

Abstract
The high prevalence and rising incidence of autoimmune diseases have become a prominent public health issue. Autoimmune disorders result from the immune system erroneously attacking the body’s own healthy cells and tissues, causing persistent inflammation, tissue injury, and impaired organ function. Existing treatments primarily rely on broad immunosuppression, leaving patients vulnerable to infections and necessitating lifelong treatments. To address these unmet needs, an emerging frontier of vaccine development aims to restore immune equilibrium by inducing immune tolerance to autoantigens, offering a potential avenue for a cure rather than mere symptom management. We discuss this burgeoning field of vaccine development against inflammation and autoimmune diseases, with a focus on common autoimmune disorders, including multiple sclerosis, type 1 diabetes, rheumatoid arthritis, inflammatory bowel disease, and systemic lupus erythematosus. Vaccine-based strategies provide a new pathway for the future of autoimmune disease therapeutics, heralding a new era in the battle against inflammation and autoimmunity.
Keywords: Vaccine, Immunotherapy, Autoimmune, Inflammation, Immune tolerance
Graphical Abstract
1. Introduction
Approximately 5 to 8 % of the United States population has been reported to have autoimmune diseases [1]. Similarly, a recent population-based study of 22 million individuals in the United Kingdom estimated that about 1 out of 10 people were affected by one of the 19 most common autoimmune disorders [2]. Despite such high prevalence and incidence rates, the race to develop the first cures for autoimmune diseases is still ongoing. General treatments for patients with autoimmune diseases include immunosuppressants, such as anti-inflammatory agents, corticosteroids, and monoclonal antibodies, which work to suppress inflammatory cells and pathways. While these approaches may delay disease progression, the broad immunosuppression or immuno-depletion can the body more vulnerable to serious infections and malignancies. Even monoclonal antibodies are not selective enough to prevent binding to cells that are not of interest but display the targeted molecule. Additionally, these therapies require prolonged periods of treatments, as they do not address the loss of immune tolerance [3]. Thus, novel approaches are needed for the treatment of inflammation and autoimmunity.
Aberrant T cell and/or B cell reactivity drives the development of autoimmunity. Naturally, the immune system maintains tolerance against the body’s own tissues as self-recognizing T cells and B cells undergo deletion, anergy, differentiation, or receptor editing in the thymus for T cells and bone marrow for B cells (central tolerance), and in the periphery (peripheral tolerance) [4–7]. Diverse regulatory T cells (Treg) and regulatory B cells (Breg) in the periphery play a critical role in restraining autoreactivity through multiple mechanisms, including production of immunosuppressive cytokines and contact-dependent interactions [8,9]. These mechanisms collectively lead to a state of immunological non-responsiveness or self-tolerance. Notably, the self-reactive cells are not completely eliminated in central tolerance and can escape into the periphery [10]. Such “leakiness” is essential to maintain host defense in that T cell and B cell repertoires need to be broad enough to respond to foreign antigens while being restricted to prevent self-reactivity. Escaped autoreactive cells can promote the development of autoimmune diseases if they erroneously mount an inflammatory response against host cells [3]. Dysregulation of immune tolerance leads to tissue destruction that begins when antigen-presenting cells (APCs) process and display antigens in the major histocompatibility complex (MHC) to T cells in the presence of inflammatory signals. Such dysregulated responses can occur through multiple mechanisms, including formation of neoepitopes, molecular mimicry, inflammatory conditions, dysregulated co-stimulation, and dysfunction of regulatory cells [11]. The ability of autoreactive T cells to induce inflammation and subsequent tissue destruction is dictated by their effector functions towards target cells, provision of T cell help to autoreactive B cells, and engagement of body’s innate immune arm [12,13]. Upon interaction with target cells, autoreactive T cells produce pro-inflammatory cytokines that initiate inflammation, and create a self-propagating cascade recruiting macrophages, neutrophils, and other immune cells [12,13]. Multiple subsets of CD4+ T helper (Th) cells and CD8+ cytotoxic T cells (CTLs) play critical roles in tissue damage, which can be counterbalanced by Treg-mediated suppression [14,15]. However, dysregulated responses, or tolerance breakdown, can occur through multiple mechanisms, including formation of neoepitopes, molecular mimicry, inflammatory conditions, dysregulated co-stimulation, and dysfunction of regulatory cells [11]. For instance, microorganisms, a notable environmental trigger of many autoimmune diseases, can provide foreign antigens with sequences homologous to human (molecular mimicry) and can stimulate the innate immune system for inflammation and thereby potentiate adaptive immune responses via pathogen-associated molecular patterns (PAMPs) [16–21]. Thus, eliminating the pathogenic microorganisms may be a potential strategy to address inflammation and autoimmunity.
To restore the body’s homeostasis, immune tolerance needs to be generated to allow recognition and initiation of a proper immune response against disease-related antigens. Therefore, vaccine-based tolerance-inducing immunotherapies against autoimmune diseases are gaining attention to accomplish those goals [22–24]. With such vaccines, the targeted autoantigen and co-loaded immune-modulating components are delivered to APCs, which present the autoantigen to T cells in the context of regulated costimulatory or coinhibitory signals. With the use of nanoparticles (NPs) or microparticles (MPs), the physicochemical properties of autoantigens and immunomodulators can be fine-tuned for optimal delivery to APCs. Other strategies include presentation of peptide-bound MHC class I or class II molecules for direct modulation of autoreactive T cells, or targeting of specific receptors or disease-related inflammatory mediators. While communication of exciting, novel ideas for antigen-specific immunotherapies are endless, the diverse pathways that are targeted commonly aim towards production of anti-inflammatory cytokines, induction of tolerogenic DCs or Treg cells, restriction of APC-T cell interaction, anergy or deletion of pathogenic T cells specific for autoantigen epitopes, reduction of infiltrating autoreactive T cells in inflamed tissues, deletion of autoantibody-producing plasma cells, and eventually, an established tolerance to autoantigens demonstrated by disease prevention or regression.
With these potential multiple mechanisms of action in mind, in this review, we discuss recent studies that aim to restore immune tolerance against inflammation and autoimmune diseases by the use of vaccine technologies that induce antigen-specific immune tolerance. In particular, we will focus on vaccine strategies in the context of the following common autoimmune disorders based on their high prevalence, our mechanistic understanding of the pathophysiology, and availability of preclinical models to evaluate drug candidates: multiple sclerosis (MS), type 1 diabetes mellitus (T1D), rheumatoid arthritis (RA), inflammatory bowel disease (IBD), and systemic lupus erythematosus (SLE).
2. Multiple sclerosis (MS)
Multiple sclerosis (MS) is the most frequent neurodegenerative disorder of the central nervous system (CNS) affecting 2.8 million people worldwide [25]. Complex gene-environment interactions contribute to this chronic, heterogenous, demyelinating disease, which can give rise to symptoms, including difficulties in movement and visual, learning, or memory impairment [26,27]. In MS, numerous immune cells, including IFN-y-producing Th1 cells, IL-17-producing Th17 cells, CD8+ T cells, B cells, and myeloid cells, are involved in the pathogenesis, breakdown of the blood brain barrier, CNS infiltration, and injury of oligodendrocytes and neurons [26,28]. Myelin proteins that are encephalitogenic and immunodominant, including myelin oligodendrocyte glycoprotein (MOG), myelin proteolipid protein (PLP), and myelin basic protein (MBP), have been identified and used to establish CD4+ T-cell-mediated animal models that resemble MS referred to as experimental autoimmune encephalomyelitis (EAE) [29–32]. In EAE, infiltrating CD4+ T cells are re-activated in the CNS by APCs, which results in monocyte recruitment and naïve CD4+ and CD8+ T cell activation through epitope spreading [33,34].
Extensive research has been performed with the EAE model to study and develop therapeutics aimed to induce antigen-specific immunological tolerance. A widely used approach for antigen-specific immunotherapeutics focuses on the delivery of antigens for antigen presentation in the absence of costimulatory or inflammatory signals or for processing of the antigen with innate mechanisms (e.g., apoptotic clearance that leads to tolerance) [23,35]. In this regard, tolerogenic APCs, particularly immature DCs, or scavengers such as Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs), and macrophage receptor with collagenous structure (MARCO)-positive macrophages, have become attractive targets for improving antigen-specific immune tolerance induction [36–38]. Stemming from studies that report polyethylene glycol (PEG) on NPs can reduce protein adsorption and complement activation, which often accompany administration of biomaterials, Li et al. prepared PEGylated poly(lactide-co-glycolide) (PLGA) NPs loaded with auto-antigen peptide MOG35–55 (Fig. 1) [39–41]. MOG-PLGA-PEG-NPs induced significant reduction in clinical scores of EAE mice as well as in expression of costimulatory molecules (i. e., CD80, CD86, and CD40) on DCs in C57BL/6 J mice compared to MOG-PLGA-NPs [41]. Furthermore, MOG-PLGA-PEG-NPs prompted significantly lower concentrations of complement byproducts C4d and C5a in vitro compared to MOG-PLGA-NPs as measured by ELISA and α2′ chain (40 kDa) of C3 in vivo as measured by Western blot analysis. Getts and colleagues decorated 500 nm polystyrene beads (PSB) with immunodominant myelin proteolipid protein PLP139–151 epitope and showed the prophylactic and therapeutic efficacy of these particles in a relapsing-remitting EAE (R-EAE) model [42]. Ag-PSB were taken up by MARCO scavenger receptor–expressing macrophages via passive targeting, and Ag-PSB induced long-term antigen-specific T-cell tolerance. The PSB MPs were succeeded by simple myelin antigen-coupled PLGA NPs (430 nm in diameter) capable of more robust therapeutic effects [43]. In accordance with complete remission and continuously reduced clinical scores in R-EAE mice, there were significantly reduced levels of encephalitogenic Th1 (IFN-γ+) and Th17(IL-17A+) CNS-infiltrating T cells. These examples have demonstrated that tolerance can be induced by particles loaded or surface-coupled with autoantigens and by harnessing tolerogenic APCs or scavengers destined for clearance of nano- and micro-particles.
Fig. 1.
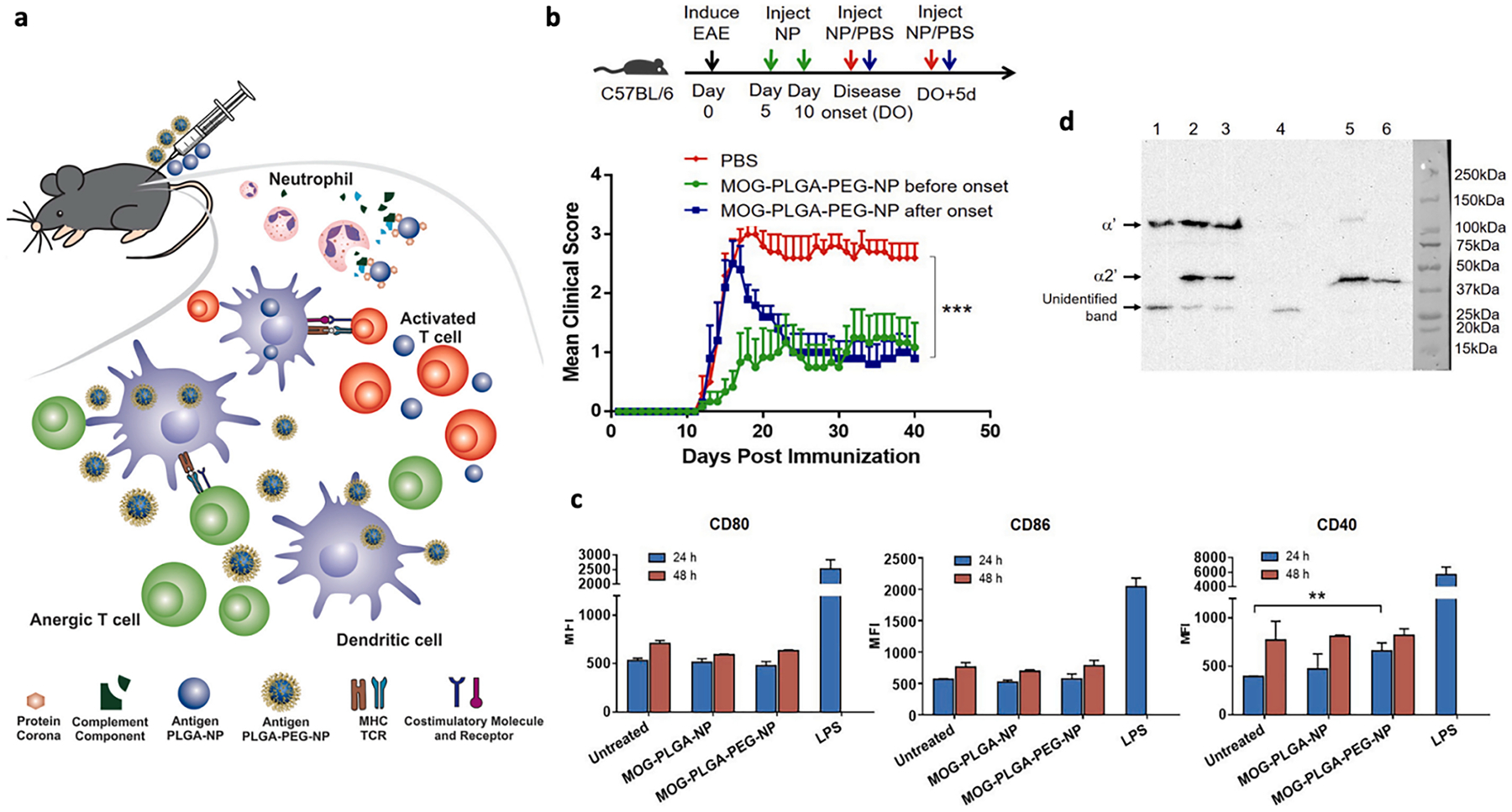
A, Subcutaneous injection of MOG-PLGA-PEG-NPs can induce antigen-specific immune tolerance in EAE. Tolerance is induced upon reduction of NP-mediated complement activation and expression of costimulatory molecules. b, Clinical scores of EAE mice comparing treatments with MOG-PLGA-PEG-NPs administered before or after onset of disease. Statistics are shown for the last data point using two-way ANOVA with post-hoc Tukey test. ***p < 0.001. c, Expression of costimulatory molecules CD80, CD86, and CD40 on BMDCs treated with PEGylated NPs in vitro. Data are represented as mean ± S.D. (n = 3). **p < 0.01. d, Complement activation in vivo (representative western blot analysis of C3 activation as determined by intensity of band representing α2′ chain, right). 1. Tissue control supernatant; 2. MOG-PLGA-NP supernatant; 3. MOG-PLGA-PEG-NP supernatant; 4. Tissue control pellet; 5. MOGPLGA-NP pellet; 6. MOG-PLGA-PEG-NP pellet.
Reproduced with permission from [41]
Other innate mechanisms have been explored to enhance active targeting to APCs. For example, mannose- or N-acetylglucosamine-decorated NPs or autoantigens that target mannose receptors on DCs have been evaluated for treating EAE [44,45]. Triantafyllakou et al. conjugated glucosamine to the C-terminal of MOG35–55 and further encapsulated the peptide into PLGA NPs [44]. The resultant NPs demonstrated a stable release over three weeks in vitro and suppressed the symptoms of the disease in a therapeutic setting. Attempts have also been made to target tissue-specific APCs. Lung APCs are highly phagocytic and may process NPs at a higher rate than liver and spleen APCs, allowing for lower doses and changing migration and activation patterns of CD4+ T cells to prevent antigen encounter in the CNS [46]. Intratracheal administration of PLP-loaded PLGA particles for direct delivery to the lung, where autoreactive T cells travel through in EAE before reaching disease sites, promoted disease amelioration, and reduced autoreactive CD4+ T cell proliferation [46,47].
While the effectiveness of therapeutic approaches was mostly evaluated by immunizing and treating the mice with the same antigen (one predominant, disease-causing autoantigen), we should note that the evolving antigen specificity during disease progression, or “epitope spreading”, is a prominent feature in both human MS patients and EAE mice [32–34,48]. For instance, in SJL/J mice immunized with PLP139–151 (spread epitope), autoreactive PLP-specific CD4+ T cells caused primary acute disease phase followed by remission, but the resultant tissue damaged promoted the activation of T cells recognizing a second PLP epitope PLP178–191 (subdominant epitope) [42]. Treatment with the spread epitope can prophylactically prevent relapsing-remitting EAE. Casella et al. showed that oligodendrocyte-derived extracellular vesicles (Ol-EVs), which naturally contained multiple myelin antigens, were effective in treating three models of EAE, including chronic (MOG35–55-immunized C57BL/6 mice and MBPAc (1–11)/B10.PL mice) and relapsing-remitting (PLP139–151/SJL) diseases upon intravenous injection [49]. Ol-EVs induced immunosuppressive monocytes as characterized by upregulation in Arg1+, PD-L1 and IL-10, which was correlated with apoptotic T cells (caspase3+ PD-1+ CD4+) but not increased Foxp3+ Treg. Moreover, tolerization with multiple myelin epitopes was proposed to have better clinical potential [50,51]. A first-in-human trial using a single infusion of autologous peripheral blood mononuclear cells coupled with seven myelin peptides (MOG1–20, MOG35–55, MBP13–32, MBP83–99, MBP111–129, MBP146–170, and PLP139–154) was shown to be effective in reducing T cell responses to four of the seven myelin epitopes three months post treatment [51]. As an alternative approach to peptide-based strategies, Krienke and colleagues utilized lipid NPs to deliver mRNAs encoding myelin antigens (called nanoparticle-formulated 1 methylpseudouridine-modified messenger RNA, or m1Ψ mRNA) (Fig. 2) [52]. Lipid NPs containing m1Ψ mRNA were able to induce bystander suppression, where m1Ψ mRNA encoding either PLP139–151 or MOG35–55 suppressed EAE in C57BL/6 × SJL mice immunized with PLP139–151 peptide. Moreover, lipid NPs carrying m1Ψ mRNA encoding four autoantigens were effective in a complex EAE model driven by multiple pathogenic T cell clones (MOG35–55, PLP139–151, PLP178–191, MBP84–104, and MOBP15–36). Interestingly, m1Ψ mRNA encoding a single MOG35–55 peptide was as effective as that encoding four epitopes, suggesting a strong bystander tolerance was induced that protected against polyclonal autoreactive T cells. Nevertheless, questions remain regarding the breadth, duration, and capacity of the induced bystander suppression.
