

Abstract
Purpose of Review
The purpose of this review is to summarize the different roles of the transcription factor SP7 in regulating bone formation and remodeling, discuss current studies in investigating the causal relationship between SP7 mutations and human skeletal disease, and highlight potential therapeutic treatments that targeting SP7 and the gene networks that it controls.
Recent Findings
Cell-type and stage-specific functions of SP7 have been identified during bone formation and remodeling. Normal bone development regulated by SP7 is strongly associated with human bone health. Dysfunction of SP7 results in common or rare skeletal diseases, including osteoporosis and osteogenesis imperfecta with different inheritance patterns. SP7-associated signaling pathways, SP7-dependent target genes, and epigenetic regulations of SP7 serve as new therapeutic targets in the treatment of skeletal disorders.
Summary
This review addresses the importance of SP7-regulated bone development in studying bone health and skeletal disease. Recent advances in whole genome and exome sequencing, GWAS, multi-omics, and CRISPR-mediated activation and inhibition have provided the approaches to investigate the gene-regulatory networks controlled by SP7 in bone and the therapeutic targets to treat skeletal disease.
Keywords: SP7, Bone development, Skeletal disease, Therapeutic approaches
Introduction
Bones are formed through either endochondral or intramembranous ossification. During early embryogenesis, migration and condensation of mesenchymal cells initiate skeletal specification. Bone remodeling is a life-long process involving osteocytes (the main orchestrator), bone-forming osteoblasts, and bone-resorbing osteoclasts [1]. As a highly dynamic tissue, both building the bone structure during embryonic development and maintaining bone homeostasis in adulthood require the regulation of transcription factors (TFs) [2]. TFs are proteins that recognize and bind to specific DNA sequences to regulate gene expression. Their activities are controlled at multiple levels, including epigenetic mechanisms, co-factor association, gene regulatory elements, and environmental cues [3, 4]. Several master TFs are identified and examined with crucial roles in regulating the bone formation, resorption, and remodeling (reviewed in [5, 6], such as SOX9, RUNX2, OSX/SP7, ATF4, NF-kB, MITF, and NFATc1 [7–13].
Osterix/SP7 (encoded by the SP7 gene) is a zinc finger-containing transcription factor that contains three C2H2-type zinc fingers as other SP/XKLF family members [14]. The protein sequence of SP7 is highly conserved between humans and mice. The human SP7 gene is consist of 3 exons and 2 introns and generates 3 alternative splicing transcripts (Type I, II, and III) [15]. The expression of SP7 is detected in chondrocytes, hypertrophic chondrocytes, osteoblasts, and osteocytes [16]. For the past decade, SP7 had been thought as the essential regulator of osteoblast differentiation and bone formation [9]. Recent studies suggest novel roles of SP7 in chondrocyte and osteocyte development and function [17, 18]. In this review, we will first summarize the function of SP7 in bone formation and remodeling (reviewed in [19]), and then will focus on recent studies on SP7 in human skeletal disease and will discuss the future therapeutic potential of SP7 for bone health.
Role OF SP7 in Different Cell Types During Bone Formation and Remodeling
The expression of Sp7 is identified in several tissues, including bone, tooth, brain, and the reproductive tract [9, 20–22]. In brain, Sp7-positive cells were reported in the mitral and granule cell layers in the olfactory bulb [20]. In bone, Sp7 is expressed at multiple stages throughout the mesenchymal bone lineage including in osteoblasts, osteocytes, pre-hypertrophic and hypertrophic chondrocytes. Since a unique suite of genes is required for the differentiation and function of each of these distinct cell types within the mesenchymal lineage, it is likely that this transcription factor utilizes unique, stage-specific mechanisms to control cell differentiation and function at each stage. Figure 1 summarizes distinct roles of Sp7 in different bone cells as evidenced by conditional deletion of this gene using Cre drivers active at different stages in mesenchymal differentiation.
Fig. 1.
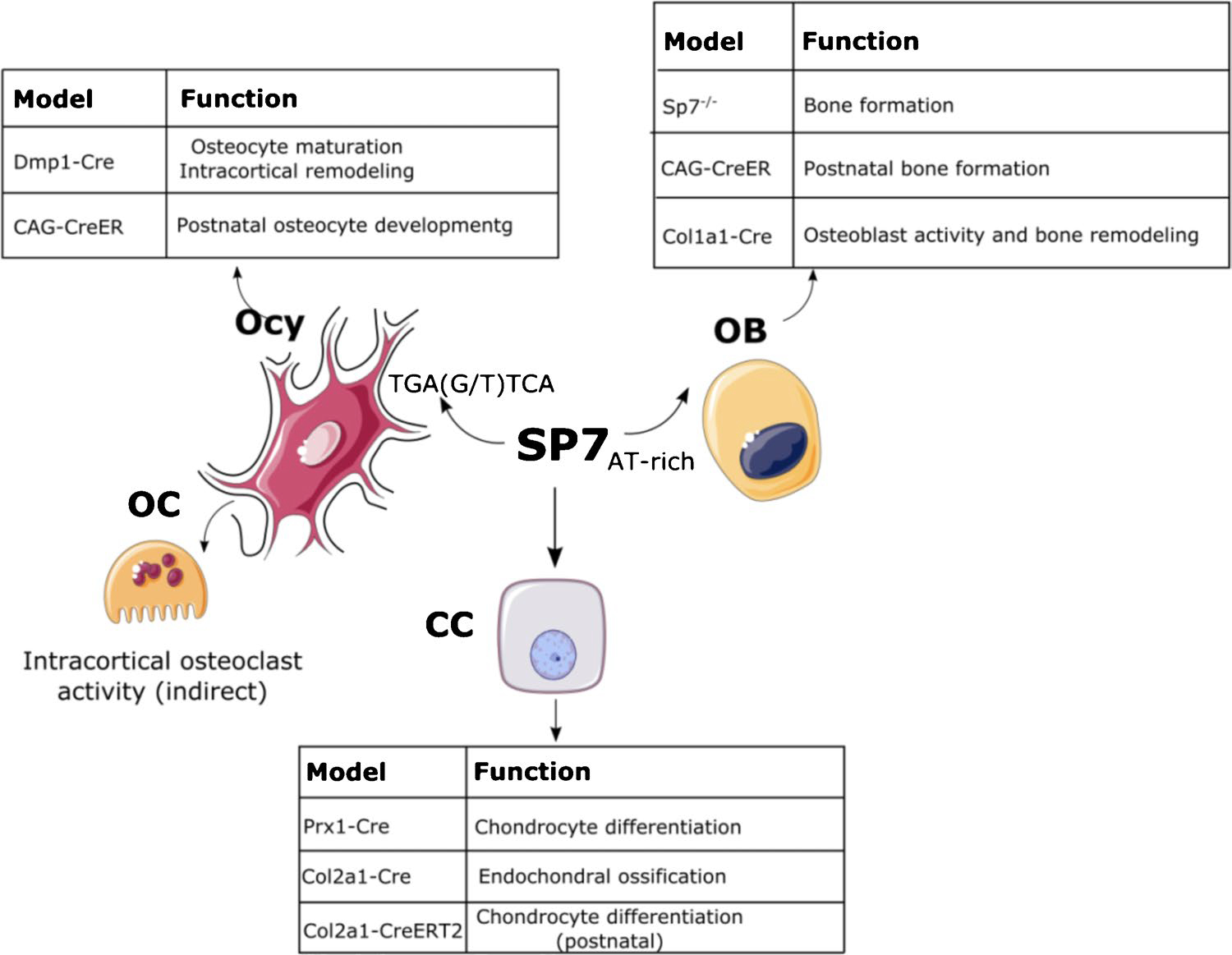
Schematic of SP7 functions in chondrocytes, osteoblasts, and osteocytes. SP7 regulates osteoblast differentiation and bone formation through osteoblast target genes by recognizing AT-rich region. In osteocytes, SP7 regulates osteocyte maturation and intracortical remodeling through osteocyte target genes by recognizing TGA(G/T) TCA motif. SP7 also regulates chondrocyte differentiation and endochondral ossification. CC, chondrocyte; OB, osteoblast; Ocy, osteocyte; OC, osteoclast. Part of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/)
Osteoblast
Runx2 and Sp7 are crucial regulators for osteoblast differentiation. Runx2-null mice completely lack ossification, which suggests that both endochondral and intramembranous ossification are blocked [8]. No bone formation and no bone matrix deposition occur in Sp7-null mice [9], which suggests that osteoblast differentiation is blocked during embryogenesis in the absence of this transcription factor. Sp7 acts downstream of Runx2 since Runx2 is expressed in the pre-osteoblasts of Sp7-null mice while Sp7 is not expressed in Runx2-null mice. When Sp7 was conditionally deleted at different time points postnatally (CAG-CreER), mutant mice showed reduced bone formation, severely altered bone structure, and accumulation of calcified cartilage. This suggests that Sp7 is required for osteoblast differentiation and bone formation in the postnatal skeleton as well [23]. Several studies reported the osteoblast-specific role of Sp7 by applying different Cre/LoxP systems. Mice exhibited osteopenia, including reduced bone formation rate and short bones, when Sp7 was conditionally deleted in osteoblasts with Col1a1-Cre [24], suggesting that Sp7 inactivation in growing bones delays osteoblast maturation. Further studies using Col1a1-CreERT2 to delete Sp7 in formed bones [25] showed that inducible Sp7 deletion leads to decreased mineralized surface and bone formation rate. These data support a role for Sp7 in maintaining osteoblast function in adult bone remodeling.
