

Summary
Sickle cell disease (SCD) is an inherited haemoglobin disorder, associated with recurrent painful episodes, ongoing haemolytic anaemia and progressive multi-organ damage. Until the early 1990s, survival beyond the fourth decade for a patient with SCD was considered unusual and prompted case reports. Nowadays, in countries with developed health care systems, more than 90 percent of newborns with SCD survive into adulthood. Nevertheless, their life expectancy is still shortened by more than two decades compared to the general population. With an increasing life expectancy, SCD has now evolved into a debilitating disorder with substantial morbidity resulting from ongoing sickle cell vasculopathy and multi-organ damage. Limited data on health care issues of older adults with SCD poses multiple challenges to patients, their families and health care providers. In this review, we will address and discuss acute and chronic complications of SCD with a special focus on the older adult.
Keywords: Sickle cell disease, older adults, vasculopathy, multi-organ damage
INTRODUCTION, BACKGROUND AND SCOPE OF THE PROBLEM
With an estimate of 275,000 newborns every year, sickle cell disease (SCD) is considered a public health burden by the World Health Organization (WHO) and United Nations (UN).1 Two-thirds of newborns with SCD occur in sub-Saharan Africa where malaria is endemic. However, due to population movements, substantial numbers of children are now being born in countries where SCD is previously considered rare (e.g., northern and western Europe).2 Although the majority of children in Africa do not reach their fifth birthday, almost all newborns in well-resourced countries can now expect to survive to adulthood due to early diagnosis and better comprehensive treatment. Median survival for SCD has dramatically improved, exceeding 45–65 years depending on the sickle genotypes.3,4 Recent data showed that median survival for HbSS was 58 years and for HbSC was 66 years in the USA5 (Fig. 1), and 67 years for adults with HbSS/HbSβ0 for a single centre in the UK.6 Nevertheless, the life expectancy of an individual with SCD lags behind that of the general population by 20–30 years.7 By their fifth decade, half of the surviving patients will have suffered documented irreversible damage to one or more organs due to ongoing vasculopathy.8
Fig. 1.

Life expectancy of SCD. Improvement in survival of patients with SCD. Reproduced from Pleasants (Nature 2014)120 with permission.
SCD was first reported in the western literature in November 1910 by Dr James Herrick in a dental student from Grenada.9 He ascribed the anaemia to peculiar elongated and sickle shaped red cells. SCD became a poster child for ‘molecular disease’ when Linus Pauling suggested that it was caused by the presence of an abnormal haemoglobin,10 which was subsequently shown to arise from a single amino acid substitution (valine for glutamic acid) in the sixth codon of β chain,11 caused by adenine to thymine nucleotide substitution.12 As early as 1949, Neel suggested that the disorder followed an autosomal recessive pattern of inheritance.13
SCD is caused by the presence of haemoglobin S (HbS, ); the syndrome comprises different genotypes that include homozygous S (HbSS), compound heterozygous forms of HbSC and β-thalassaemia (HbSβ0 thalassaemia and HbSβ+ thalassaemia). In patients of African ancestry, HbSS is the most common genotype at 65–70%, followed by HbSC (about 30%), and the rest HbSβ thalassaemia.14 SCD is remarkably variable clinically. The central mechanism underlying the pathophysiology of SCD is polymerisation of deoxy-HbS and the formation of sickled cells, which is highly dependent on the intracellular concentration of HbS. Hence, it is not surprising that the most severe genotypes are HbSS and HbSβ0 thalassaemia as these genotypes have the highest concentration of intracellular HbS. Nevertheless, clinical variability is still observed within identical genotypes. There are two key genetic modifiers: an innate ability to produce fetal haemoglobin and co-inheritance of α-thalassaemia.15 Co-inheritance of α-thalassaemia (present in about one-third of patients of African descent)16 results in some beneficial as well some harmful effects.17 Reduced intracellular HbS, RBC density and haemolysis in patients who have co-inherited a-thalassaemia leads to a concomitant increase in haematocrit with a predisposition towards complications associated with microvascular occlusion such as acute chest syndrome (ACS), acute pain, retinopathy, and osteonecrosis.18,19 Such SCD-α-thalassaemia patients also have a reduced risk of complications associated with haemolytic anaemia such as pulmonary hypertension, cardiomyopathy, kidney disease, priapism and leg ulcers.20,21 Several studies have also demonstrated association of α-thalassaemia with lower transcranial Doppler (TCD) measurements and, hence, reduced risk for stroke.22–24 Co-existing a-thalassaemia also reduces bilirubin with a quantitative effect that is independent to that of the UGT1A1 promoter polymorphism.16 Co-inheritance of α-thalassaemia blunts the response to hydroxycarbamide therapy in SCD; this may be explained by its effect on fetal haemoglobin (HbF) levels and mean corpuscular volume (MCV), two key parameters associated with hydroxycarbamide response.
HbF levels vary from 1 to >25% in HbSS patients; this innate ability to produce HbF is determined by several genetic loci.25 Presence of a HbF determinant was suggested by correlation of clinical severity and HbF level with the different HbS β haplotypes—Benin (BEN), Central African Republic (CAR-Bantu speaking), Senegal (SEN) and ArabIndia (AI)—indicative of its region of origin in Africa, Middle East and the Indian subcontinent.26 Patients with SEN or AI types have the highest HbF levels and milder disease, while those with CAR haplotypes have lower HbF levels with generally more severe disease.27,28 HbF levels impact the ‘primary’ level of disease pathology, HbS polymerisation, thus HbF levels have a global beneficial effect. Indeed, in SCD, high HbF levels are a major predictor of survival,4,6 and reduced pain;29 conversely, low levels of HbF have been associated with increased risk of brain infarcts in young children.30 In Jamaicans, higher HbF levels and the absence of α-thalassemia appeared to predict a more benign disease.31
Although substantial advances and insights on the pathophysiology of SCD have been made since the discovery of its molecular basis, translation from this knowledge into treatment has been very slow. The only available diseasemodifying therapy, hydroxyurea (hydroxycarbamide), is still under-utilised.32 Allogeneic haematopoietic stem cell transplantation (HSCT) either from bone marrow or umbilical cord blood, a potentially disease-reversing therapy, has been used in a small percentage of patients, mostly children with severe symptoms (Fig. 2). Many unmet needs remain including better management of pain (the most common complication), improving anaemia and minimising organ damage that will in turn improve quality and expectancy of life.33 As the clinical outcome is so variable, better bio- and genetic markers are needed for disease severity prediction to inform prognosis and guide management.
Fig. 2.

Important clinical milestones in SCD.
PATHOPHYSIOLOGY AND MANIFESTATIONS OF SICKLE CELL DISEASE
Polymerisation of deoxygenated HbS and the formation of irreversibly sickled red blood cells lead to a downstream cascade of vaso-occlusion, release of cytokines and inflammatory factors.34 Subsequent reperfusion of the ischaemic tissue generates free radicals and reactive oxygen species, which scavenge nitric oxide (NO).35 Chronic NO deficiency can lead to platelet activation, increased vascular resistance, and endothelial dysfunction contributing to the development of vasculopathy.36,37 Ongoing haemolytic anaemia arises from the shortened lifespan of sickled erythrocytes of 16–20 days compared to normal of 120 days.38 Patients with more severe anaemia and haemolysis have reportedly higher incidence of pulmonary hypertension, gallstones, leg ulcers, priapism, and cerebrovascular diseases, compared to patients with higher haematocrits who are more likely to suffer from more frequent acute vaso-occlusive pain, osteonecrosis and acute chest syndrome39 (Fig. 3). The latter group of SCD patients are also likely to have co-inherited α-thalassaemia.
