

Abstract
Background:
Otosclerosis is a common cause of adult-onset progressive hearing loss, affecting 0.3-0.4% of the population. It results from dysregulation of bone homeostasis in the otic capsule, most commonly leading to fixation of the stapes bone, impairing sound conduction through the middle ear. Otosclerosis has a well-known genetic predisposition including familial cases with apparent autosomal dominant mode of inheritance. While linkage analysis and genome wide association studies suggested association with several genomic loci and with genes encoding structural proteins involved in bone formation or metabolism, the molecular genetic pathophysiology of human otosclerosis is yet mostly unknown.
Methods:
Whole exome sequencing, linkage analysis, generation of CRISPR mutant mice, hearing tests, micro-CT.
Results:
Through genetic studies of a kindred with seven individuals affected by apparent autosomal dominant otosclerosis, we identified a disease-causing variant in SMARCA4, encoding a key component of the PBAF chromatin remodeling complex. We generated CRISPR-Cas9 transgenic mice carrying the human mutation in the mouse SMARCA4 ortholog. Mutant Smarca4+/E1548K mice exhibited marked hearing impairment demonstrated through acoustic startle response and auditory brainstem response tests. Isolated ossicles of the auditory bullae of mutant mice exhibited highly irregular structure of the incus bone, and their in-situ micro-CT studies demonstrated anomalous structure of the incus bone, causing disruption in the ossicular chain.
Conclusion:
We demonstrate that otosclerosis can be caused by a variant in SMARCA4, with a similar phenotype of hearing impairment and abnormal bone formation in the auditory bullae in transgenic mice carrying the human mutation in the mouse SMARCA4 ortholog.
Introduction
Otosclerosis is a common cause of adult-onset progressive hearing loss that affects 0.3-0.4% of the population[1]. The age at disease onset varies greatly, ranging between 5-55 years, with peak incidence between 15-30 years[2]. It is a disorder of bone homeostasis in the otic capsule, first documented more than 300 years ago[3]. The otic capsule, located in the temporal bone, is formed by endochondral ossification. Its formation is normally completed by one year of age, with very little further active bone remodeling throughout life. In fact, under normal conditions, osteoblast and osteoclast activity of bone turnover is almost entirely absent in the mature otic capsule[4]. In otosclerosis, through a poorly understood mechanism, osteoblasts and osteoclasts are activated and initiate abnormal bone remodeling: endochondral bone in the otic capsule is reabsorbed by osteoclasts, followed by deposition of new dense mineralized bone by osteoblasts, ultimately resulting in otosclerotic foci[5]. As these foci of poorly organized bone expand, the normal boundaries of the otic capsule gradually protrude into neighboring structures. The most common location of otosclerotic lesions in the otic capsule is the anterior part of the oval window[6,7], which upon expansion causes fixation of the adjacent stapedial footplate, impairing free motion of the stapes bone and culminating in progressive conductive hearing loss[8]. While the vast majority of otosclerosis patients present conductive hearing loss, in up to 10% of cases otosclerotic foci involve the cochlear endosteum that may also manifest as sensorineural or mixed hearing loss[9].
Genetic predisposition to otosclerosis is well-documented, characterized by an autosomal dominant mode of inheritance with incomplete penetrance, estimated at 40%[10,11]. Thus far, linkage analysis studies of families with otosclerosis identified eight disease-associated genomic loci[12–18], and genetic studies delineated polymorphisms and/or variants in several genes likely associated with the disease, including genes encoding structural proteins involved in bone formation[19,20] or proteins with regulatory roles in bone metabolism[21–25], and others[26,27] We now demonstrate that a mutation in SMARCA4, encoding a key component of the SWI/SNF chromatin remodeling complex, causes otosclerosis in humans and a similar phenotype in mice.
Results
Clinical Characterization
Seven individuals spanning two generations of a single family presented with otosclerosis (Fig. 1A). The pedigree was consistent with an autosomal dominant mode of inheritance with incomplete penetrance. The proband (IV: 1) has progressive hearing loss that began in his early 30’s, with no symptoms of vertigo or imbalance. He was diagnosed with otosclerosis following bilateral stapedectomy with minimal improvement of hearing in his right ear and is currently using hearing aids in both ears. Recent otoscopic examination was unremarkable with normal external auditory canal and tympanic membrane on both sides. His nearly 90 years old mother (III:4) reported normal hearing, as did both his siblings, their children and his own four children. On his mother’s side, his grandparents also had normal hearing; however, it is noteworthy that his grandfather passed away before 40 years of age, possibly prior to the age of initiation of symptoms. Affected individual IV:2 is a second cousin of IV: 1. She described progressive hearing loss that started in her early childhood, requiring hearing aids in both ears in her early 20’s, with further deterioration throughout the years, particularly following each of her 3 pregnancies. She was diagnosed with bilateral otosclerosis by a cranial CT (Fig. 1B), demonstrating a fenestral focus of otosclerosis with decalcification in the region of the fissula ante fenestram in the left temporal bone; there was no involvement of the round window and no evidence of pericochlear otosclerosis. Her recent otoscopic examination showed normal external auditory canals and tympanic membrane. Her father (III: 10) passed away before the age of 70 without any record of hearing loss, and this was also true for her paternal grandfather and grandmother who passed away at ages near 70 and 80 (respectively) and her 3 daughters. Patients from generation III were not available for clinical testing or questioning; all information regarding their condition was gathered through close relatives. Patient III: 1 had progressive hearing loss that started around the age of 30. She has undergone bilateral stapedectomy that was unsuccessful, and she currently has profound hearing loss. Patient III:2 had noticeable progressive hearing loss as of the age of near 30, first requiring hearing aids at nearly 40. Following diagnosis of otosclerosis he underwent stapedectomy that failed to improve his hearing. Patient III:5 started experiencing hearing loss at the age of near 65 and refused to participate in the study and provide further information. Patient III:6 reported noticing hearing deterioration near the age of 65, and is currently using hearing aids in both ears. Patient III:7 passed away during the course of the study. He suffered from progressive hearing loss that began in his late 20’s. Individual III:8 (present age above 70 years) reported normal hearing, though possibly started noticing slight deterioration in the last 3 years. Each of the individuals in generation III has 2-3 children between ages 40-57, all of whom report completely normal hearing. All affected individuals with hearing loss who participated in the study were otherwise healthy, with no chronic illnesses or any other significant health problems.
Figure 1. Pedigree and Cranial CT scan.
