

Abstract
Purpose of Review
Diabetes has a detrimental effect on bone, increasing the risk of fracture and formation of osteolytic lesions such as those seen in periodontitis. Several diabetic complications are caused by diabetes-enhanced inflammation. This review examines mechanisms by which IL-17 contributes to diabetes-enhanced periodontitis and other effects of IL-17 on bone.
Recent Findings
IL-17 upregulates anti-bacterial defenses, yet its expression is also linked to a destructive host response in the periodontium. Periodontal disease is caused by bacteria that stimulate an inflammatory response. Diabetes-enhanced IL-17 increases gingival inflammation, which alters the composition of the oral microbiota to increase its pathogenicity. In addition, IL-17 can induce osteoclastogenesis by upregulation of TNF and RANKL in a number of cell types, and IL-17 has differential effects on osteoblasts and their progenitors.
Summary
Increased IL-17 production caused by diabetes alters the pathogenicity of the oral microbiota and can promote periodontal bone resorption.
Keywords: Bacteria, Bone loss, Cytokine, Diabetic, Gingiva, Immune, Microbiome, Osteoclast, Periodontitis, Th17
Introduction
The IL-17 family consists of six members, IL-17A through IL-17F. IL-17A and IL-17F are the best studied and have the highest degree of sequence homology and signal as homodimers or heterodimers consisting of IL-17A/A, IL-17F/F, and IL-17A/F [1, 2]. IL-17 is largely produced by CD4+ T helper 17 cells (Th17) [3] and γδ T cells and also by CD8+ T cells and natural killer T (NKT) cells [4, 5]. Both Th17 and γδ T cells are found in periodontal tissues in relatively high numbers [5, 6]. IL-17 production by these cells is important in the response to bacterial infection [6, 7••, 8].
There are five distinct IL-17 receptor genes, IL-17 receptor A (IL-17 RA) through IL-17 receptor E (IL-17RE) [1]. IL-17A and IL-17F signal through a heteromeric IL-17 receptor complex (e.g., IL-17RA/IL-17RC) that forms in the presence of ligand [1]. IL-17RA is expressed ubiquitously in cells while IL-17RC is primarily expressed by resident, non-hematopoietic cells such as mesenchymal, epithelial, and endothelial cells [9, 10]. IL-17A and IL-17F are important in protecting mucosal surfaces, in part, by activating host defense mechanisms in keratinocytes through upregulation of neutrophil chemoattractants and anti-microbial peptides such as defensins [11]. Their importance in upregulating host-defenses is established in an animal model with dual IL-17A and IL17F deletion [12]. The amount of oral bacteria is increased, and the ability of bacteria to disseminate systemically from the oral cavity is increased in IL-17A/IL-17F-deleted mice compared to wild-type mice [12]. Signaling downstream of the IL-17R complex induces mitogen-activated protein kinase (MAPK) and NF-κB activation, leading to the production of pro-inflammatory cytokines and chemokines and subsequent myeloid cell recruitment to the inflamed tissue [13, 14].
IL-17 and Periodontitis
Bacteria on the tooth surface stimulate an inflammatory response that induces gingival inflammation and is referred to as gingivitis. In gingivitis, the inflammation does not spread to bone cells [15]. Once the inflammation reaches the deeper connective tissue and involves bone, RANKL is expressed by osteoblasts, osteocytes, and periodontal ligament cells to induce bone resorption [12, 16–18]. The inflammation also suppresses the production of factors that stimulate osteoblastic cells to form new bone, thereby inhibiting osseous coupling and promoting osteolytic lesion formation [19].
Numerous studies have shown that human periodontitis is associated with increased levels of locally produced IL-17 compared with healthy periodontal tissue [20]. Th17 lymphocytes and IL-17 levels are positively correlated with periodontal disease severity and with clinical parameters of periodontal destruction [21–24]. Cause and effect relationships between IL-17 and periodontitis have been carried out, but the interpretation of the results is complex since IL-17 has both important protective and destructive roles. Genetic ablation of IL-17RA leads to reduced neutrophil recruitment to periodontal tissues in response to oral infection and increased susceptibility to periodontitis, suggesting that IL-17RA signaling is needed to protect periodontal tissues from bacteria [25]. Interestingly, the production of IL-17A, which is stimulated by the minor mechanical injury that occurs during mastication in the oral cavity, contributes to maintaining homeostasis in this environment [11]. In contrast, short-term administration of an antibody to IL-17A inhibited periodontal bone loss in an acute periodontitis model, suggesting that in this model, high levels of IL-17A, per se, contributed to periodontal damage [26]. Long-term genetic deletion of IL-17A also reduces bone loss in a model of spontaneous periodontal disease associated with older mice [11]. This result is consistent with observations that, compared to young mice, aged mice exhibit a greater inflammatory and osteoclastogenic response to a bacterial stimulus [27].
Taken together, the above studies suggest that some IL-17 receptor signaling is needed to protect against bacteria in periodontal tissues since its absent increases susceptibility to bacteria-induced inflammation and periodontal bone loss [12]. However, excessive IL-17 signaling results in inflammation that leads to increased RANKL production by osteoblast-lineage and periodontal ligament fibroblasts and periodontal bone loss [12, 21, 26].
Diabetes and Bone
Over 400 million adults are estimated to have diabetes on a worldwide basis [28]. The majority (> 90%) have type-2 diabetes mellitus (T2DM), which is caused by a combination of insulin resistance and a failure to compensate by increasing insulin production [29]. T2DM is linked most closely with obesity, and susceptibility is affected by race, ethnicity, diet, exercise, smoking history, and age. Type-1 diabetes mellitus (T1DM) is due to tolerance failure, which leads to an autoimmune response that attacks the beta cells in the pancreas and is associated with inadequate insulin production [30]. Increased gingival inflammation is observed in subjects with both T1DM and T2DM [31–33]. Periodontitis, which involves inflammation - induced resorption and periodontal bone loss, is significantly increased in both T1DM and T2DM [34–38]. High glucose levels may directly contribute to the increased levels of inflammation by leading to the generation of reactive oxygen species (ROS), the nonenzymatic formation of advanced glycation end products (AGEs) that activate NF-kB, and the loss of antioxidants [35•, 39, 40]. Diabetes also affects inflammation by increasing the inflammatory activity of neutrophils [40], monocytes [41], and dendritic cells [42]. The increased risk and severity of periodontal bone loss in humans is supported by animal studies that show a comparable effect [43–45]. The latter has provided insight into mechanisms by which diabetes-enhanced inflammation affects bone and connective tissue through its effect on proteolytic enzymes, mesenchymal stem cells, osteoblasts, and fibroblasts [46, 47•].
