

XPO1 regulates the dynamic ribonucleoprotein nuclear export in response to genotoxic stress to support tolerance and can be targeted to enhance the sensitivity of cancer cells to endogenous and exogenous DNA damage.
Abstract
Exportin-1 (XPO1), the main soluble nuclear export receptor in eukaryotic cells, is frequently overexpressed in diffuse large B-cell lymphoma (DLBCL). A selective XPO1 inhibitor, selinexor, received approval as single agent for relapsed or refractory (R/R) DLBCL. Elucidating the mechanisms by which XPO1 overexpression supports cancer cells could facilitate further clinical development of XPO1 inhibitors. We uncovered here that XPO1 overexpression increases tolerance to genotoxic stress, leading to a poor response to chemoimmunotherapy. Upon DNA damage induced by MYC expression or exogenous compounds, XPO1 bound and exported EIF4E and THOC4 carrying DNA damage repair mRNAs, thereby increasing synthesis of DNA damage repair proteins under conditions of increased turnover. Consequently, XPO1 inhibition decreased the capacity of lymphoma cells to repair DNA damage and ultimately resulted in increased cytotoxicity. In a phase I clinical trial conducted in R/R DLBCL, the combination of selinexor with second-line chemoimmunotherapy was tolerated with early indication of efficacy. Overall, this study reveals that XPO1 overexpression plays a critical role in the increased tolerance of cancer cells to DNA damage while providing new insights to optimize the clinical development of XPO1 inhibitors.
Significance:
XPO1 regulates the dynamic ribonucleoprotein nuclear export in response to genotoxic stress to support tolerance and can be targeted to enhance the sensitivity of cancer cells to endogenous and exogenous DNA damage.
Graphical Abstract
Introduction
Nuclear pores (NP) are functional units comprising a large transmembrane protein complex (nucleoporins) and multiple soluble nuclear receptors (1). Soluble receptors bind cargo proteins via specific sequences and mediate their active and directional transport (2). Besides their role in cell growth and differentiation, NPs are essential components of the cellular response to several types of stresses (3, 4). Stress tolerance requires selective, rapid, and simultaneous expression of proteins involved in response pathways. NPs may modulate this response by facilitating the spatial and temporal coordination of gene expression (5, 6). Transcript-selective nuclear export, rather than, for example, increasing the number of NPs, allows for rapid modulation of gene expression (7–10). Malignant transformation and adaptation to dynamic microenvironment conditions requires cancer cells to respond to several types of stresses, thus nucleoporins and other factors involved in mRNA export are frequently overexpressed in cancer cells and their activity is required for proliferation and survival (11–13).
Exportin-1 (XPO1) is the major soluble protein export receptor in the nucleus and binds its cargo proteins via the canonical leucine-rich nuclear export signal domain (14–16). XPO1 is critical for nucleoplasm-cytoplasm partitioning after cell division and binds a broad range of proteins with diverse functions (17, 18). XPO1 locus maps within the 2p15–16 region, which is frequently amplified in diffuse large B-cell lymphoma (DLBCL; refs. 19–21). DLBCL is an aggressive disease that is curable in about two thirds of patients with chemoimmunotherapy (22, 23). Patients who are either relapsed or refractory (R/R) to first-line treatment have poor outcomes with standard therapeutic approaches (24–26). Notably, elevated XPO1 expression is associated with an advanced clinical stage in DLBCL (27), and the XPO1 inhibitor selinexor received accelerated approval as a single agent for R/R diseases to at least two lines of systemic chemoimmunotherapy (28). While clinical and preclinical data suggest that XPO1 is a therapeutic target in B-cell lymphomas, the mechanism underlying the reliance of cancer cells on XPO1 for proliferation and survival have not been fully elucidated. Proteomic studies revealed that the cargo spectrum of XPO1 in mammalian cells is broader than predicted, comprising constitutive and facultative cargoes, and including several RNA-binding proteins (ribonucleoproteins; refs. 29, 30). This suggests that XPO1 exporting activity can be modulated to maintain cellular homeostasis, an effect we postulated is linked to its oncogenic role. Elucidating these mechanisms will not only explain the “addiction” of cancer cells to XPO1 overexpression but also will facilitate further clinical development of XPO1 inhibitors by improving their therapeutic window and providing a mechanistic rationale for combinatorial treatments.