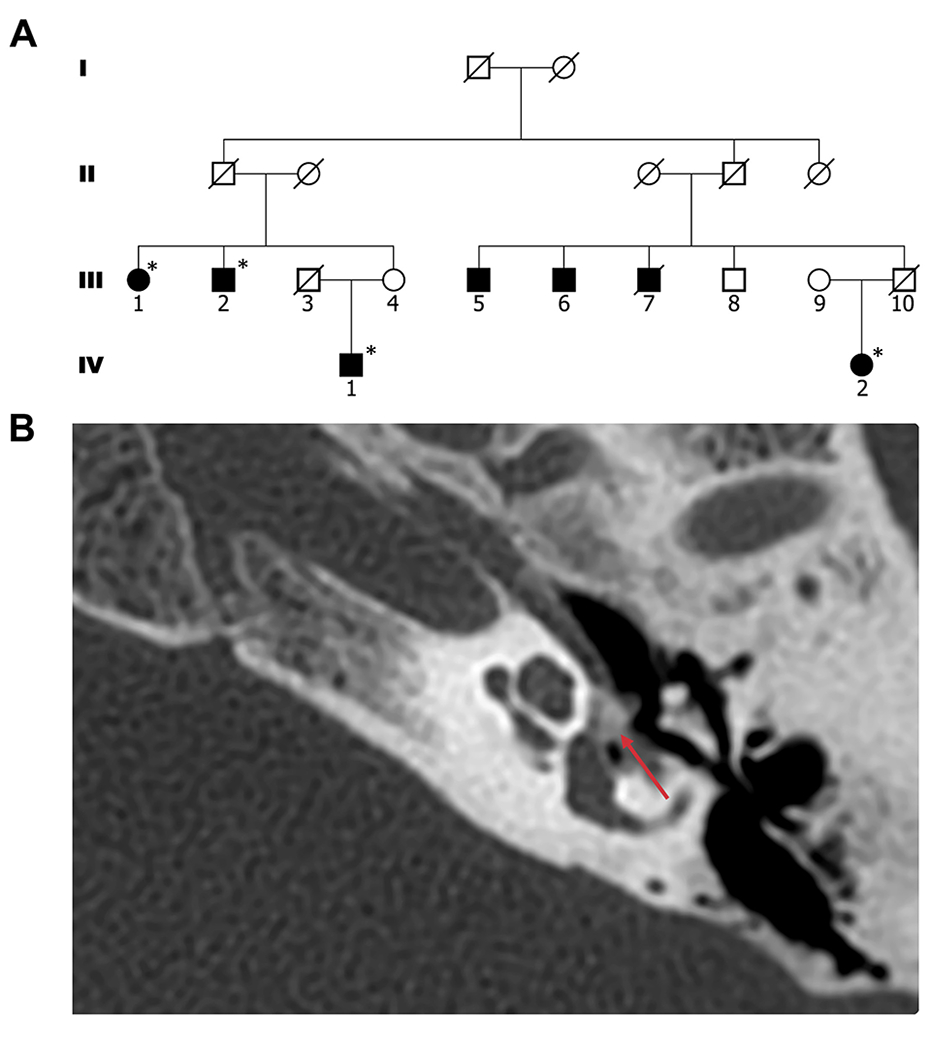
(A) The studied kindred: black-filled circles and squares denote affected individuals; * denotes affected individuals with confirmed otosclerosis diagnosed by surgery or computerized tomography; white circles and squares denote unaffected individuals. (B) Cranial CT scan of patient IV:2, showing the left temporal bone; red arrow indicates an otosclerosis focus.
Molecular genetic studies
To identify genomic loci shared between all affected individuals we performed genome wide linkage analysis. Due to the apparent incomplete penetrance of the phenotype, we only used confirmed cases (III: 1, III:2, III:6, III:7, IV:1, IV:2) in the analysis. A total of 7 heterozygous loci shared between all cases were identified (Table S1) with a maximal logarithm of odds [LOD] score of 2.3. Interestingly, none of these loci overlapped with any of the eight previously reported risk loci for otosclerosis. To identify potential disease-causing variants in these shared loci, whole-exome sequencing was performed for the two most distant relatives (IV: 1, IV:2). Following the variant filtration process (described in Methods), only 5 heterozygous variants common to both patients remained (Tables S2,3). We then further analyzed the remaining variants based on evolutionary conservation of the specific mutation site, phenotypes previously associated with the gene, tissue expression profile of the encoded protein, and the probability for loss of function intolerance characteristic of dominant disease-causing mutations using the Genome Aggregation Database (gnomAD). Following this analysis (detailed in Table S3), a single variant remained: c.4828G>A in SMARCA4 (transcript variant 1; NM_001128849.3), that was not previously reported in gnomAD. Sanger sequencing confirmed that all affected individuals were indeed heterozygous for the mutation (Fig. 2A). SMARCA4 encodes BRG1, the catalytic subunit of the PBAF complex, a member of the SWI/SNF chromatin remodeling family that regulates gene activation and repression through interaction with nucleosomes that assist in DNA packing (Fig. 2C).The mutation, altering a highly conserved Glutamate residue to Lysine in position 1610 of the protein (Fig. 2B), resides near the BROMO domain of BRG1, which mediates interaction with acetylated histones comprising nucleosomes[28](Fig. 2D).
Figure 2. Validation, conservation, and position of the SMARCA4 mutation.
