

INTRODUCTION
Sickle cell disease (SCD) is caused by the presence of hemoglobin S (HbS, α2β2s). The syndrome comprises different genotypes that include homozygous S (HbSS): compound heterozygous forms of HbS with HbC (HbSC) and β-thalassemia (HbSβ0 thalassemia and HbSβ+ thalassemia). In patients of African ancestry, HbSS is the most common genotype at 65% to 70%, followed by HbSC (about 30%) and, the rest, HbSβ thalassemia.1,2 SCD is the most common inherited blood disorder in the United States, and a major public health problem in Africa and Asia. In the United States, it affects an estimated 100,000 Americans (the vast majority are African Americans and the rest are of Hispanic descent), and about 1 in 365 African American newborns.3,4 In the past few decades, widespread implementation of newborn screening, prophylactic penicillin and vaccination, and increased health care access have contributed to improved childhood survival, now 96% to 98% in high-income countries.5–7 As a consequence, in these better-resourced countries, the burden of disease has shifted to adults, where it has evolved into a chronic disorder with multiorgan damage and substantial morbidity. Overall, life expectancy of patients with SCD is still reduced by more than 2 decades compared with the general population, but more adults are living longer. Recent studies have estimated the median survival for patients with HbSS and Sβ0 thalassemia (the most severe sickle genotypes) at 58 years in the United States,8 and 67 years in a single center in the United Kingdom.9
Older adults with SCD require greater support to address the many emerging disease complications as they age, in addition to the conditions related to physiologic aging.10 The lag in recognition of this growing adult population with SCD and resultant inadequate resource allocation pose multiple challenges for managing the older patient. A further challenge is the inequity of treatment not only between high- and low-income countries but also within well-resourced countries; in the United States, most adults with SCD fail to receive recommended treatment, whereas patients in low-income countries lack access to comprehensive care considered standard in high-income countries.11
All patients with SCD, however mild their symptoms appear to be, are continuously undergoing some degree of chronic end-organ deterioration, the rate of development of chronic disease complications being modulated by their SCD genotype (patients with HbSC and HbS/mild beta-thalassemia exhibit less deterioration than HbSS and HbS/severe beta-thalassemia), genetic background, lifestyle, health care access, and environment. Within the genotype of HbSS, observational cohorts suggested that several factors modulate the rate of development of chronic disease complications. For instance, the largest series of 102 adults older than 60 in Jamaica identified 40 adults aged 60 to 87 years of age that were still alive. Higher fetal hemoglobin level (HbF) was associated with greater survival, and although these patients experienced fewer acute painful crises, nevertheless, their anemia and renal function progressively worsened as they aged.12 Another insidious complication is silent ischemic cerebral infarcts that worsen cognitive function and contribute to morbidity in the older adult. A conglomerate of biomarkers or genetic markers13,14 has been put forward to predict disease severity and mortality. However, for the individual patient, healthy lifestyle (lack of/minimal alcohol and tobacco exposure), normal body mass index, high levels of adherence to medication, and family support remain the most important determinants of longevity.15
Based on current survival estimates and the observation that by this age cumulative organ damage has an impact on morbidity and mortality,16,17 the “older adult” is defined here as ≥40 years of age. Although sickle-related complications become more prevalent with aging, non-sickle-related causes should always be considered and appropriately treated, because these comorbidities interact and add to disease burden. Presented are 3 clinical vignettes to highlight important clinical issues frequently encountered when managing the older patient with SCD.
Case 1: Sickle Cell Anemia Patient on Exchange Transfusion with Iron Overload
T.W. is a 50-year-old woman with sickle cell anemia, history of ischemic stroke, severe carotid stenosis, atrial fibrillation, pulmonary embolism/deep vein thrombosis (DVT), chronic pain, chronic leg ulcers with osteomyelitis, and hepatic cirrhosis on liver biopsy. As active therapy for SCD, she is on a simple transfusion regimen to maintain her HbS less than 50% and also receives hydroxyurea (HU) combined with erythropoietin, but she has not been very adherent with her iron chelation therapy. On MRI screening for iron overload, she was found to have a T2* value indicating cardiac siderosis and severe hepatic iron (a liver biopsy revealed cirrhosis with increased iron). She was admitted to the hospital for aggressive intravenous iron chelation with intravenous desferal. The patient is currently under evaluation for haploidentical stem cell transplantation.
Case 2: Sickle Cell Anemia Patient with Multiple Red Cell Alloantibodies
V.A. is a 64-year-old woman with HbSS disease on active therapy with HU and erythropoietin with a history of multiple episodes of acute chest syndrome (ACS; the most recent in 2014), systemic hypertension on lisinopril, and chronic lower back pain for which she is on a stable dose of long-acting opioids. Her hemoglobin ranges from 7 to 8 g/dL; a simple transfusion regimen had been proposed, but she is noted to have developed multiple clinically significant red cell alloantibodies. In addition, she has developed transfusional hemosiderosis and is currently on deferasirox at a stable dose of 34 mg/kg/d.
Case 3: Hemoglobin SC Patient with Newly Detected Lung Infiltrate
F.E. is a 63-year-old man with hemoglobin SC disease, systemic hypertension, osteonecrosis of the hip, chronic obstructive pulmonary disease secondary to cigarette smoke exposure, bilateral pulmonary embolism with likely chronic thromboembolic pulmonary hypertension (PH), intermittent atrial fibrillation, stroke in 2007, and priapism with residual impotence. In March 2018, he was diagnosed with a new lung mass and para-aortic lymphadenopathy. Just 6 months before, he had a pneumothorax due to spontaneous rupture of bullous emphysema. During workup for his lung mass, the patient was noted to have new onset thrombocytopenia, and over the next few months of being diagnosed with lung cancer, had an anterolateral wall myocardial infarction and died.
The 3 clinical vignettes illustrate the diverse clinical complications and issues relevant to medical practitioners dealing with the older adult with SCD.
PATHOPHYSIOLOGY AND SPECIFIC MORBIDITIES OF SICKLE CELL DISEASE
The central mechanism underlying the pathophysiology of SCD is polymerization of deoxy-HbS and the formation of irreversibly sickled red blood cells that trigger a cascade of events ultimately leading to acute painful vaso-occlusive episodes that are the hallmark of SCD. All patients with SCD are chronically anemic, because of ongoing hemolysis from the shortened lifespan of sickled erythrocytes (16–20 days compared with a normal lifespan of 120 days). Subsequent reperfusion of the ischemic tissue after vaso-occlusion generates free radicals and reactive oxygen species, which scavenge nitric oxide (NO). Continuing release of cell-free hemoglobin and subsequently heme add to depletion of NO. Chronic NO deficiency leads to platelet activation, increased vascular resistance, and endothelial dysfunction contributing to the development of vasculopathy. The ongoing vasculopathy and inflammation inflict damage on various organs and impacts the patients as they live into their fourth, fifth, and even sixth decade, transforming SCD into a chronic multiorgan disorder. One of the largest and longest longitudinal studies of adult SCD showed that approximately one-half of surviving patients by their fifth decade had some form of irreversible damage of lungs, kidneys, brain, retina, or bones significantly affecting their quality of life.16 Table 1 lists the commonly recognized complications associated with SCD seen in both children and adults, although severity and clinical course may differ depending on patient’s age. One should always consider that the older adult with SCD could also have unrelated comorbidities prevalent in the general population, that is, diabetes, systemic hypertension, and connective tissue disease, that further compound or accelerate sickle-related complications.