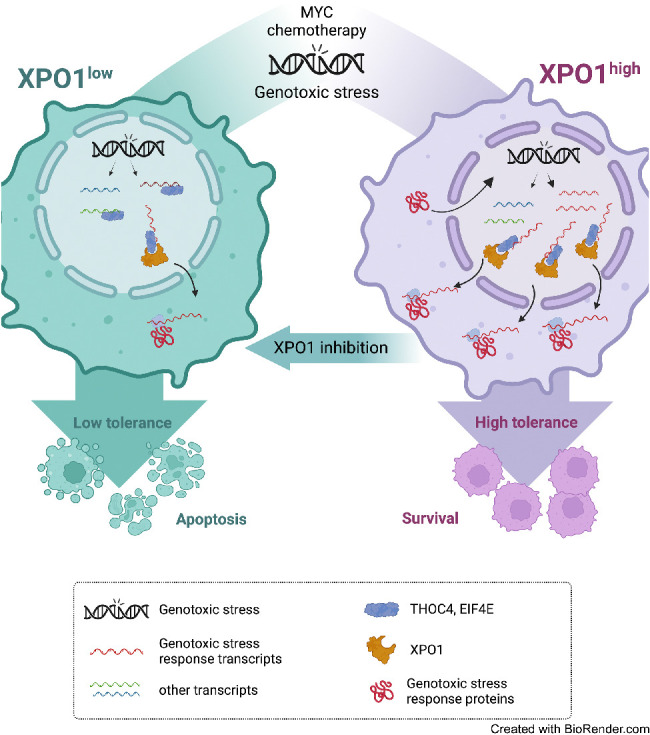
Here, we report that lymphoma cells rely on XPO1 overexpression to optimize nuclear-cytosolic mRNA trafficking in response to genotoxic stress. This results in increased genotoxic stress tolerance by facilitating timely DNA damage repair, an effect critical for DLBCL harboring elevated levels of genomic instability. Accordingly, XPO1 inhibition is sufficient to increase sensitivity to DNA-damaging agents in lymphoma cells and animal models of DLBCL. Mechanistically, we demonstrated that, in response to genotoxic stress, XPO1 preferentially exports the ribonucleoproteins THOC4 (THO complex subunit 4) and EIF4E (eukaryotic translation initiation factor 4E) carrying mRNAs that encode DNA damage repair proteins. This ultimately results in sustained expression of DNA damage repair proteins under conditions of increased turnover in cancer cells. On the basis of these findings, we designed a phase I clinical trial of selinexor in combination with chemoimmunotherapy aggressive DLBCL, demonstrating tolerability and therapeutic effect.
Patients and Methods
Clinical trial design
The clinical trial was an open-label, phase I study with a standard 3+3 dose escalation schema and was approved by the Institutional Review Board (IRB) of Weill Cornell Medicine–NewYork-Presbyterian Hospital. Registration number: NCT02471911. Written informed consent was obtained from the patients. The studies were conducted in accordance with the Declaration of Helsinki. The study's primary objective was to determine the safety and tolerability of the combination of selinexor and R-ICE. The primary endpoint was to identify the maximum tolerated dose (MTD) and recommended phase II dose (RP2D) of selinexor when combined with R-ICE. The secondary objectives of the study included determining efficacy by assessing the overall response rate and assessing the feasibility of stem cell collection in patients who received the combination treatment.
Eligible patients included those with pathologically confirmed aggressive B-cell lymphomas including DLBCL, double-hit high-grade B-cell lymphoma (DH-HGBL), indolent lymphomas transformed to DLBCL or another aggressive B-cell lymphoma, and follicular lymphoma grade 3B. A separate cohort enrolled patients with DLBCL transformed from chronic lymphocytic leukemia [CLL; i.e., Richter's transformation (RT)]. Treatment with one prior regimen administered with curative intent was required. In DLBCL transformed from CLL, there was no requirement regarding prior therapies; subjects were eligible if they were untreated or if they have received more than one line of prior treatment. Additional key inclusion criteria included adequate end-organ and bone marrow function, absence of known or suspected central nervous system (CNS) involvement, and absence of active viral hepatitis or uncontrolled HIV.