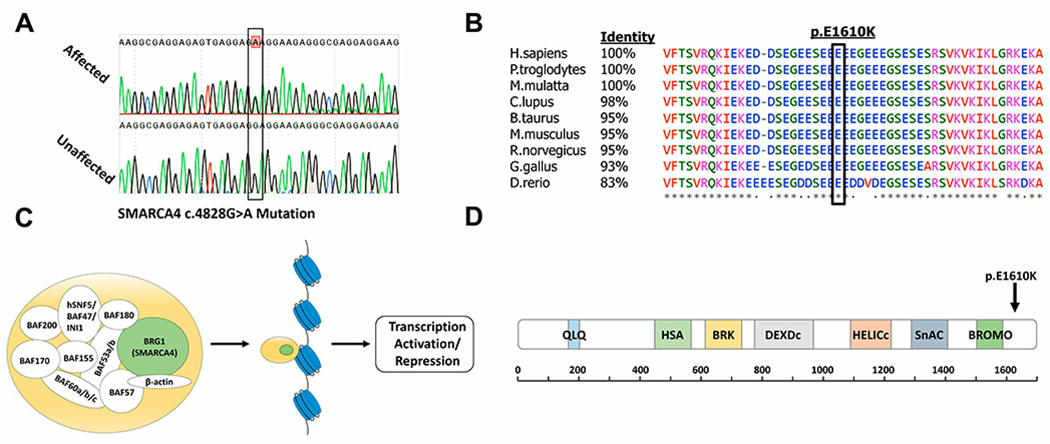
(A) Sanger sequencing results of an affected individual heterozygous for the SMARCA4 mutation and an unaffected individual with the wild-type allele. (B) Multiple sequence alignment of SMARCA4 orthologues with percentage of protein sequence identity between the human protein and several mammalian proteins. Black box marks the position of the Glutamate residue mutated to Lysine. Symbols below sequences correspond to degree of conservation: asterisk indicates identical; period indicates weakly similar; blank indicates no similarity. (C) Illustration of the various subunits composing the PBAF complex[28], a member of the human SWI/SNF family of chromatin remodelers. The catalytic subunit of the complex, BRG, is encoded by SMARCA4 (highlighted in green). (D) Schematic representation of BRG1 showing all conserved domains of the protein, with approximate corresponding positions indicated below; information obtained from SMART protein domain annotation resource[46]. Arrow marks the site of the altered amino acid in residue 1610, adjacent to the Bromodomain. Domain names: QLQ, a Gln, Leu, Gln motif; HSA, helicase/SANT-associated; BRK, brahma and kismet; DEXDc, DEAD-like helicases superfamily; HELICc, helicase superfamily c-terminal domain; SnAC, Snf2-ATP coupling, chromatin remodeling complex; BROMO, bromodomain.
Generation and analysis of CRISPR/Cas9 knock-in mice
SMARCA4 is highly conserved throughout evolution. In particular, the mouse ortholog shows 95% protein sequence identity to its human counterpart, including the specific Glutamate residue altered by the c.4828G>A mutation. Therefore, to comprehensively study the effect of this mutation on hearing and determine its role in otosclerosis, we used CRISPR/Cas9, creating knock-in mice carrying the human p.E1610K substitution in the mouse ortholog (Table S4). Several F0 chimeras were generated. A male chimera was crossed with wild-type C57BL/6J females to generate true non-chimeric F1 heterozygous Smarca4+/E1548K mice. These mice were then cross-bred to generate F2 mice of all genotypes, which were then further cross-bred according to genotype to generate heterozygous F3 mutant (Smarca4+/E1548K) and wild-type (Smarca4+/+) mice of the same genetic origin, that were used in all subsequent experiments. The mutant mice did not exhibit any overt morphological or behavioral abnormalities compared to their wild-type littermates. Furthermore, whole body X-ray scans excluded any gross anatomical anomalies in these mice (Fig. 3) and no abnormalities were found in a comprehensive histopathology analysis of tissues of the mutant mice (results not shown).
Figure 3. Mouse whole body x-ray images.
Anterior-posterior (left) and lateral (right) x-ray scans of wild-type and Smarca4+E1548K mice.
Smarca4+/E1548K mice display hearing impairment
To determine if Smarca4+/E1548K mice had a phenotype related to human otosclerosis, we performed several hearing tests. Acoustic startle response (ASR) test, assessing hearing in awake 4-month-old animals, demonstrated that the Smarca4+/E1548K mice had significantly reduced startle amplitudes in almost all tested stimulus volumes compared to their wild-type counterparts (Fig. 4A), consistent with hearing impairment. In the auditory brainstem response (ABR) test, measuring cochlear nerve response thresholds to pure tone stimuli at different frequencies in anesthetized mice, 4-month-old Smarca4+/E1548K mice showed a modest though consistent elevation in ABR threshold across all tested frequencies, with an average shift of 11.3 dB SPL (Fig. 4B). We then compared ABR threshold in response to broadband (click) stimuli and witnessed a similar trend, where Smarca4+/E1548K mice demonstrated elevated thresholds, with an average shift of 8.2 dB SPL (Fig. 4C).
Figure 4. Hearing tests performed on wild type vs. Smarca4+/E1548K mice.
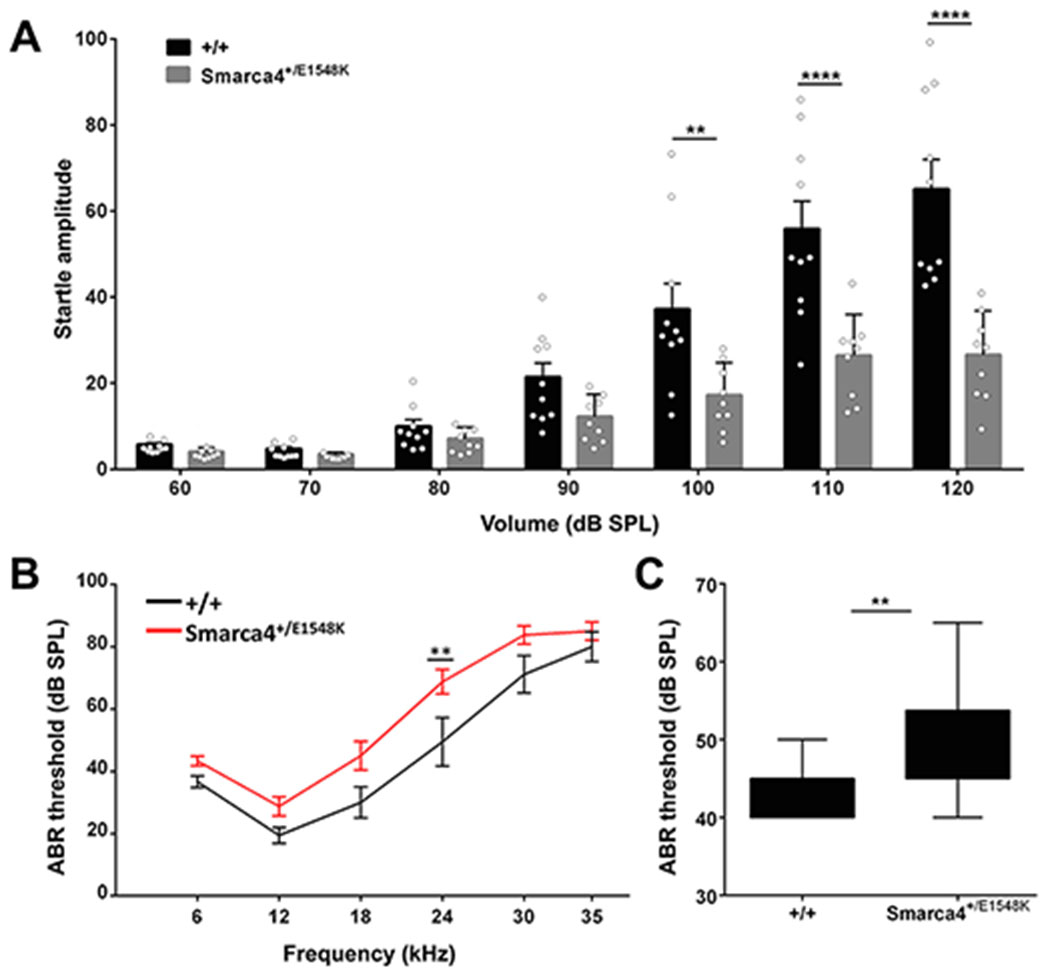
(A) Average startle amplitudes recorded in acoustic startle response tests performed on wild type (n=10) and Smarca4+/E1548K (n=9) mice between 60-120 dB SPL. Error bars indicate the SEM. **P<0.01, ****P<0.0001 (Two-way ANOVA with Holm-Sidak correction for multiple comparisons). (B) Average auditory brainstem response thresholds measured in response to pure tones at 6, 12, 18, 24, 30 and 35 kHz in wild type (n=9) and Smarca4+/E1548K (n=12) mice. Error bars indicate the SEM. *P<0.01 (Two-way ANOVA with Holm-Sidak correction for multiple comparisons). (C) Average auditory brainstem response thresholds measured in response to broadband (click) stimuli in wild type (n=9) and Smarca4+/E1548K (n=12) mice. **P<0.005 (Student’s t test).
