

Abstract
Background
Alzheimer's disease (AD) is a prevalent neurodegenerative disorder causing progressive dementia. Research suggests that microRNAs (miRNAs) could serve as biomarkers and therapeutic targets for AD. Reduced levels of miR‐137 have been observed in the brains of AD patients, but its specific role and downstream mechanisms remain unclear. This study sought to examine the therapeutic potential of miR‐137‐5p agomir in alleviating cognitive dysfunction induced in AD models and explore its potential mechanisms.
Methods
This study utilized bioinformatic analysis and a dual‐luciferase reporter assay to investigate the relationship between miR‐137‐5p and ubiquitin‐specific peptidase 30 (USP30). In vitro experiments were conducted using SH‐SY5Y cells to assess the impact of miR‐137‐5p on Aβ1–42 neurotoxicity. In vivo experiments on AD mice evaluated the effects of miR‐137‐5p on cognition, Aβ1–42 deposition, Tau hyperphosphorylation, and neuronal apoptosis, as well as its influence on USP30 levels.
Results
It was discovered that miR‐137‐5p mimics efficiently counteract Aβ1–42 neurotoxicity in SH‐SY5Y cells, a protective effect that is negated by USP30 overexpression. In vivo experiments demonstrated that miR‐137‐5p enhances the cognition and mobility of AD mice, significantly reducing Aβ1–42 deposition, Tau hyperphosphorylation, and neuronal apoptosis within the hippocampus and cortex regions. Mechanistically, miR‐137‐5p significantly suppresses USP30 levels in mice, though USP30 overexpression partially buffers against miR‐137‐5p‐induced AD symptom improvement.
Conclusion
Our study proposes that miR‐137‐5p, by instigating the downregulation of USP30, has the potential to act as a novel and promising therapeutic target for AD.
Keywords: Alzheimer's disease, Aβ1–42 , miR‐137‐5p, USP30
miRNA‐137‐5p is a regulator of cognitive decline and its potential as a therapeutic target for AD.
miRNA‐137‐5p directly targeted and downregulated USP30 expression in neuronal cells.
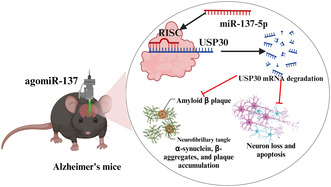
1. INTRODUCTION
Alzheimer's disease (AD) is a degenerative brain condition characterized by the progressive deterioration of cognitive function and memory loss. 1 , 2 , 3 The severity of dementia in AD patients can range from mild to severe, leading to complete dependence on others for daily activities. Research suggests that changes in the brain occur long before the onset of cognitive problems, although the exact cause of these changes remains unidentified. Over the past several decades, despite extensive research efforts, the early detection, effective treatment, and understanding of the molecular mechanisms underlying AD remain largely elusive. 4 , 5 Therefore, it is important to develop dependable diagnostic methods for the identification of AD biomarkers, particularly in the initial phases.
In recent times, microRNAs (miRNAs)—a category of small noncoding RNA molecules containing 18–22 nucleotides—have gained prominence as potential biomarkers and therapeutic targets for a wide range of human diseases, including malignant tumors, cardiovascular diseases, metabolic diseases, and neurodegenerative diseases. 6 , 7 , 8 , 9 The regulation of gene expression by miRNAs, through their binding to the 3′untranslated regions (UTR) of messenger RNA (mRNA) molecules, is suggestive of an important role in the modulation of cellular mechanisms and signaling pathways associated with the development of AD. 10 , 11 , 12 , 13 Considering the high stability of miRNAs and their detectability in various bodily fluids, a growing body of evidence suggests that distinctive patterns of miRNA expression in the brain, cerebrospinal fluid, and blood samples of AD patients may indicate their role in the initiation and progression of the disease. For instance, exosomal miR‐190a‐5p, miR‐223‐3p, and miR‐23a‐3p levels have been found to be elevated in AD patients compared to healthy controls, whereas miR‐100‐3p, miR‐451a, miR‐16‐5p, and miR‐605‐5p levels were reduced. 14 , 15 Furthermore, miRNAs such as miR‐132, miR‐34a, miR‐124, and miR‐106b have been linked to AD and are believed to play important roles in various aspects of the disease, including neuroinflammation, synaptic dysfunction, and neurodegeneration. 16 , 17 , 18 Intriguingly, studies have identified altered miRNA expression patterns in blood that could serve as potential diagnostic markers for AD. Examples of such miRNAs include miR‐134, miR‐193b, and miR‐384, which have been found to be significantly altered in AD patients compared to controls. 19 , 20 Most important, targeting miRNAs has emerged as a promising therapeutic approach for AD. For example, the intranasal delivery of miR‐132 has been demonstrated to alleviate certain AD‐related symptoms in animal models. 21 Overall, these findings highlight the potential of miRNA‐based diagnostics and therapies in the early detection and treatment of AD.
miR‐137, an miRNA that is profusely expressed in the cerebral cortex, plays an important role in determining the fate of embryonic neural stem cells. 22 The downregulation of miR‐137 in glioma stem cells, a specific type of brain cancer stem cells, is fundamental for their self‐renewal. 23 Through its interplay with the ubiquitin ligase Mind Bomb‐1, miR‐137 governs the process of neuronal maturation. Furthermore, miR‐137 is acknowledged as a significant tumor suppressor because it mediates various cancer‐related functions such as cell proliferation, migration, apoptosis, and autophagy by targeting proteins like XIAP, FUNDC1, NIX, and SRC3. 24 Recently, a gamut of brain‐specific miRNAs have been uncovered, one of which is miR‐137. It has been evidenced to aid the process of neuronal differentiation in adult subventricular neural stem cells, while simultaneously hindering neuronal maturation in adult hippocampal neurons. 25 However, the exact role of miR‐137 in AD remains to be verified. Further investigations are imperative to discern the downstream regulatory targets of miR‐137 and to truly elucidate its contribution to the pathogenesis and progression of AD.
In this research, we hypothesized that miR‐137‐5p might potentially regulate the progression of AD via its interaction with ubiquitin‐specific peptidase 30 (USP30). The aim of this study was to uncover the underlying mechanism through which miR‐137‐5p manipulates USP30, with the ultimate goal of improving neurogenesis impairment in the adult hippocampus and alleviating memory deficits in AD.
2. METHODS
2.1. Subject recruitment
In this study, we enrolled 47 patients diagnosed with AD, who were matched by gender and age, and 43 healthy control subjects. Whole blood was collected from each participant using ethylenediaminetetraacetic acid–containing tubes to obtain 250‐μL plasma aliquots. Subsequently, the blood samples were centrifuged at 2500 rpm and 4°C for 15 min and then stored at −80°C. The diagnosis of AD was based on the criteria set forth in the Diagnostic and Statistical Manual of Mental Disorders, fourth edition, revised, as well as the guidelines established by the American Academy of Neurology; Communication Disorders; and Stroke, Geriatric Dementia, and Related Diseases Working Group. Individuals with severe physical trauma or a history of mental illness, individuals with arteriosclerosis or cerebral white matter rarefaction, patients who have undergone medication treatment, and patients with severe ischemic lesions were excluded. The ethical committees of Shenyang First People's Hospital approved the study (approval number: 2021, research review number: 04), and all the participants who were enrolled provided informed consent.
