

Abstract
Human artificial chromosomes (HACs) are considered promising tools for gene delivery, functional analyses, and gene therapy. HACs have the potential to overcome many of the problems caused by the use of viral-based gene transfer systems, such as limited cloning capacity, lack of copy number control, and insertional mutagenesis during integration into host chromosomes. The recently developed alphoidtetO-HAC has an advantage over other HAC vectors because it can be easily eliminated from dividing cells by inactivation of its conditional kinetochore. This provides a unique control mechanism to study phenotypes induced by a gene or genes carried on the HAC. The alphoidtetO-HAC has a single gene acceptor loxP site that allows insertion of an individual gene of interest or a cluster of genes of up to several Mb in size in Chinese hamster ovary (CHO) hybrid cells. The HACs carrying chromosomal copies of genes can then be transferred from these donor CHO cells to different recipient cells of interest via microcell-mediated chromosome transfer (MMCT). Here, we describe a detailed protocol for loading a gene of interest into the alphoidtetO-HAC vector and for the subsequent transfer of the HAC to recipient cells using an improved MMCT protocol. The original MMCT protocol includes treatment of donor cells with colcemid to induce micronucleation, wherein the HAC becomes surrounded with a nuclear membrane. That step is followed by disarrangement of the actin cytoskeleton using cytochalasin B to help induce microcell formation. The updated MMCT protocol, described here, features the replacement of colcemid and cytochalasin B with TN16 + griseofulvin and latrunculin B, respectively, and the use of collagen/laminin surface coating to promote attachment of metaphase cells to plates during micronuclei induction. These modifications increase the efficiency of HAC transfer to recipient cells ten fold. The improved MMCT protocol has been successfully tested on several recipient cell lines, including human mesenchymal stem cells and mouse embryonic stem cells. Published 2021. This article is a U.S. Government work and is in the public domain in the USA. Current Protocols published by Wiley Periodicals LLC.
Basic Protocol 1: Insertion of a BAC containing a gene of interest into a single loxP loading site of alphoidtetO-HAC in hamster CHO cells
Basic Protocol 2: Microcell-mediated chromosome transfer from donor hamster CHO cells to mammalian cells
Keywords: gene delivery vectors, HAC, human artificial chromosome, microcell-mediated chromosome transfer, MMCT
INTRODUCTION
Human artificial chromosomes (HACs) are a new generation of gene delivery vectors that can hold megabase-size DNA sequences such as a genomic copy of a gene or a group of genes spanning an entire genomic locus (Ikeno & Hasegawa, 2020; Kouprina, Earnshaw, Masumoto, & Larionov, 2013; Kouprina et al., 2018; Kouprina, Tomilin, Masumoto, Earnshaw, & Larionov, 2014; Moralli & Monaco, 2020; Oshimura, Uno, Kazuki, Katoh, & Inoue, 2015; Satoh et al., 2018; Sinenko, Ponomartsev, & Tomilin, 2020, 2021; Wang, 2015). HACs contain a functional centromere, which allows for their long-term stable maintenance as single copy episomes without integration into host chromosomes, minimizing complications such as silencing of the incorporated gene(s).
There are two types of HACs, namely, top-down (linear) and bottom-up (circular). Construction of top-down HACs is based on telomere-directed natural chromosome truncation using p- and q-arm targeting/telomere-seeding vectors. Figure 1A depicts these vectors with their targeting and telomere sequences. The size of linear HACs ranges from 1 Mb to 10 Mb (Mills, Critcher, Lee, & Farr, 1999; Kakeda et al. 2011 and references therein). The incorporated natural centromere confers mitotic stability during cell propagation.
Figure 1.
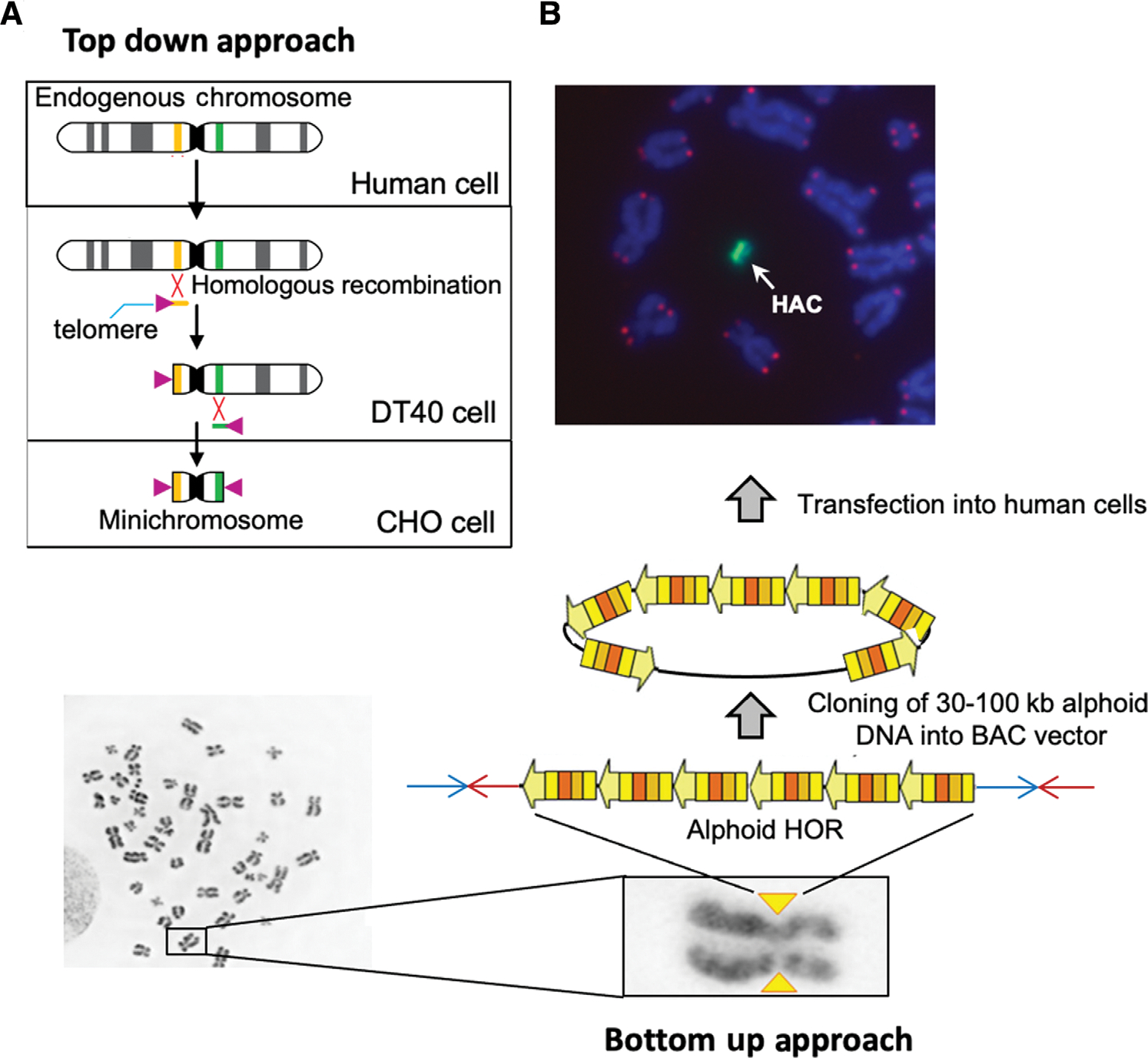
Construction of human artificial chromosomes (HACs). (A) Engineering of a HAC via a top-down approach. The HAC is generated by subsequent truncation of the p- and q-arms of the natural chromosome. As a first step, a human chromosome is transferred to homologous recombination-proficient chicken DT40 cells. The targeting linearized vectors containing human telomeric repeats (purple triangles) and sequences homologous to the q- and p-arms of the chromosome (in orange and in green) are transfected into DT40 cells. Recombinational interaction between the targeting sequences of the vectors and the targeted sequences at the chromosome seeds the formation of new telomeres, thereby truncating the distal portion of chromosomal arms and leads to formation of a mini-chromosome or a linear HAC. A gene-loading loxP cassette was included into some HACs during chromosome truncation (i.e., a cassette is included into one of the targeting vectors). Then, the HAC is transferred into Chinese hamster ovary (CHO; hprt−/−) cells. In CHO cells, a desired gene can be loaded into the loxP site of HAC by Cre/loxP-mediated recombination. From CHO cells, the HAC with a gene of interest can be transferred to desired recipient cells via a microcell mediated transfer technique (MMCT) for further gene complementation/function assays. (B) Generation of HACs via a bottom-up approach. (Left) Each human chromosome contains a centromere consisting of high-order repeats (HORs) of alpha-satellite DNA that form a discontinuous array of 1–5 Mb in size (yellow triangles). (Center) A HOR DNA array of ~30–100 kb in size cloned in a BAC vector after its transfection into human cells forms a de novo circular HAC. After transfection, the input 50-kb alphoid DNA is amplified up to 1–10 Mb. (Top) DNA fluorescence in situ hybridization (FISH) analysis using an alphoid-specific probe reveals the HAC in human cells (in green). DAPI stains the natural chromosomes (in blue). Red dots indicate telomeres. A white arrow indicates the HAC.
Bottom-up (or de novo constructed) HACs, on the other hand, are formed using alphoid DNA arrays of 30 to 100 kb in size, available in a BAC form from BAC libraries (Fig. 1B) or by using synthetic alphoid DNA arrays (Harrington, Van Bokkelen, Mays, Gustashaw, & Willard, 1997; Ikeno et al., 1998; Pesenti et al., 2018). Originally, de novo HACs were generated after transfection of the alphoid arrays into human fibrosarcoma HT1080 cells. The size of these HACs ranges from 1 to 10 Mb, depending on the extent of amplification of the input alphoid DNA. These HACs also possess a functional centromere and can, thereby, autonomously replicate and segregate in human cells. Further work has shown that tethering of histone acetyltransferases to the transforming alphoid DNA arrays eliminates the cell-type-specific barrier to the de novo assembly of centromere protein A (CENP-A) and other kinetochore proteins, enabling HAC formation in a wide range of cell types (Ohzeki et al., 2012).
For gene functional studies and potentially for gene therapy, it is important that HACs contain a single gene-loading loxP site that allows insertion of a gene or locus of interest (Hoshiya et al., 2009; Kazuki et al., 2010; Kim et al., 2011; Tedesco et al., 2012; Yakura et al., 2013; Kobayashi et al., 2017; Kononenko et al., 2015; Kazuki et al., 2019; Ohta et al., 2020; Ponomartsev et al., 2020). Recently, several HACs have been constructed that feature multiple gene-loading sites to carry several genes or to assemble a single large gene (Honma et al., 2018; Lee et al., 2018; Suzuki, Kazuki, Oshimura, & Hara, 2014; Toth et al., 2014; Yamaguchi et al., 2011; Yoshimura et al., 2015). Such HACs would allow investigation of complex biological pathways and of gene regulation, and have the potential to allow the engineering of synthetic chromosomes with a predetermined set of multiple genes.
The most advanced top-down HAC is the 21HAC vector, engineered by truncation of p- and q-arm sequences on human chromosome 21 (Kazuki et al., 2011). It was shown that this HAC contains no known gene. In addition, this HAC contains a single gene acceptor loxP site for gene loading, which makes it useful for gene delivery and gene expression in different human cells.
The most advanced de novo constructed HAC is the alphoidtetO-HAC, engineered from a 50-kb synthetic alphoid DNA array (Fig. 2A; Nakano et al., 2008). After transfection of the alphoid array into human fibrosarcoma HT1080 cells, the input DNA was amplified up to 1.1 Mb, leading to HAC formation (Fig. 2B). The alphoidtetO-HAC vector was adapted for gene functional studies by insertion of a single gene acceptor loxP site (Iida et al., 2010) and a multi-integrase cassette allowing an unlimited number of gene loadings (Lee et al., 2018). Organization of the alphoid DNA array in the alphoidtetO-HAC has been defined (Kouprina et al., 2012). This HAC contains a “conditional” kinetochore: Each repeat in the alphoid array contains the tetracycline operator (tet-O) sequence, which can be “attacked” by the binding of tet-repressor (tet-R) fusion proteins, resulting in the loss of the HAC—and of the loaded gene—from populations of dividing cells (Fig. 2C; Kononenko et al., 2015; Kouprina et al., 2018). This feature of the HAC provides a route to compare the phenotype of human cells with and without a functional copy of a gene of interest loaded into the HAC. In all, the alphoidtetO-HAC vector possesses all of the requirements for an advanced gene delivery vector.
Figure 2.