Like other Sp family members, Sp7 was previously reported to be a transcription factor that binds GC-box DNA elements in osteoblasts [26]. However, de novo motif analysis revealed that osteoblast-specific Sp7-bound enhancer regions are significantly enriched in AT-rich motifs [19]. Interestingly, Sp7 does not directly bind to AT-rich DNA sequences but rather acts with Dlx factors (that directly bind AT-rich motifs) to regulate the expression of osteoblastic genes. This suggests that Sp7 forms transcriptional complexes with other co-factors to control cell-specific gene expression. Several osteoblast-related genes have Sp7-dependent expression and show direct regulation by Sp7. One group of genes are regulated via direct binding of Sp7 to GC-rich regions, such as Bsp, Sost, Col1a1, Col1a2, Mmp13, and Enpp1 [26–31]. The other group of genes are regulated via the complex formed by Sp7 and other factors, including Runx2 (Sp7-Dlx5-Msx2), Notch2 (Sp7-Dlx5), and Col1a1 (Sp7-NFATc1) [32–34].
Osteocyte and Osteoclast
Some osteoblasts become buried within mineralized bone matrix and become osteocytes [35]. We and others have shown that Sp7 plays an important role in osteocyte differentiation and maturation. Sp7 acts upstream and directly regulates the osteocyte-specific gene Sost. Both in vivo and in vitro models showed that Sp7 specifically binds and activates the Sost promoter [23, 28]. Sp7 is required for postnatal osteocyte development. Global postnatal inactivation of Sp7 (CAG-CreER, Sp7 fl/fl) leads to deformed osteocytes and reduced expression levels of Dmp1, Phex, and Sost [23]. We deleted Sp7 using Dmp1-Cre, which targets mature osteoblasts and osteocytes [36]; these mutant mice showed increased cortical porosity, abnormal intracortical bone remodeling, osteocyte dendrite defects, and increased osteocyte apoptosis [18]. Among osteocyte-specific Sp7 target genes, we identified osteocrin (encoded by the Ostn gene) as a secreted factor that promotes osteocyte dendrite formation and maintenance. Sp7 ChIP-seq followed by de novo motif binding analysis between osteocytes and osteoblasts revealed that the osteocyte-specific Sp7 binding sites are enriched for the TGA(G/T)TCA motif bound by activator protein 1 (AP-1) family members. Therefore, Sp7 may use distinct binding factors in osteocytes versus osteoblasts. Single-cell RNA sequencing identified discrete populations of cells undergoing the osteoblast-to-osteocyte transition, and dramatic osteocyte differentiation defects in this normal process in the absence of Sp7. In addition to loss-of-function studies, overexpression of Sp7 under the control of the 2.3 kb Col1a1 promoter [37•] led to porous cortical bone, decreased number of osteocyte dendrites, disrupted lacunar-canalicular network and increased Sost expression upon skeletal unloading. Taken together, these findings raise the interesting hypothesis that proper expression level of Sp7 is required in osteocyte development.
Integrin β3 (encodes by the Itgb3 gene) associates with αV and is expressed in osteocyte dendrites where it plays an important role in mechano-transduction [38, 39]. Itgb3-null mice have significantly reduced femoral length and decreased cortical thickness [40]. Chromatin immunoprecipitation (ChIP) assays in osteoblastic MC3T3-E1 cells showed that Sp7 binds to the promoter of the Itgb3 gene (encodes integrin β3) [40]. Since both Sp7 and Itgb3 are expressed in osteocytes and Sp7 plays a critical role in osteocyte dendrite development and maintenance, more studies are needed to explore the specific role of Sp7/Itgb3 axis in dendrite formation, osteocyte maturation and mechano-sensing.
Sp7 is not expressed in osteoclasts. However, several studies showed that Sp7 may affect osteoclast activity in vivo via indirect mechanisms. Conditional deletion of Sp7 in osteoblasts and osteocytes (Dmp1-Cre; Sp7 fl/fl) does not affect osteoclasts on trabecular bone. Dmp1-Cre; Sp7 fl/fl mice, on the other hand, have increased intracortical osteoclast activity confirmed by the elevated level of TRAP staining [18]. This increased osteoclast activity may be caused by increased Rankl expression triggered by apoptotic osteocytes [41•]. When Sp7 was ablated postnatally by CAG-CreER, primary spongiosa osteoclast density is reduced [23] while more osteoclasts are noted in porous cortical bone in Sp7 mutant (CAG-CreER; Sp7 fl/fl) mice. It is possible that reduced hypertrophic chondrocyte Rankl expression drives osteoclast defects in the primary spongiosa in global/inducible Sp7 mutant mice. Thus, Sp7 likely controls bone resorption indirectly through different cell type-specific mechanisms.
Chondrocyte
There is no cartilage phenotype identified in Sp7-null embryos [9]. However, when Sp7 expression was reduced in chondrocyte-like ATDC5 cells, the expression of several chondrocyte genes is down-regulated, including Sox9, Dlx5, Alpl, and Col10a1 [42]. Chondrocyte-specific deletion of Sp7 (Col2a1-Cre; Sp7 fl/+) in mice results in impaired endochondral bone formation including delayed chondrocyte differentiation, increased hypertrophic chondrocytes, reduced formation of trabecular bone, and reduced skeletal growth [43]. Another study used both Col2a1-Cre and Prx1-Cre to ablate Sp7 expression [44]. Both models showed arrested endochondral ossification with chondrocytes blocked at the hypertrophic stage. Col2a1-Cre; Sp7 fl/fl mice also show blocked calcification of cartilage matrices and reduced Mmp13 expression. ChIP assays suggest that Sp7 regulates Mmp13 during chondrocyte maturation. Conditional deletion of Sp7 during postnatal growth (Col2a1-CreERT2; Sp7 fl/fl) results in impaired secondary ossification by delaying chondrocyte hypertrophy and conversion to osteoblasts [17]. Importantly, more studies are needed to identify the direct targets of Sp7 during chondrocyte differentiation and what co-factors are involved in this process. Ultimately, detailed analysis of how Sp7 regulates key stage-specific factors in bone lineage cells will identify novel co-factors required for this transcription factor to exert multiple roles throughout the mesenchymal lineage.
