

Abstract
BACKGROUND
Transthyretin amyloidosis, also called ATTR amyloidosis, is associated with accumulation of ATTR amyloid deposits in the heart and commonly manifests as progressive cardiomyopathy. Patisiran, an RNA interference therapeutic agent, inhibits the production of hepatic transthyretin.
METHODS
In this phase 3, double-blind, randomized trial, we assigned patients with hereditary, also known as variant, or wild-type ATTR cardiac amyloidosis, in a 1:1 ratio, to receive patisiran (0.3 mg per kilogram of body weight) or placebo once every 3 weeks for 12 months. A hierarchical procedure was used to test the primary and three secondary end points. The primary end point was the change from baseline in the distance covered on the 6-minute walk test at 12 months. The first secondary end point was the change from baseline to month 12 in the Kansas City Cardiomyopathy Questionnaire–Overall Summary (KCCQ-OS) score (with higher scores indicating better health status). The second secondary end point was a composite of death from any cause, cardiovascular events, and change from baseline in the 6-minute walk test distance over 12 months. The third secondary end point was a composite of death from any cause, hospitalizations for any cause, and urgent heart failure visits over 12 months.
RESULTS
A total of 360 patients were randomly assigned to receive patisiran (181 patients) or placebo (179 patients). At month 12, the decline in the 6-minute walk distance was lower in the patisiran group than in the placebo group (Hodges–Lehmann estimate of median difference, 14.69 m; 95% confidence interval [CI], 0.69 to 28.69; P = 0.02); the KCCQ-OS score increased in the patisiran group and declined in the placebo group (least-squares mean difference, 3.7 points; 95% CI, 0.2 to 7.2; P = 0.04). Significant benefits were not observed for the second secondary end point. Infusion-related reactions, arthralgia, and muscle spasms occurred more often among patients in the patisiran group than among those in the placebo group.
CONCLUSIONS
In this trial, administration of patisiran over a period of 12 months resulted in preserved functional capacity in patients with ATTR cardiac amyloidosis. (Funded by Alnylam Pharmaceuticals; APOLLO-B ClinicalTrials.gov number, NCT03997383.)
Transthyretin amyloidosis also called ATTR amyloidosis, is a progressive, debilitating, and fatal disease caused by misfolded transthyretin protein that accumulates as amyloid fibrils in multiple organs, including the heart, nerves, gastrointestinal tract, and musculoskeletal tissues.1–3 Patients with inherited TTR gene variants have hereditary, also known as variant, ATTR amyloidosis, which can manifest as cardiomyopathy, polyneuropathy, or a mixture of the two phenotypes.1,4,5 Patients with wild-type ATTR amyloidosis predominantly have cardiomyopathy, although polyneuropathy may coexist.2 Ongoing ATTR amyloid deposition in the heart drives the progression of infiltrative cardiomyopathy,6–8 leading to worsening heart failure, arrhythmias, and conduction disease.9–11 The median survival among patients with ATTR cardiac amyloidosis ranges from 2 to 6 years, depending on the disease stage at diagnosis.12 Current treatment options for ATTR amyloidosis with cardiomyopathy are limited. Tafamidis, which stabilizes the tetrameric transthyretin protein, was associated with reductions in death from any cause and cardiovascular-related hospitalizations after 30 months as compared with placebo in the ATTR-ACT trial; however, on average, functional capacity and quality of life declined among patients treated with tafamidis.13
Patisiran, an RNA interference therapeutic agent with a lipid nanoparticle delivery system, targets the common 3′ untranslated region of TTR messenger RNA in the liver to reduce circulating transthyretin protein levels in both variant and wild-type ATTR amyloidosis.14 Patisiran has been approved for the treatment of variant ATTR amyloidosis in patients with polyneuropathy15,16 on the basis of the results of the phase 3 APOLLO trial, which showed that patisiran halted or reversed the progression of neuropathy and improved quality of life.17,18 Furthermore, in a prespecified subgroup of patients in the APOLLO trial with evidence of cardiac amyloid involvement at baseline, measures of cardiac structure and function favored patisiran over placebo at 18 months.19 These data support the hypothesis that a reduction in amyloidogenic transthyretin protein can provide a therapeutic benefit in patients with ATTR cardiac amyloidosis. Patisiran is currently under investigation for the treatment of ATTR cardiac amyloidosis. Here, we present the primary efficacy and safety data from the 12-month double-blind period of the APOLLO-B trial, a phase 3, randomized, placebo-controlled trial involving patients with variant or wild-type ATTR cardiac amyloidosis.
METHODS
TRIAL OVERSIGHT
The APOLLO-B trial is an ongoing, international, phase 3, multicenter, double-blind, randomized, placebo-controlled trial of patisiran in patients with variant or wild-type ATTR cardiac amyloidosis. The institutional review board or independent ethics committee at each center approved the trial protocol and amendments, available with the full text of this article at NEJM.org. All patients provided written informed consent. The trial is being conducted in accordance with all applicable regulatory requirements, the International Council for Harmonisation Good Clinical Practice guidelines, and principles that have their origin in the Declaration of Helsinki. Alnylam Pharmaceuticals, the trial sponsor, designed the trial in collaboration with the principal investigators. The trial investigators collected data, which were analyzed by the sponsor and interpreted jointly by the sponsor and the authors. The authors prepared the first draft of the submitted manuscript with editorial assistance from Adelphi Communications, funded by Alnylam Pharmaceuticals. All the coauthors participated in the interpretation of the data. All the authors, who had access to the data, vouch for the accuracy and completeness of the data and for the fidelity of the trial to the protocol. All the coauthors contributed to the critical revision of the manuscript. All the investigators had confidentiality agreements with the sponsor.
PATIENTS
Key eligibility criteria included an age of 18 to 85 years; a diagnosis of ATTR cardiac amyloidosis (defined as the presence of transthyretin amyloid deposits on analysis of tissue biopsy specimens or fulfillment of validated nonbiopsy diagnostic criteria for ATTR cardiac amyloidosis20); evidence of cardiac involvement (confirmed by echocardiography), with an end-diastolic interventricular septal wall thickness exceeding 12 mm; and a history of heart failure. At baseline, some patients were receiving tafamidis, at the approved dose within each country, and others were not receiving it.
Key exclusion criteria were a New York Heart Association (NYHA) class of III and an ATTR amyloidosis stage of 3 (defined as an N-terminal pro–B-type natriuretic peptide [NT-proBNP] level of >3000 pg per milliliter occurring concomitantly with an estimated glomerular filtration rate [eGFR] of <45 ml per minute per 1.73 m2 of body surface area),12 an NYHA class of IV, a 6-minute walk distance of less than 150 m, a polyneuropathy disability score greater than II (possible scores, 0, I, II, IIIa, IIIb, and IV, with higher scores indicating more impaired walking ability), and cardiomyopathy not associated with ATTR amyloidosis (e.g., cardiomyopathy due to ischemic or valvular heart disease). Full inclusion and exclusion criteria are provided in the Supplementary Appendix (available at NEJM.org).