Table 1.
Commonly recognized sickle-related complications
| Complication | Definition | Comments |
|---|---|---|
| Pain | Acute pain, most commonly in the long bones, chest, back Chronic pain is pain lasting for>3mo | Acute episodic pain occurs throughout life. Adults with SCD experience pain on>54% of days100 Chronic pain occurs in>50% and about 40% of adults with SCD take daily opioids101 |
| Anemia | Acute anemia: A decline in hemoglobin of 2g/dL or more from steady-state values Chronic anemia: severity increases with age and major contributor to insidious organ dysfunction |
Variety of causes, including infection (transient red cell aplasia most commonly caused by acute parvo virus B19): acute hemolysis accompanying severe VOC. delayed transfusion reaction Other common causcs of anemia to consider in older patients: iron deficiency. B12 deficiency, hypothyroidism, and anemia of inflammation |
| ACS | Acute onset of respiratory symptoms with features similar to pneumonia | Although less frequent, outcome is more severe in adults30 |
| PH | Mean pulmonary artery pressure of>25mm Hg at rest, measured by right heart catheterization | 6.0%–10.4% prevalence during adulthood35, 36, 37, 102, 103 |
| Heart failure | Most commonly left ventricular failure | Universal to some degree in adults older than age 3039 |
| Chronic sickle lung disease | Progressive restrictive lung function deficit with fibrotic changes on high-rcsolution computed tomographic (CT) scan | Restrictive lung defects seen in>70% adults104 |
| VTE | DVT detected by duplex ultrasonography, ventilation perfusion scintigraphy, or CT pulmonary angiography | 11.2%−13.1% cumulative incidence rate in adulthood. High recurrence rates ~30%–40%. Contribution toward chronic thromboembolic PH. Consider indefinite anticoagulation.40, 41 Routine VTE prophylaxis during hospitalization and periods of increased thrombosis risk |
| Stroke | Acute cerebrovascular accident | Acute ischemic and hemorrhagic stroke. Hemorrhagic stroke affects both children and adults. 3-fold more in adults.105 Exclude traditional risk factors for ischemic stroke: hypertension, diabetes mellitus. hypcrlipidemia. atrial fibrillation and renal disease106 |
| SCI | Clinically silent lesions of 3 mm or more on MRI scanning | Important contributing factor to neurocognitive deficits: 53% in adults58 |
| Acute renal injury | Acute deterioration in renal function | Triggers include pain crisis, during ACS. acute drop in hemoglobin as in transient red cell aplasia. Often occurs as part of multiorgan failure |
| Renal failure | Deteriorating renal function, reduced concentrating ability, proteinuria, and progressive renal failure | Advanced disease (stage III-1V) in 4%−18% of adults48 |
| Priapism | Unwanted painful and sustained erection of the penis for more than 4h. often recurrent or persistent | 20%−89% lifetime prevalence in boys and men107 |
| AVN of bones | AVN of any bone, most commonly the femoral head and shoulder joint | >20% lifetime prevalence of symptomatic disease. Increased prevalence of asymptomatic disease108 |
| Leg ulceration | Most commonly around the malleolar regions | >14% lifetime prevalence 109, 110 |
| Cholelithiasis | Gallstones and gallbladder disease | Important genetic modifier is polymorphic (AT) repeats in promoter of UGT1AI gene.111 Gall bladder disease in 28% at median age of 28y16 |
| Retinopathy | Grade 1 —IV retinopathy | >30% of patients.112 Prevalence higher in HbSC113 |
Pain is the most common symptom for which patients seek medical attention and the most frequent complaint of the older adults with SCD (all 3 cases experience chronic pain and receive long-term pain medication). Acute painful sickle cell crisis that results from vaso-occlusion affecting bones and joints is the commonest cause of an emergency room visit and hospitalization.18,19 There are considerable differences in the frequency and intensity of sickle cell crises between adults and children; the average length of stay in adults was approximately 7.5 days18 compared with 4.4 days in children.20 In addition to vaso-occlusive crisis, osteomyelitis and septic arthritis can also give rise to acute skeletal pain, and clinical manifestations can be somewhat similar to acute painful crises. Older adults are more likely to have chronic pain, which is usually multifactorial from inflammation, central and peripheral neural sensitization, and avascular necrosis (AVN) of bone.21 A review on multiple dimensions of chronic pain in adults with SCD reported that chronic pain occurs in at least 29% of adults, most frequently in those 25 to 44 years of age.22 A recent study of chronic pain in twins showed that environmental factors are paramount in maintaining chronic pain.23 The complexity of chronic pain in adults is further illustrated by the persistence of pain despite successful reversal of sickle hematology post–stem cell transplant in a subgroup that had chronic pain pretransplant.24 Chronic pain negatively impacts quality of life, and its management can be challenging, because a significant knowledge gap exists in understanding the natural history and management of chronic pain in older adults.
Ongoing vasculopathy leads to chronic bone problems, such as osteopenia/osteoporosis, chronic arthritis, and osteonecrosis/AVN of bone.25 The incidence of AVN in SCD could vary from 3% to 50% and is highest among patients older than 45 years of age (34.9%).26 AVN most commonly affects the femoral head followed by the humeral head. Management of AVN is challenging given the limited evidence for standardized guidelines in most surgical procedures in SCD. Treatment options vary from conservative (eg, pain management, physical therapy, and decreased weight-bearing) to surgical (core decompression or arthroplasty). Early detection and intervention may delay progressive joint disease and improve quality of life. Total hip arthroplasty (THA) is usually reserved for patients with advanced AVN of the femur given its failure rate, although in recent years use of cementless prosthesis and joint management by an experienced surgeon and hematologist have improved outcomes of THA in SCD, prompting earlier intervention.27,28
Pulmonary complications account for significant morbidity and mortality in patients with SCD. Acute pulmonary complications include pneumonia, pulmonary embolism, and ACS. Although the incidence of ACS is lower in older adults compared with children (8.8 events per 100 patient-years in older adults vs 24.5 events per 100 patient-years in young children),29,30 the severity and mortality are higher in older adults, largely because of a higher incidence of bone marrow and fat emboli in adults31,32 and the presence of other organ comorbidities. Chronic pulmonary complications and sickle cell chronic lung disease are more prevalent in older adults and are characterized by impaired exercise tolerance, progressive heart failure, and impaired pulmonary function; causes include PH, pulmonary fibrosis, restrictive airway disease, and sleep-disordered breathing (nocturnal hypoxemia and obstructive sleep apnea).33,34 PH, defined as mean pulmonary artery pressure ≥25 mm Hg at rest diagnosed by right heart catheterization, affects 6% to 11% of SCD adults35–37 and is a leading cause of morbidity and early mortality.38 Transthoracic echocardiography (ECHO) has been used as a noninvasive tool for screening for PH. Elevated cardiac output and left ventricular volume overload secondary to chronic anemia are other factors underlying the cardiopulmonary complications in patients with SCD.39 As in other organ complications, deterioration in pulmonary function should prompt an appropriate screen for other causes, such as cancers, particularly in patients with a history of tobacco exposure (see case 3).