2.2. Cell culture and transfection
SH‐SY5Y cells were procured from the ATCC cell line bank (Manassas, VA) and were cultured in a humidified environment at 37°C with 5% CO2 using Dulbecco's modified Eagle's medium/F12 (Gibco) medium with 10% fetal bovine serum (FBS), 100 μg/mL of streptomycin, and 1 U/mL of penicillin. The co‐transfection process involving 3′UTR plasmids and miRNA mimics was performed using the DharmaFECT Duo transfection reagent, adhering to the manufacturer's guidelines.
2.3. Dual‐luciferase reporter assay
The target binding relationship between miR‐137‐5p and USP30 was confirmed using dual‐luciferase assays, conducted using a Promega dual‐luciferase reporter gene assay. 26 Briefly, the USP30 fragment containing the miR‐137‐5p binding site (pmirGLO‐POT1‐Wt) and the fragment with the miR‐137‐5p binding mutation site (pmirGLO‐POT1‐Mut) were cloned into the pmirGLO‐POT1‐reporter vector. These constructed vectors were then co‐transfected into 293T cells along with miR‐137‐5p mimics or mimics‐NC (miR‐137‐5p mimic negative control). The luciferase activity of the cells was measured using a multifunctional microplate reader (Biotek, USA), and the relative activity was calculated.
2.4. Western blot analysis
Total protein from both mouse hippocampal tissues and SH‐SY5Y cells was extracted using a whole cell lysis assay kit (WLA019, Wanleibio, China). The protein was then quantified following the manufacturer's instructions using a BCA protein assay kit (WLA019, Wanleibio). A 10‐μL protein sample (containing 30–40 μg of protein) was used for sodium dodecyl‐sulfate polyacrylamide gel electrophoresis (SDS‐PAGE), which was then transferred to polyvinylidene fluoride (PVDF) membranes (IPVH00010, Millipore, USA) using standard techniques, as thoroughly described in previous publications. 27 Then, the PVDF membranes were blocked using 5% skim milk at 25°C for 60 min and subsequently exposed to primary antibodies against USP30 (WLH3383, 1:1000, Wanleibio), Aβ1–42 (25524‐1‐AP, 1:500, Proteintech, China), total‐Tau (10274‐1‐AP, 1:500, Proteintech), p‐TauS396 (WL03540, 1:500, Wanleibio), Bax (WL01637, 1:1000, Wanleibio), Bcl‐2 (WL01556, 1:1000, Wanleibio), or β‐actin (WL01372, 1:1000, China) at 4°C for overnight incubation. Then, the membranes were incubated with horseradish peroxidase (HRP)‐labeled goat‐anti‐rabbit IgG (WLA023, 1: 5000, Wanleibio) for 60 min at 37°C. The signals from the Western blot bands were then visualized using enhanced chemiluminescence (WLA003, Wanleibio) with the help of an Invitrogen E‐Gel Imager system, and their quantification was carried out using a Gel‐Pro‐Analyzer. β‐Actin was used as an internal reference.
2.5. AD cell model construction and group design
In vitro AD cell models in the current study were constructed by incubating SH‐SY5Y cells with 10 μM Aβ1–42 oligomers (GL Biochem (Shanghai) Ltd) for 24 h based on previously described protocols. 28 Briefly, SH‐SY5Y cells were divided into six groups: (1) control group, SH‐SY5Y cells incubated only conventional normal medium; (2) AD group, SH‐SY5Y cells were treated with 10 μM Aβ1–42 oligomers for 24 h; (3) mimic‐NC + Aβ1–42 group, SH‐SY5Y cells were treated with 10 μM Aβ1–42 oligomers for another 24 h after transfection with mimic‐NC for 24 h; (4) miR‐137‐5p mimic + Aβ1–42 group, SH‐SY5Y cells were treated with 10 μM Aβ1–42 oligomers for another 24 h after transfection with miR‐137‐5p mimic for 24 h; (5) miR‐137 mimic + vector‐NC + Aβ1–42 group, SH‐SY5Y cells were transfected with miR‐137‐5p mimic and vector‐NC and then treated with Aβ1–42; and (6) miR‐137 mimic + USP30 vecto + Aβ1–42 group, SH‐SY5Y cells were transfected with miR‐137‐5p mimic and USP30 vector and then treated with Aβ1–42.
2.6. Cell apoptosis analysis
SH‐SY5Y cells (1 × 106/mL) were inoculated in six‐well plates and cultured for another 24 h for cell attachment. Subsequently, cells were received indicated treatment for 48 h. Subsequently, the cells were collected and rinsed thrice with cold phosphate‐buffered saline (PBS). These cells were then resuspended in 200 μL of binding buffer and incubated with both AnnexinV‐FITC (5 μL) and propidium iodide (10 μL) (WLA001, Wanleibio). The cells were incubated for 20 min at 25°C in a dark environment. Finally, apoptotic cells were determined using ACEA NovoCyte flow cytometry (NovoCyte, ACEA, USA).
2.7. Establishment of AD mouse model
Male C57BL/6J mice (aged: 6–8 weeks, body weight range: 18–22 g) were procured from Liaoning Changsheng Biotechnology Co., Ltd (license number: SCXK [Liao] 2020‐0001). The animal studies were approved by the Ethics Committee of Shenyang First People's Hospital (approval number: 2021, research review number: 04). A week before carrying out the experiment, all the animals were put on adaptive feeding. Mice had unrestricted access to both food and water, and were housed under conventional conditions: a temperature of 22 ± 1°C, a 12‐h light–dark cycle, and humidity within the range of 45%–55% with moderate ventilation. Mice were randomly assigned into four distinct groups, with each group consisting of six animals: sham group, AD group, AD + mmu‐agomiR‐137‐5p group, and AD + mmu‐agomiR‐137‐5p + USP30 vector group. The sham group mice received an intraperitoneal injection of PBS. AD model was established according to the previously described protocol. 29 , 30 In brief, mice were injected intraperitoneally with 150 mg/kg of d‐galactose and were intragastrically administered 5 mg/kg of AlCl3 once a day for 40 consecutive days. In the AD + mmu‐agomiR‐137‐5p group, 1.5 μL of mmu‐agomiR‐137 (100 μM) was stereotactically injected into the hippocampus region of mice at a rate of 0.2 μL/min. In the AD + AD + mmu‐agomiR‐137‐5p + USP30 vector group, mice received a simultaneous injection of 1.5 μL of mmu‐agomiR‐137 and 2 μL of USP30 overexpression lentivirus every 5 days for 40 days of modeling.