Odontoblast
Sp7 is expressed in many cell types during tooth development, including dental papilla, odontoblasts, alveolar bone osteoblasts, and dental follicle cells [45]. Sp7 plays an important role in tooth development including direct control of dentin sialophosphoprotein (Dspp), the key protein for postnatal odontogenesis [45]. Constitutive lineage tracing revealed that Sp7-labeled cells actively contributes to dental development, especially tooth root formation [46]. Sp7 is strongly detected in the crown in early tooth development, but then down-regulated after root formation [47]. Tamoxifen-dependent lineage-tracing further showed that Sp7 is expressed in dental mesenchymal progenitors and contributes to all relevant cell types during dental root morphogenesis. In Sp7-null embryos, mineralization was absent in the mandible, maxilla, and craniofacial bones [9, 48]. Though these mice died soon after birth, the examination of perinatal pups showed that both maxillary and mandibular incisors were smaller in Sp7 −/− mice compared to control littermates [48]. The alveolar bone that surrounds incisors was completely absent in Sp7-null mice. The formation of alveolar bone is not absolutely needed for tooth morphogenesis since initial tooth morphogenesis is normal in Sp7 −/− mice. Sp7 is crucial for the normal architecture and organization of dental tubules in the root. Sp7 −/− mice showed disorganized odontoblasts and ameloblasts around the pre-dentin-like layers in incisors, suggesting that Sp7 promotes the differentiation and maturation of odontoblasts and ameloblasts. The deletion of Sp7 specifically in odontoblasts (Col1-Cre; Sp7 fl/fl and OC-Cre; Sp7 fl/fl) showed short molar root, reduction of dentin, and malformed dentinal tubule of tooth roots [49–51]. Sp7 can also regulate cementum formation by promoting cementoblast differentiation and mineralization [51]. Overexpression of Sp7 (3.6 kb Col1a1-Sp7) accelerated the formation of cellular cementum, while deletion of Sp7 (Col1-Cre; Sp7 fl/fl and CAG-CreER; Sp7 fl/fl) dramatically reduced cementogenesis and mineralization rate. These studies stimulate much more interest in investigating the role and the mechanism of Sp7 in tooth formation and provide potential therapeutics for periodontal regeneration and alveolar bone healing.
SP7 and Human Skeletal Disease
A primary goal of human genetics is to identify DNA sequence variants that influence biomedical traits, particularly those related to the onset and progression of human disease, such as osteoporosis. Genetic discoveries have substantially improved our understanding of the molecular and genetic mechanisms responsible for many common and rare skeletal diseases and driven the development of novel preventative and therapeutic strategies [52–55]. SP7 plays fundamental roles in different cell types during bone formation and remodeling. Recently, many exciting studies have investigating connections between SP7 and skeletal disease.
Common Skeletal Disease: Osteoporosis
Genome-wide association studies (GWAS) on skeletal polygenic disorders have made great achievements in the past two decades and identified hundreds of susceptibility genes/loci for the pathophysiology of common skeletal disease [56]. Osteoporosis is a common bone disease characterized by low bone mass, deteriorative microarchitecture, loss of bone strength, and increased risk of bone fracture. Bone mineral density (BMD) is used as the diagnosis of osteoporosis and serves as a strong predictor of fracture risk [57].
Several studies demonstrated that SP7 is a locus associated with BMD at the GWS level (Table 1). GWAS analysis of BMD and related traits were performed in children (age 9) from the Avon Longitudinal Study of Parents and Children (ALSPAC) [58]. 4 polymorphisms (rs2016266, rs4759021, rs6580842, and rs10876432) residing in a linkage disequilibrium block near the SP7 gene were identified with significant associations to total body BMD. The meta-analysis of two existing studies (a European women population and an Icelandic population) [59, 60] further suggested a strong association between these SNPs and adult lumbar spine BMD. Styrkarsdottir and colleagues performed extended GWAS analysis of BMD among the European descendent subjects and identified one significant locus near the SP7 gene (rs10876432) [61]. Within the Genetic Factors of Osteoporosis (GEFOS) consortium, a large-scale GWAS meta-analysis was performed on spine and femoral neck BMD in Northern European descent subjects [62]. This confirmed SP7 (12q13, rs2016266) as a locus significantly associated with BMD. Another genome-wide meta-analysis was performed on the lumbar spine and femoral neck BMD in European and east Asian ancestry individuals [63]. SP7 (rs7108738) was further proved as a BMD-associated locus which was also associated with fracture risk. Taken together, these findings clearly demonstrate that SP7 polymorphisms in humans are associated with BMD variation and subsequent fracture susceptibility. At present, the precise mechanisms connecting SP7 polymorphisms with BMD variation remains underexplored. For example, it is possible that non-coding SP7 polymorphisms may regulate SP7 expression levels in bone cells, a possibility that requires exploration in large numbers of clinical bone biopsy samples from “genotyped” individuals and genome editing in human bone cell culture models to explore consequences of non-coding SP7 polymorphisms on SP7 mRNA levels.
Table 1.
Genome-wide association studies reported SP7 as the locus associated with BMD and fracture risk
| Studies | Traits | SNPs | Sample number and ancestry | Design |
|---|---|---|---|---|
|
| ||||
| Timpson et al. [58] | BMD (femoral head, total body) | rs2016266, rs4759021, rs6580842, rs10876432 | 1518 European children | GWAS |
| Timpson NJ, Richards J, Stykarsdottir U [58–60] | BMD (lumbar spine) | rs2016266, rs4759021, rs6580842, rs10876432 | 2094 European women; 5861 Icelandic population |
Meta-GWAS |
| Stykarsdottir et al. [61] | BMD (hip, spine) | rs10876432 | 6865 European descendants | GWAS |
| Rivadeneira et al. [62] | BMD (lumbar spine, femoral neck) | rs2016266 | 19,195 Northern European descendants | Meta-GWAS |
| Estrada et al. [63] | BMD (lumbar spine, femoral neck) | rs7108738 | 32,961 European and east Asian descendants (elderly) | Meta-GWAS |
Rare Skeletal Dysplasia: Osteogenesis Imperfecta
To date, five SP7 mutations have been reported that cause rare skeletal disease including osteogenesis imperfecta (OI) (Table 2). Although the majority of OI cases are caused by mutations in the COL1A1 and COL1A2 genes, a large number of genes that are crucial for osteoblast function, have been identified to cause skeletal fragility and a phenotype similar to collagen-mutated OI [64, 65]. Other clinical signs and symptoms may be found including dentinogenesis imperfecta (DI), blue sclera, hearing loss, growth deficiency, and joint laxity [66–68]. DI is a heredity disorder of dentin formation. DI type I, which is mostly linked to OI, is the oral symptom of inadequate collagen production [69]. In addition to DI, significant delay of tooth eruption can be found in young OI patients who had been treated with bisphosphonate [70, 71] since osteoclasts are crucial for tooth eruption and resorption of the primary teeth [72, 73].
Table 2.