Fig. 3.
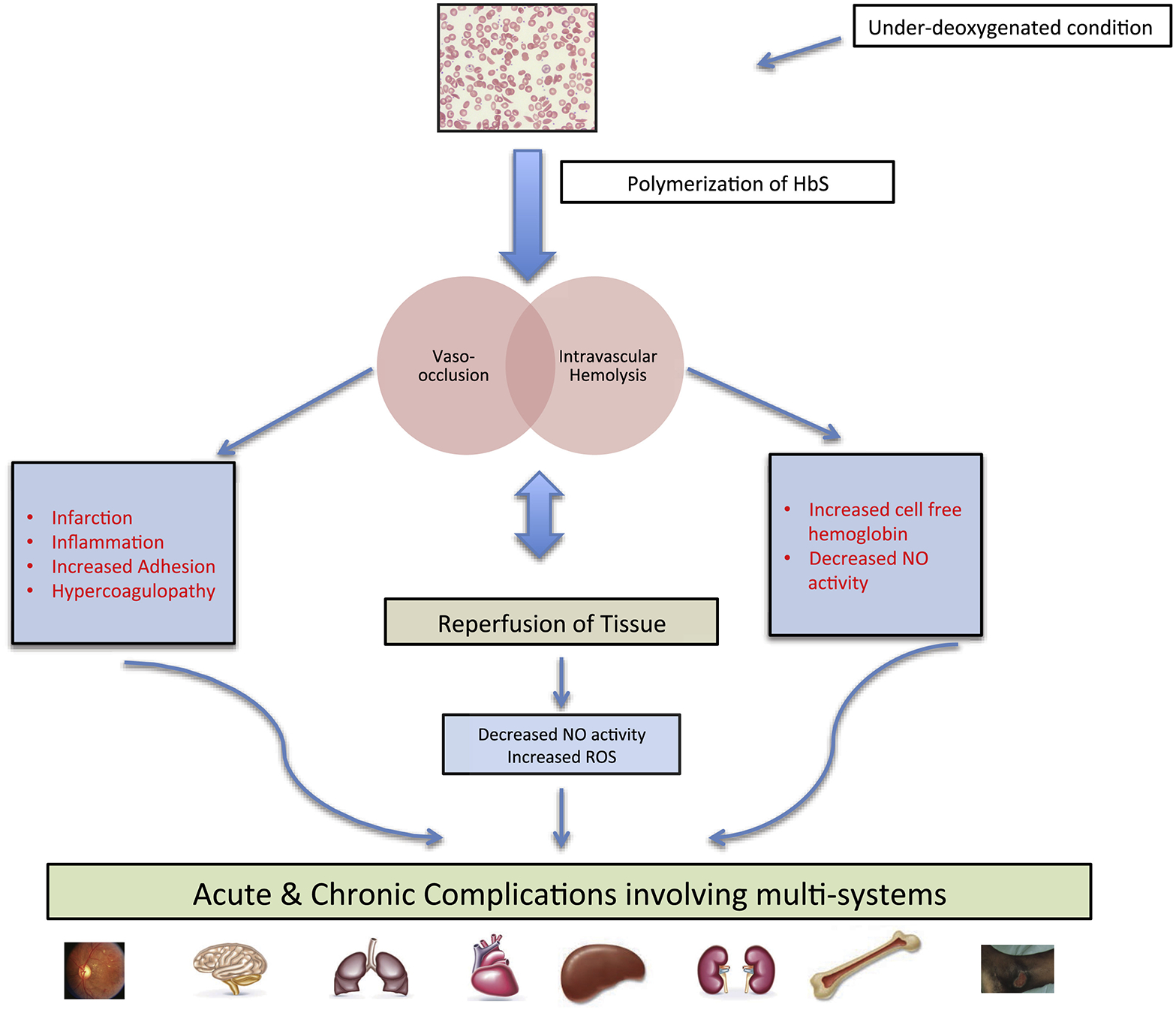
Pathophysiology and complications of SCD.
The ongoing vasoculopathy and inflammation inflicts damage on various organs, and impacts on the patients as they live into their fourth, fifth, and even sixth decade, transforming SCD into a chronic multisystem disorder. One of the largest and longest longitudinal studies of adult SCD showed that approximately one-half of surviving patients by their fifth decade had some form of irreversible damage of lungs, kidneys, brain, retina or bones, significantly affecting their quality of life.8 One should always bear in mind that the older adult with SCD could also have other co-morbidities that are not sickle-related, such as diabetes, systemic hypertension, and connective tissue disease, further compounding or accelerating the complexity of sickle-related complications.
COMPLICATIONS AND MANAGEMENT OF SICKLE CELL DISEASE IN OLDER ADULTS
Providing care for older adults with SCD can be challenging as little data or evidence-based guidelines are available. Recently, National Heart Lung and Blood Institute (NHLBI) published evidence-based guidelines that are based on pragmatic and expert consensus opinions rather than findings from randomised controlled trials.32,40 A lack of high quality evidence-based recommendations and relative rarity of the disease makes it difficult to identify health care professionals with expertise and experience to deliver care for individuals with acute and chronic complication of the disease.33
Individuals with SCD are at increased risk of acute or chronic organ damage related directly or indirectly to vaso-occlusion, haemolysis, progressive vasculopathy, and inflammation. These complications are seen in both children and adults with SCD although there are some differences in clinical course and severity depending on the patient’s age.
Pain
Acute sickle cell painful crisis is the hallmark and the most common cause of acute morbidity resulting from vaso-occlusion affecting bones and joints.30 It is the most common cause of emergency room visit and hospitalisation.41 Although the course of acute pain crisis is relatively similar in adults and children, there are considerable differences in the frequency and intensity of crisis between the two age groups. The Cooperative Study of Sickle Cell Disease (CSSCD) showed that young adults (25–29 years) have a higher frequency of crises: 1.2 pain episodes per year compared to a rate of 0.4 per year for children younger than 4 years of age.30 Healthcare Cost Utilization Project (HCUP) by Ballas and Lusardi indicated that the average length of stay in adults was approximately 7.5 days41 compared to 4.4 days in children.42 Management of acute sickle pain is supportive with hydration and analgesics. The role of hydroxyurea in prevention of painful crises will be discussed later in the review.
In addition to recurrent acute painful crises, older adults are more likely to have chronic pain which is usually multifactorial from inflammation, central and peripheral neural sensitisation and avascular necrosis of bone.43 A review on multiple dimensions of chronic pain in adults with SCD reported that chronic pain occurs in at least 29% of adults, most frequently in those 25–44 years of age.44 Chronic pain has a negative impact on quality of life45,46 and its management can be challenging, as a significant knowledge gap exists in understanding the natural history and management of chronic pain in older adults. Well-designed epidemiological and randomised control studies are needed to improve pain management.