Venous thromboembolism (VTE) occurs in both young and older SCD patients but at a higher prevalence in the latter and is likely to have a greater effect on morbidity and mortality in the older patient.40,41 The inherent hypercoagulability associated with SCD and other iatrogenic factors, such as placement of long-term indwelling catheters, recurrent hospitalization, hypoxia, inadequate VTE prophylaxis, and increasing age, influences thrombotic risk.42 Although the exact mechanisms responsible for the hypercoagulable state in SCD are unclear, they appear to be directly related to abnormal expression of procoagulant, for example, tissue factor, and proadhesive molecules, for example, P-selectin. Tissue factor expression on pulmonary endothelial cells could trigger coagulation in vivo and possibly contribute to the in situ pulmonary thrombosis observed in at least 20% of individuals with ACS.43 In addition to worsening acute sickle-related complications, thrombosis in the deep veins and pulmonary emboli contributes to chronic leg ulceration and thromboembolic PH, respectively (seen in case 3), adding to morbidity. The management of DVT and PE in SCD is similar to that of the general population and has been addressed in previous issues of the Clinics in Geriatric Medicine and more recently in clinical guidelines from the American Society of Hematology.44 For unprovoked VTE in SCD patient, the optimal duration of anticoagulation remains undefined with proponents for extension beyond the traditional 3-month treatment period based on higher incidence of recurrent VTEs in this population group (a management strategy used in both cases 1 and 3). Extended duration anticoagulation is likely to be associated with a higher bleeding risk in older SCD patients and especially those exposed to concomitant antiplatelet therapy. A deeper understanding of SCD associated thrombophilia and the efficacy and risk of anticoagulant agents promise to provide considerable benefit to the population of older adults. Until such time, the benefit of long-term/indefinite anticoagulation should be reevaluated frequently in these patients.
All patients with SCD suffer some degree of renal impairment (also referred to as sickle cell nephropathy, SCN), most commonly manifested as hyperfiltration, hyposthenuria (diminished concentrating ability), and albuminuria. Microalbuminuria (defined as a urinary albumin/creatinine ratio >4.5 mg/mmol) is an early manifestation of SCN, reaching a prevalence of greater than 50% in older patients.45 In a small number of patients (4%–12%), renal function progressively declines, leading to end-stage renal disease (ESRD) requiring renal replacement therapy.46 Approximately one-quarter of patients older than 60 years of age have stage III–IV kidney disease, and ESRD was identified as the cause of death in 45% of patients aged 60 years or older.47 It is important to note that no pathognomonic lesion defines SCN, and that other causes of renal disease, such as lupus nephritis and diabetic, hypertensive, hepatitis C virus or human immunodeficiency virus-associated nephropathy, should be considered in the differential.48 Acceleration of renal impairment should prompt investigation of common comorbidities, in particular, hypertension and diabetes.17 Observational data have shown that proteinuria responds to treatment with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers, and treatment with these agents should be considered when the urine protein/creatinine ratio is persistently elevated greater than 50 mg/mmol.49,50 Disease-modifying therapy (HU) should be considered alongside this specific treatment.48,51 ESRD eventually requires standard renal replacement therapy, including erythropoietin-stimulating agents (ESA), dialysis, or renal transplant. The use of ESA when combined with HU, as in case 2, can be effective in controlling anemia.48,52
The spectrum of hepatic dysfunction (sickle hepatopathy) ranges from mildly abnormal liver function tests, self-limited cholestasis, to severe forms of sickle cell intrahepatic cholestasis and cirrhosis.53 The overall prevalence of liver dysfunction in patients with SCD has not been well established and probably underappreciated because of the inability to discriminate abnormal liver enzymes resulting from hemolysis from those due to intrinsic liver disease. Thus, evaluating and treating any underlying and coexisting condition that can contribute to liver dysfunction is important. With the increasing use of blood transfusion, transfusional hemosiderosis is an emerging cause of liver disease (as observed in cases 1 and 2).54
Stroke, seizure, and transient ischemic attacks have been reported in more than 30% of SCD adults with SCD, and a history of these events correlates with early mortality.8 Ischemic or thrombotic stroke peaks over the age of 29 years, and hemorrhagic stroke is most frequent in the 20- to 29-year-old age group.55 Silent cerebral infarction (SCI) and incidental aneurysms are common in adults with SCD; the latter varies from 9% to 15% and tends to be multiple.56–58 Neurocognitive impairment increases with age and can be compounded by sickle-related pathologic condition, such as SCI, moya moya, overt clinical stroke, anemia, and nocturnal hypoxia, throughout the patient’s lifespan (see case 1, where deteriorating neurocognitive function could impact the informed consent process for experimental curative therapies). Non-sickle-related factors, hypertension, hyperlipidemia, cardiac disease, are likely to predispose the patient to vascular dementia, adding to the accelerative cognitive decline in older adults.59
MANAGEMENT
General Principles
Although therapeutic advances have altered patient morbidity and mortality, treatment-related (eg, secondary iron overload from blood transfusions) and physiologic aging-related complications present additional management challenges. Due consideration should be given in management of complications of pain and other acute clinical events (eg, stroke) when certain medications may require appropriate dose adjustments.10 In addition, data and evidence-based guidelines are lacking for most complications.60–62 Although treatment of the specific complications may be limited, it is prudent to optimize disease-modifying therapy (including HU or transfusion) in addition to general medical support (treatment of heart failure, correction of hypoxemia with oxygen therapy, and anticoagulation for those with thromboembolism).
When patients have a long period of stability and present with worsening of their sickle phenotype, the clinician should always be aware of new comorbidities as an explanation.17 Coexistence of other chronic diseases, especially if poorly controlled, may lead to worsening of SCD. Similarly, the higher risk of colorectal cancer among African American individuals should prompt appropriate screening, perhaps earlier than the recommended age of 50 years. Hence, a management strategy of regular comprehensive review for early recognition, prevention, and treatment of organ damage should be an essential part of routine health care in older adults with SCD (Box 1). The authors recommend a multidisciplinary approach by a team of relevant specialists with supportive input from allied health professionals, including psychology and physiotherapy and referral to specialized centers if needed.
Box1.
An outline of management strategies in the older adult with sickle cell disease
Identify a primary care physician to coordinate total patient care
- Comprehensive multisystem review to evaluate complications
- Pain: days off work due to pain
- Acute pain: Hospital admissions, frequency of pain at home
- Chronic pain: Including use of opiate analgesia
- Detailed medical history/details of comorbidities
- Sickle related:
- Renal dysfunction (proteinuria, hematuria)
- Cardiorespiratory symptoms
- Cerebrovascular: Any memory concerns
- Leg ulcers
- Visual
- History of thrombosis and anticoagulant therapy
- Nonsickle related: Diabetes, hypertension, gout
- Medication and vaccinations
- Transfusion history (to include frequency, transfusion reaction)
- Vitals (blood pressure, pulse oximetry, weight)
- Baseline laboratory testing (full blood count, biochemistry, hemolysis panel, liver panel, urinalysis)
- Investigation: ECHO, pulmonary function, sleep study
- Evaluate for evidence of organ dysfunction
- Proteinuria ± hematuria → joint renal clinic
- ECHO, tricuspid regurgitant jet velocity ≥2.5 → joint cardiopulmonary clinic
- Liver function, evidence of intrahepatic cholestasis. → joint hepatology clinic
- Avascular necrosis → joint orthopedic clinic
- Headaches, cognitive decline → joint neurology clinic/neuropsychology assessment
- Visual symptoms → yearly ophthalmology review
- Daytime or nocturnal hypoxia → joint sleep/respiratory clinic
Management of other comorbidities
Investigate/monitor therapy: HU, and similar
The 2 most widely available therapies for patients with SCD are HU and blood transfusion.
