

SUMMARY
Cyclin-dependent kinase 4 and 6 inhibitors (CDK4/6i) are key therapeutic agents in the management of metastatic hormone-receptor-positive breast cancer. However, the emergence of drug resistance limits their long-term efficacy. Here, we show that breast cancer cells develop CDK4/6i resistance via a sequential two-step process of E2F activation. This process entails retinoblastoma (Rb)-protein degradation, followed by c-Myc-mediated amplification of E2F transcriptional activity. CDK4/6i treatment halts cell proliferation in an Rb-dependent manner but dramatically reduces Rb-protein levels. However, this reduction in Rb levels insufficiently induces E2F activity. To develop CDK4/6i resistance, upregulation or activating mutations in mitogenic or hormone signaling are required to stabilize c-Myc levels, thereby augmenting E2F activity. Our analysis of pre-treatment tumor samples reveals a strong correlation between c-Myc levels, rather than Rb levels, and poor therapeutic outcomes after CDK4/6i treatment. Moreover, we propose that proteasome inhibitors can potentially reverse CDK4/6i resistance by restoring Rb levels.
In brief
Kim et al. demonstrate that breast cancer cells develop resistance to CDK4/6 inhibitors through a two-step E2F activation process involving Rb-protein degradation and c-Myc-mediated amplification of E2F activity. They suggest that proteasome inhibitors, which restore Rb levels, could potentially overcome this resistance.
Graphical Abstract
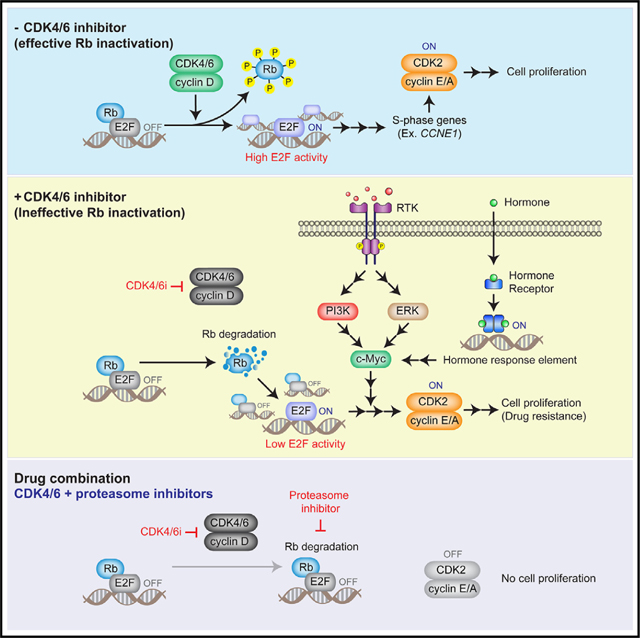
INTRODUCTION
The development and progression of cancer are closely linked to the dysregulation in cell-cycle entry, a complex and multifaceted process.1 Central to this regulation is the interaction between E2F transcription factors and the retinoblastoma (Rb) protein, which control the initiation of the cell cycle.2 During quiescence, Rb impedes cell-cycle entry by inhibiting E2Fs. This regulation is disrupted when cyclin-dependent kinases 4 and 6 (CDK4/6) become activated and phosphorylate Rb, liberating E2Fs to initiate the cell-cycle program, including CDK2 activators.3,4 In cancer, aberrant upregulation of CDK4/6 activity is frequently observed and leads to abnormal cell proliferation.5–7
CDK4/6 inhibitors (CDK4/6i) have been developed to block cell-cycle entry in tumor cells with a functional Rb pathway. Treatment with CDK4/6i in conjunction with endocrine therapy has significantly extended survival in hormone-receptor-positive (HR+)/human epidermal growth factor receptor 2-negative (HER2−) metastatic breast cancer, the most prevalent subtype.5,6,8–10 However, the efficacy of CDK4/6i is highly variable, and no biomarkers are available to predict treatment response.11,12 10%–20% of patients show primary resistance to CDK4/6i, and at least 30% of initially responsive patients develop drug resistance within 2 years.8,13 Moreover, CDK4/6i monotherapy is ineffective in treating patients with metastatic triple-negative breast cancer (TNBC), the most aggressive subtype.14 Therefore, elucidating the mechanisms of CDK4/6i resistance is essential for developing predictive biomarkers and novel therapeutic interventions.
Somatic mutations associated with CDK4/6i resistance, such as loss of FAT1 and PTEN and PIK3CA mutations, have been well characterized in patients with metastatic HR+/HER2− breast cancer.6,15–19 In addition to these genetic mechanisms, emerging evidence suggests that non-genetic mechanisms in small subsets of cancer cells induce drug adaptation, resulting in the emergence of drug-tolerant persister cells (persisters).20,21 These persisters cause residual cancer growth and the emergence of resistance mutations, contributing to major resistant clones. Furthermore, non-genetic mechanisms are clinically relevant, as 70% of patients with HR+/HER2− metastatic breast cancer who develop CDK4/6i resistance do not acquire any new somatic mutations.17 Non-genetic mechanisms of CDK4/6i resistance and the interplay between genetic and non-genetic mechanisms remain largely unknown.
Rb loss is the primary mechanism of CDK4/6i resistance documented in pre-clinical studies.5–7 However, recent studies showed that loss-of-function mutations in Rb are rare in HR+/HER2− breast cancers resistant to CDK4/6i (4.7%),17,22 raising the question of whether Rb loss is the primary mechanism of CDK4/6i resistance in the clinical setting. In addition to Rb loss, CDK4/6i resistance is frequently associated with upregulation of mitogenic signaling. Amplification of FGFR signaling23 and loss of PTEN16 are frequently observed in HR+/HER2− breast tumors resistant to CDK4/6i. Moreover, most mutations acquired in patients with breast cancer who develop CDK4/6i resistance occur in mitogenic or hormone signaling genes, such as PIK3CA, ESR1 (encoding estrogen receptor 1), FGFR1–3, and HER2.15–17,22,24 Nevertheless, patients with HR+/HER2− breast cancer respond to CDK4/6i irrespective of ESR1 mutation status,25,26 and PIK3CA mutations are present in both drug-sensitive and -resistant tumors.22 Consequently, it is unclear how alterations of mitogenic or hormone signaling factors promote CDK4/6i resistance.
In this study, we show that Rb-protein reduction and c-Myc, induced by mitogenic or hormone signaling, sequentially drive E2F transcriptional activity. This facilitates the development of persisters and ultimately CDK4/6i resistance in breast cancer. We also identify levels of c-Myc as a predictive biomarker of CDK4/6i efficacy in pre-treatment tumor samples and propose a potential therapeutic strategy to improve the long-term success of CDK4/6i in treating breast cancer.
RESULTS
CDK4/6i treatment induces Rb-protein reduction regardless of its levels
We investigated the development of CDK4/6i-tolerant persisters across four HR+/HER2− breast cancer (MCF-7, T47D, CAMA-1, and HCC1428) and three TNBC (BT-20, MDA-MB-231, and HCC1143) cell lines with a functional Rb pathway. Additionally, we used two TNBC cell lines lacking Rb expression (HCC1937 and MDA-MB-468) (Figure 1A). To evaluate persister development, we treated cells with the CDK4/6i palbociclib and monitored cell proliferation by classifying cell-cycle phases. Breast cancer cells lacking Rb expression continuously entered the cell cycle regardless of CDK4/6i treatment (Figures 1B, 1C, and S1A). In contrast, those with a functional Rb pathway nearly halted proliferation 1 day after CDK4/6i treatment (Figures 1D, 1E, S1B, and S1C). However, different levels of persisters (0%–15%) developed from Rb-intact breast cancer cell lines within 2 days of drug treatment. The variable CDK4/6i sensitivities in Rb-intact breast cancer cell lines were not correlated with expression levels of Rb protein in Figure 1A. These results indicate that while a functional Rb expression determines response to CDK4/6i, its levels do not predict the percentage of persisters in breast cancer cell lines.
Figure 1. CDK4/6i treatment reduces Rb-protein levels.

(A) Immunoblot showing Rb expression in breast cancer cell lines. Numbers represent relative Rb levels normalized by corresponding GAPDH levels and Rb levels in MCF-7 cells.
(B and D) Density scatterplots of Hoechst and EdU staining in Rb-mutant MDA-MB-468 (B) and MCF-7 (D) cells treated with palbociclib (1 μM) (n = 2,000 cells/condition).
(C and E) Percentage of S-phase cells after palbociclib (1 μM) treatment in breast cancer cells. Data are shown as means ± SD (n = 3 biological replicates).
(F) Immunoblot showing Rb expression in MCF-7 and MDA-MB-231 cells treated with palbociclib (1 μM).
(G) Percentage of S-phase cells after palbociclib (1 μM) treatment in Rb-mutant MDA-MB-468 cells with or without YFP-Rb overexpression. Data are shown as means ± SD (n = 3 biological replicates).
(H) Density scatterplots of Hoechst and EdU staining in MCF-7 cells overexpressing YFP-Rb and treated with palbociclib (1 μM) (n = 2,000 cells/condition).
(I) Percentage of S-phase cells after palbociclib (1 μM) treatment in MCF-7 and MDA-MB-231 cells with or without YFP-Rb overexpression. Data are shown as means ± SD (n = 3 biological replicates).
(J) Immunoblot showing expression of endogenous and exogenous Rb in MCF-7 and MDA-MB-231 cells treated with palbociclib (1 μM).
(K) Relative levels of endogenous and exogenous Rb protein in MCF-7 cells treated with palbociclib (1 μM). Solid lines indicate best fitted lines. The half-life of Rb protein is shown as means ± SD (n = 3 biological replicates).
(L) Relative mRNA levels of Rb and E2F target genes in MCF-7 cells treated with palbociclib (1 μM). Data are shown as means ± SD (n = 3 biological replicates).
See also Figures S1 and S2.
We examined the effect of CDK4/6 inhibition on Rb expression in MCF-7 and MDA-MB-231 cells, which effectively develop persisters. Palbociclib treatment reduced expression levels of Rb protein within 1 day in both cell lines (Figure 1F). To test whether overexpression of exogenous Rb prevents its reduction and the development of persisters, we stably introduced exogenous Rb conjugated to yellow fluorescent protein (YFP-Rb) using a lentiviral construct. Exogenous YFP-Rb was expressed 3- to 4-fold higher than endogenous Rb and did not perturb CDK4/6-dependent cell-cycle entry (Figures S2A and S2B). YFP-Rb expression restored sensitivity to CDK4/6i in Rb-mutant MDA-MB-468 cells (Figure 1G), indicating that it is functional. Despite the reduced percentage of persisters, both MCF-7 and MDA-MB-231 cells expressing YFP-Rb still developed persisters within 2 days of palbociclib treatment (Figures 1H, 1I, and S2C). Moreover, regardless of expression levels, palbociclib treatment reduced both endogenous and exogenous Rb with similar kinetics (Figures 1J, 1K, and S2D). Rb mRNA levels were constant, indicating that CDK4/6 inhibition induced Rb-protein reduction (Figure 1L). The levels of mRNAs encoded by E2F target genes were increased after the initial response to CDK4/6i treatment, suggesting that the Rb-protein reduction results in alternative E2F activation without CDK4/6 activity. However, E2F activity was not fully returned to pre-treatment levels over multiple days despite the Rb-protein reduction. These results demonstrate that CDK4/6i treatment dramatically reduces expression levels of Rb protein, but this Rb-protein reduction may lead to ineffective E2F activation.
Rb-protein reduction is associated with CDK4/6i resistance in vitro and in vivo
We next investigated the contribution of Rb-protein reduction to CDK4/6i-resistant cells established by chronic exposure to palbociclib. The half-maximal inhibitory concentrations (IC50s) of palbociclib in drug-resistant cells was higher than in drug-naive cells but similar to that observed in Rb-knockout cells generated using CRISPR-Cas9 (Figures 2A, S3A, and S3B). We found that Rb expression levels were reduced in multiple breast cancer cell lines resistant to palbociclib compared with drug-naive cells (Figures 2B and S3C). Similarly, we observed reduced Rb levels in breast cancer cell lines resistant to other CDK4/6i (abemaciclib and ribociclib) (Figure S3D). However, we found similar expression levels of Rb mRNA between drug-naive and -resistant cells but increased expression of E2F target genes in palbociclib-resistant cells (Figure 2C). These findings support the Rb-protein reduction in CDK4/6i-resistant cells and may explain results from previous studies showing high expression of the E2F target gene cyclin E in CDK4/6i-resistant cells and tissue samples.27,28
Figure 2. Reduced Rb-protein levels correlate with CDK4/6i resistance in vitro and in vivo.

(A) Dose-response curves for palbociclib treatment (48 h) in drug-naive, drug-resistant, and Rb-knockout MCF-7 cells. IC50 values are shown as means ± SD (n = 2 biological replicates).
(B) Immunoblot showing Rb expression in drug-naive and palbociclib-resistant MCF-7 and T47D cells.
(C) Relative mRNA levels of Rb and E2F target genes and MCM2 in drug-naive and palbociclib-resistant MCF-7 and T47D cells, determined by qRT-PCR. Data are shown as means ± SEM (n = 3 biological replicates).
(D) Representative immunostaining images of Hoechst, Rb, and EdU staining in drug-naive and palbociclib-resistant MCF-7 cells. Scale bar represents 50 μm.
(E) Histogram of Rb expression in drug-naive, Rb-knockout, and palbociclib-resistant MCF-7 cells (n > 15,000 cells/condition).
(F) Density scatterplots of Rb and EdU staining in drug-naive, Rb-knockout, and palbociclib-resistant MCF-7 cells (n = 5,000 cells/condition).
(G) Immunoblot showing Rb expression in drug-naive and palbociclib-resistant MCF-7 cells with continued palbociclib (1 μM) treatment and after withdrawal for over 1 week.
(H) Tumor volume over time in immunodeficient mice implanted with drug-naive MCF-7 cells. Data are shown as means ± SEM (n = 5 mice/condition).
(I) Representative images of Hoechst and Rb staining in tumor sections from MCF-7 xenograft mice. Scale bar represents 100 μm.
(J and K) Boxplots of percentage of Rb-positive cells (J) and relative Rb mRNA levels (K) in tumor sections. Blue lines indicate the median, boxes represent the 25th and 75th percentiles, and lines denote the total range for each population (n = 5 mice/condition). p values were calculated by two-tailed unpaired t tests (*p ≤ 0.05).
(L) Rb immunostaining in formalin-fixed, paraffin-embedded (FFPE) human tumors collected before and after treatment with CDK4/6i plus endocrine therapy. Scale bar represents 20 μm.
(M and N) Boxplots of Rb-protein H-scores (M) and relative Rb mRNA levels (N) in pre- and post-treatment FFPE samples. p values were calculated by two-tailed paired t tests (*p ≤ 0.05) (M: n = 4 paired samples; N: n = 3 paired samples).
See also Figure S3.
We examined the association of Rb expression with cell-cycle entry in drug-naive and -resistant as well as Rb-knockout MCF-7 cells. Rb expression in palbociclib-resistant cells showed a bimodal distribution where the lower and higher peaks were similar to its levels in Rb-knockout and drug-naive cells, respectively (Figures 2D and 2E). Moreover, while all drug-naive cells entered the cell cycle with high Rb expression, palbociclib-resistant cells mostly entered S phase (29% out of 31%) with low Rb levels (Figure 2F). We also found that the reduced expression levels of Rb in palbociclib-resistant MCF-7 and MDA-MB-231 cells were recovered to the levels in drug-naive cells by withdrawing CDK4/6i for over a week (Figures 2G and S3E). These results show that the Rb-protein reduction is induced by non-genetic mechanisms and is associated with CDK4/6i resistance in breast cancer cells.