Rare skeletal dysplasia caused by SP7 mutations
| Lapunzina et al. [74] and Tung et al. [75] | Hayat et al. [76] | Fiscaletti et al. [77•] | Ludwig et al. [78•] | Whyte et al. [79] | Lui et al. [80••] | |
|---|---|---|---|---|---|---|
|
| ||||||
| Patients (age) | Egyptian (8y) and Chinese (17y) | Pakistani (32y) | Iraq (13y) | Haitian (20y) | Race not provided (15y) | Race not provided (3y) |
| Diagnosis | Recessive OI | Recessive OI | Recessive OI | Dominant OI | Juvenile Paget’s disease | Neomorphic mutation |
| Mutations | c.1052delA (p.E351GfsX19) | c.824G > A (p.C275Y) | c.946C > T (p.R316C) | c.1019A > C (p.E340A) | c.926C > G (p.S309W) | c.926C > G (p.S309W) |
| Alterations | Absence of 3rd zinc-finger domain | No change to zinc-finger domain | Altered 1st zinc-finger domain | Altered 2nd zinc-finger domain | Altered 1st zinc-finger domain | Altered 1st zinc-finger domain |
| Height | Below normal | Short | Short | Normal | Normal | NA |
| Fractures | Yes | Yes | Yes | Yes | Yes | Yes |
| Bowing | Upper and lower limbs | Lower limbs | NA | NA | Lower limbs | Lower limbs |
| Scoliosis | Mild | Severe | Mild | Severe | Mild | Severe |
| Bone mineral density | Low at lumbar spine and femur | NA | Low at lumbar spine | Low at lumbar spine | High lumbar spine areal BMD | High lumbar BMD |
| Cortical porosity | NA | NA | High | High | NA | NA |
| Bone turnover | Normal ALP | Normal ALP | High trabecular BFR | Low BFR | High ALP and bone turnover | High ALP and bone turnover |
| Serum calcium and phosphorus | Normal | Normal | NA | Normal | Normal | Normal |
| Tooth morphology and eruption | Delayed [74, 75], DI, primary dentition, enamel hypoplasia, discoloration, obliterated pulp chamber, permanent dentition, bulbous crowns, short roots [75] | NA | Delayed, No DI | Broken teeth, No DI | DI, Short roots, Thin or no pulp | NA |
| Hearing | Normal | Normal | Loss | Loss | Loss | Normal |
| Face | Dysmorphism | Dysmorphism | Dysmorphism | Malocclusion | Dysmorphism | Craniosynostosis |
| Treatment | Bisphosphonate | NA | Zoledronate | NA | NA | Bisphosphonate |
The first SP7 mutation was reported in an 8-year-old Egyptian child [74] with recurrent fractures, below-normal height, bowing of upper and lower limbs, mild scoliosis, delayed tooth eruption, and normal hearing. Bone Densitometry (DEXA) results showed low BMD in the spine and hip. A homozygous single base pair deletion (c.1052delA) was identified in SP7 by a combination of homozygosity mapping and candidate gene approach. This deletion causes a frameshift, introduces 18 novel residues at codon 351, and results in premature termination (p.E351GfsX19). The mutant transcript is translated into a truncated SP7 protein lacking the 3rd zinc-finger domain. The altered zinc-finger structure may affect the binding affinity of SP7 to target regions. Further studies are needed to examine the mechanisms that changes the binding affinity and identify the target genes that are affected by this frameshift mutation that eventually causes the OI-like phenotypes. This same rare deletion was also reported in a 17-year-old Chinese boy with recurrent long bone fractures, vertebral collapse, tooth eruption delay, and maxillary hypoplasia [75]. He had a dental manifestation of dentinogenesis imperfecta in both primary and permanent dentition. Enamel hypoplasia, discoloration, and severely worn primary dentition were reported. His permanent dentition phenotype included bulbous crown, short roots, and impacted dentition which necessitating combined surgical and orthodontic treatment.
A 32-year-old Pakistani man with recurrent fractures, short statue, lower limb bowing, craniofacial dysmorphism, and normal hearing was found to have a SP7 homozygous missense mutation (c.824G > A) [76]. This c.824G > A mutation is located within exon 2 of SP7 and causes a cysteine to tyrosine amino acid change at position 275 (p.C275Y). At present, future studies are needed to understand how this mutation, outside the zinc finger domain of SP7, affects its function.
A 13-year-old child from Iraq with recessive pattern OI was found to have a homozygous SP7 (c.946C > T) mutation [77•]. The patient presented with short stature, low-trauma fracture, low BMD, mild scoliosis, hearing loss, facial dysmorphism, and delayed tooth eruption. The proband is from a family of eight siblings. One younger brother and one sister are homozygous for the same mutation. His younger brother was short, had low-trauma hip fracture and hearing loss. His sister had a history of low-trauma fractures, low bone density, normal height, and no hearing loss. A trans-iliac bone biopsy was obtained from the proband patient which showed high cortical porosity and high trabecular bone turnover. The c.946C > T mutation locates in exon 2 of SP7 gene and results in a substitution from arginine to cysteine (R316C). The arginine position 316 is located in the 1st zinc finger domain. The substitution of arginine with cysteine may change the affinity of SP7 to the genome or other co-factors by altering the zinc-finger cysteine-histidine ratio. More recently, we analyzed osteocyte morphology in non-decalcified iliac crest bone biopsies from two R316C patients (the proband and his younger brother) and two age- and sex-matched healthy patients. In this analysis, we noted reduced osteocyte dendrite length and number in patients versus healthy controls [18]. Sp7 binds an enhancer ~ 110 kB upstream of the Ostn transcription start site. The transcriptional activity of this putative Ostn enhancer was reduced when HEK293T cells were transfected with R316C mutated SP7 compared to wild-type SP7 [18]. As discussed above, SP7 may utilize different DNA-binding cofactors in osteocytes compared to osteoblasts. Further studies are needed to investigate whether the SP7 R316C mutation affects the binding activity and the cofactor assembly of SP7 to osteocyte-specific target genes.
One heterozygous mutation of SP7 (c.1019A > C) was identified in young adult siblings of Haitian descent (20-year-old female and 17-year-old male) who presented with dominant-pattern OI [78•]. One female patient had fractures, poor bone healing, severe scoliosis, hearing loss, normal height, and reduced cortical BMD in long bones. Though most surface parameters were within the normal range on skeletal histomorphometry, she had low mineral apposition rate and low bone turnover. The male patient shared a similar OI phenotype as his sister, including fragility fractures, low cortical volumetric BMD, and high cortical porosity. The c.1019A > C mutation is located in exon 2 of the SP7 gene and causes a glutamic acid to alanine amino acid change (E340A). The E340A mutation may alter the 2nd zinc-finger domain of SP7. When HEK293T cells were co-transfected with DLX5- and SP7-expression plasmids, the luciferase activity of SP7 promoter was significantly reduced when transfected with mutant SP7 (E340A) compared to wild-type SP7.
Two unrelated individuals were identified with the same heterozygous de novo SP7 mutation (c.926C > G) [79, 80••]. Whyte and colleagues reported a 15-year-old girl with Juvenile Paget’s disease caused by c.926C > G mutation [79]. She had recurrent fractures in lower limbs, skull deformity, scoliosis, generalized osteosclerosis, hyperostosis, short roots with very thin or no pulp teeth, and hearing loss. DXA revealed high lumbar spine areal BMD and wrist total BMD. Her serum ALP and urinary total hydroxyproline levels were elevated. Lui and colleagues reported a 3-year-old boy bearing the same SP7 c.926C > G mutation with similar phenotypes, including recurring fractures, severe scoliosis, osteosclerosis, elevated alkaline phosphatase, increased lumbar BMD, and high bone turnover [80••]. The c.926C > G mutation locates in exon 3 of SP7 gene and results in a serine to tryptophan mutation (S309W) in the 1st zinc-finger domain. Notably, knock-in mice bearing the S309W mutation were generated. Both homozygous and heterozygous knock-in mice died shortly after birth with a complex skeletal phenotype that is dissimilar to Sp7 knockout mice. The human and mouse phenotypes shared similarities and showed some differences. For example, clavicles in the knock-in mice showed decreased length and increased thickness, which resembles the clavicle osteosclerosis in the patient. Cranial hyperostosis in the patient, however, was not observed in the knock-in mouse. Further mechanistic studies revealed that the mutation alters the binding specificity of SP7 from AT-rich motifs to a GC-consensus sequence (typical of other SP family members) [33] and produces an aberrant gene expression profile, including increased expression of Col1a1 and endogenous Sp7, but decreased expression of genes involved in matrix mineralization.
In summary, there is a strong correlation between SP7 mutations and rare skeletal dysplasia (mutation locations are summarized in Fig. 2). Abnormal skeletal developmental processes caused by dysfunction of SP7 result in skeletal disease. Thus, future directions should emphasize on the establishment of both in vivo and in vitro systems to characterize these disease-causing SP7 mutations, including generating mouse models that carry SP7 mutations as well as introducing these mutations to organoids and cell lines by gene editing. These models will provide optimal systems to investigate the mechanism and the causal relationship between these mutations and the function of SP7 in bone.
Fig. 2.