Both T1DM and T2DM affect bone. There is a six-to sevenfold increased risk of fracture in T1DM and a 1.5-fold increased risk in T2DM [47•,48]. T1DM is associated with reduced bone density, and T2DM is associated with reduced mechanical strength and diminished bone quality but not a loss of bone density [47•,48]. Hyperglycemia in vivo reduces expression of transcription factors that coordinate the differentiated function of osteoblasts such as Runx-2, Dlx5, and c-fos [49]. Several diabetic complications involve upregulation of inflammatory cytokines such as TNF [35•, 48, 50–53]. When TNF is inhibited, osteoclastogenesis is reduced, and there is improved angiogenesis and increased numbers of mesenchymal stem cells [19, 51–55]. AGEs may contribute to the reduced osteoprogenitor pool seen in diabetics [55, 56]. Mesenchymal stem cell and periodontal ligament cell apoptosis are increased, and differentiation of MSC to osteoblasts is reduced by AGEs [57, 58]. Diabetes increases more than twofold the mRNA levels of 70 genes that directly or indirectly regulate apoptosis in response to bacterial challenge and leads to significantly enhanced caspase-8, −9, and −3 activities [58]. That apoptosis of matrix-producing cells is important in bone coupling following periodontal infection was shown by significant improvement by treatment with caspase-3 inhibitor that substantially reduced inflammation-induced cell death [46].
In periodontal tissues, diabetes leads to enhanced inflammation, RANKL expression, osteoclastogenesis, and periodontal bone loss [35•, 44, 50] The induction of experimental periodontal disease stimulates NF-κB activation in periodontal ligament fibroblasts, osteoblasts, and osteocytes that is associated with bone loss and reduced bone coupling, two major factors in periodontitis [16–18]. Experimental mice with osteocyte-specific ablation of RANKL through DMP-1-mediated gene deletion have significantly reduced osteoclast numbers and bone resorption in both the normoglycemic and diabetic groups, demonstrating the importance of these cells in experimental periodontitis [17]. Diabetes reduces the numbers of bone-lining cells, osteoblasts, and periodontal ligament fibroblasts that may limit bone coupling [50, 53–55]. Osseous coupling is significantly enhanced in diabetic animals by a TNF specific inhibitor that rescues bone formation and expression of bone-producing factors such as FGF-2, TGFβ-1, BMP-2, and BMP-6 [19]. The effect of diabetes on periodontitis is summarized in Fig. 1.
Fig. 1.
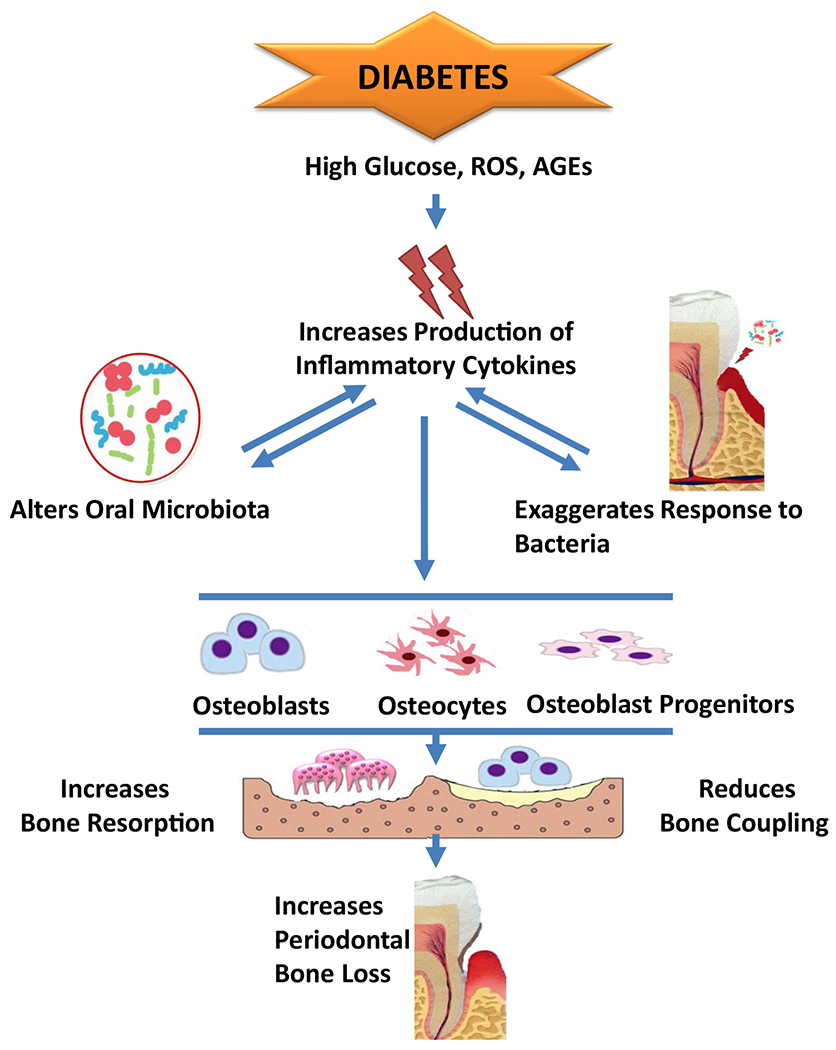
Diabetes is associated with an increased risk and severity of periodontitis. Several cytokines are elevated by diabetes and play a role in the pathogenesis of periodontal disease including IL-1, TNF, and IL-17. Conditions present in diabetes such as high glucose, ROS, and AGEs increase the inflammatory response induced by oral bacteria. Conversely, the increased inflammation alters the bacterial composition to render it more pathogenic. Theenhanced inflammation affect the key cell types, osteoblasts, osteocytes, and osteoblast progenitors, which produce RANKL and contribute to bone coupling following an episode of periodontal bone resorption.