To interrogate Rb-protein reduction in vivo, we orthotopically injected drug-naive MCF-7 cells into immunodeficient mice and treated them with either vehicle or palbociclib. Although palbociclib treatment initially decreased tumor volume, we observed tumor regrowth 15 days after drug treatment (Figure 2H). Once CDK4/6i-resistant tumors reached an average volume of 200 mm3, we examined Rb-protein and mRNA levels by immunostaining and qRT-PCR, respectively. Consistent with the in vitro results, we found a significant decrease in Rb-protein expression from mice treated with palbociclib compared with the control group (Figures 2I and 2J), while Rb mRNA levels were similar between the two conditions (Figure 2K). We also observed a similar pattern of Rb-protein reduction in paired pre- and post-treatment samples from patients with HR+/HER2− metastatic breast cancer, who developed resistance to the combined treatment of CDK4/6i and endocrine therapy. Our immunohistochemical analysis revealed a significant reduction in Rb-protein levels in post-treatment samples compared with pre-treatment samples, while Rb mRNA levels remained unchanged (Figures 2L–2N). Taken together, our results show that the Rb-protein reduction is associated with CDK4/6i resistance in breast cancer, both in vitro and in vivo, as well as in patient tumor specimens.
Targeting proteasome activity suppresses Rb-protein reduction and CDK4/6i resistance
To investigate the role of proteasome activity in Rb-protein reduction, we examined the impact of the proteasome inhibitor MG-132 on exogenous YFP-Rb levels in palbociclib-resistant MCF-7 cells. While MG-132 treatment did not alter YFP-Rb levels in drug-naive cells, it restored YFP-Rb levels in palbociclib-resistant cells (Figures 3A and 3B). Furthermore, co-treatment with palbociclib and another proteasome inhibitor, bortezomib, prevented the reduction of both endogenous and exogenous Rb protein in drug-naive MCF-7 and MDA-MB-231 cells (Figures 3C, 3D, and S4A–S4C). Inhibition of the autophagy-lysosomal pathway, another major protein degradation pathway in mammalian cells,29 with chloroquine did not prevent reduced Rb-protein levels mediated by CDK4/6i treatment. To test the potential involvement of the ubiquitin-proteasome pathway, we compared the restoration kinetics of Rb protein in palbociclib-resistant cells with a known substrate of the ubiquitin ligase anaphase-promoting complex/cyclosome (APC/C degron).30 The restoration of YFP-Rb to levels in drug-naive cells was about 12-fold slower than the increase of APC/C degron following proteasome inhibition (Figure 3B, S4D, and S4E). Our mass spectrometry analysis confirmed Rb dephosphorylation after palbociclib treatment but failed to detect any Rb ubiquitination, aligning with previously reports31,32 (Figure S4F). Moreover, we observed no difference in Rb ubiquitination between drug-naive and palbociclib-resistant MCF-7 cells (Figure S4G). These data suggest that proteasome activity may cause Rb degradation in a ubiquitin-independent manner.
Figure 3. A combination of CDK4/6 and proteasome inhibitors suppresses Rb-protein reduction and the cell growth of drug-resistant cells.

(A) Representative images of YFP-Rb in drug-naive and palbociclib-resistant MCF-7 cells before and after 48 h treatment with MG-132 (5 μM). Scale bar represents 100 μm.
(B) Averaged traces of YFP-Rb intensities following DMSO and MG-132 treatment. Data are shown as means ±95% CI (n > 4,000 cells/condition).
(C) Average traces of YFP-Rb intensities in drug-naive MCF-7 cells treated with combinations of palbociclib (1 μM), bortezomib, and chloroquine. Data are shown as means ±95% CI (n > 4,500 cells/condition).
(D) Relative endogenous Rb levels in drug-naive MCF-7 cells after treating with indicated drugs for 24 h. Data are shown as means ± SD (n = 3 biological replicates). p values were calculated by one-way ANOVA (*p ≤ 0.05; **p ≤ 0.001).
(E) YFP-Rb protein turnover. Drug-naive cells were treated with DMSO or palbociclib (1 μM) for 24 h before treatment with cycloheximide (10 μg/mL). Mean YFP-Rb levels were normalized with the initial and minimal values. Solid lines represent the best-fitted lines (n > 130 cells/condition).
(F) Heatmaps of single-cell traces for CDK2 activity and YFP-Rb intensity. Percentages indicate the proportion of proliferating (CDK2 activity >1.0 for over 2 h) and quiescent (no CDK2 activation) cells, classified based on CDK2 activity 15–30 h after treatment with cycloheximide.
(G) YFP-Rb protein turnover in proliferating and quiescent cells (n > 2,500 cells/condition).
(H) Turnover of wild-type or phosphomimetic Rb protein in Rb-knockout MCF-7. Cells were treated with palbociclib (1 μM) for 24 h before treatment with cycloheximide (10 μg/mL) (n > 130 cells/condition).
(I) Histogram of nuclear Rb, EdU, and cleaved caspase-3 staining in palbociclib-resistant MCF-7 cells 48 h after treatment with either DMSO or bortezomib (2 μM) (n > 1,100 cells/condition).
(J) Heatmaps of nuclear Rb levels, percentage of S-phase cells, and CDK2 activity. Palbociclib-resistant MCF-7 cells were treated with proteasome inhibitors (0–10 μM) for 48 h (n > 1,000 cells/condition).
(K and L) Growth curve of tumor volume in mice injected with palbociclib-resistant MCF-7 (K) or MDA-MB-231 (L) cells. Data are means ± SEM (K: control, n = 10 mice; bortezomib [bort], n = 8 mice; palbociclib [palbo] + fulvestrant [fulv], n = 9 mice; bort + palbo + fulv, n = 9 mice; L: control, n = 6 mice; bort, n = 5 mice; palbo, n = 4 mice; bort + palbo, n = 6 mice). p values were calculated by two-sided one-way ANOVA (*p ≤ 0.05; **p ≤ 0.001; ***p ≤ 0.0001).
(M) Representative images of Hoechst and Rb staining in tumor tissues. Scale bar represents 100 μm.
(N) Boxplot of Rb-positive-cell percentage from whole-tumor section (control, n = 10 mice; bort, n = 8 mice; palbo + fulv, n = 9 mice; bort + palbo + fulv, n = 8 mice). p values were calculated by two-tailed unpaired t tests (*p ≤ 0.05; ***p ≤ 0.0001).
To examine whether CDK4/6 inhibition affects Rb-protein stability, we employed the protein synthesis inhibitor cycloheximide. We observed an approximate 4-fold decrease in Rb-protein stability in MCF-7 cells treated with or resistant to CDK4/6i compared with those in the DMSO-treated condition (Figure 3E). By further classifying cells treated with cycloheximide alone into proliferation and quiescence based on CDK2 activity, we found that Rb protein was about 7-fold more stable in proliferating cells than in quiescent cells (Figures 3F and 3G). Furthermore, we alternatively inhibited CDK4/6 activity via contact inhibition and tracked CDK2 activity and YFP-Rb levels. While proliferating cells maintained high YFP-Rb levels, cells inactivating CDK2 by contact inhibition within the same population dramatically reduced YFP-Rb levels (Figures S4H and S4I). These results suggest a connection between Rb-protein stability and cell-cycle status, potentially regulated by the phosphorylation state of Rb protein. To test the impact of Rb phosphorylation on its stability, we mutated 14 CDK phosphorylation sites on Rb to glutamic acid and compared the stability of this Rb phosphomimetic protein with wild type. Prior to protein synthesis inhibition, cells were treated with CDK4/6i for 24 h to induce Rb dephosphorylation. We observed that the Rb phosphomimetic protein was approximately 4 times more stable than the wild-type Rb protein (Figure 3H). These data indicate that non-phosphorylated Rb protein in quiescent cells is unstable, leading to a proteasome-dependent reduction in Rb-protein levels.
We screened the impact of 24 proteasome inhibitors, including three FDA-approved inhibitors (bortezomib, ixazomib, and carfilzomib), on Rb-protein expression and cell proliferation using palbociclib-resistant MCF-7 cells. We measured endogenous Rb-protein expression and stained for EdU and cleaved caspase-3 to measure the percentages of S-phase cells and apoptotic cells, respectively. This analysis identified five proteasome inhibitors (bortezomib, ixazomib, carfilzomib, epoxomicin, and ONX0914) that effectively restored endogenous Rb expression, suppressed S-phase cells, and increased the apoptotic population in palbociclib-resistant MCF-7 cells (Figures 3I and S5A). Moreover, a relatively lower concentration of bortezomib in palbociclib-resistant MCF-7 cells most effectively restored endogenous Rb-protein expression, which negatively correlated with readouts of cell proliferation (percentages S-phase cells, CDK2 activity, and number of cells) (Figures 3J and S5B). CDK4/6 activity remained similar across different conditions, and proteasome inhibitors caused an accumulation of APC/C degron intensity, indicating effective inhibition of proteasome activity (Figure S5C). We next evaluated the therapeutic effect of proteasome and CDK4/6i in xenograft mouse models established by orthotopically injecting palbociclib-resistant MCF-7 and MDA-MB-231 cells into immunodeficient mice. We treated mice with palbociclib or bortezomib alone as well as in combination for 4–5 weeks. Considering that the standard treatment for HR+/HER− metastatic breast cancer is a combination of CDK4/6i with endocrine therapy, the estrogen receptor antagonist fulvestrant was incorporated into the treatment of MCF-7 xenografts. While bortezomib alone did not exhibit any anti-tumor effect, the combination of palbociclib and bortezomib significantly decreased the growth of palbociclib-resistant tumors compared with other treatments (Figures 3K, 3L, S5D, and S5E). Notably, the triple combination significantly increased the percentage of Rb-positive cells compared with the double combination of palbociclib and fulvestrant (Figures 3M and 3N). Furthermore, despite tumor growth, we observed increased percentages of Rb-expressing cells in the control (palbociclib withdrawal) and bortezomib-alone conditions compared with the double combination. These data suggest that both palbociclib withdrawal and bortezomib treatment can restore Rb levels in drug-resistant tumor cells. However, active CDK4/6 phosphorylates and inactivates Rb, thereby promoting tumor growth. This interpretation is supported by our observation of increased percentages of cells exhibiting Rb phosphorylation (S807/811) in conditions of both palbociclib withdrawal and bortezomib treatment compared with the triple-drug combination (Figure S5F and S5G). Collectively, our findings indicate that combining CDK4/6 and proteasome inhibitors suppresses the reduction of Rb protein and the growth of drug-resistant cells.
CDK4/6 inhibition generates heterogeneity in E2F and CDK2 activation kinetics
To investigate the emergence of persisters, we established MCF-7 cells that stably express sensors for CDK4/6,33 CDK2,34 and APC/C30 activities, as well as histone 2B (H2B) as a nuclear marker for cell tracking. In combination with single-cell tracing methods,34,35 these sensors allow us to monitor three distinct steps that underlie cell commitment to entering the cell cycle. The CDK4/6 and CDK2 sensors shuttle between the nucleus and cytoplasm, depending on their phosphorylation by the relevant kinase (Figure 4A). The E3 ubiquitin ligase APC/C is inactivated near the G1/S transition, resulting in the accumulation of the APC/C degron. Proliferating cells induced continuous CDK4/6 activation, followed by gradual CDK2 activation and APC/C inactivation for cell proliferation36 (Figure 4B). In contrast, cells inactivating CDK4/6 subsequently entered quiescence (G0) (Figure 4B). By tracking over 1,000 individual breast cancer cells, we found that 84% of drug-naive cells continuously activated CDK4/6 and CDK2 and inactivated APC/C (Figure 4C). Palbociclib treatment rapidly suppressed CDK4/6 activity and halted proliferation in all cells within 24 h (Figure 4D). However, when we classified cells based on CDK2 activation, 28% of MCF-7 cells adapted to CDK4/6 inhibition within 48 h of drug treatment. Given the Rb-protein reduction within 24 h of CDK4/6i treatment in Figure 1J, these results indicate that persisters arise from subsets of breast cancer cells following the Rb-protein reduction.
Figure 4. CDK4/6i treatment creates heterogeneity in E2F and CDK2 activation kinetics.
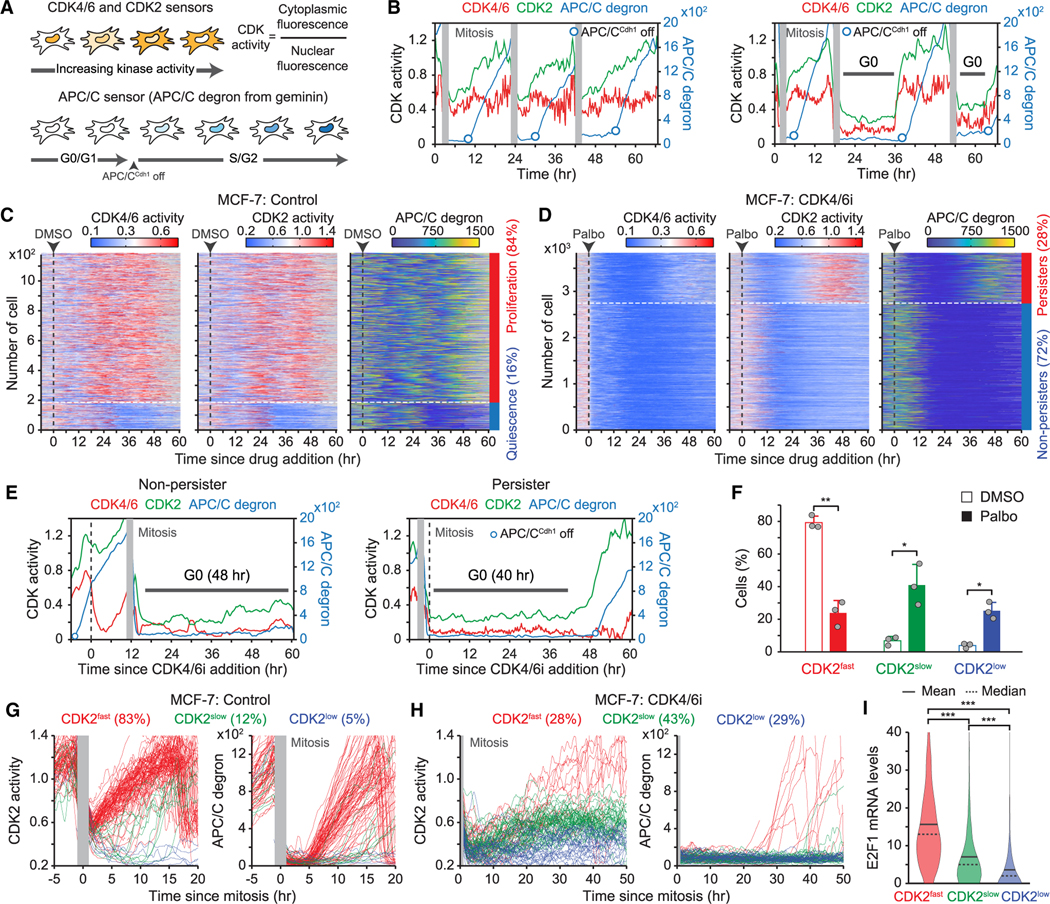
(A) Schematic diagram of live-cell sensors for CDK4/6, CDK2, and APC/C activities.
(B) Representative single-cell traces for CDK4/6 and CDK2 activities and APC/C degron intensity.
(C and D) Heatmaps of single-cell traces for CDK4/6 and CDK2 activities and APC/C degron intensity in MCF-7 cells treated with either DMSO (C) or palbociclib (1 μM) (D). Percentages indicate the proportion of persisters (CDK2 activity >1.0 for over 2 h) and non-persisters (no CDK2 activation) based on CDK2 activity 30–60 h post-treatment.
(E) Representative single-cell traces for CDK4/6 and CDK2 activities and APC/C degron intensity in MCF-7 cells treated with palbociclib (1 μM).
(F) Percentage of MCF-7 cells after DMSO or palbociclib treatment. Cells were classified into CDK2fast (CDK2 activity >1 for over 2 h), CDK2slow (CDK2 activity >0.6 for over 2 h), and CDK2low (no CDK2 activation) based on CDK2 activation kinetics 30–60 h post-treatment. Data are shown as means ± SD (n = 3 biological replicates). p values were calculated by two-tailed paired t test (*p ≤ 0.05; **p ≤ 0.001).