Smarca4+/E1548K mice have anomalous incus morphology
As otosclerosis is characterized by abnormal bone metabolism of the otic capsule in humans, we studied the parallel anatomical structure in mice, the auditory bulla. We extracted bullae from 24-month-old mice to determine if any of the bony elements comprising it were affected by the mutation. The overall structure and morphology of the bullae was similar between wild-type and mutant mice. However, upon further detailed dissection of the bullae, Smarca4+/E1548K mice displayed malformations in the auditory ossicles. While the malleus and stapes bones in mutants had morphology similar to that of the wild-type mice (Fig. 5A–D), the incus bones in Smarca4+/E1548K mice were abnormal, exhibiting malformation of the lentiform process with thickening of the distal part of the long crus, as well as a hypoplastic short crus (Fig. 5E–J).
Figure 5. Light microscopy images of isolated ossicles.
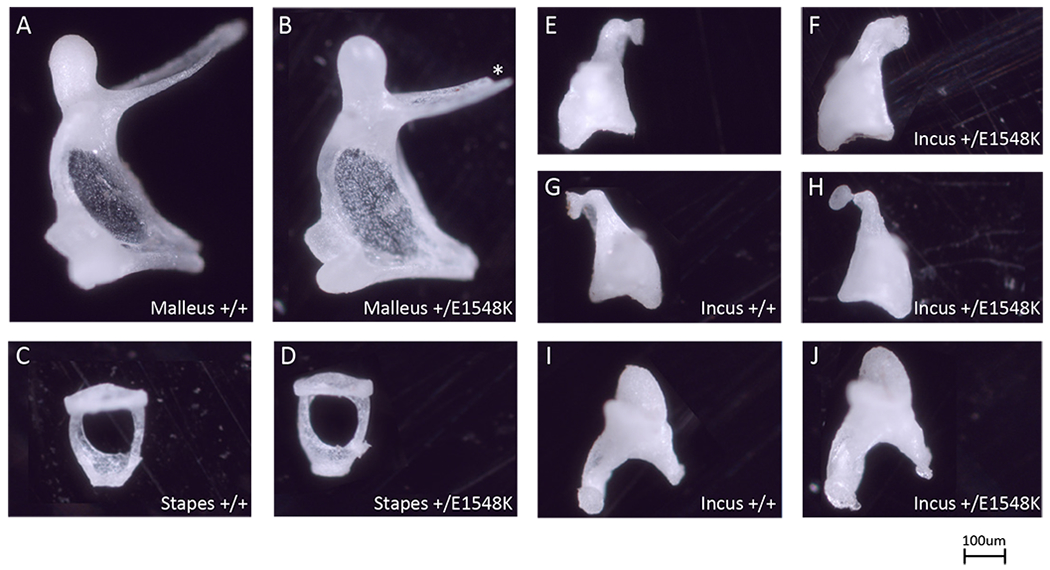
Individual ossicles isolated from wild type (A,C,E,G,I) and Smarca4+/E1548K (B,D,F,H,J) mice. Images are representative of 3 mice from each genotype. Asterisk marks technical defect.
Micro-CT images demonstrate deformity of the ossicular chain in Smarca4+/E1548K mice
To study the effect of the mutation on the 3D morphology of the ossicular chain and the interactions between the ossicles in-situ we used micro-CT scans of intact auditory bullae of aged mice. Micro-CT 3D-reconstructed images revealed normal structure and position of both the malleus and the stapes; however, there were striking structural abnormalities in the incus bone of Smarca4+/E1548K mice (Fig. 6). The bony arch connecting the distal parts of the long and short processes that was clearly visible in the wild-type mouse, was either absent (Fig. 6B,C) or showed uncharacteristic thickening (Fig. 6D) in Smarca4+/E1548K mice. Moreover, there were clear disruptions in the ossicular chain in Smarca4+/E1548K mice, in the area of the incudostapedial joint, where there is misalignment between the lentiform process of the incus and the head of the stapes. Finally, the incus in Smarca4+/E1548K mice appeared more brittle; this was demonstrated by deformities in the lentiform processes as well as fractures in parts of the bone itself (Fig. 6B), where the fracture seen in the image also appeared in the incus bone on the contralateral side (not shown).
Figure 6. Micro-CT images of mouse middle ear ossicles.