Selinexor was initially dosed at 40 mg (dose level -1), 60 mg (dose level 1), and 80 mg (dose level 2) on days -5, -3, 1, 3, and 5 and R-ICE on days 1–3 of a 21-day cycle. Because of CNS toxicity thought to be primarily related to ifosfamide, the protocol was amended so that R-ICE was given on days 1–3 and selinexor following completion of ifosfamide on days 3, 5, and 7 of each cycle. After 2 cycles, responding patients were eligible to receive SCT or chimeric antigen receptor (CAR) T-cell therapy at discretion of treating physician. Transplant-ineligible patients were allowed to complete up to 6 cycles of selinexor and RICE, and those with responses or stable disease (SD) were eligible for maintenance selinexor (60 mg weekly) at the discretion of the treating physician. The cohort of patients with CLL transformed to DLBCL were treated at dose level 1 and were not escalated.
Adverse events reporting and response evaluation
Safety was determined by continuous assessment of adverse events (AE) graded according to the NCI Common Terminology Criteria for Adverse Events, version 4.0.3. All patients were evaluated for toxicity from the time of their first treatment. Dose-limiting toxicity (DLT) was defined as any of the following treatment-related AE during the first cycle of treatment: grade 4 febrile neutropenia; grade 4 neutrophils for ≥7 days; grade 4 platelets for ≥10 days; grade 3 nausea, vomiting, diarrhea, or fatigue lasting >3 days; or grade ≥3 non-hematologic toxicities except alopecia, fatigue or electrolyte abnormalities correctable with supportive care. Tumor assessment was performed with imaging scans at baseline and after 2 cycles. Treatment responses were defined by using the revised International Workshop Criteria (IWG). If response determination was unable to be made using IWG, Lugano criteria were used. The clinical trial protocol is available in Supplementary Materials.
Analysis of DLBCL samples
Treatment-naïve DLBCL samples were obtained with the approvals of the IRBs of Weill Cornell Medicine or the University of Turin. Diagnosis was performed (and revised) by certified hemopathologists. Tumor tissues collected at these Institutions were kept at −140°C as frozen (viable) material and formalin-fixed and processed for paraffin embedding. Matched germline control DNA was obtained from saliva or peripheral lymphocytes/monocytes and stored at −20°C until use. Fifty-seven patients had material enough for protein expression and outcome analysis, and a subset of 42 patients had material enough for protein expression and genomic analysis. Copy-number alterations and other genetic alterations analyses were conducted by whole-exome sequencing (WES) and whole-genome sequencing (WGS) next generation sequencing as published (31). Additional analysis of XPO1, REL and BCL11A copy-number gains were conducted with GISTIC v2.0. XPO1 protein expression was conducted by IHC on two tissue microarray sections using a mouse monoclonal antibody against XPO1 (sc-5595, Santa Cruz Biotechnology) following standard pathology protocols. Antigen was retrieved by using 10 mmol/L sodium citrate buffer pH 6.4 and endogenous peroxidase activity was quenched by treatment with 3% hydrogen peroxide. Images were obtained using an Axioskop imaging microscope (Zeiss Inc.). Evaluation and scoring were independently made by two investigators. The percentage of positive tumor cells and intensity of staining (by an ad hoc scale from 1 to 3) were used to determine the total score of each sample. All 57 patients included for outcome analysis received R-CHOP as first-line treatment. Patients were defined as responders when achieved a complete response and remained disease-free at 5 years, and nonresponders when either a complete response was never achieved (refractory) or the disease relapsed within 2 years (relapsed).
Other DLBCL samples containing RNA sequencing (RNA-seq), WES and WGS were complied, collected, and re-analyzed in a database totalizing 4,655 cases as published in (31). Clinical outcome analysis, cell-of-origin classification and pathway analysis of these samples were conducted as published in (31). For analysis that required specific molecular and clinical annotations, we used the largest possible subset of the data that had the required information and the number of patients included in each of these analyses is indicated in the respective figure panel.
Cell lines and compounds
DLBCL cell lines were obtained from the ATCC, DMSZ, or the Ontario Cancer Institute. P493–6 B cells were obtained from the original developer Dr. Dirk Eick. Cell lines identification was performed once a year by short tandem repeat analysis at the University of Arizona Genetics Core. Mycoplasma contamination was tested quarterly by PCR. DLBCL cell lines OCI-Ly1, OCI-Ly18, OCI-Ly8, and OCI-Ly7 were cultured in IMDM supplemented with 10% FCS and 1% penicillin G and streptomycin. DLBCL cell lines Toledo, DoHH2, TMD8, Karpas422, SU-DHL4, SU-DHL6, SU-DHL8, WSU-DLBCL-2, Farage, HBL-1, and MD901 were cultured in RPMI supplemented with 10% FCS, 1% penicillin G and streptomycin, and 1% HEPES. P493–6 B cells were cultured in RPMI supplemented with 10% FCS, 1% penicillin G and streptomycin, 1% HEPES and 1% pyruvate. Selinexor was obtained from Karyopharm. Cyclophosphamide, mechlorethamine, doxorubicin, and vincristine were purchased from Sigma-Aldrich, and dexamethasone and etoposide were purchased from SelleckChem.