2.8. Water maze test
To confirm the successful creation of the AD model, a water maze test was carried out to evaluate the spatial learning and memory skills of mice across all groups. In particular, the mice engaged in adaptive training involving a concealed platform. This training occurred once per day, with the number of dead ends escalating daily (starting at one dead end for the initial session, followed by two dead ends for the subsequent session, and so forth, ultimately reaching four dead ends by the fourth session). The training program offered a full‐path length for the mice to traverse. After 4 days of adaptive training, all mice participated in a consecutive 2‐day testing phase, with each test lasting for 5 min. If a mouse was unable to complete the swim within the allocated time, the recorded time was set at 5 min. The water maze employed for the experiment was a black circular tank (diameter: 190 cm, height: 55 cm). It was filled with tap water to 30 cm. The tank was segmented into four equal regions, each having a depth of 30 cm. In one of these quadrants, a platform (diameter: 10 cm, height: 29 cm) was constructed 35 cm from the pool's edge. Throughout the acquisition trial phase (days 1–5), mice were given a time frame of 60 s to locate the platform. If they could not do so within 120 s, the experimenter guided them to the platform, where they were permitted to remain for 10 s. The time required for each mouse to reach the platform was documented as escape latency, which was determined by averaging the results of the four daily trials. On the last day of the acquisition trial, a memory assessment (probe test) was performed by removing the platform and allowing the mice to swim freely for 120 s. The number of times they crossed the platform's former location and the duration spent in the target quadrant were recorded automatically.
2.9. TdT‐mediated dUTP nick‐end labeling assay
The TdT‐mediated dUTP nick‐end labeling (TUNEL) assay, utilizing a commercially available kit (C1091, Beyotime, China), was applied to assess cell apoptosis in rat hippocampal tissue. To begin with, 5‐μm‐thick paraffin sections of the hippocampus were treated with 50 μL of 0.1% Triton X‐100 for 8 min at room temperature. Then, the sections were rinsed thrice with PBS and then blocked using 50 μL of 3% H2O2 for 10 min. Next, the sections were incubated with 50 μL of the TUNEL reaction mixture for 60 min at 37°C in a dark setting. Finally, the specimens were counterstained with hematoxylin and examined under an optical microscope (BX53, OLYMPUS, Japan).
2.10. Hematoxylin–eosin staining
The morphological alterations in the cortical tissues and hippocampus of mice were evaluated through pathological examination. Upon conclusion of the Morris Water Maze (MWM) experiment, mice were dissected, and their brain tissue was instantly positioned on an ice tray after the sagittal suture. Then, the brain tissues were preserved in a 4% formalin solution at 4°C for 8 h. Then, they were transferred to a solution of 70% ethanol for 5 min followed by a sequence of dehydration stages using 80%, 90%, and 95% ethanol, and absolute ethanol lasting for 4 h each. Subsequently, the tissues were bathed in xylene for 30 min and encapsulated in paraffin. Coronal sections, 3 μm thick, were procured from the optic chiasma region—inclusive of the hippocampus. For hematoxylin–eosin (H&E) tests, three sequential slices were obtained from each sample.
2.11. Nissl staining
The process of preparing mice hippocampus and cerebral cortex tissue slices is identical to the H&E staining section. Then, the slices were stained with a 0.5% cresyl violet solution (Sinopharm, China) by immersing them at room temperature for 10 min. The stained slices were then immediately rinsed with 0.25% cold glacial acetic acid solution (Tianjin Comeo) for 3 s. The sections were later rinsed, dried, and coated with a neutral resin to enable microscopic analysis.
2.12. RNA extraction and quantitative reverse transcription polymerase chain reaction
Cells and serum were subjected to RNA extraction utilizing the miRCURY isolation kit (Exiqon, Denmark) in compliance with the manufacturer's guidelines. Then, complementary DNA conversion and miRNA or mRNA quantification were performed using quantitative reverse transcription polymerase chain reaction (qRT‐PCR) on a StepOnePlus real‐time PCR system (Applied Biosystems, Thermo Fischer Scientific, USA). To quantify miRNA, the miRCURY LNA universal RT miRNA PCR system (Exiqon) was employed in conjunction with predesigned LNA primers (Exiqon). U6 and β‐actin functioned as reference genes for miRNA‐137‐5p and USP30, respectively. The iQ SYBR Green Supermix kit (Bio‐Rad, USA), together with predesigned primers (GeneGlobe, QIAGEN), was utilized. Livak method (2−ΔΔCT) was applied to determine miRNA and mRNA fold changes. The primer sequences for the relevant genes employed in qRT‐PCR are presented in Table 1.
TABLE 1.
Primer sequences for qRT‐PCR.
| Primer name | Primer sequences (5′‐3′) |
|---|---|
| miR‐137‐5p F | GGACACGCTTATTGCTTAAGAATA |
| miR‐137‐5p R | GTGCAGGGTCCGAGGTATTC |
| U6 F | CTCGCTTCGGCAGCACA |
| U6 R | AACGCTTCACGAATTTGCGT |
| USP30 F | CTTCTGAAAGCCTTGTCC |
| USP30 R | CGGTCTCGCTCATCTTC |
| β‐Actin‐F | GGAGATTACTGCCCTGGCTCCTAGC |
| β‐Actin‐R | GGCCGGACTCATCGTACTCCTGCTT |
Abbreviation: USP30, ubiquitin‐specific peptidase 30.
2.13. Statistical analysis
All quantitative data were represented using mean ± standard deviation, with analysis performed using GraphPad Prism (version 8.0) and SPSS (version 22.0). To differentiate between two or numerous groups, Student's t‐test or analysis of variance (ANOVA), followed by multiple comparisons, was conducted. A p‐value <0.05 was deemed to exhibit statistical significance.
3. RESULTS
3.1. miR‐137‐5p was significantly decreased in AD patient serum and can directly target USP30
First, the expression level of miR‐137 and USP30 in human serum of AD patients and healthy controls was evaluated using qRT‐PCR analysis. Figure 1A,B shows that the miR‐137‐5p level decreased 1.8‐fold in AD patients relative to the healthy control group (p < 0.001), whereas USP30 levels increased 1.6‐fold (p < 0.001). Next, we also analyzed the correlations of miR‐137‐5p and USP30 in AD patient serum by performing Pearson's correlation analysis, and the results, as expected, showed that miR‐137‐5p was negatively correlated with USP30 (Figure 1C) (p < 0.001). Based on these findings, we speculated that USP30 might function as a downstream gene of miR‐137‐5p. To confirm our hypothesis, we consequently identified the putative miRNAs that could bind to USP30 3′UTR using TargetScan (http://www.targetscan.org/) and starBase (http://starbase.sysu.edu.cn/) databases. As expected, results revealed the 3′UTR of USP30 contains putative binding sites (positions 2061–2068) for miR‐137‐5p (Figure 1D). In addition, we validated the existence of a targeted binding relationship between miR‐137‐5p and USP30 using a dual‐luciferase reporter system. The dual‐luciferase reporter gene assay (Figure 1E) revealed miR‐137‐5p mimic led to a significant decrease in luciferase activity of USP30 3′UTR wild type (p < 0.001); conversely, no noticeable change was observed in the luciferase activity of the mutant type (MUT) of USP 30 3′UTR (p > 0.05), suggesting miR‐137‐3p can specifically bind to USP30 mRNA 3′UTR and cause its transcriptional repression.
FIGURE 1.