Fig. 2.
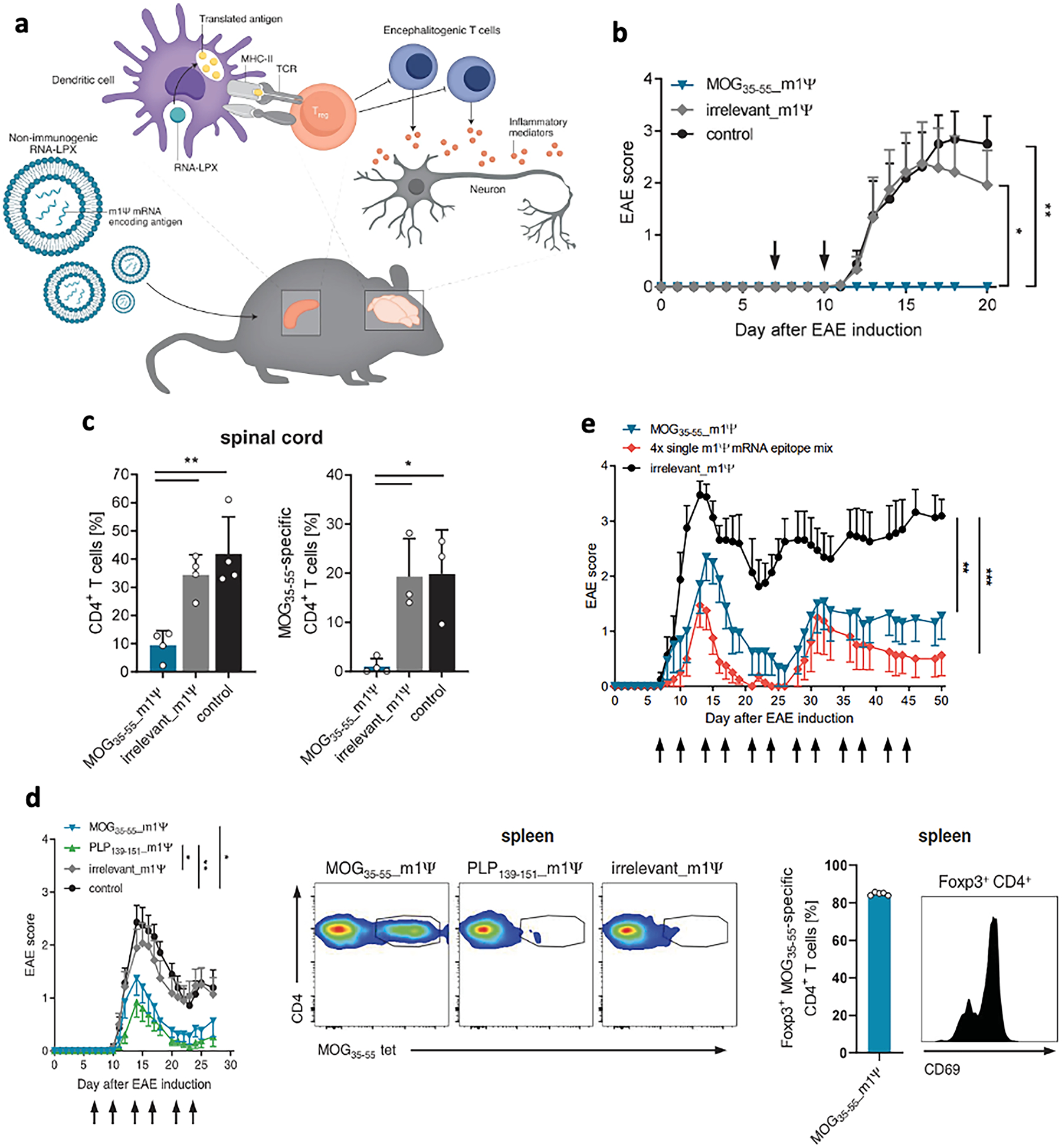
A, Schematic illustration of anti-inflammatory lipid nanoparticles containing myelin oligodendrocyte glycoprotein (MOG) m1Ψ mRNA. b, Disease severity in MOG35–55–induced EAE (n = 6 to 8 C57BL/6mice per group) treated with m1YmRNA or saline (control) on days 7 and 10 after disease induction. c, Frequency of CD4+ T cell and MOG35–55–specific CD4+ T cells in the spinal cord of mice as treated in b.Thy1.1+ 2D2 CD4+ T cells were transferred 1 day before EAE induction into Thy1.2+ recipient mice and analyzed on day 16 after disease induction. d, Treatment with m1Ψ mRNA leads to therapeutically effective bystander tolerance. PLP139–151–induced EAE mice (C57Bl/6xSJL mice) were treated with MOG35–55 m1Ψ,PLP139–151 m1Ψ, irrelevant m1Ψ mRNA, or saline (control) twice per week starting on days 7 and 10 after disease induction with 40 mg of m1Ψ mRNA. Expansion of endogenous MOG35–55–specific CD4+ T cells and frequency of tetramer+ Foxp3+ Treg cells in spleen on day 28 were shown. e, Induction and maintenance of antigen-encoding m1Ψ mRNA-induced tolerance is independent of the EAE disease model and mediated by bystander immunosuppression. EAE model based on autoreactivity concurrently against MOG35–55, PLP139–151, PLP178–191, MBP84–104 and MOBP15–36 induced in F1 C57BL/6 × SJL mice. Disease was scored after treatment with either MOG35–55 m1Ψ mRNA alone, with a mixture of m1Ψ mRNA encoding MOG35–55, PLP139–151, PLP178–191 and MBP84–104 autoantigen epitopes or irrelevant m1Ψ mRNA. Panel a and b-g are reproduced with permission from [196] and [52], respectively.
To address the concern that antigen delivery alone to an inflammatory microenvironment found in MS could adversely trigger an immune response that augments the disease, antigens have also been co-delivered with immunomodulators for antigen presentation in a tolerogenic microenvironment in animal models of MS. Rapamycin is one such immunomodulator, more specifically, an inhibitor of the mechanistic target of rapamycin (mTOR). Rapamycin has been shown to affect the immune system through various pathways, including induction of tolerogenic DCs and Tregs, T cell anergy or inhibition of proliferation, and CD8+ T cell differentiation [53–56]. Maldonado and colleagues developed tolerogenic NPs (tNPs) composed of PLGA, pegylated polylactic acid (PLA-PEG), rapamycin, and protein or peptide antigens that modify adaptive immune responses and produce a lasting tolerance able to protect against multiple immunogenic challenges with antigen [57]. They first showed that OVA323–339-loaded tNPs were able to target and block humoral immunity in mice immunized with OVA and CpG by inhibiting anti-OVA antibody responses upon challenge with OVA. tNPs also prevented antibody hypersensitivity responses previously seen in mice that received multiple intravenous injections of an immunogenic particulate form of OVA (pOVA). The encapsulation of rapamycin was crucial in these studies as free rapamycin did not affect the anti-OVA response. Mechanistically, tNPs were shown to inhibit the proliferation of Ag-specific effector CD4+ T cells while expanding the proportion and number of Ag-specific Foxp3+ CD4+ T cells [58]. Treatment of PLP139–151-loaded tNPs at the peak of disease led to complete remission and inhibited disease relapse in a R-EAE model. Furthermore, mice treated with tNPs on days −14 and −21 prior to adoptive transfer of encephalitogenic T cells from PLP139–151/CFA immunized mice were completely protected from disease, showing a durable endogenous regulatory response.
The aryl hydrocarbon receptor (AhR) is a transcription factor reported to promote Treg differentiation by augmenting TGF-β signaling or inducing anti-inflammatory DCs that secrete retinoic acid [59,60]. Mitigation of disease in preclinical MS and T1D studies have demonstrated the therapeutic potential of AhR-targeting ligands [60,61]. For induction of antigen-specific tolerance, Kenison et al., evaluated nano-liposomes co-loaded with an AhR agonist 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) and MOG35–55 peptide (NLPITE+MOG) in mouse models of EAE [62]. A single IV injection of NLPITE+MOG 15 d after disease induction effectively suppressed disease development. Notably, in C57Bl/6 × SJL F1 mice immunized with PLP139–151 peptide, subcutaneous treatment with either NLPITE+PLP or NLPITE+MOG ameliorated EAE, reduced proliferative recall response to PLP139–151 and increased IL-10+ CD4+ T cells in CNS, suggesting NLPs induce bystander suppression against pathogenic immune responses towards other autoantigens expressed in the same tissue. The authors also showed that NLPITE+MOG suppressed chronic progressive EAE in NOD mice immunized with MOG35–55, which was associated with increased MOG-specific IL-10+ CD4 T cells and downregulated proinflammatory activities in astrocytes, microglia, and monocytes in the CNS.
Several groups have investigated the co-loading of autoantigen and T cell-modulating agents. Wang et al. designed PLGA NPs encapsulating inhibitory peptide LABL, which binds with ICAM-1 to block signal-2 in T cell activation and decorated the surface with antigenic peptide MOG35–55 for signal-1 [63]. Inhibition of symptoms in EAE mice, reduced expression of MHC-II and CD86 on DCs, and diminished levels of infiltrating Th1 and Th17 cells in the CNS indicated successful antigen-specific immune tolerance via abortive T-cell activation. Casey and colleagues conjugated tolerogenic cytokine transforming growth factor beta 1 (TGF-β) onto the surface of Ag-loaded PLGA NPs, which enhanced Ag-specific tolerance with lower doses and enabled subcutaneous injection, which is more clinically accessible than intravenous injection [64]. Cappellano and colleagues encapsulated PLGA NPs with either MOG35–55 (MOG-PLGA) or rhIL-10 (IL10-PLGA), which showed sustained in vitro release for several weeks with similar kinetics [65]. Subcutaneous vaccination with both MOG-PLGA and IL10-PLGA ameliorated the course of EAE in MOG35–55-immunized C57Bl/6 mice. While the approach deviates pathogenic T cell activation, it remains to be elucidated whether the approach generates antigen-specific Treg cells in the inflamed CNS.
Multiple particle systems have also been examined. For instance, Keselowsky’s group investigated the efficacy of dual-sized PLGA-based MP MPs (dMPs) for delivery of MOG35–55 in addition to TGF-β1, GM-CSF, and vitamin D3. The rationale behind the design was that non-phagocytosable MPs (~30 μm) lead to the extracellular release of DC chemokines (GM-CSF) and protolerogenic cytokines (TGF-β1) in subcutaneous tissue whereas phagocytosable MPs (~1 μm) are engulfed by locally recruited APCs for antigen presentation and for vitamin D3 delivery to its nuclear receptor. Early treatment with dMPs with MOG35–55 prevented disease progression, which was associated with decreased Th1/Th17 cells and MHCII+ CD86+ DCs [66]. The same group also systematically evaluated the design principles of dMP system in treating EAE and found combined FMS-like tyrosine kinase 3 ligand (FLT3L, for DC differentiation) and stromal cell derived factor 1 (SDF-1, for immune cell recruitment) is equivalent to GM-CSF to achieve maximal efficacy, highlighting the multifaceted role of GM-CSF in both DC recruitment and differentiation [67]. Interestingly, the group also showed that reversing the sizes of phagocytosable and non-phagocytosable dMPs abrogated treatment effects.
Reactive oxygen species (ROS) have been implicated as mediators of inflammatory cytokine production and inflammation-induced tissue injury and has been explored as a therapeutic target for many inflammatory diseases. However, its modulation for reestablishing antigen-specific immune tolerance is still emerging [68]. Nguyen et al. prepared an immunosuppressive therapeutic vaccine with mesoporous silica nanoparticles (MSN) loaded with MOG35–55 and decorated on the surface with ROS-scavenging cerium oxide nanoparticles (CeNP) [69]. Upon intravenous injection with MSN-MOG (without CeNP), EAE mice had significantly increased the frequency and number of Tregs, diminished levels of CNS-infiltrating APCs and autoreactive CD4+ T cells, and suppressed the disease in EAE mice. However, after Treg depletion, the vaccinated mice lost protection against EAE, shown by a rebound in clinical scores as well as levels of CNS-infiltrating APCs and CD4+ T cells. CeNPs, which help reduce intracellular ROS of APCs and suppress maturation, were able to enhance the therapeutic efficacy of MSN-MOG and treat mice in the chronic phase of EAE.
3. Type 1 diabetes mellitus (T1D)
T1D is an autoimmune disorder characterized by immune-mediated destruction of insulin-producing β cells in the pancreas [70]. The resultant dysregulation of insulin secretion and blood glucose control facilitates complications, including diabetic retinopathy, and kidney and cardiovascular diseases [71]. The current treatment involves lifelong exogenous insulin supplementation and delays onset of diabetes-associated complications but does not address the underlying autoimmune pathophysiology. In November 2022, a century after the first use of insulin, the US FDA approved teplizumab as the first disease-modifying therapy in T1D, highlighting the potential of immunotherapy in treating T1D. An alternative to broad T cell modulation is antigen-specific immunotherapy. Multiple T1D-specific autoantigen candidates have been tested in clinical trials, yet limited efficacy has been reported [72–74]. Applying the wealth of knowledge gained from neo-epitope discovery (i.e., hybrid insulin peptide), biomaterials and micro/nanoparticle delivery, various research groups have sought to improve antigen delivery to induce antigen-specific immune tolerance [75–77].
In order to provide proof-of-concept of new therapies and to optimize delivery platform, studies typically resort to transferred T1D models initially where β-cell specific, diabetogenic CD4+ or CD8+ T cells from TCR-transgenic mice are transferred to immunodeficient hosts (i.e., the NOD–severe combined immunodeficiency (NOD–SCID) strain) [78]. These mono-/oligo-clonal T cell-transferred T1D models provide a useful tool to validate new therapeutic approaches that modify disease progression in an antigen-specific manner. Moreover, the delivery platform may be tailored for individual antigenic epitopes, given different requirements for CD4+ and CD8+ T cell modulation [79]. For example, Prasad and colleagues formulated PLGA NPs that encapsulated either a CD4 (p31) or a CD8 (NRPA7) peptide epitope (Ag-PLGA) [80]. Intravenous delivery of Ag-PLGA blunted diabetogenic activities of the p31-recognizing BDC2.5 CD4+ and NRPA7-recognizing NY8.3 CD8+ T cells in NOD.SCID recipients, respectively. The therapeutic efficacy was mediated by antigen-specific Tregs and depended on PD-1 and CTLA-4, as antibody blockade of either molecule during induction or maintenance phase abrogated the protective effects of p31-PLGA. Jamison et al. encapsulated a hybrid insulin peptide (2.5HIP), consisting of an insulin C-peptide fragment fused to a peptide from chromogranin A (ChgA), into PLGA NPs [77]. Intravenous injection of the 2.5HIP-PLGA prevented disease progression in about 80 % of NOD.SCID mice transferred with BDC2.5 T cells and significantly reduced islet infiltration. 2.5HIP-PLGA treatment resulted in a higher frequency of Ag-specific Treg in the pancreas that downregulated CD127 and upregulated CD103 at a late time point. Yet, we should note that these mono-/oligo-clonal T cell-transferred T1D models do not faithfully reflect the complexity in the pathogenesis of human T1D, such as epitope spreading that expands the pathogenic T cell repertoire [81].
On the other hand, the NOD mouse model, which is poly-antigenic and exhibits spontaneous autoimmune-mediated diabetes, closely recapitulates human T1D [82]. For achieving a broader coverage of autoantigens or islet-reactive T cell clones, delivering disease initiating or perpetuating peptide or delivering multiple antigens appear to be an attractive strategy. For example, Liu et al. reported subcutaneous delivery of insulin B chain 9–23 peptide (insB9–23) autoantigen loaded PLGA MPs suppressed islet inflammation and diabetes incidence in NOD mice, which were associated with decreased splenic bulk CD4+ T cells and increased Tregs [83]. Interestingly, the authors also reported an increase in the size of the thymic CD4+ T cell pool. Considering the low frequency of insB9–23-specific T cells, it remains to be elucidated whether these MPs can have a direct impact on negative selection in central tolerance. Qu et al. loaded insB9–23 peptide into mannosylated sodium alginate NPs via Fe2+-Fe3+ coprecipitation (MAN-ALG(PEP)) [84]. The resultant NPs (~300 nm) led to 60 % of T1D protection when subcutaneously administered into 4-week-old NOD mice. The author also noted a similar increase in total CD4+ and total CD8+ T cells as well as Tregs after MAN-ALG(PEP) treatment compared to control. Notably, the Miller group has developed PLGA-based NPs (COUR CNPs) that encapsulate multiple diabetogenic proteins (recombinant insulin, GAD65, and ChgA) [85]. The authors noted targeting individual peptide epitopes (p31, p31-NRPA7-insulinB9:23, ProinsC19-A3, or intact insulin) failed to inhibit T1D development in NOD mice, whereas CNPs with all three diabetogenic proteins significantly inhibited T1D progression. In line with these in vivo results, an ex vivo recall culture experiment with various peptides revealed that CNPs with all three components induced the broadest tolerogenic phenotype in the self-reactive T cells, increasing IL-10 and decreasing IFN-γ secretion.