TRIAL DESIGN
Patients were randomly assigned (in a 1:1 ratio) to receive patisiran (at a dose of 0.3 mg per kilogram of body weight; maximum dose, 30 mg) or placebo intravenously once every 3 weeks for 12 months. Randomization was stratified according to the use of tafamidis at baseline (yes vs. no), genotype (variant vs. wild-type ATTR), and NYHA class and age at baseline (NYHA class I or II and age <75 years vs. all others). All patients received standard premedication to reduce the risk of infusion-related reactions with patisiran (see the Supplementary Appendix for additional details). Patients were instructed to take the recommended daily oral dose of vitamin A during the trial. At the end of the 12-month doubleblind period, patients were eligible to be enrolled in the ongoing open-label extension period (ClinicalTrials.gov number, NCT03997383).
END POINTS
For all efficacy end points, we assessed the change from baseline to month 12 in the patisiran group as compared with the placebo group. The primary end point was the change in functional capacity, as measured by the distance covered on the 6-minute walk test. The first secondary end point was the change in health status and quality of life, as measured by the Kansas City Cardiomyopathy Questionnaire–Overall Summary (KCCQ-OS) score (scores range from 0 to 100, with a score of 0 to 24 indicating very poor to poor quality of life, 25 to 49 poor to fair, 50 to 74 fair to good, and 75 to 100 good to excellent).21 The second secondary end point was a composite of death from any cause, cardiovascular events, and change from baseline in the 6-minute walk test distance at 12 months. The third secondary end point was a composite of death from any cause, hospitalizations for any cause, and urgent visits for heart failure over 12 months. Exploratory end points included the change in cardiac biomarkers NT-proBNP and troponin I, modified body-mass index (modified BMI, defined as the conventional BMI [weight in kilograms divided by square of height in meters] multiplied by the albumin level in grams per liter, with lower values indicating worse nutritional status), and echocardiographic measures of cardiac structure and function, which were assessed in a core laboratory. The pharmacodynamic effect of transthyretin protein reduction was assessed by measurement of the change in serum transthyretin levels from baseline to month 12. Safety was monitored throughout the trial and included assessments of adverse events, clinical laboratory variables, and vital signs. Further details on the end points and safety assessments are provided in the Supplementary Appendix.
STATISTICAL ANALYSIS
Assuming a treatment difference of approximately 29 m, with a standard deviation of 75 m, between the patisiran and placebo groups for the change from baseline to month 12 in the distance covered on the 6-minute walk test, we estimated that a sample size of 300 patients would be needed for the trial to have at least 90% power to detect a between-group treatment difference at a two-sided alpha level of 0.05. The primary end point was analyzed with the use of the stratified Wilcoxon rank-sum test and the stratified Hodges–Lehmann estimate of median difference, with stratification according to baseline use of tafamidis (yes vs. no). To control the overall type I error rate, the primary and secondary end points were tested in a prespecified hierarchical order. The statistical analysis plan (available with the protocol) includes additional details on the primary and sensitivity analyses for the efficacy end points, the hierarchical testing procedure, the analyses for exploratory end points, and the handling of missing data. The reported confidence intervals have not been adjusted for multiplicity and may not be used in place of hypothesis testing. For the analysis of deaths (efficacy analysis only), heart transplantation and the implantation of left ventricular assist devices were counted as deaths. Data for patients who died from coronavirus disease 2019 (Covid-19) were censored at the date of death, and the deaths were not counted as events in the efficacy analysis.
Safety data were summarized with the use of descriptive statistics. Efficacy and safety were analyzed in the full analysis set, which included all patients who underwent randomization and received any amount of patisiran or placebo. Pharmacodynamic assessments were conducted in all patients who received at least one complete dose of patisiran or placebo and who had an evaluable measurement of serum transthyretin levels at baseline and at least one evaluable measurement after baseline.
RESULTS
PATIENTS
From October 2019 through May 2021, a total of 360 patients at 69 sites in 21 countries were enrolled in the trial and randomly assigned to receive patisiran (181 patients) or placebo (179 patients) (Fig. S1 in the Supplementary Appendix). One patient, who was randomly assigned to the placebo group but did not receive the assigned placebo because of difficulty with intravenous access, was excluded from the efficacy and safety analysis. A total of 172 patients (95.0%) in the patisiran group and 167 patients (93.3%) in the placebo group completed the month 12 visit. The demographic and clinical characteristics of the patients at baseline were balanced between the two groups and were generally representative of the global population of patients with ATTR cardiac amyloidosis (Table 1, and Tables S1 and S2). The median age of the patients was 76 years, most patients were men, and 25% of the patients were receiving tafamidis at baseline. Approximately 80% of the patients had wild-type ATTR amyloidosis. Among the patients with variant ATTR amyloidosis, there were 16 different pathogenic transthyretin protein variants; 41% (29 patients) had V122I, which was the most common variant in this subgroup (Table S3). The baseline demographic and disease characteristics of patients in this trial with variant ATTR amyloidosis and evidence of polyneuropathy are shown in Table S4. Information about the cardiac medications patients received during the trial are provided in Table S5.
Table 1.
Demographic and Clinical Characteristics of the Patients at Baseline.*
| Characteristic | Patisiran (N = 181) | Placebo (N = 178) |
|---|---|---|
| Median age at screening (range) — yr | 76 (47–85) | 76 (41–85) |
| Male sex — no. (%) | 161 (89) | 160 (90) |
| Race — no. (%)† | ||
| White | 138/180 (77) | 140/174 (80) |
| Asian | 23/180 (13) | 15/174 (9) |
| Black | 16/180 (9) | 15/174 (9) |
| Other | 3/180 (2) | 4/174 (2) |
| Wild-type ATTR amyloidosis — no. (%) | 144 (80) | 144 (81) |
| Median time since diagnosis of ATTR amyloidosis (range) — yr | 0.8 (0.0–6.0) | 0.4 (0.0–10.0) |
| Treatment with tafamidis — no. (%) | ||
| At baseline | 46 (25) | 45 (25) |
| Started during 12-month double-blind period | 5 (3) | 3 (2) |
| ATTR amyloidosis stage — no. (%)‡ | ||
| Stage 1 | 124 (69) | 120 (67) |
| Stage 2 | 46 (25) | 45 (25) |
| Stage 3 | 11 (6) | 13 (7) |
| Polyneuropathy disability score — no. (%) | ||
| 0: no impairment | 96 (53) | 109 (61) |
| I: preserved walking, sensory disturbances | 63 (35) | 55 (31) |
| II: impaired walking without need for a stick or crutches | 22 (12) | 14 (8) |
| NYHA class — no. (%) | ||
| Class I | 10 (6) | 15 (8) |
| Class II | 156 (86) | 150 (84) |
| Class III | 15 (8) | 13 (7) |
| Median 6-minute walk distance (IQR) — m | 358.0 (295.0–420.0) | 367.7 (300.0–444.3) |
| Score on the KCCQ-OS§ | 69.8+21.2 | 70.3+20.7 |
| Laboratory values | ||
| Median NT-proBNP level (IQR) — pg/ml | 2008.0 (1135.0–2921.0) | 1813.0 (952.0–3079.0) |
| Median high-sensitivity troponin I level (IQR) — pg/ml¶ | 64.0 (38.6–92.0) | 60.2 (38.2–103.1) |
| Median mBMI (IQR)‖ | 1147.0 (988.4–1273.8) | 1134.0 (1018.7–1259.1) |
| Median eGFR (IQR) — ml/min/1.73 m2 | 71 (58–83) | 67 (51–84) |
| Median creatinine (IQR) — mg/dl | 1.0 (0.9–1.2) | 1.0 (0.8–1.4) |
| Coexisting conditions — no. (%) | ||
| Diabetes mellitus | 30 (17) | 25 (14) |
| Hypertension | 84 (46) | 101 (57) |
| Concomitant medication — no. (%) | ||
| Diuretic | 168 (93) | 164 (92) |
| Mineralocorticoid receptor antagonist | 92 (51) | 74 (42) |
| Beta-blocker | 73 (40) | 77 (43) |
| ACEI, ARB, or ARNI | 82 (45) | 71 (40) |
| SGLT2 inhibitor | 8 (4) | 7 (4) |
Plus-minus values are means ±SD. Percentages may not total 100 because of rounding. ACEI denotes angiotensinconverting-enzyme inhibitor, ARB angiotensin-receptor blocker, ARNI angiotensin receptor-neprilysin inhibitor, ATTR transthyretin-mediated, eGFR estimated glomerular filtration rate, IQR interquartile range, NT-proBNP N-terminal pro- B-type natriuretic peptide, NYHA New York Heart Association, and SGLT2 sodium-glucose cotransporter-2.