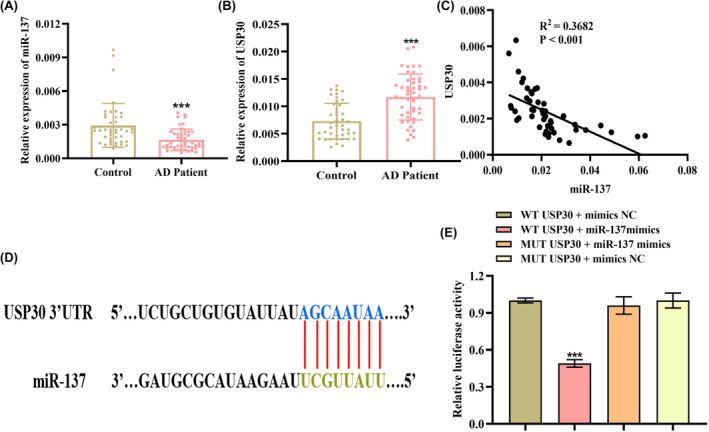
miR‐137‐5p was significantly downregulated in AD (Alzheimer's disease) patient serum. qRT‐PCR analysis of (A) miR‐137‐5p and (B) USP30 (ubiquitin‐specific peptidase 30) levels in serum samples from AD patients (n = 47) and healthy controls (n = 43). (C) The correlation between miR‐137‐5p and USP30 levels in AD patients was evaluated using Spearman's correlation analysis. (D) The predicted binding site in the 3′UTR (untranslated region) of USP30 for miR‐137‐5p. (E) Dual‐luciferase reporter system analyzing the targeting relationship between miR‐137‐5p and USP30. ***p < 0.001.
3.2. Restoring miR‐137‐5p level rescues Aβ1–42‐induced apoptosis in vitro
We then assessed the impact of altered miR‐137‐5p expression on cell apoptosis in the immortalized human neuroblastoma cell line SH‐SY5Y induced by Aβ1–42 exposure. First, we manipulated the expression of miR‐137‐5p in SH‐SY5Y cells through transfection miR‐137‐5p mimics. As shown in Figure 2A, the expression level of miR‐221 exhibited a substantial increase when compared to the control group (p < 0.001), indicating the successful transfection. It has been well established that neuronal apoptosis‐related phenotypes are the leading cause of Aβ‐induced cytotoxicity. Figure 2B,C shows that Aβ1–42 administration could trigger SH‐SY5Y apoptotic cell death when compared with controls (apoptosis rate: 30.14 ± 1.74% vs. 3.31 ± 0.65%) (p < 0.001), whereas miR‐137‐5p mimics effectively alleviated Aβ‐induced neuronal apoptosis compared to mimics control (apoptosis rate: 9.59 ± 1.49% vs. 30.48 ± 2.00%) (p < 0.001). Moreover, we further confirm whether USP30 was upregulated in Aβ‐induced AD cell model. As expected, USP30 protein level was obviously increased in Aβ1–42‐treated SH‐SY5Y cells (Figure 2D).
FIGURE 2.
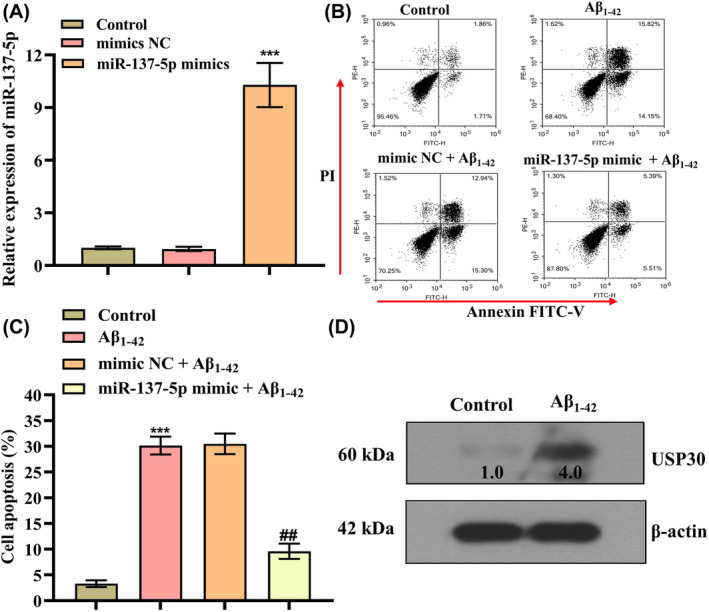
miR‐137‐5p mimics rescues Aβ1–42‐induced apoptosis in vitro. (A) qRT‐PCR analysis of miR‐137‐5p level in SH‐SY5Y cells after transfection of miR‐137‐5p mimics and mimics NC for 24 h, ***p < 0.001. (B, C) Flow cytometry analysis of cell apoptosis after the treatment of 10 μM Aβ1–42 for 24 h; * represents compared with control group, ***p < 0.001; @ represents compared with mimics NC + Aβ1–42 group, @@ p < 0.01. (D) Western blotting analysis of the protein level of USP30 (ubiquitin‐specific peptidase 30) in Aβ1–42‐treated cells.
3.3. USP30 overexpression partially abolished the effect of miR‐137‐5p on Aβ1–42‐treated SH‐SY5Y cells
Additionally, we explore whether the mitigating impacts of miR‐137‐5p on Aβ1–42‐induced apoptosis were accomplished at least partially via the downregulation of USP30. We first transfected cells with USP30 vector to upregulate its level in cells, and qRT‐PCR and Western blotting results revealed that both mRNA and protein levels of USP30 were remarkably elevated in SH‐SY5Y cells (Figure 3A,B) (p < 0.001). Figure 2C,D shows that the apoptotic cells were significantly increased in the Aβ1–42 treatment group compared with controls, whereas miR‐137‐5p mimics could effectively reduce cell apoptosis. In addition, USP30 overexpression alleviated the antiapoptotic effect of miR‐137‐5p mimics in cells (Figure 3C,D). Collectively, these data suggest miR‐137‐5p might at least partially exert ameliorating effects on Aβ1–42‐induced apoptosis through downregulating USP30.
FIGURE 3.