In addition to the PLGA-based platform, other biopolymers have been used for the delivery of autoantigens. For example, Firdessa-Fite et al. prepared soluble antigen arrays (SAgAs) using either stable click chemistry (cSAgA) or hydrolysable linker (hSAgA) to attach multiple peptide copies on hyaluronan backbone [86]. SAgAs were loaded with the hybrid peptide 2.5HIP, a hybrid insulin peptide and natural ligand of BDC2.5 CD4 T cells, or with its more potent p79 mimotope [76]. SAgAs loaded with both epitopes delayed diabetes in 8-week NOD mice upon subcutaneous injection whereas SAgAs loaded with individual epitope did not. Interestingly, SAgAs delivering 2.5HIP expanded Foxp3+ Treg whereas SAgAs delivering its more potent p79 ligand induced IL-10+ Tr1. In addition, tetramer staining revealed few cross-reactive T cells recognizing both epitopes. These results suggest 2.5HIP-reactive and p79-reactive T cells were distinct populations and SAgAs treatment triggered divergent differentiation programs in them. The cSAgAs formulation afforded stronger induction of CD73/FR4, PD-1, and IL-10 and better protection against diabetes compared to hSAgAs. In an elegant strategy, Wilson et al., attached β-linked polymeric N-acetylgalactosamine or N-acetylglucosamine to antigens for targeting multiple tolerogenic cells upon intravenous injection, including hepatic DCs, KCs, LSECs, and hepatocytes via C-type lectin receptors [87]. The p(GalNAc) and p(GluNAc) polymers were attached to antigens via a reduction-sensitive self-immolating linker, which allowed for liberation of the antigen for antigen presentation upon endocytosis. In a BDC2.5 transferred T1D model, p31-p(GluNAc) treatment protected mice from diabetes and increased Foxp3+ Treg in the spleen compared to free p31 peptide. This antigen delivery platform was applied to other autoimmune diseases, as the same group has shown the efficacy of pGalNAc-antigen in encephalitogenic T cell transferred EAE and relapsing-remitting EAE [88]. Another example of novel polymer being used for the delivery of antigens is acetalated dextran (Ac-Dex) [89–91]. Ac-Dex is prepared by modifying the hydroxyl groups with pendant acetals, which are sensitive to acidic conditions [92]. Therefore, Ac-Dex MPs degrade in low pH or enzymatic conditions in the lysosomes of APCs and release antigenic proteins or peptides. Chen et al. co-loaded p31 peptide and rapamycin into Ac-Dex MPs and demonstrated its efficacy in a BDC2.5 transferred T1D model [90]. Mechanistically, Ac-Dex MPs delayed effector CD4 T cells expansion in periphery and increased Foxp3+ Treg: IFN-g+ Teff ratio in pancreatic lymph nodes. Notably, the same group also showed utility of the particle formulations in encapsulating both rapamycin and PLP or dexamethasone and MOG for the treatment of EAE [89,91].
As an alternative to delivering antigens to APCs, direct modulation of autoreactive T cells can be achieved using peptide-bound MHC class I- or class II-decorated NPs (termed pMHCI-NPs or pMHCII-NPs) [93,94]. The NP matrices were composed of crosslinked dextran or pegylated iron oxide (CLIO– or PFM–NPs, respectively) and surface-decorated with pMHCII molecules. For T1D models, IAg7-NPs containing 2.5mi, IGRP4–22 or IGRP128–145 and Kd-NPs containing IGRP206–214 or NRP-V7 were used [93,94]. pMHCII-NPs exhibited an extremely short half-life, cleared by phagocytes within 1–5 min depending on the organs, or captured by cognate T cells within 10 min upon injection [95]. However, the pMHC-NPs also showed extremely fast pharmacokinetics, leading to assembly of antigen receptor microclusters, sustained T cell signaling and reprogramming [95,96]. Systemic treatment triggered expansion of cognate TR1 cells from antigen-experienced, pathogenic follicular T helper cells (Tfh), and reversed disease in all three models without compromising systemic immunity [93,97,98]. The authors highlighted the potential of this approach for inducing immune tolerance in multiple autoimmune diseases.
Co-delivery of tolerogenic agents with autoantigen is another appealing approach to re-establish immune tolerance and achieve maximal efficacy. For example, Phillips et al. incorporated human insulin B9–23 peptide, all-trans retinoic acid (RA) and transforming growth factor beta 1 (TGF-β1) into PLGA NPs and showed a single subcutaneous injection of NPs prevented disease onset in 11-week-old NOD mice in 65 % of animals [99]. Interestingly, the authors did not detect an upregulation of Treg but instead observed an increase in regulatory B cells in mesenteric lymph nodes, suggesting a possible involvement of regulatory B cells in tolerance induction. Alternatively, Yeste and colleagues designed PEG-coated gold NPs that co-delivered proinsulin and an Ahr ligand 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) [100]. The resultant NPs induced a tolerogenic phenotype in mouse and human DCs by inducing AhR-dependent Socs2 expression and prevented diabetes in more than 70 % of treated 8-week-old NOD mice. Nucleic acid delivery and genome editing can also be useful tools to precisely manipulate APCs, compared to small molecules [101]. For example, a CRISPR-Cas9 plasmid and three guide RNAs targeting costimulatory molecules (CD40, CD80, and CD86) were encapsulated along with 2.5mi, a T1D-related peptide, into cationic lipid-containing PEGPLGA NPs [102]. The resultant NPs genetically edited DCs into tolerogenic phenotype and expanded antigen-specific Tregs and prevented diabetes incidence in NOD mice when administered at a late prediabetic stage.
Modular design strategies have been developed to further incorporate chemotactic agents together with pro-tolerogenic and antigen components. Keselowsky’s group evaluated the effectiveness of dual-sized PLGA-based MP (dMP), which was composed of two ~ 1 μm phagocytosable MPs (loaded with vitamin V3 and insulin peptide) and two ~30 μm nonphagocytosable MPs (loaded with TGFβ1 and GM-CSF) [103,104]. In vitro studies confirmed the dMPs reduced maturation markers on DCs [103,104]. The original formulation, which contained insB9–23 peptide, protected 40 % of the 4-week-old NOD mice from diabetes up to 32 weeks of age after subcutaneous injection [103]. More recently, the same group re-engineered the dMPs to encapsulate denatured insulin in place of insB9–23 peptide with the goal of expanding the presented peptide library [104]. The re-engineered dMPs treatment protected against disease onset in 60 % of 8-week-old NOD mice, and the reversal of diabetes was observed in 36 % of the mice by 8 weeks. The efficacy was associated with increased tolerogenic DCs in the injection site draining lymph nodes and increased frequency of Treg in the pancreatic LN and spleen. In another example, Chen et al. designed a modular immune-homeostatic microparticle (IHM) that rapidly released monocyte chemotactic protein-1 (MCP-1) for T cell migration (tmax = 5 h) and induced T cell apoptosis via surface immobilized Fas ligand (FasL) (Fig. 3) [105]. The authors further loaded diabetes-related glutamic acid decarboxylase 524–543 (GAD524–543). Intravenous injection of GAD peptide loaded IHM (IHMGAD) increased Treg frequency in pancreatic lymph nodes, restored islet numbers and reversed hyper-glycemia in NOD mice for up to 12 weeks. It remains to be elucidated how the MP coordinated activated T cell apoptosis and Treg formation and its effects on normal immunity.
Fig. 3.
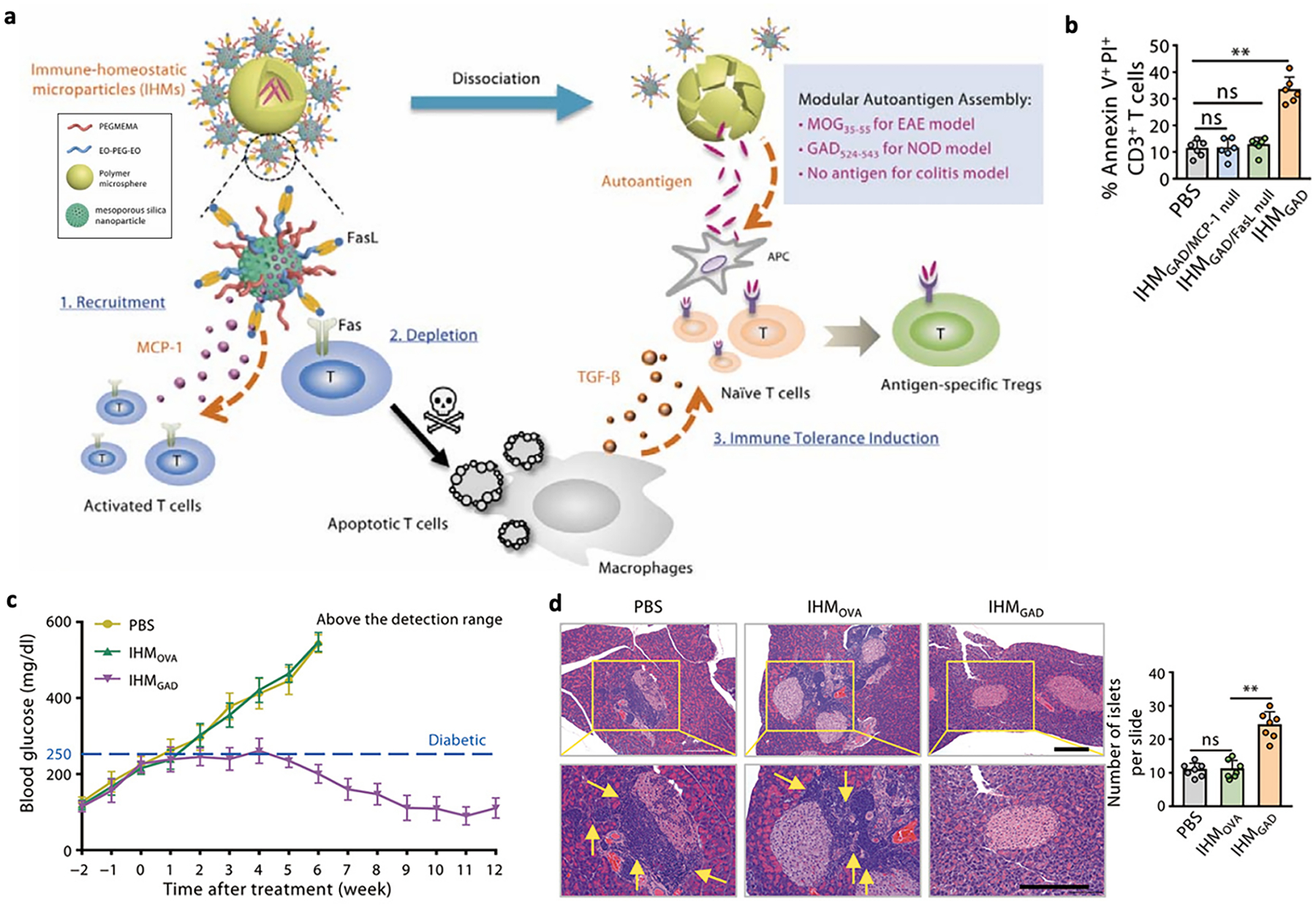
A, Schematic illustration of IHMs induced antigen-specific immune tolerance via an apoptosis and reestablishment strategy. b, Frequencies of apoptotic CD3+ T cells in the blood 6 h after injection of PBS or IHMs in NOD mice. c, Blood glucose was measured to monitor diabetes progression in the NOD mice treated with PBS, IHMOVA and IHMGAD. d, Insulitis as measured by H&E staining. Three sections were taken from each tissue for H&E staining, and the average number of islets on each slide was calculated.
Reproduced with permission from [105]
The route of administration plays an important role in immune tolerance induction [106]. From a standpoint of immune modulation, tolerogenic tissue environment can be harnessed or remodeled by targeting therapeutics to specific areas. For example, attempts have been made to modify the antigen presentation process directly in lymph nodes. Gammon et al. reported an intra-lymph node injected micro-particle that co-encapsulated either p31 or NRPV7 with rapamycin into PLGA MP [107]. The MP was designed as a diffusion limited depot because it was sufficiently large not to drain out of lymph nodes. The authors showed both the antigen and rapamycin are needed for maximal efficacy, and p31/rapamycin MP expanded significantly higher ratios of antigen-specific Treg in the treated lymph nodes and early draining lymph nodes, compared with p31 MP or rapamycin MP. Interestingly, the authors showed that MP treatment promoted tolerogenic micro-domains in LN stroma, which were characterized as an increased ratio of laminin α4 to laminin α5 in areas surrounding high endothelial venules and cortical ridge. Tolerogenic stromal architecture may also be modeled and applied in vivo using hydrogel network. Tegou et al. tethered secondary lymphoid chemokine CCL21 and p31 peptide to PEG hydrogel via click-chemistry and further incorporated IGRP206–214 via PEG-poly(propylene sulfide) (PEG-PPS) nanovesicles [108]. The loading of IGRP peptide into nanovesicles preserved its bioactivity compared to tethering it onto the gel. Interestingly, implantation of p31 and CCL21 combination gel under the kidney capsule of NOD mice promoted formation of LN-like stromal networks in the proximity containing gp38+ fibroblastic reticular cells. Tolerance may also be induced by skin resident APCs. Arikat et al. fabricated steel microneedle arrays consisting of thirty 500 μm projections with > 30 μg of proinsulin coating, with the aim of targeting Langerhans cells residing in the epidermis and dermal dendritic cells [109]. Upon intradermal injection, the proinsulin-coated microneedles rapidly release proinsulin (77.5 % released in 1 min) and efficiently engaged adoptively transferred G9 CD8+ T cells, which recognize the InsB15–23 fragment of proinsulin. It remains to be seen whether the technology can desirably deliver immunological signals that induce tolerance in sites of inflammation.
Oral administration may present an alternative route of immunotherapy for T1D, as oral mucosa is populated with tolerogenic DCs that can promote Treg differentiation [110]. Chen et al encapsulated the antigen heat shock protein 65–6Xp277 (H6P) into RGD- and mannose-modified chitosan NPs [111]. Oral vaccination of NOD mice increased uptake of H6P by DCs in the Peyer’s patches, compared with the solution form and prevented diabetes in NOD mice when treated at 4 weeks of age. More recently, Mao and colleagues designed oral vaccines using lactic acid bacteria bacterium-like particles that deliver either a single-chain insulin analog (insulin B- and A-chain with a linker peptide), an intracellular domain of insulinoma-associated protein 2, or both [112–114]. The authors showed the antigen-carrying bacterium-like particles induced Th2 and Treg differentiation and led to reduced disease incidence and islet protection in NOD mice.
4. Rheumatoid arthritis (RA)
Rheumatoid arthritis is an autoimmune disease driven by Th1 or Th17 pro-inflammatory T cell responses and mediated by various antibodies against autoantigens, such as rheumatoid factors, citrullinated proteins, and heat shock proteins (HSPs) [115]. Pathogenesis is unclear, but it likely starts upon a divergence in thymic selection or peripheral tolerance, and with infiltration of inflammatory cells into affected tissues, including the synovial membrane, cartilage, and bones, it leads to the destruction of joints and bones [115–117]. In RA patients, anti-citrullinated protein antibodies that recognize post-translationally modified citrullinated host proteins are often produced, followed by immune reactions and infiltration of immune cells into the joints [118]. Additionally, patients have high incidence rates of antibodies and T cells specific for type II collagen (CII), a protein solely expressed in the articular cartilage of joints, designating CII as one of the major autoantigens of human RA [119]. Inducing anti-CII immunity in animals leads to a collagen-induced arthritis (CIA) model that has a pathogenesis and symptoms that resemble human RA, including inflammation and bone erosion in the arthritic joints. Thus, this CIA model has been widely used for the evaluation of RA therapeutics.
While current treatments include disease-modifying antirheumatic drugs, like nonsteroidal anti-inflammatory drugs (NSAIDs) and methotrexate, which can mitigate symptoms, these drugs cannot completely abolish symptoms and they may come with severe side effects, including infection or gastrointestinal problems. The goal to reestablish immunologic tolerance in patients remains unmet, and exploration of antigen-specific immunotherapies that can immunologically reset the body to a healthy state is underway [120]. One approach to induce antigen-specific immune tolerance is to deliver antigens to APCs with immune-modifying agents. Chen et al. prepared a tolerogenic polypeptide vaccine (tPvax) by loading a multiepitope citrullinated peptide (Cit-ME) and rapamycin onto lipid-coated calcium phosphate NPs with core–shell structures [121]. Upon administration of tPvax to Wistar rats induced with CIA, CII-specific antibody titers and IFN-y levels were significantly reduced, Treg cell and IL-10 levels were significantly increased, and paw thicknesses and clinical scores were notably decreased, showing clear therapeutic effects. Loading rapamycin provided a tolerogenic environment for DCs and promoted downregulation of costimulatory molecules and inflammatory cytokines, while Cit-ME, which consists of major epitopes of citrullinated type II collagen, fibrinogen, vimentin, and fibronectin, targeted the epitope spreading tendency of experimental arthritis to citrullinated antigens. In another example, rapamycin and CitP, a citrullinated multiepitope autoantigen derived from citrullinated filaggrin, β-fibrinogen, vimentin, and collagen type II, were loaded onto nanoemulsions (NEs) [122]. The NEs were targeted to ectopic lymphoid structures, which consist of T cells, B cells, and follicular dendritic cells that are formed with the help of APCs and are often present in the inflamed synovium of RA, to promote direct interaction with the immune cells. Administration of the NEs resulted in protection of CIA and adjuvant-induced arthritis (AIA) mice from chronic and acute arthritis, respectively, as shown by significantly reduced paw thicknesses and clinical scores. Additionally, proinflammatory cytokine levels (i.e., tumor necrosis factor alpha (TNF-α), IL-17A, and IL-1β in CIA, and IFN-γ, TNF-α, IL-1β, and IL-6 in AIA) were significantly reduced, while anti-inflammatory cytokine levels (e.g., TGF-β1 in CIA and IL-10 in AIA) were notably increased. Thus, targeting ectopic lymphoid structures with NEs carrying rapamycin and CitP significantly alleviated RA symptoms, demonstrating the potential of their delivery system co-deliverubg multiepitope self-antigen and rapamycin. As an alternative strategy, Srivastava and colleagues coadministered rapamycin-loaded PLGA NPs (PLGA-R) with Siglec-engaging tolerance-inducing antigenic liposomes, which are liposomal NPs decorated with a target antigen and ligands of the B cell Siglec, CD22 [123]. These NPs were previously engineered to induce antigen-specific B cell tolerance, and co-engagement of CD22 and the B cell receptor would send an apoptotic signal promoting antigen-specific B cell deletion and tolerance [124–126]. Upon coadministration, naïve mice exhibited strong tolerance to multiple antigen challenges, and the onset of disease in K/BxN mice, which develop spontaneous arthritis to glucose-6-phosphate-isomerase (GPI), was delayed, with some mice being disease free [123]. Later on, the two NPs were combined to form a hybrid delivery system with a rapamycin-loaded PLGA core and a lipid monolayer with protein antigen and CD22L on the surface (Fig. 4) [127]. Hybrid NPs loaded with GPI as the target antigen significantly delayed the disease onset and prevented arthritis in K/BxN mice. Successful induction of tolerance to self-antigen was shown by decreased levels of anti-GPI IgG1, increased levels of Treg cells, and diminished levels of antibody secreting plasma cells and GPI-specific plasma cells. Overall, codelivery of rapamycin and autoantigens induced immune tolerance and ameliorated symptoms in RA animal models in addition to the EAE and T1D models mentioned previously, further showing the therapeutic potential and universality of combining immunomodulatory signals and autoantigens for antigen-specific immunotherapy.