Skeletal complications (sickle cell bone disease)
Patients with SCD are at risk of developing acute and chronic disorders of bone and joints. Bone pain arising from vaso-occlusive crisis, osteomyelitis and septic arthritis is the most common acute skeletal complication. Clinical manifestation of osteomyelitis and septic arthritis can be somewhat similar to acute painful crises. Therefore, it is sometimes difficult to make an appropriate diagnosis. The providers should have a high index of suspicion for this serious complication. The general principles for management of osteomyelitis and septic arthritis in SCD are similar to those in the general population. To date, no randomised controlled studies are available for the management of osteomyelitis and septic arthritis in both children and adults with SCD.47
Ongoing vasculopathy leads to chronic bone problems such as chronic arthritis, osteonecrosis/avascular necrosis (AVN) of bone, decreased bone growth and decreased bone mineral density (osteopenia/osteoporosis).48 While there is marrow hyperplasia and expansion compensating for haemolytic anaemia, repeated vaso-occlusion can result in infarction of bone marrow. This process can lead to replacement of red marrow with bone trabeculation and thinning of the cortex.48
AVN or aseptic necrosis arises from complete occlusion of circulation to the bones and bone infarction, although the pathogenesis is not clearly understood. Multiple factors such as vascular blockade, vessel occlusion, thrombosis, oedema, and progressive ischaemia of the microvasculature by sickled cells play a role.49 The femoral head is the most commonly affected site for AVN followed by the humeral head. The Cooperative Study of SCD reported that the estimated age at diagnosis of AVN was 28 years and age-specific prevalence rate was highest in patients who were over 45 years of age(34.9%).50 In contrast, the prevalence among patients under 25 years of age was approximately 6%.50 A recent study reported that the incidence of AVN in SCD could vary from 3% to 50%.51 Management of AVN can be challenging given limited evidence for standardised guidelines for most surgical procedures in SCD. Options include conservative measures (e.g., pain management and decreased weight bearing, physical therapy) or surgical management (surgical core decompression or arthroplasty). Early detection and intervention at an early stage is needed to delay progressive joint disease and improve quality of life. Total hip arthroplasty (THA) is usually reserved for patients with advanced AVN of the femur, given its failure rate. However, outcomes of THA in SCD have improved with optimisation of medical management, use of cementless prosthesis, and co-management by an experienced surgeon and haematologist during the preoperative and postoperative period.51,52
Pulmonary complications
Pulmonary complications account for significant morbidity and mortality in patients with SCD. Acute pulmonary complications include pneumonia, pulmonary embolism and acute chest syndrome (ACS). ACS, defined as a new radiodensity on chest radiograph accompanied by fever and/or respiratory symptoms, can affect all age groups and is the leading cause of death in patients with SCD.4 Although the incidence of ACS is lower in older adults compared to children (8.8 events/100 patient years in older adults versus 24.5 events/100 patient years in young children),53,54 the severity and mortality are higher in older adults, largely due to a higher incidence of bone marrow and fat emboli in adults55,56 and other co-morbidities.
Chronic pulmonary complications or sickle cell chronic lung disease (SCCLD), are more prevalent in older adults, and are characterised by impaired exercise tolerance, progressive heart failure, and impaired pulmonary function.57,58 Pulmonary hypertension (PH) defined by a mean arterial pressure ≥25 mmHg at rest, is the most common chronic pulmonary complication and a major cause of morbidity and mortality among older adults.59,60 Other forms of SCCLD include pulmonary fibrosis, restrictive airway disease and sleep disordered breathing (nocturnal hypoxaemia and obstructive sleep apnoea).61
Tricuspid regurgitant jet velocity (TRV) has been used as a non-invasive predictive measure of PH. TRV ≥2.5 m/s measured by 2D echocardiogram are suggestive of PH and ≥3 m/s are highly predictive of PH.62 PH as defined by echo studies ≥2.5 m/s) has a prevalence of 30–40% in HbSS and 10–28% in HbSC genotype.63–65 Three studies utilising right heart catheterisation have shown that the prevalence of PH in SCD is between 6% and 10%.64,66,67 Approximately 50% of patients with SCD-related PH have pre-capillary PH that is multifactorial in aetiology: endothelial injury from recurrent sickling, inflammation, chronic thromboembolism, and haemolysis-induced nitric oxide depletion.68–70 The rest have post-capillary PH arising from left ventricular dysfunction. Patients with pulmonary arterial hypertension, as defined by right heart catheterisations, tend to be older with poor functional capacity and higher levels of N-terminal pro-brain natriuretic peptide.64 The patients with signs and symptoms suggestive of PH should be screened with 2D echocardiogram and it is suggested that those with TRV 2.9 ≥m/s should be evaluated with a right heart catheterisation to confirm the diagnosis.
More recently, a prospective study of 122 consecutive stable adults with SCD using comprehensive 2D and 3D echocardiography suggest that the elevated cardiac output and left ventricular volume overload secondary to chronic anaemia may be the dominant factor responsible for abnormal cardiopulmonary haemodynamics in patients with SCD.71
There are limited data for optimal treatment of PH. Recommended management includes treatment of heart failure, correction of hypoxaemia with oxygen therapy, anticoagulation for those with thromboembolism, hydroxyurea or transfusion, multidisciplinary approach with haematologists and pulmonologists, and referral to specialised centres.70 Well-designed epidemiological studies are needed to study the incidence, prevalence and natural history of chronic pulmonary complications among older adults with SCD.