Diabetes, IL-17 and Periodontal Bone Loss
Diabetes increases periodontal bone loss in animals and in humans [19, 59–63]. Until recently, there was little evidence shown that diabetes causes the change in bacterial composition to render it more pathogenic [63, 64]. Human studies have reported variable findings on the effect of hyperglycemia on the oral microbiota. For example, the effect of diabetes on the level of oral bacteria such as Capnocytophaga, Porphyromonas gingivalis, or Tannerella forsythia has been shown to vary widely [65–69]. Even with more robust techniques such as DNA-seq, there is little consensus on the specific changes in oral bacterial compositions caused by diabetes [65, 70–72]. Several factors may play into this variability including confounding factors such as the degree and duration of hyperglycemia, environmental factors such as diet and oral hygiene, and medications. Just as importantly, there is no consensus of the specific bacterial composition that causes periodontitis. In addition, it is possible that the major change induced by diabetes is the response to bacteria rather than the change in bacterial pathogenicity. Diabetes increases mRNA levels of host defense, pro-apoptotic, inflammatory, and coagulation/hemostasis/complement regulating genes [60, 61]. In controlled experiments where bacteria are injected into connective tissue, diabetic animals exhibit greater inflammation compared to normoglycemic controls [73,74]. Therefore, it is necessary to test whether diabetes alters the pathogenicity of the oral microbiota rather than just change its composition.
To address the issue of the impact of diabetes on the overall microbial pathogenicity, our lab undertook experiments on examined db/db mice that spontaneously develop T2DM and their lean control littermates that are normoglycemic [75••]. Prior to the development of hyperglycemia, both groups of mice had a similar oral microbiota as determined by beta- and alpha-diversity measurements. The onset of hyperglycemia in the db/db group caused a decrease in microbial diversity and a higher proportion of Proteobacteria (Enterobacteriaceae) and Firmicutes (Enterococcus, Staphylococcus, and Aerococcus). The increase in these bacterial taxa has been associated with changes in other diabetic pathologies [69, 76]. Similarly, reduced bacterial diversity occurs in other sites caused by diabetes or in the oral microbiota associated with aging and is thought to render the bacterial community more susceptible to disruption [27, 77, 78].
To test the impact of diabetes on bacterial pathogenicity, transfer experiments were carried out in which the oral microbiota was collected from normoglycemic and diabetic mice and used to inoculate normoglycemic germ-free mice [75••]. The oral microbiota from diabetic mice was more pathogenic than that of normoglycemic mice. In these studies, inflammation was measured by increased neutrophil accumulation and IL-6 expression. Diabetic oral microbiota also induced more bone loss as reflected by a greater induction of RANKL and osteoclastogenesis with a resultant increase in osteolysis. Since inflammation is greater in the diabetic periodontal tissues, we examined whether the enhanced inflammation of diabetes contributed to the bacterial changes. A pilot study determined that IL-17 exhibited the greatest change when inflammatory mediators were compared between diabetic and normoglycemic mice. To experimentally determine whether high levels of IL-17 induced a change in bacterial composition, we examined one group of diabetic mice treated by microinjection of IL-17 antibody into the gingiva and compared their responses to mice that had microinjection of control IgG. Treatment with IL-17 blocking antibody altered the oral microbiota in the diabetic mice. Interestingly, the antibody treatment made the composition of the oral microbiota in the diabetic group closer to that in the normoglycemic mice. The blocking antibody also diminished the pathogenicity of the oral microbiota in the diabetic group as determined by bacterial transfer experiments. The microbiota from the oral cavities of the diabetic mice treated with the IL-17 antibody stimulated less recruitment of neutrophils, had reduced IL-6 and RANKL expression, and diminished osteoclastogenesis and bone loss compared to microbiota transfer from diabetic mice, treated with control IgG. These results indicate that IL-17 induced inflammation alters the bacterial composition in the oral cavity and renders it more pathogenic (Fig. 2). This may be due to the generation of inflammatory products that serve as bacterial substrates and favor the growth of pathogenic species. It is also possible that the host-response is skewed by high levels of IL-17 in the diabetic group so that it is less effective in dealing with bacteria that are pathogenic, as shown in studies with leukocyte adhesion deficiency [79, 80]. Both scenarios point toward the importance of controlling inflammation in order to reduce oral microbial pathogenicity. We propose that diabetes causes an increase in IL-17 and inflammation, which, in turn, alters the microbial composition of the oral cavity in a pro-inflammatory direction. This enhances the level of inflammation, which increases osteoclastogenesis and limits osseous coupling, producing more bone loss.
Fig. 2.
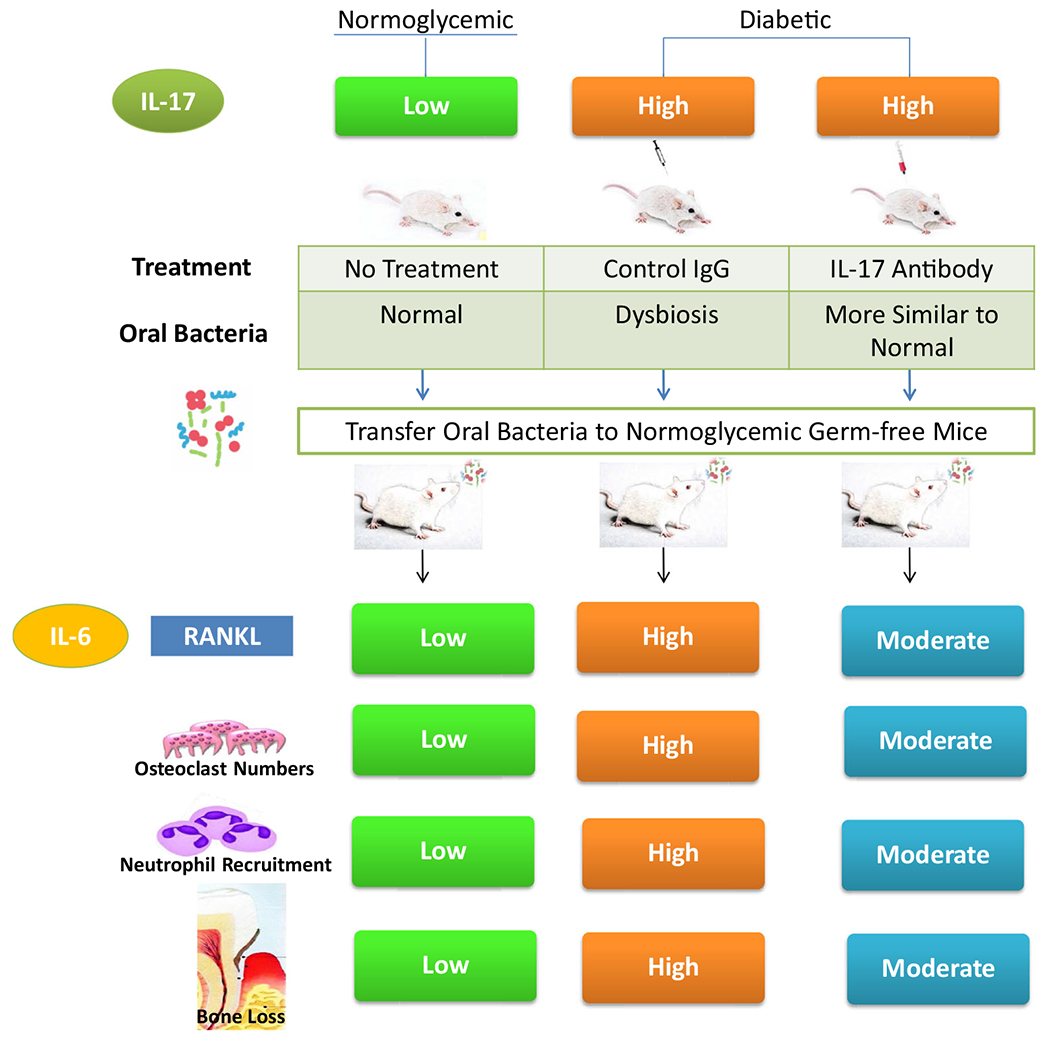
