gene product in lymphoid cell lines and oral hairy leukoplakia lesions. J. Med. Virol. 72:102-111. [DOI] [PubMed] [Google Scholar]
- 14.Feederle, R., M. Kost, M. Baumann, A. Janz, E. Drouet, W. Hammerschmidt, and H. J. Delecluse. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 19:3080-3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuchs, W., B. G. Klupp, H. Granzow, N. Osterreider, and T. C. Mettenleiter. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary envelopes but not of mature virions. J. Virol. 71:364-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gardella, T., P. Medveczky, T. Sairenji, and C. Mulder. 1984. Detection of circular and linear herpesvirus DNA molecules in mammalian cells by gel electrophoresis. J. Virol. 50:248-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong, M., and E. Kieff. 1990. Intracellular trafficking of two major Epstein-Barr virus glycoproteins, gp 350/220 and gp110. J. Virol. 64:1507-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonnella, R., A. Farina, R. Santarelli, S. Raffa, R. Feederle, R. Bei, M. Granato, A. Modesti, L. Frati, H.-J. Delecluse, M. R. Torrisi, A. Angeloni, and A. Faggioni. 2005. Characterization and intracellular localization of the Epstein-Barr virus protein BFLF2: interactions with BFRF1 and with the nuclear lamina. J. Virol. 79:3713-3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham, F. L., W. C. Smiley, W. Russell, and R. Nairn. 1977. Characteristics of human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59-74. [DOI] [PubMed] [Google Scholar]
- 20.Granzow, H., F. Weiland, A. Jons, B. G. Klupp, A. Karger, and T. C. Mettenleiter. 1997. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: a reassessment. J. Virol. 71:2072-2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffin, B. E., E Bijorck, G. Bjursell, and T. Lindhal. 1981. Sequence complexity of circular Epstein-Bar virus DNA in transformed cells. J. Virol. 40:11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 23.Janz, A., M. Oezel, C. Kurzeder, J. Mautner, D. Pich, M. Kost, W. Hammerschmidt, and H. J. Delecluse. 2000. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 74:10142-10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenney, S., E. Holley-Guthrie, E. C. Mar, and M. Smith. 1989. The Epstein-Barr virus BMLF1 promoter contains an enhancer element that is responsive to the BZLF1 and BRLF1 transactivators. J. Virol. 63:3878-3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kieff, E. 1996. Epstein-Barr virus and its replication, p. 1109-1161. In B. N. Fields and D. Knipe (ed.), Fundamental virology. Raven Press, New York, N.Y.
- 26.Klupp, B. G., H. Granzow, and T. C. Mettenleiter. 2000. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 74:10063-10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lake, C. M., and L. M. Hutt-Fletcher. 2000. Epstein-Barr virus that lacks glycoprotein gN is impaired in assembly and infection. J. Virol. 74:11162-11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lake, C. M., and L. M. Hutt-Fletcher. 2004. The Epstein-Barr virus BFRF1 and BFLF2 proteins interact and coexpression alters their cellular localization. Virology 320:99-106. [DOI] [PubMed] [Google Scholar]
- 29.Lee, S. K., and R. Longnecker. 1997. The Epstein-Barr virus glycoprotein 110 carboxy-terminal tail domain is essential for lytic virus replication. J. Virol. 71:4092-4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin, J. C., J. E. Shaw, M. C. Smith, and J. S. Pagano. 1979. Effect of 12-O-tetradecanoyl-phorbol-13-acetate on the replication of Epstein-Barr virus. I. Characterization of viral DNA. Virology 99:183-187. [DOI] [PubMed] [Google Scholar]
- 31.Lin, J. C., N. D. Sista, F. Besencon, J. Kamine, and J. S. Pagano. 1991. Identification and functional characterization of Epstein-Barr virus DNA polymerase by in vitro transcription-translation of a cloned gene. J. Virol. 65:2728-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luka, J., B. Kallin, and G. Klein. 1979. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 94:228-231. [DOI] [PubMed] [Google Scholar]
- 33.Mackett, M., M. J. Conway, J. R. Arrand, R. S. Haddad, and L. M. Hutt-Fletcher. 1990. Characterization and expression of a glycoprotein encoded by the Epstein-Barr virus BamHI 1 fragment. J. Virol. 64:2545-2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mettenleiter, T. C. 2002. Herpesvirus assembly and egress. J. Virol. 76:1537-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muranyi, W., J. Haas, M. Wagner, G. Krohne, and U. H. Koszinowski. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854-857. [DOI] [PubMed] [Google Scholar]