(G and H) Single-cell traces for CDK2 activity and APC/C degron intensity aligned to the end of mitosis in the control (G) and the palbociclib (1 μM)-treated (H) conditions.
(I) E2F1 mRNA level in MCF-7 cells classified based on CDK2 activation kinetics after palbociclib (1 μM) treatment (n > 800 cells/condition). p values were calculated by one-way ANOVA (***p ≤ 0.0001).
See also Figure S6.
Since CDK4/6 is required for initiation of the cell cycle in the G1 phase, our single-cell trace showed that CDK4/6 inhibition in proliferating cells with high CDK2 activity led to cell-cycle arrest after mitosis (Figure 4E). However, persisters exhibited CDK2 activation and subsequent APC/C inactivation without CDK4/6 reactivation after a period of quiescence. Moreover, we observed increased heterogeneity in CDK2 activation kinetics upon CDK4/6 inhibition. With further classification of cells based on kinetics of CDK2 activation as fast (CDK2fast), slow (CDK2slow), or none (CDK2low), we found that CDK2slow cells were rare (9%) in drug-naive cells but that they significantly increased (44%) with CDK4/6i treatment (Figures 4F, S6A, and S6B). Both CDK2fast and CDK2slow cells had fast CDK2 activation kinetics before drug treatment (Figure S6B), suggesting that CDK4/6 inhibition increased the percentage of CDK2slow cells. We next in-silico-aligned single-cell traces by the end of mitosis to compare CDK2 activation kinetics between control- and CDK4/6i-treated conditions. After mitosis, 83% of drug-naive cells immediately induced CDK4/6 and CDK2 activation, followed by APC/C inactivation, and completed cell-cycle progression within 20 h (Figures 4G and S6C). In contrast, CDK4/6-treated breast cancer cells exhibited heterogeneous CDK2 activation kinetics and timing of APC/C inactivation following an initial period of quiescence (Figure 4H). CDK2 activation kinetics and cell-cycle progression in CDK4/6i-treated cells were notably slower than in drug-naive cells. Given that CDK2 activators are regulated by E2F activity, we tested whether the variability in CDK2 activation kinetics might be due to heterogeneity in E2F activity. By performing live-cell followed by fixed-cell analysis, we monitored CDK2 activity and the corresponding mRNA levels of E2F target genes, E2F1 and Cdc25A, using mRNA fluorescence in situ hybridization. Upon categorizing cells based on their CDK2 activation kinetics, we found a significant correlation between the expression levels of E2F1 and Cdc25A and the classification of CDK2 activation kinetics (Figures 4I, S6D, and S6E). Our data demonstrate that CDK4/6i treatment leads to slow and variable kinetics for E2F and CDK2 activation.
Following Rb-protein reduction, other mechanisms are required to develop persisters
We hypothesized the possible existence of two distinct pathways that inactivate Rb (Figure 5A). The first pathway involves active CDK4/6, which initiates effective Rb inactivation through phosphorylation, thereby triggering high E2F activity. The second pathway, which becomes evident in the presence of CDK4/6i, leads to a reduction in Rb-protein levels. However, this Rb-protein reduction may cause incomplete inactivation, resulting in either low or variable E2F activity. We first examined whether variable CDK2 activation kinetics are caused by different levels of Rb-protein reduction. To monitor Rb levels and CDK2 activation during persister development, we stably introduced YFP-Rb and the CDK2 reporter into Rb-knockout MCF-7 cells. Additionally, we specifically measured the post-translational regulation of Rb by linking YFP-Rb to iRFP-tagged H2B using a 2A site (Figure 5B). This 2A site mediates the separation of H2B-iRFP from YFP-Rb during protein translation, resulting in a strong correlation between H2B-iRFP and YFP-Rb expression levels in individual MCF-7 cells (Figure S7A). Upon classifying cells based on CDK2 activation kinetics, we observed that 97% of drug-naive cells, identified as CDK2fast cells, maintained constant H2B-iRFP and YFP-Rb expression levels (Figure 5C). However, palbociclib treatment halted proliferation and dramatically reduced YFP-Rb levels without affecting H2B-iRFP levels within 24 h (Figure 5D). This led to a decreased correlation between H2B-iRFP and YFP-Rb intensities (Figure 5E). Furthermore, CDK4/6i treatment initially decreased YFP-Rb levels across CDK2fast, CDK2slow, and CDK2low cells with similar kinetics (Figure 5F). These results suggest that rather than different levels of Rb-protein reduction, other factors cause variable CDK2 activation kinetics and the fates of persisters versus non-persisters. Single-cell measurements further revealed the restoration of YFP-Rb in CDK2fast cells, indicating that Rb protein can be stabilized through CDK2-mediated Rb phosphorylation. In contrast, reduced YFP-Rb levels in CDK2low cells were not restored and decreased even further. Moreover, cells entering a quiescent state reduced YFP-Rb levels even in the absence of CDK4/6i (Figure S7B). This further supports that non-phosphorylated Rb is unstable and leads to Rb-protein reduction.
Figure 5. Breast cancer cells entering quiescence induces Rb-protein reduction.
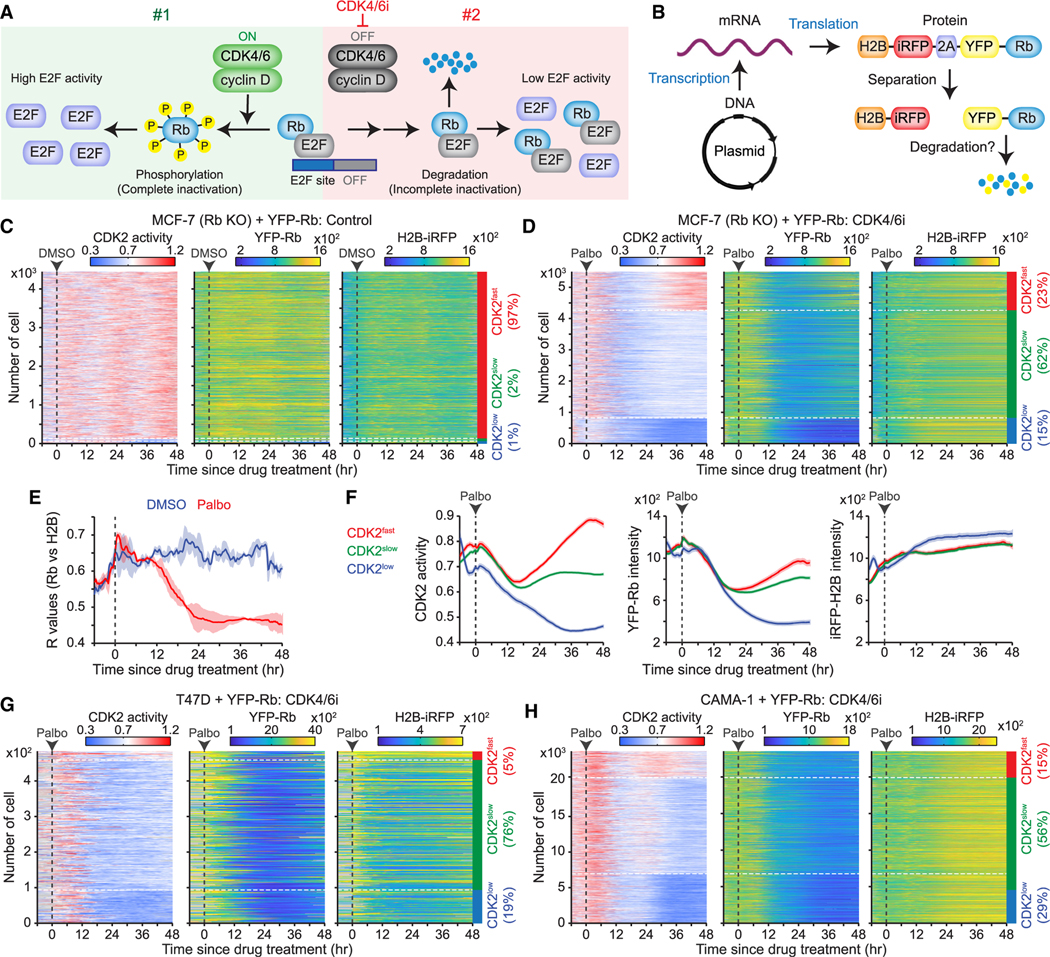
(A) Schematic representation of our hypothesis showing two Rb inactivation pathways by active CDK4/6 (#1) and proteolytic degradation of Rb (#2).
(B) Schematic illustration of a plasmid-encoded construct designed to assess post-translational Rb degradation.
(C and D) Heatmaps of single-cell traces for CDK2 activity and YFP-Rb and H2B-iRFP intensities over time. Rb-knockout MCF-7 cells expressing the H2B-iRFP-p2a-YFP-Rb construct treated with either DMSO (C) or palbociclib (1 μM) (D). CDK2fast (CDK2 activity >0.8 for over 2 h), CDK2slow (CDK2 activity >0.6 for over 2 h), and CDK2low (no CDK2 activation) cells were classified based on CDK2 activity 30–48 h post-treatment.
(E) Averaged correlation (R) values between YFP-Rb and H2B-iRFP intensities following DMSO or palbociclib (1 μM) treatment. Data are shown as means ± SD (n = 2 biological replicates).
(F) Average traces of CDK2 activity and YFP-Rb and H2B-iRFP intensities, based on the CDK2 classification after palbociclib (1 μM) treatment (n > 800 cells/condition).
(G and H) Heatmaps of single-cell traces for CDK2 activity and YFP-Rb and H2B-iRFP intensities in T47D (G) and CAMA-1 (H) cells treated with palbociclib (1 μM).
See also Figure S7.
We found similar results that palbociclib treatment initially halted cell proliferation and reduced YFP-Rb levels with similar kinetics regardless of the persister fate in MDA-MB-231, T47D, and CAMA-1 cells (Figures 5G, 5H, and S7C–S7F). This reduction in Rb-protein levels, followed by drug adaptation and variable CDK2 activation kinetics, is a general feature of breast cancer cells with different sensitivities to CDK4/6i. Together, these data suggest that following Rb-protein reduction, other factors may enhance E2F activity to promote the development of CDK4/6i-tolerant persisters.
In the presence of CDK4/6i, mitogenic signaling regulates downstream of the Rb pathway to promote persisters
We next examined the role of mitogenic signaling, such as the PI3K and ERK pathways, in the development of CDK4/6i-tolerant persisters. We assessed responses to the combined targeting of the PI3K or the ERK pathway with CDK4/6i in wild-type and Rb-knockout MCF-7 cells expressing sensors for CDK2 and CDK4/6 activities and cell-cycle phases using a Cdt1 degron.37 The Cdt1 degron is rapidly degraded during the S phase, thereby marking G1/S and S/G2 transitions (Figure S8A and S8B). Most wild-type (95%) and Rb-knockout (98%) MCF-7 cells continuously activated CDK4/6 and CDK2, initiating the S phase (Figure S8C and S8D). Treatment with palbociclib halted cell proliferation in wild-type, but not in Rb-knockout, cells (Figure 6A, S8E, and S8F), confirming that Rb knockout alone is sufficient to induce CDK4/6i adaptation. However, the combination of palbociclib with either the PIK3CA inhibitor alpelisib (PIK3CAi) or the panPI3K inhibitor pictilisib (PI3Ki) significantly suppressed the development of persisters in both cell types (Figures 6A–6C). The MEK inhibitor trametinib (MEKi) was less effective in MCF-7 cells that harbor the activating PIK3CA mutation (E545K).38 Additionally, the activation profiles of the PI3K and ERK pathways varied across breast cancer cell lines (Figure S8G). For instance, while MCF-7 cells primarily activated the PI3K pathway, MDA-MB-231 cells predominantly induced ERK pathway activation (Figure 6D). Consequently, PI3Ki effectively suppressed persister development in MCF-7 cells, whereas MEKi was more efficient in MDA-MB-231 cells (Figure 6E). We also observed a correlation between reduced c-Myc levels, induced by mitogenic signaling39,40 and associated with CDK4/6i resistance,41,42 and the therapeutic efficacy in MCF-7 and MDA-MB-231 cells (Figure 6F). Moreover, drug combinations led to a pronounced reduction in Rb protein, despite the synergistic inhibition of persisters (Figure 6G). Our results demonstrate that the PI3K and/or ERK pathways regulate downstream of the Rb pathway and c-Myc levels to promote CDK4/6i-tolerant persisters.
Figure 6. Under treatment with CDK4/6i, mitogenic signaling facilitates persister development by regulating downstream of the Rb pathway.

(A) Percentage of persisters treated with indicated drugs in wild-type and Rb-knockout MCF-7 cells. Data are shown as means ± SD (n = 3 biological replicates). p values were calculated by one-way ANOVA (*p ≤ 0.05; **p ≤ 0.001; ***p ≤ 0.0001).
(B and C) Heatmaps of single-cell traces for CDK4/6 and CDK2 activities in wild-type (B) and Rb-knockout (C) MCF-7 cells treated with palbociclib (1 μM) in combination with pictilisib (250 nM). The percentages mark the proportion of persisters (CDK2 activity >1.0 for over 2 h) and non-persisters (no CDK2 activation) cells classified based on CDK2 activity during 30–48 h after drug treatment.
(D) Immunoblot showing phosho-AKT, total-AKT, phospho-ERK, and total-ERK expression in MCF-7 and MDA-MB-231 cells.
(E) Percentage of S-phase cells. Cells were treated with palbociclib (1 μM) alone or in combination with trametinib (100 nM) or pictilisib (250 nM) for 2 days. Data are shown as means ± SD (n = 3 biological replicates).
(F) Violin plots of c-Myc levels in cells treated with indicated drugs for 2 days (n > 6,000 cells/condition).
(G) Immunoblot showing Rb and GAPDH expression in MCF-7 and MDA-MB-231 cells treated with indicated drugs for 2 days.
(K) Density scatterplot of Hoechst and EdU staining. Wild-type and Rb-knockout MCF-7 cells resistant to palbociclib were treated with DMSO and pictilisib (250 nM) for 2 days (n = 1,000 cells/condition).
(I–K) Percentage of S-phase cells (I), c-Myc levels (J), and EU levels (K) in wild-type and Rb-knockout MCF-7 cells resistant to palbociclib. Data are shown as means ± SD (n = 3 biological replicates).
(E, F, and I–K) p values were calculated by one-way ANOVA (*p ≤ 0.05; **p ≤ 0.001; ***p ≤ 0.0001).
See also Figures S8 and S9.
We investigated the impact of endocrine therapy on c-Myc levels and persister development. Given that c-Myc augments the expression of actively transcribed genes,43–45 we also measured the global mRNA transcription rate using 5-ethynyl uridine (EU). In combination with palbociclib, fulvestrant treatment showed a synergistic effect, significantly reducing the development of persisters in HR+/HER2− breast cancer cell lines (Figure S9A). Additionally, fulvestrant treatment led to a decrease in c-Myc levels and the global mRNA transcription rate (Figures S9B and S9C). These results indicate that the addition of endocrine therapy could effectively impede the development of CDK4/6i-tolerant persisters by regulating c-Myc levels.
We evaluated the effects of PI3K inhibition on the growth of CDK4/6i-resistant MCF-7 cells. Additionally, we employed Rb-knockout cells to test whether Rb expression is required for the synergistic effect of PI3Ki. Consistent with the therapeutic effects of drug combinations on persister development, PI3Ki treatment effectively suppressed cell proliferation in both wild-type and Rb-knockout MCF-7 cells resistant to palbociclib (Figures 6H, 6I, and S9D). Furthermore, PI3K inhibition significantly reduced c-Myc levels and the global mRNA transcription rate in both cell types (Figures 6J, 6K, S9E, and S9F). These results suggest that targeting mitogenic signaling inhibits CDK4/6i resistance by regulating c-Myc and mRNA transcription downstream of the Rb pathway.