Race was reported by the patients.
Risk stratification was based on the levels of the serum biomarkers NT-proBNP and an eGFR. Stage 1 (lower risk) was defined by an NT-proBNP level of 3000 pg per milliliter or less and eGFR of at least 45 ml per minute per 1.73 m2. Stage 2 (intermediate risk) included all patients who did not meet the criteria for stages 1 or 3. Stage 3 (higher risk) was defined by an NT-proBNP greater than 3000 pg per milliliter and an eGFR of less than 45 ml per minute per 1.73 m2.
Kansas City Cardiomyopathy Questionnaire-Overall Summary (KCCQ-OS) scores range from 0 to 100, with a score of 0 to 24 indicating very poor to poor quality of life, 25 to 49 poor to fair, 50 to 74 fair to good, and 75 to 100 good to excellent.
Troponin I levels were assessed at baseline in a total of 174 patients in the patisiran group and in 172 patients in the placebo group.
The modified body-mass index (mBMI) was calculated as the conventional BMI (weight in kilograms divided by the square of the height in meters) multiplied by the serum albumin level in grams per liter.
EffICACY
Primary End Point
The magnitude of decline from baseline in the 6-minute walk distance at 12 months was significantly lower in the patisiran group than in the placebo group, with a median change from baseline of −8.15 m (95% confidence interval [CI], −16.42 to 1.50) in the patisiran group and −21.35 m (95% CI, −34.05 to −7.52) in the placebo group; the Hodges–Lehmann estimate of the median difference was 14.69 m (95% CI, 0.69 to 28.69; P = 0.02) (Fig. 1A). Results from the prespecified sensitivity analyses of the 6-minute walk test were consistent with those of the primary analysis (Fig. S2 and Table S6). A consistent difference in the 6-minute walk distance was observed with patisiran as compared with placebo across prespecified subgroups defined according to baseline demographic and disease characteristics, including variant and wild-type ATTR amyloidosis (Fig. S3A). In a post hoc analysis, the results for 6-minute walk distance among 205 patients who did not have walking impairment due to neuropathy (polyneuropathy disability score, 0) at baseline were consistent with those of the primary analysis (Table S7).
Figure 1. Changes over Time between the Patisiran Group and the Placebo Group.
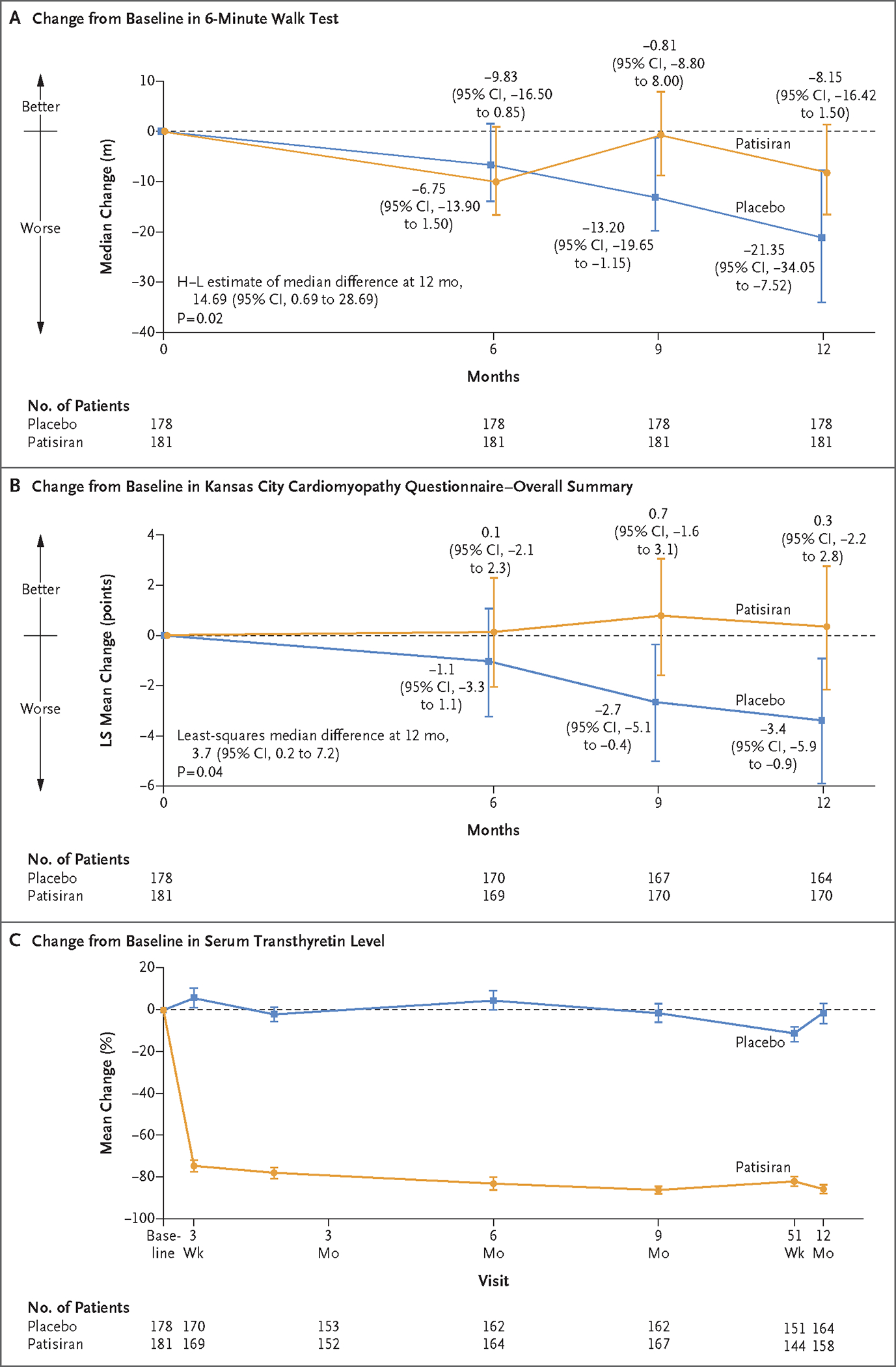
Panel A shows the median change from baseline in the distance covered on the 6-minute walk test (primary end point) with 95% confidence intervals. A farther distance walked indicates better function. At baseline, the median distance covered on the 6-minute walk test was 358.00 m (interquartile range, 295.00 to 420.00) in the patisiran group and 367.74 m (interquartile range, 300.00 to 444.25) in the placebo group. Missing assessments owing to deaths not caused by coronavirus disease 2019 [Covid-19] or owing to the inability to walk because of progression of transthyretin amyloidosis (also known as ATTR amyloidosis) disease were imputed as −115 meters (the worst 10th percentile change observed across all patients in the double-blind period). Other missing data (e.g., assessments obtained after a serious adverse event due to Covid-19, which were counted as missing) were multiply imputed assuming that data were missing at random (see the statistical analysis plan, available with the protocol, for details). Missing data were imputed 100 times to create 100 complete data sets. All data sets were analyzed with the use of the stratified Wilcoxon rank-sum test and the stratified Hodges–Lehmann (H–L) estimate, and the results were combined according to Rubin’s rules. The 6-minute walk test assessments were conducted in all patients at baseline; 6% and 8% of patients in the patisiran and placebo