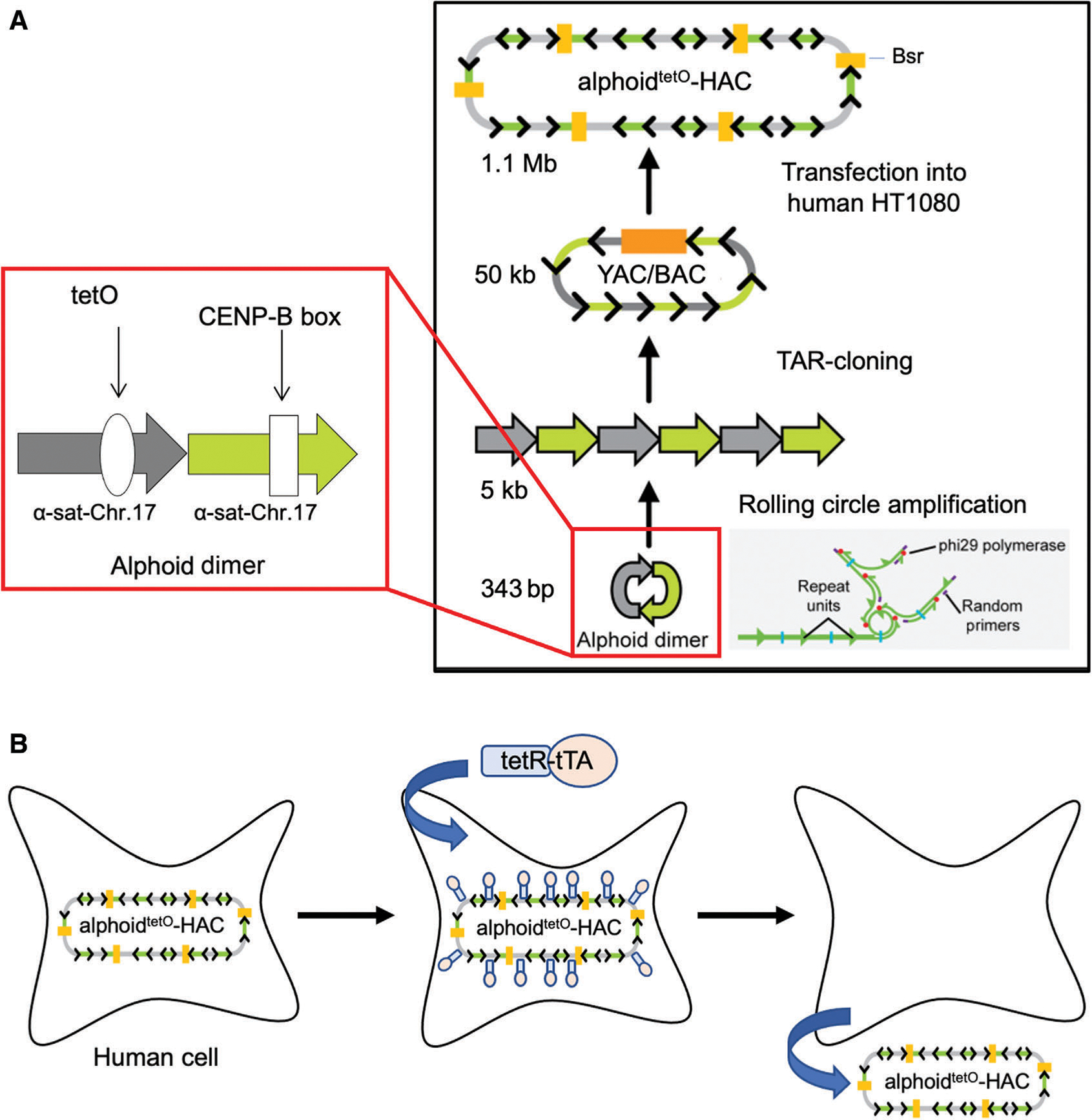
Generation of the alphoidtetO-HAC via a bottom-up approach using a synthetic alphoid DNA array. (A) Schematic representation of construction of a synthetic tetO-containing DNA tandem repeats array of ~50 kb in size by rolling circle amplification (RCA) in vitro and transformation-associated recombination (TAR) cloning in yeast cells (Ebersole et al., 2005). The 343 bp synthetic dimer is amplified up to ~5 kb in size fragments by RCA. One monomer of the dimer is derived from a chromosome 17 alphoid type I 16-mer unit and contains a CENP-B box. The second monomer is a wholly synthetic sequence derived from alphoid DNA consensus, with sequences corresponding to the CENP-B box replaced by a 42-bp tetO motif. Then, the RCA-amplified 5-kb fragments are assembled in yeast by TAR cloning, leading to formation of an ~50-kb synthetic array cloned in a YAC/BAC vector that is moved from yeast to bacterial cells for further BAC DNA isolation. After transfection of 50 kb input BAC DNA into human cells, the alphoidtetO-HAC (tetO-HAC) is formed. Formation of the HAC is accompanied by multimerization of input DNA up to 1.1 Mb. The HAC is selected in human cells on blasticidin-containing medium. (B) Loss of the alphoidtetO-HAC from cells is induced by the transcriptional activator (tTA) fused with the tet-repressor (tetR) targeting the HAC kinetochore.
A genomic copy of a gene or locus of interest can be directly isolated from a genome as a circular yeast artificial chromosome (YAC) by transformation-associated recombination (TAR) in the yeast Saccharomyces cerevisiae (Fig. 3A; Kouprina & Larionov, 2008; Current Protocols article: Kouprina, Kim, & Larionov, 2021). Such YAC gene isolates may then be retrofitted into a BAC version using the yeast-bacteria-mammalian cell shuttle vector pJBRV1 (Fig. 3B and 3C; Kim et al., 2011), which contains a 3′ HPRT-loxP-eGFP-F′ cassette; this permits gene loading into a loxP site of the HAC in HPRT-deficient Chinese hamster ovary (CHO) hybrid cells. The vector also features an F′ factor origin of replication, which allows YAC propagation as a BAC molecule. Conversion of the YAC into a BAC is advantageous because purification of circular molecules is much easier from Escherichia coli compared to yeast cells. The eGFP transgene allows easy identification of cells that carry the HAC with the loaded gene (Kim et al., 2011; Kononenko et al., 2014).
Figure 3.
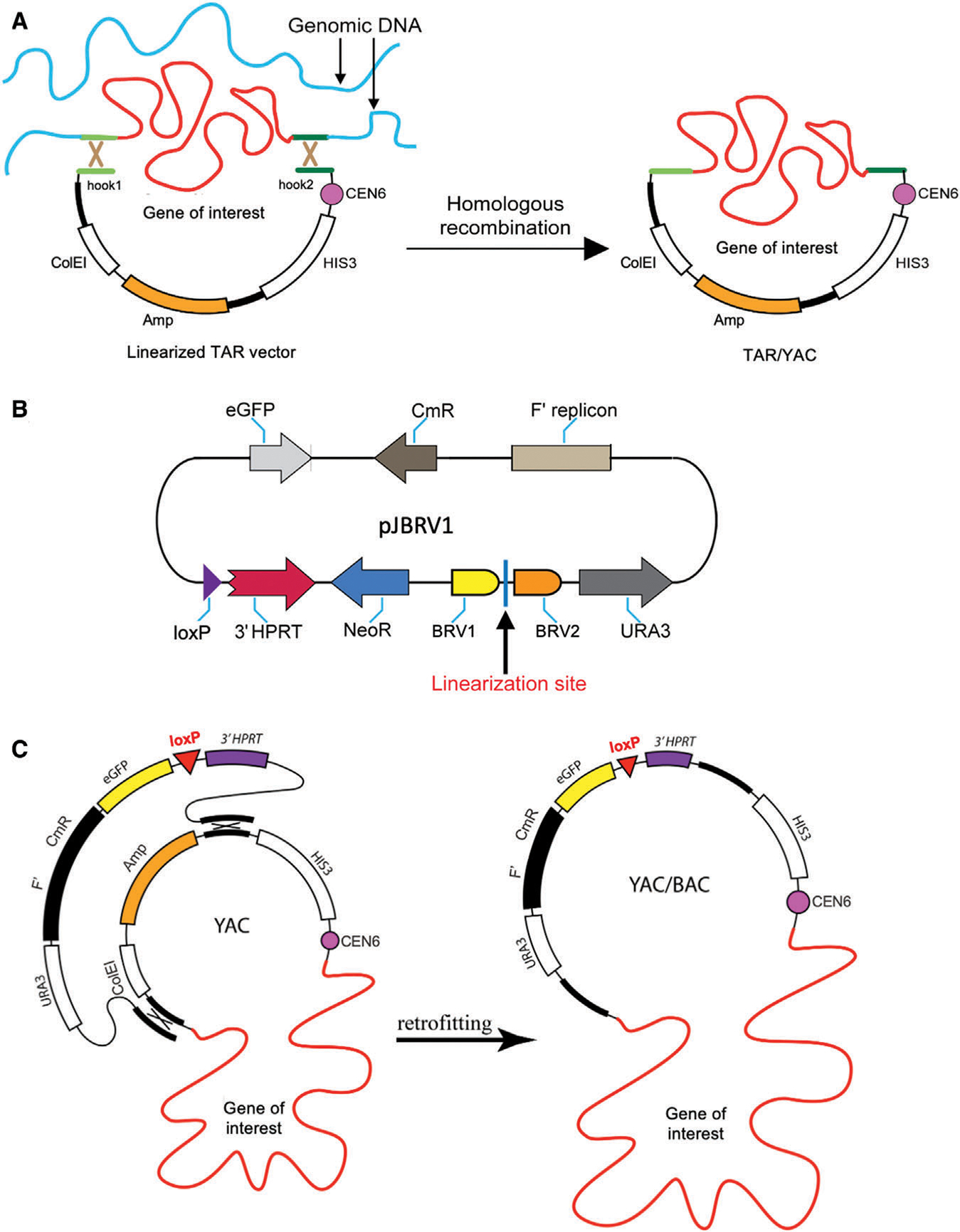
Selective gene isolation from human genome by transformation-associated recombination (TAR) cloning. (A) The diagram shows TAR cloning of a gene of interest from total human genomic DNA with a linearized TAR vector containing two unique targeting sequences (hook 1 and hook 2 in green) homologous to the 5′ and 3′ ends of a gene of interest (also in green; Kouprina, & Larionov, 2008; Kouprina et al., 2021). The TAR vector DNA is linearized at a unique restriction site located between the hooks to expose targeting sequences. After transformation into the yeast Saccharomyces cerevisiae, recombination between targeting sequences in the TAR vector and the targeted sequences of the genomic DNA fragment carrying a gene of interest results in the rescue of the gene/region of interest as a circular TAR/YAC molecule. (B) Scheme of the pJBRV1 vector (Kim et al., 2011). This vector contains the URA3 yeast selectable marker, a BAC cassette containing the F′ factor origin of replication (F′), the chloramphenicol acetyltransferase (CmR) gene, and a 3′ HPRT-loxP-eGFP-NeoR cassette allowing gene loading into a unique loxP site of the alphoidtetO-HAC in Hprt-deficient Chinese hamster ovary (CHO) cells by Cre-loxP mediated recombination. The vector is linearized between two sequences, BRV1 and BRV2 (in yellow and in brown), which have homology to vector sequences in a TAR/YAC molecule. (C) Schematic representation of retrofitting a circular TAR/YAC carrying a gene of interest into a YAC/BAC. Recombination between targeting sequences in the pJBRV1 vector and homologous regions in the TAR/YAC molecule replaces the ColEI origin of replication by the F′ -factor origin of replication that allows propagation of a molecule in a BAC form in bacterial cells. Then, the YAC/BAC molecules along with a gene of interest can be moved to Escherichia coli by electroporation for further BAC DNA isolation. Figures 3A and 3C are adapted from Kononenko et al. (2014).
HACs carrying a gene or a cluster of genes can be moved from a CHO donor cell line to recipient target cells via microcell-mediated chromosome transfer (MMCT; Ege & Ringertz, 1974; Liskovykh, Lee, Larionov, & Kouprina, 2016). MMCT is based on the fusion of appropriate recipient cells and microcells derived from hamster CHO donor cells. Microcells consist of micronuclei surrounded by plasma membrane and are formed by treatment of cells with mitotic spindle inhibitors. Each generated micronuclei contains a single chromosome (Ege, & Ringertz, 1974). Because the chromosomes are maintained inside microcells during transfer, they are protected from physical damage. The advantage of MMCT transfer of artificial chromosomes versus entire chromosomes is that HAC vectors can carry an individual gene or a gene cluster of interest, whereas natural chromosomes contain thousands of genes, which can confound the study of a single, specific gene or cluster. The efficiency of MMCT, however, is low and can benefit from further improvement.
In this article, we describe two basic protocols for the efficient delivery of a gene of interest into target cells via HAC vectors using MMCT. Basic Protocol 1 describes insertion of a BAC containing a gene/locus of interest (which can be obtained by TAR cloning; see Fig. 2) into a single loxP loading site of the alphoidtetO-HAC in donor hamster CHO cells (Fig. 4). Basic Protocol 2 describes an improved MMCT protocol for HAC transfer from CHO cells to target recipient cells (Liskovykh et al., 2016). In this updated protocol, we have replaced the use of colcemid and cytochalasin B with TN-16 + griseofulvin and latrunculin B, respectively, (Fig. 5; Liskovykh et al., 2016). We have also optimized the surface coating of culture flasks to promote attachment of the donor cells, in order to retain metaphase cells on their substrate during micronuclei induction. In all, we have found that these modifications increase the efficiency of MMCT transfer about ten fold and reduce MMCT-associated DNA damage.