The sequential induction of E2F activity by Rb-protein reduction and c-Myc promotes the development of CDK4/6i-tolerant persisters
We hypothesized that after the decrease in Rb-protein levels, mitogenic or hormone signaling stabilizes c-Myc levels to enhance E2F transcriptional activity, thereby promoting persister development. Using a doxycycline-inducible approach, we evaluated the effect of c-Myc induction on Rb-protein reduction, E2F activity, and the development of CDK4/6i-tolerant persisters. After treating MCF-7 cells with palbociclib combined with either DMSO or doxycycline, we monitored CDK4/6 and CDK2 activity as well as Cdt1 degron. While palbociclib treatment still led to cell-cycle arrest within 24 h, the induction of c-Myc significantly increased the percentage of persisters (Figures 7A–7C). Conversely, knockdown of c-Myc or cyclin E1/2 decreased the percentage of persisters (Figures S10A–S10D). Moreover, we found that palbociclib treatment initially caused cell-cycle arrest and reduced Rb-protein levels and E2F activity within 24 h regardless of c-Myc induction, despite an elevated global transcription rate (Figures 7D–7F, S10E, and S10F). However, c-Myc induction significantly increased the percentage of persisters and the expression of E2F target genes following the initial response to CDK4/6i treatment (Figures 7E, 7F, and S10F). To further investigate whether heterogeneity in c-Myc levels correlates with CDK2 activation kinetic, we performed live-cell imaging followed by fixed-cell analysis. After classifying palbociclib-treated cells based on CDK2 activation kinetics (Figure S10G), we found a significant correlation between these classifications and levels of c-Myc protein (Figure S10H). As a control, we also confirmed that the expression levels of the E2F target PCNA protein, but not β-actin, positively correlated with the classification of CDK2 activation kinetics. Additionally, we observed elevated c-Myc levels in palbociclib-resistant cells compared with drug-naive cells (Figure S10I). These results demonstrate that the sequential induction of E2F activity by Rb-protein reduction and c-Myc-mediated amplification of transcriptional activity facilitates the development of CDK4/6i-tolerant persisters.
Figure 7. c-Myc enhances E2F activity to facilitate persister development following Rb-protein reduction.

(A and B) Heatmaps of single-cell traces for CDK4/6 and CDK2 activities and Cdt1 degron intensity. MCF-7 cells expressing the c-Myc inducible construct were treated with palbociclib (1 μM) plus either DMSO (A) or doxycycline (5 μM) (B).
(C) Percentage of persisters. Data are shown as means ± SD (n = 3 biological replicates). p values were calculated using two-tailed paired t test (**p ≤ 0.001).
(D‒F) Rb and GAPDH expression (D), percentage of S-phase (E), and relative mRNA expression of E2F target genes (F) in MCF-7 cells expressing an inducible c-Myc construct. Cells were treated with palbociclib (1 μM) plus either DMSO or doxycycline (5 μM) for indicated time. Data are shown as means ± SD (n = 3 biological replicates). p values were calculated using two-tailed paired t test (*p ≤ 0.05; **p ≤ 0.001).
(G) Kaplan-Meier plots of PFS based on c-Myc genetic amplification status. The p value was calculated by the log-rank test.
(H) Representative images of pre-treatment tissue samples showing high c-Myc expression (H-score > 130) or low c-Myc expression (H-score ≤ 130). Scale bar represents 50 μm.
(I) Kaplan-Meier plots evaluating PFS in relation to c-Myc protein expression levels. p value was calculated by the log-rank test.
See also Figure S10 and S11.
Genetic amplification and levels of c-Myc are predictive biomarkers of CDK4/6i efficacy
We next investigated the potential of c-Myc as a predictive biomarker for CDK4/6i efficacy in patients with HR+/HER2− metastatic breast cancer. We analyzed the relationship between c-Myc genetic amplification in pre-treatment tumor samples and progression-free survival (PFS) among 468 patients who received first-line CDK4/6i and endocrine therapy with available MSK-IMPACT data. Of these patients, 56 (12%) exhibited c-Myc amplifications. Our analysis revealed that c-Myc amplifications in pre-treatment tumor samples were associated with poor PFS (univariate hazard ratio 1.50, 95% confidence interval [CI] 1.08–2.07, p = 0.015) (Figure 7G). This association remained significant after adjustment for endocrine therapy partner (multivariate hazard ratio 1.53, 95% CI 1.11–2.13, p = 0.009) (Figure S11A). Since mitogenic signaling exclusively regulates c-Myc levels post-translationally,39,40 we also assessed the relationship between c-Myc protein expression and PFS. We obtained pre-treatment tissues samples from patients with HR+/HER2− metastatic breast cancer who received CDK4/6i and endocrine therapy as first-line (n = 33) or second- or later-line (n = 15) treatment, along with matched clinical data from a separate patient cohort at Columbia University Medical Center. We calculated H-scores for c-Myc expression and established 130 as the cutoff value based on the H-score distribution. Samples were classified into high (H-score > 130) and low (H-score ≤ 130) c-Myc expression (Figure 7H). We found that high c-Myc expression in pre-treatment tumor samples was significantly associated with poor PFS (univariate hazard ratio 2.76, 95% CI 1.32–5.78, p = 0.0005) (Figure 7I). Additionally, we found no significant correlation between Rb levels and CDK4/6i-treatment outcomes (univariate hazard ratio 0.8, 95% CI 0.41–1.56, p = 0.49) (Figure S11B and S11C). These findings show that both DNA amplification status and protein expression levels of c-Myc are predictive of CDK4/6i response in patients with HR+/HER2− metastatic breast cancer.
DISCUSSION
Our results establish a fundamental mechanistic role for post-translational regulation of Rb-protein levels and c-Myc-mediated transcriptional activity in the development of CDK4/6i tolerance and resistance in breast cancer. Previous studies showed that CDK2 activation drives CDK4/6i resistance in breast cancer cells.27,42,46,47 However, the alternative inactivation of Rb, leading to E2F and CDK2 activation in the absence of CDK4/6 activity, remained elusive. Our study reveals that although functional Rb expression is required for CDK4/6i treatment to halt cell proliferation, the treatment results in a substantial loss of Rb protein. This provides an alternative means to initiate Rb inactivation and E2F activation without CDK4/6 activity. Nonetheless, these cells retain low levels of E2F transcriptional activity, suggesting that the Rb-protein reduction alone is insufficient for fully unleashing E2F function. Instead, amplification or acquisition of activating mutations in mitogenic or hormone signaling is necessary to overcome low E2F activity and develop CDK4/6i resistance in HR+ breast cancer. These findings elucidate how amplification or acquisition of activating mutations in mitogenic and hormone signaling genes facilitates CDK4/6i resistance.15–17,22–24 Moreover, these non-genetic mechanisms of CDK4/6i resistance may explain how 70% of patients with HR+/HER− metastatic breast cancer develop resistance to CDK4/6i without acquiring any new driver mutations.17 The Rb-protein reduction may also support pre-clinical studies indicating that Rb loss is the primary mechanism of CDK4/6i resistance5–7 and clinical studies revealing the rare occurrence of Rb mutations in patients with HR+/HER2− breast cancer (4.7%).17,22
Numerous potential biomarkers of CDK4/6i resistance have been evaluated in preclinical and clinical studies, including Rb loss, p16 loss, PIK3CA mutation, FAT1 mutation, FGFR activity, CDK6 expression, cyclin D1 expression, and CCNE amplification.11,12,48 However, reliable biomarkers for CDK4/6i efficacy remain unavailable for patients with breast and other cancers.11,12 Our results indicate that after Rb-protein reduction, mitogenic signaling is necessary to upregulate c-Myc expression, which globally enhances transcriptional activity, including E2F activity. Since breast cancer cells equally reduce Rb-protein levels following CDK4/6i treatment, the development of persisters depends on c-Myc levels. Thus, we propose that c-Myc is a biomarker capable of predicting CDK4/6i efficacy. Considering that c-Myc stability is exclusively regulated at the post-translational level,39,40 its protein levels may be a more accurate predictor than genetic amplification status or mRNA levels. Our findings align with a recent study demonstrating a correlation between phosphorylated AKT (p-AKT) levels and poor PFS in patients with HR+/HER2− metastatic breast cancer treated with combined CDK4/6i and endocrine therapy.36 However, as breast cancer cells activate PI3K and/or ERK pathways, c-Myc protein may predict CDK4/6i efficacy more reliably than either p-AKT or p-ERK alone. According to the Cancer Genome Atlas, TNBC expresses higher levels of c-Myc than other breast cancer subtypes.41 This may also explain why CDK4/6i monotherapy is ineffective in TNBC treatment.
Approximately 40% of patients with HR+/HER2− breast cancer have activating mutations in PIK3CA.49 Co-targeting the PI3K pathway and CDK4/6 synergistically prevents drug resistance in HR+/HER2− breast cancer and TNBC.36,50 However, unexpected toxicities with combinations of CDK4/6i and PI3Ki or mTOR inhibitors severely limit clinical success.5 A key question is how to maximize the effects of CDK4/6i-based combinations while minimizing their toxicities. Proteasome inhibitors have shown promising pre-clinical and clinical outcomes in treating various types of cancer, particularly multiple myeloma.51 Our pre-clinical data suggest that the combination of proteasome and CDK4/6i can restore Rb expression and suppress the persistence of CDK4/6i resistance in breast cancer xenograft mouse models. Since c-Myc is an estrogen-responsive gene,52 our findings indicate that endocrine therapy suppresses CDK4/6i resistance by downregulating c-Myc levels. Consequently, we anticipate that a combination of proteasome and CDK4/6i may effectively block the growth of drug-resistant cells without the need for endocrine therapy. In support of this, our experiments with MDA-MB-231 xenografts showed that the combination of proteasome and CDK4/6i effectively blocked the persistence of CDK4/6i resistance. This combination may be valuable as a second-line therapy following the emergence of CDK4/6i resistance in HR+/HER2− breast cancer. Additionally, a previous clinical trial in mantle cell lymphoma reported promising clinical outcomes and tolerability for the combination of palbociclib and bortezomib.53
Limitations of the study
Our investigation sheds light on the molecular mechanism underlying CDK4/6i resistance in breast cancer cells. We found that breast cancer cells alternatively induce Rb inactivation through proteasome-dependent degradation. However, in line with previous research,31,32 our study was unable to detect Rb ubiquitination. These findings led us to propose that non-p-Rb is intrinsically unstable and undergoes proteasome-dependent degradation. Despite these contributions, our study does not conclusively establish how proteasomes target non-p-Rb protein for degradation. Recent studies have unveiled ubiquitin-independent pathways for proteasome degradation,54,55 suggesting potential avenues for further investigation intothe mechanisms of Rb degradation. Furthermore, it is important to consider that other studies have implicated specific E3 ligases, NRBE3,56 TRIM28,57 and β-TRCP1,58 in both Rb ubiquitination and degradation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hee Won Yang (hy2602@cumc.columbia.edu).
Materials availability
Plasmids generated in this study are currently not available at Addgene; however, they can be provided upon request.
Data and code availability
Raw data were deposited on Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
Imaging analysis code is available at https://github.com/tjdtn5160/image-analysis-kim-2023.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit mAb anti-phospho-Rb (Ser807/811) | Cell Signaling Technology | Cat# 8516; RRID: AB_11178658 |
| Mouse mAb anti-Rb (4H1) | Cell Signaling Technology | Cat# 9309; RRID: AB_823629 |
| Rabbit mAb anti-cleaved caspase3 (Asp175) | Cell Signaling Technology | Cat# 9579; RRID: AB_10897512 |
| Rabbit mAb anti-c-Myc (D84C12) | Cell Signaling Technology | Cat# 5605; RRID: AB_1903938 |
| Mouse mAb anti-β-Actin (8H10D10) | Cell Signaling Technology | Cat# 3700; RRID: AB_2242334 |
| Rabbit mAb anti-Phospho-Akt (Ser473) | Cell Signaling Technology | Cat# 9271; RRID: AB_329825 |
| Mouse mAb anti-Akt (pan) (40D4) | Cell Signaling Technology | Cat# 2920; RRID: AB_1147620 |
| Rabbit mAb anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling Technology | Cat# 4370; RRID: AB_2315112 |
| Mouse mAb anti-p44/42 MAPK (Erk1/2) (L34F12) | Cell Signaling Technology | Cat# 4696; RRID: AB_390780 |
| Rabbit polyclonal Ab anti-GAPDH | Santa Cruz Biotechnology | Cat# sc-25778; RRID: AB_10167668 |
| Mouse mAb anti-PCNA (PC10) | Santa Cruz Biotechnology | Cat# sc-56; RRID: AB_628110 |
| Rabbit mAb anti-Rb | Abcam | Cat# ab181616; RRID: AB_2848193 |
| Rabbit mAb anti-c-Myc (Y69) | Abcam | Cat# ab32072; RRID: AB_731658 |
| IgG (H + L) Highly Cross-Adsorbed Goat anti-Rabbit, Alexa Fluor™ Plus 488 | Invitrogen | Cat# A32731; RRID: AB_2633280 |
| IgG (H + L) Highly Cross-Adsorbed Goat anti-Mouse, Alexa Fluor™ Plus 488 | Invitrogen | Cat# A32723; RRID: AB_2633275 |
| IgG (H + L) Highly Cross-Adsorbed Goat anti-Mouse, Alexa Fluor™ 568 | Invitrogen | Cat# A11031; RRID: AB_144696 |
| IgG (H + L) Cross-Adsorbed Goat anti-Rabbit Alexa Fluor™ 488 | Invitrogen | Cat# A11008; RRID: AB_143165 |
| IgG (H + L) Highly Cross-Adsorbed Goat anti-Mouse, Alexa Fluor™ 568 | Invitrogen | Cat# A11031; RRID: AB_144696 |
| Goat-anti-mouse IR Dye 800CW | LI-COR Biosciences | Cat# 926-32210; RRID: AB_621842 |
| Goat-anti-rabbit IR Dye 680RD | LI-COR Biosciences | Cat# 926-68071; RRID: AB_10956166 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| 5-Ethynyl-2′-deoxyuridine | Sigma-Aldrich | Cat# 900584 |