Fig. 4.
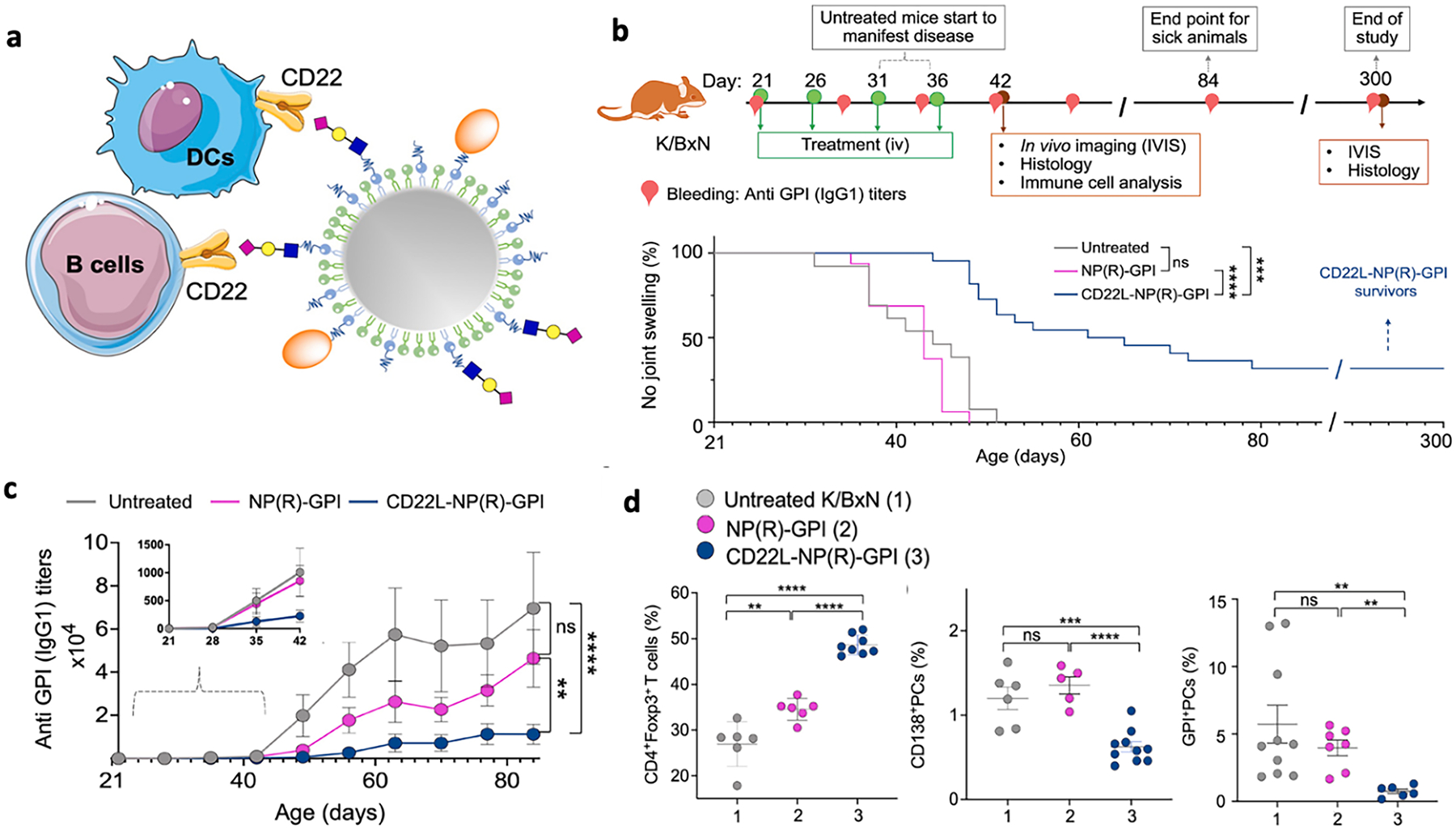
A, Schematic illustration of IHMs induced antigen-specific immune tolerance via an apoptosis and reestablishment strategy. b, Frequencies of apoptotic CD3+ T cells in the blood 6 h after injection of PBS or IHMs in NOD mice. c, Blood glucose was measured to monitor diabetes progression in the NOD mice treated with PBS, IHMOVA and IHMGAD. d, Insulitis as measured by H&E staining. Three sections were taken from each tissue for H&E staining, and the average number of islets on each slide was calculated. Reprinted (adapted) with permission from Brzezicka et al. [127] Copyright 2022 American Chemical Society.
Moreover, antigen-presenting DCs can be modulated by targeted delivery of inhibitors. Galea et al., synthesized liposomes encapsulating an antigen and a NF-κB inhibitor, calcitriol, to exploit DC antigen presentation and target antigen-specific memory T cells [128]. The liposomes reduced DC MHC class II expression, suppressed the differentiation and function of Teff cells, induced antigen-specific Treg cells in a programmed cell death ligand 1 (PD-L1)-dependent manner, and expanded programmed cell death 1+ (PD-1+) antigen-specific T cells in vivo [128,129]. Therapeutic efficacy in a RA mouse model was shown by a significant reduction in disease severity [128]. Recently, the same group loaded liposomes with calcitriol and RA joint-specific self-peptide collagen II259–273 and treated anti-citrullinated protein antibody-positive RA patients on methotrexate [130]. Dose-associated immunoregulatory effects and remission of disease were observed in this phase I trial. Specifically, antigen-specific and bystander immune responses were displayed by increased levels of type II collagen-specific and bystander Cit64vimentin59–71-specific T cells, decreased levels of memory B cells and inflammatory myeloid populations, reduced citrullinated protein antibody V domain glycosylation, and upregulation of T cell transcripts associated with tolerogenic TCR signaling and exhaustion. Mangal et al. metabolically reprogrammed pro-inflammatory DCs in the presence of self-antigens with MPs loaded with PFK15 (a glycolytic inhibitor), alpha-ketoglutarate (aKG, an immunosuppressive Krebs cycle metabolite), and bovine collagen type II (bc2, a CIA-specific antigen) [131]. These MPs promoted antigen presentation along with inhibition of DC glycolysis, which is necessary for pro-inflammatory antigen-specific immune responses. Treated CIA mice had significantly reduced paw thickness and arthritis scores along with lower levels of activated DCs in draining lymph nodes and proliferation of bc2-specific Tregs in joint-associated popliteal lymph nodes. These studies have shown that inhibiting the development of pro-inflammatory DCs generated anti-inflammatory T cell responses in CIA mice.
A different immunization strategy utilizing natural, self-assembling nanostructures was demonstrated by Zampieri and colleagues, who engineered immunomodulatory self-assembling plant virus nanoparticles (pVNPs) comprised of tomato bushy stunt virus to function as both a peptide scaffold and an adjuvant [132]. Tomato bushy stunt virus was genetically engineered to express immunodominant peptides for RA. Upon administration, CIA mice had a significant reduction in the severity of disease symptoms, with increased serum levels of IL-10. The strategy was also applicable to the T1D model, as NPs prepared from cowpea mosaic virus expressing the immunodominant peptide p524 were capable of inducing partial protection from the onset of diabetes in NOD mice. The genetically encoded peptides can be modified for incorporation of new sequences associated with different autoimmune diseases although the characteristics and underlying mechanisms of the different plant virus systems may be more suitable for specific disease models and would need to be further considered in designing pVNPs for other therapeutic applications.
TNF-α is a proinflammatory cytokine that acts against the suppressive activity of Treg cells and plays a key role in RA pathogenesis, and anti-TNF-α antibodies, like infliximab, have been shown to induce Treg cell populations in RA patients [133]. However, extended anti-TNF-α therapies may lead to opportunistic infections and malignancies’. Buanec et al. sought to design a safer alternative and engineered human TNF-α kinoid (hTNF-K) to target TNF-α through the novel approach of inducing autoimmune protection against a human cytokine [134]. hTNF-K is a heterocomplex of keyhole limpet hemocyanin and immunogenic hTNF-α derivative, and immunization in hTNF-α transgenic mice mimicking RA led to a safe induction of anti-TNF-α Abs that neutralized hTNF-α bioactivities but did not result in significant T cell responses. Kinoid-immunized hTNF-α transgenic mice were protected in both an acute model of hTNF-α-galactosamine lethal shock and a chronic model of arthritis. The studies highlighted the potential of active immunization against an endogenous cytokine for therapeutic application in high-TNF-α producing diseases.
Notably, there are a number of promising RA-associated antigen peptides that are yet to be explored in the context of vaccine delivery systems. For instance, HSPs are highly expressed proteins at sites of inflammation, like inflamed synovium, and are presented by MHC class II molecules [135]. Immunization with mycobacterial HSP70 was shown to induce an anti-inflammatory HSP-specific T-cell response, and B29, a HSP70-derived peptide, induced antigen-specific Tregs expressing Lymphocyte Activation Gene-3 (LAG-3), a molecule that binds MHCII and can send inhibitory signals to APCs [136]. When LAG-3+ B29-induced Tregs were adoptively transferred into mice with proteoglycan-induced arthritis, an experimental RA model, it was found that the transferred Tregs migrated to sites of Inflammation, remained active for up to 50 days and suppressed the disease activity. In HLA–DQ8–transgenic mice, which can develop arthritis upon immunization with human proteoglycan, immunization with B29 and restimulation with proteoglycan in combination with B29 ex vivo, led to a suppressed response towards proteoglycan [137]. When naïve human T cells were primed and restimulated with B29, there was a significant increase in B29-specific CD4+ T cells, showing the clinical potential of this peptide. Another example of a therapeutic HSP-derived peptide is dnaJP1, a synthetic peptide that mimics a “shared epitope” common to HLA class II alleles associated with RA [138]. Immune tolerization to this proinflammatory T cell epitope was induced in patients, resulting in a phenotypic change in effector T cells, from a proinflammatory to a tolerogenic phenotype. Oral administration in RA patients reduced TNF-α-producing T cells, increased IL-10-secreting T cells, and led to clinical improvements as assessed by ACR20 and ACR50 responses. RA patients with increased levels of PD-1 (programmed death 1), CTLA-4, and their ligands had successful antigenic tolerization, suggesting that PD-1 and CTLA-4-expressing T cells may be regulating the epitope-specific effector T cells involved in pathogenic inflammatory responses. While other immune-tolerizing studies have utilized disease-stimulating antigens as the tolerogen, such as MOG in EAE models, HSP is not linked to any initial cause of autoimmune inflammation. Therefore, these studies also shed light upon targeting components that amplify rather than trigger the disease in antigen-specific immunotherapy although comparison of the proportion of responsive patients would need to be further assessed.
5. Inflammatory bowel disease (IBD)
Inflammatory bowel disease is characterized by the chronic inflammation of the gastrointestinal tract. It can be subdivided into two categories: Crohn’s disease (CD) and ulcerative colitis (UC). While they share many of the same symptoms, they have unique pathologies and clinical signs. The exact pathogenesis of IBD is unknown, but many factors are involved such as diet, genetics, dysregulated immune system, and the gut microbiome [139]. Research for treatments of IBD has been growing as the prevalence of the disease has increased by 85.1 % from 1990 to 2017, reaching 1.6 million Americans and 6.8 million cases globally [140,141]. Current treatments have focused on general immunosuppression, such as aminosalicylates, corticosteroids, and anti-inflammatory cytokine antibodies, such as infliximab (anti-TNF-α) or ustekinumab (anti-IL-12/23). More recently, engineered nanoparticle carriers that can deliver drugs locally to inflamed regions in IBD have gained interests [142,143].
Antibodies against inflammatory cytokines, such as anti-TNF-α, require frequent dosing and may trigger anti-drug antibodies. Thus, as an alternative approach, vaccination against inflammatory cytokines have been explored. One such vaccine was against IL-12 and IL-23 by immunizing against the shared p40 subunit using the peptide 38TPDAPGETV46 and was shown to result in high titers of IgG that were also highly specific [144,145]. This vaccine was tested in the 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced actuate and chronic colitis models. Treatment for the acute model increased the production of immunosuppressive cytokine IL-10 and suppressed inflammatory cytokines, such as IFN-γ, IL-17, TNF-α, p40, and IL-12. For the chronic model, the levels of p40, IL-23, IL-17, IL-10, TNF-α, and TGF-β1 decreased compared to controls, inhibiting intestinal inflammation and fibrosis, and improving animal body weight. The percentages of CD4+ IFN-γ+ Th1 and CD4+ IL-17+ Th17 cells decreased in the mesenteric lymph nodes, indicating that the vaccine was also effective at inhibiting the differentiation of Th1 and Th17 cells. The vaccine was capable of inducing antibodies that lasted for 4 months, and this phenomenon was reversible and adjustable based on the immunization frequency. Additionally, while the cytokine levels were reduced, they did not fall below the baseline, allowing cytokines to function normally. The research group also engineered a vaccine against the unshared subunit IL-23 p19 to neutralize IL-23 and not IL-12 and still achieved similar results [146], suggesting IL-23 can be exclusively targeted as it plays a larger role in IBD pathogenesis. Kinoid immunization for the induction of anti-TNF-α antibodies previously discussed in RA can also be applied to IBD and would likely yield similar results. Another example is the vaccine against IL-18 [147]. Guan and colleagues selected antigenic peptides from mouse IL-18 and formulated virus-like particle (VLP) vaccines for individual peptides based on hepatitis B core antigen. Immunization of Balb/c mice with the VLP encoding the sequence EMDPPENIDDIQS (84–96) elicited high levels of serum IL-18 specific IgG, and sera harvested from the mice partially inhibited IFN-γ secretion in splenocytes after in vitro stimulation. In addition, mice immunized with the VLP ameliorated both acute and chronic colitis after repeated TNBS challenges.
Another vaccine that has been studied for IBD is against the mucin MUC1, a glycoprotein that is expressed on the colonic epithelium and is responsible for defending against pathogenic bacteria while also hydrating and lubricating the epithelia [148]. MUC1 contains an extracellular domain composed of a variable number of tandem repeat (VNTR) region with 80–200 repeats of the 20 amino acid sequence HGVTSAPDTRPAPGSTAPPA which have O-glycosylation sites that are significantly glycosylated. However, in IBD and adenocarcinomas, MUC1 is overexpressed and hypoglycosylated, exposing the protein backbone [149]. Additionally, the chronic inflammation from IBD can also lead to colitis-associated colon cancer. Since mouse MUC1 only has 34 % homology to the human protein, the human MUC1 transgene needed to be expressed in mice, with similar low and high expression in healthy and unhealthy cells, respectively. Beatty et al. looked at two common C57BL/6 mouse models of IBD with the new human MUC1 expression: IL-10 knockout (IL-10−/−) and dextran sulfate sodium (DSS)-induced colitis [150,151]. They administered glycopeptide TnMUC1–100mer, a peptide to elicit immunity against the abnormal MUC1, along with adjuvant E6020 via the intranasal route and boosted twice after 2 weeks. The anti-MUC1 vaccine was shown to develop a MUC1-specific CD8+ T-cell response, production of anti-MUC1 IgG, reduce colonic inflammation, and prevent colitis-associated colon cancer in both mouse models without inducing autoimmune side effects. We envision that targeting tissue-specific antigens can be a promising strategy for IBD.
Although not using vaccines, some pilot studies have shown promising results using chimeric antigen receptor-Treg (CAR-Treg) or antigen-specific Treg for treating murine colitis. A pioneering study designed a CAR specific for 2,4,6-trinitrophenol (TNP) model antigen, which is commonly used for the induction of mouse colitis [152]. Using a colitis model induced by cyclophosphamide and 2,4,6-trinitrobenzene sulfonic acid (TNBS), the authors showed transfer of 1×105 TNP-CAR Tregs conferred protection from colitis compared with wild-type Tregs. CAR-Tregs directed against carcinoembryonic antigen (CEA), an over-expressed tissue antigen in both colitis and colorectal cancer, was later developed [153]. Infused CEA-specific CAR-Tregs accumulated in the inflamed colon and suppressed disease severity in T cell-transferred colitis compared with irrelevant CAR Tregs. There has recently been evidence that CAR-T cells can be boosted with vaccines against cancer. They achieved this by linking albumin-binding phospholipid polymers, which traffic to lymph nodes, to a CAR ligand, and these “amph-ligands” are taken up by resident APCs and serve as a booster for the CAR-T cells [154,155]. A similar booster approach may be applied towards the CAR-Treg therapy described. Open questions remain as to identifying candidate antigens for human IBD and effectively eliciting gut targeting, antigen-specific Tregs.
Antibodies against gut microbial antigens are common in IBD patients [156–160]. It is predominantly accepted that chronic intestinal inflammation in the gut is at least in part due to the breakdown of immune tolerance of commensal bacteria. For example, they may develop increased antibody titers against E. coli membrane porin C (OmpC), which is also associated with increased disease severity [161]. Interestingly, most UC patients (~70 %) and some CD patients (~25 %) develop peri-nuclear anti-neutrophil cytoplasmic antibodies, which is cross-reactive with OmpC [162–165]. This indicates that antigens from the gut microbiome can lead to autoreactivity and be involved in the pathogenesis of the disease. Glycoprotein-2 is a receptor on M cells involved in translocating FimH bacteria across the intestinal epithelium [166–168]. Additionally, glycoprotein-2 is a known antigen for pancreatic autoantibodies [169]. While anti-neutrophil cytoplasmic antibodies appear to be a unique characteristic in identifying UC, anti-glycoprotein-2 antibody levels are significantly higher in CD patients compared to UC, irritable bowel syndrome, and healthy controls [166,168]. Vermeulen et al. have also screened for autoantigens in IBD patients and identified FAM84A (family with sequence similarity 84, member A) to be found in 20 % of CD patients, 11 % of UC patients, and 0 % of the healthy controls [165]. Antibodies against FAM84A have also been identified in MS patients, indicating potential overlap in autoimmunity. While vaccines have not been developed for these autoantigens in the treatment of IBD, they may function as potential targets for certain manifestations of the disease.