Figure 4.
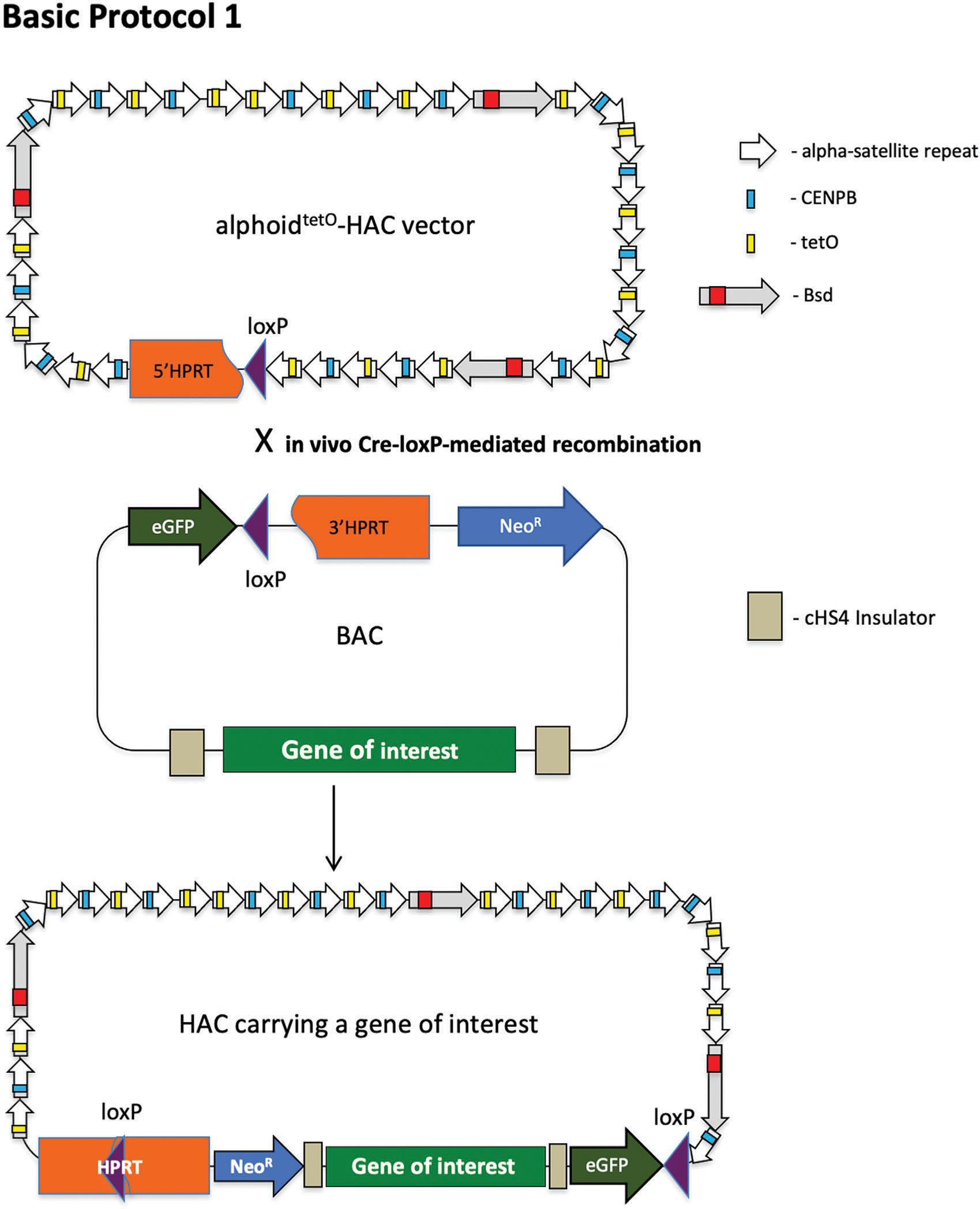
Scheme of Basic Protocol 1, describing the uploading of a full-size genomic copy of a gene of interest into the alphoidtetO-HAC vector. The HAC contains a 5 HPRT-loxP cassette. A gene of interest is cloned by TAR cloning in yeast as a TAR/YAC molecule, retrofitted by pJBRV1 vector, and transferred to bacterial cells as a BAC/YAC molecule for further BAC DNA isolation (see Fig. 3). A BAC molecule contains a 3′ HPRT-loxP-eGFP -NeoR cassette. A gene of interest protected by insulators (Lee et al., 2013) is uploaded into the loxP site of the HAC by Cre-loxP mediated recombination followed by reconstitution of the HPRT gene, which allows cells harboring the gene of interest-containing HAC to grow on HAT-containing medium. HAC, human artificial chromosome; TAR, transformation-associated recombination.
Figure 5.
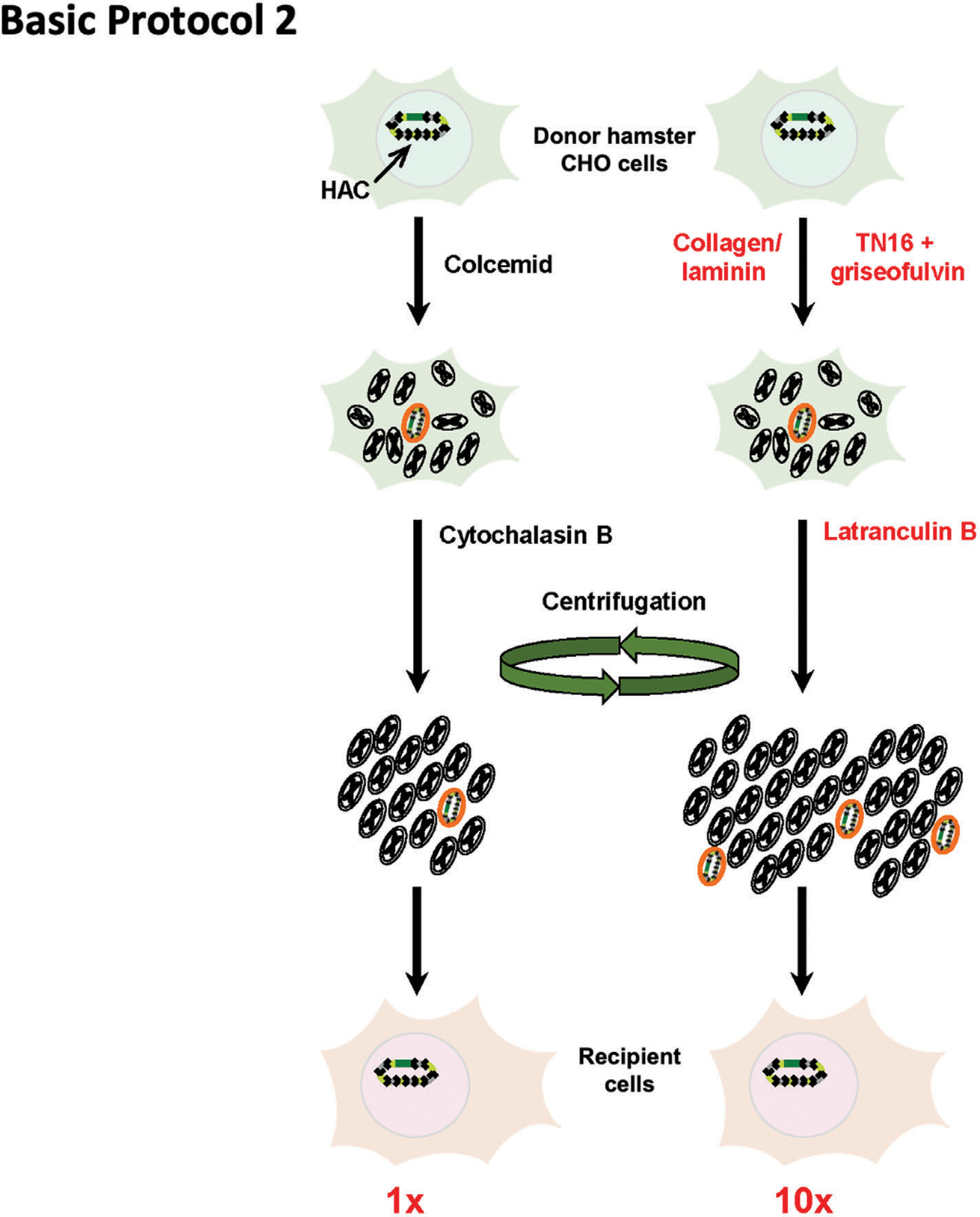
Scheme of Basic Protocol 2. (Left) Original microcell-mediated chromosome transfer (MMCT) technique. (Right) The modified MMCT transfer includes replacement of two key chemicals, colcemid and cytochalasin, by TN16 + griseofulvin and latrunculin B, respectively. In addition, collagen/laminin surface coating promotes attachment of the metaphase cells to plates during micronuclei induction. Efficiency of the new protocol is ~10 times higher compared to the original one (red numbers at the bottom). The figure is adapted from Kouprina et al. (2018).
This improved MMCT technique for gene transfer via HAC vectors has the potential to facilitate gene functional studies and has already been successfully used to transfer HACs to embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), mesenchymal stem cells (MSCs), and embryonic fibroblasts, and to immortalized cell lines such us human fibrosarcoma HT1080, cervical cancer HeLa, and human embryonic kidney HEK293 cells (Fig. 6).
Figure 6.
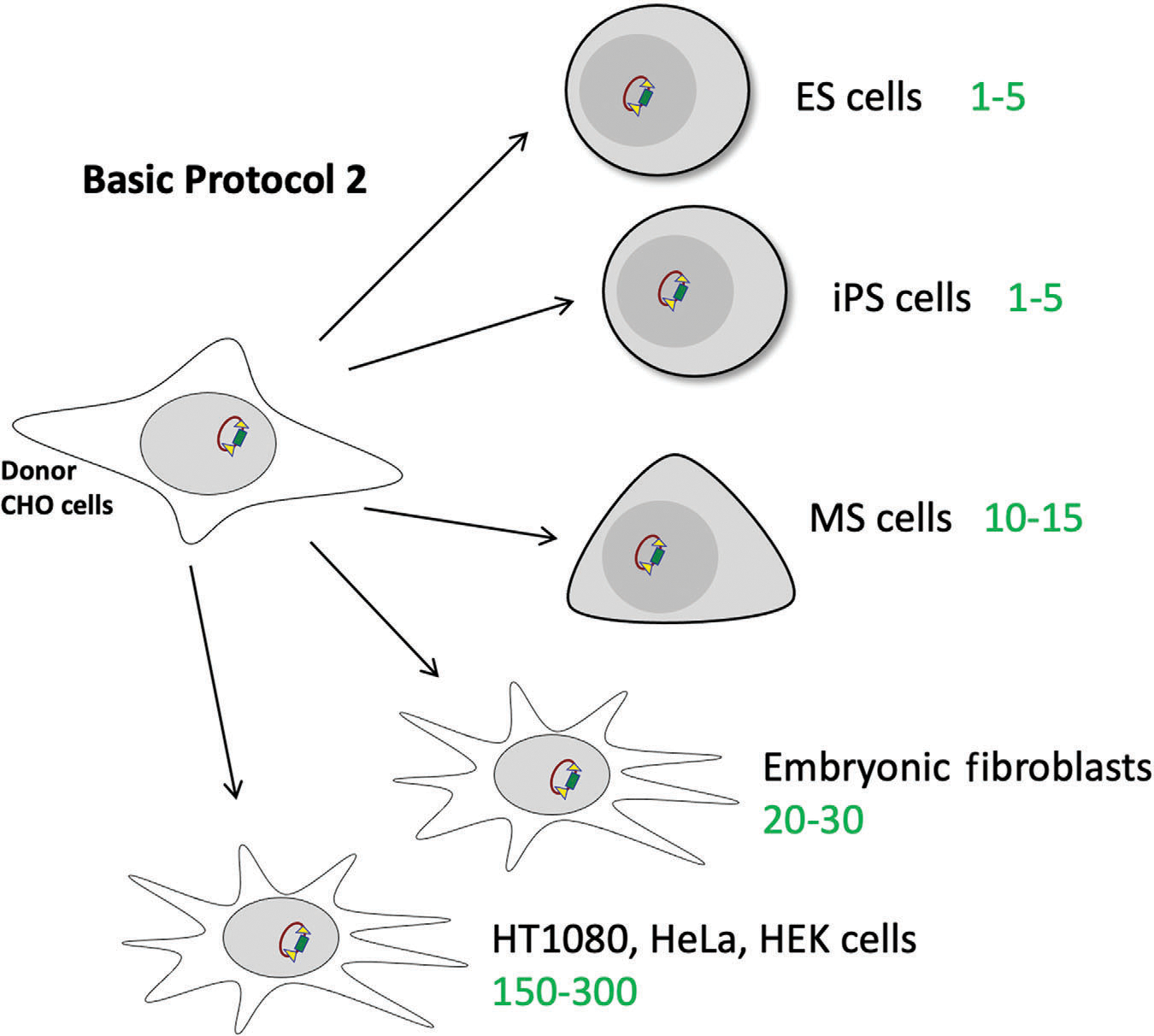
Expected efficiency of Basic Protocol 2 for different cell lines, including primary cell cultures like ES, iPS, MS cells, embryonic fibroblasts, and immortalized cell lines such as HT1080, HeLa, and HEK. The expected number of colonies for each type of cells is marked in green. ES, embryonic stem; iPS, induced pluripotent stem; MS, mesenchymal stem.