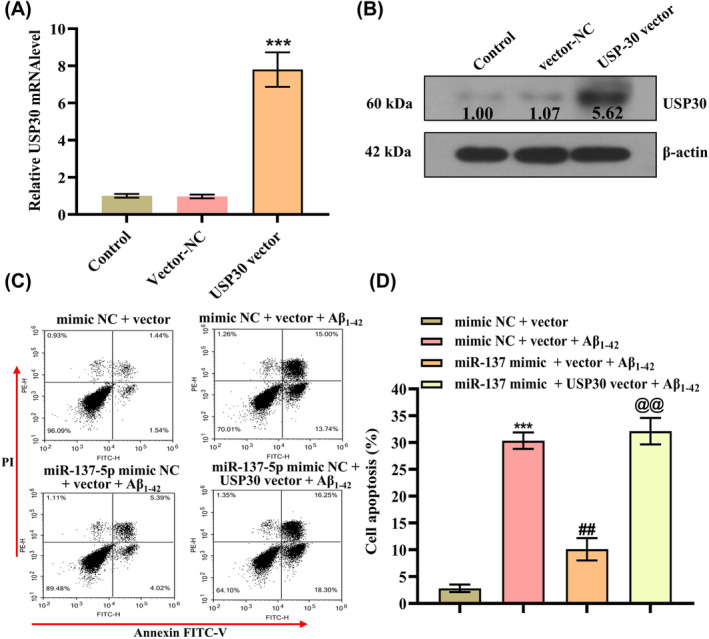
USP30 (ubiquitin‐specific peptidase 30) overexpression impeded the protective effect of miR‐137‐5p on Aβ1–42‐induced cytotoxicity. (A) qRT‐PCR and (B) Western blot analysis of USP30 mRNA (messenger RNA) and protein levels in SH‐SY5Y cells after transfection with USP30 vector or vector control, ***p < 0.001. (C, D) Flow cytometry analysis of cell apoptosis in each group, * represents compared with mimic NC + vector, ***p < 0.001; # represents compared with mimic NC + vector + Aβ1–42, ## p < 0.01; @ represents compared with miR‐137‐5p mimic + mimics NC + Aβ1–42 group, @@ p < 0.01.
3.4. Improved cognition and Aβ deposition by miR‐137‐5p through targeting USP30 in AD mice
Inspired by the in vitro results, we also validated the effect of miR‐137‐5p/USP30 axis in AD models. Figure 4A shows that the wild C57 mice were randomly assigned into four groups, namely the normal controls, the d‐galactose and aluminum chloride–treated mice as the AD models, the miR‐137‐5p agomir‐treated group, and the miR‐137‐5p agomir + USP30‐treated group. During the 5‐day acquisition testing period, the control group animals exhibited a gradual decrease in latencies when locating the concealed platform, indicating repeated training had a positive impact (Figure 4B). The AD model mice exhibited significantly higher latencies, lower platform crossing time, and time in the target quadrant than those in the control group, indicating the learning‐ and memory‐impaired model was effectively established. Conversely, miR‐137‐5p agomir could lead to a decrease in escape latency and increase in platform crossing time, and the time in the target quadrant when compared with the model group, while overexpression USP30 could prevent these improvements in mice (Figure 4B,D). Similarly, Figure 4E shows the sparse distribution of hippocampal and cortex neurons of mice in the AD model group, along with a notable reduction in the number of cell layers. Additionally, the cells appeared blurred, atrophied, and fragmented, indicating a substantial impairment. Encouragingly, the administration of miR‐137‐5p agomir successfully improved these pathological alterations, indicating a beneficial protective effect against hippocampal and cortex neuron damage in AD mice. Meanwhile, overexpression of USP30 alleviated the neuroprotection effect of miR‐137‐5p agomir on neuron damage (Figure 4E).
FIGURE 4.
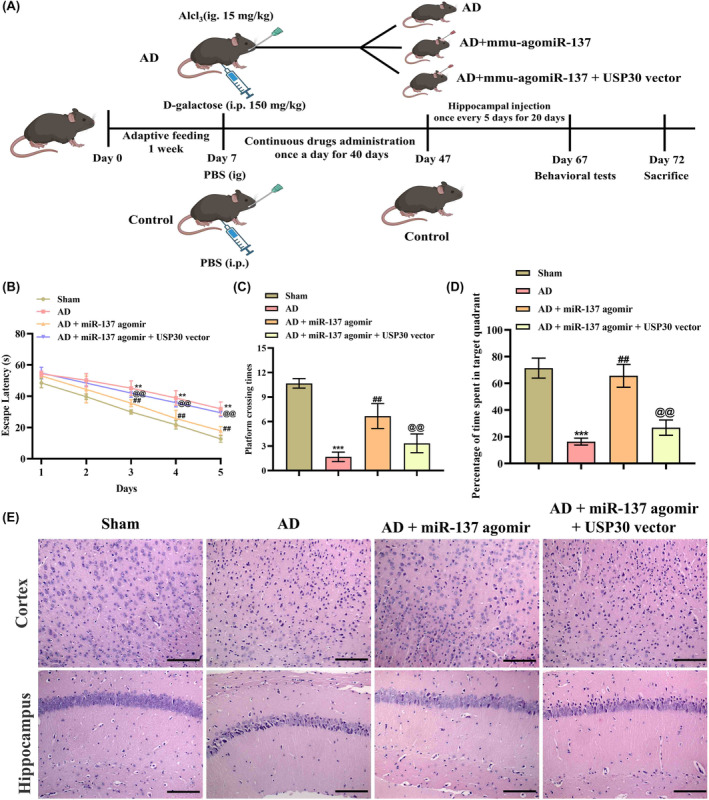
miR‐137‐5p improves spatial memory and cognitive deficit in AD (Alzheimer's disease) mice by downregulating USP30 (ubiquitin‐specific peptidase 30). (A) Schematic diagram of interventions in animal experiments. Morris water maze test analysis of (B) movement time, (C) platform crossing time, and (D) target quadrant dwell time of mice in water maze (n = 6). (E) H&E (hematoxylin–eosin) staining analysis of the pathological changes in mice hippocampus and cortex tissues (magnification: ×200, scale bar: 100 μm). * represents compared with sham group, **p < 0.01, ***p < 0.001. # represents compared with AD group, ## p < 0.01. @ represents compared with AD + miR‐137‐5p agomir group, @@ p < 0.01.
Additionally, Nissl staining of mice hippocampal and cortex tissues evidenced that miR‐137‐5p agomir can lead to the restoration of nerve cell morphology and volume, in addition to the increased number of cytoplasmic Nissl body compared with AD mice, whereas overexpression of USP30 abrogated these pathological changes to a certain extent (Figure 5A). In line with the aforementioned findings, TUNEL assay results also demonstrated miR‐137‐5p agomir could inhibit neuronal apoptosis and USP30 rescued this effect (Figure 5B,D). Western blotting analysis indicated that USP30, Aβ1–42, and Tau hyperphosphorylation levels were increased in AD mice compared with the sham group, and miR‐137‐5p agomir effectively alleviated this phenomenon (Figure 5E,F). Collectively, these data suggested miR‐137‐5p could protect against neuron damage in mice through targeting USP30.
FIGURE 5.