Renal complications (sickle cell nephropathy, SCN)
Patients with SCD are at risk of developing a spectrum of acute and chronic renal complications. Acute manifestations include microscopic or gross haematuria, urinary tract infection and acute kidney injury. One French study reported that acute kidney injury occurs in 4–10% of hospitalised patients with SCD and is more frequent in patients with acute chest syndrome and pulmonary hypertension.72
Common sickle-related renal complications include hyperfiltration, hyposthenuria (diminished concentrating ability), and albuminuria. In a small number of patients, renal function declines progressively leading to end-stage renal disease.73,74 Microalbuminuria is age-dependent and occurs in more than one-third of adults.21 One study showed that by the age of 55 years, 60% of patients with SCD were found to have microalbuminuria.21 Advanced chronic kidney disease (stage III–IV, defined as eGFR 15–60 mL/min per 1.73m2) occurs in 4–18% of adults with SCD.8,75 In 2009, Serjeant et al. reported that the prevalence of advanced kidney disease in patients over 60 years of age is approximately 24% and end-stage renal failure (ESRF) was the cause of death in 45% of patients aged 60 years or older.76
Few options are available for treatment of SCN. Current therapeutic recommendations are largely adopted from expert or consensus opinion. One should bear in mind that the older adult with SCD is also at risk of other diseases such as hypertension, diabetes, connective tissue disorder and other forms of glomerulonephritis. All of these conditions can have a significant impact on renal function. Early recognition of albuminuria is essential as it is a sensitive marker of glomerulopathy and chronic renal impairment. We suggest regular monitoring for proteinuria, haematuria and renal function during clinic attendance. Once patients develop proteinuria with a urinary protein to creatinine ratio (uPCR) of >50 mg/mmol (442 mg/g), persistent microscopic haematuria, or declining renal function (>10% fall in eGFR per year), they should be referred for joint specialist care (nephrologist and haematologist). When to start on angiotensin-converting enzyme (ACE) inhibitors or angiotensin receptor blockers (ARBs) is debatable. Some use a cutoff of uPCR ratio >50 mg/mmol while others prefers to wait until uPCR is >100 mg/mmol74 before commencing ACE inhibitors. Hydroxyurea therapy should be considered as recent studies indicate that it can decrease glomerular hyperfiltration and microalbuminuria, subsequently preventing the progression of renal dysfunction in SCD.77,78
Management of ESRF includes erythropoietin-stimulating agents (ESA), dialysis or renal transplant. The use of ESA can be effective, particularly in combination with hydroxyurea, in correcting anaemia.74,79 Ten-year survival for patients on dialysis is 14% compared to 56% for SCD after renal transplantation.80 Despite the poor prognosis of ESRF secondary to SCD, patients are less likely to receive a renal transplant.81 Exchange transfusion is recommended in preventing intra-renal sickling and promoting preservation of the transplanted kidney.73
Hepatobiliary complications
Sickle hepatopathy is heterogeneous in nature and has been used to describe the overlapping causes of liver dysfunction. Sickle hepatopathy can vary from mildly abnormal liver function tests and self-limited cholestasis to severe forms of sickle cell intrahepatic cholestasis (SCIC) and cirrhosis.82 It is important to recognise SCIS as it can be life threatening. It was first described in 195383 and clinically it comprises severe right upper quadrant pain, acute hepatomegaly, coagulopathy, extreme hyperbilirubinaemia (predominantly conjugated in contrast to unconjugated in haemolytic anaemia), and moderately elevated liver enzymes with occasional progression to acute hepatic failure.84
The overall prevalence of liver dysfunction in patients with SCD has not been well established. It is difficult to differentiate abnormal liver enzymes due to intrinsic liver disease from those resulting from haemolysis. One study reported that 10% of patients have abnormal liver dysfunction85 but another study by Johnson et al. showed that 30% of patients had abnormal liver biochemical tests during a 5-year follow-up period.86 The clinicopathological features can be further complicated by increased iron deposition in the reticuloendothelial system and Kupffer cells and/or viral hepatitis as a complication of cumulative red cell transfusions.87
Data on management of sickle hepatopathy is limited and not evidence based. The critical management is accurate identification and treatment of every underlying and coexisting condition that can contribute to liver dysfunction. Although limited treatment options are available, the exclusion of other causes of liver dysfunction, the use of iron chelating agents for patients with iron overload and treatment of underlying viral hepatitis are recommended.87 Use of hydroxyurea may improve the clinical course of SCD in general, thus reducing the need for blood transfusion. For patients with severe acute liver dysfunction syndromes, and progressive liver cholestasis, the use of exchange blood transfusion (EBT) should be considered along with supportive care to treat intrahepatic cholestasis and its associated coagulopathy.84 There are several case reports showing that prompt EBT can reverse a clinical course of acute SCIC. Liver transplantation may play a role in a subset of patients with acute hepatic crisis, liver failure or cirrhosis.87 We recommend that patients with evidence of hepatic damage be referred to centres where they can be co-managed by a haematologist and hepatologist. More importantly, future multi-centre collaborative studies are needed to better understand and define the natural history of hepatic complications of sickle cell disease and its management.
Neurological complications
Cerebrovascular accidents (CVA) are devastating complications affecting both children and older adults with SCD, and one of the leading causes of death in both children and adults.4,88 The 1978–1988 epidemiological study by the Cooperative Study of Sickle Cell Disease (CSSCD) reported that the incidence of stroke for HbSS disease was 0.61 per 100 patient.89 The study also showed that 24% of individuals with SCD experience a clinical stroke by age 45 years.89 The type of stroke varies with age. Ischaemic or thrombotic stroke is most common in children between the age of 2 and 9 years with a second peak in adults over the age of 29 years.89 Haemorrhagic stroke accounts for approximately one-third of the CVAs in SCD, being most frequent in the 20–29 age group.89 A recent study based on 1998–2007 California discharge statistics estimated that the rate of stroke in young adults (13–34 years) was 360/100,000 person years compared to 1160/100,000 among middle ages (35–64 years) and 4700/100,000 person years among elderly adults (65 years and older) with SCD.90
The majority of studies for CVAs in SCD are restricted to children and the evidence-based data for older adults are limited. The Stroke Prevention in Sickle Cell Anaemia (STOP) study demonstrated that continuous blood transfusion is effective in reducing CVA in children at high risk as identified by abnormal transcranial Doppler (TCD) ultrasonography.91 A follow-up study (STOP II) subsequently showed that discontinuing blood transfusion results in a high frequency of reversion to abnormal TCD velocities and stroke.92 Thus, the recommendation is to continue blood transfusion indefinitely as primary stroke prevention in children at risk as identified by TCD screening. As for management of acute ischaemic strokes in adult patients with SCD, the evidence is very limited. Older adults with acute ischaemic stroke should also be evaluated for other risk factors for stroke such diabetes mellitus, hyperlipidaemia, hypertension, atrial fibrillation, and cardiovascular anomaly (patent foramen ovale). Our preferred initial treatment for adult SCD patients presenting with ischaemic stroke is an exchange blood transfusion.93 The use of thrombolytics should be individualised and considered in older adults with ischaemic stroke if they meet the strict eligibility criteria. Secondary preventive strategies for strokes and silent cerebral infarcts include a regular monthly transfusion (preferably an exchange transfusion)93 and modification of non-sickle related risk factors. In younger adults, HSCT should be considered if suitable donors are available.
Neurocognitive impairment, whilst well established in children with SCD, also increases with age94 and can be compounded by other pathology such as silent cerebral infarcts, moya moya and overt clinical stroke throughout the SCD lifespan.95 Although neuropsychological data in adults with SCD is limited, abnormalities in frontal lobe blood flow, dementia and progressive encephalopathy with age have been well documented.96–98
Silent infarcts and endothelial dysfunction of SCD, in combination with ageing, are likely to predispose patients with SCD, particularly to vascular dementia.97 Treatment for these chronic neurocognitive issues is not so well established, however limited evidence suggests that hydroxyurea therapy in children can improve general cognitive ability, verbal reasoning and fluid reasoning.99
Venous thromboembolism (VTE)
Haemostatic abnormalities seen in SCD such as coagulation activation, endothelial dysfunction and vaso-occlusion can increase the risk of VTE.100–102 Recent studies by Naik et al. showed that the incidence rate of first VTE among patients aged ≥15 years with SCD was 5.2 events/1000 person years with a cumulative incidence rate of 7.4% by the age of 30 years and 11.3% by age of 40 years.103 Age in itself is considered an independent risk factor for VTE. Therefore, as patients with SCD live longer, we expect to see an increased incidence of VTE in SCD. There is still ongoing debate on the duration of treatment with anticoagulation for VTE and benefits of prophylactic anticoagulation in patients with SCD.104
IMPORTANCE OF HEALTH MAINTENANCE AND AVAILABLE TREATMENT OPTIONS
Health maintenance encompasses early recognition, prevention and treatment of organ damage, and should be an essential part of routine health care in older adults with SCD. Box 1 lists the recommended health maintenance for older adults with SCD.