STRATEGIC PLANING
Before uploading the gene of interest (or any target DNA fragment, such as a chromosomal locus) into the HAC, a TAR/YAC molecule containing the region of interest should be prepared and then retrofitted by the shuttle vector pJBRV1 into a BAC form (Fig. 2B). The underlying principle of TAR cloning is the capture of the DNA fragment of interest via homologous recombination in yeast, followed by its transfer to E. coli cells. In E. coli cells, the TAR/BAC molecules can be amplified to provide enough material for transfections to donor hamster CHO cells carrying the alphoidtetO-HAC gene delivery vector. Several examples of the use of this procedure (TAR cloning and retrofitting) are described in detail elsewhere (Kim et al., 2011; Kononenko et al., 2014; Kouprina et al., 2021). The pJBRV1 vector is available from the Larionov’s laboratory upon request.
For Basic Protocol 1, the user should employ BAC DNA of transfection-suitable quality containing the gene or region of interest. BAC DNA may be isolated using a Large-Construct Kit (Qiagen, cat. no. 12462). Before transfection, BAC DNA should be checked by contour-clamped homogeneous electric field (CHEF; Current Protocols article: Finney, 2001). This step is necessary to confirm that the BAC DNA is intact and has been isolated predominantly as covalently closed circular molecules (Fig. 7). In addition, to exclude deletions and/or rearrangements, the BAC DNA should be checked by digestion with several restriction enzymes to confirm the expected restriction patterns. If there is any doubt, the BAC DNA should be sequenced before insertion into the HAC (see Kouprina et al., 2021).
Figure 7.
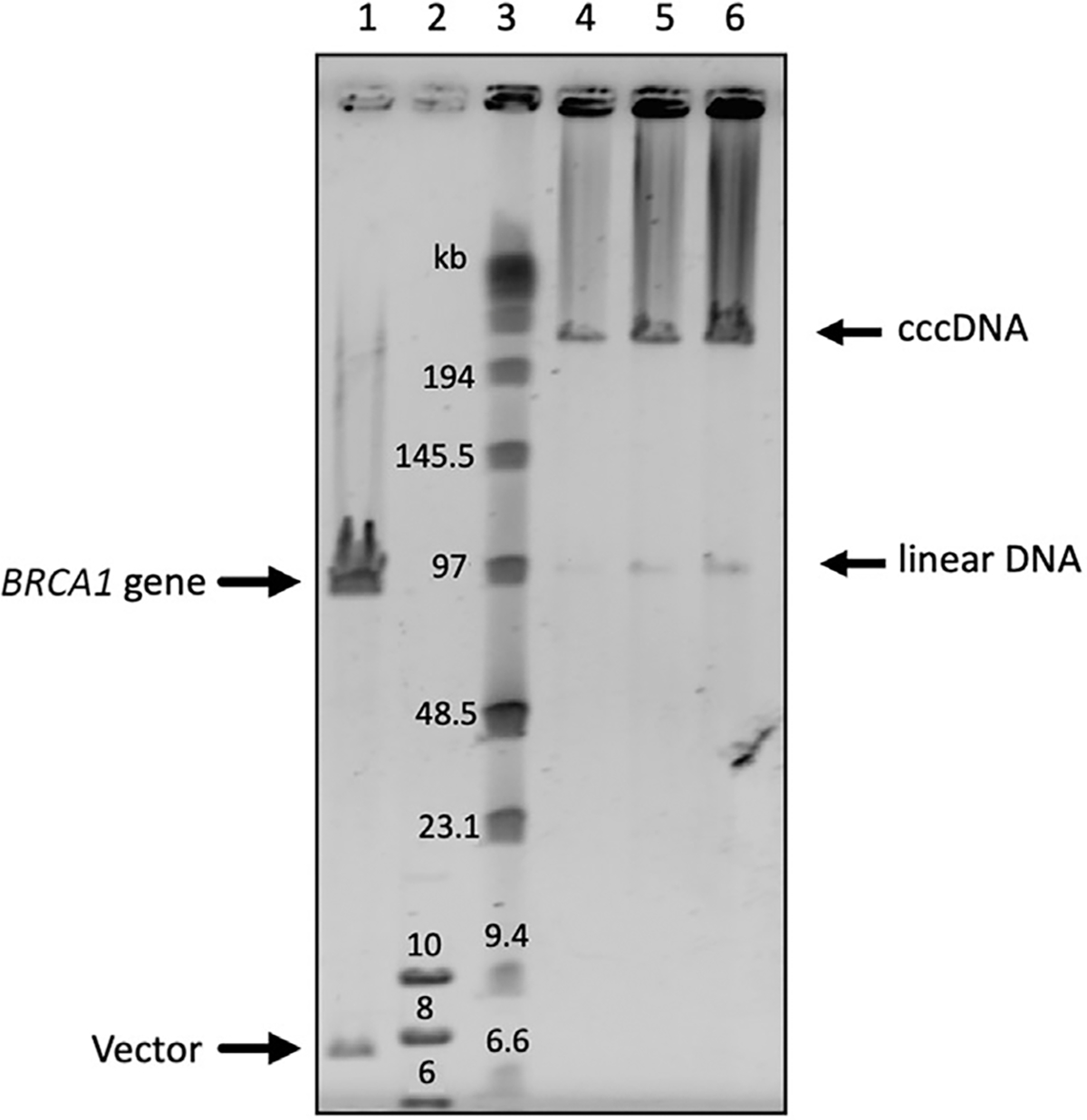
CHEF gel to verify the quality and integrity of BAC DNA preparation. Here, BAC DNA contains the human BRCA1 gene isolated by TAR cloning in yeast (Kononenko et al., 2014). Lane 1 is BRCA1/BAC DNA linearized by NotI. Lane 2 is GeneRuler 1 kb plus DNA ladder (Thermo Fisher Scientific, cat. no. SM1331). Lane 3 is Pulse Marker™ 0.1–200 kb (MilliporeSigma, cat. no. D2291). Lanes 4, 5, and 6 are 0.25, 0.5, and 1 μg of uncut BRCA1/BAC DNA loaded onto the gel, respectively. As seen, the purified BRCA1/BAC DNA is predominately presented as covalently closed circular (ccc) molecules. TAR, transformation-associated recombination.
Hamster CHO cells containing the alphoid-tetOHAC vector should be grown in exponential phase, usually to about 70% to 80% of confluency. The cells are available from the Larionov’s laboratory upon request. The cells should be grown in F12 medium supplemented with Glutamax (Thermo Fisher Scientific, cat. no. 31765035) and FBS (Thermo Fisher Scientific, cat. no. 26140), plus 1% PenStrep (Thermo Fisher Scientific, cat. no. 15070063) and 8 μg/ml blasticidin S (Thermo Fisher Scientific, cat. no. A1113903). If the cells reach full confluence, the culture should be diluted 1:20 and regrown. Each replicate of Basic Protocol 1 requires approximately one to two million cells to perform Cre/loxP loading of 1 μg of the BAC containing a gene of interest into the single loxP site of the HAC.
INSERTION OF A BAC CONTAINING A GENE OF INTEREST INTO A SINGLE LOXP LOADING SITE OF ALPHOIDtetO-HAC IN HAMSTER CHO CELLS
Here, the user will insert a TAR/BAC containing a gene or genomic locus of interest into the alphoidtetO-HAC backbone via Cre/loxP-mediated recombination. There are three major components: CHO cells carrying alphoidtetO-HAC (available upon request from the authors), the TAR/BAC with a gene of interest (see Fig. 2 for details), and the plasmid encoding Cre-recombinase. Cre-mediated recombination (Abremski, & Hoess, 1984) is based on Cre-recombinase recognition of a specific DNA sequence (loxP site), followed by recombination between two loxP sites. The alphoidtetO-HAC contains one recombination site, and the TAR/BAC, another. When the TAR/BAC DNA is transfected along with the Cre-encoding plasmid, the Cre-recombinase expressed from the plasmid carries out recombination between the two loxP sites, inserting the TAR/BAC molecule into the alphoidtetO-HAC. A scheme of the uploading of the TAR/BAC molecule into the loxP site of the alphoidtetO-HAC is shown in Figure 4.
To confirm TAR/BAC uploading into the alphoidtetO-HAC backbone, the user will then exploit the feature that CHO cells are HPRT-deficient and cannot survive when sodium hypoxanthine, aminopterin, and thymidine (HAT) are added to the culture medium. The alphoidtetO-HAC backbone contains a segment of the HPRT gene, and the TAR/BAC vector contains the rest. Each of these parts is nonfunctional by itself, and only when Cre/loxP recombination occurs correctly, the active HPRT gene is reconstituted. This then allows cells to survive and form clones in the presence of HAT supplementation (Fig. 4). Basic Protocol 1 is applicable for HACs containing a single loxP gene-loading site.
Materials
Cre-plasmid (Addgene, cat. no. 31132)
F12 growth medium (see recipe)
2× cell culture freezing medium (CFM; see recipe)
F12-HAT medium (see recipe)
Opti-MEM (Thermo Fisher Scientific, cat. no. 51985034)
Viafect (Promega, cat. no. E4981)
PBS (Thermo Fisher Scientific, cat. no. 10010023)
0.25% trypsin (Thermo Fisher Scientific, cat. no. 25200056)
GeneRuler 1 kb plus DNA ladder (Thermo Fisher Scientific, cat. no. SM1331)
Pulse Marker™ 0.1–200 kb (MilliporeSigma, cat. no. D2291)
Nuclease-free water (Quality Biological, cat. no. 351-029-721)
ReadyMix™ Taq PCR Reaction Mix (MilliporeSigma, cat. no. P4600)
Diagnostic primers to confirm HPRT reconstitution:
F1: 5′-AGCCTTCTGTACACATTTCTTCTC-3′
R1: 5′-GCTCTACTAAGCAGATGGCCACAGAACTAG-3′
Diagnostic primers for gene or region of interest
Large-Construct Kit (Qiagen, cat. no. 12462)
DNA Purification Kit (Qiagen, cat. no. 69506)
6-well culture plates (Thermo Fisher Scientific, cat. no. 140675)
96-well culture plates (Thermo Fisher Scientific, cat. no. 167008)
24-well culture plates (Thermo Fisher Scientific, cat. no. 142475)
Cloning cylinders (Thermo Fisher Scientific, cat. no. 09-552-20)
Cryovial (Thermo Fisher Scientific, cat. no. 5012–0012)
1.7-ml microcentrifuge tubes (Thomas Scientific, cat. no. 1159M35)
2-ml collection tubes (Qiagen QIAquick PCR Purification Kit, cat. no. 28104)
15-ml centrifuge tubes (Falcon, Corning, cat. no. 352196)
50-ml centrifuge tubes (Falcon, Corning, cat. no. 352070)
10-ml disposable pipets (Falcon, Corning, cat. no. 356551)
10-cm culture dishes (Thermo Fisher Scientific cat. no. 174902)
Cell culture incubator
−80°C freezer
Liquid nitrogen tank
PCR thermocycler
NanoDrop spectrophotometer or equivalent
Refrigerated centrifuge
Refrigerated microcentrifuge
Transfection
Approximate time for this part of the protocol is 3 weeks.
Purify BAC DNA containing a gene of interest and Cre-plasmid from the bacterial stock with large-construct kit.
One day before transfection, plate 3.5 × 105 CHO cells per well in a 6-well plate in 2 ml F12 growth medium so that the cells are ~70%−80% confluent at the time of transfection.
-
The next day, mix 1 μg BAC (30–150 kb in size) and 0.1 μg Cre-plasmid in 200 μl Opti-MEM medium without serum in a 1.7-ml tube. Mix gently by tapping.
Note that this and all the calculations below are for one well of a standard 6-well plate. Increase accordingly.
Mix Viafect transfection reagent gently before use and then add 10 μl directly to the mix from step 3.
Mix gently and incubate 5 min at room temperature.
-
After incubation, add mixture from step 5 directly to one well with the cells (from step 2).
Do not mix by pipetting, just add drop by drop into the well.
Changing the medium before transfection is not necessary.
-
Shake plate vigorously, backward and forward, and left to right, before placing back in the incubator.
Optional: Change the medium 6 hr after transfection.
Incubate cells overnight (typically 16–18 hr) at 37°C.
Wash cells with PBS once, add 300 μl of 0.25% trypsin, and incubate 5 min at 37°C.