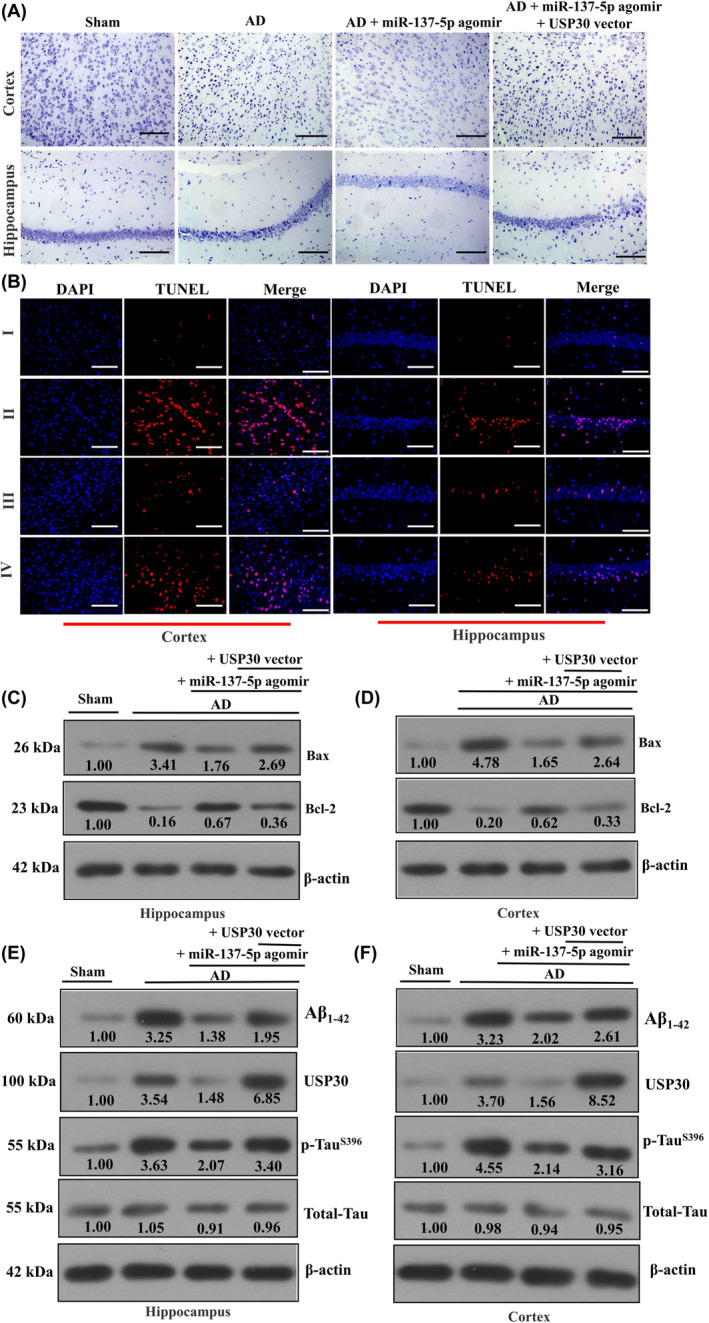
miR‐137‐5p rescues cortex and hippocampus neuron damage. (A) Representative images of Nissl staining for cortex and hippocampus neurons (magnification: ×200, scale bar: 100 μm). (B) TUNEL (TdT‐mediated dUTP nick‐end labeling) assay analysis of neuronal apoptosis in cortex and hippocampus regions of AD (Alzheimer's disease) mice (magnification: ×400, scale bar: 50 μm). (C–F) Western blot analysis of the protein levels of Bax, Bcl‐2, USP30 (ubiquitin‐specific peptidase 30), Aβ1–42, and p‐TauS396 in cortex and hippocampus regions of AD mice.
4. DISCUSSION
In recent decades, miRNAs have significant potential in the diagnosis and treatment of AD due to their crucial role in posttranscriptional gene regulation. 31 These small noncoding RNA molecules can serve as biomarkers for AD if abnormalities or alterations are detected. During the early stages of AD development, specific miRNAs undergo significant changes in expression, presenting potential early warning signs. Notably, increased expression of miRNA‐206 has been associated with cognitive decline and pathological progression in AD. 32 Conversely, miRNA‐29b and miRNA‐181c expression levels are already downregulated in the early stages of AD, offering new avenues for AD screening. 33 Moreover, miRNAs hold remarkable potential in AD treatment as they function as therapeutic targets, regulating gene expression to alter the neurodegeneration process. For instance, miRNA‐132 plays a neuroprotective role by suppressing neuroinflammation when its expression is increased, thereby delaying disease progression. 34 Research has indicated that the utilization or precise manipulation of endogenous miRNAs offers a potentially advantageous substitute for current RNA‐based treatments. 35 Research has uncovered significant alterations in the expression levels of numerous miRNAs. This powerful evidence underscores their influence on cellular aging and neurodegeneration. Similarly, several previous studies have indicated a decrease in miR‐137‐5p expression in the blood of individuals with AD. Consequently, we sought to investigate the intricate molecular mechanisms that underlie the involvement of miR‐137‐5p during AD progression. Our study provides compelling evidence supporting the notion that AD patients exhibit decreased expression of miR‐137‐5p and increased expression of USP30. Furthermore, we have identified USP30 as a direct target of miR‐137‐5p. Intriguingly, our findings suggest that the restoration of miR‐137‐5p expression contributes to the toxicity and apoptosis induced by Aβ1–42 through its targeting of USP30.
Recent research has shown substantial deviations in the expression levels of miR‐137 in AD, linking it to pathological progressions and cognitive downturns. Notably concentrated in the dentate gyrus—a subsection of the hippocampal structure—miR‐137 demonstrates vital involvement in neuronal growth and disease manifestation. 36 miR‐137 has been documented to exhibit downregulation in the hippocampus after excessive ketamine treatment, and its overexpression was shown to safeguard against hippocampal neurodegeneration and memory loss induced by ketamine. 37 Moreover, miR‐137 was found to be linked with susceptibility to schizophrenia through the modulation of networks implicated in neural growth and cognitive function, which frequently coincided with Parkinson's disease. 38 Our previous studies also evidenced miR‐137 could reduce Tau hyperphosphorylation in AD mice and cells. 39 Therefore, further research is necessary to understand the specific role of miR‐137 in the pathogenesis of AD and the potential effects of targeted therapy against miR‐137. In our investigation, we conducted gain‐of‐function experiments, which revealed that miR‐137 overexpression could counteract Aβ1–42‐induced cell damage and apoptosis in SH‐SY5Y cells by modulating USP30.
USP30, a deubiquitinase localized in mitochondria, antagonizes mitophagy that is triggered by the ubiquitin ligase parkin (also referred to as PARK2) and protein kinase PINK1. 40 , 41 Overexpression of USP30 can inhibit parkin's propensity to prompt mitophagy by detaching the ubiquitin that parkin appends to damaged mitochondria. Conversely, a diminution in USP30 activity furthers the disintegration of mitochondria in neuronal cells. 42 Several research investigations propose that the expression levels of USP30 experience a notable augmentation in diverse neurodegenerative disorders, including AD, Parkinson's disease, and Huntington's disease. 43 In addition, several inhibitors targeting USP30 can reduce mitochondrial stress response, decrease neuronal death, and improve behavioral deficits in animal models of neurodegenerative diseases. USP30 has been implicated in the regulation of mitophagy, an essential cellular process that governs the quality control of mitochondria, which are often found compromised in individuals with AD. 44 Furthermore, perturbations caused by USP30 in the nuclear‐encoded mitochondrial proteins are associated with the pathophysiology of AD, suggesting their potential as effective therapeutic targets. Consistently, our data also demonstrated USP30 was significantly upregulated in AD patients and associated with neuronal damage. However, there is still a need for an in‐depth understanding of the function and regulatory mechanisms of USP30 in the progression of AD.