Cell-cycle distribution and γH2AX level
Thirty minutes following doxycycline withdrawal (to induce MYC expression), P493–6 B-cells were exposed to vehicle or selinexor (1 μmol/L) for 6 hours and cellular DNA content (for cell-cycle analysis) and γH2AX levels (for DNA damage analysis) were simultaneously assessed by flow cytometry. Cells were fixed and permeabilized by using the FIX & PERM Cell Permeabilization Kit (ThermoFisher Scientific) according to the manufacturer's protocol. Cells were then stained with FITC anti-γH2AX (BioLegend) and DAPI (1 μg/mL). Labeled cells were analyzed by using a MACSQuant flow cytometer (Miltenyi Biotech) and analyzed using FlowJo software (TreeStar Inc.).
Growth inhibition and apoptosis analysis
Lymphoma cells were grown on U-bottom 96-well plates at respective concentrations sufficient to keep untreated cells under exponential growth over the complete drug exposure time. For single drug experiments, cells were exposed to 5 concentrations of selinexor for 72 hours. For combinatorial experiments, cells were exposed concurrently or sequentially to 4 concentrations of selinexor and a chemotherapy drug for 72 hours. Cell viability was analyzed by using a fluorometric reduction method (CellTiter-Blue, Promega) and fluorescence (560Ex/590Em) was determined with the Synergy4 microplate reader (Biotek). The fluorescence was determined for three replicates per treatment condition and normalized to their respective controls (vehicle-treated). CompuSyn software (Biosoft) was used to plot dose–effect curves and determine the drug concentrations that inhibit the growth of the cell lines by 50% (GI50) compared with the control. To quantify the chemosensitization effect, the Dose Reduction Index (DRI) was calculated with CompuSyn software (Biosoft). The DRI represents how many folds a dose of a drug can be reduced by the addition of another drug to achieve the same efficacy. For apoptosis analysis DLBCL cells were grown on U-bottom 96-well plates at 25,000 cells per well, sufficient to keep untreated cells under exponential growth over the complete drug exposure time. Cells were exposed to 1 μmol/L of Selinexor for 72 hours for each cell line. Caspase-3/7 activity was measured using a specific luminescent method (Caspase-Glo 3/7 assay) and luminescence was determined by Spectramax Id5 plate reader (Molecular Device). The luminescence was determined for three replicates per treatment condition and normalized to their respective vehicle controls. Senescence was determined by activation of β-galactosidase enzyme (CellEvent Senescence Green Detection Kit). Cells were exposed to 0.3 μmol/L of selinexor (or vehicle) for up to 120 hours, washed with PBS and fixated using a 2% paraformaldehyde solution for 15 minutes at room temperature. After fixation, cells were incubated with the kit reagents for 2 hours and analyzed in a MACSQuant flow cytometer (Miltenyi Biotech).
Single cell gel electrophoresis assays
An alkaline comet assay was used to assess DNA damage as described previously (32, 33). Cells were exposed to vehicle or selinexor (1 μmol/L) for 6 hours followed by etoposide (3 μmol/L), and DNA damage was assessed after 4 hours of continuous exposure and after 4 hours of recovery (where etoposide was removed). Two slides per condition were imaged for each experiment with an Axiovert 200M fluorescent microscope (Zeiss Inc.), and approximately 70 comets per slide were scored with Comet Score (TriTek).