groups, respectively, had missing assessments at month 12. Panel B shows the leastsquares mean change from baseline in the Kansas City Cardiomyopathy Questionnaire–Overall Summary (KCCQ-OS) score (secondary end point), analyzed with the use of a mixed model with repeated measures. Lower values indicate worse quality of life. The KCCQ-OS assessments were conducted in all patients at baseline; 5% and 6% of patients in the patisiran and placebo groups, respectively, had missing assessments at month 12. Panel C shows the mean percentage change from baseline in serum transthyretin levels (measured with the use of a proprietary enzyme-linked immunosorbent assay [ELISA]). At baseline, the mean (±SD) serum transthyretin level was 235.32±68.05 mg per liter in the patisiran group and 244.77±73.17 mg per liter in the placebo group. At month 12, the mean serum transthyretin level was 30.93±33.60 mg per liter in the patisiran group and 229.40±77.15 mg per liter in the placebo group. Transthyretin levels were measured in all patients at baseline; 13% and 8% of patients in the patisiran and placebo groups, respectively, had missing measurements at month 12. The widths of the confidence intervals have not been adjusted for multiplicity and may not be used in place of hypothesis testing. I bars indicate confidence intervals.
Secondary End Points
The KCCQ-OS score decreased from baseline in the placebo group (least-squares mean change, −3.4 points [95% CI, −5.9 to −0.9]) and increased slightly from baseline in the patisiran group (least-squares mean change, 0.3 points [95% CI, −2.2 to 2.8]), representing a leastsquares mean difference of 3.7 points (95% CI, 0.2 to 7.2; P = 0.04) (Fig. 1B). The between-group difference was consistent across prespecified subgroups (Fig. S3B) and across all KCCQ domains (Fig. S4).
For the composite end point of death from any cause, cardiovascular events, and change from baseline in the distance covered on the 6-minute walk test, the win ratio over the 12-month double-blind period was 1.27 (95% CI, 0.99 to 1.61) and was not significant (Table S8). The estimated hazard ratio (patisiran-to-placebo) for the composite end point of death from any cause, hospitalizations for any cause, and urgent visits for heart failure was 0.88 (95% CI, 0.58 to 1.34) in the overall trial population and 0.10 (95% CI, 0.62 to 1.60) in the patients who were not receiving tafamidis at baseline (Table S8). In the 12-month double-blind period, 4 deaths (2.2%) occurred in the patisiran group and 10 deaths (5.6%) occurred in the placebo group. The estimated hazard ratio (patisiran-to-placebo) was 0.36 (95% CI, 0.11 to 1.14). In the placebo group, two patients (1.1%) received heart transplants, which were counted as deaths in the efficacy analysis. Data for a patient who died from Covid-19 (one patient [0.56%] in the patisiran group) were censored at the date of death and this death was not included as an event in the efficacy analysis.
Exploratory End Points
At 12 months, the median change from baseline in the NT-proBNP level was 131 pg per milliliter (interquartile range, −280 to 817) in the patisiran group and 518 pg per milliliter (interquartile range, 51 to 1544) in the placebo group. The median change in the troponin I level from baseline to month 12 was 3.8 pg per milliliter (interquartile range, −7.1 to 19.9) in the patisiran group and 14.5 pg per milliliter (interquartile range, 0.0 to 32.2) in the placebo group. The ratios (patisiran-to-placebo) of the adjusted geometric mean factor increase from baseline in the levels of NT-proBNP and troponin I were 0.80 (95% CI, 0.73 to 0.89) and 0.87 (95% CI, 0.80 to 0.95), respectively (Fig. 2A and 2B).
Figure 2. Change in Cardiac Biomarker Levels (Exploratory End Points).