There has been evidence that exposure to helminths can attenuate IBD and other autoimmune diseases [170]. Researchers have used molecules such as P28GSTs (schistosome enzymes with glutathione S-transferase activity) from multiple sources to create vaccines against schistosomiasis, a disease caused by the worms [171]. A P28GST vaccine from Schistosoma haematobium (Sh28GST) has been shown to promote Th2 cells and prevent intestinal inflammation in TNBS-induced colitis model as seen in Fig. 5a, and completed Phase 2a clinical trials [172,173]. They prepared multiple W/O/W formulations of PLGA-based MPs loaded with ShP28GST (P28GST-PLGA) and tested the optimized formulation that provided constant release over several weeks in vivo [174]. Their P28GST-PLGA formulation had lower Wallace scores (measure of colitis severity based on macroscopic lesions) compared to P28GST-free PLGA MPs with as low as one injection. Surprisingly, it required 3 injections of their formulation resulted in significantly elevated levels of anti-P28GST IgG antibodies but had no effect on the Wallace score, indicating that the vaccination was likely not reliant on an IgG response, shown in Fig. 5b [174]. They did not look at the IgE response, which may have played a role in this anti-inflammatory effect. This strategy presents a unique approach of vaccination for IBD by taking advantage of the immune response against helminths as opposed to any involved disease-causing antigens.
Fig. 5.
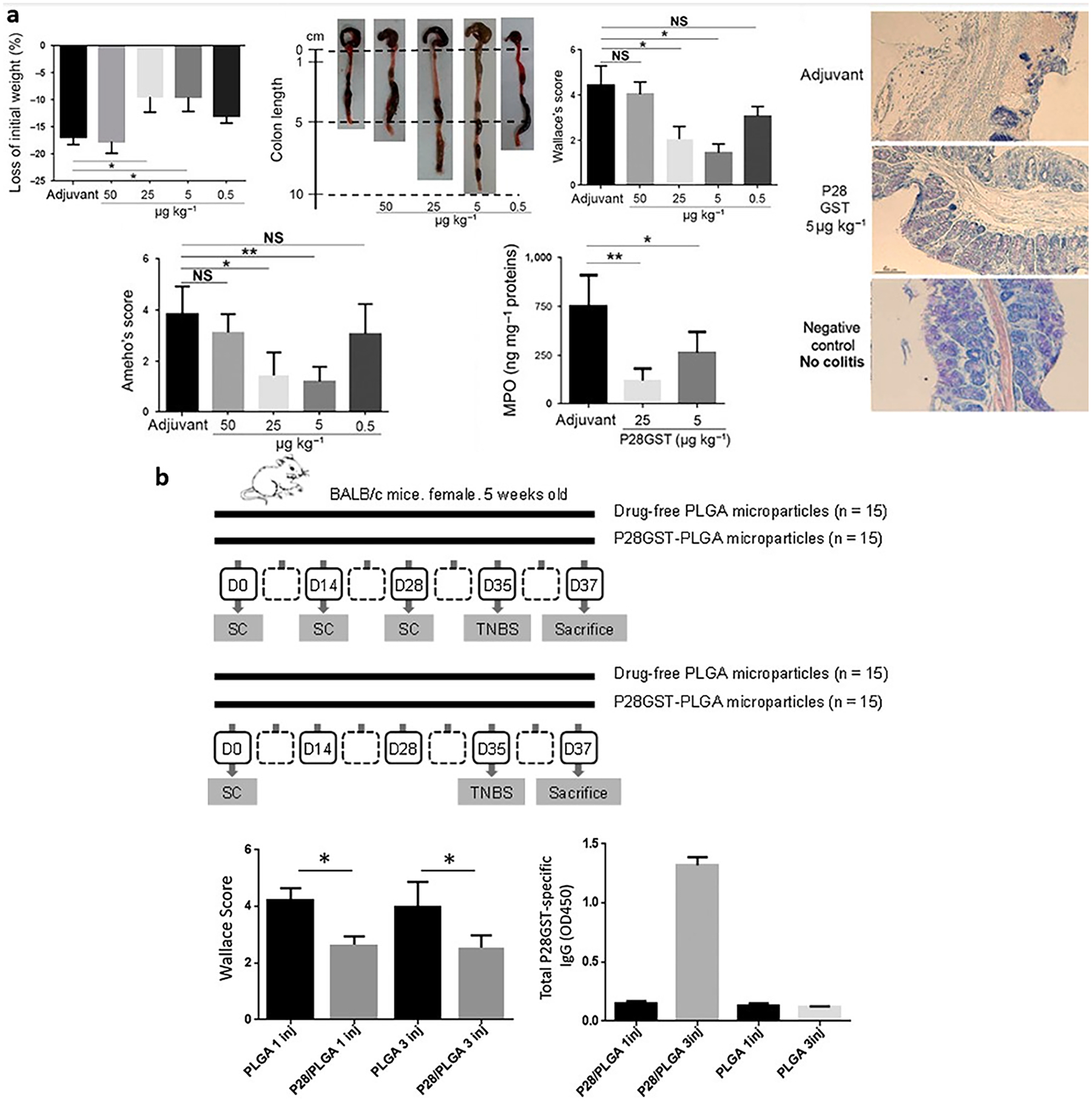
Immunization with a helminth protein can reduce inflammation through a Th2-type response. a, BALB/c mice with acute TNBS colitis were protected by P28GST immunization by decreasing weight loss, restoring colon length, decreased clinical/histological scores, and MPO. Immunizations of mice by three subcutaneous injections were administered 2 weeks apart, with the last injection occurring 1 week before colitis induction. b, Performance of P28GST-PLGA MPs in vivo, including Wallace Score and anti-P28GST IgG production. MPs were prepared with recombinant P28GST and lactose, with 11.9 % theoretic drug loading, 0.6/2/200 mL of W/O/W phases, and 1500/500 rpm primary/secondary emulsion stirring speeds. BALB/c mice were split into four treatment groups: 3 injections of drug-free PLGA MPs, 3 injections of P28GST-PLGA MPs, 1 injection of drug-free PLGA MPs, or 1 injection of P28GST-PLGA MPs, with TNBS induction 35 days after the first injection and sacrificing the mice on day 37. Panel a and b are reproduced with permission from [172] and [174], respectively.
Alternatively, Mycobacterium avium ss. Paratuberculosis (MAP) has been considered a somewhat controversial suspect for causing Crohn’s disease in humans [175]. This was linked due to a similar disease in cattle, Johne’s disease, which has similar symptoms, results in ~$250 million annually in losses due to the associated reduction of milk yield, and is estimated to affect about 90 % of the herds in the US [176]. Vaccines for Johne’s disease have been developed but with limited effectiveness. Additionally, MAP infection has been proposed to be an environmental risk factor for other autoimmune diseases like MS, RA, lupus, T1D, and Parkinson’s [175]. It has been challenging to study MAP due to the difficulty in diagnosing infection of the disease because like Mycobacterium tuberculosis (TB), it is typically a latent infection that evades an immune response. There has been some evidence of cross-reactivity between MAP and intestinal proteins, as one study found 30 % of a cohort of 50CD patients had significant double reactivity against human gastrointestinal glutathione perocidase (GPg)111–125 and MAP glycosyl transferase d (gsd)230–244 [177]. They determined that the antibodies had greater affinity towards the MAP mimic than human sequence, indicating that MAP likely initiated immunity towards the autoantigen. Therefore, although MAP is an infectious disease, it appears to be involved in tolerance breakdown for CD and could benefit from the development of a vaccine. Researchers have found that the deletion of relA in MAP prevented it from having a persistent infection in cattle and goats [178]. To develop an effective attenuated vaccine against MAP, researchers compared this live ΔrelA mutant with the 35 kDa membrane protein MMP, a virulence protein that enhances epithelial cell invasion in the intestines. They encapsulated MMPs into a PLGA and monophosphoryl lipid A (PLGA/MPLA) NP vector ex vivo and in vivo in steers, with MPLA being used to stimulate CTLA-4 for antigen uptake [179]. They found that ΔrelA, MMP alone, and PLGA/MPLA-MMP NPs were all able to elicit a proliferative response of CD4 and CD8 T cells, with greater CTL activity with PLGA/MPLA-MMP NPs. While this study focused on creating a new peptide-based NP MAP vaccine for Johne’s disease, these strategies can be further investigated in models of Crohn’s disease to determine its effectiveness. However, improvements in technology for diagnosing MAP will better elucidate its clinical therapeutic efficacy.
6. Systemic lupus erythematosus (SLE)
SLE is a complex disease with symptoms varying from patient to patient, but a common characteristic is high titers of autoantibodies reactive to molecules found in the nucleus like ds-DNA and extractable nuclear antigens, such as Ro, La, Smith, and ribonucleoprotein [180,181]. Anti-dsDNA antibodies bind to DNA structures and form immune complexes, which activate dendritic cells and consequently B cells and T cells. This leads to overexpression of IFN-1, which promotes autoreactive B-cell and Th17 differentiation and Treg suppression [181]. Current treatments for SLE include non-steroidal immunosuppressants like cyclophosphamide, which depletes less mature B-cells, and biologics like belimumab, which bind to soluble B lymphocyte stimulator (BlyS), a costimulator for B-cell survival and function [182]. However, the available treatments are intended for initiation and maintenance of therapy rather than complete remission of disease, and they may come with adverse events (e.g., gastrointestinal or hematological problems and infections). While belimumab was a major breakthrough for lupus treatments as it the first drug FDA-approved specifically for lupus, it is administered to patients in combination with standard therapy [182,183]. Thus, there is high demand for more efficacious and safer lupus therapeutics with long-term protection.
Therapeutic development for lupus has been challenging due to its complexity and the variability of symptoms among lupus patients. While there are less accounts on antigen-specific immunotherapies for lupus models, compared to the aforementioned autoimmune diseases, there have been a couple propitious reports on the therapeutic efficacy of immunogenic SLE-related peptides. P140, a peptide derived from an epitope of the small nuclear ribonucleoprotein U1–70 K, can be recognized by IgG antibodies and CD4+ T cells in SLE mouse models [184]. P140 has been shown to reduce proteinuria and anti-dsDNA IgG titers and increase the survival rate in lupus-prone MRL/lpr mice [185]. As this immunomodulating peptide reduces autophagic flux, hinders MHCII stability, and weakens T cell responses against certain peptides that encompass T cell autoepitopes, it may be inhibiting presentation of autoantigens and interfering with the activation of autoreactive T cells. This leads to reduced T cell help for autoreactive B cell differentiation and autoantibody production. The peptide sequence is identical in mice and humans, so therapeutic application was easily transferred to humans. In vitro P140 studies with cells from healthy donors and patients resulted in dose-dependent downregulation of HLA class II expression in human lupus B cells, hampered proliferation of CD4+ T cells, prevented the differentiation of B cells, and reduced IgG levels (Fig. 6) [186]. Promising results in early clinical trials, including reduction of marker anti-native DNA antibodies and disease regression as defined by the Systemic Lupus Erythematosus Disease Activity Index, has led to FDA approval for phase III clinical trials with a “Fast Track” designation [185,187,188].
Fig. 6.
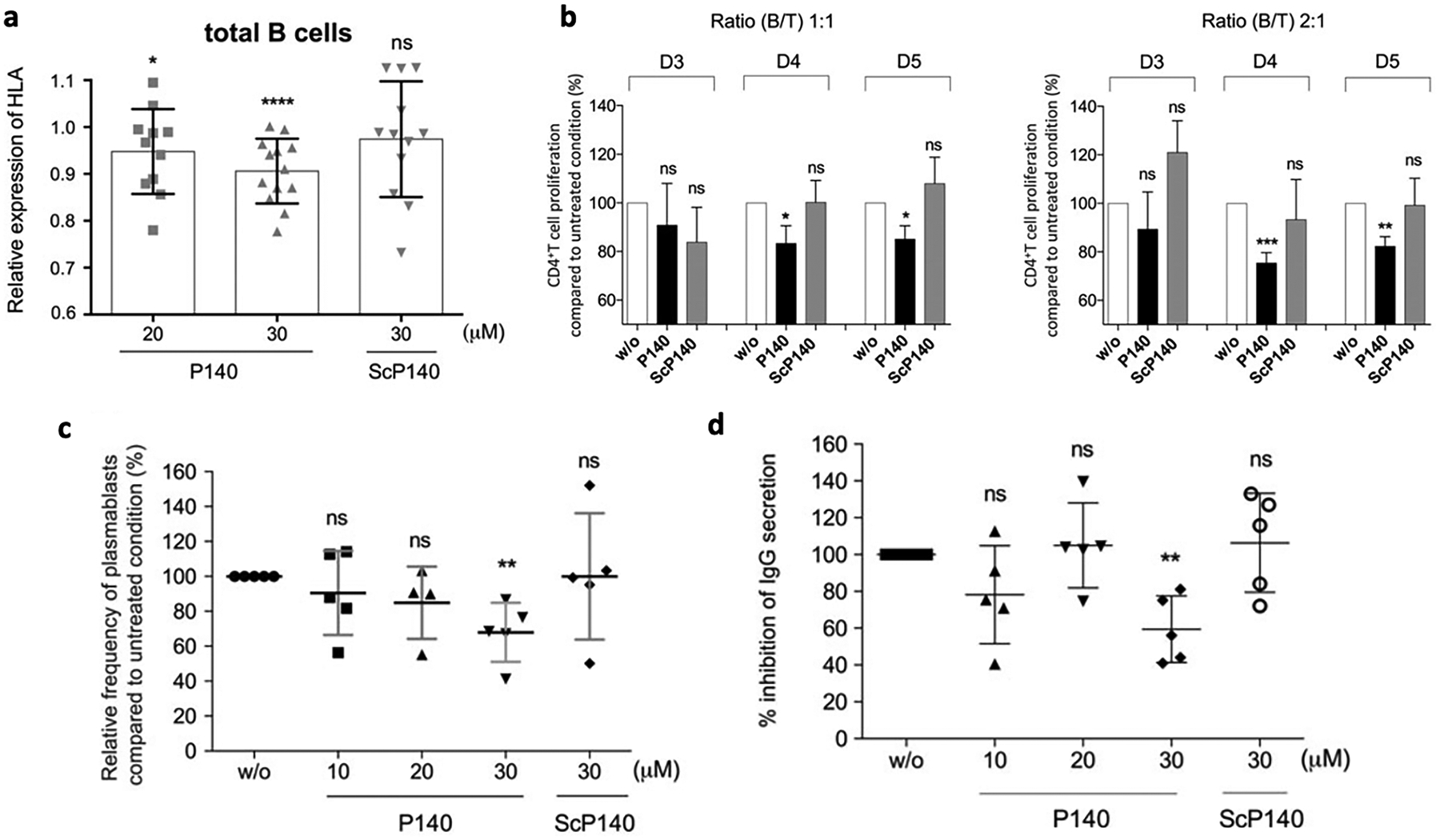
A, CD19+ B cells from SLE patients were treated with increasing concentrations of P140 or control peptide ScP140. Expression of HLA–DR/DP/DQ was measured by flow cytometry. Relative expression values were normalized to that of untreated cells. * = P < 0.05; *** = P < 0.001; **** = P < 0.0001, by unpaired t-test. b, Varying ratios of B and T cells from 2 different healthy donors were mixed to induce an allogeneic reaction. Proliferation of CD4+ T cells cocultured without (w/o) peptide or with peptide P140 or ScP140 was assessed over 3–5 days. Results are the mean SEM from 5 independent experiments. * = P < 0.05; ** = P < 0.01; *** = P < 0.001, by Mann-Whitney U test. NS = not significant. (c-d) B cells from healthy donors were cultured in the presence of anti-CD40/anti-IgM antibodies and interleukin-21 without (w/o) or with increasing concentrations of P140 peptide or ScP140 in vitro. B cell differentiation was assessed after 6 days by evaluating expression of CD19, CD27, and CD38 using flow cytometry. c, Relative frequency of plasmablasts (CD19+ CD27highCD38high cells) compared to untreated cells. d, Percentage of inhibition of IgG secretion in supernatant after 8 days of culture measured by enzyme-linked immunosorbent assay. Each symbol represents an individual healthy donor; bars show the mean SD (n = 5 independent experiments). ** = P < 0.01 by Mann-Whitney U test. NS = not significant.
Reproduced with permission from [186]
In lupus patients, Th cells that drive production of anti-DNA autoantibodies can recognize immunodominant epitopes of nucleosomal histones [189–192]. Major epitopes of lupus-nephritis-inducing Th cells and autoimmune B cells were reported to be localized in histone regions, and tolerization with these peptides led to protection against disease. Interestingly, while low-dose tolerance therapy with nucleosomal histone peptide epitopes H1′22–42, H416–39, and H471–94 can control disease in lupus-prone SNF1 mice [193], H471–94 monotherapy was found to be more effective than a cocktail of the three epitopes in delaying the onset of severe nephritis and prolonging survival of mice [189]. H471–94 single-peptide treatment resulted in reduced titers of IgG class anti-ssDNA, anti-nucleosome, and anti-histone autoantibodies, suppressed IFN-y and IL-17 responses to nucleosomal histone peptide epitopes, induced CD8+ Tregs, and promoted DCs to produce increased levels of TGF-β while inhibiting IL-6 production upon stimulation. Favorable traits of H471–94 include high potency at low doses; the capacity to suppress responses against other pathogenic epitopes, whole nucleosomes, and ribonucleoprotein; and recognition by autoimmune T and B cells of most lupus patients [189,194,195]. However, more in vivo studies are needed to assess its therapeutic efficacy for clinical application.
7. Perspectives and conclusions
Autoimmune diseases are unmistakably complex. In such circumstances, trying to target a single autoantigen may seem irrational, but many studies have shown that there are promising vaccine candidates that can induce antigen-specific immune tolerance along with bystander suppression. These disease-specific vaccines aim to restore the body’s homeostasis and potentially offer a cure for autoimmune diseases. This is a drastically different approach than simply treating the symptoms, such as in the case of immunosuppressants that suppress the immune system as a whole and need to be administered for life.