Disease-Modifying Therapies
Hydroxyurea
The main benefits of HU therapy are thought to accrue from induction of HbF that has an inhibitory effect on the rate of HbS polymerization. The efficacy of HU is also thought to arise from other effects, for example, reducing inflammation, suppressing leukocytosis and thrombocytosis, diminishing reactive oxygen species, and possibly by serving as an NO donor. HU is a disease-modifying treatment that reduced acute SCD complications in a placebo controlled randomized trial63 and whose long-term use in nonrandomized studies demonstrated enhanced survival in patients with HbSS disease.64,65 Because HU has a long-term safety profile with minimal side effects, the authors would argue that SCD patients should be offered the option of early rather than late HU treatment. HU has also recently been found to be relatively safe in settings with high infectious disease burden, increasing confidence in broadening the clinical use of this drug.66,67
It is therefore rather surprising that despite its clear benefits, HU therapy remains underutilized, probably because of a reluctance on the part of both patients and clinicians. Moreover, even when HU is prescribed, adherence can be below par, suggesting an important role for adherence counseling by the physician, which is often neglected. These considerations are compounded in the older adult with SCD with cognitive impairment. HU therapy in the older patient deserves a special mention: the older patient is likely to be more sensitive to HU dosage and easily susceptible to developing neutropenia, thrombocytopenia, and reticulocytopenia. HU treatment thus requires careful and more frequent monitoring in older adults compared with younger patients. The reasons for the relative sensitivity to HU myelotoxicity are not well known, but suggestions include repeated bone marrow infarcts and reduced hemopoietic reserve. In the older adult, the authors routinely initiate HU at 7.5 mg/kg/d and gently escalate the dose while carefully monitoring the absolute neutrophil, reticulocyte, and the platelet counts until reaching a maximum tolerated dose. Neutropenia (<1500/mL) and reticulocytopenia (<90,000/mL) would trigger withholding the drug and reinitiation at a lower dose. Combining HU with erythropoietin allows more aggressive HU dosing, which is required in high-risk disease and renal insufficiency both frequently encountered in the older adult with SCD (see case 1). In extreme cases, even a minimal dose of 5 mg/kg/d is not tolerated, requiring a search for alternative therapies.
Transfusion
Red blood cell transfusion of is one of the most effective therapies for patients with SCD. The rationale of blood transfusion in SCD is to (a) Improve oxygen carrying capacity of blood to tissues, and (b) Dilute concentration of circulating sickled erythrocytes to improve microvascular circulation.68,69 Blood transfusion can be given as simple (or top-up) or as an exchange transfusion, either manually or automated using an apheresis machine. Intermittent blood transfusion may be used for treatment of acute complications or preparation of surgery, or long term to reduce the incidence and severity of sickle-related organ damage.
Randomized trials have demonstrated the efficacy of blood transfusion for primary and secondary prevention of stroke in children with SCD. Although stroke pathophysiology is likely to be different among adults and children, it seems reasonable to extrapolate these findings at least to ischemic stroke in adults. Many adults with a history of stroke in childhood or adulthood remain on exchange transfusion as secondary prevention albeit with a less stringent target HbS (<50% as opposed to <30%). It should be noted that although older adults with acute ischemic stroke may benefit from exchange transfusion (standard practice in children), thrombolysis may also be in order, particularly if there are traditional risk factors (eg, hypertension and hyperlipidemia); appropriate treatment and order of treatment require careful discussion with a stroke physician.
Given the clinical benefits, not surprisingly the use of blood transfusion therapy in the management of SCD is increasing. Clinical decisions to use transfusion for prevention of recurrent acute pain, priapism, leg ulcers, renal dysfunction and PH are based more on observation and clinical experience than evidence. Although very effective in preventing the several complications of SCD, transfusion therapy carries the risk of secondary iron overload, alloimmunization (both observed in these 3 cases), and transmission of blood-borne diseases, such as hepatitis C.69,70
Alloimmunization to RBC antigens is relatively more common in patients with SCD71–73 and can lead to difficulties in sourcing appropriately matched blood products, delayed transfusion reactions triggering more vaso-occlusive crises, significant morbidities, and fatalities in some cases.74–76 Alloimmunization to multiple RBC antigens often prompts the development of autoantibodies posing challenges of sourcing appropriately matched blood products; in many cases, the “least incompatible” blood is transfused under immunologic cover. As prevention, many blood transfusion centers use extended red cell phenotyping, with some having adopted molecular genotyping using microarray chips for red blood cell phenotype prediction, and some proposing genome sequencing.77–79
Recurrent blood transfusion, whether sporadic or long term,80 also leads to iron overload that most commonly affects the liver; cardiac and endocrine siderosis are unusual in SCD.81 MRI is a noninvasive method of measuring body iron load and should be used in those with a high annual blood transfusion rate and raised serum ferritin.82 Iron chelation therapy should be considered in those with serum ferritin greater than 1000 mg/L and liver iron concentration of greater than 7 mg/g dry weight and chelation efficacy monitored with serial serum ferritin and MRI measurements.83,84
Newer treatments
In 2017, the Food and Drug Administration approved pharmacy grade L-glutamine (Endari)85 for the reduction of acute complications of SCD in both adults and children. Being only the second drug approved for SCD treatment in the last 30 years, this news was met with much enthusiasm from the broader hematology community. However, the failure to include patients with mild renal impairment and one-third of the study population discontinuing the study drug raise concerns about the generalizability of these findings to older adults. Nevertheless, for older adults with normal kidney function experiencing acute crises ineffectively controlled by HU, addition of L-glutamine appears justifiable. Other agents currently in phase 3 clinical trials include Crizanlizumab (a P-selectin inhibitor),86 small molecule anti-sickling agents, for example, GBT440 (Voxelotor),87 and anti-inflammatory agents, for example, rivipansel.88 These agents hold promise for patients failing to benefit from HU or those having severe symptoms despite HU therapy. Inducing HbF expression by targeting the DNA methyltransferase 1 with an oral combination of small molecules, decitabine and tetrahydrouridine, was safe and is now being evaluated for efficacy in a phase 2 study.89,90 The development of novel agents and consideration of multimodality treatments raise several questions and possible concerns. One of these is whether combination therapy with a variety of agents targeting different pathophysiologic events will become the new standard, and will this approach have any impact on the chronic vascular pathobiology of SCD. Another concern is whether these novel therapies will be cost-effective in low- and middle-resource settings, and if their efficacy end points are cost-effective to justify their use in that particular setting.