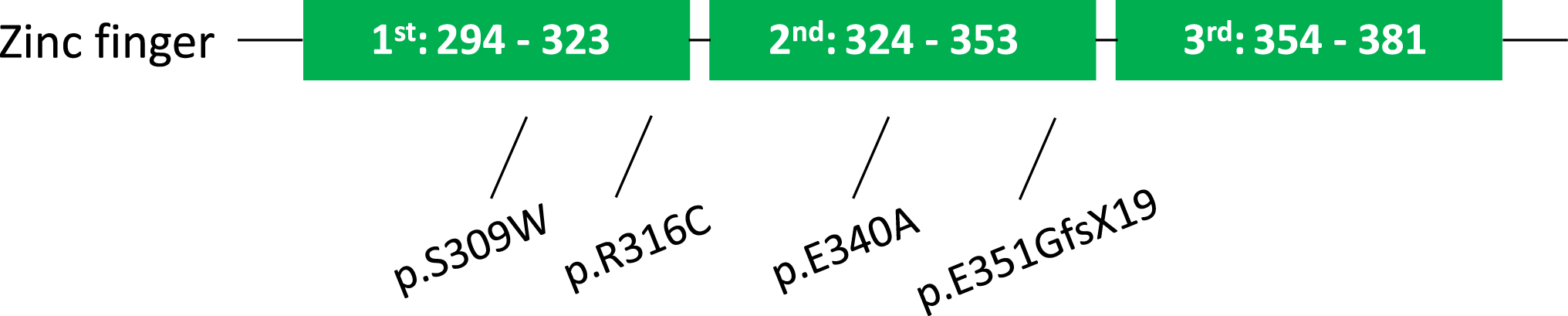
The sequence of human SP7 zinc finger domains. Four SP7 mutations located within zinc finger domains are marked. Green boxes: 3 zinc finger domains
Bone Caner: Osteosarcoma
SP7 is expressed in nearly all osteoblastic tumors but not expressed in giant cell tumor, chondroblastoma, and chondromyxoid fibroma [81]. Osteosarcoma is the most common bone sarcoma and the 3rd most common malignancy in children and adolescents [82]. Not knowing the exact cause, osteosarcoma could be due to DNA mutations of oncogenes and tumor suppressor genes in the primitive mesenchymal bone-forming cells and produces malignant osteoid. SP7 expression is significantly reduced in both murine and human osteosarcoma cells (murine: K7, K7M2; human: HOS, Krib, MG) compared with its expression in normal murine and human osteoblastic cells (MC3T3 and HOB) [83]. Overexpression of SP7, on the other hand, can inhibit tumor cell growth, suppress osteolytic lesions, and suppress lung metastasis. SP7 down-regulates expression of the cytokine IL-1α and reduces lytic activity in osteosarcoma cells by targeting the GC-rich region of the IL-1α promoter [84]. Suppression of SP7 resulted in up-regulation of both IL-1α promoter activity and IL-1α protein level. Site-directed mutagenesis and chromatin immunoprecipitation showed that SP7 was unable to repress the IL-1α promoter activity in osteosarcoma cells after mutating the GC-rich SP1-binding site. p53 is a tumor suppressor regulating cell proliferation and apoptosis. Trp53 (encodes p53) knockout mice were characterized by a denser skeleton compared to the wild-type littermates and the Trp53-deficient bone marrow-derived mesenchymal progenitor cells had a higher capacity to differentiate towards the osteoblastic fate [85, 86]. p53 represses the osteogenic transcriptional network of SP7 by blocking SP7-DNA binding and SP7-DLX5 interaction [87]. In summary, SP7 regulates the normal formation of mature osteoblasts and abnormal SP7 expression may trigger mesenchymal stem/stromal cells and/or osteoblastic precursors to form osteosarcoma microenvironment by producing excessive osteoid matrix. Future studies are necessary to examine the important roles of SP7 with oncogenes and tumor suppressor genes in osteosarcomas, and evaluate the correlation between SP7 to growth factors, cytokines, chemokines, and metalloproteinases in osteosarcoma microenvironment.
Bone Metastasis
Skeletal metastases are common in patients with advanced breast cancer. Interestingly, SP7 expression is elevated in human breast cancer cell lines and tumors of some breast cancer patients [88, 89]. This finding suggests that epithelial breast cancer cells may adapt an osteogenic SP7-dependent gene expression program to promote skeletal metastasis. Patients with high SP7 expression had poorer survival rates and SP7 expression was significantly associated with lymph node metastasis. SP7 knockdown inhibited breast cancer invasion and osteolytic metastasis, whereas overexpression of SP7 promoted the invasiveness. SP7 facilitates bone metastasis of breast cancer by upregulating the expression of genes (MMP9, MMP13, VEGF, IL-8, and PTHrP) that contribute to the metastatic cascade. Particularly, SP7 directly targets the GC-rich promoter of MMP9 and mediates the SP7-driven breast cancer invasion in the bone metastatic niche.
Cancer-associated fibroblasts (CAFs) are cells of the mesenchymal lineage involved in supporting tumorigenesis from growth to metastasis [90]. SP7 expression in CAFs with pro-tumorigenic characteristics was reported using Osx-Cre; TdTomato (TdTOsx) reporter mice [91•]. Most of these TdTomato-positive cells were positive for CD45 and had tumor-infiltrating immune cell-related genes expressed. SP7 is expressed in a subset of hematopoietic stem cells (HSC) that give rise to TdTOsx+;CD45+ tumor-infiltrating immune populations. This study supports that a subset of CAFs, derived from Osx + cells in the bone marrow, contributes to extracellular matrix (ECM) production at the tumor site, thereby creating a tumor-supporting stroma. These findings emphasize the importance of SP7 in the tumor microenvironment and progression. Future studies are needed to identify the mechanisms that regulate Osx + mesenchymal cells during extracellular matrix remodeling and examine how SP7 contributes to this process.
Therapeutic Approaches
Targeting SP7-Dependent Genes: Ostn
Long-term glucocorticoid (GC) treatment is associated with skeletal side effects including bone loss, fracture, osteoporosis, and osteonecrosis [92]. GCs induce osteocyte apoptosis and cause loss of osteocyte dendrites [93]. Our previous work demonstrated that the Sp7 target gene osteocrin (Ostn) regulates osteocyte dendrite formation and maintenance [18]. Therefore, we tested whether Ostn overexpression might restore osteocyte defects in prednisolone-treated mice [94]. Though not able to rescue the reduced cortical thickness caused by prednisolone, Ostn treatment led to modest protection at the level of osteocyte morphology from the deleterious effects of GC treatment. These findings support a modest therapeutic potential for systemic osteocrin delivery in preserving osteocyte morphology during disease. Since SP7 level is reduced in the setting of GC treatment, it remains possible that other cell type-specific SP7 target genes may represent attractive therapeutic targets for bone loss due to GC excess.
Targeting Post-Transcriptional Modifications of SP7: Ubiquitination
Melatonin is a neurohormone mainly secreted by pinealocytes in the pineal gland and plays an important role in bone-related diseases by promoting bone formation and preventing bone destruction [95, 96]. Melatonin stabilized the SP7 protein production by blocking the ubiquitin–proteasome pathway and promoted osteoblast differentiation via the PKA and PKC signaling pathways [97]. The same group previously showed that the E3 ligases Cbl-b and c-Cbl induced SP7 ubiquitination and degradation [98]. Melatonin treatment partially prevented SP7 degradation by inhibiting these ubiquitin ligases. This suggests that melatonin may be a potent osteogenic agent targeting mature osteoblast differentiation and bone formation in the treatment of osteoporosis. Future research identifying targeted approaches to manipulate SP7 degradation may be beneficial in the setting of osteoporosis and metastatic bone diseases.