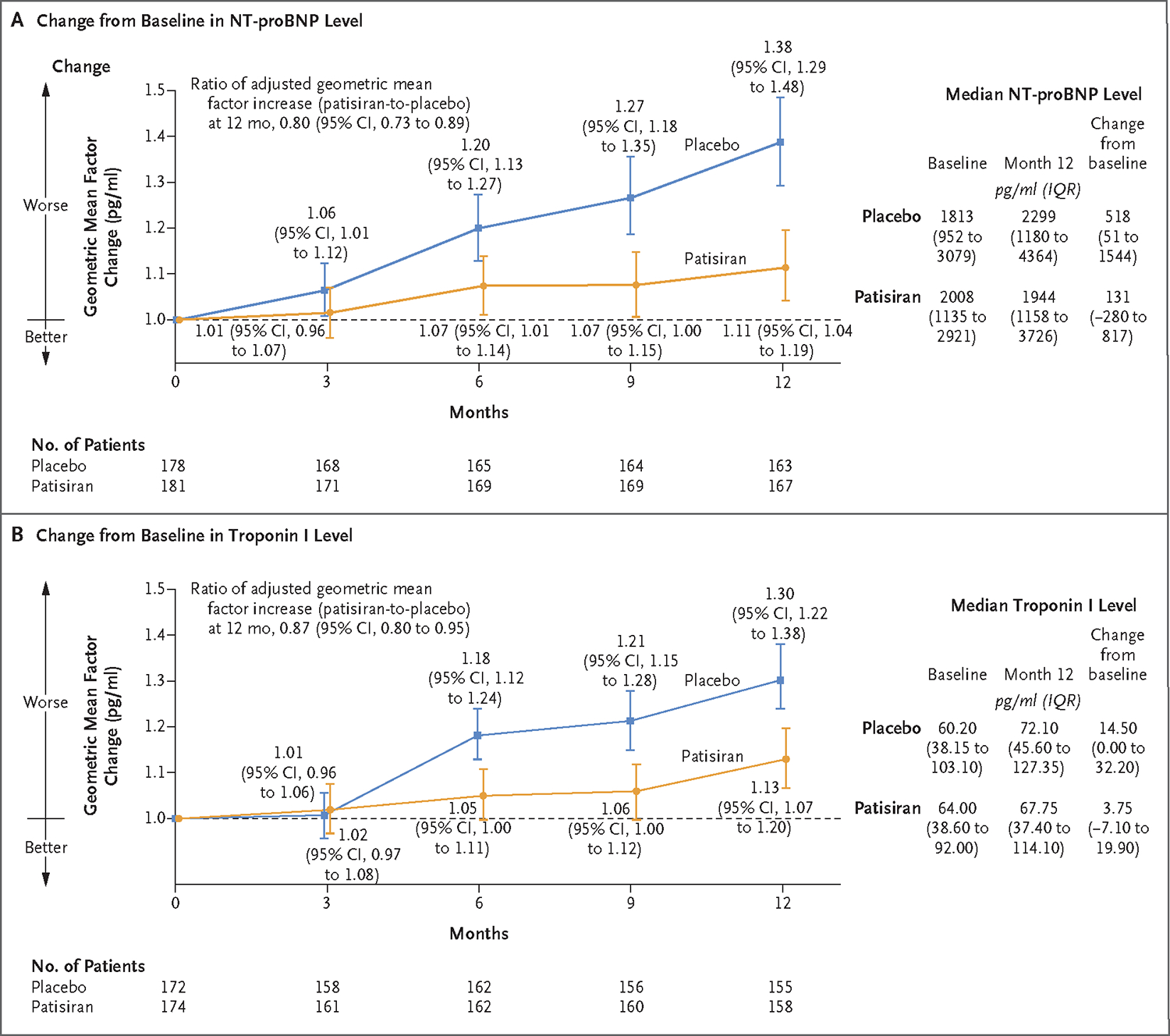
Panel A shows the adjusted geometric mean factor change from baseline to month 12 in the N-terminal pro–B-type natriuretic peptide (NT-proBNP) level, a cardiac biomarker, with higher values indicating a greater level of cardiac stress. All patients had an NT-proBNP measurement obtained at baseline; assessments were missing for 8% of patients in both the patisiran and placebo groups at month 12. Panel B shows the adjusted geometric mean factor change from baseline in the high-sensitivity troponin I level, a measure of myocardial wall injury, with higher values indicating a greater level of myocardial injury. Troponin I measurements were missing for 4% of patients in the patisiran group and 3% of patients in the placebo group at baseline; at month 12, measurements were missing for 9% and 10% of patients in the patisiran and placebo groups, respectively. The number of patients assessed at each time point is shown for both biomarkers. The median levels of each cardiac biomarker measured at baseline and 12 months and the change from baseline to month 12 are also shown. I bars indicate confidence intervals. The widths of the confidence intervals have not been adjusted for multiplicity and may not be used in place of hypothesis testing. IQR denotes interquartile range.
Echocardiographic analyses were performed to assess the change from baseline to month 12 in left ventricular structure and function. The change in left ventricular mass was −1.00 g (95% CI, −7.31 to 5.32) in the patisiran group and 8.45 g (95% CI, 2.02 to 14.89) in those who received placebo, with a between-group leastsquares mean difference of −9.45 g (95% CI, −18.48 to −0.42). The change in left ventricular global longitudinal strain (least-squares mean change from baseline) was 0.36 percentage points (95% CI, 0.01 to 0.71) in the patisiran group and 0.90 percentage points (95% CI, 0.55 to 1.26) in the placebo group, with a between-group least-squares mean difference of −0.54 percentage points (95% CI, −1.04 to −0.05). The change in left ventricular stroke volume was 0.48 ml (95% CI, −1.19 to 2.15) in the patisiran group and −2.52 ml (95% CI, −4.21 to −0.82) in the placebo group, with a between-group leastsquares mean difference of 3.00 ml (95% CI, 0.61 to 5.38) (Fig. S5). In addition to the echocardiographic results, investigators observed a least-squares mean change from baseline to week 48 in the modified BMI (calculated as the BMI × g of albumin per liter) of −34.5 (95% CI, −50.6 to −18.4) in the patisiran group as compared with −44.7 (95% CI, −60.9 to −28.6) in the placebo group, for a least-squares mean difference of 10.3 (95% CI, −12.5 to 33.0) (Fig. S6).
Pharmacodynamics
A rapid and sustained reduction in the serum transthyretin level was observed in the patisiran group, with a mean (±SD) percent reduction of 86.8±13.6% at 12 months (Fig. 1C). The reduction in the transthyretin level among patients receiving patisiran was similar across subgroups at month 12, regardless of whether patients had been receiving tafamidis treatment at baseline (83.7±16.3% reduction in those who were receiving tafamidis and 87.9±12.3% in those who were not) and whether patients had variant ATTR amyloidosis (85.2±12.7% reduction) or wild-type ATTR amyloidosis (87.2±13.9% reduction).
SAFETY
Adverse events occurred in 91% of patients in the patisiran group and in 94% of patients in the placebo group (Table 2); the majority of events were mild or moderate in severity. The frequency of severe and serious adverse events was similar in the two groups. Adverse events that occurred in at least 5% of patients treated with patisiran and that were at least 3 percentage points more common in the patisiran group than in the placebo group included infusion-related reactions, arthralgia, and muscle spasms. All infusion-related reactions were mild or moderate in severity, and one patient in the patisiran group discontinued the trial regimen owing to a mild infusion-related reaction. Serious adverse events reported in at least 2% of patients in either group were cardiac failure, atrial fibrillation, complete atrioventricular block, amyloidosis, and syncope (Table 2 and Table S9). Adverse events leading to discontinuation of the trial regimen occurred in five patients (3%) in each group. In the safety analysis, five deaths (3%) occurred in the patisiran group, and eight deaths (4%) occurred in the placebo group. No clinically relevant changes in laboratory measures (including hematologic measures, blood chemical measures, liver-function tests, and renal measures), vital signs, or electrocardiograms were observed in either group during the trial.
Table 2.