Add 4 ml F12-HAT medium, re-suspend cells by pipetting up and down five to seven times, and transfer suspension to a 10-cm culture dish containing 6 ml F12-HAT medium. Place plate in the incubator.
Let cells grow until individual colonies appear. This usually takes 2–3 weeks.Change F12-HAT medium every 2–3 days.
Pick colonies. To do this, wash cells once with PBS, apply a cloning cylinder around a colony, and add 30 μl 0.25% trypsin into the cylinder’s well. Incubate cells 5 min. Add 150 μl F12-HAT medium into the cylinder. Re-suspend cells well by pipetting five to seven times and transfer suspension into one well of a 96-well plate.
Continue to grow cells and then transfer them as described from one well of a 96-well plate to one well of a 24-well plate for further colony expansion.
Continue to expand each colony until a confluent 10-cm culture plate is obtained.
-
Collect cells from the 10-cm culture plate. To do this, wash cells once with 10 ml PBS, add 2 ml 0.25% trypsin, incubate 5 min at 37°C, add 8 ml F12 growth medium, and re-suspend well. Use 5 × 105 cells for further genomic DNA extraction. Prepare frozen stocks for the remaining cells.
It is important not to cross-contaminate culture. Use separate pipet tips for each individual clone.
To prepare frozen stocks, centrifuge the cells at 1,200 rpm (1,000 × g) for 3 min at room temperature. Discard the supernatant. Re-suspend the cell pellet in 1 ml of F12 growth medium and add 1 ml of 2× CFM (see Reagents and Solutions). Mix well and add 0.5 ml of the mixture to four cryovials. Place cryovials into the freezing box and place it at −80°C. For long-term storage, place the cells in the liquid nitrogen tank the next day.
-
To check whether the DNA segment of interest has been inserted into the HAC intact, isolate genomic DNA using a genomic DNA purification kit and then use 1 μl of the DNA elution in a 50-μl PCR reaction with diagnostic primers to confirm HPRT gene reconstitution. Expected fragment size is 1,600 bp.
PCR confirmation of the HPRT gene reconstitution is a critical step, informing the user that the desired gene of interest was correctly uploaded into the HAC backbone. In addition, it is also important to confirm that the gene of interest is intact, at least by PCR analysis, using diagnostic primers for exons. The user should also evaluate whether the gene is expressed, either by reverse transcription PCR (RT-PCR) or Western blot.
Additionally, it is advisable to perform fluorescence in situ hybridization (FISH) analysis on a metaphase spread to confirm that alphoidtetO-HAC remains autonomous and has not integrated into the chromosomes. For more detail, see previously published papers
MICROCELL-MEDIATED CHROMOSOME TRANSFER FROM DONOR HAMSTER CHO CELLS TO MAMMALIAN CELLS
Microcell-mediated chromosome transfer (MMCT) allows individual chromosomes, megabase-sized chromosome fragments, or artificial chromosomes to be transferred from donor cells to a cell of choice (Ege & Ringertz, 1974; Liskovykh et al., 2016).
Here, the user will prepare microcells from the donor hamster CHO cells containing the alphoidtetO-HAC vector carrying the gene of interest (obtained in Basic Protocol 1).
Basic Protocol 2 includes prolonged incubation of the CHO cells in the presence of drugs (TN16 and griseofulvin) that block cell cycle progression in metaphase and induce formation of a nuclear envelope around individual condensed chromosomes, also known as micronuclei. The next step of the procedure involves centrifugation in the presence of an actin cytoskeleton disruptive drug (latranculin B). This stage leads to formation of a cytoplasmic membrane around each micronucleus. Thus, centrifugation yields a population of microcells: A HAC surrounded by two membranes, nuclear and cytoplasmic. The cytoplasmic membrane of the microcells allows them to fuse with recipient cells.
The fusion procedure is similar to the protocol to form a hybridoma cell. There are several methods to induce fusion between two different cell types, but in our protocol we have found the HVJ Envelope Cell Fusion Kit to work well.
Because the alphoidtetO-HAC backbone contains a blasticidin resistance gene, selection should be done with this antibiotic. At the end of the protocol, the user will have several clones of cells carrying the alphoidtetO-HAC bearing the gene of interest. A scheme of the MMCT protocol is presented on Figure 5. Basic Protocol 2 is applicable to any type of HAC.
Additional Materials (see also Basic Protocol 1)
Donor cells from Basic Protocol 1
Target recipient cells
F12 growth medium (see recipe)
DMEM medium (Thermo Fisher Scientific, cat. no. 10566016)
Collagen solution (Thermo Fisher Scientific, cat. no. A1048301; prepare dilution according to the manufacturer’s protocol)
PBS (Thermo Fisher Scientific, cat. no. 10010023)
Laminin solution (Thermo Fisher Scientific, cat. no. 23017015; prepare dilution according to the manufacturer’s protocol)
Griseofulvin (Santa Cruz, cat. no. sc-202171)
TN-16 (Santa Cruz, cat. no. sc-204347)
Latranculin B (Santa Cruz, cat. no. sc-203318)
HVJ Envelope Cell Fusion Kit (Cosmo Bio, cat. no. ISK-CF-001-EX)
Endotoxin-free water (Thermo Fisher Scientific, cat. no. 10977023)
~2 L of distilled sterile water (37°C pre-warmed)
T25 culture flasks (Thermo Fisher Scientific, T25, cat. no. 156340)
8-μm filters (Millipore, cat. no. TETP02500)
5-μm filters (Millipore, cat. no. TMTP02500)
3-μm filters (Millipore, cat. no. TSTP02500)
Swinnex filter holders (Millipore, cat. no. SX0002500)
10-cm culture dishes (Thermo Fisher Scientific, cat. no. 174902)
0.5-L spin bottles
0.5-L sterile bottles
Parafilm
50-ml centrifuge tubes (Corning, Falcon, cat. no. 352070)
15-ml centrifuge tubes (Corning, Falcon, cat. no. 352196)
1.7-ml microcentrifuge tubes (Thomas Scientific, cat. no. 1159M35)
2-ml collection tubes (Qiagen QIAquick PCR Purification Kit, cat. no. 28104)
Autoclave
Centrifuge for 0.5-L bottles (Sorvall RC 5B or equivalent)
Centrifuge for 1.7–2 ml tubes
Water bath (37°C pre-warmed)
Hemocytometer
Refrigerated centrifuge
Fluorescent light microscope
Microcell-mediated chromosome transfer
Steps 1–8 are preparational steps. Estimated time for these steps is 2 days.
-
1
Prepare medium for donor CHO cells: F12, 10% FBS, 1% PenStrep, 10 μg/ml blasticidin.
-
2
Prepare the appropriate medium for the recipient cell line of choice.
-
3
Incubate six T25 flasks, each with 2 ml collagen solution (final concentration: 50 μg/ml), overnight at room temperature.
-
4
Remove collagen solution.
Optional: Collagen solution can be stored for further use at 4°C for a month.
-
5
Gently wash flasks with 3 ml PBS, three times.
-
6
Add laminin solution (final concentration: 25 μg/ml in PBS) to each flask, 2 ml per flask, and incubate overnight at room temperature.
Optional: Laminin solution can be stored for further use at 4°C for a month.
-
7
Wash flasks with 3 ml PBS, three times.
Optional: Collagen/laminin-treated flasks can be stored at 4°C for up to 4 weeks. Be sure to seal the flasks carefully to avoid contamination.
-
8
Assemble four 8-μm, four 5-μm, and four 3-μm filters in Swinnex filter holders. Wrap them with aluminum foil, autoclave 40 min, and then dry 2–3 hr.
The filters can be prepared in advance and stored at room temperature.
Prepare cell culture
Steps 9–12 are cell culture preparation steps. Estimated time for these steps is 3 days.
-
9
Grow up to 3 × 106 of the recipient cells in a 10-cm culture dish in proper medium until they reach 70%−80% of confluency.
Users will need the cells ready in step 33, so plan accordingly.
-
10
Add 3 ml culture medium to each of the six flasks from step 7 and seed donor CHO cells from Basic Protocol 1 until they reach 80% confluency.
Typically, to have 80% of culture confluency on the next day, the user should seed 3 × 105 CHO cells per flask. Sometimes, however, it takes 2 days to reach 80% confluency. Users should check culture confluency before proceeding to the next step.
-
11
When the CHO donor cells reach 80% confluency, change culture medium to F12 growth medium containing a cytostatic cocktail: 160 μM TN-16 and 50 μM griseofulvin. Change medium with fresh F12 growth medium plus cytostatic cocktail every 24 hr.
-
12
Grow cells for 72 hr.
After 72 hr, proceed immediately to microcell preparation.
Prepare microcells and recipient cells
Approximate time for this part of the protocol is 2–4 hr.
-
13
Prepare the latranculin B medium by mixing 400 ml DMEM and 40 μl 2 mM latranculin B in a sterile 0.5-L bottle.
-
14
Warm latranculin B-containing medium to 37°C.
-
15
Collect cell culture medium from all T25 culture flasks with the CHO donor cells (from step 12), centrifuge at 1,200 rpm (1,000 × g) for 3 min at room temperature, and discard supernatant. Re-suspend cells in 6 ml latranculin B-containing medium and put cells back into the flasks.
-
16
Fill each T25 flask until it is full with latranculin B-containing medium (typically, 60–61 ml of the medium per flask).
Fill the flask as much as possible with the medium.
-
17
Close cap of the flask tightly, and closely wrap each neck and cap of the flask with Parafilm three times to protect flasks from contamination with water.
-
18
Place each T25 flask from step 16 into a 0.5-L spin bottle with a wide neck and attach them with an adhesive tape to a bottle wall from the inside (see Fig. 8). The side where the cells are seating should face the inside (center) of the bottle. Fill each of the spin bottles with the attached flask with pre-warmed (37°C) distilled water (130 ml per each bottle). Each bottle (with the flask and the water) should weigh ~280 g. Check that they are balanced before placing bottles into the centrifuge.
-
19
19. Centrifuge water-filled bottles containing the flasks for 60 min at 34°C at 8,000 rpm(10,800 × g).
-
20
After centrifuging, remove latranculin B-containing medium.
Microcell pellets become visible at the angles of the flask.
Do not aspirate the microcell pellets off.
-
21
Add 1 ml of warm serum-free DMEM medium to each flask and re-suspend microcell pellets completely.
-
22
Transfer re-suspended microcells into a 50-ml Falcon tube.
-
23
Wash flasks once again with 1 ml DMEM medium and collect microcells in the same tube.
-
24
Centrifuge microcell suspension at 3,000 rpm (900 × g) for 5 min.
-
25
Re-suspend microcell pellets in 1 ml DMEM medium.
-
26
Adjust volume to 48 ml with DMEM medium.
-
27
Divide the combined microcell suspension into four equal portions using 15-ml Falcon tubes.
-
28
Filter each of the microcell portions sequentially through the 8-, 5-, and 3-μm filters prepared in step 8.
-
29
After filtration, centrifuge tubes at 3,000 rpm (900 × g) for 5 min.
-
30
After centrifugation, re-suspend each pellet completely in 1 ml serum-free DMEM medium.
-
31
Combine all pellets together in a 15-ml Falcon tube and adjust volume to 10 ml with DMEM medium. Then, re-suspend the pellet carefully and centrifuge again at 3,000 rpm (900 × g) for 5 min.
-
32
After centrifugation, place tube with the collected microcells under the hood.
Clean microcells are now ready for fusion. A microcell pellet should be visible. If necessary, the microcells can be kept on ice for up to 2 hr.
-
33
Aspirate medium from the recipient cells from step 9.
Cells should be at 70%−80% confluency.
-
34
Wash once with 10 ml PBS, add 2 ml 0.25% trypsin, incubate for 5 min at 37°C, and then add 8 ml of the recipient cell’s culture medium. Re-suspend cells well.
-
35
Count cells using a hemocytometer and transfer 1.5 × 106 cells into two separate 15-ml Falcon tubes. One tube will be used for fusion and the other will serve as a negative control. Treat both tubes equally in the following steps.
-
36
Centrifuge cells at 1,200 rpm (1,000 × g) for 3 min.
-
37
Re-suspend cells in 10 ml serum-free DMEM medium and spin them down at 1,200 rpm (1,000 × g) for 3 min. Repeat.
The clean recipient cells are now ready for fusion.
Users should be aware that steps 13–37 are time consuming, especially for the inexperienced user. Average time for this part of the protocol (steps 13–37) is 4–6 hr and it should be followed immediately by a fusion procedure, i.e., steps 38–49 of this protocol.
Figure 8.

T25 flask position in the centrifuge bottle. (A) Position of the adhesive tape on the flask. (B) Position of the flask inside the centrifuge bottle (upper view). (C) Position of the flask inside the centrifuge bottle (lateral view).
Fusion of microcells and recipient cells
Approximate time for this part of the protocol is 30–45 min.
-
38
Use the HVJ envelope cell fusion kit. Dilute cell fusion buffer (20× concentrate; stored at 4°C, part of the HVJ envelope cell fusion kit) 1:20 with sterile, pure ice-cold water (e.g., endotoxin-free water).