Diabetes increases IL-17 expression, leading to increased periodontal inflammation and dysbiosis. Treatment with IL-17 antibody reduces dysbiosis in diabetic animals. When transferred to germ-free mice, the oral microbiota from diabetic mice induce more periodontal inflammation and bone loss than microbiota from normal mice. Treatment of diabetic donor mice with IL-17 antibody reduces the inflammation and bone loss stimulated by the oral microbiota when it was transferred to germ-free recipients. Thus, diabetes increases the pathogenicity of the oral microbiota through an IL-17-mediated mechanism
Although T1DM and T2DM have different etiologies and their impact on bone is not identical, they share many of the same complications that result from the common thread of increased inflammation. Both animal studies and human studies confirm that both forms of diabetes increase inflammatory events in periodontal tissue, impair new bone formation, and increase expression of RANKL in response to a bacteria-induced challenge. Studies in animals, moreover, suggest that there are multiple mechanisms affected by diabetes that impact a large number of cell types. As in humans, poorly controlled diabetes in rodents accelerates the process of periodontal disease and causes it to occur at a younger age. The etiology of periodontal disease involves the host response to bacterial challenge. Animal studies indicate that the host response triggers activation of NF-κB and inflammation in periodontal ligament cells, osteoblasts, and osteocytes. This then affects the expression of RANKL and coupled bone formation, which are both further exacerbated by the presence of diabetes. In addition to affecting cells which are in close proximity to bone, the inflammatory response to bacteria also modifies the oral microbiota to render it more pathogenic. Thus, animal studies have provided new insights into the pathogenic mechanisms of diabetes and merit further investigation to unravel the cause and effect relationships between diabetes and periodontitis/peri-implantitis. However, the results of these experiments will need to be confirmed in human studies, which will present new challenges.
IL-17 and Its Effect on Osteoclasts and Osteoblasts
IL-17 can induce osteoclastogenesis, inhibit bone formation, and lead to bone loss in osteoporosis [81–83]. IL-17 induces bone loss by increasing pro-osteoclastogenic cytokines including TNF-α and RANKL from osteoblasts. IL-17 promotes osteoclastogenesis by stimulating RANKL expression by osteoblastic cells, through interactions with IL-17RA. Deletion of the IL-17RA protects mice from ovariectomy-induced bone loss. Estrogen deprivation promotes RANKL expression and bone resorption in association with upregulation of the IL-17 effector Act1 [84]. Anti-IL-17 suppresses ovariectomy-induced CD4+ T cell proliferation, pro-inflammatory cytokine production, and B cell lymphopoiesis with reduced Treg formation [85]. This treatment restores trabecular microarchitecture and improves bone biomechanical properties [86]. IL-17A inhibits late osteoblast differentiation by downregulation of genes involved in the Wnt pathway, including Axin2, Wisp1, and Bmp4 [87]. In addition, IL-17 induces NF-κB activity in osteoblasts, which can inhibit the differentiated function of these cells [16].
IL-17Aaccelerates bone formation in early bone fracture healing by stimulating the proliferation and osteoblastic differentiation of mesenchymal progenitor cells [88, 89]. In a fracture healing model, loss of IL-17A in IL-17A-deficient mice, disrupted proliferation, and differentiation of MSCs, resulting in delayed callus formation and lower bone mineral density [89, 90]. Moreover, IL-17 acts in a synergistic manner with BMP-2 to enhance new bone formation [91].
In spondyloarthritis or rheumatoid arthritis, IL-17 promotes osteoclastogenesis through induction of RANKL [92]. In rheumatoid arthritis, IL-17 has a pathogenic role in cartilage destruction by inhibiting matrix synthesis by chondrocytes and promoting MMP production [93]. It also contributes to uncoupling by inhibition of bone formation [94]. IL-17 blockade has therapeutic value by reducing joint inflammation and articular bone erosion and diminishing systemic bone loss by suppressing RANKL, IL-1, and TNFα production [95, 96]. Phase I trials indicate that signs and symptoms of rheumatoid arthritis are significantly suppressed following treatment with anti-IL-17 antibodies, without notable adverse effects [97, 98]. Targeting IL-17 with antibodies has been shown to improve rheumatoid disease activity in patients and reduce bone loss [99–101]. Thus, IL-17 produced by Th17 cells can induce bone loss in estrogen deficiency associated osteoporosis and rheumatoid arthritis. In contrast, IL-17 produced by γδ T cells appears to accelerate bone formation in early bone fracture healing by stimulating the proliferation and differentiation of mesenchymal progenitor cells.
Conclusions
In conclusion, IL-17 receptor signaling is needed to protect against bacteria in periodontal tissues. However, excessive IL-17 signaling results in periodontal inflammation and bone loss. The host response to oral microbiota in diabetic conditions triggers inflammation that is more severe than in the euglycemic state. Additionally, the inflammatory response to bacteria also modifies the oral microbiota to render it more pathogenic. Importantly, the pathogenicity of the oral microbiota from a diabetic mouse was more pathogenic compared to microbiota from a normal mouse when it was transferred to a germ-free host. Therefore, further animal studies could potentially reveal the relationships between diabetes and periodontitis/peri-implantitis. Finally, human studies will be needed in the future to confirm the results in the animal models.
Footnotes
Conflict of Interest The authors have no conflicts of interest.