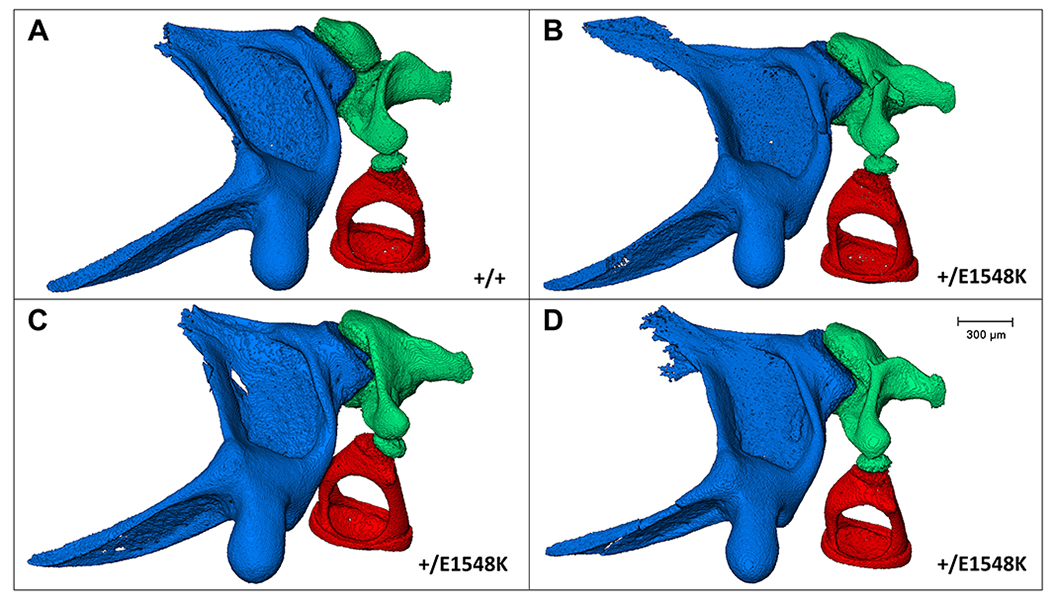
In-situ structure and interactions between the middle ear ossicles of wild type (A) and Smarca4+E1548K (B-D) mice following left auditory bullae micro-CT scanning, segmentation, and image processing. Blue, malleus; green, incus; red, stapes.
Discussion
We studied a family affected with otosclerosis spanning 2 generations (Fig. 1). In line with previous genetic studies of otosclerosis, the pedigree showed autosomal-dominant inheritance with incomplete penetrance[10,11]. The disease-causing mutation we identified in this family is a heterozygous c.4828G>A variant in SMARCA4, causing a p.E1610K substitution in a highly conserved residue in the encoded BRG1 protein (Figs. 2A,B). BRG1 is an ATP-dependent catalytic subunit of the BAF/PBAF complexes that belong to the SWI/SNF family of chromatin remodelers that regulate gene expression (Fig. 2C)[28]. Mutations in genes encoding components of the SWI/SNF complex, including SMARCA4, were previously reported to cause Coffin-Siris syndrome (CSS, MIM 135900), an autosomal dominant congenital multiple malformation syndrome characterized by severe developmental delay, intellectual disability, hypotonia and facial dysmorphism[29]. Interestingly, 34% of CSS patients carrying a mutation in SMARCA4 also had hearing loss[30]. It is noteworthy that all previously reported mutations in SMARCA4 causing CSS were located in more proximal regions of the protein, particularly in and around the two helicase domains of BRG1[31], whereas in our patients the mutation occurred near the BROMO domain, close to the c-terminus of the protein (Fig. 2D). This may explain the absence of any of the severe clinical features of CSS in our patients who presented with isolated otosclerosis.
To ascertain the pathogenicity of the SMARCA4 mutation and comprehensively study its effects we created transgenic mice carrying the human mutation in the mouse ortholog using CRISPR/Cas9. Similar to the human patients, the mutant Smarca4+/E1548K mice did not exhibit any gross morphological, anatomical, or behavioral abnormalities compared to their wild-type counterparts (Fig. 3). To determine if Smarca4+/E1548K mice recapitulate any elements of the phenotype observed in our patients, we first performed several hearing tests. On the acoustic startle response (ASR) test, Smarca4+/E1548K mice exhibited markedly lower responses to sound stimuli compared to wild-type mice, clearly demonstrating hearing impairment (Fig. 4A). We then used an auditory brainstem response (ABR) test to measure cochlear nerve response thresholds to pure tone stimuli at different frequencies and to broadband (click) stimuli. In both tests we observed elevated thresholds in Smarca4+/E1548K mice compared to the thresholds in wild-type mice (Figs. 4B,C); although the differences were modest, they too support hearing impairment in the mutant mice.
Next, we isolated auditory bullae from 24-month-old mice in order to study the long-term effect of the mutation on structures involved in hearing. Although there were no apparent differences in the overall morphology or in initial histological sections of the bulla itself, upon further detailed dissection of the bulla we observed anomalies in the structure of the incus bone in Smarca4+/E1548K mice (Figs. 5E–J). This was supported by 3D reconstructed micro-CT images of auditory ossicles in-situ, demonstrating similar results, where the structure of the incus bone was highly irregular, affecting both the incudomalleolar and incudostapedial joints (Fig. 6). These abnormalities likely interrupt the ossicular chain and interfere with proper sound conduction and are most probably responsible for the hearing impairment seen in Smarca4+/E1548K mice.
In line with these results, it was previously shown that several mouse models for CHARGE syndrome caused by mutations in a different chromatin remodeler protein (Chd7), also display ossicle malformation and hearing impairment[32–34]. Interestingly, in humans, CHD7 and BRG1 have been previously shown to occupy similar regulatory elements in the genome interchangeably and demonstrate a synergistic effect in regulating neural crest formation during embryogenesis[35]. Although the phenotypes seen in CHARGE syndrome that are recapitulated in Smarca4+/E1548K mice most likely result from an aberrant developmental process rather than a progressive postnatal one as in human otosclerosis, it clearly suggests that BRG1 plays a critical role in proper ossicle development. In the same context, while a FOXL1 variant was implicated in human otosclerosis, abnormal craniofacial skeleton development with atypical cartilage formation and mineralization were observed in foxl1-knockout zebrafish, implicating developmental abnormalities in otosclerosis[36].
Notably, it was previously reported that BRG1 is necessary for transcription of osteocalcin, a bone-specific gene that is expressed in late stages of osteoblast differentiation. This occurs through a BMP2-induced interaction between BRG1 and Runx2, a key transcription factor regulating osteoblast differentiation, which recruits BRG1 and mediates its binding to the promoter region of osteocalcin, facilitating transcription activation[37,38]. This suggests that aside from its role in ossicle formation during embryogenesis, BRG1 may also play a role in maintaining postnatal bone homeostasis in the otic capsule through regulating osteoblast differentiation.
It is noteworthy that BRG1 is a well-known tumor suppressor gene implicated in many types of cancer in humans[39–41]. In fact, it has been shown that BRG1 acts in this regard mostly through repression of the expression of most of its target genes[42]. As otosclerosis results from dysregulation of bone-turnover in the otic capsule, it is plausible that the molecular mechanism underlying the disease in our studied family is based upon loss of BRG1-mediated repressed expression of genes active in osteoblast differentiation due to the mutation in SMARCA4. This, in turn, might lead to a gradual increase in osteoblast and osteoclast activity that results in abnormal deposition of bone, eventually leading to otosclerosis. The precise downstream molecular mechanisms through which the SMARCA4 mutation causes otosclerosis are yet to be elucidated.