Box 1. Recommended health maintenance and outpatient management of SCD in older adults.
Education for patients and care givers
Administration of appropriate immunisations
Annual screening for blood borne diseases (hepatitis C and HIV) in transfusion dependent patients
Screening and prevention of chronic complications including iron overload, cerebrovascular disease, pulmonary hypertension, hepatopathy, and nephropathy
Annual ophthalmology evaluation for retinopathy
Assessment of bone health and leg ulcers
Assessment of stress and depression
Assessment of pain and narcotic use
Assessment of steady-state blood results and physiological measurements (oxygen saturation and blood pressure)
The two most widely available therapies for patients with SCD are hydroxyurea and blood transfusion.
Hydroxyurea
As HbF is a potent inhibitor of intracellular polymerisation, therapeutic induction of HbF has been a major objective in the management of SCD. Pharmacological agents include cytotoxic drugs such as hypomethylating agents (5-azacytidine or decitabine), Ara-C, hydroxyurea, short chain amino acids and their analogues, erythropoietin and more recently histone deacetylase (HDAC) inhibitors such as vorinostat. Of these agents, hydroxyurea has the best safety profile with minimal side-effects105 and was approved by the USA Food and Drug Administration (FDA) in 1998 for treatment of adults with severe SCA. The primary mechanism of action of hydroxyurea in vivo is the reversible inhibition of ribonucleotide reductase (RR), a critical enzyme for DNA synthesis during S phase of the cell cycle.106 The exact molecular mechanism by which hydroxyurea increases HbF level is still not fully understood. In addition to increasing HbF levels, the clinical efficacy of hydroxyurea for SCD includes decreasing white blood cells, decreasing endothelial adhesion, decreasing degree of chronic inflammation and improving cell hydration.107–109 Despite its safety and efficacy, it is still underutilised among patients with SCD. In 2014, the NHLBI published new evidence-based guidelines for the management of patients with SCA and Box 2 lists the evidenced-based recommendations for hydroxyurea therapy.32
Box 2. Evidence-based recommendations for use of hydroxyurea in adults with SCD32.
Two or more moderate to severe crises during a 12-month period
Sickle cell-associated pain that interferes with daily activitiesand quality of life
History of recurrent or severe acute chest syndrome
Severe symptomatic anaemia that interferes with daily activity or quality of life
Patients with chronic kidnev disease who are on erythropoietin (adding hydroxyurea improves anaemia)
Note: Recommend discontinuing hydroxyurea in women who are pregnant or breast-feeding.
Transfusion therapy
Transfusion of red blood cells is one of the most effective therapies for patients with SCD. The rationale of blood transfusion in SCD is to: (a) improve oxygen carrying capacity of blood to tissues, and (b) dilute concentration of circulating sickled erythrocytes to improve microvascular circulation.110 Blood transfusion can be given intermittently for treatment of acute complications or in preparation for surgery, or long term to reduce the incidence and severity of sickle-related organ damage. Transfusion can be categorised as simple (or top-up) versus exchange transfusion, in which blood is removed either manually or by automated apheresis machine. Exchange transfusion is preferred to simple transfusion in clinical situations where the concentration of HbS needs to be reduced quickly without increasing blood viscosity, such as acute stroke or acute chest syndrome or preparation of major surgery.14 The evidence is limited in terms of the HbS% or number of transfusions needed for clinical efficacy. The decision whether to transfuse should be based on risks versus benefits of transfusion of red blood cells depending on the clinical situation, and availability. Commonly accepted indications for intermittent transfusion include severe symptomatic anaemia, acute multi-organ failure and severe vaso-occlusive crisis including acute chest syndrome.14 Although it is very effective in preventing several complications of SCD, transfusion therapy carries the risk of secondary iron overload, alloimmunisation and transmission of blood borne diseases such as hepatitis C. Therefore, long term transfusion therapy is usually reserved for stroke prevention and management, severe anaemia, and progressive/recurrent organ damage.14,111,112 A multicentre trial has been conducted to determine the role of short-term transfusion therapy in improving cognitive ability of older adults, however the results are awaiting.
EMERGING THERAPIES
To date, allogeneic HSCT is the only disease-reversing treatment modality for SCD.113 Although human leukocyte antigen (HLA)-matched related HSCT is widely accepted as a standard of care for children, it is still investigational for older adults. The commonly accepted indications for HSCT in adults include a history of cerebrovascular accident, recurrent vaso-occlusive crisis despite hydroxyurea therapy, osteonecrosis, red cell immunisation and recurrent acute chest syndrome. Experience with myeloablative HSCT in older adults with SCD is limited, as patients older than 16 years old were usually excluded given its treatment related toxicity. Unlike haematological malignancies, achieving complete myeloablation is not essential as mixed chimaerism of donor and recipient is sufficient to reverse clinical complications of the underlying genetic defect.114,115 Hence, investigators have explored non-myeloablative and less toxic conditioning regimens for allogeneic HSCT in older adults, and results are very positive and encouraging.116 However, allogeneic HSCT is still limited by lack of HLA-matched related or unrelated donors. Currently, the use of alternative donors such as haploidentical donors (siblings or parents) and cord blood are under investigation. Autologous HSCT after correction of genetic defect using genome-editing technique is another potential approach in curing sickle cell disease. It can overcome the limitation of finding a matched donor with a lower risk of morbidity such as graft versus host disease and graft rejection. Other gene therapy approaches include increasing fetal haemoglobin by genome-editing of γ globin gene117 or repressors of its expression, such as BCL11A.118,119
CONCLUSION
There has been a significant understanding of the pathophysiology and factors contributing to the severity of SCD over the past decade. Yet, many patients still face a lifetime of complications and management remains a challenge. As the life expectancy of patients with SCD has increased compared to their historical peers, there is an urgent need for largescaled randomised clinical controlled trials for older adults to obtain evidence-based management and effective therapeutic agents. Establishing an international database for SCD will be of great importance to understand the natural history and diverse heterogeneity of the disease.
Footnotes
Conflicts of interest and sources of funding: The authors state that there are no conflicts of interest to disclose.