Curative therapies
When neither HU nor regular blood transfusion is effective in limiting disease progression, other interventions should be considered even in the older adult with SCD.10 These interventions include fully matched or haploidentical hematopoietic stem cell transplant (HSCT), gene therapy, or experimental drug treatment.91–93
The favorable outcome of HLA-identical sibling HSCT in children94 prompts allogeneic transplantation sooner rather than later in young patients with symptomatic SCD.95 However, outcomes are poorer in those older than 16 years,94 using standard myeloablative conditioning. Reduction of the intensity of conditioning (nonmyeloablative) has expanded allogeneic transplantation as a treatment option for adult patients with preexisting organ dysfunction.96 The HLA-matched sibling donors can be HbAS or HbAA, successful transplantation results in mixed chimerism with remarkably little graft-versus-host disease, and reversal of sickle hematology. The nonmyeloablative conditioning regimen developed at the authors’ institution has now been adopted by several institutions.91 However, although successful HSCT leads to resolution of pain in most adult SCD patients, those with chronic pain and no identifiable contributory sickle complications pre-HSCT were more likely to experience persistent pain post-HSCT.24
Although HLA-matched sibling transplantation has been very successful for adults and children, most patients with SCD do not have an HLA-matched sibling.97 Alternative donor options include haploidentical, unrelated umbilical cord blood, and matched unrelated donors, most promising of which is haploidentical family members (a treatment strategy under consideration for case 1).92 Another experimental option is gene addition using the anti-sickling β-globin vector containing the HbAT87Q mutation (Bluebird Bio) that recently reported encouraging results in patients with severe beta-thalassemia.98 This treatment is still in its early stages in SCD, but preliminary reports have revealed therapeutic expression in all 7 patients in which it was tested.99 The relative lack of toxicity of this treatment and the ability to induce one’s own hematopoietic precursors ex vivo are distinct advantages when managing the older adult with SCD.
SUMMARY AND FUTURE DIRECTIONS
Therapeutic advances and improved access to health care have contributed to greater life expectancy and consequently a growing population of older adults with SCD. The older adult with SCD is likely to have multiple aging comorbidities added to the cumulative complications of a chronic disease that largely affects the central nervous, cardiopulmonary, and renal systems. A background of chronic pain and chronic anemia not only impairs activities of daily living but also potentially worsens cognitive decline and impacts every component of their health care management and delivery. The older adult with SCD tends to have a complex medication regimen that needs careful consideration and appropriate dosage adjustments given impairment of multiple organs. Moreover, cognitive dysfunction impacts adherence requiring involvement of family members or reminders that assist in compliance with the prescribed drug regimen. The scarcity of data from clinical trials specifically aimed at treating SCD in older adults limits an evidence-based approach to treatment. In the absence of such evidence, data from randomized trials in children and younger adults with SCD are routinely extrapolated to guide management of the older adult with SCD. Thus, the basis of most management decisions for these patients reflects expert opinion or the consensus of domain experts. In recent years, great strides have been made in reducing traditional sickle-related complications, such as proteinuria, blood pressure, VTE, and cardiopulmonary disease, particularly evident in older age groups. Overall mortalities have improved, even though these are still elevated compared with adults without SCD. These trends indicate that the time has come for a fundamental shift in traditional care from single disease practices to a more patient-centered framework in a multidisciplinary team with a greater focus on geriatric conditions (Fig. 1).
Fig. 1.
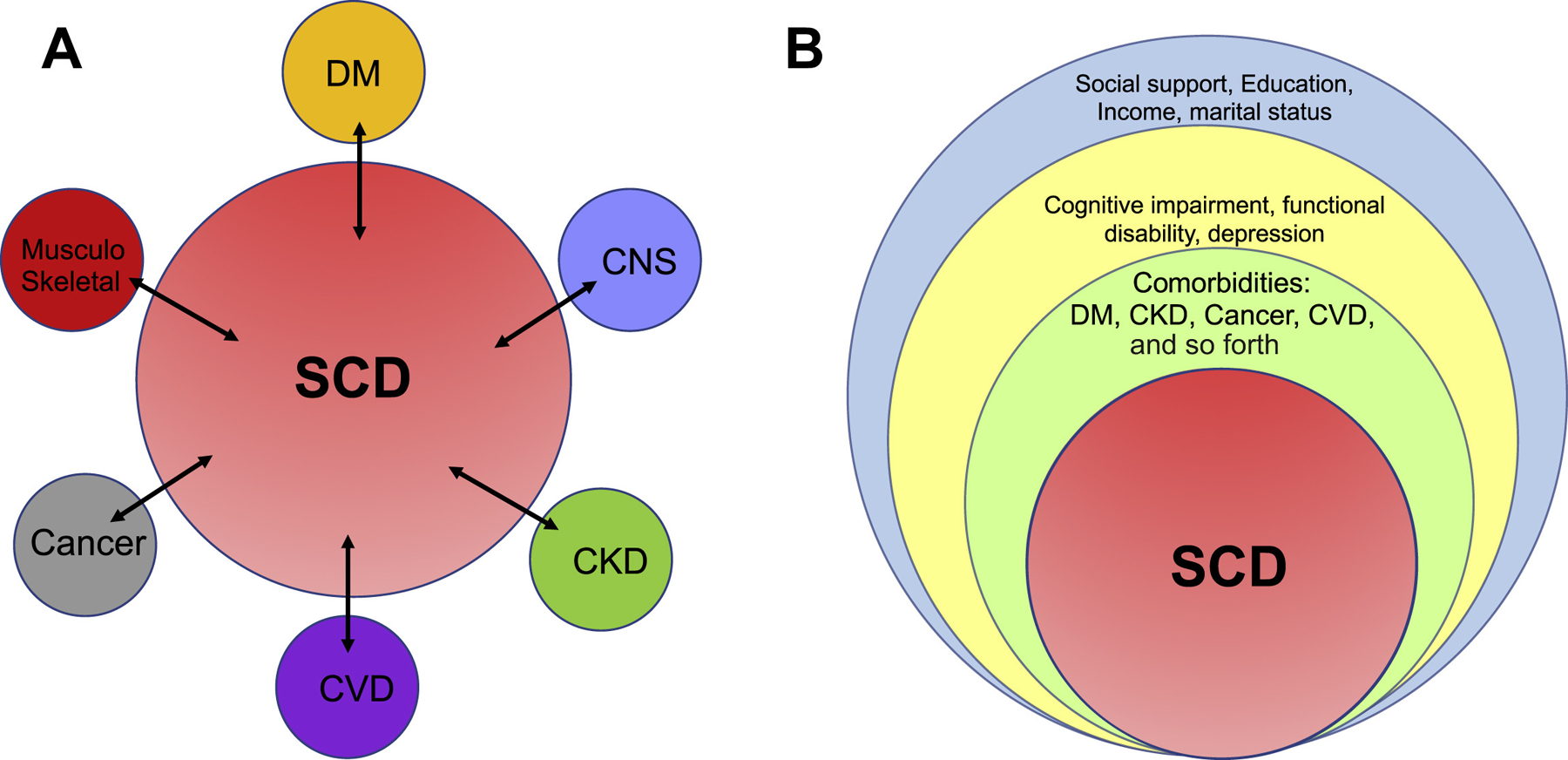
A proposed model of managing SCD in the older adult. (A) The traditional single disease focused framework of comorbidity where comorbid conditions are considered as disease pairs, such as SCD and diabetes mellitus, SCD and cardiovascular disease, and SCD and dementia. Most clinical practice guidelines are based on this framework. (B) Multimorbid conceptual framework demonstrating a more patient-centric approach to managing the older adult with SCD in the context of multiple chronic conditions, geriatric syndromes, functional status, and social determinants of health. CKD, chronic kidney disease; CNS, central nervous system; CVD, cardiovascular disease; DM, diabetes mellitus.