- 36.Neubauer, A., J. Rudolph, C. Brandmuller, F. T. Just, and N. Osterrieder. 2002. The equine herpesvirus 1 UL34 gene product is involved in an early step in virus egress and can be efficiently replaced by a UL34-GFP fusion protein. Virology 300:189-204. [DOI] [PubMed] [Google Scholar]
- 37.Neuhierl, B., R. Feederle, W. Hammerschmidt, and H. J. Delecluse. 2002. Glycoprotein gp 110 of Epstein-Barr virus determines viral tropism and efficiency of infection. Proc. Natl. Acad. Sci. USA 99:15036-15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearson, G. R., J. Luka, L. Petti, J. Sample, M. Birkenbach, D. Braun, and E. Kieff. 1987. Identification of an Epstein-Barr virus early gene encoding a second component of the restricted early antigen complex. Virology 160:151-161. [DOI] [PubMed] [Google Scholar]
- 39.Penaranda, M. E., L. A. Lagenaur, L. T. Pierek, J. W. Berline, L. A. MacPhail, D. Greenspan, J. Greenspan, and J. M. Palefsky. 1997. Expression of Epstein-Barr virus BMFR-2 and BDLF-3 genes in hairy leukoplakia. J. Gen. Virol. 78:3361-3370. [DOI] [PubMed] [Google Scholar]
- 40.Pulvertaft, J. V. 1964. Cytology of Burkitt tumor (African lymphoma). Lancet i:238-240. [DOI] [PubMed] [Google Scholar]
- 41.Ragoczy, T., and G. Miller. 1999. Role of the Epstein-Barr virus RTA protein in activation of distinct classes of viral lytic cycle genes. J. Virol. 73:9858-9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reynolds, A. E., B. J. Ryckman, J. D. Baines, Y. Zhou, L. Liang, and R. J. Roller. 2001. UL31 and UL34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75:8803-8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reynolds, A. E., E. G. Wills, R. J. Roller, B. J. Ryckman, and J. D. Baines. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939-8952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roller, R., Y. Zhou, R. Schnetzer, J. Ferguson, and D. DeSalvo. 2000. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J. Virol. 74:117-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sanchez, V., and D. H. Spector. 2002. CMV makes a timely exit. Science 297:778-779. [DOI] [PubMed] [Google Scholar]
- 46.Scott, E. S., and P. O'Hare. 2001. Fate of the inner nuclear membrane protein lamin B receptor and nuclear lamins in herpes simplex virus type 1 infection. J. Virol. 75:8818-8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shiba, C., T. Daikoku, F. Goshima, H. Takakuwa, Y. Yamauchi, O. Koiwai, and Y. Nishiyama. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for viral envelopment. J. Gen. Virol. 81:2397-2405. [DOI] [PubMed] [Google Scholar]
- 48.Simpson-Holley, M., J. Baines, R. Roller, and D. M. Knipe. 2004. Herpes simplex virus UL31 and UL34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J. Virol. 78:5591-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takada, K. 1984. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int. J. Cancer 33:27-32. [DOI] [PubMed] [Google Scholar]
- 50.Tanner, J., J. Weis, D. Fearon, Y. Whang, and E. Kieff. 1987. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 50:203-213. [DOI] [PubMed] [Google Scholar]
- 51.Torrisi, M. R., M. Cirone, A. Pavan, C. Zompetta, G. Barile, L. Frati, and A. Faggioni. 1989. Localization of Epstein-Barr virus envelope glycoproteins on the inner nuclear membrane of virus-producing cells. J. Virol. 63:828-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trivedi, P., K. Takazawa C. Zompetta, L. Cuomo, E. Anastasiadou, A. Carbone, S. Uccini, F. Belardelli, K. Takada, L. Frati, and A. Faggioni. 2004. Infection of HHV-8+ primary effusion lymphoma cells with a recombinant Epstein-Barr virus leads to restricted EBV latency, altered phenotype, and increased tumorigenicity without affecting TCL1 expression. Blood 103:313-316. [DOI] [PubMed] [Google Scholar]
- 53.Yamauchi, Y., C. Shiba, F. Goshima, A. Nawa, T. Murata, and Y. Nishiyama. 2001. Herpes simplex virus type 2 UL34 protein requires UL31 protein for its relocation to the internal nuclear membrane in transfected cells. J. Gen. Virol. 82:1423-1428. [DOI] [PubMed] [Google Scholar]
- 54.Ye, G. J., K. T. Vaughan, R. B. Vallee, and B. Roizman. 2000. The herpes simplex virus 1 UL34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 74:1355-1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.zur Hausen, H., F. J. O'Neill, U. K. Freese, and E. Hecker. 1978. Persisting oncogenic herpesvirus induced by the tumour promotor TPA. Nature 272:373-375. [DOI] [PubMed] [Google Scholar]