| 5-Ethynyl-Uridine | Click Chemistry Tools | Cat# 1261 |
| Palbociclib | Selleck Chemicals | Cat# S1116 |
| Abemaciclib | Selleck Chemicals | Cat# S7158 |
| Ribociclib | Selleck Chemicals | Cat# S7440 |
| Trametinib | Selleck Chemicals | Cat# S2673 |
| EN4 | Selleck Chemicals | Cat# S9807 |
| Bortezomib | MedChem Express | Cat# HY-10227 |
| Ixazomib | MedChem Express | Cat# HY-10453 |
| Carfilzomib | MedChem Express | Cat# HY-10455 |
| Alpelisib | MedChem Express | Cat# HY-15244 |
| Fulvestrant | MedChem Express | Cat# HY-13636 |
| Pictilisib | MedChem Express | Cat# HY-50094 |
| Palbociclib (mice experiment) | MedChem Express | Cat# HY-50767 |
| Chloroquine | MedChem Express | Cat# HY-17589A |
| MG-132 | Calbiochem | Cat# 474790 |
| Epoxomicin | Cayman Chemical | Cat# 134381-21-8 |
| ONX0914 | Cayman Chemical | Cat# 960374-59-8 |
| Celastrol | Cayman Chemical | Cat# 34157-83-0 |
| Omarigliptin | Cayman Chemical | Cat# 1226781-44-7 |
| PMSF | Cayman Chemical | Cat# 329-98-6 |
| Maribavir | Cayman Chemical | Cat# 176161-24-3 |
| MLN9708 (Ixazomib citrate) | Cayman Chemical | Cat# 1201902-80-8 |
| Nafamostat mesylate | Cayman Chemical | Cat# 82956-11-4 |
| N-ethylmaleimide | Cayman Chemical | Cat# 128-53-0 |
| Camostat mesylate | Cayman Chemical | Cat# 59721-29-8 |
| Leupeptin hemisulfate | Cayman Chemical | Cat# 103476-89-7 |
| MG101 | Cayman Chemical | Cat# 110044-82-1 |
| Loxistatin acid | Cayman Chemical | Cat# 76684-89-4 |
| PI1840 | Cayman Chemical | Cat# 1401223-22-0 |
| Aloxistatin | Cayman Chemical | Cat# 88321-09-9 |
| VR23 | Cayman Chemical | Cat# 1624602-30-7 |
| Paritaprevir | Cayman Chemical | Cat# 1221573-85-8 |
| Delanzomib | Cayman Chemical | Cat# 847499-27-8 |
| Oprozomib | Cayman Chemical | Cat# 935888-69-0 |
| Z-FA-FMK | Cayman Chemical | Cat# 197855-65-5 |
| 17β-estradiol pellet (0.36 mg) | Innovative Research of America | Cat# SE-121 |
| 16% Paraformaldehyde | Ted Pella Inc | Cat# NC1537886 |
| Fetal Bovine Serum (FBS) | Gibco | Cat# 10-437-028 |
| Puromycin | Invivogen | Cat# ant-pr-1 |
| Blasticidin | Invivogen | Cat# ant-bl-1 |
| DPBS without Ca2+, Mg2+ | Genesee | Cat# 25-508 |
| Triton X-100 | Millipore Sigma | Cat# T8787 |
| Hoechst 33342 | Thermo Scientific | Cat# 62249 |
| Dimethyl sulfoxide (DMSO) | Millipore Sigma | Cat# D2438 |
| Trypsin-EDTA, 0.25% 1X, phenol red, without Calcium and Magnesium | Genesee | Cat# 25-510 |
| Bovine Serum Albumin (BSA) Protease-free powder | Millipore Sigma | Cat# A3311 |
| Protein G Agarose Beads | Cell Signaling Technology | Cat# 37478 |
| SimplyBlue™ SafeStain | Invitrogen | Cat# LC6060 |
| AFDye 647 Picolyl Azide | Click Chemistry Tools | Cat# 1300 |
| (+)-SODIUM L-ASCORBATE | Millipore Sigma | Cat# A4034 |
| COPPER(II) SULFATE (CuSO4) | Millipore Sigma | Cat# C1297 |
| Halt protease inhibitor cocktail | Thermo Scientific | Cat# 1861279 |
| PhosSTOP | Millipore Sigma | Cat# 4906845001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| GeneJET Plasmid Miniprep Kit | Thermo Fisher | Cat# K0503 |
| NucleoBond Xtra Midi prep kit | Macherey-Nagel | Cat# 740410.100 |
| PureLink™ RNA Mini Kit | Invitrogen | Cat# 12183018A |
| SuperScript IV VILO Master Mix | Invitrogen | Cat# 11756050 |
| PowerTrack SYBR Green Master Mix | Thermo Fisher | Cat# A46109 |
| ViewRNA ISH Cell Assay | Invitrogen | Cat# QVC0001 |
| ViewRNA Cell Probe Set: E2F1 | Invitrogen | Cat# VA1-12108-VC |
| ViewRNA Cell Probe Set: Cdc25A | Invitrogen | Cat# VA1-15814-VC |
| ViewRNA Cell Probe Set: RB1 | Invitrogen | Cat# VA1-3000151-VC |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| MCF-7 | ATCC | Cat# HTB-22 |
| CAMA-1 | ATCC | Cat# HTB-21 |
| MDA-MB-157 | ATCC | Cat# HTB-24 |
| MDA-MB-231 | ATCC | Cat# CRM-HTB-26 |
| MDA-MB-468 | ATCC | Cat# HTB-132 |
| T47D | ATCC | Cat# CRL-2865 |
| HCC1428 | ATCC | Cat# CRL-2327 |
| HCC1143 | ATCC | Cat# CRL-2321 |
| HCC1937 | ATCC | Cat# CRL-2336 |
| BT-483 | ATCC | Cat# HTB-121 |
| BT-20 | ATCC | Cat# HTB-19 |
|
| ||
| Deposited Data | ||
|
| ||
| Raw data | Mendeley Data | Mendeley: http://doi.org/10.17632/c47xz5mgp3.1 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| J:NU mice | The Jackson Laboratory | Cat#007850 |
| Experimental models: Organisms/strains | ||
| gRNA targeting Rb (GUUCGAGGUGAACCAUUAAUGUUUUAGAGCUAUGCU) | Integrated DNA Technologies | N/A |
| gRNA targeting Rb (AAGUGAACGACAUCUCAUCUGUUUUAGAGCUAUGCU) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE1, sense sequence (GAUCAGCACUUUCUUGAGCAACACC) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE1, anti-sense sequence (CCCUAGUCGUGAAAGAACUCGUUGUGG) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE1, sense sequence (AUGCAAAAGGUUUCAGGGUAUCAGT) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE1, anti-sense sequence (ACUACGUUUUCCAAAGUCCCAUAGUCA) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE2, sense sequence (CAUUCUGACUUGGAACCACAGAUGA) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE2, anti-sense sequence (ACGUAAGACUGAACCUUGGUGUCUACU) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE2, sense sequence (ACGUAAGACUGAACCUUGGUGUCUACU) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE2, anti-sense sequence (AGUCAGGAACGUAAUAGUAACUUUGUG) | Integrated DNA Technologies | N/A |
| siRNA targeting c-Myc, sense sequence (AUCAUUGAGCCAAAUCUUAAAAAAA) | Integrated DNA Technologies | N/A |
| siRNA targeting c-Myc, anti-sense sequence (UAUAGUAACUCG GUUUAGAAUUUUUUU) | Integrated DNA Technologies | N/A |
| siRNA targeting c-Myc, sense sequence (CGACGAGACCUUCAUCAAAAACATC) | Integrated DNA Technologies | N/A |
| siRNA targeting c-Myc, anti-sense sequence (CUGCUGCUCUGGAAGUAGUUUUUGUAG) | Integrated DNA Technologies | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLV-DHB (a.a.995–1087)-mVenus-p2a-mCherry-Rb (a.a.886–928)-IRES-blasticidin | Yang et al.33 | RRID: Addgene_126679 |
| pLV-H2B-iRFP670-p2a-mCerulean-Geminin (1–110)-IRES-Neomycin | This paper | |
| pLV-YFP-Rb-IRES-blasticidin | This paper | |
| pLV-YFP-Rb (14 Ser/Thr to Glu)-IRES-puromycin | This paper | |
| pLV-H2B-iRFP670-p2a-YFP-Rb-IRES-blasticidin | This paper | |
| pCW57.1-Cyclin D1-IRES-puromycin | This paper | |
| pCW57.1-Cyclin E1-IRES-puromycin | This paper | |
| pCW57.1-c-Myc-IRES-puromycin | This paper | |
| pCMV-VSV-G | Stewart et al.59 | RRID: Addgene_8454 |
| pRSV-rev | Dull et al.60 | RRID: Addgene_12253 |
| pMDLg/pRRE | Dull et al.60 | RRID: Addgene_12251 |
|
| ||
| Software and algorithms | ||
|
| ||
| MATLAB | MathWorks | https://www.mathworks.com/ |
| MATLAB Scripts | MathWorks | http://doi.org/10.5281/zenodo.8310175 |
| Image Studio Lite | LI-COR Biosciences | https://www.licor.com/bio/image-studio-lite/ |
| Image Lab 5.2.1 | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |
EXPERIMENTAL MODEL AND SUBJECT PARTICIPANT DETAILS
Cell lines
MCF-7, CAMA-1, MDA-MB-231, MDA-MB-468, T47D, HCC1428, HCC1143, HCC1937, BT-483, and BT-20 cells were obtained from ATCC. MCF-7, CAMA-1, MDA-MB-231, and MDA-MB-468 cells were cultured in DMEM media (Genesee Scientific) supplemented with 10% fetal bovine serum (FBS). T47D, HCC1428, HCC1143, and HCC1937 cells were cultured in RPMI-1640 media (Genesee Scientific) supplemented with 10% FBS. BT-483 cells were grown in RPMI-1640 media supplemented with 20% FBS. BT-20 cells were cultured in MEM (Gibco) supplemented with 10% FBS. All cell lines were maintained at 37°C in a 5% CO2 atmosphere and tested negative for mycoplasma.
Animals
All experiments were conducted following NIH guidelines and protocols approved by the IACUC at Columbia University Irving Medical Center. 7–8-week-old female J:NU mice (The Jackson Laboratory) were housed in a barrier facility with ad libitum access to food pellets and water on a 12-h light-dark cycle. Mice were regularly monitored to follow institutional guidelines for ethical endpoints. All experimental endpoints were at 14–16 weeks of age unless otherwise specified.
Human samples
For c-Myc and Rb protein staining, FFPE metastatic lesions from HR+/HER2− breast cancer patients treated with combined CDK4/6i and endocrine therapy in the advanced setting were selected retrospectively by database extraction from the archives of the Tissue Bank at Columbia University Irving Medical Center. All clinical samples were coded to maintain patient confidentiality. This study was approved by the Columbia University Institutional Review Board and conformed to HIPAA regulations.
To investigate the correlation between c-Myc amplification status and PFS, the study cohort comprised samples from 4,640 patients. All patients underwent prospective clinical sequencing as part of their clinical care from April 2015 to August 2019. All patients provided written informed consent for tumor sequencing and review of medical records for demographic, clinical, and pathological information. Genomic sequencing was performed on tumor DNA extracted from formalin-fixed, paraffin-embedded tissue and normal DNA extracted from mononuclear cells from peripheral blood. Patient samples were sequenced in an improvement amendments-compliant clinical laboratory using one of the three versions of the MSK-IMPACT targeted sequencing panel (including 341-, 410-, and 468-genes, respectively).61 All versions of the panel contained coverage within exonic regions in MYC.
METHOD DETAILS
Plasmid cloning and viral production
Using Gibson cloning, DHB (a.a.994–1087)-mCherry, H2B-iRFP670-p2a-mCerulean-Cdt1 (a.a.1–100), YFP-Rb, and H2B-iRFP670-p2a-YFP-Rb were inserted into pLenti-IRES vectors tagged with puromycin, blasticidin, or neomycin. Human phosphomimetic Rb by converting 14 Serine/Threonine sites to Glutamic acid (T5E, S230E, S249E, T252E, T356E, T373E, S612E, S780E, S788E, S795E, S807E, S811E, T821E, and T826E) was synthesized and inserted into a pLenti-IRES-puromycin vector using Gibson cloning. Cyclin D1, Cyclin E1, or c-Myc were inserted into the pCW57.1 vector (Addgene, #50661) using NheI and BamHI sites. Stable cell lines were generated by introducing lentiviral constructs into cells via viral transduction. Lentiviruses were produced in HEK-293T cells co-transfected them with pCMV-VSV-G (Addgene, #8454), pRSV-rev (Addgene, #12253), and pMDLg/pRRE (Addgene, #12251) using the PEI transfection reagent. 3-day after transfection, the supernatant containing virus was collected, centrifuged for 5 min, and filtered through 0.45 μM filtration unit (Millipore). The filtered supernatant was concentrated using 100 kDa centrifugal filters (Millipore) and stored at −80°C. Cells were transduced with the virus in growth media and selected using appropriate antibiotics: blasticidin (10 μg/mL), puromycin (1 μg/mL), or neomycin (400 μg/mL).
Rb-knockout cell line
To generate Rb-knockout cells, MCF-7 cells were transfected with a ribonucleoprotein complex containing a CRISPR-Cas9 guide RNA (crRNA:tracrRNA-ATTO550 duplex) and a Cas9 enzyme protein. Transfected cells with ATTO550 fluorescent signal were then selected and separated into single cells by fluorescence-activated cell sorting. Knockout cells were confirmed by immunoblotting. We used two pooled CRISPR-Cas9 guide RNAs to knockout Rb: 5′-GUUCGAGGUGAACCAUUAAUGUUUUAGAGCUAUGCU-3′, 5′-AAGUGAAC GACAUCUCAUCUGUUUUAGAGCUAUGCU-3’.
Drug-resistant cells
To establish CDK4/6i resistance, breast cancer cells were chronically exposed to palbociclib (1 μM), abemaciclib (1 μM), or ribociclib (1 μM) for over a month. Cell culture media with drugs were replaced every 2–3 days. CDK4/6i resistance was confirmed by measuring IC50 and the percentage of S-phase cells.
siRNA transfection
siRNA duplexes were purchased from Integrated DNA Technologies. These were: Negative control, CCNE1 siRNA1: sense sequence 5′-GAUCAGCACUUUCUUGAGCAACACC −3′, anti-sense sequence 3′- CCCUAGUCGUGAAAGAACUCGUUGUGG −5’; CCNE1 siRNA 2: sense sequence 5′- AUGCAAAAGGUUUCAGGGUAUCAGT −3′, anti-sense sequence 3′- ACUACGUUUUCCAAAGUCCCAUAGUCA −5′, CCNE2 siRNA 1: sense sequence 5′- CAUUCUGACUUGGAACCACAGAUGA −3′, anti-sense sequence 3′- ACGUAAGACU GAACCUUGGUGUCUACU −5’; CCNE2 siRNA 2: sense sequence 5′- ACGUAAGACUGAACCUUGGUGUCUACU −3′, anti-sense sequence 3′- AGUCAGGAACGUAAUAGUAACUUUGUG −5′ and c-Myc siRNA 1: sense sequence 5′- AUCAUUGAGCCAAAUCUUA AAAAAA −3′, anti-sense sequence 3′- UAUAGUAACUCGGUUUAGAAUUUUUUU −5’; c-Myc siRNA 2: sense sequence 5′- CGAC GAGACCUUCAUCAAAAACATC −3′, anti-sense sequence 3′- CUGCUGCUCUGGAAGUAGUUUUUGUAG −5’. For knockdown experiments, MCF-7 cells mixed with a combination of 20 nM siRNA duplexes were transfected 24 h before live-cell experiments.
Immunoblots
Cells were plated in either 6-well plates or 10 cm dishes. After washing with ice-cold PBS, cells were lysed in CHAPS buffer (150 mM NaCl, 1mM EDTA, 0.3% CHAPS, 10 mM NEM, 50 mM Tris, pH 8.0) supplemented with 1× Halt Protease Inhibitor Cocktail (Thermo Scientific, #1861279) and Phosphatase Inhibitor Cocktail (Sigma-Aldrich, #4906845001) was used to obtain whole-cell lysates. Lysates were collected in microcentrifuge tubes and centrifuged at 12,000 rpm for 15 min at 4°C to remove cell debris. Proteins (10–20 μg) were mixed with LDS sample buffer (Invitrogen, #NP0007) containing 2% 2-mercaptoethanol and heated at 70°C for 5 min. Samples were then separated using SDS-PAGE on a NuPAGE 4–12% Bis-Tris protein gel (Invitrogen) in MOPS SDS running buffer. Subsequently, semi-dry transfer was performed using the Bio-Rad Trans-Blot Turbo system onto 0.45 μm PVDF membranes with Tris/Glycine buffer (Bio-Rad, #1610734) + 10% MeOH. Membranes were washed in TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween 20), blocked for 1 h in blocking buffer (LI-COR, #927–60001), and then incubated overnight with primary antibodies diluted in blocking buffer as indicated in the STAR Methods. Afterward, membranes were incubated for 2 h with secondary antibodies (goat-anti-mouse IR Dye 800CW [LI-COR, 1:2000, #926–32210] or goat-anti-rabbit IR Dye 680RD [LI-COR, 1:2000, #926–68071]) diluted in blocking buffer. Blots were detected using an Odyssey CLx Infrared Imaging system (LI-COR).