Targeting at the Transcriptional and Translational Level: lncRNAs
Recent study showed that inflammatory osteoclasts (iOCLs) exosomes specifically target osteoblasts via ephrinA2/EphA2 [99]. Exosomal lncRNA LIOCE enriched in iOCL exosomes promoted osteogenic activity by being incorporated into osteoblasts. lncRNA LIOCE directly binds and stabilizes SP7 protein and reduces its ubiquitination, and thus, exosomes bearing lncRNA LIOCE may be an effective strategy to increase bone formation in osteoporosis and other bone metabolic disorders [99]. The metastasis-associated lung adenocarcinoma transcript-1 (MALAT1) noncoding RNA is induced during osteoblast differentiation and is expressed at lower levels in cells from osteoporosis patients versus controls [100]. MALAT1 positively regulates the expression of SP7 by binding to miR-143 and miR-196 and inhibiting the binding of these inhibitory miRNAs to the SP7 mRNA [101]. Therefore, multiple non-coding RNAs (LIOCE, MALAT, miR-143, and miR-196) all control SP7 at the post-transcriptional level. These non-coding RNAs provide an additional opportunity for future diagnostics and therapeutics to modulate SP7 activity for diseases including osteoporosis and metastatic bone disease.
Future Directions and Conclusions
SP7 plays important regulatory roles in different mesenchymal lineage bone cell types during bone formation and remodeling, including osteoblasts, osteocytes, and chondrocytes. As discussed in this review, common SP7 polymorphisms are linked to BMD variation and fracture risk, rare SP7 mutations cause skeletal dysplasia, and SP7 may contribute to bone metastasis. Novel genomic and proteomic methods should enable the discovery of cell type-specific gene regulatory elements and cofactors for SP7 to help explain how this single transcription factor accomplishes so many important roles throughout the mesenchymal bone cell lineage. Improved understanding of the mechanisms underlying SP7-causing skeletal diseases will provide novel therapeutic targets for the treatment of skeletal disorders.
Acknowledgements
J.S.W. acknowledges funding support from the National Institute of Health (T32DK007028, K99AR081897). M.N.W. acknowledges funding support from the National Institute of Health (P01DK011794, R01DK116716).
Footnotes
Compliance with Ethical Standards
Conflict of interest MNW receives research funding from Radius Health and holds equity in and is a scientific advisory board member of Relation Therapeutics.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Berendsen AD, Olsen BR. Bone development. Bone. 2015;80:14–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan WCW, Tan Z, To MKT, Chan D. Regulation and role of transcription factors in osteogenesis. Int J Mol Sci. 2021;22:5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell. 2018;172:650–65. [DOI] [PubMed] [Google Scholar]
- 4.Mitsis T, Efthimiadou A, Bacopoulou F, Vlachakis D, Chrousos G, Eliopoulos E. Transcription factors and evolution: an integral part of gene expression (review). World Acad Sci J. 2020;2:3–8. [Google Scholar]
- 5.Long F, Ornitz DM. Development of the endochondral skeleton. Csh Perspect Biol. 2013;5:a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rolph D, Das H. Transcriptional regulation of osteoclastogenesis: the emerging role of KLF2. Front Immunol. 2020;11:937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B. Sox9 is required for cartilage formation. Nat Genet. 1999;22:85–9. [DOI] [PubMed] [Google Scholar]
- 8.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–64. [DOI] [PubMed] [Google Scholar]
- 9.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. [DOI] [PubMed] [Google Scholar]
- 10.Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, et al. Requirement for NF-κB in osteoclast and B-cell development. Gene Dev. 1997;11:3482–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology implication for Coffin-Lowry syndrome. Cell. 2004;117:387–98. [DOI] [PubMed] [Google Scholar]
- 12.Lu S-Y, Li M, Lin Y-L. Mitf induction by RANKL is critical for osteoclastogenesis. Mol Biol Cell. 2010;21:1763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. [DOI] [PubMed] [Google Scholar]
- 14.Philipsen S, Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 1999;27:2991–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao Y, Jheon A, Nourkeyhani H, Kobayashi H, Ganss B. Molecular cloning, structure, expression, and chromosomal localization of the human Osterix (SP7) gene. Gene. 2004;341:101–10. [DOI] [PubMed] [Google Scholar]
- 16.Nakashima K, de Crombrugghe B. Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet. 2003;19:458–66. [DOI] [PubMed] [Google Scholar]
- 17.Xing W, Godwin C, Pourteymoor S, Mohan S. Conditional disruption of the osterix gene in chondrocytes during early postnatal growth impairs secondary ossification in the mouse tibial epiphysis. Bone Res. 2019;7:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang JS, Kamath T, Mazur CM, Mirzamohammadi F, Rotter D, Hojo H, et al. Control of osteocyte dendrite formation by Sp7 and its target gene osteocrin. Nat Commun. 2021;12:6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hojo H, Ohba S. Sp7 Action in the skeleton: its mode of action, functions, and relevance to skeletal diseases. Int J Mol Sci. 2022;23:5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park J-S, Park G-I, Kim J-E. Osterix is dispensable for the development of the mouse olfactory bulb. Biochem Bioph Res Co. 2016;478:110–5. [DOI] [PubMed] [Google Scholar]
- 21.Aguilar R, Bustos FJ, Nardocci G, Zundert B, Montecino M. Epigenetic silencing of the osteoblast-lineage gene program during hippocampal maturation. J Cell Biochem. 2021;122:367–84. [DOI] [PubMed] [Google Scholar]
- 22.Mullen RD, Wang Y, Liu B, Moore EL, Behringer RR. Osterix functions downstream of anti-Müllerian hormone signaling to regulate Müllerian duct regression. Proc National Acad Sci. 2018;115:8382–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou X, Zhang Z, Feng JQ, Dusevich VM, Sinha K, Zhang H, et al. Multiple functions of Osterix are required for bone growth and homeostasis in postnatal mice. Proc National Acad Sci. 2010;107:12919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baek W, Lee M, Jung JW, Kim S, Akiyama H, de Crombrugghe B, et al. Positive regulation of adult bone formation by osteoblast-specific transcription factor osterix. J Bone Miner Res. 2009;24:1055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baek W-Y, de Crombrugghe B, Kim J-E. Postnatally induced inactivation of osterix in osteoblasts results in the reduction of bone formation and maintenance. Bone. 2010;46:920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ortuño MJ, Susperregui ARG, Artigas N, Rosa JL, Ventura F. Osterix induces Col1a1 gene expression through binding to Sp1 sites in the bone enhancer and proximal promoter regions. Bone. 2013;52:548–56. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y, Huang Y, Zhang L, Zhang C. Transcriptional regulation of bone sialoprotein gene expression by Osx. Biochem Bioph Res Co. 2016;476:574–9. [DOI] [PubMed] [Google Scholar]
- 28.Yang F, Tang W, So S, de Crombrugghe B, Zhang C. Sclerostin is a direct target of osteoblast-specific transcription factor osterix. Biochem Bioph Res Co. 2010;400:684–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yano H, Hamanaka R, Nakamura-Ota M, Adachi S, Zhang JJ, Matsuo N, et al. Sp7/Osterix induces the mouse pro-α2(I) collagen gene (Col1a2) expression via the proximal promoter in osteoblastic cells. Biochem Bioph Res Co. 2014;452:531–6. [DOI] [PubMed] [Google Scholar]
- 30.Zhang C, Tang W, Li Y. Matrix metalloproteinase 13 (MMP13) is a direct target of osteoblast-specific transcription factor osterix (Osx) in osteoblasts. Plos One. 2012;7:e50525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.China GPKL of O and TRI The First Affiliated Hospital of Sun Yat-sen University, Guangzhou Guangdong, Gao M, Su Q, Liang T, Ma J, Stoddart M, et al. Transcriptional activation of ENPP1 by osterix in osteoblasts and osteocytes. Eur Cells Mater. 2018;36:1–14. [DOI] [PubMed] [Google Scholar]
- 32.Kawane T, Komori H, Liu W, Moriishi T, Miyazaki T, Mori M, et al. Dlx5 and Mef2 regulate a novel Runx2 enhancer for osteoblast-specific expression. J Bone Miner Res. 2014;29:1960–9. [DOI] [PubMed] [Google Scholar]
- 33.Hojo H, Ohba S, He X, Lai LP, McMahon AP. Sp7/osterix is restricted to bone-forming vertebrates where it acts as a Dlx cofactor in osteoblast specification. Dev Cell. 2016;37:238–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koga T, Matsui Y, Asagiri M, Kodama T, de Crombrugghe B, Nakashima K, et al. NFAT and Osterix cooperatively regulate bone formation. Nat Med. 2005;11:880–5. [DOI] [PubMed] [Google Scholar]
- 35.Franz-Odendaal TA, Hall BK, Witten PE. Buried alive: how osteoblasts become osteocytes. Dev Dynam. 2005;235:176–90. [DOI] [PubMed] [Google Scholar]
- 36.Lu Y, Xie Y, Zhang S, Dusevich V, Bonewald LF, Feng JQ. DMP1-targeted cre expression in odontoblasts and osteocytes. J Dent Res. 2007;86:320–5. [DOI] [PubMed] [Google Scholar]
- 37. Moriishi T, Ito T, Fukuyama R, Qin X, Komori H, Kaneko H, et al. Sp7 transgenic mice with a markedly impaired lacunocanalicular network induced sost and reduced bone mass by unloading. Int J Mol Sci. 2022;23:3173. • A novel study reported that overexpression of Sp7 in vivo impaired the lacunar-canalicular network and failed to restore bone formation during unloading. It demonstrated the importance of proper Sp7 expression level in osteocytes.