Safety during the 12-Month Double-Blind Treatment Period.
| Event | Patisiran (N = 181) | Placebo (N = 178) |
|---|---|---|
| no. of patients (%) | ||
| Any adverse event | 165 (91) | 168 (94) |
| Adverse events occurring in ≥10% of patients in either group | ||
| Cardiac failure | 54 (30) | 68 (38) |
| Infusion-related reaction | 22 (12) | 16 (9) |
| Constipation | 20 (11) | 19 (11) |
| Atrial fibrillation | 16 (9) | 26 (15) |
| Covid-19* | 16 (9) | 25 (14) |
| Adverse events occurring in ≥5% of patients treated with patisiran and at least 3 percentage points more common in the patisiran group | ||
| Infusion-related reaction | 22 (12) | 16 (9) |
| Arthralgia | 14 (8) | 8 (4) |
| Muscle spasm | 12 (7) | 4 (2) |
| Any serious adverse event† | 61 (34) | 63 (35) |
| Any severe adverse event† | 47 (26) | 52 (29) |
| Serious adverse events occurring in ≥2% of patients in either group | ||
| Cardiac failure | 15 (8) | 13 (7) |
| Atrial fibrillation | 5 (3) | 4 (2) |
| Atrioventricular block complete | 2 (1) | 4 (2) |
| Amyloidosis‡ | 1 (1) | 4 (2) |
| Syncope | 2 (1) | 4 (2) |
| Cardiac adverse events§ | 82 (45) | 100 (56) |
| Cardiac serious adverse events§ | 32 (18) | 28 (16) |
| Any adverse event leading to treatment discontinuation | 5 (3) | 5 (3) |
| Death: safety analysis¶ | 5 (3) | 8 (4) |
Included are all patients with Covid-19 of any severity, including two patients in the patisiran group who had asymptomatic Covid-19.
Serious adverse events were defined as adverse events that resulted in death, were life-threatening, resulted in inpatient hospitalization or prolongation of existing hospitalization, resulted in persistent or clinically significant disability or incapacity, were a congenital anomaly or birth defect, or were important medical events as determined by the investigators. All adverse events (including serious adverse events) were graded for severity. Severe events were defined as adverse events for which more than minimal, local, or noninvasive intervention was received; which had a severe effect on limiting self-care activities of daily living; or which had the potential for life-threatening consequences or death.
Amyloidosis includes adverse events that were reported as worsening amyloidosis, ATTR disease progression, worsening of amyloid polyneuropathy, and amyloidosis in the bladder.
Cardiac adverse events and serious adverse events included all events selected according to the Medical Dictionary for Regulatory Activities, version 23.0, system organ class: Cardiac disorders.
Death in the patisiran group included sudden cardiac death (1 patient) and death from heart failure, pancreatitis, Covid-19, and undetermined cause (1 patient each). Death in the placebo group included death from heart failure (3 patients), undetermined cause (2 patients), and cholangitis, infection, and pancreatic cancer (1 patient each). Although heart transplantation and the implantation of left ventricular assist devices were counted as deaths in the efficacy analysis, they were not counted as deaths in the safety analysis.
DISCUSSION
ATTR cardiac amyloidosis is characterized by unrelenting disease progression due to ongoing amyloid deposition in the heart.6–8,13 The present trial showed that treatment with patisiran suppressed the level of circulating transthyretin protein by silencing messenger RNA and resulted in preserved functional capacity, health status, and quality of life, as measured by the 6-minute walk test and the KCCQ-OS score after 12 months of treatment. These results are consistent with the effects of transthyretin reduction observed in trials of variant ATTR amyloidosis with polyneuropathy17,22,23 and add to a growing body of evidence that supports the benefit of the therapeutic reduction of the circulating amyloidogenic protein, transthyretin, in ATTR amyloidosis, a finding previously established in systemic amyloidoses caused by serum amyloid A or immunoglobulin light chain.24–27
Over the 12-month double-blind period, disease progression was observed more often in the placebo group than in the patisiran group, as measured by the 6-minute walk test and the KCCQ-OS score. In addition, the patients who received patisiran showed a stable health status and quality of life, as measured by the KCCQ-OS score. The magnitude of the differences measured between the trial groups for these end points were determined by the rate of disease progression in the placebo group, with between-group differences starting to appear by 6 to 9 months.
The overall incidence and type of adverse events were similar in the patisiran and placebo groups, with a small percentage of adverse events (3% in each group) leading to discontinuation of the trial regimen. All patients received premedication before each injection of patisiran or placebo to reduce the risk of infusion-related reactions.
The APOLLO-B trial enrolled patients at 69 sites in 21 countries, with a trial population that is generally representative of the global population of patients with ATTR cardiac amyloidosis; however, this trial has certain limitations. The size of the trial precluded formal assessment of the efficacy of patisiran in certain subgroups, such as patients receiving tafamidis at baseline. The trial also excluded patients who are the most severely affected by ATTR amyloidosis (i.e., patients with both NYHA class III and ATTR amyloidosis stage 3 or with NYHA Class IV heart failure). However, recent studies have shown that the clinical phenotype of patients who receive a diagnosis of ATTR cardiac amyloidosis is changing, which is likely to be related, in part, to an increase in disease awareness and improvements in diagnostic pathways; patients in whom ATTR cardiac amyloidosis has been diagnosed in recent years have had less severe disease and a shorter duration of symptoms before diagnosis.28 This trial population may be more representative of the current demographic characteristics of patients than the population in previous studies, thus precluding any direct comparison of outcomes. Another limitation, owing to the duration of the trial, is that the 12-month randomized trial period did not provide sufficient power to assess the effect of treatment on end points related to mortality and hospitalization. Finally, specific measures of polyneuropathy were not assessed in this trial. Although the patient population had disease manifestations that were predominantly cardiac in nature, it is possible for patients with variant ATTR amyloidosis to have polyneuropathy. The improvement or worsening of polyneuropathy can influence performance on the 6-minute walk test. However, patients with substantially limited ambulation owing to neuropathy (patients requiring one or two canes to walk or those who were wheelchair users) were excluded from the trial, and patients with wild-type ATTR amyloidosis, who rarely have such limiting neuropathy, comprised the majority of the trial population. The results of a post hoc analysis of the primary end point that was limited to the evaluation of patients without impaired walking ability due to neuropathy (i.e., polyneuropathy disability score, 0) at baseline suggest that the treatment difference in the 6-minute walk test distance was unrelated to an effect of patisiran on neuropathy.
In the context of a disease characterized by progression, treatment with patisiran in patients with variant or wild-type ATTR cardiac amyloidosis over 12 months preserved functional capacity, health status, and quality of life. Studies of longer duration are needed to determine whether the benefits observed with the reduction of transthyretin mediated by RNA interference are associated with a reduction in mortality and improved hospitalization outcomes in patients with ATTR cardiac amyloidosis.
Supplementary Material
Acknowledgments
Supported by Alnylam Pharmaceuticals. During the Covid-19 pandemic, infusions at Columbia University Medical Center were performed in the Irving Institute for Clinical and Translational Research, which is supported by the National Center for Advancing Translational Sciences, National Institutes of Health (UL1TR001873).
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
We thank all the patients and their families for their participation in the APOLLO-B trial, Xingyu Li (Alnylam Pharmaceuticals), and Juanjuan Li (Alnylam Pharmaceuticals) for their contributions, and Kristen Brown, Ph.D., of Adelphi Communications (Macclesfield, United Kingdom) for medical writing assistance with an earlier version of the manuscript, funded by Alnylam Pharmaceuticals.