Materials and methods
Animal use and maintenance
Animal experiments were approved by the Ben-Gurion University (BGU) Committee for the Ethical Care and Use of Laboratory Animals. The experiments, carried out in the BGU rodent facility, were performed according to the Israel Animal Welfare Law (1994) and the NRC Guidelines for the Care and Use of Laboratory Animals (2011). BGU’s Animal Care and Use Program is approved by the Association for the Assessment and Accreditation of Laboratory Animal Care International. At Tel Aviv University, all procedures with animals met the guidelines described in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the Animal Care and Use Committees of Tel Aviv University (01-19-093)
Linkage analysis
Genotyping of affected individuals was performed using Illumina Human Omni Express Bead Chip (Illumina Omni Express, 654,062 SNPs). For linkage analysis and multipoint logarithm of odds (LOD) score calculation we used SUPERLINK ONLINE SNP 1.1 (http://cbl-hap.cs.technion.ac.il/superlink-snp), assuming an autosomal dominant mode of inheritance. Given the variability in age of disease onset and the incomplete penetrance of otosclerosis, only confirmed affected individuals were included in analyses (III: 1, III:2, III:6, III:7, IV: 1, IV:2).
Exome sequencing analysis
Whole exome sequencing of the two most genetically distant affected individuals (IV: 1, IV: 2) was performed as previously described[43]. Sequencing data were analyzed using Ingenuity Variant Analysis software (http://www.qiagen.com/ingenuity) by QIAGEN (Redwood City, CA). Variant filtration started with 187,356 variants spanning 28,956 genes and consisted of several filters: first, a biological context filter was used to determine if variants in any of the genes previously associated with otosclerosis exist in our test subjects, based on The Human Gene Mutation Database (HGMD). Next, we excluded all variants that are observed with an allele frequency greater than or equal to 0.01% in the genomes in the 1000 genomes project, NHLBI ESP exomes, or the ExAC and gnomAD databases. Next, we kept only variants predicted to affect protein coding sequences (missense, stop codon loss or gain, frameshift, in-del mutations, variants located within 10 bases into an intron predicted to disrupt splicing by MaxEntScan or predicted deleterious by having CADD score >11.0). Lastly, we kept only variant that are in the genomic loci found to be shared between all affected individuals in our linkage analysis.
Multiple sequence alignment
Nine orthologues of SMARCA4 were selected to represent protein conservation using multiple sequence alignment. Protein sequences were acquired from the National Center for Biotechnology Information GenBank (http://www.ncbi.nlm.nih.gov). RefSeq sequence accession numbers used for the analysis were: NP_001122321.1 (H.sapiens), XP_512384.4 (P.troglodytes), XP_002808217.1 (M.mulatta), XP_005632936.1 (C.lupus), NP_001099084.1 (B.taurus), NP_001167549.1 (M.musculus), NP_599195.1 (R.norvegicus), NP_990390.1 (G.gallus), NP_853634.1 (D.rerio). Percentage of identity and similarity were acquired from the Basic Local Alignment Search Tool (Blast) (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Protein sequence alignment was carried out using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/).
Generation of Smarca4+/E1548K knock-in mice using CRISPR/Cas9
Smarca4+/E1548K knock-in mice carrying the human c.4828G>A (p.E1610K) mutation in SMARCA4 were generated by altering the Glu codon GAG in position 1548 to a AAG Lys codon using the CRISPR/Cas9 system according to protocols previously described[44,45]. In brief, single-guide RNA (sgRNA) and single-stranded DNA oligonucleotide (ssODN) targeting the designated locus were designed using the DESKGEN online tool (www.deskgen.com; oligo sequences are available in table S4). sgRNA and Cas9 mRNA were then generated using the mMESSAGE mMACHINE T7 Ultra kit (Life Technologies, cat. AM1345) and MEGAshortscript T7 kit (Life Technologies, cat. AM1354), respectively. Following RNA purification, the sgRNA and Cas9 mRNA were injected into C57BL/6JRcc zygotes using a microinjection system under standard conditions. Zygotes were then cultured until the stage of two-cell embryos and transferred into the oviduct of the recipient females. The resulting F0 mice were genotyped at the age of 3-4 weeks by Sanger sequencing genomic DNA from tail samples using TaKaRa Ex Taq kit (Takara Bio Inc.). Following genotyping, a selected F0 chimera male was crossed with wild-type C57BL/6J females (Envigo, Jerusalem, Israel) to produce true non-chimeric heterozygous F1 mice. These heterozygotes were then inbred to produce wild-type, heterozygous or homozygous F2 littermates. These littermates were then inbred according to genotype to create all-wild-type or all-heterozygous mice which were used in all subsequent experimentations. All tested mice were genotyped using Sanger sequencing prior to experimentation (primer sequences are available in table S4).
Mouse whole body X-ray scans
To obtain whole body X-ray scans adult mice were anesthetized by intraperitoneal injection of 100mg/kg ketamine and 10mg/kg xylazine and X-ray images were acquired using a digitized CR30-X Agfa platform (Agfa Healthcare).
Acoustic startle response
Acoustic startle response test was used to assess for conductive hearing loss in Smarca4+/E1548K mice. To test the startle response mice were placed in a size-fitted holder with an open bottom, allowing them to hear all sounds in their surroundings while keeping them from moving freely. The bottom of the holder was attached to a platform that was placed on top of a piezoelectric motion detector. The platform with the attached restrainer were placed inside a sound isolated chamber (Panlab) where mice were being exposed to auditory stimulation in varying intensities, measuring, and recording the amount of force exerted from their feet onto the platform in response to these stimuli. Sound stimuli intensities were calibrated using a sound level meter (SOUND BEE, TFA) by placing the device in the same location on the platform where mice were to be tested. To generate the auditory stimuli and record the startle responses we used the PACKWIN 2.0 software (Panlab). The auditory stimulus protocol consisted of 50- ms broadband white-noise pulses from 60-120 dB in 10 dB steps, presented at a pseudo-random order of 8-22 seconds between them where no inter-trial interval repeated more than three times. Each sound intensity was repeated 6 times per protocol, and response amplitudes were normalized to weight of each tested mouse. Mice were habituated inside the holder for 10 minutes prior to testing, with a background broadband white noise at the level of 60 dB which was kept throughout the experiment. All mice were tested by the same operator on the same day.