By binding to the 3′UTR of target mRNA molecules, miRNAs control gene expression at the posttranscriptional phase, leading to gene suppression and hindered protein synthesis. 45 miR‐137 is a versatile miRNA that plays an important role in various biological processes (neuronal development, neuroprotection, immune response, and tumor suppression) by regulating different downstream genes. 46 , 47 , 48 For example, miR‐137 minimizes Aβ‐triggered neurotoxicity by inactivating the NF‐κB pathway via targeting TNFAIP1 in Neuro2a cells. 49 Serum‐secreted exosomes containing miR‐137 impact the oxidative stress in neurons by controlling OXR1 within the framework of Parkinson's disease. 50 MiR‐137‐5p has been found to relieve inflammation by increasing the expression of IL‐10R1 in rats suffering from spinal cord injuries. 51 In this study, we first showed that miR‐137‐5p can alleviate the symptoms of AD in mice and cells, primarily through the direct downregulation of USP30.
In conclusion, our study revealed that Aβ1–42 treatment notably downregulated miR‐137‐5p and upregulated USP30 in cortical neurons and SH‐SY5Y cells. We also demonstrated that miR‐137‐5p counteracted Aβ1–42‐induced neurotoxicity by targeting USP30 in SH‐SY5Y cells, elucidating the molecular mechanism of miR‐137 in relation to Aβ‐induced neurotoxicity. Thus, miR‐137 may serve as a potential therapeutic target for AD treatment.
AUTHOR CONTRIBUTIONS
Yang Jiang carried out experiments and wrote the manuscript; Wei Bian, Jing Chen, and Xiao Pan Cao contributed to some experiments and analyzed data; Chun Yao Dong and Ying Xiao were responsible for clinical sample collection and processing; Xiao Hong Sun and Bing Xu supervised and designed the research. All the authors approved the final version of the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare in the current work.
ETHICS STATEMENT
All the animal experiments were approved by ethical committees of Shenyang First People's Hospital (approval number: 2021, research review number: 04).
ACKNOWLEDGMENTS
We thank the patient consent giving samples for research purposes.
Jiang Y, Bian W, Chen J, et al. miRNA‐137‐5p improves spatial memory and cognition in Alzheimer's mice by targeting ubiquitin‐specific peptidase 30. Anim Models Exp Med. 2023;6:526‐536. doi: 10.1002/ame2.12368
Funding information
Liaoning Province Science and Technology Project (grant/award number: 2019‐BS‐221) and Shenyang Science and Technology Project (grant/award number: 19‐112‐4‐040).
Contributor Information
Bing Xu, Email: xb1968131@163.com.
XiaoHong Sun, Email: xhsun@cmu.edu.cn.
REFERENCES
- 1. Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet. 2021;397(10284):1577‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ossenkoppele R, van der Kant R, Hansson O. Hansson tau biomarkers in Alzheimer's disease: towards implementation in clinical practice and trials. Lancet Neurol. 2022;21(8):726‐734. [DOI] [PubMed] [Google Scholar]
- 3. Passeri E, Elkhoury K, Morsink M, et al. Alzheimer's disease: treatment strategies and their limitations. Int J Mol Sci. 2022;23(22):13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang G, Li R, Bai Q, Alty J. Multimodal learning of clinically accessible tests to aid diagnosis of neurodegenerative disorders: a scoping review. Health Inf Sci Syst. 2023;11(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Banerjee R, Rai A, Iyer SM, Narwal S, Tare M. Animal models in the study of Alzheimer's disease and Parkinson's disease: a historical perspective. Animal Model Exp Med. 2022;5(1):27‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gowda P, Reddy PH, Kumar S. Deregulated mitochondrial microRNAs in Alzheimer's disease: focus on synapse and mitochondria. Ageing Res Rev. 2022;73:101529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Su L, Li R, Zhang Z, Liu J, du J, Wei H. Identification of altered exosomal microRNAs and mRNAs in Alzheimer's disease. Ageing Res Rev. 2022;73:101497. [DOI] [PubMed] [Google Scholar]
- 8. Nunomura A, Perry G. Perry RNA and oxidative stress in Alzheimer's disease: focus on microRNAs. Oxid Med Cell Longev. 2020;2020:2638130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu X, Wang Q, Zhao H, et al. Role of miR‐21‐5p/FilGAP axis in estradiol alleviating the progression of monocrotaline‐induced pulmonary hypertension. Animal Model Exp Med. 2022;5(3):217‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saliminejad K, Khorram Khorshid HR, Soleymani Fard S, Ghaffari SH. An overview of microRNAs: biology, functions, therapeutics, and analysis methods. J Cell Physiol. 2019;234(5):5451‐5465. [DOI] [PubMed] [Google Scholar]
- 11. Lu TX, Rothenberg ME. MicroRNA. J Allergy Clin Immunol. 2018;141(4):1202‐1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pozniak T, Shcharbin D, Bryszewska M. Circulating microRNAs in medicine. Int J Mol Sci. 2022;23(7):3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Song Z, Qu Y, Xu Y, et al. Microarray microRNA profiling of urinary exosomes in a 5XFAD mouse model of Alzheimer's disease. Animal Model Exp Med. 2021;4(3):233‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vandendriessche C, Bruggeman A, Van Cauwenberghe C, et al. Extracellular vesicles in Alzheimer's and Parkinson's disease: small entities with large consequences. Cell. 2020;9(11):2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Serpente M, Fenoglio C, D'Anca M, et al. MiRNA profiling in plasma neural‐derived small extracellular vesicles from patients with Alzheimer's disease. Cell. 2020;9(6):1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tian M, Shen J, Qi Z, Feng Y, Fang P. Bioinformatics analysis and prediction of Alzheimer's disease and alcohol dependence based on ferroptosis‐related genes. Front Aging Neurosci. 2023;15:1201142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chum PP, Bishara MA, Solis SR, Behringer EJ. Cerebrovascular miRNAs track early development of Alzheimer's disease and target molecular markers of angiogenesis and blood flow regulation. J Alzheimers Dis. 2023;1‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pereira RL, Oliveira D, Pêgo AP, Santos SD, Moreira FTC. Electrochemical miRNA‐34a‐based biosensor for the diagnosis of Alzheimer's disease. Bioelectrochemistry. 2023;154:108553. [DOI] [PubMed] [Google Scholar]
- 19. Li Y, Meng S, Di W, et al. Amyloid‐β protein and MicroRNA‐384 in NCAM‐labeled exosomes from peripheral blood are potential diagnostic markers for Alzheimer's disease. CNS Neurosci Ther. 2022;28(7):1093‐1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu CG, Zhao Y, Lu Y, et al. ABCA1‐labeled exosomes in serum contain higher microRNA‐193b levels in Alzheimer's disease. Biomed Res Int. 2021;2021:5450397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walgrave H, Penning A, Tosoni G, et al. microRNA‐132 regulates gene expression programs involved in microglial homeostasis. iScience. 2023;26(6):106829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yin J, Lin J, Luo X, et al. miR‐137: a new player in schizophrenia. Int J Mol Sci. 2014;15(2):3262‐3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vazifehmand R, Ali DS, Othman Z, et al. The evaluation expression of non‐coding RNAs in response to HSV‐G47∆ oncolytic virus infection in glioblastoma multiforme cancer stem cells. J Neurovirol. 2022;28(4–6):566‐582. [DOI] [PubMed] [Google Scholar]