APPENDIX
The authors’ full names and academic degrees are as follows: Mathew S. Maurer, M.D., Parag Kale, M.D., Marianna Fontana, M.D., Ph.D., John L. Berk, M.D., Martha Grogan, M.D., Finn Gustafsson, M.D., Ph.D., Rebecca R. Hung, M.D., Ph.D., Robert L. Gottlieb, M.D., Ph.D., Thibaud Damy, M.D., Ph.D., Alejandra González-Duarte, M.D., Ph.D., Nitasha Sarswat, M.D., Yoshiki Sekijima, M.D., Ph.D., Nobuhiro Tahara, M.D., Ph.D., Mark S. Taylor, M.B., B.S., Ph.D., Milos Kubanek, M.D., Ph.D., Erwan Donal, M.D., Ph.D., Tomas Palecek, M.D., Ph.D., Kenichi Tsujita, M.D., Ph.D., W.H. Wilson Tang, M.D., Wen-Chung Yu, M.D., Laura Obici, M.D., Marcus Simões, M.D., Ph.D., Fábio Fernandes, M.D., Ph.D., Steen Hvitfeldt Poulsen, D.M.Sci., Igor Diemberger, M.D., Ph.D., Federico Perfetto, M.D., Ph.D., Scott D. Solomon, M.D., Marcelo Di Carli, M.D., Prajakta Badri, Ph.D., Matthew T. White, Ph.D., Jihong Chen, Ph.D., Elena Yureneva, M.D., Marianne T. Sweetser, M.D., Ph.D., Patrick Y. Jay, M.D., Ph.D., Pushkal P. Garg, M.D., John Vest, M.D., and Julian D. Gillmore, M.D., Ph.D.
The authors’ affiliations are as follows: Columbia University Irving Medical Center (M.S.M.) and Grossman School of Medicine, NYU Langone (A.G.-D.) — both in New York; the Center for Advanced Heart and Lung Disease, Baylor University Medical Center (P.K., R.L.G.), Baylor Scott & White Research Institute, and Texas A&M Health Science Center, Dallas (R.L.G.), and TCU School of Medicine, Fort Worth (R.L.G.) — all in Texas; the National Amyloidosis Centre, UCL, Division of Medicine, Royal Free Hospital, London (M.F., J.D.G.); Boston University School of Medicine (J.L.B.), the Cardiovascular Division, Brigham and Women’s Hospital (S.D.S., M.D.C.), and the Division of Nuclear Medicine and Molecular Imaging, Brigham and Women’s Hospital, Harvard Medical School (M.D.C.), Boston, and Alnylam Pharmaceuticals, Cambridge (P.B., M.T.W., J.C., E.Y., M.T.S., P.Y.J., P.P.G., J.V.) — all in Massachusetts; the Department of Cardiovascular Diseases, Mayo Clinic College of Medicine, Rochester, MN (M.G.); the Department of Cardiology, Rigshospitalet, University of Copenhagen, Copenhagen (F.G.), and the Department of Cardiology, Aarhus University Hospital, Aarhus (S.H.P.) — both in Denmark; the Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville (R.R.H.); the Cardiology Department and French National Reference Center for Cardiac Amyloidosis, GRC Amyloid Research Institute and Clinical Investigation Centre 1430 at Hôpitaux Universitaires Henri-Mondor Assistance Publique–Hôpitaux de Paris, and IMRB, INSERM, Université Paris Est Creteil, Creteil (T.D.), and INSERM, LTSI UMR 1099, Centre Hospitalier Universitaire de Rennes, Rennes (E.D.) — both in France; Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, México City (A.G.-D.); the Department of Medicine, University of Chicago, Chicago (N.S.); the Department of Medicine (Neurology and Rheumatology), Shinshu University School of Medicine, Matsumoto (Y.S.), the Division of Cardiovascular Medicine, Department of Medicine, Kurume University School of Medicine, Kurume (N.T.), and the Department of Cardiovascular Medicine, Graduate School of Medical Sciences, Kumamoto University, Kumamoto (K.T.) — all in Japan; Westmead Amyloidosis Service, Westmead Hospital, Sydney (M.S.T.); the Department of Cardiology, Institute for Clinical and Experimental Medicine (M.K.), and the 2nd Department of Medicine, Department of Cardiovascular Medicine, First Faculty of Medicine, Charles University in Prague and General University Hospital in Prague (T.P.) — both in Prague, Czech Republic; the Heart Vascular and Thoracic Institute, Cleveland Clinic, Cleveland (W.H.W.T.); Taipei Veterans General Hospital and National Yang Ming Chiao Tung University, Taipei, Taiwan (W.-C.Y.); Amyloidosis Research & Treatment Center, Fondazione IRCCS Policlinico San Matteo di Pavia, Pavia (L.O.), the Department of Medical and Surgical Sciences, University of Bologna, and the Cardiology Unit, IRCCS Azienda Ospedaliero–Universitaria di Bologna, Bologna (I.D.), and the Department of Clinical and Experimental Medicine, Careggi University Hospital, Florence (F.P.) — all in Italy; and Unidade de Pesquisa Clínica–UPC, Hospital Das Clinicas da Faculdade de Medicina de Ribeirão Preto–USP (M.S.), and Instituto do Coração–HCFMUSP (F.F.) — both in São Paulo.
Footnotes
A complete list of the APOLLO-B trial investigators and collaborators is provided in the Supplementary Appendix, available at NEJM.org.
Contributor Information
M.S. Maurer, Columbia University Irving Medical Center, New York
P. Kale, Center for Advanced Heart and Lung Disease, Baylor University Medical Center, Texas
M. Fontana, National Amyloidosis Centre, UCL, Division of Medicine, Royal Free Hospital, London
J.L. Berk, Boston University School of Medicine, Massachusetts
M. Grogan, Department of Cardiovascular Diseases, Mayo Clinic College of Medicine, Rochester, MN
F. Gustafsson, Department of Cardiology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark
R.R. Hung, Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University Medical Center, Nashville
R.L. Gottlieb, Center for Advanced Heart and Lung Disease, Baylor University Medical Center, Texas Baylor Scott & White Research Institute, and Texas A&M Health Science Center, Dallas, Texas; TCU School of Medicine, Fort Worth, Texas.
T. Damy, Cardiology Department and French National Reference Center for Cardiac Amyloidosis, GRC Amyloid Research Institute and Clinical Investigation Centre 1430 at Hôpitaux Universitaires Henri-Mondor Assistance Publique–Hôpitaux de Paris, and IMRB, INSERM, Université Paris Est Creteil, Creteil, France
A. González-Duarte, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, México City Grossman School of Medicine, NYU Langone, New York.