Auditory brainstem response
To assess for sensorineural hearing loss in Smarca4+/E1548K mice we performed auditory brainstem response tests, by exposing anesthetized mice to various sound stimuli and measuring their responses using subdermal needle electrodes introduced underneath both ears and above the vertex of the skull. Stimuli were generated using MF1 speakers (Tucker-Davis Technologies) calibrated by an ER-10b+ microphone (Etymotic Research) and consisted of click stimuli as well as pure tones at 6, 12, 18, 24, 30 and 35 kHz. Auditory thresholds were determined by increasing sound intensities from 10-90 dB-SPL in 5 dB increment steps at each frequency until reproducible waveforms were observed; a total of 512 responses per frequency-intensity pair were recorded using the RZ6 multiprocessor and analyzed using the BioSigRZ software (Tucker-Davis Technologies). All recordings were conducted in an acoustic chamber (MAC-1, Industrial Acoustic Company). Mice were anesthetized using intra-peritoneal injections of ketamine and xylazine (100; 10 mg/kg, respectively) and kept on a heating pad at 37°C during the entire experiment.
Light microscopy
To study the long-term effect of the mutation on individual auditory ossicles we extracted auditory bullae from 20–24-month-old mice following anesthesia using isoflurane followed by cervical dislocation. Each ossicle was then carefully extracted from the bulla, cleaned in PBS and photographed on a standard light microscope with 4x magnification.
X-ray Micro-Computer Tomography
Micro-CT was used to study the long-term effect of the mutation on in-situ interactions and overall morphology of the ossicular chain in 3D. We extracted auditory bullae from 20–24-month-old mice following anesthesia using isoflurane and subsequent cervical dislocation. After cleaning the bullae from surrounding tissue, samples were kept in 4% formaldehyde. Micro-CT scans were performed using an Xradia Versa 520 instrument (Zeiss X-ray Microscopy, Pleasanton, CA, USA), with emission source set at 50kV and 4W. Samples were scanned under two resolutions using the same parameters described above. First, a low-resolution scan was used to image the entire auditory bulla; samples were placed 80 mm from the source and 15 mm from the detector. Then, a high-resolution scan was used to observe small structural details and determine the relative position of each ossicle. In the second scan, samples were placed 14 mm from the source and 27 mm from the detector. In both cases samples were scanned under 4X magnification lens using 1601 projections acquired during the course of a 360° rotation with 1-4sec exposure time per projection. Volume reconstruction was performed with the Xradia reconstruction software using a standard back projection algorithm, with a total reconstructed volume of 1024x1024x1024 voxels in both scans, that translated to a final voxel size of 5.7 and 2.3 μm in the low-resolution and high-resolution scans, respectively. Image analyses were performed using the Amira 2019.3 software (Thermo Fisher Scientific, USA).
Statistics
Statistical analyses were done using GraphPad Prism software. Statistical significance of differences between groups was calculated using a two-way ANOVA test with Holm-Sidak correction for multiple comparisons or Student’s two-tailed t test where applicable.
Study approval
The study was approved by the Soroka Medical Center Institutional Review Board (approval #5071G) and the Israel Ministry of Health National Helsinki committee (approval #920100319). DNA was extracted from peripheral blood or saliva following written informed consent by all individuals studied.
Supplementary Material
Key messages:
What is already known on this topic:
Otosclerosis, a common cause of adult-onset progressive hearing loss, is known to have familial disposition. However, its genetic determinants and molecular pathways are poorly understood.
What this study adds:
We demonstrate that otosclerosis can be caused by dominant mutation in SMARCA4, encoding a key component of the PBAF chromatin remodeling complex, and generate a mouse model for the disease.
How this study might affect research, practice or policy:
The study opens new venues for understanding molecular pathogenicity of otosclerosis and its pre-implantation / pre-symptomatic diagnosis and prevention.
Acknowledgements / Funding
The research was funded by the Morris Kahn Family Foundation (O.S.B.) and by the National Knowledge Center for Rare/Orphan Diseases of the Israel Ministry of Science, Technology and Space, Ben-Gurion University of the Negev, Beer-Sheva, Israel (O.S.B.) and the National Institutes of Health /NIDCD grant R01DC011835 (K.B.A.)
Footnotes
The authors have declared that no conflict of interest exists.
References
- 1.Declau F, Van Spaendonck M, Timmermans JP, et al. Prevalence of otosclerosis in an unselected series of temporal bones. Otol Neurotol 2001;22:596–602. [DOI] [PubMed] [Google Scholar]
- 2.Gordon MA. The genetics of otosclerosis: A review. Otol Neurotol 1989;10:426–38. [DOI] [PubMed] [Google Scholar]
- 3.Quesnel AM, Ishai R, McKenna MJ. Otosclerosis: Temporal Bone Pathology. Otolaryngol Clin North Am 2018;51:291–303. [DOI] [PubMed] [Google Scholar]
- 4.Chole RA, McKenna M. Pathophysiology of Otosclerosis. Otol Neurotol 2001;22:249–57. [DOI] [PubMed] [Google Scholar]
- 5.Rudic M, Keogh I, Wagner R, et al. The pathophysiology of otosclerosis: Review of current research. Hear Res 2015;330:51–6. [DOI] [PubMed] [Google Scholar]
- 6.Hueb MM, Goycoolea MV., Paparella MM, et al. Otosclerosis: The University of Minnesota temporal bone collection. Otolaryngol - Head Neck Surg 1991;105:396–405. [DOI] [PubMed] [Google Scholar]
- 7.Schuknecht HF, Barber W. Histologic Variants in Otosclerosis. Laryngoscope 1985;95:1307–17. [DOI] [PubMed] [Google Scholar]
- 8.Ealy M, Smith RJH. Otosclerosis. In: Medical Genetics in the Clinical Practice of ORL. Basel: : KARGER; 2011. 122–9. [Google Scholar]
- 9.Browning GG, Gatehouse S. Sensorineural hearing loss in stapedial otosclerosis. Ann Otol Rhinol Laryngol 1984;93:13–6. [DOI] [PubMed] [Google Scholar]
- 10.Ealy M, Smith RJH. The Genetics of otosclerosis. Hear Res 2010;266:70–4. [DOI] [PubMed] [Google Scholar]
- 11. Morrison AW. Genetic factors in otosclerosis. Ann R Coll Surg Engl 1967;41:202–37. [PMC free article] [PubMed] [Google Scholar]
- 12. Tomek MS, Brown MR, Mani SR, et al. Localization of a gene for otosclerosis to chromosome 15q25-26. Hum Mol Genet 1998;7:285–90. [DOI] [PubMed] [Google Scholar]