-
39
Prepare 2 ml of 1× cell fusion buffer on ice.
-
40
Suspend microcells (from step 32) and recipient cells (from step 37) pellets separately in 0.5 ml ice-cold cell fusion buffer (from step 39) using 2-ml tubes (preferably round-bottom tubes).
-
41
Centrifuge both suspensions at 3,000 rpm (900 × g) for 3 min at 4°C using a centrifuge in a cold-room or a pre-cooled centrifuge.
-
42
Re-suspend recipient cells to a concentration of 8 × 103 cells/μl in cell fusion buffer.
For example, re-suspend 1.5 × 106 recipient cells in 188 μl of cell fusion buffer.
-
43
Re-suspend microcell pellet in cell fusion buffer using the same volume used to re-suspend the recipient cells.
It is impossible to count the microcells. Therefore, use the same volume as that used for the recipient cells.
-
44
Mix both suspensions together (from steps 42 and 43) by pipetting.
-
45
Place a 10-μl aliquot of the HVJ-E suspension (part of the HVJ envelope cell fusion kit) from −80°C on ice and then add to the mixed cells suspension from step 44. Mix by tapping.
-
46
Leave mixture on ice for 5 min to allow HVJ-E to be adsorbed on the cell surface.
-
47
Place mixture into a water bath at 37°C for 15 min (mix by tapping every 5 min to induce cell fusion).
-
48
After 15 min, centrifuge mixture at room temperature at 3,000 rpm (900 × g) for 3 min to remove residual viral proteins.
-
49
Re-suspend fused cells in 1 ml of the recipient cell medium and transfer mix to a 10-cm culture dish containing 10 ml of the same medium. Place dish in the incubator for 24 hr.
-
50
Start blasticidin S selection after 24 hr of incubation by replacing culture medium with medium containing 10 μg/ml of blasticidin S. Change medium every 2–3 days.
-
51
After 2–3 weeks, pick colonies using cloning cylinders and 0.25% trypsin.
After 2–3 weeks of selection, the user will have individual clones of the recipient cells containing the alphoidtetO-HAC along with the loaded gene of interest. It is important to confirm that the gene of interest remained intact after MMCT transfer. Use PCR analysis with diagnostic primers for exons. The user should also evaluate whether the gene is expressed, either by RT-PCR or Western blot. It is advisable to perform FISH analysis on a metaphase spread to confirm that the alphoidtetO-HAC remained autonomous after MMCT transfer and has not integrated into the chromosomes.
REAGENTS AND SOLUTIONS
Cell culture freezing medium (CFM), 2×
F12 growth medium, supplemented with 20% FBS (Thermo Fisher Scientific, cat. no. 26140) and 20% dimethyl sulfoxide (DMSO; MilliporeSigma, cat. no. D2650–100ML).
Store at 4°C for 2 weeks.
F12 growth medium
F12 medium (Thermo Fisher Scientific, cat. no. 31765035) supplemented with 10% FBS (Thermo Fisher Scientific, cat. no. 26140), 1% PenStrep (Thermo Fisher Scientific, cat. no. 15070063), and 8 μg/ml blasticidin S HCl (10 mg/ml; Thermo Fisher Scientific, cat. no. A1113903).
Store at 4°C up to 2 months.
F12-HAT medium
F12 growth medium (see recipe) with 1× HAT supplement (Thermo Fisher Scientific, cat. no. 21060017).
Store at 4°C up to 2 months.
COMMENTARY
Background Information
Adenovirus-, lentivirus-, and retrovirus-derived vectors that employ complementary DNA (cDNA) or “minigene” constructs have been widely used for decades (Buchholz, Muhlebach, & Cichutek, 2009; Epstein, 2009; Lufino, Edser, & Wade-Martins, 2008; Maier, von Kalle, & Laufs, 2010; Matrai, Chuah, & VandenDriessche, 2010; Mingozzi, & High, 2011). Viral episomal vectors carrying herpes simplex virus type 1 (HSV-1) and Epstein-Barr virus (EBV) amplicons can deliver and allow expression of even full-length genes of up to 150 kb in size (Lufino et al., 2008). However, HSV-1 and EBV viral vectors lack copy number control, which affects the physiological regulation of the expression of the endogenous loci (Odom, Gregorevic, & Chamberlain, 2007). In addition, the use of viral vectors may induce undesired immunological responses and may occasionally integrate into the host genome, which can result in detrimental insertional mutagenesis and gene silencing (Cavazzana-Calvo et al., 2010; Odom et al., 2007). Consequently, there is a need for high capacity, non-integrating vectors that can stably maintain long-term expression of a gene of interest in quiescent as well as dividing cells. None of the currently used viral vectors, however, meet these requirements.
Human artificial chromosomes (HACs) represent an alternative system for gene delivery and expression in gene function studies and gene therapy applications. HAC-based vectors overcome many of the aforementioned problems faced by viral-based gene transfer systems (Basu & Willard, 2006; Kazuki & Oshimura, 2011; Kouprina et al., 2013). First, the presence of a functional centromere enables the long-term stable maintenance of HACs as single copy episomes, without integration into the host chromosomes, thereby minimizing complications like silencing of a therapeutic gene. Second, there is no upper size limit to DNA cloned in a HAC: Entire genomic loci or large gene clusters can be incorporated and faithfully reproduce their normal pattern of gene expression.
Several studies have demonstrated the use of top-down and de novo generated HACs for delivery and expression of genes in human gene-deficient cell lines and in animal transgenesis. An impressive list of genes loaded into HAC vectors has been compiled by Oshimura and co-authors (Oshimura et al., 2015). An illustrative example of correction of gene deficiency i n a m odel m ouse and patient-derived cells by a top-down generated HAC is the use of the 21HAC carrying the dystrophin (DYS) gene, mutations in which lead to Duchenne muscular dystrophy (DMD; Hoshiya et al., 2009). This gene is extremely large, 2.4 Mb. Complete correction of genetic deficiency w as s hown i n a h uman DMD patient-derived cell line and in iPS cells derived from DMD model (mdx) mice (Kazuki et al., 2010). Other examples of the application of de novo constructed HACs to correct genetic deficiencies in human patient-derived cells include the use of the alphoidtetO-HAC with a regulated centromere (Kouprina et al., 2018) carrying entire genomic copies of NBS1, VHL, and BRCA1 genes (Kim et al., 2011; Kononenko et al., 2014). It is important that the phenotypes arising from stable gene expression were reversed when cells were “cured” of the HAC, by inactivating its kinetochore in proliferating cell populations. Other de novo-constructed HACs carrying human beta-globin, GCH1, or CSN2 genes have been successfully used for the creation of transgenic mice (Suzuki et al., 2014; Suzuki et al., 2016). In those studies, the authors demonstrated that the de novo HACs were transmitted through the mouse germline, thereby providing evidence of the meiotic stability of HACs in vivo. Figure 9 illustrates multiple applications of the most advanced HACs, 21HAC and alphoidtetO−HAC.
Figure 9.
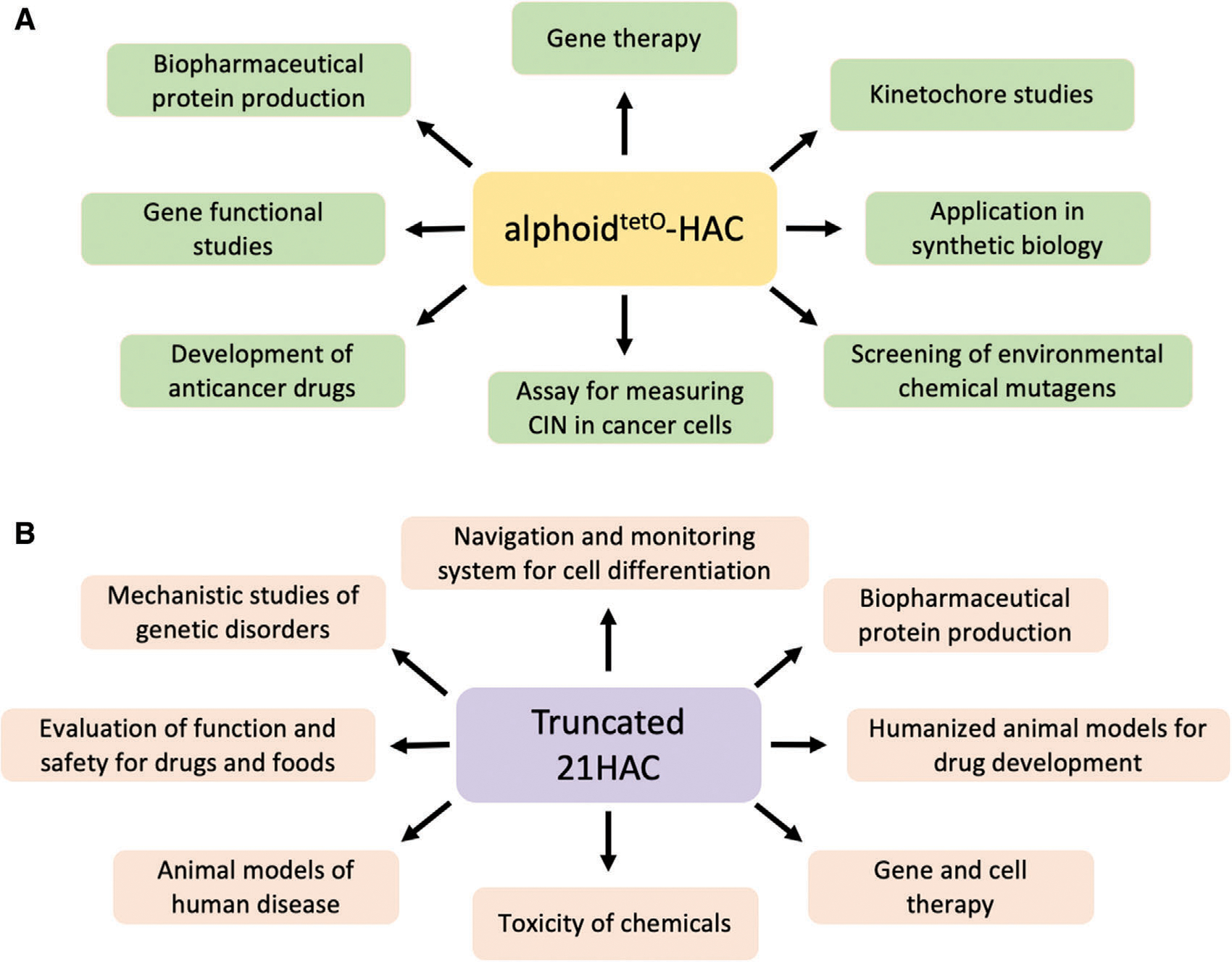
Multiple applications of (A) alphoidtetO-HAC and (B) 21HAC. CIN, chromosome instability.
It is important that gene transfer by HAC vectors avoids a potentially mutagenic step of DNA transfection into target human cells. Instead, a gene of interest inserted into a gene-loading loxP site of a HAC is transferred into target cells using microcell-mediated chromosome transfer (MMCT). During transfer, HACs are maintained inside microcells that protect them from physical damage during the procedure (Suzuki, Kazuki, Oshimura, & Hara, 2016).
The MMCT method was developed in the 1970s (Ege & Ringertz, 1974; Fournier & Ruddle, 1977) and was applied in the past to transfer entire natural chromosomes between cells lines for various studies. For example, MMCT has contributed to mapping genes through functional complementation (Horibata et al., 2004; Kurimasa, Ohno, & Oshimura, 1993; Matsuura et al., 2006; Matsuura et al., 1998; Matsuura et al., 1997; Oshimura & Barrett, 1997; Seyda et al., 2001). MMCT has been also applied for the analysis of chromosome status, such as aneuploidy and epigenetics (Kazuki et al., 2014; O’Doherty et al., 2005). So far, MMCT is the only method available to transfer natural or artificial chromosomes into recipient cells. As mentioned above, one of the advantages of artificial chromosomes over entire chromosomes is that HAC vectors can carry an individual gene or a gene cluster of interest, while natural chromosomes contain thousands of genes, which would complicate the study of a single, specific gene or a gene cluster.
A limitation of chromosome transfer from donor cells into recipient cells by the original MMCT protocol, however, is its low efficiency. Thus, several laboratories have worked to optimize the MMCT protocol. The Oshimura group has mainly focused on the last step of MMCT, i.e., the fusion of microcells with the recipient cells. In one study to increase chromosome and HAC transfer efficiency, the authors proposed the use of the lipid envelope of an inactivated hemagglutinating virus of Japan (HVJ), which has an effect on membrane fusion similar to polyethylene glycol (PEG) for microcell-cell fusion (Yamaguchi et al., 2011). Another MMCT improvement involves the expression of hemagglutinin (H) and fusion (F) measles virus envelope proteins in the donor cells (i.e., hamster CHO; Katoh et al., 2004). The H and F proteins mediate both viral attachment and membrane fusion by binding to the human CD46 surface receptor. However, this modified MMCT protocol is limited to recipient cell lines that express the CD46 receptor.