Human and Animal Rights All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11(10):763–76. 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 2.Patel DD, Kuchroo VK. Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity. 2015;43(6):1040–51. 10.1016/j.immuni.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–32. 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 4.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10(7):479–89. 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 5.Sutton CE, Mielke LA, Mills KH. IL-17-producing gammadelta T cells and innate lymphoid cells. Eur J Immunol. 2012;42(9):2221–31. 10.1002/eji.201242569. [DOI] [PubMed] [Google Scholar]
- 6.Hamada S, Umemura M, Shiono T, Tanaka K, Yahagi A, Begum MD, et al. IL-17A produced by gammadelta T cells plays a critical role in innate immunity against listeria monocytogenes infection in the liver. J Immunol. 2008;181(5):3456–63. 10.4049/jimmunol.181.5.3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.••.Dutzan N, Kajikawa T, Abusleme L, Greenwell-Wild T, Zuazo CE, Ikeuchi T, et al. A dysbiotic microbiome triggers TH17 cells to mediate oral mucosal immunopathology in mice and humans. Sci Transl Med. 2018;10(463). 10.1126/scitranslmed.aat0797 [DOI] [PMC free article] [PubMed] [Google Scholar]; Demonstrates that TH17 cells promote periodontal tissue destruction and pharmacological inhibition of TH17 cell differentiation can reduce periodontal bone loss.
- 8.Wilharm A, Tabib Y, Nassar M, Reinhardt A, Mizraji G, Sandrock I, et al. Mutual interplay between IL-17-producing gammadeltaT cells and microbiota orchestrates oral mucosal homeostasis. Proc Natl Acad Sci U S A. 2019;116(7):2652–61. 10.1073/pnas.1818812116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu S. Structural insights into the interleukin-17 family cytokines and their receptors. Adv Exp Med Biol. 2019;1172:97–117. 10.1007/978-981-13-9367-9_5 [DOI] [PubMed] [Google Scholar]
- 10.Ho AW, Gaffen SL. IL-17RC: a partner in IL-17 signaling and beyond. Semin Immunopathol. 2010;32(1):33–42. 10.1007/s00281-009-0185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dutzan N, Abusleme L, Bridgeman H, Greenwell-Wild T, Zangerle-Murray T, Fife ME, et al. On-going mechanical damage from mastication drives homeostatic Th17 cell responses at the oral barrier. Immunity. 2017;46(1): 133–47. 10.1016/j.immuni.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsukasaki M, Komatsu N, Nagashima K, Nitta T, Pluemsakunthai W, Shukunami C, et al. Host defense against oral microbiota by bone-damaging T cells. Nat Commun. 2018;9(1):701. 10.1038/s41467-018-03147-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Awane M, Andres PG, Li DJ, Reinecker HC. NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. J Immunol. 1999;162(9):5337–44. [PubMed] [Google Scholar]
- 14.Shalom-Barak T, Quach J, Lotz M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J Biol Chem. 1998;273(42):27467–73. 10.1074/jbc.273.42.27467. [DOI] [PubMed] [Google Scholar]
- 15.Graves DT, Li J, Cochran DL. Inflammation and uncoupling as mechanisms of periodontal bone loss. J Dent Res. 2011;90(2):143–53. 10.1177/0022034510385236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pacios S, Xiao W, Mattos M, Lim J, Tarapore RS, Alsadun S, et al. Osteoblast lineage cells play an essential role in periodontal bone loss through activation of nuclear factor-kappa B. Sci Rep. 2015;5:16694. 10.1038/srep16694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graves DT, Alshabab A, Albiero ML, Mattos M, Correa JD, Chen S, et al. Osteocytes play an important role in experimental periodontitis in healthy and diabetic mice through expression of RANKL. J Clin Periodontol. 2018;45(3):285–92. 10.1111/jcpe.12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zheng J, Chen S, Albiero ML, Vieira GHA, Wang J, Feng JQ, et al. Diabetes activates periodontal ligament fibroblasts via NF-kappaB in vivo. J Dent Res. 2018;97(5):580–8. 10.1177/0022034518755697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pacios S, Kang J, Galicia J, Gluck K, Patel H, Ovaydi-Mandel A, et al. Diabetes aggravates periodontitis by limiting repair through enhanced inflammation. FASEB J. 2012;26(4):1423–30. 10.1096/fj.11-196279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zenobia C, Hajishengallis G. Basic biology and role of interleukin-17 in immunity and inflammation. Periodontol 2000. 2015;69(1):142–59. 10.1111/prd.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson RB, Wood N, Serio FG. Interleukin-11 and IL-17 and the pathogenesis of periodontal disease. J Periodontol. 2004;75(1):37–43. 10.1902/jop.2004.75.1.37. [DOI] [PubMed] [Google Scholar]
- 22.Chen XT, Chen LL, Tan JY, Shi DH, Ke T, Lei LH. Th17 and Th1 Lymphocytes Are Correlated with Chronic Periodontitis. Immunol Invest. 2016;45(3):243–54. 10.3109/08820139.2016.1138967. [DOI] [PubMed] [Google Scholar]
- 23.Dutzan N, Vernal R, Vaque JP, Garcia-Sesnich J, Hernandez M, Abusleme L, et al. Interleukin-21 expression and its association with proinflammatory cytokines in untreated chronic periodontitis patients. J Periodontol. 2012;83(7):948–54. 10.1902/jop.2011.110482. [DOI] [PubMed] [Google Scholar]
- 24.Lester SR, Bain JL, Johnson RB, Serio FG. Gingival concentrations of interleukin-23 and −17 at healthy sites and at sites of clinical attachment loss. J Periodontol. 2007;78(8):1545–50. 10.1902/jop.2007.060458. [DOI] [PubMed] [Google Scholar]
- 25.Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, et al. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor-dependent signals. Blood. 2007;109(9):3794–802. 10.1182/blood-2005-09-010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eskan MA, Jotwani R, Abe T, Chmelar J, Lim JH, Liang S, et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat Immunol. 2012;13(5):465–73. 10.1038/ni.2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y, Dong G, Xiao W, Xiao E, Miao F, Syverson A, et al. Effect of aging on periodontal inflammation, microbial colonization, and disease susceptibility. J Dent Res. 2016;95(4):460–6. 10.1177/0022034515625962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.IDF Diabetes Atlas. 8th ed. Brussels, Belgium: International Diabetes Federation; 2017. [Google Scholar]
- 29.Sami W, Ansari T, Butt NS, Hamid MRA. Effect of diet on type 2 diabetes mellitus: a review. Int J Health Sci (Qassim). 2017;11(2):65–71. [PMC free article] [PubMed] [Google Scholar]
- 30.Boldison J, Wong FS. Immune and pancreatic beta cell interactions in type 1 diabetes. Trends Endocrinol Metab. 2016;27(12):856–67. 10.1016/j.tem.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Xiao E, Wu Y, Graves DT. Impact of diabetes on periodontal disease. In: Diabetic bone disease: Springer; 2016. p. 95–112. [Google Scholar]
- 32.Nelson RG, Shlossman M, Budding LM, Pettitt DJ, Saad MF, Genco RJ, et al. Periodontal disease and NIDDM in Pima Indians. Diabetes Care. 1990;13(8):836–40. 10.2337/diacare.13.8.836. [DOI] [PubMed] [Google Scholar]
- 33.Novotna M, Podzimek S, Broukal Z, Lencova E, Duskova J. Periodontal diseases and dental caries in children with type 1 diabetes mellitus. Mediat Inflamm. 2015;2015:379626. 10.1155/2015/379626. [DOI] [PMC free article] [PubMed]
- 34.Shlossman M, Knowler WC, Pettitt DJ, Genco RJ. Type 2 diabetes mellitus and periodontal disease. J Am Dent Assoc. 1990;121(4):532–6. 10.14219/jada.archive.1990.0211. [DOI] [PubMed] [Google Scholar]
- 35.•.Graves DT, Corrêa JD, Silva TA. The Oral Microbiota Is Modified by Systemic Diseases. J Dent Res. 2019;98:148–156. 10.1177/0022034518805739. [DOI] [PMC free article] [PubMed] [Google Scholar]; This review discusses the oral microbiome and how it is modified by systemic diseases such as diabetes, rheumatoid arthritis and lupus erythematosus.