Nevertheless, the capacity, breadth, and durability of immune tolerance by antigen-specific vaccines have yet to be thoroughly investigated. Timing and dosing of the autoantigen delivery may possibly require individual evaluation for different autoreactive T cells, which can be activated and expanded at different stages or flare-ups of the disease. Disease perpetuating and spread epitopes are attractive candidates and have shown great potential in treating experimental autoimmune models with adoptive transfer or active immunization. However, in spontaneous autoimmune diseases, the presence of other autoantigen-reactive T cells, emergence of hybrid peptide neoepitopes, and multiple mechanisms of tolerance breakdown may probably compromise the therapeutic efficacy. In this regard, approaches capable of inducing multiple Treg clones that elicit strong bystander suppression will be beneficial. Study of combinatorial regimens, including combinatorial epitope library and combined immunomodulator, may lead to better therapeutic outcomes. In addition, prolonged treatment or multiple cycles of vaccine administration is likely to be required to achieve sustained response; thus, potential safety and side effects such as anaphylactic response also need to be evaluated. These are important challenges that need to be addressed to enhance clinical translation.
Nonetheless, the expanding expertise in vaccine and nanoparticle development and GMP-level, large-scale manufacturing in the setting of cancer immunotherapy is likely to benefit the field of antigen-specific tolerogenic immunotherapies, especially those with complex compositions. Overall, vaccine-based approaches to induce antigen-specific Tregs and immune tolerance have great promise to lead the next wave of novel therapeutics against inflammation and autoimmunity.
Acknowledgments
This work was supported in part by NIH (R01DE030691, R01DE031951, R01DK125087, R01CA271799, R01NS122536, R01DE026728, R44DK135218). A.K. is supported by a predoctoral fellowship from the Cellular Biotechnology Training Program (T32GM008353). O.A.A. is supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE 1841052. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Declaration of Competing Interest
JJM declares financial interests as board membership, a paid consultant, research funding, and/or equity holder in EVOQ Therapeutics and Saros Therapeutics. The University of Michigan has a financial interest in EVOQ Therapeutics.
Data availability
No data was used for the research described in the article.
References
- [1].Sener AG, Afsar I, Infection and autoimmune disease, Rheumatol. Int 32 (2012) 3331–3338. [DOI] [PubMed] [Google Scholar]
- [2].Conrad N, Misra S, Verbakel JY, Verbeke G, Molenberghs G, Taylor PN, Mason J, Sattar N, McMurray JJV, McInnes IB, Khunti K, Cambridge G, Incidence, prevalence, and co-occurrence of autoimmune disorders over time and by age, sex, and socioeconomic status: a population-based cohort study of 22 million individuals in the UK, Lancet 401 (2023) 1878–1890. [DOI] [PubMed] [Google Scholar]
- [3].Gammon JM, Jewell CM, Engineering Immune Tolerance with Biomaterials, Adv. Healthc. Mater 8 (2019) e1801419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].ElTanbouly MA, Noelle RJ, Rethinking peripheral T cell tolerance: checkpoints across a T cell’s journey, Nat. Rev. Immunol 21 (2021) 257–267. [DOI] [PubMed] [Google Scholar]
- [5].Goldrath AW, Bevan MJ, Selecting and maintaining a diverse T-cell repertoire, Nature 402 (1999) 255–262. [DOI] [PubMed] [Google Scholar]
- [6].Xing Y, Hogquist KA, T-cell tolerance: central and peripheral, Cold Spring Harb. Perspect. Biol 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Basten A, Silveira PA, B-cell tolerance: mechanisms and implications, Curr. Opin. Immunol 22 (2010) 566–574. [DOI] [PubMed] [Google Scholar]
- [8].Schmidt A, Oberle N, Krammer PH, Molecular mechanisms of treg-mediated T cell suppression, Front. Immunol 3 (2012) 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Catalán D, Mansilla MA, Ferrier A, Soto L, Oleinika K, Aguillón JC, Aravena O, Immunosuppressive mechanisms of regulatory B cells, Front. Immunol 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yu W, Jiang N, Ebert PJ, Kidd BA, Müller S, Lund PJ, Juang J, Adachi K, Tse T, Birnbaum ME, Newell EW, Wilson DM, Grotenbreg GM, Valitutti S, Quake SR, Davis MM, Clonal deletion prunes but does not eliminate self-specific αβ CD8(+) T lymphocytes, Immunity 42 (2015) 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Theofilopoulos AN, Kono DH, Baccala R, The multiple pathways to autoimmunity, Nat. Immunol 18 (2017) 716–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rock KL, Latz E, Ontiveros F, Kono H, The sterile inflammatory response, Annu. Rev. Immunol 28 (2010) 321–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Choi B, Lee C, Yu JW, Distinctive role of inflammation in tissue repair and regeneration, Arch. Pharm. Res 46 (2023) 78–89. [DOI] [PubMed] [Google Scholar]
- [14].Jäger A, Kuchroo VK, Effector and regulatory T-cell subsets in autoimmunity and tissue inflammation, Scand. J. Immunol 72 (2010) 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sun L, Su Y, Jiao A, Wang X, Zhang B, T cells in health and disease, Signal Transduct. Target. Ther 8 (2023) 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chervonsky AV, Influence of microbial environment on autoimmunity, Nat. Immunol 11 (2010) 28–35. [DOI] [PubMed] [Google Scholar]
- [17].Bjornevik K, Münz C, Cohen JI, Ascherio A, Epstein-Barr virus as a leading cause of multiple sclerosis: mechanisms and implications, Nature Reviews, Neurology 19 (2023) 160–171. [DOI] [PubMed] [Google Scholar]
- [18].Soldan SS, Lieberman PM, Epstein-Barr virus and multiple sclerosis, Nat. Rev. Microbiol 21 (2023) 51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hyöty H, Viruses in type 1 diabetes, Pediatr. Diabetes 17 (2016) 56–64. [DOI] [PubMed] [Google Scholar]
- [20].Krutyhołowa A, Strzelec K, Dziedzic A, Bereta GP, Łazarz-Bartyzel K, Potempa J, Gawron K, Host and bacterial factors linking periodontitis and rheumatoid arthritis, Front. Immunol 13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Qiu P, Ishimoto T, Fu L, Zhang J, Zhang Z, Liu Y, The gut microbiota in inflammatory bowel disease, Front. Cell. Infect. Microbiol 12 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Anderson RP, Jabri B, Vaccine against autoimmune disease: antigen-specific immunotherapy, Curr. Opin. Immunol 25 (2013) 410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kishimoto TK, Maldonado RA, Nanoparticles for the induction of antigen-specific immunological tolerance, Front. Immunol 9 (2018) 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Park J, Wu Y, Li Q, Choi J, Ju H, Cai Y, Lee J, Oh YK, Nanomaterials for antigen-specific immune tolerance therapy, Drug Deliv. Transl. Res 13 (2023) 1859–1881. [DOI] [PubMed] [Google Scholar]
- [25].Federation MSI, Atlas of MS, 3rd Edition 2020. [Google Scholar]
- [26].Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, Rocca MA, Multiple sclerosis, Nat. Rev. Dis. Primers 4 (2018) 43. [DOI] [PubMed] [Google Scholar]
- [27].Ghasemi N, Razavi S, Nikzad E, Multiple sclerosis: pathogenesis symptoms, diagnoses and cell-based therapy, Cell J 19 (2017) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Attfield KE, Jensen LT, Kaufmann M, Friese MA, Fugger L, The immunology of multiple sclerosis, Nat. Rev. Immunol 22 (2022) 734–750. [DOI] [PubMed] [Google Scholar]
- [29].Glatigny S, Bettelli E, Experimental autoimmune encephalomyelitis (EAE) as animal models of multiple sclerosis (MS), Cold Spring Harb. Perspect. Med. 8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kurschus F, T cell mediated pathogenesis in EAE: molecular mechanisms, Biomed. J 38 (2015). [DOI] [PubMed] [Google Scholar]
- [31].Baxter AG, The origin and application of experimental autoimmune encephalomyelitis, Nat. Rev. Immunol 7 (2007) 904–912. [DOI] [PubMed] [Google Scholar]
- [32].Dendrou CA, Fugger L, Friese MA, Immunopathology of multiple sclerosis, Nat. Rev. Immunol 15 (2015) 545–558. [DOI] [PubMed] [Google Scholar]
- [33].McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD, Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis, Nat. Med 11 (2005) 335–339. [DOI] [PubMed] [Google Scholar]
- [34].Ji Q, Castelli L, Goverman JM, MHC class I–restricted myelin epitopes are cross-presented by Tip-DCs that promote determinant spreading to CD8+ T cells, Nat. Immunol 14 (2013) 254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kontos S, Grimm AJ, Hubbell JA, Engineering antigen-specific immunological tolerance, Curr. Opin. Immunol 35 (2015) 80–88. [DOI] [PubMed] [Google Scholar]
- [36].Carambia A, Freund B, Schwinge D, Bruns OT, Salmen SC, Ittrich H, Reimer R, Heine M, Huber S, Waurisch C, Eychmüller A, Wraith DC, Korn T, Nielsen P, Weller H, Schramm C, Lüth S, Lohse AW, Heeren J, Herkel J, Nanoparticle-based autoantigen delivery to Treg-inducing liver sinusoidal endothelial cells enables control of autoimmunity in mice, J. Hepatol 62 (2015) 1349–1356. [DOI] [PubMed] [Google Scholar]
- [37].Casey LM, Hughes KR, Saunders MN, Miller SD, Pearson RM, Shea LD, Mechanistic contributions of Kupffer cells and liver sinusoidal endothelial cells in nanoparticle-induced antigen-specific immune tolerance, Biomaterials 283 (2022), 121457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hess KL, Oh E, Tostanoski LH, Andorko JI, Susumu K, Deschamps JR, Medintz IL, Jewell CM, Engineering immunological tolerance using quantum dots to tune the density of self-antigen display, Adv. Funct. Mater 27 (2017) 1700290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Suk JS, Xu Q, Kim N, Hanes J, Ensign LM, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery, Adv. Drug Deliv. Rev 99 (2016) 28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD, The role of complement in biomaterial-induced inflammation, Mol. Immunol 44 (2007) 82–94. [DOI] [PubMed] [Google Scholar]
- [41].Li PY, Bearoff F, Zhu P, Fan Z, Zhu Y, Fan M, Cort L, Kambayashi T, Blankenhorn EP, Cheng H, PEGylation enables subcutaneously administered nanoparticles to induce antigen-specific immune tolerance, J. Control. Release 331 (2021) 164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, Miller SD, Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis, Nat. Biotechnol 30 (2012) 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, Miller SD, A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease, ACS Nano 8 (2014) 2148–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Triantafyllakou I, Clemente N, Khetavat RK, Dianzani U, Tselios T, Development of PLGA Nanoparticles with a Glycosylated Myelin Oligodendrocyte Glycoprotein Epitope (MOG(35–55)) against Experimental Autoimmune Encephalomyelitis (EAE), Mol. Pharm 19 (2022) 3795–3805. [DOI] [PubMed] [Google Scholar]
- [45].Belogurov AA Jr, Stepanov AV, Smirnov IV, Melamed D, Bacon A, Mamedov AE, Boitsov VM, Sashchenko LP, Ponomarenko NA, Sharanova SN, Liposome-encapsulated peptides protect against experimental allergic encephalitis, FASEB J. 27 (2013) 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Saito E, Gurczynski SJ, Kramer KR, Wilke CA, Miller SD, Moore BB, Shea LD, Modulating lung immune cells by pulmonary delivery of antigen-specific nanoparticles to treat autoimmune disease, Sci. Adv 6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Odoardi F, Sie C, Streyl K, Ulaganathan VK, Schläger C, Lodygin D, Heckelsmiller K, Nietfeld W, Ellwart J, Klinkert WE, T cells become licensed in the lung to enter the central nervous system, Nature 488 (2012) 675–679. [DOI] [PubMed] [Google Scholar]
- [48].Quintana FJ, Patel B, Yeste A, Nyirenda M, Kenison J, Rahbari R, Fetco D, Hussain M, O’Mahony J, Magalhaes S, Epitope spreading as an early pathogenic event in pediatric multiple sclerosis, Neurology 83 (2014) 2219–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Casella G, Rasouli J, Boehm A, Zhang W, Xiao D, Ishikawa LLW, Thome R, Li X, Hwang D, Porazzi P, Molugu S, Tang H-Y, Zhang G-X, Ciric B, Rostami A, Oligodendrocyte-derived extracellular vesicles as antigen-specific therapy for autoimmune neuroinflammation in mice, Sci. Transl. Med 12 (2020) eaba0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Smith CE, Miller SD, Multi-peptide coupled-cell tolerance ameliorates ongoing relapsing EAE associated with multiple pathogenic autoreactivities, J. Autoimmun 27 (2006) 218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lutterotti A, Yousef S, Sputtek A, Stürner KH, Stellmann J-P, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, Schippling S, Miller SD, Sospedra M, Martin R, Antigen-specific tolerance by autologous myelin peptide–coupled cells: A phase 1 trial in multiple sclerosis, Sci. Transl. Med 5 (2013) 188ra175–188ra175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Krienke C, Kolb L, Diken E, Streuber M, Kirchhoff S, Bukur T, Akilli-Öztürk Ö, Kranz LM, Berger H, Petschenka J, Diken M, Kreiter S, Yogev N, Waisman A, Karikó K, Türeci Ö, Sahin U, A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis, Science 371 (2021) 145–153. [DOI] [PubMed] [Google Scholar]
- [53].Fischer R, Turnquist HR, Taner T, Thomson AW, Use of rapamycin in the induction of tolerogenic dendritic cells, Handb. Exp. Pharmacol (2009) 215–232. [DOI] [PubMed] [Google Scholar]
- [54].Chen X, Li S, Long D, Shan J, Li Y, Rapamycin facilitates differentiation of regulatory T cells via enhancement of oxidative phosphorylation, Cell. Immunol 365 (2021), 104378. [DOI] [PubMed] [Google Scholar]
- [55].Powell JD, Lerner CG, Schwartz RH, Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation, J. Immunol 162 (1999) 2775–2784. [PubMed] [Google Scholar]
- [56].Araki K, Youngblood B, Ahmed R, The role of mTOR in memory CD8 T-cell differentiation, Immunol. Rev 235 (2010) 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O’Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH, Kishimoto TK, Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance, in: Proceedings of the National Academy of Sciences of the United States of America, 112 (2015) E156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].LaMothe RA, Kolte PN, Vo T, Ferrari JD, Gelsinger TC, Wong J, Chan VT, Ahmed S, Srinivasan A, Deitemeyer P, Maldonado RA, Kishimoto TK, Tolerogenic nanoparticles induce antigen-specific regulatory T cells and provide therapeutic efficacy and transferrable tolerance against experimental autoimmune encephalomyelitis, Front. Immunol 9 (2018) 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Stevens EA, Mezrich JD, Bradfield CA, The aryl hydrocarbon receptor: a perspective on potential roles in the immune system, Immunology 127 (2009) 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah A-M, Burns EJ, Weiner HL, An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis, in: Proceedings of the National Academy of Sciences, 107 (2010) 20768–20773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kerkvliet NI, Steppan LB, Vorachek W, Oda S, Farrer D, Wong CP, Pham D, Mourich DV, Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes, Immunotherapy 1 (2009) 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kenison JE, Jhaveri A, Li Z, Khadse N, Tjon E, Tezza S, Nowakowska D, Plasencia A, Stanton VP Jr., Sherr DH, Quintana FJ, Tolerogenic nanoparticles suppress central nervous system inflammation, in: Proceedings of the National Academy of Sciences of the United States of America, 117 (2020) 32017–32028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wang H, Shang J, He Z, Zheng M, Jia H, Zhang Y, Yang W, Gao X, Gao F, Dual peptide nanoparticle platform for enhanced antigen-specific immune tolerance for the treatment of experimental autoimmune encephalomyelitis, Biomater. Sci 10 (2022) 3878–3891. [DOI] [PubMed] [Google Scholar]
- [64].Casey LM, Pearson RM, Hughes KR, Liu JMH, Rose JA, North MG, Wang LZ, Lei M, Miller SD, Shea LD, Conjugation of Transforming Growth Factor Beta to Antigen-Loaded Poly(lactide- co-glycolide) Nanoparticles Enhances Efficiency of Antigen-Specific Tolerance, Bioconjug. Chem 29 (2018) 813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, Gigliotti CL, Boggio E, Clemente N, Comi C, Dianzani C, Boldorini R, Chiocchetti A, Reno F, Dianzani U, Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis, Vaccine 32 (2014) 5681–5689. [DOI] [PubMed] [Google Scholar]
- [66].Cho JJ, Stewart JM, Drashansky TT, Brusko MA, Zuniga AN, Lorentsen KJ, Keselowsky BG, Avram D, An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis, Biomaterials 143 (2017) 79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kwiatkowski AJ, Helm EY, Stewart J, Leon J, Drashansky T, Avram D, Keselowsky B, Design principles of microparticle size and immunomodulatory factor formulation dictate antigen-specific amelioration of multiple sclerosis in a mouse model, Biomaterials 294 (2023), 122001. [DOI] [PubMed] [Google Scholar]