Recently, another group suggested an alternative chromosome transfer method called retro-MMCT (Suzuki, Kazuki, Oshimura, & Hara, 2016). This method is based on the use of donor CHO cells expressing envelope proteins derived from ecotropic or amphotropic murine leukemia viruses. Using this method, the authors transferred the 21HAC to NIH3T3 cells with an approximately twenty-fold higher efficiency than that obtained using the conventional MMCT method. Potentially, retro-MMCT is applicable to a variety of recipient cells. However, the proposed protocol includes additional time-consuming steps to modify donor cells, with the risk of unexpected consequences. Thus, a safe and efficient MMCT technique has remained an important challenge.
In our recent work, we developed an improved MMCT protocol that has been applied to transfer the alphoidtetO-HAC to different cell types (Liskovykh et al., 2016). Contrary to other researchers, we focused more on steps of the MMCT protocol that have previously been neglected (micronucleation and microcells formation), and found that replacement of the key reagents, colcemid (a microtubule inhibitor that arrests cells at metaphase) and cytochalasin B (an actin inhibitor that induces actin cytoskeleton disassembling), by TN16 + griseofulvin and latrunculin B, in combination with collagen/laminin surface coating to optimize cell adherence to the culture flask surface, increases the efficiency of alphoidtetO−HAC transfer to recipient cells about ten fold. In addition, the modified MMCT transfer technique is less damaging to the HAC than the standard MMCT method. The improved MMCT protocol was tested on different types of recipient cells, including human mesenchymal stem cells and mouse embryonic stem cells, and could facilitate cell engineering by HACs.
Critical Parameters
High-quality BAC DNA and cell culture are critical for Basic Protocol 1. Before loading BAC DNA into the HAC, the user must check DNA by CHEF before and after linearization with a DNA restriction enzyme with a unique restriction site. It is important that most of the DNA is isolated as covalently closed circular molecules (cccDNA; Fig. 7). Also, hamster CHO cells must be actively growing and in adequate numbers to optimize transfection efficiency.
One of the critical parameters for any cell culture discussed in these protocols is mycoplasma contamination. The presence of mycoplasma in cell cultures can slow cell cycle progression, and alter the cytoplasmic membrane, which can result in low penetration by transfection reagents or low fusion efficacy. It is strongly recommended that all the cells are checked for mycoplasma contamination before starting any experiments.
For Basic Protocol 1, transfection conditions are very important. It is critical to maintain the ratio of DNA-to-transfection reagent to get adequate efficiency (the ratio depends on the size of a BAC molecule and is determined experimentally). Also, the incubation time of DNA and transfection reagent is critical. Too long an incubation will result in a low yield of colonies after transfection.
For Basic Protocol 2, the time to start cytostatic treatment is a key parameter. In addition to having an appropriate number of cells seeded, the user must check the confluency of the culture in the microscope. It is critically important to start treatment at 80% of confluency, which occurs likely 1 to 2 days after seeding them (variation is likely a result of the batch of serum used in the culture medium).
It is also critical to attach flasks to the centrifugation tube exactly as described in the protocol (see Fig. 8). Failure to do so may result in cracking the flasks and losing the microcells or dilution of the sample with water. In that case, the entire experiment must be repeated.
The efficiency of the MMCT procedure strongly correlates with the recipient cell type used. Usually, easily transfected cell types like HT1080, HeLa, or HEK293 cells show great efficiency with MMCT (Fig. 6). By contrast, primary cultures like mouse embryonic fibroblasts (MEFs), human-induced mesenchymal stromal cells (hiMSC), and pluripotent cells (embryonic stem cells, iPS cells) show low efficiency. The user should calibrate the required number of colonies to the recipient cell type.
Troubleshooting
Transfection issues
Most problems the users will face are related to the efficiency of transfection. To estimate the efficiency of transfection, we recommend using a BAC containing a region of interest with a fluorescent marker like enhanced green fluorescent p rotein (EGFP; available upon request from the Larionov group). Note that the bigger the BAC, the lower the transfection efficiency. If no transformants appear, keep the following things in mind:
Preparation of large-size BAC molecules may induce nicks or double strand breaks (DSBs) in BAC DNA that would lead to a reduction in the number of transformants and affect proper Cre/loxP recombination. For high DNA quality, use the proper kits for DNA extraction.
BAC DNA integrity and quality should be checked by CHEF electrophoresis, to be sure that the DNA was isolated predominantly as covalently closed circular molecules (Fig. 7), and by a spectrophotometer. Usually by using a spectrophotometer, we expect the A260/A280 ratio to be between 1.7 and 1.9.
Note that the amount of the Viafect transfection reagent was optimized and provided for donor CHO cells. For other cell lines, it may have to be modified.
Density of cells: Be sure that at the time of transfection, the cells are at 80% of confluency. Using cells at lower or higher confluence levels can reduce the efficiency of transfection.
Note that the presence of mycoplasma can greatly reduce transfection efficiency. Check that cells are mycoplasma free using a LookOut® Mycoplasma PCR Detection Kit (MilliporeSigma, cat. no. MP0035–1KT).
MMCT issues
The cells do not stop in metaphase and continue to proliferate as normal
Check the expiration dates of cytostatic reagents. CHO cells usually form a poorly attached round mitotic cell population in the flask during cytostatic treatment. Care must thus be taken in collecting this cell population because the cells are easily washed out.
Flasks are cracked or broken after centrifugation
Check the level of water in the flasks. During centrifugation, the flasks should be completely embedded in water to prevent damage.
The final microcell pellet is invisible or extremely tiny
Be sure that cytostatic treatment begins when donor cells are ~80% confluent on a flask. Fewer initially treated cells will result in very low yield of microcells.
Be sure that in step 17 of Basic Protocol 2 the flask does not have large air bubbles. Such bubbles can prevent formation of microcells during centrifugation. Usually after centrifugation, only extracellular matrix and cell residue are visible on the flask under the microscope. If cells are still attached to the surface of the flask after centrifugation, then very likely the flask was not filled enough with the latranculin-containing DMEM medium.
No colonies visible after blasticidin selection
Check steps 1 to 3 of Troubleshooting, Transfection issues section.
Low efficiency of fusion: In rare cases, it may be necessary to try different proportions of HVJ-E. Follow the manufacturer’s instructions.
Low efficiency of fusion: It is important to check that donor and recipient cells are mycoplasma free. Presence of mycoplasma can greatly reduce the efficiency of fusion.
Understanding Results
Typically, the yield of colonies for Basic Protocol 1 depends on the quality of BAC DNA and the quality of the cell culture. Usually the average number of colonies after transfection with ~2 μg of BAC DNA of 30 to 100 kb in size and ~3.5 × 105 CHO cells is 10 to 50. Users should expect to obtain one to five colonies if transfection was performed using a BAC >100 kb. In rare cases, Basic Protocol 1 must be repeated several times to obtain one to five colonies.
Typically, the yield of colonies for Basic Protocol 2 depends on the quality of the cell culture and the type of recipient cells (Fig. 6). Usually, with cancer cell lines like HT1080, HeLa, or HEK, the user should expect an average of 150 to 300 colonies. With primary cell cultures like MEF or MSC, the user should expect an average of 10 to 30 colonies. With ESCs and iPSCs, the expected number is, on average, one to five colonies. In rare cases, Basic Protocol 2 must be repeated several times to obtain one to five colonies.
Time Considerations
The full procedure described in Basic Protocol 1 can be completed within 3 weeks. Starting with exponentially growing cells, the transfection itself takes 1 day to perform. The rest of the time is required for colony selection and growth. Users should anticipate that if additional selective agents (e.g., another antibiotic) are used together with those described in the protocol, colony formation and growth may be significantly slower.
The full procedure described in Basic Protocol 2 can be completed within 4 weeks. Preparation steps for the protocol take 2 days. The rest of the protocol takes 1 day, with the remaining time required for colony formation and growth. The speed of colony formation and growth, however, depends on the characteristics of the recipient cells. Generally, rapidly proliferative cells form colonies faster.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute, Center for Cancer Research, US (M.L., V.L., N.K.; ZIA BC010413)
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Literature Cited
- Abremski K, & Hoess R (1984). Bacteriophage P1 site-specific recombination. Purification and properties of the Cre recombinase protein. Journal of Biological Chemistry, 259(3), 1509–1514. [PubMed] [Google Scholar]
- Basu J, & Willard HF (2006). Human artificial chromosomes: Potential applications and clinical considerations. Pediatric Clinics of North America, 53(5), 843–853, viii. doi: 10.1016/j.pcl.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Buchholz CJ, Muhlebach MD, & Cichutek K (2009). Lentiviral vectors with measles virus glycoproteins - dream team for gene transfer? Trends in Biotechnology, 27(5), 259–265. doi: 10.1016/j.tibtech.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, … Leboulch P (2010). Transfusion independence and HMGA2 activation after gene therapy of human betathalassaemia. Nature, 467(7313), 318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole T, Okamoto Y, Noskov VN, Kouprina N, Kim JH, Leem SH, … Larionov V (2005). Rapid generation of long synthetic tandem repeats and its application for analysis in human artificial chromosome formation. Nucleic Acids Research, 33(15), e130. doi: 10.1093/nar/gni129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ege T, & Ringertz NR (1974). Preparation of microcells by enucleation of micronucleate cells. Experimental Cell Research, 87(2), 378–382. doi: 10.1016/0014-4827(74)90494-7. [DOI] [PubMed] [Google Scholar]
- Epstein AL (2009). Progress and prospects: Biological properties and technological advances of herpes simplex virus type 1-based amplicon vectors. Gene Therapy, 16(6), 709–715. doi: 10.1038/gt.2009.42. [DOI] [PubMed] [Google Scholar]
- Finney M (2000). Pulsed-field gel electrophoresis. Current Protocols in Molecular Biology, 2.5B.1-2.5B.9. doi: 10.1002/0471142727. mb0205bs51. [DOI] [PubMed] [Google Scholar]
- Fournier RE, & Ruddle FH (1977). Microcell-mediated transfer of murine chromosomes into mouse, Chinese hamster, and human somatic cells. Proceedings of the National Academy of Sciences of the United States of America, 74(1), 319–323. doi: 10.1073/pnas.74.1.319. first-generation human artificial microchromosomes. Nature Genetics, 15(4), 345–355. doi: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honma K, Abe S, Endo T, Uno N, Oshimura M, Ohbayashi T, & Kazuki Y (2018). Development of a multiple-gene-loading method by combining multi-integration system-equipped mouse artificial chromosome vector and CRISPR-Cas9. PLoS One, 13(3), e0193642. doi: 10.1371/journal.pone.0193642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, … Tanaka K (2004). Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proceedings of the National Academy of Sciences of the United States of America, 101(43), 15410–15415. doi: 10.1073/pnas.0404587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiya H, Kazuki Y, Abe S, Takiguchi M, Kajitani N, Watanabe Y, … Oshimura M (2009). A highly stable and nonintegrated human artificial chromosome (HAC) containing the 2.4 Mb entire human dystrophin gene. Molecular Therapy, 17(2), 309–317. doi: 10.1038/mt.2008.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida Y, Kim JH, Kazuki Y, Hoshiya H, Takiguchi M, Hayashi M, … Oshimura M (2010). Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Research, 17(5), 293–301. doi: 10.1093/dnares/dsq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeno M, Grimes B, Okazaki T, Nakano M, Saitoh K, Hoshino H, … Masumoto H (1998). Construction of YAC-based mammalian artificial chromosomes. Nature Biotechnology, 16(5), 431–439. doi: 10.1038/nbt0598-431. [DOI] [PubMed] [Google Scholar]
- Ikeno M, & Hasegawa Y (2020). Applications of bottom-up human artificial chromosomes in cell research and cell engineering. Experimental Cell Research, 390(1), 111793. doi: 10.1016/j.yexcr.2019.111793. [DOI] [PubMed] [Google Scholar]
- Kakeda M, Nagata K, Osawa K, Matsuno H, Hiratsuka M, Sano A, … Tomizuka K (2011). A new chromosome 14-based human artificial chromosome (HAC) vector system for efficient transgene expression in human primary cells. Biochemical and Biophysical Research Communications, 415(3), 439–444. doi: 10.1016/j.bbrc.2011.10.088. [DOI] [PubMed] [Google Scholar]