- 36.Taylor G, Burt B, Becker M, Genco R, Shlossman M, Knowler W, et al. Non-insulin dependent diabetes mellitus and alveolar bone loss progression over 2 years. J Periodontol. 1998;69:76–83. 10.1902/jop.1998.69T.76. [DOI] [PubMed] [Google Scholar]
- 37.Safkan-Seppala B, Sorsa T, Tervahartiala T, Beklen A, Konttinen YT. Collagenases in gingival crevicular fluid in type 1 diabetes mellitus. J Periodontol. 2006;77(2):189–94. 10.1902/jop.2006.040322. [DOI] [PubMed] [Google Scholar]
- 38.Patil VS, Patil VP, Gokhale N, Acharya A, Kangokar P. Chronic periodontitis in type 2 diabetes mellitus: oxidative stress as a common factor in periodontal tissue injury. J Clin Diagn Res. 2016;10(4):Bc12–6. 10.7860/jcdr/2016/17350.7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Albert DA, Ward A, Allweiss P, Graves DT, Knowler WC, Kunzel C et al. Diabetes and oral disease: implications for health professionals. Ann N Y Acad Sci. 2012;1255:1–15. 10.1111/j.1749-6632.2011.06460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karima M, Kantarci A, Ohira T, Hasturk H, Jones VL, Nam BH, et al. Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: association with periodontitis. J Leukoc Biol. 2005;78(4):862–70. 10.1189/jlb.1004583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salvi GE, Beck JD, Offenbacher S. PGE2, IL-1 beta, and TNF-alpha responses in diabetics as modifiers of periodontal disease expression. Ann Periodontol. 1998;3(1):40–50. 10.1902/annals.1998.3.1.40. [DOI] [PubMed] [Google Scholar]
- 42.Song L, Dong G, Guo L, Graves DT. The function of dendritic cells in modulating the host response. Mol Oral Microbiol. 2018;33(1):13–21. 10.1111/omi.12195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JH, Lee DE, Choi SH, Cha JH, Bak EJ, Yoo YJ. Diabetic characteristics and alveolar bone loss in streptozotocin- and streptozotocin-nicotinamide-treated rats with periodontitis. J Periodontal Res. 2014;49(6):792–800. 10.1111/jre.12165. [DOI] [PubMed] [Google Scholar]
- 44.Wu YY, Xiao E, Graves DT. Diabetes mellitus related bone metabolism and periodontal disease. Int J Oral Sci. 2015;7(2):63–72. 10.1038/ijos.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lalla E, Lamster IB, Feit M, Huang L, Spessot A, Qu W, et al. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 2000;105(8):1117–24. 10.1172/JCI8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pacios S, Andriankaja O, Kang J, Alnammary M, Bae J, de Brito BB, et al. Bacterial infection increases periodontal bone loss in diabetic rats through enhanced apoptosis. Am J Pathol. 2013;183(6):1928–35. 10.1016/j.ajpath.2013.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.•.Napoli N, Chandran M, Pierroz DD, Abrahamsen B, Schwartz AV, Ferrari SL, et al. Mechanisms of diabetes mellitus-induced bone fragility. Nat Rev Endocrinol. 2017;13(4):208–19. 10.1038/nrendo.2016.153 [DOI] [PubMed] [Google Scholar]; This review discusses how conditions present in diabetes, increased inflammation, hyperglycaemia, oxidative stress and advanced glycation endproducts affect bone formation and resorption in type-1 and type-2 diabetes.