- 38.McNamara LM, Majeska RJ, Weinbaum S, Friedrich V, Schaffler MB. Attachment of osteocyte cell processes to the bone matrix. Anatomical Rec. 2009;292:355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thi MM, Suadicani SO, Schaffler MB, Weinbaum S, Spray DC. Mechanosensory responses of osteocytes to physiological forces occur along processes and not cell body and require αVβ3 integrin. Proc National Acad Sci. 2013;110:21012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moon YJ, Yun C-Y, Choi H, Kim JR, Park B-H, Cho E-S. Osterix regulates corticalization for longitudinal bone growth via integrin β3 expression. Exp Mol Medicine. 2018;50:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCutcheon S, Majeska RJ, Spray DC, Schaffler MB, Vazquez M. Apoptotic osteocytes induce RANKL production in bystanders via purinergic signaling and activation of pannexin channels. J Bone Miner Res. 2020;35:966–77. • A recent study showed that extracellular ATP, released from apoptotic osteocytes via Panx1 channels, is a major signal for triggering bystander osteocyte to express RANKL.
- 42.Omoteyama K, Takagi M. The effects of Sp7/Osterix gene silencing in the chondroprogenitor cell line, ATDC5. Biochem Bioph Res Co. 2010;403:242–6. [DOI] [PubMed] [Google Scholar]
- 43.Oh J-H, Park S-Y, de Crombrugghe B, Kim J-E. Chondrocyte-specific ablation of osterix leads to impaired endochondral ossification. Biochem Bioph Res Co. 2012;418:634–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishimura R, Wakabayashi M, Hata K, Matsubara T, Honma S, Wakisaka S, et al. Osterix regulates calcification and degradation of chondrogenic matrices through matrix metalloproteinase 13 (MMP13) expression in association with transcription factor Runx2 during endochondral ossification*. J Biol Chem. 2012;287:33179–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen S, Gluhak-Heinrich J, Wang YH, Wu YM, Chuang HH, Chen L, et al. Runx2, Osx, and Dspp in tooth development. J Dent Res. 2009;88:904–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ono W, Sakagami N, Nishimori S, Ono N, Kronenberg HM. Parathyroid hormone receptor signalling in osterix-expressing mesenchymal progenitors is essential for tooth root formation. Nat Commun. 2016;7:11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miyazaki T, Inoue M, Baba TT, Komori T. Overexpression of Sp7 in odontoblasts results in dentinogenesis imperfecta due to the inhibition of odontoblast maturation. J Oral Biosci. 2017;59:113–20. [Google Scholar]
- 48.Bae J, Clarke JC, Rashid H, Adhami MD, McCullough K, Scott JS, et al. Specificity protein 7 is required for proliferation and differentiation of ameloblasts and odontoblasts. J Bone Miner Res. 2018;33:1126–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Jiang Y, Qin C, Liu Y, Ho SP, Feng JQ. Essential role of osterix for tooth root but not crown dentin formation. J Bone Miner Res. 2015;30:742–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim TH, Bae CH, Lee JC, Kim JE, Yang X, de Crombrugghe B, et al. Osterix regulates tooth root formation in a site-specific manner. J Dent Res. 2015;94:430–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cao Z, Zhang H, Zhou X, Han X, Ren Y, Gao T, et al. Genetic evidence for the vital function of osterix in cementogenesis. J Bone Miner Res. 2012;27:1080–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Abood A, Farber CR. Using “-omics” Data to inform genome-wide association studies (GWASs) in the Osteoporosis Field. Curr Osteoporos Rep. 2021;19:369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang T-L, Shen H, Liu A, Dong S-S, Zhang L, Deng F-Y, et al. A road map for understanding molecular and genetic determinants of osteoporosis. Nat Rev Endocrinol. 2019;16:91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hannan FM, Newey PJ, Whyte MP, Thakker RV. Genetic approaches to metabolic bone diseases. Brit J Clin Pharmaco. 2019;85:1147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179:2393–419. [DOI] [PubMed] [Google Scholar]
- 56.Zhu X, Bai W, Zheng H. Twelve years of GWAS discoveries for osteoporosis and related traits: advances, challenges and applications. Bone Res. 2021;9:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanis JA, Delmas P, Burckhardt P, Cooper C, Torgerson D. Guidelines for diagnosis and management of osteoporosis. Osteoporosis Int. 1997;7:390–406. [DOI] [PubMed] [Google Scholar]
- 58.Timpson NJ, Tobias JH, Richards JB, Soranzo N, Duncan EL, Sims A-M, et al. Common variants in the region around osterix are associated with bone mineral density and growth in childhood. Hum Mol Genet. 2009;18:1510–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Richards J, Rivadeneira F, Inouye M, Pastinen T, Soranzo N, Wilson S, et al. Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet. 2008;371:1505–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, et al. Multiple genetic loci for bone mineral density and fractures. New Engl J Medicine. 2008;358:2355–65. [DOI] [PubMed] [Google Scholar]
- 61.Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, et al. New sequence variants associated with bone mineral density. Nat Genet. 2009;41:15–7. [DOI] [PubMed] [Google Scholar]
- 62.Rivadeneira F, Styrkársdottir U, Estrada K, Halldórsson BV, Hsu Y-H, Richards JB, et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat Genet. 2009;41:1199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Estrada K, Styrkarsdottir U, Evangelou E, Hsu Y-H, Duncan EL, Ntzani EE, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Forlino A, Marini JC. Osteogenesis imperfecta. Lancet. 2016;387:1657–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marini JC, Forlino A, Bächinger HP, Bishop NJ, Byers PH, Paepe AD, et al. Osteogenesis imperfecta. Nat Rev Dis Primers. 2017;3:17052. [DOI] [PubMed] [Google Scholar]
- 66.Majorana A, Bardellini E, Brunelli PC, Lacaita M, Cazzolla AP, Favia G. Dentinogenesis imperfecta in children with osteogenesis imperfecta: a clinical and ultrastructural study. Int J Paediatr Dent. 2010;20:112–8. [DOI] [PubMed] [Google Scholar]
- 67.Biria M, Abbas FM, Mozaffar S, Ahmadi R. Dentinogenesis imperfecta associated with osteogenesis imperfecta. Dent Res J. 2012;9:489–94. [PMC free article] [PubMed] [Google Scholar]
- 68.Abukabbos H, Al-Sineedi F. Clinical manifestations and dental management of dentinogenesis imperfecta associated with osteogenesis imperfecta: case report. Saudi Dent J. 2013;25:159–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim J-W, Simmer JP. Hereditary dentin defects. J Dent Res. 2007;86:392–9. [DOI] [PubMed] [Google Scholar]
- 70.Malmgren B, Tsilingaridis G, Monsef-Johansson N, Qahtani ZHA, Dahllöf G, Åström E. Bisphosphonate therapy and tooth development in children and adolescents with osteogenesis imperfecta. Calcif Tissue Int. 2020;107:143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dwan K, Phillipi CA, Steiner RD, Basel D. Bisphosphonate therapy for osteogenesis imperfecta. Cochrane Db Syst Rev. 2014;10:CD005088. [DOI] [PubMed] [Google Scholar]
- 72.Marks SC, Schroeder HE. Tooth eruption: theories and facts. Anat Rec. 1996;245:374–93. [DOI] [PubMed] [Google Scholar]
- 73.Wise G. Cellular and molecular basis of tooth eruption. Orthod Craniofac Res. 2009;12:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lapunzina P, Aglan M, Temtamy S, Caparrós-Martín JA, Valencia M, Letón R, et al. Identification of a frameshift mutation in osterix in a patient with recessive osteogenesis imperfecta. Am J Hum Genet. 2010;87:110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tung JY, Ho JL, Wong R, Fung S. Dental phenotype in an adolescent with osteogenesis imperfecta type XII. Bmj Case Rep. 2022;15:e246554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hayat A, Hussain S, Bilal M, Kausar M, Almuzzaini B, Abbas S, et al. Biallelic variants in four genes underlying recessive osteogenesis imperfecta. Eur J Med Genet. 2020;63:103954. [DOI] [PubMed] [Google Scholar]
- 77. Fiscaletti M, Biggin A, Bennetts B, Wong K, Briody J, Pacey V, et al. Novel variant in Sp7/Osx associated with recessive osteogenesis imperfecta with bone fragility and hearing impairment. Bone. 2018;110:66–75. • This study reported the first missense mutation identified within the zinc-finger domain of SP7 that causes osteogenesis imperfecta.