Animal experiments
All animal procedures followed NIH protocols and were approved by the Animal Institute Committee of the Weill Cornell Medicine. A patient-derived tumor xenograft (PDTX) of a chemoresistant MYC-driven XPO1 amplified DLBCL was established as previously described (34). Briefly, NSG mice housed in a barrier environment were subcutaneously injected in both flanks with primary human DLBCL cells. When tumors reached a palpable size (approximately 75–100 mm3), mice were first randomized into two treatment arms to receive either vehicle or CHOP on day 1. On the second day, each arm was further randomized into two arms, generating four treatment groups: A, vehicle; B, selinexor; C, CHOP; D, the combination of selinexor and CHOP. Two groups (A and B) received vehicle and two groups (C and D) received selinexor at 7.5 mg/kg orally according to the schedule shown in Fig. 3B. Tumor volume was monitored every other day using electronic digital calipers in two dimensions. Tumor volume was calculated using the following formula: tumor volume (mm3) = (smallest diameter2 × largest diameter)/2. The AUC was calculated for each individual tumor growth curve to reflect the entire tumor growth (from randomization to sacrifice). Mice were weighed twice a week, and liver and kidney chemistry panels were run at sacrifice. Mice were sacrificed when at least 2 tumors of each group reached 20 mm in any dimension or when they showed signs of severe distress or toxicity.
Figure 3.

XPO1 inhibition chemosensitizes high-grade B-cell lymphomas. A, Top, table of MYC and BCL2 statuses of the cell lines used in the subsequent experiments. Bottom, DRI of doxorubicin, etoposide, mechlorethamine, vincristine, gemcitabine, or carboplatin in combination with selinexor in a panel of 5 MYC-driven high-grade B-cell lymphoma cell lines including double-hit (i.e., concurrent MYC and BCL2 aberrations). B, Schematic of dosing and scheduling of a R/R DLBCL (carrying copy gains of XPO1, MYC and BCL2) PDTX experiment. XPO1 immunobloting from the primary sample before establishment of the PDTX (PDX-G0), after two passages in mice (PDX-G2) and after four passages in mice (PDX-G4). C, Area under the curve or tumor growth of a R/R DLBCL PDTX treated with vehicle, selinexor, CHOP, or the combination of selinexor + CHOP. D, TUNEL staining quantification of apoptotic index in residual R/R DLBCL PDTX tissue from C. E, Gene set enrichment analysis of genes differentially expressed in R/R DLBCL PDTX treated with CHOP + selinexor versus CHOP alone. NES, normalized enriched score.
Immunoblots
Whole-cell lysates were prepared using RIPA lysis buffer (20 mmol/L Tris pH 8.0, 150 mmol/L NaCl, 5 mmol/L EDTA, 1% Triton X-100) supplemented with a fresh protease inhibitor cocktail (Roche). Protein concentration was determined by BCA assay (Pierce Biotechnology) according to the manufacturer's instructions. Protein lysates were resolved by SDS-PAGE, transferred to PVDF membranes, and probed with the following primary antibodies: anti-RAD51 (8875, Cell Signaling Technology), anti-WEE1 (13084, Cell Signaling Technology), anti-RPA1 (2267, Cell Signaling Technology), anti-CHEK1 (2360, Cell Signaling Technology), anti-KU70 (4588, Cell Signaling Technology), anti-XPO1 (sc-5595 or sc-74454, Santa Cruz Biotechnology), anti-MYC (9402, Cell Signaling Technology), anti-β-actin (A3854, Sigma). Membranes were then incubated with horseradish peroxidase–conjugated secondary antibodies (Santa Cruz) and detected with enhanced chemiluminescence (Pierce). Quantitative densitometry analysis of Western blot bands was performed employing Image J.
Protein decay determination
To inhibit protein translation, cells were cultured in a complete medium supplemented with cycloheximide at 300 μg/mL (Sigma) immediately followed by treatment with vehicle or selinexor (1 μmol/L) for 1 hour, 2 hours, 4 hours, 8 hours, and 24 hours. Cells were harvested at each time point and baseline and immediately lysed and processed for Western blotting as described above.
Quantitative real-time PCR
DLBCL cells were exposed to vehicle, Selinexor (1 μmol/L), etoposide (3 μmol/L) or their combination for 6 hours, and RNA was extracted by TRizol reagent (ThermoFisher). Residual PDTX tissue was harvested after mice were sacrificed and RNA isolated by using the RNeasy Plus Mini kit (Qiagen) according to the manufacturers’ protocol. 1 (DLBCL cells) or 0.5 (PDTX) μg of RNA for each condition was converted to cDNA using the Verso cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer's protocol. Quantitative reverse transcription PCR was carried out in 384-well plates using the 7900HT Fast Real-Time PCR System (Applied Biosystems). A dissociation curve analysis was performed for each sample to verify PCR specificity, and no template samples were used as negative control. Messenger RNA fold change was calculated by the ΔΔCt method by using the average of 2 housekeeping genes as internal control and vehicle-treated cells as a calibrator.