- 48.Jiao H, Xiao E, Graves DT. Diabetes and Its Effect on Bone and Fracture Healing. Curr Osteoporos Rep. 2015;13(5):327–35. 10.1007/s11914-015-0286-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu H, Kraut D, Gerstenfeld LC, Graves DT. Diabetes interferes 559 with the bone formation by affecting the expression of transcrip- 560 tion factors that regulate osteoblast differentiation. Endocrinology. 5612003;144(1):346–52. 10.1210/en.2002-220072. [DOI] [PubMed] [Google Scholar]
- 50.Liu R, Bal H, Desta T, Krothapalli N, Alyassi M, Luan Q, et al. Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. J Dent Res. 2006;85(6):510–4. 10.1177/154405910608500606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ko KI, Coimbra LS, Tian C, Alblowi J, Kayal RA, Einhorn TA, et al. Diabetes reduces mesenchymal stem cells in fracture healing through a TNFalpha-mediated mechanism. Diabetologia. 2015;58(3):633–42. 10.1007/s00125-014-3470-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim JC, Ko KI, Mattos M, Fang M, Zhang C, Feinberg D, et al. TNFalpha contributes to diabetes impaired angiogenesis in fracture healing. Bone. 2017;99:26–38. 10.1016/j.bone.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu J, Jiang Y, Mao J, Gu B, Liu H, Fang B. High levels of glucose induces a dose-dependent apoptosis in human periodontal ligament fibroblasts by activating caspase-3 signaling pathway. Appl Biochem Biotechnol. 2013;170(6):1458–71. 10.1007/s12010-013-0287-y. [DOI] [PubMed] [Google Scholar]
- 54.Behl Y, Krothapalli P, Desta T, Roy S, Graves DT. FOXO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes. 2009;58(4):917–25. 10.2337/db08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinberg E, Maymon T, Moses O, Weinreb M. Streptozotocin-induced diabetes in rats diminishes the size of the osteoprogenitor pool in bone marrow. Diabetes Res Clin Pract. 2014;103(1):35–41. 10.1016/j.diabres.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 56.Kanazawa I, Sugimoto T. Diabetes mellitus-induced bone fragility. Intern Med. 2018;57(19):2773–85. 10.2169/internalmedicine.0905-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li DX, Deng TZ, Lv J, Ke J. Advanced glycation end products (AGEs) and their receptor (RAGE) induce apoptosis of periodontal ligament fibroblasts. Braz J Med Biol Res. 2014;47(12):1036–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Al-Mashat HA, Kandru S, Liu R, Behl Y, Desta T, Graves DT. Diabetes enhances mRNA levels of proapoptotic genes and caspase activity, which contribute to impaired healing. Diabetes. 2006;55(2):487–95. 10.2337/diabetes.55.02.06.db05-1201. [DOI] [PubMed] [Google Scholar]
- 59.Cianciola LJ, Park BH, Bruck E, Mosovich L, Genco RJ. Prevalence of periodontal disease in insulin-dependent diabetes mellitus (juvenile diabetes). J Am Dent Assoc. 1982;104(5):653–60. 10.14219/jada.archive.1982.0240. [DOI] [PubMed] [Google Scholar]
- 60.Andriankaja OM, Galicia J, Dong G, Xiao W, Alawi F, Graves DT. Gene expression dynamics during diabetic periodontitis. J Dent Res. 2012;91(12):1160–5. 10.1177/0022034512465292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lalla E, Papapanou PN. Diabetes mellitus and periodontitis: a tale of two common interrelated diseases. Nat Rev Endocrinol. 2011;7(12):738–48. 10.1038/nrendo.2011.106. [DOI] [PubMed] [Google Scholar]
- 62.Loe H. Periodontal disease. The sixth complication of diabetes mellitus. Diabetes Care. 1993;16(1):329–34. [PubMed] [Google Scholar]
- 63.Casarin RC, Barbagallo A, Meulman T, Santos VR, Sallum EA, Nociti FH, et al. Subgingival biodiversity in subjects with uncontrolled type-2 diabetes and chronic periodontitis. J Periodontal Res. 2013;48(1):30–6. 10.1111/j.1600-0765.2012.01498.x. [DOI] [PubMed] [Google Scholar]
- 64.Mashimo PA, Yamamoto Y, Slots J, Park BH, Genco RJ. The periodontal microflora of juvenile diabetics. Culture, immunofluorescence, and serum antibody studies. J Periodontol. 1983;54(7):420–30. 10.1902/jop.1983.54.7.420. [DOI] [PubMed] [Google Scholar]
- 65.Campus G, Salem A, Uzzau S, Baldoni E, Tonolo G. Diabetes and periodontal disease: a case-control study. J Periodontol. 2005;76(3):418–25. 10.1902/jop.2005.763.418. [DOI] [PubMed] [Google Scholar]
- 66.da Cruz GA, de Toledo S, Sallum EA, Sallum AW, Ambrosano GM, de Cassia Orlandi Sardi J, et al. Clinical and laboratory evaluations of non-surgical periodontal treatment in subjects with diabetes mellitus. J Periodontol. 2008;79(7):1150–7. 10.1902/jop.2008.070503. [DOI] [PubMed] [Google Scholar]
- 67.Sastrowijoto SH, Hillemans P, van Steenbergen TJ, Abraham-Inpijn L, de Graaff J. Periodontal condition and microbiology of healthy and diseased periodontal pockets in type 1 diabetes mellitus patients. J Clin Periodontol. 1989;16(5):316–22. 10.1111/j.1600-051x.1989.tb01662.x. [DOI] [PubMed] [Google Scholar]
- 68.Aemaimanan P, Amimanan P, Taweechaisupapong S. Quantification of key periodontal pathogens in insulin-dependent type 2 diabetic and non-diabetic patients with generalized chronic periodontitis. Anaerobe. 2013;22:64–8. 10.1016/j.anaerobe.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 69.Demmer RT, Breskin A, Rosenbaum M, Zuk A, LeDuc C, Leibel R, et al. The subgingival microbiome, systemic inflammation and insulin resistance: the oral infections, glucose intolerance and insulin resistance study. J Clin Periodontol. 2017;44(3):255–65. 10.1111/jcpe.12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Merchant AT, Shrestha D, Chaisson C, Choi YH, Hazlett LJ, Zhang J. Association between serum antibodies to oral microorganisms and hyperglycemia in adults. J Dent Res. 2014;93(8):752–9. 10.1177/0022034514538451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sakalauskiene J, Kubilius R, Gleiznys A, Vitkauskiene A, Ivanauskiene E, Saferis V Relationship of clinical and microbiological variables in patients with type 1 diabetes mellitus and periodontitis. Med Sci Monit. 2014;20:1871–7. 10.12659/MSM.890879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou M, Rong R, Munro D, Zhu C, Gao X, Zhang Q, et al. Investigation of the effect of type 2 diabetes mellitus on subgingival plaque microbiota by high-throughput 16S rDNA pyrosequencing. PLoS One. 2013;8(4):e61516. 10.1371/journal.pone.0061516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Graves DT, Naguib G, Lu H, Leone C, Hsue H, Krall E. Inflammation is more persistent in type 1 diabetic mice. J Dent Res. 2005;84:324–8. [DOI] [PubMed] [Google Scholar]
- 74.Naguib G, Al-Mashat H, Desta T, Graves DT. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J Invest Dermatol. 2004;123:87–92. 10.1111/j.0022-202X.2004.22711.x. [DOI] [PubMed] [Google Scholar]
- 75.••.Xiao E, Mattos M, Vieira GHA, Chen S, Correa JD, Wu Y, et al. Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe. 2017;22(1):120–8 e4. 10.1016/jxhom.2017.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that IL-17 alters the oral bacterial composition in diabetic mice and that the oral bacteria from diabetic mice are more pathogenic when transferred to germ free hosts than bacteria from normoglycemic controls. The increased pathogencity of oral bacteria in diabetic mice is largely reversed by inhibiton of IL-17.