- 13. Van Den Bogaert K, Govaerts PJ, Schatteman I, et al. A second gene for otosclerosis, OTSC2, maps to chromosome 7q34-36. Am J Hum Genet 2001;68:495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen W, Campbell CA, Green GE, et al. Linkage of otosclerosis to a third locus (OTSC3) on human chromosome 6p21.3-22.3. J Med Genet 2002;39:473–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brownstein Z, Goldfarb A, Levi H, et al. Chromosomal mapping and phenotypic characterization of hereditary otosclerosis linked to the OTSC4 locus. Arch Otolaryngol - Head Neck Surg 2006;132:416–24. [DOI] [PubMed] [Google Scholar]
- 16. Van Den Bogaert K, De Leenheer EMR, Chen W, et al. A fifth locus for otosclerosis, OTSC5, maps to chromosome 3q22-24. J Med Genet 2004;41:450–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thys M, Van Den Bogaert K, Iliadou V, et al. A seventh locus for otosclerosis, OTSC7, maps to chromosome 6q13-16.1. Eur J Hum Genet 2007;15:362–8. [DOI] [PubMed] [Google Scholar]
- 18. Bel Hadj Ali I, Thys M, Beltaief N, et al. A new locus for otosclerosis, OTSC8, maps to the pericentromeric region of chromosome 9. Hum Genet 2008;123:267–72. [DOI] [PubMed] [Google Scholar]
- 19. McKenna MJ, Kristiansen AG, Bartley ML, et al. Association of COL1A1 and otosclerosis: evidence for a shared genetic etiology with mild osteogenesis imperfecta. Am J Otol 1998;19:604–10. [PubMed] [Google Scholar]
- 20. Ziff JL, Crompton M, Powell HRF, et al. Mutations and altered expression of SERPINF1 in patients with familial otosclerosis. Hum Mol Genet 2016;25:2393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schrauwen I, Valgaeren H, Tomas-Roca L, et al. Variants affecting diverse domains of MEPE are associated with two distinct bone disorders, a craniofacial bone defect and otosclerosis. Genet Med 2019;21:1199–208. [DOI] [PubMed] [Google Scholar]
- 22. Bouzid A, Tekari A, Jbeli F, et al. Osteoprotegerin gene polymorphisms and otosclerosis: An additional genetic association study, multilocus interaction and meta-analysis. BMC Med Genet 2020;21:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schrauwen I, Thys M, Vanderstraeten K, et al. Association of bone morphogenetic proteins with otosclerosis. J Bone Miner Res 2008;23:507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thys M, Schrauwen I, Vanderstraeten K, et al. The coding polymorphism T263I in TGF-β1 is associated with otosclerosis in two independent populations. Hum Mol Genet 2007;16:2021–30. [DOI] [PubMed] [Google Scholar]
- 25. Abdelfatah N, Mostafa AA, French CR, et al. A pathogenic deletion in Forkhead Box L1 (FOXL1) identifies the first otosclerosis (OTSC) gene. Hum Genet 2022;141:965–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schrauwen I, Ealy M, Huentelman MJ, et al. A Genome-wide Analysis Identifies Genetic Variants in the RELN Gene Associated with Otosclerosis. Am J Hum Genet 2009;84:328–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Imauchi Yutaka, Xavier Jeunemaître Magali Boussion, et al. Relation between renin-angiotensin-aldosterone system and otosclerosis: A genetic association and in vitro study. Otol Neurotol 2008;29:295–301. [DOI] [PubMed] [Google Scholar]
- 28. Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem 2009;78:273–304. [DOI] [PubMed] [Google Scholar]
- 29. Tsurusaki Y, Okamoto N, Ohashi H, et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet 2012;44:376–8. [DOI] [PubMed] [Google Scholar]
- 30. Vasko A, Drivas TG, Schrier Vergano SA. Genotype-phenotype correlations in 208 individuals with coffin-siris syndrome. Genes (Basel) 2021;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li D, Ahrens-Nicklas RC, Baker J, et al. The variability of SMARCA4-related Coffin–Siris syndrome: Do nonsense candidate variants add to milder phenotypes? Am J Med Genet Part A 2020;182:2058–67. [DOI] [PubMed] [Google Scholar]
- 32. Pau H, Hawker K, Fuchs H, et al. Characterization of a new mouse mutant, flouncer, with a balance defect and inner ear malformation. Otol Neurotol 2004;25:707–13. [DOI] [PubMed] [Google Scholar]
- 33. Ogier JM, Carpinelli MR, Arhatari BD, et al. CHD7 deficiency in ‘ Looper’, a new mouse model of CHARGE syndrome, results in ossicle malformation, otosclerosis and hearing impairment. PLoS One 2014;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ogier JM, Arhatari BD, Carpinelli MR, et al. An intronic mutation in Chd7 creates a cryptic splice site, causing aberrant splicing in a mouse model of CHARGE syndrome. Sci Rep 2018;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bajpai R, Chen DA, Rada-Iglesias A, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 2010;463:958–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hawkey-Noble A, Pater JA, Kollipara R, et al. Mutation of foxl1 Results in Reduced Cartilage Markers in a Zebrafish Model of Otosclerosis. Genes (Basel) 2022;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Villagra A, Cruzat F, Carvallo L, et al. Chromatin Remodeling and Transcriptional Activity of the Bone-specific Osteocalcin Gene Require CCAAT/Enhancer-binding Protein β-dependent Recruitment of SWI/SNF Activity. J Biol Chem 2006;281:22695–706. [DOI] [PubMed] [Google Scholar]
- 38. Young DW, Pratap J, Javed A, et al. SWI/SNF chromatin remodeling complex is obligatory for BMP2-induced, Runx2-dependent skeletal gene expression that controls osteoblast differentiation. J Cell Biochem 2005;94:720–30. [DOI] [PubMed] [Google Scholar]
- 39. Wong AKC, Shanahan F, Chen Y, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res 2000;60:6171–7. [PubMed] [Google Scholar]
- 40. Shain AH, Pollack JR. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PLoS One 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 2013;45:592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res 2011;21:396–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Drabkin M, Zilberberg N, Menahem S, et al. Nocturnal Atrial Fibrillation Caused by Mutation in KCND2 , Encoding Pore-Forming (α) Subunit of the Cardiac Kv4.2 Potassium Channel. Circ Genomic Precis Med 2018;11:1–9. [DOI] [PubMed] [Google Scholar]
- 44. Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 2014;9:1956–68. [DOI] [PubMed] [Google Scholar]
- 46. Letunic I, Bork P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res 2018;46:D493–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.