- [68].Tomíc S, Janjetovíc K, Mihajlovíc D, Milenkovíc M, Kravíc-Stevovíc T, Markovíc Z, Todorovíc-Markovíc B, Spitalsky Z, Micusik M, Vučevíc D, Čolić M, Trajkovíc V, Graphene quantum dots suppress proinflammatory T cell responses via autophagy-dependent induction of tolerogenic dendritic cells, Biomaterials 146 (2017) 13–28. [DOI] [PubMed] [Google Scholar]
- [69].Nguyen TL, Choi Y, Im J, Shin H, Phan NM, Kim MK, Choi SW, Kim J, Immunosuppressive biomaterial-based therapeutic vaccine to treat multiple sclerosis via re-establishing immune tolerance, Nat. Commun 13 (2022) 7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Eizirik DL, Colli ML, Ortis F, The role of inflammation in insulitis and beta-cell loss in type 1 diabetes, Nat. Rev. Endocrinol 5 (2009) 219–226. [DOI] [PubMed] [Google Scholar]
- [71].Melendez-Ramirez LY, Richards RJ, Cefalu WT, Complications of type 1 diabetes, Endocrinol. Metab. Clin. North Am 39 (2010) 625–640. [DOI] [PubMed] [Google Scholar]
- [72].Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Effects of oral insulin in relatives of patients with type 1 diabetes: The Diabetes Prevention Trial-Type 1, Diabetes Care 28 (2005) 1068. [DOI] [PubMed] [Google Scholar]
- [73].Näntö-Salonen K, Kupila A, Simell S, Siljander H, Salonsaari T, Hekkala A, Korhonen S, Erkkola R, Sipilä JI, Haavisto L, Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial, Lancet 372 (2008) 1746–1755. [DOI] [PubMed] [Google Scholar]
- [74].Ludvigsson J, Krisky D, Casas R, Battelino T, Castaño L, Greening J, Kordonouri O, Otonkoski T, Pozzilli P, Robert J-J, GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus, N. Engl. J. Med 366 (2012) 433–442. [DOI] [PubMed] [Google Scholar]
- [75].Alhadj Ali M, Liu Y-F, Arif S, Tatovic D, Shariff H, Gibson VB, Yusuf N, Baptista R, Eichmann M, Petrov N, Heck S, Yang JHM, Tree TIM, Pujol-Autonell I, Yeo L, Baumard L, Stenson R, Howell A, Clark A, Boult Z, Powrie J, Adams L, Wong FS, Luzio S, Dunseath G, Green K, O’Keefe A, Bayly G, Thorogood N, Andrews R, Leech N, Joseph F, Nair S, Seal S, Cheung H, Beam C, Hills R, Peakman M, Dayan CM, Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes, Sci. Transl. Med 9 (2017) eaaf7779. [DOI] [PubMed] [Google Scholar]
- [76].Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, Armstrong M, Powell RL, Reisdorph N, Kumar N, Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion, Science 351 (2016) 711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Jamison BL, Neef T, Goodspeed A, Bradley B, Baker RL, Miller SD, Haskins K, Nanoparticles containing an insulin–ChgA hybrid peptide protect from transfer of autoimmune diabetes by shifting the balance between effector T cells and regulatory T cells, J. Immunol 203 (2019) 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Van Belle T, Taylor P, Von Herrath M, Mouse models for type 1 diabetes, Drug Discov. Today Dis. Model 6 (2009) 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Shen H, Ackerman AL, Cody V, Giodini A, Hinson ER, Cresswell P, Edelson RL, Saltzman WM, Hanlon DJ, Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles, Immunology 117 (2006) 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Prasad S, Neef T, Xu D, Podojil JR, Getts DR, Shea LD, Miller SD, Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells, J. Autoimmun 89 (2018) 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Prasad S, Kohm AP, McMahon JS, Luo X, Miller SD, Pathogenesis of NOD diabetes is initiated by reactivity to the insulin B chain 9–23 epitope and involves functional epitope spreading, J. Autoimmun 39 (2012) 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Pearson JA, Wong FS, Wen L, The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes, J. Autoimmun 66 (2016) 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Liu M, Feng D, Liang X, Li M, Yang J, Wang H, Pang L, Zhou Z, Yang Z, Kong D, Li C, Old dog new tricks: PLGA microparticles as an adjuvant for insulin peptide fragment-induced immune tolerance against type 1 diabetes, Mol. Pharm 17 (2020) 3513–3525. [DOI] [PubMed] [Google Scholar]
- [84].Qu Y, Shi H, Liu M, Zhang M, Wang H, Pang L, Zhang C, Kong D, Li C, In vivo insulin peptide autoantigen delivery by mannosylated sodium alginate nanoparticles delayed but could not prevent the onset of type 1 diabetes in nonobese diabetic mice, Mol. Pharm 18 (2021) 1806–1818. [DOI] [PubMed] [Google Scholar]
- [85].Podojil JR, Genardi S, Chiang M-Y, Kakade S, Neef T, Murthy T, Boyne MT II, Elhofy A, Miller SD, Tolerogenic immune-modifying nanoparticles encapsulating multiple recombinant pancreatic β cell proteins prevent onset and progression of type 1 diabetes in nonobese diabetic mice, J. Immunol 209 (2022) 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Firdessa-Fite R, Johnson SN, Leon MA, Khosravi-Maharlooei M, Baker RL, Sestak JO, Berkland C, Creusot RJ, Soluble antigen arrays efficiently deliver peptides and arrest spontaneous autoimmune diabetes, Diabetes 70 (2021) 1334–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wilson DS, Damo M, Hirosue S, Raczy MM, Brünggel K, Diaceri G, Quaglia-Thermes X, Hubbell JA, Synthetically glycosylated antigens induce antigen-specific tolerance and prevent the onset of diabetes, Nature, Biomed. Eng 3 (2019) 817–829. [DOI] [PubMed] [Google Scholar]
- [88].Tremain AC, Wallace RP, Lorentz KM, Thornley TB, Antane JT, Raczy MR, Reda JW, Alpar AT, Slezak AJ, Watkins EA, Synthetically glycosylated antigens for the antigen-specific suppression of established immune responses, Nat. Biomed. Eng (2023) 1–14. [DOI] [PubMed] [Google Scholar]
- [89].Chen N, Peine KJ, Collier MA, Gautam S, Jablonski KA, Guerau-de-Arellano M, Ainslie KM, Bachelder EM, Co-delivery of disease associated peptide and rapamycin via acetalated dextran microparticles for treatment of multiple sclerosis, Adv. Biosyst 1 (2017) 1700022. [Google Scholar]
- [90].Chen N, Kroger CJ, Tisch RM, Bachelder EM, Ainslie KM, Prevention of type 1 diabetes with acetalated dextran microparticles containing rapamycin and pancreatic peptide P31, Adv. Healthc. Mater 7 (2018) 1800341. [DOI] [PubMed] [Google Scholar]
- [91].Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, Peng H, Yang Y, Vangundy Z, Papenfuss TL, Treatment of experimental autoimmune encephalomyelitis by codelivery of disease associated Peptide and dexamethasone in acetalated dextran microparticles, Mol. Pharm 11 (2014) 828–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Bachelder EM, Pino EN, Ainslie KM, Acetalated dextran: A tunable and acid-labile biopolymer with facile synthesis and a range of applications, Chem. Rev 117 (2017) 1915–1926. [DOI] [PubMed] [Google Scholar]
- [93].Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C, Agrawal S, Keough MB, Yong VW, James E, Moore A, Yang Y, Stratmann T, Serra P, Santamaria P, Expanding antigen-specific regulatory networks to treat autoimmunity, Nature 530 (2016) 434–440. [DOI] [PubMed] [Google Scholar]
- [94].Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, Yang Y, Medarova Z, Moore A, Santamaria P, Reversal of autoimmunity by boosting memory-like autoregulatory T cells, Immunity 32 (2010) 568–580. [DOI] [PubMed] [Google Scholar]
- [95].Yang Y, Ellestad KK, Singha S, Uddin MM, Clarke R, Mondal D, Garabatos N, Soĺe P, Fandos C, Serra P, Santamaria P, Extremely short bioavailability and fast pharmacodynamic effects of pMHC-based nanomedicines, J. Control. Release 338 (2021) 557–570. [DOI] [PubMed] [Google Scholar]
- [96].Singha S, Shao K, Yang Y, Clemente-Casares X, Solé P, Clemente A, Blanco J, Dai Q, Song F, Liu SW, Peptide–MHC-based nanomedicines for autoimmunity function as T-cell receptor microclustering devices, Nat. Nanotechnol 12 (2017) 701–710. [DOI] [PubMed] [Google Scholar]
- [97].Solé P, Yamanouchi J, Garnica J, Uddin MM, Clarke R, Moro J, Garabatos N, Thiessen S, Ortega M, Singha S, Mondal D, Fandos C, Saez-Rodriguez J, Yang Y, Serra P, Santamaria P, A T follicular helper cell origin for T regulatory type 1 cells, Cell. Mol. Immunol 20 (2023) 489–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Umeshappa CS, Singha S, Blanco J, Shao K, Nanjundappa RH, Yamanouchi J, Paŕes A, Serra P, Yang Y, Santamaria P, Suppression of a broad spectrum of liver autoimmune pathologies by single peptide-MHC-based nanomedicines, Nat. Commun 10 (2019) 2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Phillips BE, Garciafigueroa Y, Engman C, Liu W, Wang Y, Lakomy RJ, Meng WS, Trucco M, Giannoukakis N, Arrest in the progression of type 1 diabetes at the mid-stage of insulitic autoimmunity using an autoantigen-decorated all-trans retinoic acid and transforming growth factor beta-1 single microparticle formulation, Front. Immunol 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, Tukpah A-M, Babon JAB, DeNicola M, Kent SC, Tolerogenic nanoparticles inhibit T cell–mediated autoimmunity through SOCS2, Sci. Signal 9 (2016) ra61–ra61. [DOI] [PubMed] [Google Scholar]
- [101].Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, Gillis K, Brown L, Lafreniere D, Gallo M, Knox J, Hogeland K, Trucco M, Giannoukakis N, A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes, Diabetes 57 (2008) 1544–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Luo Y-L, Liang L-F, Gan Y-J, Liu J, Zhang Y, Fan Y-N, Zhao G, Czarna A, Lu Z-D, Du X-J, Shen S, Xu C-F, Lian Z-X, Wang J, An all-in-one nanomedicine consisting of CRISPR-Cas9 and an autoantigen peptide for restoring specific immune tolerance, ACS Appl. Mater. Interfaces 12 (2020) 48259–48271. [DOI] [PubMed] [Google Scholar]
- [103].Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG, A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice, Clin. Immunol 160 (2015) 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, Dolgova NV, Xia C, Brusko TM, Wasserfall CH, Clare-Salzler MJ, Atkinson MA, Keselowsky BG, Dual-sized microparticle system for generating suppressive dendritic cells prevents and reverses type 1 diabetes in the nonobese diabetic mouse model, ACS Biomater Sci. Eng 5 (2019) 2631–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chen X, Yang X, Yuan P, Jin R, Bao L, Qiu X, Liu S, Liu T, Gooding JJ, Chen W, Liu G, Bai Y, Liu S, Jin Y, Modular immune-homeostatic microparticles promote immune tolerance in mouse autoimmune models, Sci. Transl. Med 13 (2021) eaaw9668. [DOI] [PubMed] [Google Scholar]
- [106].Cremel M, Gúerin N, Horand F, Banz A, Godfrin Y, Red blood cells as innovative antigen carrier to induce specific immune tolerance, Int. J. Pharm 443 (2013) 39–49. [DOI] [PubMed] [Google Scholar]
- [107].Gammon JM, Carey ST, Saxena V, Eppler HB, Tsai SJ, Paluskievicz C, Xiong Y, Li L, Ackun-Farmmer M, Tostanoski LH, Gosselin EA, Yanes AA, Zeng X, Oakes RS, Bromberg JS, Jewell CM, Engineering the lymph node environment promotes antigen-specific efficacy in type 1 diabetes and islet transplantation, Nat. Commun 14 (2023) 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Zisi Tegou F, Velluto D, Stock AA, Fitzgerald SN, Stealey S, Zustiak SP, Bayer AL, Tomei AA, CCL21 and beta-cell antigen releasing hydrogels as tolerance-inducing therapy in Type I diabetes, J. Control. Release 348 (2022) 499–517. [DOI] [PubMed] [Google Scholar]
- [109].Arikat F, Hanna SJ, Singh RK, Vilela L, Wong FS, Dayan CM, Coulman SA, Birchall JC, Targeting proinsulin to local immune cells using an intradermal microneedle delivery system; a potential antigen-specific immunotherapy for type 1 diabetes, J. Control. Release 322 (2020) 593–601. [DOI] [PubMed] [Google Scholar]
- [110].Hovav AH, Dendritic cells of the oral mucosa, Mucosal Immunol. 7 (2014) 27–37. [DOI] [PubMed] [Google Scholar]
- [111].Chen Y, Wu J, Wang J, Zhang W, Xu B, Xu X, Zong L, Targeted delivery of antigen to intestinal dendritic cells induces oral tolerance and prevents autoimmune diabetes in NOD mice, Diabetologia 61 (2018) 1384–1396. [DOI] [PubMed] [Google Scholar]
- [112].Mao R, Yang M, Yang R, Chen Y, Diao E, Zhang T, Li D, Chang X, Chi Z, Wang Y, Oral delivery of the intracellular domain of the insulinoma-associated protein 2 (IA-2ic) by bacterium-like particles (BLPs) prevents type 1 diabetes mellitus in NOD mice, Drug Deliv. 29 (2022) 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Mao R, Wang J, Xu Y, Wang Y, Wu M, Mao L, Chen Y, Li D, Zhang T, Diao E, Chi Z, Wang Y, Chang X, Oral delivery of bi-autoantigens by bacterium-like particles (BLPs) against autoimmune diabetes in NOD mice, Drug Deliv. 30 (2023) 2173339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Mao R, Chen Y, Wu Q, Zhang T, Diao E, Wu D, Wang M, Liu Y, Lu L, Chang X, Zheng Y, Wang Y, Oral delivery of single-chain insulin (SCI-59) analog by bacterium-like particles (BLPs) induces oral tolerance and prevents autoimmune diabetes in NOD mice, Immunol. Lett 214 (2019) 37–44. [DOI] [PubMed] [Google Scholar]
- [115].McInnes IB, Schett G, The pathogenesis of rheumatoid arthritis, N. Engl. J. Med 365 (2011) 2205–2219. [DOI] [PubMed] [Google Scholar]
- [116].Li C, Zheng X, Hu M, Jia M, Jin R, Nie Y, Recent progress in therapeutic strategies and biomimetic nanomedicines for rheumatoid arthritis treatment, Expert Opin. Drug Deliv 19 (2022) 883–898. [DOI] [PubMed] [Google Scholar]
- [117].McInnes IB, Schett G, Cytokines in the pathogenesis of rheumatoid arthritis, Nat. Rev. Immunol 7 (2007) 429–442. [DOI] [PubMed] [Google Scholar]
- [118].Catrina AI, Ytterberg AJ, Reynisdottir G, Malmstrom V, Klareskog L, Lungs, joints and immunity against citrullinated proteins in rheumatoid arthritis, Nat. Rev. Rheumatol 10 (2014) 645–653. [DOI] [PubMed] [Google Scholar]
- [119].Cho Y-G, Cho M-L, Min S-Y, Kim H-Y, Type II collagen autoimmunity in a mouse model of human rheumatoid arthritis, Autoimmun. Rev 7 (2007) 65–70. [DOI] [PubMed] [Google Scholar]
- [120].Meehan GR, Thomas R, Al Khabouri S, Wehr P, Hilkens CM, Wraith DC, Sieghart D, Bonelli M, Nagy G, Garside P, Tough DF, Lewis HD, Brewer JM, Preclinical models of arthritis for studying immunotherapy and immune tolerance, Ann. Rheum. Dis 80 (2021) 1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Chen X, Du G, Bai S, Dijia L, Li C, Hou Y, Zhang Y, Zhang Z, Gong T, Fu Y, Bottini M, Restoring immunological tolerance in established experimental arthritis by combinatorial citrullinated peptides and immunomodulatory signals, Nano Today 41 (2021), 101307. [Google Scholar]
- [122].Li C, Chen X, Luo X, Wang H, Zhu Y, Du G, Chen W, Chen Z, Hao X, Zhang Z, Sun X, Nanoemulsions target to ectopic lymphoids in inflamed joints to restore immune tolerance in rheumatoid arthritis, Nano Lett. 21 (2021) 2551–2561. [DOI] [PubMed] [Google Scholar]
- [123].Srivastava A, Arlian BM, Pang L, Kishimoto TK, Paulson JC, Tolerogenic nanoparticles impacting B and T lymphocyte responses delay autoimmune arthritis in K/BxN mice, ACS Chem. Biol 16 (2021) 1985–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Macauley MS, Pfrengle F, Rademacher C, Nycholat CM, Gale AJ, von Drygalski A, Paulson JC, Antigenic liposomes displaying CD22 ligands induce antigen-specific B cell apoptosis, J. Clin. Invest 123 (2013) 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Duong BH, Tian H, Ota T, Completo G, Han S, Vela JL, Ota M, Kubitz M, Bovin N, Paulson JC, Nemazee D, Decoration of T-independent antigen with ligands for CD22 and Siglec-G can suppress immunity and induce B cell tolerance in vivo, J. Exp. Med 207 (2010) 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Macauley MS, Paulson JC, Siglecs induce tolerance to cell surface antigens by BIM-dependent deletion of the antigen-reactive B cells, J. Immunol 193 (2014) 4312–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Brzezicka KA, Arlian BM, Wang S, Olmer M, Lotz M, Paulson JC, Suppression of autoimmune rheumatoid arthritis with hybrid nanoparticles that induce B and T Cell tolerance to self-antigen, ACS Nano 16 (2022) 20206–20221. [DOI] [PubMed] [Google Scholar]