- 76.Grice EA, Snitkin ES, Yockey LJ, Bermudez DM, Program NCS, Liechty KW, et al. Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc Natl Acad Sci U S A. 2010;107(33):14799–804. 10.1073/pnas.1004204107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patterson E, Marques TM, O’Sullivan O, Fitzgerald P, Fitzgerald GF, Cotter PD, et al. Streptozotocin-induced type-1-diabetes disease onset in Sprague-Dawley rats is associated with an altered intestinal microbiota composition and decreased diversity. Microbiology. 2015;161(Pt 1):182–93. 10.1099/mic.0.082610-0 [DOI] [PubMed] [Google Scholar]
- 78.Ussar S, Fujisaka S, Kahn CR. Interactions between host genetics and gut microbiome in diabetes and metabolic syndrome. Mol Metab. 2016;5(9):795–803. 10.1016/j.molmet.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Curtis MA, Zenobia C, Darveau RP. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe. 2011;10(4):302–6. 10.1016/jxhom.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abusleme L, Moutsopoulos NM. IL-17: overview and role in oral immunity and microbiome. Oral Dis. 2017;23(7):854–65. 10.1111/odi.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Won HY, Lee JA, Park ZS, Song JS, Kim HY, Jang SM, et al. Prominent bone loss mediated by RANKL and IL-17 produced by CD4+ T cells in TallyHo/JngJ mice. PLoS One. 2011;6(3):e18168. 10.1371/journal.pone.0018168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao R, Wang X, Feng F. Upregulated cellular expression of IL-17 by CD4+ T-cells in osteoporotic postmenopausal women. Ann Nutr Metab. 2016;68(2):113–8. 10.1159/000443531. [DOI] [PubMed] [Google Scholar]
- 83.Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PLoS One. 2012;7(9):e44552. 10.1371/joumal.pone.0044552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.DeSelm CJ, Takahata Y, Warren J, Chappel JC, Khan T, Li X, et al. IL-17 mediates estrogen-deficient osteoporosis in an Act1-dependent manner. J Cell Biochem. 2012;113(9):2895–902. 10.1002/jcb.24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tyagi AM, Mansoori MN, Srivastava K, Khan MP, Kureel J, Dixit M, et al. Enhanced immunoprotective effects by anti-IL-17 antibody translates to improved skeletal parameters under estrogen deficiency compared with anti-RANKL and anti-TNF-alpha antibodies. J Bone Miner Res. 2014;29(9):1981–92. 10.1002/jbmr.2228. [DOI] [PubMed] [Google Scholar]
- 86.Shukla P, Mansoori MN, Singh D. Efficacy of anti-IL-23 monotherapy versus combination therapy with anti-IL-17 in estrogen deficiency induced bone loss conditions. Bone. 2018;110:84–95. 10.1016/j.bone.2018.01.027. [DOI] [PubMed] [Google Scholar]
- 87.Uluckan O, Jimenez M, Karbach S, Jeschke A, Grana O, Keller J, et al. Chronic skin inflammation leads to bone loss by IL-17-mediated inhibition of Wnt signaling in osteoblasts. Sci Transl Med. 2016;8(330):330ra37. 10.1126/scitranslmed.aad8996. [DOI] [PubMed] [Google Scholar]
- 88.Ono T, Takayanagi H. Osteoimmunology in bone fracture healing. Curr Osteoporos Rep. 2017;15(4):367–75. 10.1007/s11914-017-0381-0. [DOI] [PubMed] [Google Scholar]
- 89.Ono T, Okamoto K, Nakashima T, Nitta T, Hori S, Iwakura Y, et al. IL-17-producing gammadelta T cells enhance bone regeneration. Nat Commun. 2016;7:10928. 10.1038/ncomms10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bahney CS, Zondervan RL, Allison P, Theologis A, Ashley JW, Ahn J, et al. Cellular biology of fracture healing. J Orthop Res. 2019;37(1):35–50. 10.1002/jor.24170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Croes M, Kruyt MC, Groen WM, van Dorenmalen KMA, Dhert WJA, Oner FC, et al. Interleukin 17 enhances bone morphogenetic protein-2-induced ectopic bone formation. Sci Rep. 2018;8(1):7269. 10.1038/s41598-018-25564-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, et al. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. J Immunol. 2003;170(5):2655–62. 10.4049/jimmunol.170.5.2655. [DOI] [PubMed] [Google Scholar]
- 93.Reynolds G, Cooles FA, Isaacs JD, Hilkens CM. Emerging immunotherapies for rheumatoid arthritis. Hum Vaccin Immunother. 2014;10(4):822–37. 10.4161/hv.27910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li X, Yuan FL, Lu WG, Zhao YQ, Li CW, Li JP, et al. The role of interleukin-17 in mediating joint destruction in rheumatoid arthritis. Biochem Biophys Res Commun. 2010;397(2):131–5. 10.1016/j.bbrc.2010.05.111. [DOI] [PubMed] [Google Scholar]
- 95.Koenders MI, Lubberts E, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Di Padova FE, et al. Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol. 2005;167(1):141–9. 10.1016/S0002-9440(10)62961-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Karmakar S, Kay J, Gravallese EM. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheum Dis Clin N Am. 2010;36(2):385–404. 10.1016/j.rdc.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van den Berg WB, Miossec P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):549–53. 10.1038/nrrheum.2009.179. [DOI] [PubMed] [Google Scholar]
- 98.Daoussis D, Andonopoulos AP, Liossis SN. Wnt pathway and IL-17: novel regulators of joint remodeling in rheumatic diseases. Looking beyond the RANK-RANKL-OPG axis. Semin Arthritis Rheum. 2010;39(5):369–83. 10.1016/j.semarthrit.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 99.Kampylafka E, d’Oliveira I, Linz C, Lerchen V, Stemmler F, Simon D, et al. Resolution of synovitis and arrest of catabolic and anabolic bone changes in patients with psoriatic arthritis by IL-17A blockade with secukinumab: results from the prospective PSARTROS study. Arthritis Res Ther. 2018;20(1):153. 10.1186/s13075-018-1653-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van der Heijde D, Gladman DD, Kishimoto M, Okada M, Rathmann SS, Moriarty SR, et al. Efficacy and safety of ixekizumab in patients with active psoriatic arthritis: 52-week results from a phase III study (SPIRIT-P1). J Rheumatol. 2018;45(3):367–77. 10.3899/jrheum.170429. [DOI] [PubMed] [Google Scholar]
- 101.Blanco FJ, Moricke R, Dokoupilova E, Codding C, Neal J, Andersson M, et al. Secukinumab in active rheumatoid arthritis: a phase III randomized, double-blind, active comparator-and placebo-controlled study. Arthritis Rheumatol. 2017;69(6):1144–53. 10.1002/art.40070. [DOI] [PubMed] [Google Scholar]