N. Sarswat, Department of Medicine, University of Chicago, Chicago
Y. Sekijima, Department of Medicine (Neurology and Rheumatology), Shinshu University School of Medicine, Matsumoto, Japan
N. Tahara, Division of Cardiovascular Medicine, Department of Medicine, Kurume University School of Medicine, Kurume, Japan
M.S. Taylor, Westmead Amyloidosis Service, Westmead Hospital, Sydney
M. Kubanek, Department of Cardiology, Institute for Clinical and Experimental Medicine, Prague, Czech Republic
E. Donal, INSERM, LTSI UMR 1099, Centre Hospitalier Universitaire de Rennes, Rennes, France
T. Palecek, 2nd Department of Medicine, Department of Cardiovascular Medicine, First Faculty of Medicine, Charles University in Prague and General University Hospital in Prague, Prague, Czech Republic
K. Tsujita, Department of Cardiovascular Medicine, Graduate School of Medical Sciences, Kumamoto University, Kumamoto, Japan
W.H.W. Tang, Heart Vascular and Thoracic Institute, Cleveland Clinic, Cleveland
W.-C. Yu, Taipei Veterans General Hospital and National Yang Ming Chiao Tung University, Taipei, Taiwan
L. Obici, Amyloidosis Research & Treatment Center, Fondazione IRCCS Policlinico San Matteo di Pavia, Pavia, Italy
M. Simões, Unidade de Pesquisa Clínica–UPC, Hospital Das Clinicas da Faculdade de Medicina de Ribeirão Preto–USP, São Paulo.
F. Fernandes, Instituto do Coração–HCFMUSP, São Paulo.
S.H. Poulsen, Department of Cardiology, Aarhus University Hospital, Aarhus, Denmark
I. Diemberger, Department of Medical and Surgical Sciences, University of Bologna, and the Cardiology Unit, IRCCS Azienda Ospedaliero–Universitaria di Bologna, Bologna, Italy
F. Perfetto, Department of Clinical and Experimental Medicine, Careggi University Hospital, Florence, Italy
S.D. Solomon, Cardiovascular Division, Brigham and Women’s Hospital, Massachusetts
M. Di Carli, Cardiovascular Division, Brigham and Women’s Hospital, Massachusetts Division of Nuclear Medicine and Molecular Imaging, Brigham and Women’s Hospital, Harvard Medical School, Massachusetts.
P. Badri, Boston, and Alnylam Pharmaceuticals, Cambridge, Massachusetts
M.T. White, Boston, and Alnylam Pharmaceuticals, Cambridge, Massachusetts
J. Chen, Boston, and Alnylam Pharmaceuticals, Cambridge, Massachusetts
E. Yureneva, Boston, and Alnylam Pharmaceuticals, Cambridge, Massachusetts
M.T. Sweetser, Boston, and Alnylam Pharmaceuticals, Cambridge, Massachusetts
P.Y. Jay, Boston, and Alnylam Pharmaceuticals, Cambridge, Massachusetts
P.P. Garg, Boston, and Alnylam Pharmaceuticals, Cambridge, Massachusetts
J. Vest, Boston, and Alnylam Pharmaceuticals, Cambridge, Massachusetts
J.D. Gillmore, National Amyloidosis Centre, UCL, Division of Medicine, Royal Free Hospital, London
REFERENCES
- 1.Maurer MS, Hanna M, Grogan M, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (transthyretin amyloid outcome survey). J Am Coll Cardiol 2016;68:161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol 2019;73:2872–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol 2019;15:387–404. [DOI] [PubMed] [Google Scholar]
- 4.Rapezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J 2013;34:5 20–8. [DOI] [PubMed] [Google Scholar]
- 5.Coelho T, Maurer MS, Suhr OB. THAOS — the Transthyretin Amyloidosis Outcomes Survey: initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr Med Res Opin 2013;29:63–76. [DOI] [PubMed] [Google Scholar]
- 6.Fontana M, Pica S, Reant P, et al. Prognostic value of late gadolinium enhancement cardiovascular magnetic resonance in cardiac amyloidosis. Circulation 2015;132:1570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martinez-Naharro A, Kotecha T, Norrington K, et al. Native T1 and extracellular volume in transthyretin amyloidosis. JACC Cardiovasc Imaging 2019;12:810–9. [DOI] [PubMed] [Google Scholar]
- 8.Chacko L, Karia N, Venneri L, et al. Progression of echocardiographic parameters and prognosis in transthyretin cardiac amyloidosis. Eur J Heart Fail 2022; 24:1700–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane T, Fontana M, Martinez-Naharro A, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation 2019;140:16–26. [DOI] [PubMed] [Google Scholar]
- 10.Nativi-Nicolau J, Judge DP, Hoffman JE, et al. Natural history and progression of transthyretin amyloid cardiomyopathy: insights from ATTR-ACT. ESC Heart Fail 2021;8:3875–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez-Lopez E, Escobar-Lopez L, Obici L, et al. Prognosis of transthyretin cardiac amyloidosis without heart failure symptoms. JACC CardioOncol 2022;4:442–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J 2018;39:2799–806. [DOI] [PubMed] [Google Scholar]
- 13.Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med 2018;379:1007–16. [DOI] [PubMed] [Google Scholar]
- 14.Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 2013;369:819–29. [DOI] [PubMed] [Google Scholar]
- 15.Alnylam Pharmaceuticals. Prescribing information. Onpattro (patisiran) lipid complex injection, for intravenous use. 2022. (https://www.alnylam.com/sites/default/files/pdfs/ONPATTRO-Prescribing-Information.pdf ).
- 16.European Medicines Agency. Summary of product characteristics. Onpattro 2 mg/ml concentrate for solution for infusion. 2018. (https://www.ema.europa.eu/documents/product-information/onpattro-epar-product-information_en.pdf ).
- 17.Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med 2018;379: 11–21. [DOI] [PubMed] [Google Scholar]
- 18.Obici L, Berk JL, González-Duarte A, et al. Quality of life outcomes in APOLLO, the phase 3 trial of the RNAi therapeutic patisiran in patients with hereditary transthyretin-mediated amyloidosis. Amyloid 2020;27:153–62. [DOI] [PubMed] [Google Scholar]
- 19.Solomon SD, Adams D, Kristen A, et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation 2019;139:431–43. [DOI] [PubMed] [Google Scholar]
- 20.Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 2016;133:2404–12. [DOI] [PubMed] [Google Scholar]
- 21.Green CP, Porter CB, Bresnahan DR, Spertus JA. Development and evaluation of the Kansas City Cardiomyopathy Questionnaire: a new health status measure for heart failure. J Am Coll Cardiol 2000;35:1245–55. [DOI] [PubMed] [Google Scholar]
- 22.Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 2018;379:22–31. [DOI] [PubMed] [Google Scholar]
- 23.Adams D, Tournev IL, Taylor MS, et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid 2023;30:1–9. [DOI] [PubMed] [Google Scholar]
- 24.Lachmann HJ, Goodman HJ, Gilbert-son JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med 2007;356:2361–71. [DOI] [PubMed] [Google Scholar]
- 25.Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol 2012;30:4541–9. [DOI] [PubMed] [Google Scholar]
- 26.Kastritis E, Palladini G, Minnema MC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med 2021;385:46–58. [DOI] [PubMed] [Google Scholar]
- 27.Martinez-Naharro A, Patel R, Kotecha T, et al. Cardiovascular magnetic resonance in light-chain amyloidosis to guide treatment. Eur Heart J 2022;43:4722–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ioannou A, Patel RK, Razvi Y, et al. Impact of earlier diagnosis in cardiac ATTR amyloidosis over the course of 20 years. Circulation 2022;146:1657–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.