- [128].Galea R, Nel HJ, Talekar M, Liu X, Ooi JD, Huynh M, Hadjigol S, Robson KJ, Ting YT, Cole S, Cochlin K, Hitchcock S, Zeng B, Yekollu S, Boks M, Goh N, Roberts H, Rossjohn J, Reid HH, Boyd BJ, Malaviya R, Shealy DJ, Baker DG, Madakamutil L, Kitching AR, O’Sullivan BJ, Thomas R, PD-L1- and calcitriol-dependent liposomal antigen-specific regulation of systemic inflammatory autoimmune disease, JCI Insight 4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Bergot A-S, Buckle I, Cikaluru S, Naranjo JL, Wright CM, Zheng G, Talekar M, Hamilton-Williams EE, Thomas R, Regulatory T cells induced by single-peptide liposome immunotherapy suppress islet-specific T cell responses to multiple antigens and protect from autoimmune diabetes, J. Immunol 204 (2020) 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Sonigra A, Nel HJ, Wehr P, Ramnoruth N, Patel S, van Schie KA, Bladen MW, Mehdi AM, Tesiram J, Talekar M, Rossjohn J, Reid HH, Stuurman FE, Roberts H, Vecchio P, Gourley I, Rigby M, Becart S, Toes RE, Scherer HU, Le Cao KA, Campbell K, Thomas R, Randomized phase I trial of antigen-specific tolerizing immunotherapy with peptide/calcitriol liposomes in ACPA+ rheumatoid arthritis, JCI Insight 7 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mangal JL, Inamdar S, Le T, Shi X, Curtis M, Gu H, Acharya AP, Inhibition of glycolysis in the presence of antigen generates suppressive antigen-specific responses and restrains rheumatoid arthritis in mice, Biomaterials 277 (2021), 121079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Zampieri R, Brozzetti A, Pericolini E, Bartoloni E, Gabrielli E, Roselletti E, Lomonosoff G, Meshcheriakova Y, Santi L, Imperatori F, Merlin M, Tinazzi E, Dotta F, Nigi L, Sebastiani G, Pezzotti M, Falorni A, Avesani L, Prevention and treatment of autoimmune diseases with plant virus nanoparticles, Sci. Adv 6 (2020) eaaz0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Farrugia M, Baron B, The role of TNF-α in rheumatoid arthritis: A focus on regulatory T cells, J. Clin. Transl. Res 2 (2016) 84. [PMC free article] [PubMed] [Google Scholar]
- [134].Le Buanec H, Delavallee L, Bessis N, Paturance S, Bizzini B, Gallo R, Zagury D, Boissier MC, TNFalpha kinoid vaccination-induced neutralizing antibodies to TNFalpha protect mice from autologous TNFalpha-driven chronic and acute inflammation, PNAS 103 (2006) 19442–19447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].van Eden W, van der Zee R, Prakken B, Heat-shock proteins induce T-cell regulation of chronic inflammation, Nat. Rev. Immunol 5 (2005) 318–330. [DOI] [PubMed] [Google Scholar]
- [136].van Herwijnen MJ, Wieten L, van der Zee R, van Kooten PJ, Wagenaar-Hilbers JP, Hoek A, den Braber I, Anderton SM, Singh M, Meiring HD, van Els CA, van Eden W, Broere F, Regulatory T cells that recognize a ubiquitous stress-inducible self-antigen are long-lived suppressors of autoimmune arthritis, PNAS 109 (2012) 14134–14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].De Wolf C, Van Der Zee R, Den Braber I, Glant T, Maillère B, Favry E, van Lummel M, Koning F, Hoek A, Ludwig I, An arthritis-suppressive and treg cell-inducing CD4+ T cell epitope is functional in the context of HLA-restricted T cell responses, Arthritis Rheumatol. 68 (2016) 639–647. [DOI] [PubMed] [Google Scholar]
- [138].Koffeman EC, Genovese M, Amox D, Keogh E, Santana E, Matteson EL, Kavanaugh A, Molitor JA, Schiff MH, Posever JO, Bathon JM, Kivitz AJ, Samodal R, Belardi F, Dennehey C, van den Broek T, van Wijk F, Zhang X, Zieseniss P, Le T, Prakken BA, Cutter GC, Albani S, Epitope-specific immunotherapy of rheumatoid arthritis: clinical responsiveness occurs with immune deviation and relies on the expression of a cluster of molecules associated with T cell tolerance in a double-blind, placebo-controlled, pilot phase II trial, Arthritis Rheum. 60 (2009) 3207–3216. [DOI] [PubMed] [Google Scholar]
- [139].Baumgart DC, Sandborn WJ, Inflammatory bowel disease: clinical aspects and established and evolving therapies, Lancet 369 (2007) 1641–1657. [DOI] [PubMed] [Google Scholar]
- [140].Kappelman MD, Moore KR, Allen JK, Cook SF, Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population, Dig. Dis. Sci 58 (2013) 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Jairath V, Feagan BG, Global burden of inflammatory bowel disease, Lancet, Gastroenterol. Hepatol 5 (2020) 2–3. [DOI] [PubMed] [Google Scholar]
- [142].Nedelcu A, Mosteanu O, Pop T, Mocan T, Mocan L, Recent advances in nanoparticle-mediated treatment of inflammatory bowel diseases, Appl. Sci 11 (2021) 438. [Google Scholar]
- [143].Abed OA, Attlassy Y, Xu J, Han K, Moon JJ, Emerging nanotechnologies and microbiome engineering for the treatment of inflammatory bowel disease, Mol. Pharm 19 (2022) 4393–4410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Guan Q, Ma Y, Hillman C-L, Ma A, Zhou G, Qing G, Peng Z, Development of recombinant vaccines against IL-12/IL-23 p40 and in vivo evaluation of their effects in the downregulation of intestinal inflammation in murine colitis, Vaccine 27 (2009) 7096–7104. [DOI] [PubMed] [Google Scholar]
- [145].Guan Q, Ma Y, Hillman C-L, Qing G, Ma AG, Weiss CR, Zhou G, Bai A, Warrington RJ, Bernstein CN, Targeting IL-12/IL-23 by employing a p40 peptide-based vaccine ameliorates TNBS-induced acute and chronic murine colitis, Mol. Med 17 (2011) 646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Guan Q, Burtnick HA, Qing G, Weiss CR, Ma AG, Ma Y, Warrington RJ, Peng Z, Employing an IL-23 p19 vaccine to block IL-23 ameliorates chronic murine colitis, Immunotherapy 5 (2013) 1313–1322. [DOI] [PubMed] [Google Scholar]
- [147].Guan Q, Warrington R, Moreno S, Qing G, Weiss C, Peng Z, Sustained suppression of IL-18 by employing a vaccine ameliorates intestinal inflammation in TNBS-induced murine colitis, Future Sci. OA 5 (2019) FSO405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Brayman M, Thathiah A, Carson DD, MUC1: a multifunctional cell surface component of reproductive tissue epithelia, Reprod. Biol. Endocrinol 2 (2004) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Ryan SO, Vlad AM, Islam K, Garíepy J, Finn OJ, Tumor-associated MUC1 glycopeptide epitopes are not subject to self-tolerance and improve responses to MUC1 peptide epitopes in MUC1 transgenic mice, Biol. Chem 390 (2009) 611–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Beatty PL, Narayanan S, Garíepy J, Ranganathan S, Finn OJ, Vaccine against MUC1 antigen expressed in inflammatory bowel disease and cancer lessens colonic inflammation and prevents progression to colitis-associated colon cancer, Cancer Prev. Res 3 (2010) 438–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Beatty P, Ranganathan S, Finn OJ, Prevention of colitis-associated colon cancer using a vaccine to target abnormal expression of the MUC1 tumor antigen, Oncoimmunology 1 (2012) 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Elinav E, Waks T, Eshhar Z, Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice, Gastroenterology 134 (2008) 2014–2024. [DOI] [PubMed] [Google Scholar]
- [153].Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z, Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells, Mol. Ther 22 (2014) 1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Ma L, Hostetler A, Morgan DM, Maiorino L, Sulkaj I, Whittaker CA, Neeser A, Pires IS, Yousefpour P, Gregory J, Vaccine-boosted CAR T crosstalk with host immunity to reject tumors with antigen heterogeneity, Cell 186 (2023) 3148–3165.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Ma L, Dichwalkar T, Chang JY, Cossette B, Garafola D, Zhang AQ, Fichter M, Wang C, Liang S, Silva M, Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor, Science 365 (2019) 162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Bourgonje AR, Roo-Brand G, Lisotto P, Sadaghian Sadabad M, Reitsema RD, de Goffau MC, Faber KN, Dijkstra G, Harmsen HJ, Patients with inflammatory bowel disease show IgG immune responses towards specific intestinal bacterial genera, Front. Immunol 13 (2022), 842911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Shome M, Song L, Williams S, Chung Y, Murugan V, Park JG, Faubion W, Pasha SF, Leighton JA, LaBaer J, Serological profiling of Crohn’s disease and ulcerative colitis patients reveals anti-microbial antibody signatures, World J. Gastroenterol 28 (2022) 4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Armstrong H, Alipour M, Valcheva R, Bording-Jorgensen M, Jovel J, Zaidi D, Shah P, Lou Y, Ebeling C, Mason AL, Host immunoglobulin G selectively identifies pathobionts in pediatric inflammatory bowel diseases, Microbiome 7 (2019) 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Hevia A, López P, Suárez A, Jacquot C, Urdaci MC, Margolles A, Sànchez B, Association of levels of antibodies from patients with inflammatory bowel disease with extracellular proteins of food and probiotic bacteria, Biomed Res. Int 2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Garabatos N, Santamaria P, Gut microbial antigenic mimicry in autoimmunity, Front. Immunol 13 (2022), 873607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Papp M, Altorjay I, Dotan N, Palatka K, Foldi I, Tumpek J, Sipka S, Udvardy M, Dinya T, Lakatos L, New serological markers for inflammatory bowel disease are associated with earlier age at onset, complicated disease behavior, risk for surgery, and NOD2/CARD15 genotype in a Hungarian IBD cohort, Off. J. Am. Coll. Gastroenterol. ACG 103 (2008) 665–681. [DOI] [PubMed] [Google Scholar]
- [162].Olives J-P, Breton A, Hugot J-P, Oksman F, Johannet C, Ghisolfi J, Navarro J, Ćezard J-P, Antineutrophil cytoplasmic antibodies in children with inflammatory bowel disease: prevalence and diagnostic value, J. Pediatr. Gastroenterol. Nutr 25 (1997) 142–148. [DOI] [PubMed] [Google Scholar]
- [163].Targan SR, Landers CJ, Cobb L, MacDermott RP, Vidrich A, Perinuclear anti-neutrophil cytoplasmic antibodies are spontaneously produced by mucosal B cells of ulcerative colitis patients, J. Immunol. (baltimore, Md.) 155 (1995) (1950) 3262–3267. [PubMed] [Google Scholar]
- [164].Cohavy O, Bruckner D, Gordon L, Misra R, Wei B, Eggena M, Targan S, Braun J, Colonic bacteria express an ulcerative colitis pANCA-related protein epitope, Infect. Immun 68 (2000) 1542–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Vermeulen N, de Beeck KO, Vermeire S, Steen KV, Michiels G, Ballet V, Rutgeerts P, Bossuyt X, Identification of a novel autoantigen in inflammatory bowel disease by protein microarray, Inflamm. Bowel Dis 17 (2011) 1291–1300. [DOI] [PubMed] [Google Scholar]
- [166].Werner L, Paclik D, Fritz C, Reinhold D, Roggenbuck D, Sturm A, Identification of pancreatic glycoprotein 2 as an endogenous immunomodulator of innate and adaptive immune responses, J. Immunol 189 (2012) 2774–2783. [DOI] [PubMed] [Google Scholar]
- [167].Hase K, Kawano K, Nochi T, Pontes GS, Fukuda S, Ebisawa M, Kadokura K, Tobe T, Fujimura Y, Kawano S, Uptake through glycoprotein 2 of FimH+ bacteria by M cells initiates mucosal immune response, Nature 462 (2009) 226–230. [DOI] [PubMed] [Google Scholar]
- [168].Shpoliansky M, Roggenbuck D, Pinsker M, Salamon N, Weiss B, Shouval DS, Werner L, Antibodies against glycoprotein 2 are specific biomarkers for pediatric Crohn’s disease, Dig. Dis. Sci 66 (2021) 2619–2626. [DOI] [PubMed] [Google Scholar]
- [169].Derer S, Brethack A-K, Pietsch C, Jendrek ST, Nitzsche T, Bokemeyer A, Hov JR, Schäffler H, Bettenworth D, Grassl GA, Inflammatory bowel disease–associated GP2 autoantibodies inhibit mucosal immune response to adherent-invasive bacteria, Inflamm. Bowel Dis 26 (2020) 1856–1868. [DOI] [PubMed] [Google Scholar]
- [170].Smallwood TB, Giacomin PR, Loukas A, Mulvenna JP, Clark RJ, Miles JJ, Helminth immunomodulation in autoimmune disease, Front. Immunol 8 (2017) 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Tebeje BM, Harvie M, You H, Loukas A, McManus DP, Schistosomiasis vaccines: where do we stand? Parasit. Vectors 9 (2016) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Driss V, El Nady M, Delbeke M, Rousseaux C, Dubuquoy C, Sarazin A, Gatault S, Dendooven A, Riveau G, Colombel J, The schistosome glutathione S-transferase P28GST, a unique helminth protein, prevents intestinal inflammation in experimental colitis through a Th2-type response with mucosal eosinophils, Mucosal Immunol. 9 (2016) 322–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Capron M, Béghin L, Leclercq C, Labreuche J, Dendooven A, Standaert A, Delbeke M, Porcherie A, Nachury M, Boruchowicz A, Safety of P28GST, a protein derived from a schistosome helminth parasite, in patients with Crohn’s disease: a pilot study (ACROHNEM), J. Clin. Med 9 (2019) 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Thi THH, Priemel PA, Karrout Y, Driss V, Delbeke M, Dendooven A, Flament MP, Capron M, Siepmann J, Preparation and investigation of P28GST-loaded PLGA microparticles for immunomodulation of experimental colitis, Int. J. Pharm 533 (2017) 26–33. [DOI] [PubMed] [Google Scholar]
- [175].Dow CT, Proposing BCG Vaccination for Mycobacterium avium ss. paratuberculosis (MAP) associated autoimmune diseases, Microorganisms 8 (2020) 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Phanse Y, Wu C-W, Venturino AJ, Hansen C, Nelson K, Broderick SR, Steinberg H, Talaat AM, A protective vaccine against Johne’s disease in cattle, Microorganisms 8 (2020) 1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Polymeros D, Bogdanos DP, Day R, Arioli D, Vergani D, Forbes A, Does cross-reactivity between mycobacterium avium paratuberculosis and human intestinal antigens characterize Crohn’s disease? Gastroenterology 131 (2006) 85–96. [DOI] [PubMed] [Google Scholar]
- [178].Park KT, Allen AJ, Bannantine JP, Seo KS, Hamilton MJ, Abdellrazeq GS, Rihan HM, Grimm A, Davis WC, Evaluation of two mutants of Mycobacterium avium subsp. paratuberculosis as candidates for a live attenuated vaccine for Johne’s disease, Vaccine 29 (2011) 4709–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Abdellrazeq GS, Elnaggar MM, Bannantine JP, Schneider DA, Souza CD, Hwang J, Mahmoud AH, Hulubei V, Fry LM, Park K-T, A peptide-based vaccine for Mycobacterium avium subspecies paratuberculosis, Vaccine 37 (2019) 2783–2790. [DOI] [PubMed] [Google Scholar]
- [180].Riemekasten G, Humrich JY, Hiepe F, Antibodies Against ENA (Sm, RNP, SSA, SSB), Elsevier, 2019. [Google Scholar]
- [181].Wang X, Xia Y, Anti-double stranded DNA antibodies: origin, pathogenicity, and targeted therapies, Front. Immunol 10 (2019) 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Katarzyna P-B, Wiktor S, Ewa D, Piotr L, Current treatment of systemic lupus erythematosus: a clinician’s perspective, Rheumatol. Int (2023) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Chao Y-S, Adcock L, Belimumab treatment for adults with systemic lupus erythematosus: a review of clinical effectiveness, cost-effectiveness, and guidelines, (2018). [PubMed]
- [184].Schall N, Page N, Macri C, Chaloin O, Briand JP, Muller S, Peptide-based approaches to treat lupus and other autoimmune diseases, J. Autoimmun 39 (2012) 143–153. [DOI] [PubMed] [Google Scholar]
- [185].Schall N, Muller S, Resetting the autoreactive immune system with a therapeutic peptide in lupus, Lupus 24 (2015) 412–418. [DOI] [PubMed] [Google Scholar]
- [186].Wilhelm M, Wang F, Schall N, Kleinmann JF, Faludi M, Nashi EP, Sibilia J, Martin T, Schaeffer E, Muller S, Lupus regulator peptide P140 represses B cell differentiation by reducing HLA class II molecule overexpression, Arthritis Rheumatol. 70 (2018) 1077–1088. [DOI] [PubMed] [Google Scholar]
- [187].Muller S, Monneaux F, Schall N, Rashkov RK, Oparanov BA, Wiesel P, Geiger JM, Zimmer R, Spliceosomal peptide P140 for immunotherapy of systemic lupus erythematosus: results of an early phase II clinical trial, Arthritis Rheum. 58 (2008) 3873–3883. [DOI] [PubMed] [Google Scholar]
- [188].Zimmer R, Scherbarth HR, Rillo OL, Gomez-Reino JJ, Muller S, Lupuzor/P140 peptide in patients with systemic lupus erythematosus: a randomised, double-blind, placebo-controlled phase IIb clinical trial, Ann. Rheum. Dis 72 (2013) 1830–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Kang HK, Chiang MY, Liu M, Ecklund D, Datta SK, The histone peptide H4 71–94 alone is more effective than a cocktail of peptide epitopes in controlling lupus: immunoregulatory mechanisms, J. Clin. Immunol 31 (2011) 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Kaliyaperumal A, Mohan C, Wu W, Datta SK, Nucleosomal peptide epitopes for nephritis-inducing T helper cells of murine lupus, J. Exp. Med 183 (1996) 2459–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Kaliyaperumal A, Michaels MA, Datta SK, Naturally processed chromatin peptides reveal a major autoepitope that primes pathogenic T and B cells of lupus, J. Immunol 168 (2002) 2530–2537. [DOI] [PubMed] [Google Scholar]
- [192].Kaliyaperumal A, Michaels MA, Datta SK, Antigen-specific therapy of murine lupus nephritis using nucleosomal peptides: tolerance spreading impairs pathogenic function of autoimmune T and B cells, J. Immunol 162 (1999) 5775–5783. [PubMed] [Google Scholar]
- [193].Kang H-K, Michaels MA, Berner BR, Datta SK, Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsets, J. Immunol 174 (2005) 3247–3255. [DOI] [PubMed] [Google Scholar]
- [194].Shi Y, Kaliyaperumal A, Lu L, Southwood S, Sette A, Michaels MA, Datta SK, Promiscuous presentation and recognition of nucleosomal autoepitopes in lupus: role of autoimmune T cell receptor α chain, J. Exp. Med 187 (1998) 367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Lu L, Kaliyaperumal A, Boumpas DT, Datta SK, Major peptide autoepitopes for nucleosome-specific T cells of human lupus, J. Clin. Invest 104 (1999) 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Wardell CM, Levings MK, mRNA vaccines take on immune tolerance, Nat. Biotechnol 39 (2021) 419–421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No data was used for the research described in the article.