References
- 1.Aygun B, Odame I. A global perspective on sickle cell disease. Pediatr Blood Cancer 2012; 59: 386–90. [DOI] [PubMed] [Google Scholar]
- 2.Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet 2013; 381: 142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamideh D, Alvarez O. Sickle cell disease related mortality in the United States (1999–2009). Pediatr Blood Cancer 2013; 60: 1482–6. [DOI] [PubMed] [Google Scholar]
- 4.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med 1994; 330: 1639–44. [DOI] [PubMed] [Google Scholar]
- 5.Elmariah H, Garrett ME, De Castro LM, et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol 2014; 89: 530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gardner K, Douiri A, Drasar E, et al. Survival in adults with sickle cell disease in a high-income setting. Blood 2016; 128: 1436–8. [DOI] [PubMed] [Google Scholar]
- 7.Lanzkron S, Carroll CP, Haywood C Jr. Mortality rates and age at death from sickle cell disease: US, 1979–2005. Public Health Rep 2013; 128: 110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powars DR, Chan LS, Hiti A, et al. Outcome of sickle cell anemia: a 4decade observational study of 1056 patients. Medicine 2005; 84: 363–76. [DOI] [PubMed] [Google Scholar]
- 9.Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Intern Med 1910; 6: 517–21. [PMC free article] [PubMed] [Google Scholar]
- 10.Pauling L, Itano HA, Singer SJ, et al. Sickle cell anemia: a molecular disease. Science 1949; 110: 543–8. [DOI] [PubMed] [Google Scholar]
- 11.Ingram VM. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature 1957; 180: 326–8. [DOI] [PubMed] [Google Scholar]
- 12.Goldstein J, Konigsberg W, Hill RJ. The structure of human hemoglobin: VI. The sequence of amino acids in the tryptic peptides of the b chain. J Biol Chem 1963; 238: 2016–27. [PubMed] [Google Scholar]
- 13.Neel JV. The inheritance of sickle cell anemia. Science 1949; 110: 64–6. [DOI] [PubMed] [Google Scholar]
- 14.Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet 2010; 376: 2018–31. [DOI] [PubMed] [Google Scholar]
- 15.Gardner K, Thein SL. Genetic factors modifying sickle cell disease severity. In: Costa FF, Conran N, editors. Sickle Cell Anemia – From Basic Science to Clinical Practice. Switzerland: Springer International, 2016; 371–97. [Google Scholar]
- 16.Vasavda N, Menzel S, Kondaveeti S, et al. The linear effects of alphathalassaemia, the UGT1A1 and HMOX1 polymorphisms on cholelithiasis in sickle cell disease. Br J Haematol 2007; 138: 263–70. [DOI] [PubMed] [Google Scholar]
- 17.Ballas SK. Effect of alpha-globin genotype on the pathophysiology of sickle cell disease. Pediatr Pathol Mol Med 2001; 20: 107–21. [PubMed] [Google Scholar]
- 18.Embury SH, Dozy AM, Miller J, et al. Concurrent sickle-cell anemia and alpha-thalassemia: effect on severity of anemia. N Engl J Med 1982; 306: 270–4. [DOI] [PubMed] [Google Scholar]
- 19.Embury SH, Clark MR, Monroy G, et al. Concurrent sickle cell anemia and alpha-thalassemia. Effect on pathological properties of sickle erythrocytes. J Clin Invest 1984; 73: 116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Embury SH, Hebbel RP, Mohandas N, et al. , editors. Sickle Cell Disease: Basic Principles and Clinical Practice. New York: Raven Press, 1994. [Google Scholar]
- 21.Day TG, Drašar ER, Fulford T, et al. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica 2012; 97: 201–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernaudin F, Verlhac S, Chevret S, et al. G6PD deficiency, absence of alpha-thalassemia, and hemolytic rate at baseline are significant independent risk factors for abnormally high cerebral velocities in patients with sickle cell anemia. Blood 2008; 112: 4314–7. [DOI] [PubMed] [Google Scholar]
- 23.Flanagan JM, Frohlich DM, Howard TA, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood 2011; 117: 6681–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cox SE, Makani J, Soka D, et al. Haptoglobin, alpha-thalassaemia and glucose-6-phosphate dehydrogenase polymorphisms and risk of abnormal transcranial Doppler among patients with sickle cell anaemia in Tanzania. Br J Haematol 2014; 165: 699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thein SL. Genetic modifiers of sickle cell disease. Hemoglobin 2011; 35: 589–606. [DOI] [PubMed] [Google Scholar]
- 26.Pagnier J, Mears JG, Dunda-Belkhodja O, et al. Evidence for the multicentric origin of the sickle cell hemogloin gene in Africa. Proc Natl Acad Sci USA 1984; 81: 1771–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagel RL, Rao SK, Dunda-Belkhodja O, et al. The hematologic characteristics of sickle cell anemia bearing the Bantu haplotype: the relationship between G gamma and HbF level. Blood 1987; 69: 1026–30. [PubMed] [Google Scholar]
- 28.Labie D, Pagnier J, Lapoumeroulie C, et al. Common haplotype dependency of high G gamma-globin gene expression and high Hb F levels in beta-thalassemia and sickle cell anemia patients. Proc Natl Acad Sci USA 1985; 82: 2111–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease: rates and risk factors. N Engl J Med 1991; 325: 11–6. [DOI] [PubMed] [Google Scholar]
- 30.Wang WC, Pavlakis SG, Helton KJ, et al. MRI abnormalities of the brain in one-year-old children with sickle cell anemia. Pediatr Blood Cancer 2008; 51: 643–6. [DOI] [PubMed] [Google Scholar]
- 31.Thomas PW, Higgs DR, Serjeant GR. Benign clinical course in homozygous sickle cell disease: a search for predictors. J Clin Epidemiol 1997; 50: 121–6. [DOI] [PubMed] [Google Scholar]
- 32.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA 2014; 312: 1033–48. [DOI] [PubMed] [Google Scholar]
- 33.Savage WJ, Buchanan GR, Yawn BP, et al. Evidence gaps in the management of sickle cell disease: a summary of needed research. Am J Hematol 2015; 90: 273–5. [DOI] [PubMed] [Google Scholar]
- 34.Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med 1997; 337: 762–9. [DOI] [PubMed] [Google Scholar]
- 35.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation 2004; 11: 129–51. [PubMed] [Google Scholar]
- 36.Reiter CD, Wang X, Tanus-Santos JE, et al. Cell-free hemoglobin limits nitric oxide bioavailability in sickle cell disease. Nat Med 2002; 8: 1383–9. [DOI] [PubMed] [Google Scholar]
- 37.Moncada S, Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol 2006; 176: 213–54. [DOI] [PubMed] [Google Scholar]
- 38.McCurdy PR, Sherman AS. Irreversibly sickled cells and red cell survival in sickle cell anemia: a study with both DF32P and 51CR. Am J Med 1978; 64: 253–8. [DOI] [PubMed] [Google Scholar]
- 39.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 2007; 21: 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.National Institutes of Health: National Heart Lung and Blood Institute. Evidence-Based Management of Sickle Cell Disease: NIH Expert Panel Report. Vol 161. Bethesda: US Department of Health and Human Services, 2014. [Google Scholar]