KEY POINTS.
Survival of patients with sickle cell disease (SCD) in well-resourced countries has improved greatly in the last 60 years; a newborn in the United States and United Kingdom can now expect to live to adulthood.
Although survival has improved, life expectancy of SCD patients is still 20 to 30 years less than that of the general population.
In well-resourced countries, the burden of disease has now shifted to adults, in which SCD has evolved into a debilitating disorder with complications associated with long-term chronic illness, in addition to those due to aging.
The lag in providing resources and adequately equipping health care professionals with appropriate skills poses multiple challenges for effective management of this growing population of older adults with SCD.
Patient outcomes are optimal when managed by a core of multidisciplinary specialists.
Disclosure Statement:
This research was funded by Intra-mural grant support.
REFERENCES
- 1.Williams TN, Thein SL. Sickle cell anemia and its phenotypes. Annu Rev Genomics Hum Genet 2018;19:113–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ware RE, de Montalembert M, Tshilolo L, et al. Sickle cell disease. Lancet 2017; 390(10091):311–23. [DOI] [PubMed] [Google Scholar]
- 3.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med 2010;38(4 Suppl):S512–21. [DOI] [PubMed] [Google Scholar]
- 4.Hassell KL. Sickle cell disease: a continued call to action. Am J Prev Med 2016; 51(1 Suppl 1):S1–2. [DOI] [PubMed] [Google Scholar]
- 5.Quinn CT, Rogers ZR, McCavit TL, et al. Improved survival of children and adolescents with sickle cell disease. Blood 2010;115(17):3447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica 2007;92(7):905–12. [DOI] [PubMed] [Google Scholar]
- 7.Paulukonis ST, Eckman JR, Snyder AB, et al. Defining sickle cell disease mortality using a population-based surveillance system, 2004 through 2008. Public Health Rep 2016;131(2):367–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elmariah H, Garrett ME, De Castro LM, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol 2014;89(5): 530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gardner K, Douiri A, Drasar E, et al. Survival in adults with sickle cell disease in a high-income setting. Blood 2016;128(10):1436–8. [DOI] [PubMed] [Google Scholar]
- 10.Thein SL, Tisdale J. Sickle cell disease—unanswered questions and future directions in therapy. Semin Hematol 2018;55(2):51–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGann PT, Hernandez AG, Ware RE. Sickle cell anemia in sub-Saharan Africa: advancing the clinical paradigm through partnerships and research. Blood 2017;129(2):155–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Serjeant GR, Chin N, Asnani MR, et al. Causes of death and early life determinants of survival in homozygous sickle cell disease: the Jamaican cohort study from birth. PLoS One 2018;13(3):e0192710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rees DC, Gibson JS. Biomarkers in sickle cell disease. Br J Haematol 2012; 156(4):433–45. [DOI] [PubMed] [Google Scholar]
- 14.Kalpatthi R, Novelli EM. Measuring success: utility of biomarkers in sickle cell disease clinical trials and care. Hematol Am Soc Hematol Educ Program 2018;2018(1):482–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ballas SK, Pulte ED, Lobo C, et al. Case series of octogenarians with sickle cell disease. Blood 2016;128(19):2367–9. [DOI] [PubMed] [Google Scholar]
- 16.Powars DR, Chan LS, Hiti A, et al. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine 2005;84(6):363–76. [DOI] [PubMed] [Google Scholar]
- 17.Sandhu MK, Cohen A. Aging in sickle cell disease: co-morbidities and new issues in management. Hemoglobin 2015;39(4):221–4. [DOI] [PubMed] [Google Scholar]
- 18.Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol 2005; 79(1):17–25. [DOI] [PubMed] [Google Scholar]
- 19.Lanzkron S, Little J, Field J, et al. Increased acute care utilization in a prospective cohort of adults with sickle cell disease. Blood Adv 2018;2(18):2412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Panepinto JA, Brousseau DC, Hillery CA, et al. Variation in hospitalizations and hospital length of stay in children with vaso-occlusive crises in sickle cell disease. Pediatr Blood Cancer 2005;44(2):182–6. [DOI] [PubMed] [Google Scholar]
- 21.Lanzkron S, Haywood C Jr. The five key things you need to know to manage adult patients with sickle cell disease. Hematol Am Soc Hematol Educ Program 2015;2015:420–5. [DOI] [PubMed] [Google Scholar]
- 22.Taylor LE, Stotts NA, Humphreys J, et al. A review of the literature on the multiple dimensions of chronic pain in adults with sickle cell disease. J Pain Symptom Manage 2010;40(3):416–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burri A, Ogata S, Rice D, et al. Twelve-year follow-up of chronic pain in twins: changes in environmental and genetic influence over time. Eur J Pain 2018. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 24.Darbari DS, Liljencrantz J, Ikechi A, et al. Pain and opioid use after reversal of sickle cell disease following HLA-matched sibling haematopoietic stem cell transplant. Br J Haematol 2019;184(4):690–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almeida A, Roberts I. Bone involvement in sickle cell disease. Br J Haematol 2005;129(4):482–90. [DOI] [PubMed] [Google Scholar]
- 26.Milner PF, Kraus AP, Sebes JI, et al. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med 1991;325(21):1476–81. [DOI] [PubMed] [Google Scholar]
- 27.Issa K, Naziri Q, Maheshwari AV, et al. Excellent results and minimal complications of total hip arthroplasty in sickle cell hemoglobinopathy at mid-term follow-up using cementless prosthetic components. J Arthroplasty 2013;28(9):1693–8. [DOI] [PubMed] [Google Scholar]
- 28.Jack CM, Howard J, Aziz ES, et al. Cementless total hip replacements in sickle cell disease. Hip Int 2016;26(2):186–92. [DOI] [PubMed] [Google Scholar]
- 29.Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood 1997;89(5):1787–92. [PubMed] [Google Scholar]
- 30.Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994;84(2):643–9. [PubMed] [Google Scholar]
- 31.Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med 2000;342(25):1855–65. [DOI] [PubMed] [Google Scholar]
- 32.Dang NC, Johnson C, Eslami-Farsani M, et al. Bone marrow embolism in sickle cell disease: a review. Am J Hematol 2005;79(1):61–7. [DOI] [PubMed] [Google Scholar]
- 33.Khan AB, Kesse-Adu R, Breen C, et al. A descriptive study of the characteristics of older adults with sickle cell disease. Am J Hematol 2018;93(2):E38–40. [DOI] [PubMed] [Google Scholar]
- 34.Sharma S, Efird JT, Knupp C, et al. Sleep disorders in adult sickle cell patients. J Clin Sleep Med 2015;11(3):219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parent F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med 2011;365(1):44–53. [DOI] [PubMed] [Google Scholar]
- 36.Fonseca GH, Souza R, Salemi VM, et al. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J 2012;39(1):112–8. [DOI] [PubMed] [Google Scholar]