Quantitative real-time PCR
Total RNA was isolated using the PureLink RNA purification kit (Invitrogen, #12183018A) and reverse transcribed using the SuperScript IV VILO Master Mix (Invitrogen, #11756050) according to the manufacturers’ instruction. Real-time PCR was performed using the PowerTrack SYBR Green Master Mix (Thermo Fisher, #A46109) on a QuantStudio 6 Flex real-time PCR system (Applied Biosystems, #4485691). The reaction was initiated with a hot-start Taq DNA polymerase activation step at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 5 s, and annealing and extension at 60C for 30 s. A melting curve stage was run at 95°C for 15 s, 60°C for 1 min, and 95°C for 15 s. All reactions were performed in triplicate. Relative mRNA levels were calculated using the 2−ΔΔCt method by normalizing the expression to a housekeeping gene, RSP23. The primers for the real-time PCR reactions were procured from Integrated DNA Technologies and were indicated in the Table S1.
Immunofluorescence and RNA fluorescence in situ hybridization (FISH)
Cells were stained and imaged in 96-well plates (Cellvis, #P96–1.5H-N). To measure the percentage of cells in the S phase, cells were incubated either with 10 μM EdU (Sigma-Aldrich, #900584) for 15 min, or as otherwise indicated, at 37°C prior to fixation. To evaluate the mRNA transcription rate, cells were treated with 1 mM EU (Click Chemistry Tools, #1261) for 30 min at 37°C before fixation. Cells were fixed using 4% paraformaldehyde (Ted Pella, #NC1537886) in PBS at a ratio of 1:1 with culture medium for 15 min at room temperature, followed by PBS wash. Cells were permeabilized in 0.2% Triton X-100 in PBS for 15 min. The incorporation of EdU or EU was detected using a click reaction in 2 mM CuSO4, 20 mg/mL sodium ascorbate in TBS with 3 μM AFDye 647 picolyl azide (Click Chemistry Tools, #1300) for 15 min, and PBS wash. Cells were incubated in a blocking solution (consisting of 0.1% Triton X-100, 10% FBS, 1% BSA, and 0.01% NaN3) for 1 h. Following the blocking step, cells were incubated with primary antibodies diluted in the same blocking solution overnight at 4°C. Cells were washed with PBS and treated with Alexa-labeled secondary antibodies. Cell nuclei were stained with Hoechst 33342 dye (10 mg/mL), diluted in PBS (1:2,000), prior to imaging. All washes were performed with an automated plate washer (aspirate to 50 μL, dispense 250 μL, repeated 3 times, BioTek 405 TS).
The mRNA FISH procedure was performed using the Affymetrix Quantigene ViewRNA ISH Cell Assay Kit, with custom probes targeting E2F1 and CDC25A purchased from Invitrogen. Briefly, after live cell imaging, cells fixation and permeabilization were carried out as described in the immunofluorescence section. The fixed cells were then photobleached the fluorescent protein using 3% H2O2 + 20 mM HCl in PBS for 2 h at room temperature. Probe hybridization, amplification, and Alexa Fluor 555 labeling were conducted following the manufacturer’s protocols.
Immunohistochemistry
Upon completion of drug treatment, the animals underwent vascular perfusion with 1% paraformaldehyde in PBS to ensure tissue preservation. Subsequent to extraction, tissues were stored in 1% paraformaldehyde for 2 h at 4°C. This was followed by an overnight incubation in 30% sucrose in PBS after washing with PBS. Tissue sections, each of 40-μm thickness, were procured using a cryostat after embedding in optimal cutting temperature compound. To perform staining of tissue sections, they were initially rinsed with PBS-T (0.3% Triton X-100) and subsequently incubated with 5% normal goat serum in PBS-T for an hour. The sections were then treated with either rabbit anti-retinoblastoma protein (anti-Rb) (Abcam, #ab181616) or rabbit anti-phospho-Rb (Ser807/811) (Cell Signaling Technology, #8516) antibodies, with an overnight incubation. Following a wash with PBS-T, the sections were incubated for 3 h with goat Alexa Fluor 488 anti-rabbit secondary antibodies (Invitrogen, #A-11008), diluted in PBS-T (1:2,000). The sections were mounted and sealed with DAPI (Sigma-Aldrich) introduced at a concentration of 2 μg/mL, 5 min before mounting. The H-scores were determined by multiplying the percentage of positive cells (ranging from 0 to 100%) by the staining intensity (ranging from 0 to 3).
FFPE tissue staining
FFPE slides were initially incubated at 60°C for 1 h. Subsequently, the slides were placed on a glass slide rack and immersed in xylene in a glass staining dish for 5 min, followed by a transfer to another dish containing fresh xylene for an additional 5 min. For rehydration, the slides were sequentially immersed in staining dishes filled with 100%, 90%, and 70% ethanol for 3 min each, followed by transitioning the slides to two successive PBS solutions. After rehydration, slides were placed in a plastic Coplin jar containing 1x citric acid (pH 6.0, Sigma-Aldrich, #C9999). Subsequently, they were transferred to a pre-heated epitope retrieval steamer (IHC world, #IW-1102) for 40 min to facilitate antigen retrieval. After cooling to room temperature, the slides underwent two washes with distilled water and a subsequent wash with PBS-T (0.3% Triton X-100). The dewaxed slides were then incubated at room temperature with a blocking solution (10% Normal Goat Serum, and 1% BSA in PBS-T) for 1 h. Following the blocking step, the slides were incubated overnight at room temperature with primary antibodies diluted in the same blocking solution. Slides were then washed with PBS-T and treated with HRP-conjugated secondary antibodies (SignalStain Boost IHC Detection Reagent, Cell Signaling Technology, #8125) for 1 h at room temperature. For signal detection, slides were exposed to a substrate buffer (immPACT DAB Substrate Kit, Vector Laboratories, #SK-4105) and subsequently washed three times with distilled water. The cell nuclei were stained using DAPI (1:500) diluted in PBS-T at room temperature for 15 min. Slides were rinsed with PBS-T and mounted using immuno-mount (Thermo Scientific, #9990402) prior to imaging.
Live- and fixed-cell imaging and tumor tissue imaging
Cells were seeded and maintain as 30–80% confluency during experiments in 96-well glass-like polymer bottom plates (Cellvis, #P96–1.5P). Either a Nikon microscope mounted onto an inverted Eclipse Ti-2 body (Nikon) or an Axio Observer 7 microscope (Zeiss) were used to image cells. Multichannel fluorescent images were captured using either a Hamamatsu ORCA-Flash4.0 V3 digital CMOS camera or a Hamamatsu ORCA-Fusion digital CMOS camera. Images were acquired with 10× (Nikon CFI Plan Apo Lambda, 0.45 NA, no binning), 203 (Nikon CFI Plan Apo Lambda, 0.75 NA, 2-by-2-pixel binning), 10× (Zeiss Plan-Apochromat, 0.45 NA, no binning), or 203 (Zeiss Plan-Apochromat, 0.8 NA, 2-by-2-pixel binning) objectives. Live-cell imaging was performed in a humidified chamber at 37°C with 5% CO2, with 3–6 sites imaged in each well every 12 min. Fixed-cell imaging involved imaging 25–32 sites in each well. Total light exposure time was kept under 500 ms for each time point. mRNA FISH images were acquired with a z stack interval of 1.5 μm. Whole tumor cross-sections and representative regions were imaged using an Axio Observer 7 microscope with Apotome2 (Zeiss) equipped with a Hamamatsu ORCA-Fusion digital CMOS camera (Hamamatsu photonics K.K.). Images were acquired using a 10× (Zeiss Plan-Apochromat, 0.45 NA, no binning), or 40× (Zeiss Plan-Apochromat, 1.3 NA, 2-by-2 pixel binning) objective, with a z stack interval of 1.5 μm. Image analysis was performed using MATLAB and ImageJ software.
Live- and fixed-cell image analysis
Automated image analysis was performed using a custom MATLAB pipeline. To correct uneven illumination, raw images were flat-filed corrected. Nuclei were segmented using a nucleus marker either H2B or Hoechst staining with a Laplacian of Gaussian blob detector. A threshold for nuclear segmentation was defined based on histogram curvature. If the size of the detected nuclei exceeded the median object size, a curvature-based object splitting algorithm was utilized to split cells at two points of high perimeter curvature. To subtract background signal, the 50th percentile of pixels outside of a dilated nuclear mask (7.8 μm for nuclear signals and 15.6 μm for cytoplasmic signals) was calculated. After background subtraction, the nuclear median signal was computed based on nuclear segmentation. The cytoplasmic signal was determined by calculating the median signal within a ring outside the nucleus, ranging from 0.65 μm to 3.25 μm outside the edge of the nucleus. Overlapping cells with adjacent cell nuclei were excluded from the analysis. Mitosis was identified by evaluating the H2B signals of both nearest daughter cells, which were expected to be between 45 and 55% of the mother cell’s signal.
Measurements for CDK2 and CDK4/6 activities
Kinase Translocation Reporters (KTRs) contain a degenerate CDK2 substrate motif derived from the CDK2 sensor and thus detect activities measured by the CDK2 sensor during S and G2 phases as shown in previous studies.33,62,63 Consequently, we used a linear regression to calculate the correction factor based on the CDK2 sensor in the presence of palbociclib (1 μM). This analysis revealed that applying a correction factor of around 0.41 to the measured CDK2 activity leads to a nearly consistent derived reporter signal throughout the cell cycle following CDK4/6 inhibition. The contributions of CDK2 activities to CDK4/6 sensor was adjusted by subtracting a calculated fraction of the CDK2 sensor signal as follows: CDK4/6 activity = CDK4/6 sensor activity - 0.41 3 CDK2 sensor activity.
Analysis of c-Myc–amplification status and PFS
To determine copy number amplification, normalized coverage values (normalized for overall coverage of sample, adjusted for GC content of each target region) of each tumor sample are divided by normalized coverage values from the corresponding normal control and then log-transformed. Log-ratio values are then segmented using circular binary segmentation algorithm; segmented values are then grouped into clusters. p values are then generated for each segment, using a null distribution parameterized by probe regions with segments belonging to segment cluster where mean log ratio is closest to zero, as described in Ross et al. p values were then adjusted for false-discovery rate using the Benjamini Hochberg procedure. c-Myc amplifications were called based on fold change ≥2.0 with p < 0.05. PFS was defined as the time from initiation of combined CDK4/6i and endocrine therapy until disease progression or death.
We determined the association between c-Myc amplifications (as called from a pre-treatment sample) and disease progression on therapy with first line CDK4/6i inhibitors. We categorized CDK4/6i regimens based on their endocrine therapy backbone as follows: i) CDK4/6i plus aromatase inhibitors, ii) CDK4/6i plus selective estrogen receptor degraders including fulvestrant. We used univariate and multivariate Cox proportional hazard models (adjusted for endocrine therapy back bone) to determine association between c-Myc amplifications and PFS.
Immunoprecipitation and in-gel digestion for mass spectrometry
After washing with ice-cold PBS, cells were lysed in a buffer containing 0.5% Nonidet-40 (Sigma-Aldrich, #74385), 150 mM NaCl (Sigma-Aldrich, #746398), 10 mM KCl (Sigma-Aldrich, #P9333), 1.5 mM MgCl2 (Sigma-Aldrich, #M2670) and 10 mM Tris-HCl (Millipore Sigma, #C4706). The supernatants were collected and incubated overnight at 4°C with mouse anti-Rb (1:100; Cell Signaling Technology, #9309). Samples were then pulled down by rotation with Protein G Agarose Beads (Cell Signaling Technology, #37478) at 4°C for 2 h. The supernatants were collected and separated on a NuPAGE 4–12% Bis-Tris protein gel (1.0 mm, Invitrogen). For LC-MS/MS analysis, the protein gel was stained with SimplyBlue SafeStain (Invitrogen, #LC6065). Gel slices containing the protein bands of interest were excised and subjected to in-gel digestion following a previously described protocol with minor modifications.64 The gel slices were washed with 1:1 Acetonitrile and 100 mM ammonium bicarbonate for 30 min, followed by dehydration with 100% acetonitrile for 10 min until shrunk. Excess acetonitrile was removed, and the slices were dried in a speed vacuum at room temperature for 10 min. The gel slices were reduced with 5 mM DTT for 30 min at 56C in an air thermostat, cooled down to room temperature, and alkylated with 11 mM IAA for 30 min in the dark. The gel slices were then washed with 100 mM of ammonium bicarbonate and 100% acetonitrile for 10 min each. Excess acetonitrile was removed and dried in a speed-vacuum for 10 min at room temperature and the gel slices were re-hydrated in a solution of 25 ng/μL trypsin in 50 mM ammonium bicarbonate for 30 min on ice and digested overnight at 37°C in an air thermostat. Digested peptides were collected and further extracted from gel slices in an extraction buffer (1:2 ratio by volume of 5% formic acid: acetonitrile) at high speed, shaking in an air thermostat. The supernatants from both extractions were combined and dried in a speed vacuum. Peptides were dissolved in 3% acetonitrile/0.1% formic acid.
LC-MS/MS analysis
Thermo Scientific UltiMate 3000 RSLCnano system and Thermo Scientific EASY Spray source with Thermo Scientific Acclaim PepMap100 2 cm × 75 μm trap column and Thermo Scientific EASY-Spray PepMap RSLC C18 50 cm × 75 μm ID column were used to separate desalted peptides with a 5–30% acetonitrile gradient in 0.1% formic acid over 100 min at a flow rate of 250 nL/min. The column temperature was maintained at a constant 50°C during all experiments. Thermo Scientific Orbitrap Fusion Tribrid mass spectrometer was used for peptide MS/MS analysis. Survey scans of peptide precursors were performed from 350 to 1500 m/z at 120K FWHM resolution (at 200 m/z) with a 4 × 105 ion count target and a maximum injection time of 50 ms. The instrument was set to run in top speed mode with 3 s cycles for the survey and the MS/MS scans. After a survey scan, tandem MS was performed on the most abundant precursors exhibiting a charge state from 2 to 6 of greater than 5 × 103 intensity by isolating them in the quadrupole at 1.6 Th. HCD fragmentation was applied with 30% collision energy and resulting fragments were detected using the rapid scan rate in the ion trap. The AGC target for MS/MS was set to 5 × 104 and the maximum injection time was 22 ms. The dynamic exclusion was set to 60 s with a 10-ppm mass tolerance around the precursor and its isotopes. Monoisotopic precursor selection was enabled.
Raw mass spectrometric data were analyzed using the MaxQuant environment (2) v. 2.2.0.0 and employed Andromeda for database search (3) at default settings with a few modifications. The default was used for first search tolerance and main search tolerance: 20 ppm and 6 ppm, respectively. MaxQuant was set up to search with the reference human proteome database downloaded from UniProt. MaxQuant performed the search trypsin digestion with up to 2 missed cleavages. Peptide, Site, and protein FDR were all set to 1% with a minimum of 1 peptide needed for identification, and label-free quantitation (LFQ) was performed with a minimum ratio count of 1. The following modifications were used as variable modifications for identifications and included for protein quantification: Oxidation of methionine (M), Acetylation of the protein N terminus, and Deamination for asparagine or glutamine (NQ).
Xenograft experiments
Female J:NU mice (The Jackson Laboratory), aged 7–8 weeks, were anesthetized using ketamine (90 mg/kg) and xylazine (10 mg/kg) by intraperitoneal injection. Mice were injected with drug-naïve MCF-7 cells (5×106 cells per mouse), palbociclib-resistant MCF-7 cells (1×106 cells per mouse), or palbociclib-resistant MDA-MB-231 cells (2×106 cells per mouse) embedded in Matrigel (Corning) directly into the abdominal mammary fat pad. Mice injected with MCF-7 cells received a 17β-estradiol pellet (0.36 mg) (Innovative Research of America, #SE-121) in the right side of their necks one week prior to cell injection. Tumor size was monitored twice a week using a digital caliper (volume = [width2 × length] × 1/2) and drug treatment started when the average tumor volume reached 100 mm3.