- 24. Li W, Zhang X, Zhuang H, et al. MicroRNA‐137 is a novel hypoxia‐responsive microRNA that inhibits mitophagy via regulation of two mitophagy receptors FUNDC1 and NIX. J Biol Chem. 2014;289(15):10691‐10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanooghi D, Lotfi A, Bagher Z, et al. Large‐scale analysis of microRNA expression in motor neuron‐like cells derived from human umbilical cord blood mesenchymal stem cells. Sci Rep. 2022;12(1):5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang B, Tang Y, Wang H, et al. Down‐regulation of long non‐coding RNA HOTAIR promotes angiogenesis via regulating miR‐126/SCEL pathways in burn wound healing. Cell Death Dis. 2020;11(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kurien BT, Scofield RH. Western blotting. Methods. 2006;38(4):283‐293. [DOI] [PubMed] [Google Scholar]
- 28. Pan Q, Guo K, Xue M, Tu Q. Estrogen protects neuroblastoma cell from amyloid‐β 42 (Aβ42)‐induced apoptosis via TXNIP/TRX axis and AMPK signaling. Neurochem Int. 2020;135:104685. [DOI] [PubMed] [Google Scholar]
- 29. Li X, Zhang J, Yang Y, Wu Q, Ning H. MicroRNA‐340‐5p increases telomere length by targeting telomere protein POT1 to improve Alzheimer's disease in mice. Cell Biol Int. 2021;45(6):1306‐1315. [DOI] [PubMed] [Google Scholar]
- 30. Cai Y, Chai Y, Fu Y, et al. Salidroside ameliorates Alzheimer's disease by targeting NLRP3 inflammasome‐mediated pyroptosis. Front Aging Neurosci. 2021;13:809433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rezaee D, Saadatpour F, Akbari N, et al. The role of microRNAs in the pathophysiology of human central nervous system: a focus on neurodegenerative diseases. Ageing Res Rev. 2023;92:102090. [DOI] [PubMed] [Google Scholar]
- 32. Peng D, Wang Y, Xiao Y, et al. Extracellular vesicles derived from astrocyte‐treated with haFGF(14‐154) attenuate Alzheimer phenotype in AD mice. Theranostics. 2022;12(8):3862‐3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou H, Zhang R, Lu K, et al. Deregulation of miRNA‐181c potentially contributes to the pathogenesis of AD by targeting collapsin response mediator protein 2 in mice. J Neurol Sci. 2016;367:3‐10. [DOI] [PubMed] [Google Scholar]
- 34. Wei Z, Meng X, El Fatimy R, et al. Environmental enrichment prevents Aβ oligomer‐induced synaptic dysfunction through mirna‐132 and hdac3 signaling pathways. Neurobiol Dis. 2020;134:104617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xia P, Chen J, Liu Y, et al. MicroRNA‐22‐3p ameliorates Alzheimer's disease by targeting SOX9 through the NF‐κB signaling pathway in the hippocampus. J Neuroinflammation. 2022;19(1):180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kong Y, Liang X, Liu L, et al. High throughput sequencing identifies microRNAs mediating α‐synuclein toxicity by targeting neuroactive‐ligand receptor interaction pathway in early stage of drosophila Parkinson's disease model. PLoS ONE. 2015;10(9):e0137432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang C, Zhang X, Zheng J, et al. Upregulation of miR‐137 protects anesthesia‐induced hippocampal neurodegeneration. Int J Clin Exp Pathol. 2014;7(8):5000‐5007. [PMC free article] [PubMed] [Google Scholar]
- 38. Wright C, Turner JA, Calhoun VD, et al. Potential impact of miR‐137 and its targets in schizophrenia. Front Genet. 2013;4:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jiang Y, Xu B, Chen J, et al. Micro‐RNA‐137 inhibits tau hyperphosphorylation in Alzheimer's disease and targets the CACNA1C gene in transgenic mice and human neuroblastoma SH‐SY5Y cells. Med Sci Monit. 2018;24:5635‐5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bingol B, Tea JS, Phu L, et al. The mitochondrial deubiquitinase USP30 opposes parkin‐mediated mitophagy. Nature. 2014;510(7505):370‐375. [DOI] [PubMed] [Google Scholar]
- 41. Wang F, Gao Y, Zhou L, et al. USP30: structure, emerging physiological role, and target inhibition. Front Pharmacol. 2022;13:851654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ordureau A, Paulo JA, Zhang J, et al. Global landscape and dynamics of parkin and USP30‐dependent ubiquitylomes in iNeurons during mitophagic signaling. Mol Cell. 2020;77(5):1124‐1142.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luo H, Krigman J, Zhang R, Yang M, Sun N. Pharmacological inhibition of USP30 activates tissue‐specific mitophagy. Acta Physiol (Oxf). 2021;232(3):e13666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sanchez‐Martinez A, Martinez A, Whitworth AJ. FBXO7/ntc and USP30 antagonistically set the ubiquitination threshold for basal mitophagy and provide a target for Pink1 phosphorylation in vivo. PLoS Biol. 2023;21(8):e3002244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martinez B, Peplow PV. MicroRNAs in mouse and rat models of experimental epilepsy and potential therapeutic targets. Neural Regen Res. 2023;18(10):2108‐2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu J, Xu Y, Tang H, et al. miR‐137 is a diagnostic tumor‐suppressive miRNA that targets SPHK2 to promote M1‐type tumor‐associated macrophage polarization. Exp Ther Med. 2023;26(2):397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pimenta R, Mioshi CM, Gonçalves GL, et al. Intratumoral restoration of miR‐137 plus cholesterol favors homeostasis of the miR‐137/coactivator p160/AR axis and negatively modulates tumor progression in advanced prostate cancer. Int J Mol Sci. 2023;24(11):9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chmielewska N, Wawer A, Wicik Z, Osuch B, Maciejak P, Szyndler J. miR‐9a‐5p expression is decreased in the hippocampus of rats resistant to lamotrigine: a behavioural, molecular and bioinformatics assessment. Neuropharmacology. 2023;227:109425. [DOI] [PubMed] [Google Scholar]
- 49. He D, Tan J, Zhang J. Zhang miR‐137 attenuates Aβ‐induced neurotoxicity through inactivation of NF‐κB pathway by targeting TNFAIP1 in Neuro2a cells. Biochem Biophys Res Commun. 2017;490(3):941‐947. [DOI] [PubMed] [Google Scholar]
- 50. Jiang Y, Liu J, Chen L, et al. Serum secreted miR‐137‐containing exosomes affects oxidative stress of neurons by regulating OXR1 in Parkinson's disease. Brain Res. 2019;1722:146331. [DOI] [PubMed] [Google Scholar]
- 51. Lv ZC, Cao XY, Guo YX, et al. MiR‐137‐5p alleviates inflammation by upregulating IL‐10R1 expression in rats with spinal cord injury. Eur Rev Med Pharmacol Sci. 2019;23(11):4551‐4557. [DOI] [PubMed] [Google Scholar]