- 41.Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol 2005; 79: 17–25. [DOI] [PubMed] [Google Scholar]
- 42.Panepinto JA, Brousseau DC, Hillery CA, et al. Variation in hospitalizations and hospital length of stay in children with vaso-occlusive crises in sickle cell disease. Pediatr Blood Cancer 2005; 44: 182–6. [DOI] [PubMed] [Google Scholar]
- 43.Lanzkron S, Haywood C Jr. The five key things you need to know to manage adult patients with sickle cell disease. Hematology Am Soc Hematol Educ Program 2015; 2015: 420–5. [DOI] [PubMed] [Google Scholar]
- 44.Taylor LE, Stotts NA, Humphreys J, et al. A review of the literature on the multiple dimensions of chronic pain in adults with sickle cell disease. J Pain Symptom Manage 2010; 40: 416–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anie KA, Steptoe A, Bevan DH. Sickle cell disease: pain, coping and quality of life in a study of adults in the UK. Br J Health Psychol 2002; 7: 331–44. [DOI] [PubMed] [Google Scholar]
- 46.Ballas SK, Barton FB, Waclawiw MA, et al. Hydroxyurea and sickle cell anemia: effect on quality of life. Health Qual Life Outcomes 2006; 4: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marti-Carvajal AJ, Agreda-Perez LH. Antibiotics for treating osteomyelitis in people with sickle cell disease. Cochrane Database Syst Rev 2012; 12: CD007175. [DOI] [PubMed] [Google Scholar]
- 48.Almeida A, Roberts I. Bone involvement in sickle cell disease. Br J Haematol 2005; 129: 482–90. [DOI] [PubMed] [Google Scholar]
- 49.Osunkwo I An update on the recent literature on sickle cell bone disease. Curr Opin Endocrinol Diabetes Obesity 2013; 20: 539–46. [DOI] [PubMed] [Google Scholar]
- 50.Milner PF, Kraus AP, Sebes JI, et al. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med 1991; 325: 1476–81. [DOI] [PubMed] [Google Scholar]
- 51.Issa K, Naziri Q, Maheshwari AV, et al. Excellent results and minimal complications of total hip arthroplasty in sickle cell hemoglobinopathy at mid-term follow-up using cementless prosthetic components. J Arthroplasty 2013; 28: 1693–8. [DOI] [PubMed] [Google Scholar]
- 52.Jack CM, Howard J, Aziz ES, et al. Cementless total hip replacements in sickle cell disease. Hip Int 2016; 26: 186–92. [DOI] [PubMed] [Google Scholar]
- 53.Vichinsky EP, Styles LA, Colangelo LH, et al. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood 1997; 89: 1787–92. [PubMed] [Google Scholar]
- 54.Castro O, Brambilla DJ, Thorington B, et al. The acute chest syndrome in sickle cell disease: incidence and risk factors. The Cooperative Study of Sickle Cell Disease. Blood 1994; 84: 643–9. [PubMed] [Google Scholar]
- 55.Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med 2000; 342: 1855–65. [DOI] [PubMed] [Google Scholar]
- 56.Dang NC, Johnson C, Eslami-Farsani M, et al. Bone marrow embolism in sickle cell disease: a review. Am J Hematol 2005; 79: 61–7. [DOI] [PubMed] [Google Scholar]
- 57.Knight-Madden JM, Forrester TS, Lewis NA, et al. The impact of recurrent acute chest syndrome on the lung function of young adults with sickle cell disease. Lung 2010; 188: 499–504. [DOI] [PubMed] [Google Scholar]
- 58.Miller AC, Gladwin MT. Pulmonary complications of sickle cell disease. Am J Respir Crit Care Med 2012; 185: 1154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Castro LM, Jonassaint JC, Graham FL, et al. Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. Am J Hematol 2008; 83: 19–25. [DOI] [PubMed] [Google Scholar]
- 60.Castro O, Gladwin MT. Pulmonary hypertension in sickle cell disease: mechanisms, diagnosis, and management. Hematol Oncol Clin North Am 2005; 19: 881–96. vii. [DOI] [PubMed] [Google Scholar]
- 61.Sharma S, Efird JT, Knupp C, et al. Sleep disorders in adult sickle cell patients. J Clin Sleep Med 2015; 11: 219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aldrich TK, Dhuper SK, Patwa NS, et al. Pulmonary entrapment of sickle cells: the role of regional alveolar hypoxia. J Appl Physiol 1996; 80: 531–9. [DOI] [PubMed] [Google Scholar]
- 63.Gladwin MT, Sachdev V, Jison ML, et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N Engl J Med 2004; 350: 886–95. [DOI] [PubMed] [Google Scholar]
- 64.Parent F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med 2011; 365: 44–53. [DOI] [PubMed] [Google Scholar]
- 65.Ataga KI, Moore CG, Jones S, et al. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. Br J Haematol 2006; 134: 109–15. [DOI] [PubMed] [Google Scholar]
- 66.Mehari A, Gladwin MT, Tian X, et al. Mortality in adults with sickle cell disease and pulmonary hypertension. JAMA 2012; 307: 1254–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fonseca GH, Souza R, Salemi VM, et al. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J 2012; 39: 112–8. [DOI] [PubMed] [Google Scholar]
- 68.Bunn HF, Nathan DG, Dover GJ, et al. Pulmonary hypertension and nitric oxide depletion in sickle cell disease. Blood 2010; 116: 687–92. [DOI] [PubMed] [Google Scholar]
- 69.Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med 2008; 359: 2254–65. [DOI] [PubMed] [Google Scholar]
- 70.Gordeuk VR, Castro OL, Machado RF. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood 2016; 127: 820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mushemi-Blake S, Melikian N, Drasar E, et al. Pulmonary haemodynamics in sickle cell disease are driven predominantly by a high-output state rather than elevated pulmonary vascular resistance: a prospective 3-dimensional echocardiography/Doppler study. PloS One 2015; 10: e0135472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Audard V, Homs S, Habibi A, et al. Acute kidney injury in sickle patients with painful crisis or acute chest syndrome and its relation to pulmonary hypertension. Nephrol Dial Transplant 2010; 25: 2524–9. [DOI] [PubMed] [Google Scholar]
- 73.Sharpe CC, Thein SL. Sickle cell nephropathy a practical approach. Br J Haematol 2011; 155: 287–97. [DOI] [PubMed] [Google Scholar]
- 74.Sharpe CC, Thein SL. How I treat renal complications in sickle cell disease. Blood 2014; 123: 3720–6. [DOI] [PubMed] [Google Scholar]
- 75.Powars DR, Elliott-Mills DD, Chan L, et al. Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann Intern Med 1991; 115: 614–20. [DOI] [PubMed] [Google Scholar]
- 76.Serjeant GR, Serjeant BE, Mason KP, et al. The changing face of homozygous sickle cell disease: 102 patients over 60 years. Int J Lab Hematol 2009; 31: 585–96. [DOI] [PubMed] [Google Scholar]
- 77.Laurin LP, Nachman PH, Desai PC, et al. Hydroxyurea is associated with lower prevalence of albuminuria in adults with sickle cell disease. Nephrol Dial Transplant 2014; 29: 1211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aygun B, Mortier NA, Smeltzer MP, et al. Hydroxyurea treatment decreases glomerular hyperfiltration in children with sickle cell anemia. Am J Hematol 2013; 88: 116–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Steinberg MH. Erythropoietin for anemia of renal failure in sickle cell disease. N Engl J Med 1991; 324: 1369–70. [DOI] [PubMed] [Google Scholar]