Mice injected with drug-naïve MCF-7 cells were treated with either control (50 mM sodium D-lactate buffer [pH 4.0], daily by oral gavage) or palbociclib (50 mg/kg dissolved in 50 mM sodium D-lactate buffer [pH 4.0], daily by oral gavage). Mice injected with palbociclib-resistant MCF-7 cells were treated with (1) control (50 mM sodium D-lactate buffer [pH 4.0], daily by oral gavage), (2) bortezomib (0.5 mg/kg dissolved in corn oil, twice a week by IP), (3) palbociclib (50 mg/kg dissolved in 50 mM sodium D-lactate buffer [pH 4.0], daily by oral gavage) + fulvestrant (3 mg/kg dissolved in corn oil, twice a week by IP), or (4) bortezomib + palbociclib + fulvestrant. Mice injected with palbociclib-resistant MDA-MB-231 cells were administered (1) control (50 mM sodium D-lactate buffer [pH 4.0], daily by oral gavage), (2) bortezomib (0.5 mg/kg dissolved in corn oil, twice a week by IP), (3) palbociclib (50 mg/kg dissolved in 50 mM sodium D-lactate buffer [pH 4.0], daily by oral gavage), or (4) bortezomib + palbociclib.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were conducted using either t-tests or one-way ANOVA. Statistical significance was defined as a p value of 0.05 or lower, using the appropriate statistical test method. Details regarding statistics, number of biological replicates, and sample sizes are reported in each figure legend.
Supplementary Material
Highlights.
Breast cancer cells develop CDK4/6i resistance via sequential E2F activation
Rb-protein degradation and c-Myc-mediated amplification of E2F activity are key mechanisms
c-Myc levels, not Rb levels, predict poor outcomes after CDK4/6i treatment
Proteasome inhibitors restoring Rb levels can potentially overcome CDK4/6i resistance
ACKNOWLEDGMENTS
We thank Richard Baer, Howard Worman, Sean Collins, Neil Vasan, and Markus Siegelin for helpful comments, Alejandro Chavez for providing the proteasome inhibitor library, and Kevin Gardner for offering MDA-MB-231 cells. We thank Kaitlin Abrantes, Jiamiao Chen, and Caitlin O’Neil for assisting with tissue imaging. This work was supported by the Neuroendocrine Tumor Research Foundation (M.K., 855538); the Melanoma Research Foundation (M.K. and H.W.Y.); Research Scholar Grant (M.K., RSG-22-167-01-MM; H.W.Y., RSG-22-101-01-CDP); V Scholar (H.W.Y., V2023-017); NIH grants R37-CA266270 (M.K.), R03-AG073833 (M.K.), and R01-GM145884 (H.W.Y.); HICCC Grant P30-CA013696 (M.K. and H.W.Y.); MSK Cancer Center Support Grant P30-CA008748(S.C.); and the Breast Cancer Research Foundation (S.C.). These studies used the resources of HICCC Flow Cytometry Shared Resources (P30-CA013696).
Footnotes
DECLARATION OF INTERESTS
S.C. reports consulting fees from Novartis, AstraZeneca, and Boxer Capital; shares in Totus Medicines; and research support from Daiichi-Sankyo, Ambrx, and AstraZeneca.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113198.
REFERENCES
- 1.Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- 2.Nevins JR (2001). The Rb/E2F pathway and cancer. Hum. Mol. Genet. 10, 699–703. 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- 3.Kim S, Leong A, Kim M, and Yang HW (2022). CDK4/6 initiates Rbinactivation and CDK2 activity coordinates cell-cycle commitment and G1/S transition. Sci. Rep. 12, 16810. 10.1038/s41598-022-20769-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher RP (2016). Getting to S: CDK functions and targets on the path to cell-cycle commitment. F1000Res. 5, 2374. 10.12688/f1000research.9463.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goel S, Bergholz JS, and Zhao JJ (2022). Targeting CDK4 and CDK6 in cancer. Nat. Rev. Cancer 22, 356–372. 10.1038/s41568-022-00456-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Álvarez-Fernández M, and Malumbres M. (2020). Mechanisms of Sensitivity and Resistance to CDK4/6 Inhibition. Cancer Cell 37, 514–529. 10.1016/j.ccell.2020.03.010. [DOI] [PubMed] [Google Scholar]
- 7.Fassl A, Geng Y, and Sicinski P. (2022). CDK4 and CDK6 kinases: Frombasic science to cancer therapy. Science 375, eabc1495. 10.1126/science.abc1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnston S, Martin M, Di Leo A, Im SA, Awada A, Forrester T, Frenzel M, Hardebeck MC, Cox J, Barriga S, et al. (2019). MONARCH 3 final PFS: a randomized study of abemaciclib as initial therapy for advanced breast cancer. NPJ Breast Cancer 5, 5. 10.1038/s41523-018-0097-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO,Ettl J, Patel R, Pinter T, Schmidt M, et al. (2015). The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol. 16, 25–35. 10.1016/S1470-2045(14)71159-3. [DOI] [PubMed] [Google Scholar]
- 10.Turner NC, Ro J, André F, Loi S, Verma S, Iwata H, Harbeck N, Loibl S, Huang Bartlett C, Zhang K, et al. (2015). Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 373, 209–219. 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- 11.Schoninger SF, and Blain SW (2020). The Ongoing Search for Bio-markers of CDK4/6 Inhibitor Responsiveness in Breast Cancer. Mol. Cancer Therapeut. 19, 3–12. 10.1158/1535-7163.MCT19-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anurag M, Haricharan S, and Ellis MJ (2020). CDK4/6 Inhibitor Biomarker Research: Are We Barking Up the Wrong Tree? Clin. Cancer Res. 26, 3–5. 10.1158/1078-0432.CCR-19-3119. [DOI] [PubMed] [Google Scholar]
- 13.Hortobagyi GN, Stemmer SM, Burris HA, Yap YS, Sonke GS,Paluch-Shimon S, Campone M, Blackwell KL, André F, Winer EP, et al. (2016). Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 375, 1738–1748. 10.1056/NEJMoa1609709. [DOI] [PubMed] [Google Scholar]
- 14.Bianchini G, Balko JM, Mayer IA, Sanders ME, and Gianni L(2016). Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 13, 674–690. 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mao P, Cohen O, Kowalski KJ, Kusiel JG, Buendia-Buendia JE,Cuoco MS, Exman P, Wander SA, Waks AG, Nayar U, et al. (2020). Acquired FGFR and FGF Alterations Confer Resistance to Estrogen Receptor (ER) Targeted Therapy in ER(+) Metastatic Breast Cancer. Clin. Cancer Res. 26, 5974–5989. 10.1158/1078-0432.CCR-19-3958. [DOI] [PubMed] [Google Scholar]
- 16.Costa C, Wang Y, Ly A, Hosono Y, Murchie E, Walmsley CS,Huynh T, Healy C, Peterson R, Yanase S, et al. (2020). PTEN Loss Mediates Clinical Cross-Resistance to CDK4/6 and PI3Kalpha Inhibitors in Breast Cancer. Cancer Discov. 10, 72–85. 10.1158/2159-8290.CD-18-0830. [DOI] [PubMed] [Google Scholar]
- 17.O’Leary B, Cutts RJ, Liu Y, Hrebien S, Huang X, Fenwick K, André F, Loibl S, Loi S, Garcia-Murillas I, et al. (2018). The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 8, 1390–1403. 10.1158/2159-8290.CD-18-0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, Razavi P, Li Q, Toy W, Liu B, Ping C, Hsieh W, Sanchez-Vega F, Brown DN, Da Cruz Paula AF, et al. (2018). Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 34, 893–905.e8. 10.1016/j.ccell.2018.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Q, Jiang B, Guo J, Shao H, Del Priore IS, Chang Q, Kudo R, Li Z, Razavi P, Liu B, et al. (2022). INK4 Tumor Suppressor Proteins Mediate Resistance to CDK4/6 Kinase Inhibitors. Cancer Discov. 12, 356–371. 10.1158/2159-8290.CD-20-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marine JC, Dawson SJ, and Dawson MA (2020). Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 20, 743–756. 10.1038/s41568-020-00302-4. [DOI] [PubMed] [Google Scholar]
- 21.Leonce C, Saintigny P, and Ortiz-Cuaran S. (2022). Cell-Intrinsic Mechanisms of Drug Tolerance to Systemic Therapies in Cancer. Mol. Cancer Res. 20, 11–29. 10.1158/1541-7786.MCR-21-0038. [DOI] [PubMed] [Google Scholar]
- 22.Wander SA, Cohen O, Gong X, Johnson GN, Buendia-Buendia JE, Lloyd MR, Kim D, Luo F, Mao P, Helvie K, et al. (2020). The Genomic Landscape of Intrinsic and Acquired Resistance to Cyclin-Dependent Kinase 4/6 Inhibitors in Patients with Hormone Receptor-Positive Metastatic Breast Cancer. Cancer Discov. 10, 1174–1193. 10.1158/2159-8290.CD-19-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA,Sudhan DR, Guerrero-Zotano AL, Croessmann S, Guo Y, et al. (2019). Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun. 10, 1373. 10.1038/s41467-019-09068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, Jansen VM, Estrada MM, Hutchinson KE, Giltnane JM, Schwarz LJ, et al. (2017). Association of FGFR1 with ERalpha Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER(+) Breast Cancer. Clin. Cancer Res. 23, 6138–6150. 10.1158/1078-0432.CCR-17-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cristofanilli M, Rugo HS, Im SA, Slamon DJ, Harbeck N, Bondarenko I, Masuda N, Colleoni M, DeMichele A, Loi S, et al. (2022). Overall Survival with Palbociclib and Fulvestrant in Women with HR+/HER2- ABC: Updated Exploratory Analyses of PALOMA-3, a Double-blind, Phase III Randomized Study. Clin. Cancer Res. 28, 3433–3442. 10.1158/1078-0432.CCR-22-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fribbens C, O’Leary B, Kilburn L, Hrebien S, Garcia-Murillas I, Beaney M, Cristofanilli M, Andre F, Loi S, Loibl S, et al. (2016). Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. J. Clin. Oncol. 34, 2961–2968. 10.1200/JCO.2016.67.3061. [DOI] [PubMed] [Google Scholar]
- 27.Herrera-Abreu MT, Palafox M, Asghar U, Rivas MA, Cutts RJ, Garcia-Murillas I, Pearson A, Guzman M, Rodriguez O, Grueso J, et al. (2016). Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor–Positive Breast Cancer. Cancer Res. 76, 2301–2313. 10.1158/0008-5472.CAN-15-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turner NC, Liu Y, Zhu Z, Loi S, Colleoni M, Loibl S, DeMichele A, Harbeck N, André F, Bayar MA, et al. (2019). Cyclin E1 Expression and Palbociclib Efficacy in Previously Treated Hormone Receptor-Positive Metastatic Breast Cancer. J. Clin. Oncol. 37, 1169–1178. 10.1200/JCO.18.00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varshavsky A. (2017). The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem. 86, 123–128. 10.1146/annurev-biochem-061516-044859. [DOI] [PubMed] [Google Scholar]
- 30.Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H,Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- 31.Sdek P, Ying H, Chang DLF, Qiu W, Zheng H, Touitou R, Allday MJ, and Xiao ZXJ (2005). MDM2 promotes proteasome-dependent ubiquitin-independent degradation of retinoblastoma protein. Mol. Cell 20, 699–708. 10.1016/j.molcel.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 32.Hsieh J, Molusky MM, McCabe KM, Fotakis P, Xiao T, Tascau L, Zeana-Schliep L, DaSilva-Jardine P, and Tall AR (2022). TTC39B destabilizes retinoblastoma protein promoting hepatic lipogenesis in a sex-specific fashion. J. Hepatol. 76, 383–393. 10.1016/j.jhep.2021.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang HW, Cappell SD, Jaimovich A, Liu C, Chung M, Daigh LH, Pack LR, Fan Y, Regot S, Covert M, and Meyer T. (2020). Stress-mediated exit to quiescence restricted by increasing persistence in CDK4/6 activation. Elife 9, e44571. 10.7554/eLife.44571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, and Meyer T. (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. 10.1016/j.cell.2013.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang HW, Chung M, Kudo T, Meyer T, Yang HW, Chung M, Kudo T, and Meyer T. (2017). Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 549, 404–408. 10.1038/nature23880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alves CL, Ehmsen S, Terp MG, Portman N, Tuttolomondo M, Gammelgaard OL, Hundebøl MF, Kaminska K, Johansen LE, Bak M, et al. (2021). Co-targeting CDK4/6 and AKT with endocrine therapy prevents progression in CDK4/6 inhibitor and endocrine therapy-resistant breast cancer. Nat. Commun. 12, 5112. 10.1038/s41467-021-25422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakaue-Sawano A, Yo M, Komatsu N, Hiratsuka T, Kogure T, Hoshida T, Goshima N, Matsuda M, Miyoshi H, and Miyawaki A. (2017). Genetically Encoded Tools for Optical Dissection of the Mammalian Cell Cycle. Mol. Cell 68, 626–640.e5. 10.1016/j.molcel.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Hollestelle A, Nagel JHA, Smid M, Lam S, Elstrodt F, Wasielewski M, Ng SS, French PJ, Peeters JK, Rozendaal MJ, et al. (2010). Distinct gene mutation profiles among luminal-type and basal-type breast cancer cell lines. Breast Cancer Res. Treat. 121, 53–64. 10.1007/s10549-009-0460-8. [DOI] [PubMed] [Google Scholar]
- 39.Zhu J, Blenis J, and Yuan J. (2008). Activation of PI3K/Akt and MAPKpathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc. Natl. Acad. Sci. USA 105, 6584–6589. 10.1073/pnas.0802785105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, and Kuo MT(2012). Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 72, 2622–2633. 10.1158/0008-5472.CAN-11-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ji W, Zhang W, Wang X, Shi Y, Yang F, Xie H, Zhou W, Wang S,and Guan X. (2020). c-myc regulates the sensitivity of breast cancer cells to palbociclib via c-myc/miR-29b-3p/CDK6 axis. Cell Death Dis. 11, 760. 10.1038/s41419-020-02980-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freeman-Cook K, Hoffman RL, Miller N, Almaden J, Chionis J,Zhang Q, Eisele K, Liu C, Zhang C, Huser N, et al. (2021). Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell 39, 1404–1421.e11. 10.1016/j.ccell.2021.08.009. [DOI] [PubMed] [Google Scholar]
- 43.Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young R.a. (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. (2012). c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79. 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB,Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pandey K, Park N, Park KS, Hur J, Cho YB, Kang M, An HJ, Kim S, Hwang S, and Moon YW (2020). Combined CDK2 and CDK4/6 Inhibition Overcomes Palbociclib Resistance in Breast Cancer by Enhancing Senescence. Cancers 12, 3566. 10.3390/cancers12123566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida A,Bu Y, Qie S, Wrangle J, Camp ER, Hazard ES,Hardiman G, de Leeuw R, Knudsen KE, and Diehl JA (2019). SLC36A1-mTORC1 signaling drives acquired resistance to CDK4/6 inhibitors. Sci. Adv. 5, eaax6352. 10.1126/sciadv.aax6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finn RS, Liu Y, Zhu Z, Martin M, Rugo HS, Diéras V, Im SA, Gelmon KA, Harbeck N, Lu DR, et al. (2020). Biomarker Analyses of Response to Cyclin-Dependent Kinase 4/6 Inhibition and Endocrine Therapy in Women with Treatment-Naive Metastatic Breast Cancer. Clin. Cancer Res. 26, 110–121. 10.1158/1078-0432.CCR-19-0751. [DOI] [PubMed] [Google Scholar]
- 49.Cancer Genome Atlas Network; Fulton RS, McLellan MD, Schmidt H,Kalicki-Veizer J, McMichael JF, Fulton LL, Dooling DJ, Ding L, Mardis ER, et al. (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Teo ZL, Versaci S, Dushyanthen S, Caramia F, Savas P, Mintoff CP, Zethoven M, Virassamy B, Luen SJ, McArthur GA, et al. (2017). Combined CDK4/6 and PI3Kalpha Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res. 77, 6340–6352. 10.1158/0008-5472.CAN-17-2210. [DOI] [PubMed] [Google Scholar]
- 51.Manasanch EE, and Orlowski RZ (2017). Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 14, 417–433. 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shang Y, Hu X, DiRenzo J, Lazar MA, and Brown M. (2000). Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103, 843–852. 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 53.Martin P, Ruan J, Furman R, Rutherford S, Allan J, Chen Z, Huang X, DiLiberto M, Chen-Kiang S, and Leonard JP (2019). A phase I trial of palbociclib plus bortezomib in previously treated mantle cell lymphoma. Leuk. Lymphoma 60, 2917–2921. 10.1080/10428194.2019.1612062. [DOI] [PubMed] [Google Scholar]
- 54.Makaros Y, Raiff A, Timms RT, Wagh AR, Gueta MI, Bekturova A,Guez-Haddad J, Brodsky S, Opatowsky Y, Glickman MH, et al. (2023). Ubiquitin-independent proteasomal degradation driven by C-degron pathways. Mol. Cell 83, 1921–1935.e7. 10.1016/j.molcel.2023.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gu X, Nardone C, Kamitaki N, Mao A, Elledge SJ, and Greenberg ME (2023). The midnolin-proteasome pathway catches proteins for ubiquitination-independent degradation. Science 381, eadh5021. 10.1126/science.adh5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Zheng Z, Zhang J, Wang Y, Kong R, Liu J, Zhang Y, Deng H, Du X, and Ke Y. (2015). A Novel Retinoblastoma Protein (RB) E3 Ubiquitin Ligase (NRBE3) Promotes RB Degradation and Is Transcriptionally Regulated by E2F1 Transcription Factor. J. Biol. Chem. 290, 28200–28213. 10.1074/jbc.M115.655597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang Z, Li X, Tang B, Li H, Zhang J, Sun R, Ma J, Pan Y, Yan B, Zhou Y, et al. (2023). SETDB1 Modulates Degradation of Phosphorylated RB and Anticancer Efficacy of CDK4/6 Inhibitors. Cancer Res. 83, 875–889. 10.1158/0008-5472.CAN-22-0264. [DOI] [PubMed] [Google Scholar]
- 58.Dang F, Nie L, Zhou J, Shimizu K, Chu C, Wu Z, Fassl A, Ke S,Wang Y, Zhang J, et al. (2021). Inhibition of CK1epsilon potentiates the therapeutic efficacy of CDK4/6 inhibitor in breast cancer. Nat. Commun. 12, 5386. 10.1038/s41467-021-25700-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L. (1998). A third-generation lentivirus vector with a conditional packaging system. J. Virol. 72, 8463–8471. 10.1128/JVI.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. (2015). Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 17, 251–264. 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gerosa L, Chidley C, Fröhlich F, Sanchez G, Lim SK, Muhlich J, Chen JY, Vallabhaneni S, Baker GJ, Schapiro D, et al. (2020). Receptor-Driven ERK Pulses Reconfigure MAPK Signaling and Enable Persistence of Drug-Adapted BRAF-Mutant Melanoma Cells. Cell Syst. 11, 478–494.e9. 10.1016/j.cels.2020.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim S, Carvajal R, Kim M, and Yang HW (2023). Kinetics of RTK activation determine ERK reactivation and resistance to dual BRAF/MEK inhibition in melanoma. Cell Rep. 42, 112570. 10.1016/j.celrep.2023.112570. [DOI] [PubMed] [Google Scholar]
- 64.Shevchenko A, Tomas H, Havlis J, Olsen JV, and Mann M. (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860. 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data were deposited on Mendeley and are publicly available as of the date of publication. The DOI is listed in the key resources table.