- 37.Mehari A, Gladwin MT, Tian X, et al. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA 2012;307(12):1254–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gordeuk VR, Castro OL, Machado RF. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood 2016;127(7):820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mushemi-Blake S, Melikian N, Drasar E, et al. Pulmonary haemodynamics in sickle cell disease are driven predominantly by a high-output state rather than elevated pulmonary vascular resistance: a prospective 3-dimensional echocardiography/Doppler study. PLoS One 2015;10(8):e0135472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naik RP, Streiff MB, Haywood C Jr, et al. Venous thromboembolism incidence in the cooperative study of sickle cell disease. J Thromb Haemost 2014;12(12): 2010–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brunson A, Lei A, Rosenberg AS, et al. Increased incidence of VTE in sickle cell disease patients: risk factors, recurrence and impact on mortality. Br J Haematol 2017;178(2):319–26. [DOI] [PubMed] [Google Scholar]
- 42.Shet AS, Wun T. How I diagnose and treat venous thromboembolism in sickle cell disease. Blood 2018;132(17):1761–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mekontso Dessap A, Deux JF, Abidi N, et al. Pulmonary artery thrombosis during acute chest syndrome in sickle cell disease. Am J Respir Crit Care Med 2011;184(9):1022–9. [DOI] [PubMed] [Google Scholar]
- 44.Witt DM, Nieuwlaat R, Clark NP, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: optimal management of anticoagulation therapy. Blood Adv 2018;2(22):3257–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Day TG, Drašar ER, Fulford T, et al. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica 2012;97(2):201–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drawz P, Ayyappan S, Nouraie M, et al. Kidney disease among patients with sickle cell disease, hemoglobin SS and SC. Clin J Am Soc Nephrol 2016; 11(2):207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serjeant GR, Serjeant BE, Mason KP, et al. The changing face of homozygous sickle cell disease: 102 patients over 60 years. Int J Lab Hematol 2009;31(6): 585–96. [DOI] [PubMed] [Google Scholar]
- 48.Sharpe CC, Thein SL. How I treat renal complications in sickle cell disease. Blood 2014;123(24):3720–6. [DOI] [PubMed] [Google Scholar]
- 49.Aoki RY, Saad ST. Enalapril reduces the albuminuria of patients with sickle cell disease. Am J Med 1995;98(5):432–5. [DOI] [PubMed] [Google Scholar]
- 50.Falk RJ, Scheinman J, Phillips G, et al. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. N Engl J Med 1992;326(14):910–5. [DOI] [PubMed] [Google Scholar]
- 51.Laurin LP, Nachman PH, Desai PC, et al. Hydroxyurea is associated with lower prevalence of albuminuria in adults with sickle cell disease. Nephrol Dial Transplant 2014;29(6):1211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steinberg MH. Erythropoietin for anemia of renal failure in sickle cell disease. N Engl J Med 1991;324(19):1369–70. [DOI] [PubMed] [Google Scholar]
- 53.Berry PA, Cross TJ, Thein SL, et al. Hepatic dysfunction in sickle cell disease: a new system of classification based on global assessment. Clin Gastroenterol Hepatol 2007;5(12):1469–76 [quiz: 1369]. [DOI] [PubMed] [Google Scholar]
- 54.Drasar E, Igbineweka N, Vasavda N, et al. Blood transfusion usage among adults with sickle cell disease—a single institution experience over ten years. Br J Haematol 2011;152(6):766–70. [DOI] [PubMed] [Google Scholar]
- 55.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998;91(1):288–94. [PubMed] [Google Scholar]
- 56.Birkeland P, Gardner K, Kesse-Adu R, et al. Intracranial aneurysms in sickle-cell disease are associated with the hemoglobin SS genotype but not with Moyamoya syndrome. Stroke 2016;47(7):1710–3. [DOI] [PubMed] [Google Scholar]
- 57.Nabavizadeh SA, Vossough A, Ichord RN, et al. Intracranial aneurysms in sickle cell anemia: clinical and imaging findings. J Neurointerv Surg 2016;8(4):434–40. [DOI] [PubMed] [Google Scholar]
- 58.Kassim AA, Pruthi S, Day M, et al. Silent cerebral infarcts and cerebral aneurysms are prevalent in adults with sickle cell anemia. Blood 2016;127(16): 2038–40. [DOI] [PubMed] [Google Scholar]
- 59.Vermeer SE, Prins ND, den Heijer T, et al. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med 2003;348(13):1215–22. [DOI] [PubMed] [Google Scholar]
- 60.National Institutes of Health: National Heart Lung and Blood Institute. Evidence-based management of sickle cell disease. In: NIH expert panel report. Bethesda (MD): U.S Department of Health and Human Services; 2014. Available at: http://www.nhlbi.nih.gov/sites/www.nhlbi.nih.gov/files/sickle-cell-disease-report.pdf. [Google Scholar]
- 61.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312(10):1033–48. [DOI] [PubMed] [Google Scholar]
- 62.Savage WJ, Buchanan GR, Yawn BP, et al. Evidence gaps in the management of sickle cell disease: a summary of needed research. Am J Hematol 2015;90(4): 273–5. [DOI] [PubMed] [Google Scholar]
- 63.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995;332(20):1317–22. [DOI] [PubMed] [Google Scholar]
- 64.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol 2010;85(6):403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS). Blood 2010; 115(12):2354–63. [DOI] [PubMed] [Google Scholar]
- 66.Opoka RO, Ndugwa CM, Latham TS, et al. Novel use of Hydroxyurea in an African Region with Malaria (NOHARM): a trial for children with sickle cell anemia. Blood 2017;130(24):2585–93. [DOI] [PubMed] [Google Scholar]
- 67.Tshilolo L, Tomlinson G, Williams TN, et al. Hydroxyurea for children with sickle cell anemia in sub-saharan Africa. N Engl J Med 2019;380(2):121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ballas SK, Kesen MR, Goldberg MF, et al. Beyond the definitions of the phenotypic complications of sickle cell disease: an update on management. Scientific-WorldJournal 2012;2012:949535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chou ST, Fasano RM. Management of patients with sickle cell disease using transfusion therapy: guidelines and complications. Hematol Oncol Clin North Am 2016;30(3):591–608. [DOI] [PubMed] [Google Scholar]
- 70.Chou ST. Transfusion therapy for sickle cell disease: a balancing act. Hematol Am Soc Hematol Educ Program 2013;2013:439–46. [DOI] [PubMed] [Google Scholar]
- 71.Vichinsky EP, Earles A, Johnson RA, et al. Alloimmunization in sickle cell anemia and transfusion of racially unmatched blood. N Engl J Med 1990;322(23): 1617–21. [DOI] [PubMed] [Google Scholar]
- 72.O’Suoji C, Liem RI, Mack AK, et al. Alloimmunization in sickle cell anemia in the era of extended red cell typing. Pediatr Blood Cancer 2013;60(9):1487–91. [DOI] [PubMed] [Google Scholar]
- 73.Chou ST, Jackson T, Vege S, et al. High prevalence of red blood cell alloimmunization in sickle cell disease despite transfusion from Rh-matched minority donors. Blood 2013;122(6):1062–71. [DOI] [PubMed] [Google Scholar]