- Katoh M, Ayabe F, Norikane S, Okada T, Masumoto H, Horike S, … Oshimura M (2004). Construction of a novel human artificial chromosome vector for gene delivery. Biochemical and Biophysical Research Communications, 321(2), 280–290. doi: 10.1016/j.bbrc.2004.06.145. [DOI] [PubMed] [Google Scholar]
- Kazuki Y, Hiratsuka M, Takiguchi M, Osaki M, Kajitani N, Hoshiya H, … Oshimura M (2010). Complete genetic correction of ips cells from Duchenne muscular dystrophy. Molecular Therapy, 18(2), 386–393. doi: 10.1038/mt.2009.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki Y, Hoshiya H, Takiguchi M, Abe S, Iida Y, Osaki M, … Oshimura M (2011). Refined human artificial chromosome vectors for gene therapy and animal transgenesis. Gene Therapy, 18(4), 384–393. doi: 10.1038/gt.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki Y, Kobayashi K, Hirabayashi M, Abe S, Kajitani N, Kazuki K, … Oshimura M (2019). Humanized UGT2 and CYP3A transchromosomic rats for improved prediction of human drug metabolism. Proceedings of the National Academy of Sciences of the United States of America, 116(8), 3072–3081. doi: 10.1073/pnas.1808255116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki Y, & Oshimura M (2011). Human artificial chromosomes for gene delivery and the development of animal models. Molecular Therapy, 19(9), 1591–1601. doi: 10.1038/mt.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazuki Y, Yakura Y, Abe S, Osaki M, Kajitani N, Kazuki K, … Oshimura M (2014). Down syndrome-associated haematopoiesis abnormalities created by chromosome transfer and genome editing technologies. Scientific Reports, 4, 6136. doi: 10.1038/srep06136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Kononenko A, Erliandri I, Kim TA, Nakano M, Iida Y, … Kouprina N (2011). Human artificial chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proceedings of the National Academy of Sciences of the United States of America, 108(50), 20048–20053. doi: 10.1073/pnas.1114483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Abe C, Endo M, Kazuki Y, Oshimura M, & Chiba K (2017). Gender difference of hepatic and intestinal CYP3A4 in CYP3AHumanized mice generated by a human chromosome-engineering technique. Drug Metabolism Letters, 11(1), 60–67. doi: 10.2174/1872312811666170404153804. [DOI] [PubMed] [Google Scholar]
- Kononenko AV, Bansal R, Lee NC, Grimes BR, Masumoto H, Earnshaw WC, … Kouprina N (2014). A portable BRCA1-HAC (human artificial chromosome) module for analysis of BRCA1 tumor suppressor function. Nucleic Acids Research, 42(21), e164. doi: 10.1093/nar/gku870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kononenko AV, Lee NC, Liskovykh M, Masumoto H, Earnshaw WC, Larionov V, & Kouprina N (2015). Generation of a conditionally self-eliminating HAC gene delivery vector through incorporation of a tTAVP64 expression cassette. Nucleic Acids Research, 43(9), e57. doi: 10.1093/nar/gkv124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N, Earnshaw WC, Masumoto H, & Larionov V (2013). A new generation of human artificial chromosomes for functional genomics and gene therapy. Cellular and Molecular Life Sciences, 70(7), 1135–1148. doi: 10.1007/s00018-012-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N, & Larionov V (2008). Selective isolation of genomic loci from complex genomes by transformation-associated recombination cloning in the yeast Saccharomyces cerevisiae. Nature Protocols, 3(3), 371–377. doi: 10.1038/nprot.2008.5. [DOI] [PubMed] [Google Scholar]
- Kouprina N, Petrov N, Molina O, Liskovykh M, Pesenti E, Ohzeki JI, … Larionov V (2018). Human artificial chromosome with regulated centromere: A tool for genome and cancer studies. ACS Synthetic Biology, 7(9), 1974–1989. doi: 10.1021/acssynbio.8b00230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N, Samoshkin A, Erliandri I, Nakano M, Lee HS, Fu H, … Larionov V (2012). Organization of synthetic alphoid DNA array in human artificial chromosome (HAC) with a conditional centromere. ACS Synthetic Biology, 1(12), 590–601. doi: 10.1021/sb3000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N, Tomilin AN, Masumoto H, Earnshaw WC, & Larionov V (2014). Human artificial chromosome-based gene delivery vectors for biomedicine and biotechnology. Expert Opinion on Drug Delivery, 11(4), 517–535. doi: 10.1517/17425247.2014.882314. [DOI] [PubMed] [Google Scholar]
- Kouprina N, Kim JH, & Larionov V (2021). Highly selective, CRISPR/Cas9-mediated isolation of genes and genomic loci from complex genomes by TAR cloning in yeast. Current Protocols, 1(8), e207. doi: 10.1002/cpz1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimasa A, Ohno K, & Oshimura M (1993). Restoration of the cholesterol metabolism in 3T3 cell lines derived from the sphingomyelinosis mouse (spm/spm) by transfer of a human chromosome 18. Human Genetics, 92(2), 157–162. doi: 10.1007/BF00219684. [DOI] [PubMed] [Google Scholar]
- Lee NC, Kononenko AV, Lee HS, Tolkunova EN, Liskovykh MA, Masumoto H, … Kouprina N (2013). Protecting a transgene expression from the HAC-based vector by different chromatin insulators. Cellular and Molecular Life Sciences, 70(19), 3723–3737. doi: 10.1007/s00018-013-1362-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NCO, Kim JH, Petrov NS, Lee HS, Masumoto H, Earnshaw WC, … Kouprina N (2018). Method to assemble genomic DNA fragments or genes on human artificial chromosome with regulated kinetochore using a multiintegrase system. ACS Synthetic Biology, 7(1), 63–74. doi: 10.1021/acssynbio.7b00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liskovykh M, Lee NC, Larionov V, & Kouprina N (2016). Moving toward a higher efficiency of microcell-mediated chromosome transfer. Molecular Therapy. Methods & Clinical Development, 3, 16043. doi: 10.1038/mtm.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lufino MM, Edser PA, & Wade-Martins R (2008). Advances in high-capacity extrachromosomal vector technology: Episomal maintenance, vector delivery, and transgene expression. Molecular Therapy, 16(9), 1525–1538. doi: 10.1038/mt.2008.156. [DOI] [PubMed] [Google Scholar]
- Maier P, von Kalle C, & Laufs S (2010). Retroviral vectors for gene therapy. Future Microbiology, 5(10), 1507–1523. doi: 10.2217/fmb.10.100. [DOI] [PubMed] [Google Scholar]
- Matrai J, Chuah MK, & VandenDriessche T (2010). Recent advances in lentiviral vector development and applications. Molecular Therapy, 18(3), 477–490. doi: 10.1038/mt.2009.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura S, Matsumoto Y, Morishima K, Izumi H, Matsumoto H, Ito E, … Kajii T (2006). Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. American Journal of Medical Genetics. Part A, 140(4), 358–367. doi: 10.1002/ajmg.a.31069. [DOI] [PubMed] [Google Scholar]
- Matsuura S, Tauchi H, Nakamura A, Kondo N, Sakamoto S, Endo S, … Komatsu K (1998). Positional cloning of the gene for Nijmegen breakage syndrome. Nature Genetics, 19(2), 179–181. doi: 10.1038/549. [DOI] [PubMed] [Google Scholar]
- Matsuura S, Weemaes C, Smeets D, Takami H, Kondo N, Sakamoto S, … Komatsu K (1997). Genetic mapping using microcell-mediated chromosome transfer suggests a locus for Nijmegen breakage syndrome at chromosome 8q21–24. American Journal of Human Genetics, 60(6), 1487–1494. doi: 10.1086/515461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills W, Critcher R, Lee C, & Farr CJ (1999). Generation of an approximately 2.4 Mb human X centromere-based minichromosome by targeted telomere-associated chromosome fragmentation in DT40. Human Molecular Genetics, 8(5), 751–761. doi: 10.1093/hmg/8.5.751. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, & High KA (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nature Reviews Genetics, 12(5), 341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- Moralli D, & Monaco ZL (2020). Gene expressing human artificial chromosome vectors: Advantages and challenges for gene therapy. Experimental Cell Research, 390(1), 111931. doi: 10.1016/j.yexcr.2020.111931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano M, Cardinale S, Noskov VN, Gassmann R, Vagnarelli P, Kandels-Lewis S, … Masumoto H (2008). Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Developmental Cell, 14(4), 507–522. doi: 10.1016/j.devcel.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, … Fisher EM (2005). An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science, 309(5743), 2033–2037. doi: 10.1126/science.1114535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom GL, Gregorevic P, & Chamberlain JS (2007). Viral-mediated gene therapy for the muscular dystrophies: Successes, limitations and recent advances. Biochimica et Biophysica Acta, 1772(2), 243–262. doi: 10.1016/j.bbadis.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta Y, Kazuki K, Abe S, Oshimura M, Kobayashi K, & Kazuki Y (2020). Development of Caco-2 cells expressing four CYPs via a mammalian artificial chromosome. BMC Biotechnology, 20(1), 44. doi: 10.1186/s12896-020-00637-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohzeki J, Bergmann JH, Kouprina N, Noskov VN, Nakano M, Kimura H, … Masumoto H (2012). Breaking the HAC barrier: Histone H3K9 acetyl/methyl balance regulates CENP-A assembly. EMBO Journal, 31(10), 2391–2402. doi: 10.1038/emboj.2012.82. 33(5), 710–715. doi: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshimura M, Uno N, Kazuki Y, Katoh M, & Inoue T (2015). A pathway from chromosome transfer to engineering resulting in human and mouse artificial chromosomes for a variety of applications to bio-medical challenges. Chromosome Research, 23(1), 111–133. doi: 10.1007/s10577-014-9459-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesenti E, Kouprina N, Liskovykh M, AurichCosta J, Larionov V, Masumoto H, … Molina O (2018). Generation of a synthetic human chromosome with two centromeric domains for advanced epigenetic engineering studies. ACS Synthetic Biology, 7(4), 1116–1130. doi: 10.1021/acssynbio.8b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomartsev SV, Sinenko SA, Skvortsova EV, Liskovykh MA, Voropaev IN, Savina MM, … Tomilin AN (2020). Human alphoid(tetO) artificial chromosome as a gene therapy vector for the developing hemophilia a model in mice. Cells, 9(4), 879. doi: 10.3390/cells9040879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh D, Abe S, Kobayashi K, Nakajima Y, Oshimura M, & Kazuki Y (2018). Human and mouse artificial chromosome technologies for studies of pharmacokinetics and toxicokinetics. Drug Metabolism and Pharmacokinetics, 33(1), 17–30. doi: 10.1016/j.dmpk.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Seyda A, Newbold RF, Hudson TJ, Verner A, MacKay N, Winter S, … Robinson BH (2001). A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14–2p13. American Journal of Human Genetics, 68(2), 386–396. doi: 10.1086/318196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinenko SA, Ponomartsev SV, & Tomilin AN (2020). Human artificial chromosomes for pluripotent stem cell-based tissue replacement therapy. Experimental Cell Research, 389(1), 111882. doi: 10.1016/j.yexcr.2020.111882. [DOI] [PubMed] [Google Scholar]
- Sinenko SA, Ponomartsev SV, & Tomilin AN (2021). Pluripotent stem cell-based gene therapy approach: Human de novo synthesized chromosomes. Cellular and Molecular Life Sciences, 78(4), 1207–1220. doi: 10.1007/s00018-020-03653-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Kazuki Y, Oshimura M, & Hara T (2014). A novel system for simultaneous or sequential integration of multiple gene-loading vectors into a defined site of a human artificial chromosome. PLoS One, 9(10), e110404. doi: 10.1371/journal.pone.0110404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Kazuki Y, Oshimura M, & Hara T (2016). Highly efficient transfer of chromosomes to a broad range of target cells using Chinese hamster ovary cells expressing murine leukemia virus-derived envelope proteins. PLoS One, 11(6), e0157187. doi: 10.1371/journal.pone.0157187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedesco FS, Gerli MF, Perani L, Benedetti S, Ungaro F, Cassano M, … Cossu G (2012). Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Science Translational Medicine, 4(140), 140ra189. doi: 10.1126/scitranslmed.3003541. [DOI] [PubMed] [Google Scholar]
- Toth A, Fodor K, Praznovszky T, Tubak V, Udvardy A, Hadlaczky G, & Katona RL (2014). Novel method to load multiple genes onto a mammalian artificial chromosome. PLoS One, 9(1), e85565. doi: 10.1371/journal.pone.0085565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z(2015).Genome engineering in cattle: Recent technological advancements. Chromosome Research, 23(1), 17–29. doi: 10.1007/s10577-014-9452-6. [DOI] [PubMed] [Google Scholar]
- Yakura Y, Ishihara C, Kurosaki H, Kazuki Y, Komatsu N, Okada Y, … Oshimura M (2013). An induced pluripotent stem cell-mediated and integration-free factor VIII expression system. Biochemical and Biophysical Research Communications, 431(2), 336–341. doi: 10.1016/j.bbrc.2012.12.096. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Kazuki Y, Nakayama Y, Nanba E, Oshimura M, & Ohbayashi T (2011). A method for producing transgenic cells using a multi-integrase system on a human artificial chromosome vector. PLoS One, 6(2), e17267. doi: 10.1371/journal.pone.0017267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura Y, Nakamura K, Endo T, Kajitani N, Kazuki K, Kazuki Y, … Ohbayashi T (2015). Mouse embryonic stem cells with a multi-integrase mouse artificial chromosome for transchromosomic mouse generation. Transgenic Research, 24(4), 717–727. doi: 10.1007/s11248-015-9884-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.