Imaging analysis code is available at https://github.com/tjdtn5160/image-analysis-kim-2023.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit mAb anti-phospho-Rb (Ser807/811) | Cell Signaling Technology | Cat# 8516; RRID: AB_11178658 |
| Mouse mAb anti-Rb (4H1) | Cell Signaling Technology | Cat# 9309; RRID: AB_823629 |
| Rabbit mAb anti-cleaved caspase3 (Asp175) | Cell Signaling Technology | Cat# 9579; RRID: AB_10897512 |
| Rabbit mAb anti-c-Myc (D84C12) | Cell Signaling Technology | Cat# 5605; RRID: AB_1903938 |
| Mouse mAb anti-β-Actin (8H10D10) | Cell Signaling Technology | Cat# 3700; RRID: AB_2242334 |
| Rabbit mAb anti-Phospho-Akt (Ser473) | Cell Signaling Technology | Cat# 9271; RRID: AB_329825 |
| Mouse mAb anti-Akt (pan) (40D4) | Cell Signaling Technology | Cat# 2920; RRID: AB_1147620 |
| Rabbit mAb anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling Technology | Cat# 4370; RRID: AB_2315112 |
| Mouse mAb anti-p44/42 MAPK (Erk1/2) (L34F12) | Cell Signaling Technology | Cat# 4696; RRID: AB_390780 |
| Rabbit polyclonal Ab anti-GAPDH | Santa Cruz Biotechnology | Cat# sc-25778; RRID: AB_10167668 |
| Mouse mAb anti-PCNA (PC10) | Santa Cruz Biotechnology | Cat# sc-56; RRID: AB_628110 |
| Rabbit mAb anti-Rb | Abcam | Cat# ab181616; RRID: AB_2848193 |
| Rabbit mAb anti-c-Myc (Y69) | Abcam | Cat# ab32072; RRID: AB_731658 |
| IgG (H + L) Highly Cross-Adsorbed Goat anti-Rabbit, Alexa Fluor™ Plus 488 | Invitrogen | Cat# A32731; RRID: AB_2633280 |
| IgG (H + L) Highly Cross-Adsorbed Goat anti-Mouse, Alexa Fluor™ Plus 488 | Invitrogen | Cat# A32723; RRID: AB_2633275 |
| IgG (H + L) Highly Cross-Adsorbed Goat anti-Mouse, Alexa Fluor™ 568 | Invitrogen | Cat# A11031; RRID: AB_144696 |
| IgG (H + L) Cross-Adsorbed Goat anti-Rabbit Alexa Fluor™ 488 | Invitrogen | Cat# A11008; RRID: AB_143165 |
| IgG (H + L) Highly Cross-Adsorbed Goat anti-Mouse, Alexa Fluor™ 568 | Invitrogen | Cat# A11031; RRID: AB_144696 |
| Goat-anti-mouse IR Dye 800CW | LI-COR Biosciences | Cat# 926-32210; RRID: AB_621842 |
| Goat-anti-rabbit IR Dye 680RD | LI-COR Biosciences | Cat# 926-68071; RRID: AB_10956166 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Doxycycline | Sigma-Aldrich | Cat# D9891 |
| 5-Ethynyl-2′-deoxyuridine | Sigma-Aldrich | Cat# 900584 |
| 5-Ethynyl-Uridine | Click Chemistry Tools | Cat# 1261 |
| Palbociclib | Selleck Chemicals | Cat# S1116 |
| Abemaciclib | Selleck Chemicals | Cat# S7158 |
| Ribociclib | Selleck Chemicals | Cat# S7440 |
| Trametinib | Selleck Chemicals | Cat# S2673 |
| EN4 | Selleck Chemicals | Cat# S9807 |
| Bortezomib | MedChem Express | Cat# HY-10227 |
| Ixazomib | MedChem Express | Cat# HY-10453 |
| Carfilzomib | MedChem Express | Cat# HY-10455 |
| Alpelisib | MedChem Express | Cat# HY-15244 |
| Fulvestrant | MedChem Express | Cat# HY-13636 |
| Pictilisib | MedChem Express | Cat# HY-50094 |
| Palbociclib (mice experiment) | MedChem Express | Cat# HY-50767 |
| Chloroquine | MedChem Express | Cat# HY-17589A |
| MG-132 | Calbiochem | Cat# 474790 |
| Epoxomicin | Cayman Chemical | Cat# 134381-21-8 |
| ONX0914 | Cayman Chemical | Cat# 960374-59-8 |
| Celastrol | Cayman Chemical | Cat# 34157-83-0 |
| Omarigliptin | Cayman Chemical | Cat# 1226781-44-7 |
| PMSF | Cayman Chemical | Cat# 329-98-6 |
| Maribavir | Cayman Chemical | Cat# 176161-24-3 |
| MLN9708 (Ixazomib citrate) | Cayman Chemical | Cat# 1201902-80-8 |
| Nafamostat mesylate | Cayman Chemical | Cat# 82956-11-4 |
| N-ethylmaleimide | Cayman Chemical | Cat# 128-53-0 |
| Camostat mesylate | Cayman Chemical | Cat# 59721-29-8 |
| Leupeptin hemisulfate | Cayman Chemical | Cat# 103476-89-7 |
| MG101 | Cayman Chemical | Cat# 110044-82-1 |
| Loxistatin acid | Cayman Chemical | Cat# 76684-89-4 |
| PI1840 | Cayman Chemical | Cat# 1401223-22-0 |
| Aloxistatin | Cayman Chemical | Cat# 88321-09-9 |
| VR23 | Cayman Chemical | Cat# 1624602-30-7 |
| Paritaprevir | Cayman Chemical | Cat# 1221573-85-8 |
| Delanzomib | Cayman Chemical | Cat# 847499-27-8 |
| Oprozomib | Cayman Chemical | Cat# 935888-69-0 |
| Z-FA-FMK | Cayman Chemical | Cat# 197855-65-5 |
| 17β-estradiol pellet (0.36 mg) | Innovative Research of America | Cat# SE-121 |
| 16% Paraformaldehyde | Ted Pella Inc | Cat# NC1537886 |
| Fetal Bovine Serum (FBS) | Gibco | Cat# 10-437-028 |
| Puromycin | Invivogen | Cat# ant-pr-1 |
| Blasticidin | Invivogen | Cat# ant-bl-1 |
| DPBS without Ca2+, Mg2+ | Genesee | Cat# 25-508 |
| Triton X-100 | Millipore Sigma | Cat# T8787 |
| Hoechst 33342 | Thermo Scientific | Cat# 62249 |
| Dimethyl sulfoxide (DMSO) | Millipore Sigma | Cat# D2438 |
| Trypsin-EDTA, 0.25% 1X, phenol red, without Calcium and Magnesium | Genesee | Cat# 25-510 |
| Bovine Serum Albumin (BSA) Protease-free powder | Millipore Sigma | Cat# A3311 |
| Protein G Agarose Beads | Cell Signaling Technology | Cat# 37478 |
| SimplyBlue™ SafeStain | Invitrogen | Cat# LC6060 |
| AFDye 647 Picolyl Azide | Click Chemistry Tools | Cat# 1300 |
| (+)-SODIUM L-ASCORBATE | Millipore Sigma | Cat# A4034 |
| COPPER(II) SULFATE (CuSO4) | Millipore Sigma | Cat# C1297 |
| Halt protease inhibitor cocktail | Thermo Scientific | Cat# 1861279 |
| PhosSTOP | Millipore Sigma | Cat# 4906845001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| GeneJET Plasmid Miniprep Kit | Thermo Fisher | Cat# K0503 |
| NucleoBond Xtra Midi prep kit | Macherey-Nagel | Cat# 740410.100 |
| PureLink™ RNA Mini Kit | Invitrogen | Cat# 12183018A |
| SuperScript IV VILO Master Mix | Invitrogen | Cat# 11756050 |
| PowerTrack SYBR Green Master Mix | Thermo Fisher | Cat# A46109 |
| ViewRNA ISH Cell Assay | Invitrogen | Cat# QVC0001 |
| ViewRNA Cell Probe Set: E2F1 | Invitrogen | Cat# VA1-12108-VC |
| ViewRNA Cell Probe Set: Cdc25A | Invitrogen | Cat# VA1-15814-VC |
| ViewRNA Cell Probe Set: RB1 | Invitrogen | Cat# VA1-3000151-VC |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| MCF-7 | ATCC | Cat# HTB-22 |
| CAMA-1 | ATCC | Cat# HTB-21 |
| MDA-MB-157 | ATCC | Cat# HTB-24 |
| MDA-MB-231 | ATCC | Cat# CRM-HTB-26 |
| MDA-MB-468 | ATCC | Cat# HTB-132 |
| T47D | ATCC | Cat# CRL-2865 |
| HCC1428 | ATCC | Cat# CRL-2327 |
| HCC1143 | ATCC | Cat# CRL-2321 |
| HCC1937 | ATCC | Cat# CRL-2336 |
| BT-483 | ATCC | Cat# HTB-121 |
| BT-20 | ATCC | Cat# HTB-19 |
|
| ||
| Deposited Data | ||
|
| ||
| Raw data | Mendeley Data | Mendeley: http://doi.org/10.17632/c47xz5mgp3.1 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| J:NU mice | The Jackson Laboratory | Cat#007850 |
| Experimental models: Organisms/strains | ||
| gRNA targeting Rb (GUUCGAGGUGAACCAUUAAUGUUUUAGAGCUAUGCU) | Integrated DNA Technologies | N/A |
| gRNA targeting Rb (AAGUGAACGACAUCUCAUCUGUUUUAGAGCUAUGCU) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE1, sense sequence (GAUCAGCACUUUCUUGAGCAACACC) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE1, anti-sense sequence (CCCUAGUCGUGAAAGAACUCGUUGUGG) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE1, sense sequence (AUGCAAAAGGUUUCAGGGUAUCAGT) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE1, anti-sense sequence (ACUACGUUUUCCAAAGUCCCAUAGUCA) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE2, sense sequence (CAUUCUGACUUGGAACCACAGAUGA) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE2, anti-sense sequence (ACGUAAGACUGAACCUUGGUGUCUACU) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE2, sense sequence (ACGUAAGACUGAACCUUGGUGUCUACU) | Integrated DNA Technologies | N/A |
| siRNA targeting CCNE2, anti-sense sequence (AGUCAGGAACGUAAUAGUAACUUUGUG) | Integrated DNA Technologies | N/A |
| siRNA targeting c-Myc, sense sequence (AUCAUUGAGCCAAAUCUUAAAAAAA) | Integrated DNA Technologies | N/A |
| siRNA targeting c-Myc, anti-sense sequence (UAUAGUAACUCG GUUUAGAAUUUUUUU) | Integrated DNA Technologies | N/A |
| siRNA targeting c-Myc, sense sequence (CGACGAGACCUUCAUCAAAAACATC) | Integrated DNA Technologies | N/A |
| siRNA targeting c-Myc, anti-sense sequence (CUGCUGCUCUGGAAGUAGUUUUUGUAG) | Integrated DNA Technologies | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLV-DHB (a.a.995–1087)-mVenus-p2a-mCherry-Rb (a.a.886–928)-IRES-blasticidin | Yang et al.33 | RRID: Addgene_126679 |
| pLV-H2B-iRFP670-p2a-mCerulean-Geminin (1–110)-IRES-Neomycin | This paper | |
| pLV-YFP-Rb-IRES-blasticidin | This paper | |
| pLV-YFP-Rb (14 Ser/Thr to Glu)-IRES-puromycin | This paper | |
| pLV-H2B-iRFP670-p2a-YFP-Rb-IRES-blasticidin | This paper | |
| pCW57.1-Cyclin D1-IRES-puromycin | This paper | |
| pCW57.1-Cyclin E1-IRES-puromycin | This paper | |
| pCW57.1-c-Myc-IRES-puromycin | This paper | |
| pCMV-VSV-G | Stewart et al.59 | RRID: Addgene_8454 |
| pRSV-rev | Dull et al.60 | RRID: Addgene_12253 |
| pMDLg/pRRE | Dull et al.60 | RRID: Addgene_12251 |
|
| ||
| Software and algorithms | ||
|
| ||
| MATLAB | MathWorks | https://www.mathworks.com/ |
| MATLAB Scripts | MathWorks | http://doi.org/10.5281/zenodo.8310175 |
| Image Studio Lite | LI-COR Biosciences | https://www.licor.com/bio/image-studio-lite/ |
| Image Lab 5.2.1 | Bio-Rad | https://www.bio-rad.com/en-us/product/image-lab-software?ID=KRE6P5E8Z |