- 74.Vidler JB, Gardner K, Amenyah K, et al. Delayed haemolytic transfusion reaction in adults with sickle cell disease: a 5-year experience. Br J Haematol 2015; 169(5):746–53. [DOI] [PubMed] [Google Scholar]
- 75.de Montalembert M, Dumont MD, Heilbronner C, et al. Delayed hemolytic transfusion reaction in children with sickle cell disease. Haematologica 2011;96(6): 801–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pirenne F, Yazdanbakhsh K. How I safely transfuse patients with sickle-cell disease and manage delayed hemolytic transfusion reactions. Blood 2018;131(25): 2773–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chou ST, Evans P, Vege S, et al. RH genotype matching for transfusion support in sickle cell disease. Blood 2018;132(11):1198–207. [DOI] [PubMed] [Google Scholar]
- 78.Hendrickson JE, Tormey CA, Shaz BH. Red blood cell alloimmunization mitigation strategies. Transfus Med Rev 2014;28(3):137–44. [DOI] [PubMed] [Google Scholar]
- 79.Hendrickson JE, Tormey CA. Rhesus pieces: genotype matching of RBCs. Blood 2018;132(11):1091–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Drasar E, Vasavda N, Igbineweka N, et al. Serum ferritin and total units transfused for assessing iron overload in adults with sickle cell disease. Br J Haematol 2012;157(5):645–7. [DOI] [PubMed] [Google Scholar]
- 81.Porter J, Garbowski M. Consequences and management of iron overload in sickle cell disease. Hematol Am Soc Hematol Educ Program 2013;2013:447–56. [DOI] [PubMed] [Google Scholar]
- 82.St Pierre TG, Clark PR, Chua-anusorn W, et al. Non-invasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood 2005;105(2):855–61. [DOI] [PubMed] [Google Scholar]
- 83.Davis BA, Allard S, Qureshi A, et al. Guidelines on red cell transfusion in sickle cell disease. Part I: principles and laboratory aspects. Br J Haematol 2017; 176(2):179–91. [DOI] [PubMed] [Google Scholar]
- 84.Davis BA, Allard S, Qureshi A, et al. Guidelines on red cell transfusion in sickle cell disease Part II: indications for transfusion. Br J Haematol 2017;176(2): 192–209. [DOI] [PubMed] [Google Scholar]
- 85.Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med 2018;379(3):226–35. [DOI] [PubMed] [Google Scholar]
- 86.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med 2017;376(5):429–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blyden G, Bridges KR, Bronte L. Case series of patients with severe sickle cell disease treated with voxelotor (GBT440) by compassionate access. Am J Hematol 2018. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 88.Telen MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 2015;125(17):2656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Molokie R, Lavelle D, Gowhari M, et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: a randomized phase 1 study. PLoS Med 2017;14(9):e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lavelle D, Engel JD, Saunthararajah Y. Fetal hemoglobin induction by epigenetic drugs. Semin Hematol 2018;55(2):60–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Guilcher GMT, Truong TH, Saraf SL, et al. Curative therapies: allogeneic hematopoietic cell transplantation from matched related donors using myeloablative, reduced intensity, and nonmyeloablative conditioning in sickle cell disease. Semin Hematol 2018;55(2):87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Joseph JJ, Abraham AA, Fitzhugh CD. When there is no match, the game is not over: alternative donor options for hematopoietic stem cell transplantation in sickle cell disease. Semin Hematol 2018;55(2):94–101. [DOI] [PubMed] [Google Scholar]
- 93.Esrick EB, Bauer DE. Genetic therapies for sickle cell disease. Semin Hematol 2018;55(2):76–86. [DOI] [PubMed] [Google Scholar]
- 94.Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017;129(11):1548–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fitzhugh CD, Walters MC. The case for HLA-identical sibling hematopoietic stem cell transplantation in children with symptomatic sickle cell anemia. Blood Adv 2017;1(26):2563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 2014;312(1):48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Walters MC, De Castro LM, Sullivan KM, et al. Indications and results of HLA-identical sibling hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant 2016;22(2):207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion-dependent beta-thalassemia. N Engl J Med 2018;378(16):1479–93. [DOI] [PubMed] [Google Scholar]
- 99.Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med 2017;376(9):848–55. [DOI] [PubMed] [Google Scholar]
- 100.Smith WR, Penberthy LT, Bovbjerg VE, et al. Daily assessment of pain in adults with sickle cell disease. Ann Intern Med 2008;148(2):94–101. [DOI] [PubMed] [Google Scholar]
- 101.Smith WR, McClish DK, Dahman BA, et al. Daily home opioid use in adults with sickle cell disease: the PiSCES project. J Opioid Manag 2015;11(3):243–53. [DOI] [PubMed] [Google Scholar]
- 102.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004;350(9):886–95. [DOI] [PubMed] [Google Scholar]
- 103.Gladwin MT, Barst RJ, Gibbs JS, et al. Risk factors for death in 632 patients with sickle cell disease in the United States and United Kingdom. PLoS One 2014; 9(7):e99489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Klings ES, Wyszynski DF, Nolan VG, et al. Abnormal pulmonary function in adults with sickle cell anemia. Am J Respir Crit Care Med 2006;173(11):1264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.DeBaun MR, Kirkham FJ. Central nervous system complications and management in sickle cell disease. Blood 2016;127(7):829–38. [DOI] [PubMed] [Google Scholar]
- 106.Strouse JJ, Jordan LC, Lanzkron S, et al. The excess burden of stroke in hospitalized adults with sickle cell disease. Am J Hematol 2009;84(9):548–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Adeyoju AB, Olujohungbe AB, Morris J, et al. Priapism in sickle-cell disease; incidence, risk factors and complications—an international multicentre study. BJU Int 2002;90(9):898–902. [DOI] [PubMed] [Google Scholar]
- 108.Hernigou P, Habibi A, Bachir D, et al. The natural history of asymptomatic osteonecrosis of the femoral head in adults with sickle cell disease. J Bone Joint Surg Am 2006;88(12):2565–72. [DOI] [PubMed] [Google Scholar]
- 109.Serjeant GR. Leg ulceration in sickle cell anemia. Arch Intern Med 1974;133(4): 690–4. [PubMed] [Google Scholar]
- 110.Koshy M, Entsuah R, Koranda A, et al. Leg ulcers in patients with sickle cell disease. Blood 1989;74(4):1403–8. [PubMed] [Google Scholar]
- 111.Vasavda N, Menzel S, Kondaveeti S, et al. The linear effects of alpha-thalassaemia, the UGT1A1 and HMOX1 polymorphisms on cholelithiasis in sickle cell disease. Br J Haematol 2007;138(2):263–70. [DOI] [PubMed] [Google Scholar]
- 112.Mathew R, Bafiq R, Ramu J, et al. Spectral domain optical coherence tomography in patients with sickle cell disease. Br J Ophthalmol 2015;99(7):967–72. [DOI] [PubMed] [Google Scholar]
- 113.Downes SM, Hambleton IR, Chuang EL, et al. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmology 2005;112(11):1869–75. [DOI] [PubMed] [Google Scholar]