- 80.Scheinman JI. Sickle cell disease and the kidney. Nat Clin Pract Nephrol 2009; 5: 78–88. [DOI] [PubMed] [Google Scholar]
- 81.Abbott KC, Hypolite IO, Agodoa LY. Sickle cell nephropathy at end-stage renal disease in the United States: patient characteristics and survival. Clin Nephrol 2002; 58: 9–15. [DOI] [PubMed] [Google Scholar]
- 82.Berry PA, Cross TJ, Thein SL, et al. Hepatic dysfunction in sickle cell disease: a new system of classification based on global assessment. Clin Gastroenterol Hepatol 2007; 5: 1469–76. [DOI] [PubMed] [Google Scholar]
- 83.Green TW, Conley CL, Berthrong M. The liver in sickle cell anemia. Bull Johns Hopkins Hosp 1953; 92: 99–127. [PubMed] [Google Scholar]
- 84.Gardner K, Suddle A, Kane P, et al. How we treat sickle hepatopathy and liver transplantation in adults. Blood 2014; 123: 2302–7. [DOI] [PubMed] [Google Scholar]
- 85.Schubert TT. Hepatobiliary system in sickle cell disease. Gastroenterology 1986; 90: 2013–21. [DOI] [PubMed] [Google Scholar]
- 86.Johnson CS, Omata M, Tong MJ, et al. Liver involvement in sickle cell disease. Medicine 1985; 64: 349–56. [DOI] [PubMed] [Google Scholar]
- 87.Ebert EC, Nagar M, Hagspiel KD. Gastrointestinal and hepatic complications of sickle cell disease. Clin Gastroenterol Hepatol 2010; 8: 483–9. [DOI] [PubMed] [Google Scholar]
- 88.Leikin SL, Gallagher D, Kinney TR, et al. Mortality in children and adolescents with sickle cell disease. Cooperative Study of Sickle Cell Disease. Pediatrics 1989; 84: 500–8. [PubMed] [Google Scholar]
- 89.Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998; 91: 288–94. [PubMed] [Google Scholar]
- 90.Strouse JJ, Jordan LC, Lanzkron S, et al. The excess burden of stroke in hospitalized adults with sickle cell disease. Am J Hematol 2009; 84: 548–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Adams RJ, McKie VC, Hsu L, et al. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial doppler ultrasonography. N Engl J Med 1998; 339: 5–11. [DOI] [PubMed] [Google Scholar]
- 92.Adams RJ. TCD in sickle cell disease: an important and useful test. Pediatr Radiol 2005; 35: 229–34. [DOI] [PubMed] [Google Scholar]
- 93.Kassim AA, Galadanci NA, Pruthi S, et al. How I treat and manage strokes in sickle cell disease. Blood 2015; 125: 3401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Steen RG, Fineberg-Buchner C, Hankins G, et al. Cognitive deficits in children with sickle cell disease. J Child Neurol 2005; 20: 102–7. [DOI] [PubMed] [Google Scholar]
- 95.Debaun MR, Armstrong FD, McKinstry RC, et al. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood 2012; 119: 4587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mackin RS, Insel P, Truran D, et al. Neuroimaging abnormalities in adults with sickle cell anemia: associations with cognition. Neurology 2014; 82: 835–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vermeer SE, Prins ND, den Heijer T, et al. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med 2003; 348: 1215–22. [DOI] [PubMed] [Google Scholar]
- 98.Anie KA. Psychological complications in sickle cell disease. Br J Haematol 2005; 129: 723–9. [DOI] [PubMed] [Google Scholar]
- 99.Puffer E, Schatz J, Roberts CW. The association of oral hydroxyurea therapy with improved cognitive functioning in sickle cell disease. Child Neuropsychol 2007; 13: 142–54. [DOI] [PubMed] [Google Scholar]
- 100.Ataga KI, Key NS. Hypercoagulability in sickle cell disease: new approaches to an old problem. Hematology Am Soc Hematol Educ Program 2007: 91–6. [DOI] [PubMed] [Google Scholar]
- 101.Lim MY, Ataga KI, Key NS. Hemostatic abnormalities in sickle cell disease. Curr Opin Hematol 2013; 20: 472–7. [DOI] [PubMed] [Google Scholar]
- 102.Naik RP, Streiff MB, Haywood C Jr, et al. Venous thromboembolism in adults with sickle cell disease: a serious and under-recognized complication. Am J Med 2013; 126: 443–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Naik RP, Streiff MB, Haywood C Jr, et al. Venous thromboembolism incidence in the Cooperative Study of Sickle Cell Disease. J Thromb Haemost 2014; 12: 2010–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.van Hamel Parsons V, Gardner K, Patel R, et al. Venous thromboembolism in adults with sickle cell disease: experience of a single centre in the UK. Ann Hematol 2016; 95: 227–32. [DOI] [PubMed] [Google Scholar]
- 105.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med 1995; 332: 1317–22. [DOI] [PubMed] [Google Scholar]
- 106.Elford HL. Effect of hydroxyurea on ribonucleotide reductase. Biochem Biophys Res Commun 1968; 33: 129–35. [DOI] [PubMed] [Google Scholar]
- 107.Bridges KR, Barabino GD, Brugnara C, et al. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood 1996; 88: 4701–10. [PubMed] [Google Scholar]
- 108.Benkerrou M, Delarche C, Brahimi L, et al. Hydroxyurea corrects the dysregulated L-selectin expression and increased H(2)O(2) production of polymorphonuclear neutrophils from patients with sickle cell anemia. Blood 2002; 99: 2297–303. [DOI] [PubMed] [Google Scholar]
- 109.Cokic VP, Smith RD, Beleslin-Cokic BB, et al. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J Clin Invest 2003; 111: 231–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ballas SK, Kesen MR, Goldberg MF, et al. Beyond the definitions of the phenotypic complications of sickle cell disease: an update on management. ScientificWorld Journal 2012; 2012: 949535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Josephson CD, Su LL, Hillyer KL, et al. Transfusion in the patient with sickle cell disease: a critical review of the literature and transfusion guidelines. Transfus Med Rev 2007; 21: 118–33. [DOI] [PubMed] [Google Scholar]
- 112.Quirolo K How do I transfuse patients with sickle cell disease? Transfusion 2010; 50: 1881–6. [DOI] [PubMed] [Google Scholar]
- 113.Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood 2007; 110: 2749–56. [DOI] [PubMed] [Google Scholar]
- 114.Walters MC, Patience M, Leisenring W, et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant 2001; 7: 665–73. [DOI] [PubMed] [Google Scholar]
- 115.Gaziev J, Lucarelli G. Stem cell transplantation for hemoglobinopathies. Curr Opin Pediatr 2003; 15: 24–31. [DOI] [PubMed] [Google Scholar]
- 116.Hsieh MM, Fitzhugh CD, Weitzel RP, et al. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA 2014; 312: 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Traxler EA, Yao Y, Wang YD, et al. A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 2016; 22: 987–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hoban MD, Orkin SH, Bauer DE. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood 2016; 127: 839–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mansilla-Soto J, Riviere I, Sadelain M. Genetic strategies for the treatment of sickle cell anaemia. Br J Haematol 2011; 154: 715–27. [DOI] [PubMed] [Google Scholar]
- 120.Pleasants S Epidemiology: a moving target. Nature 2014; 515: S2–3. [DOI] [PubMed] [Google Scholar]