- 78. Ludwig K, Ward LM, Khan N, Robinson M-E, Miranda V, Bardai G, et al. Dominant osteogenesis imperfecta with low bone turnover caused by a heterozygous SP7 variant. Bone. 2022;160:116400. • A recent study reported a novel SP7 variant (E340A) that causes dominant osteogenesis imperfecta.
- 79.Whyte MP, Campeau PM, McAlister WH, Roodman GD, Kurihara N, Nenninger A, et al. Juvenile Paget’s Disease from heterozygous mutation of SP7 encoding osterix (specificity protein 7, transcription factor SP7). Bone. 2020;137:115364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lui JC, Raimann A, Hojo H, Dong L, Roschger P, Kikani B, et al. A neomorphic variant in SP7 alters sequence specificity and causes a high-turnover bone disorder. Nat Commun. 2022;13:700. •• This study generated the mouse model that carries the neomorphic (S309W) variant. SP7 S309W losses its affinity for AT-rich motifs, which is critical for normal osteoblast differentiation.
- 81.Horvai AE, Roy R, Borys D, O’Donnell RJ. Regulators of skeletal development: a cluster analysis of 206 bone tumors reveals diagnostically useful markers. Modern Pathol. 2012;25:1452–61. [DOI] [PubMed] [Google Scholar]
- 82.Wittig JC, Bickels J, Priebat D, Jelinek J, Kellar-Graney K, Shmookler B, et al. Osteosarcoma: a multidisciplinary approach to diagnosis and treatment. Am Fam Physician. 2002;65:1123–32. [PubMed] [Google Scholar]
- 83.Cao Y, Zhou Z, de Crombrugghe B, Nakashima K, Guan H, Duan X, et al. Osterix, a transcription factor for osteoblast differentiation, mediates antitumor activity in murine osteosarcoma. Cancer Res. 2005;65:1124–8. [DOI] [PubMed] [Google Scholar]
- 84.Cao Y, Jia S-F, Chakravarty G, de Crombrugghe B, Kleinerman ES. The osterix transcription factor down-regulates interleukin-1α expression in mouse osteosarcoma cells. Mol Cancer Res. 2008;6:119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang X, Kua H-Y, Hu Y, Guo K, Zeng Q, Wu Q, et al. p53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J Cell Biol. 2006;172:115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.He Y, de Castro LF, Shin MH, Dubois W, Yang HH, Jiang S, et al. p53 loss increases the osteogenic differentiation of bone marrow stromal cells. Stem Cells. 2015;33:1304–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Artigas N, Gámez B, Cubillos-Rojas M, Diego CS, Valer JA, Pons G, et al. p53 inhibits SP7/osterix activity in the transcriptional program of osteoblast differentiation. Cell Death Differ. 2017;24:2022–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dai Q-S, Zhou H-Y, Wu Z-H, Long J-T, Shao N, Cheang T-Y, et al. Osterix transcriptional factor is involved in the metastasis of human breast cancers. Oncol Lett. 2015;10:1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yao B, Wang J, Qu S, Liu Y, Jin Y, Lu J, et al. Upregulated osterix promotes invasion and bone metastasis and predicts for a poor prognosis in breast cancer. Cell Death Dis. 2019;10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. [DOI] [PubMed] [Google Scholar]
- 91. Ricci B, Tycksen E, Celik H, Belle JI, Fontana F, Civitelli R, et al. Osterix-Cre marks distinct subsets of CD45- and CD45+ stromal populations in extra-skeletal tumors with pro-tumorigenic characteristics. Elife. 2020;9:e54659. • A paper reported Osx+ mesenchymal cells during extracellular matrix remodeling and contributes to the tumor microenvironment.
- 92.Weinstein RS. Glucocorticoids, osteocytes, and skeletal fragility: the role of bone vascularity. Bone. 2010;46:564–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fowler TW, Acevedo C, Mazur CM, Hall-Glenn F, Fields AJ, Bale HA, et al. Glucocorticoid suppression of osteocyte perilacunar remodeling is associated with subchondral bone degeneration in osteonecrosis. Sci Rep-uk. 2017;7:44618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mazur CM, Andrade CDC, Tokavanich N, Sato T, Bruce M, Brooks DJ, et al. Partial prevention of glucocorticoid-induced osteocyte deterioration in young male mice with osteocrin gene therapy. Iscience. 2022;25:105019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sánchez-Barceló EJ, Mediavilla MD, Tan DX, Reiter RJ. Scientific basis for the potential use of melatonin in bone diseases: osteoporosis and adolescent idiopathic scoliosis. J Osteoporos. 2010;2010:830231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lu X, Yu S, Chen G, Zheng W, Peng J, Huang X, et al. Insight into the roles of melatonin in bone tissue and bone-related diseases (Review). Int J Mol Med. 2021;47:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Han Y, Kim Y-M, Kim HS, Lee KY. Melatonin promotes osteoblast differentiation by regulating Osterix protein stability and expression. Sci Rep-uk. 2017;7:5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Choi YH, Han Y, Lee SH, Jin Y-H, Bahn M, Hur KC, et al. Cbl-b and c-Cbl negatively regulate osteoblast differentiation by enhancing ubiquitination and degradation of Osterix. Bone. 2015;75:201–9. [DOI] [PubMed] [Google Scholar]
- 99.Ren L, Zeng F, Deng J, Bai Y, Chen K, Chen L, et al. Inflammatory osteoclasts-derived exosomes promote bone formation by selectively transferring lncRNA LIOCE into osteoblasts to interact with and stabilize Osterix. Faseb J. 2022;36:e22115. [DOI] [PubMed] [Google Scholar]
- 100.Gao Y, Xiao F, Wang C, Wang C, Cui P, Zhang X, et al. Long noncoding RNA MALAT1 promotes osterix expression to regulate osteogenic differentiation by targeting miRNA-143 in human bone marrow-derived mesenchymal stem cells. J Cell Biochem. 2018;119:6986–96. [DOI] [PubMed] [Google Scholar]
- 101.Zang L-Y, Yang X-L, Li W-J, Liu G-L. Long noncoding rna metastasis-associated lung adenocarcinoma transcript 1 promotes the osteoblast differentiation of human bone marrow-derived mesenchymal stem cells by targeting the microRNA-96/osterix axis. J Craniofac Surg. 2022;33:956–61. [DOI] [PubMed] [Google Scholar]