Cellular fractionation and RNA export assay
DLBCL cell lines were exposed to either vehicle or selinexor (1 μmol/L) for 6 hours and cellular fractionation was performed as previously described (34). Briefly, 3 × 107 cells were washed twice with cold PBS, resuspended with slow pipetting in lysis Buffer (10 mmol/L Tris, 140 mmol/L NaCl, 1.5 mmol/L MgCl2, 0.5% NP40, 1 mmol/L DTT and 200 U/mL RNAase inhibitor) and centrifuged at 1,000 g for 5 minutes. The supernatant, containing the cytosolic fraction, was transferred to a fresh tube. The pellet, containing the nuclear fraction, was first resuspended in lysis buffer and then 1/10 volume of detergent solution (3.3% sodium deoxycholate, 6.6% Tween 40) was added under slow vortexing. After 5 minutes of incubation on ice, the solution was centrifuged and the supernatant, containing the post-nuclear fraction, was added to the cytoplasmic fraction. RNA from cytoplasmic and nuclear fractions was extracted by Trizol reagent (ThermoFisher). The expression of specific transcripts was determined by qRT-PCR, as described above.
RNA immunoprecipitation
RNA immunoprecipitation (RIP) was performed on nuclei isolated using the cellular fractionation protocol described above as previously reported (34). Briefly, 1 μg of nuclei lysate was precleared with 50 μL of protein G conjugated magnetic beads (Dynabeads Protein G, ThermoFisher Scientific) for 1h. Precleared lysates were incubated with 6 μmol/L of anti-eIF4E antibody (RN001P, MBL) or 1 μg of anti-THOC4 antibody (12655, Cell Signaling Technology) overnight at 4°C with rotation. After overnight incubation, 50 μL of Dynabeads were added to each sample and incubated for 2 hours with rotation. To isolate RNAs from immunoprecipitated reactions, beads were resuspended in Elution Buffer (100 mmol/L Tris-HCl, 4% SDS, 20% glycerol, 12% β-mercaptoethanol, and incubated for 5 minutes at 98°C. RNA was isolated using TRIzol reagent (ThermoFisher).
Labeling and capture of newly synthesized protein
Newly synthesized proteins were labeled using the Click-iT Protein Labeling Kit (Invitrogen) as previously described (34). 30 × 106 OCI-Ly1 cells were pretreated with either vehicle or selinexor (1 μmol/L) for 2 hours. After one wash, cells were resuspended in methionine-free RPMI1640 medium (Gibco) supplemented with the methionine analog L-azidohomoalanine (AHA; 50 μmol/L). Each flask was then divided into two flasks (15 × 106 cells each) and cells were exposed to vehicle, etoposide (3 μmol/L), selinexor (1 μmol/L), or the combination of selinexor and etoposide for 6 hours. Cells were then harvested and resuspended in lysis buffer (50 mmol/L Tris-HCl, pH 8.0, 1% SDS) containing a fresh protease inhibitor cocktail. 150 μg of protein was used to crosslink the AHA-labeled nascent proteins to alkyne-derivatized biotin in the Click-iT Protein Reaction Buffer (Invitrogen) according to the manufacturer's instructions. The resulting protein pellet was resolubilized in 1% SDS, followed by quenching of the SDS with 6% NP-40, both supplemented with a protease inhibitor cocktail. Biotin-crosslinked nascent proteins were then captured overnight with streptavidin-coated Dynabeads M-280 (Invitrogen) and then eluted from the beads by boiling the samples for 5 min in 2% SDS loading buffer for Western blot analysis.
Immunofluorescence and proximity ligation assay
OCI-Ly1 and Toledo cells were treated with vehicle (DMSO) or selinexor (1 μmol/L) for 6 hours. Following doxycycline withdrawal (to induce MYC expression), P493–6 cells were exposed to vehicle or selinexor (1 μmol/L) for 6 hours. At the end of treatment, cells were centrifuged at 350 g for 5 minutes, plated on 8 mm coverslips previously coated with Cell-Tak, and incubated for 10 minutes at 37°C to adhere. Cells were fixed for 40 minutes (4% paraformaldehyde, 4% sucrose in PBS) at room temperature and permeabilized (Triton X-100 0.1% in PBS) for 5 minutes. For immunofluorescence, fixed cells were quenched for 5 minutes (glycine 0.1 mol/L in PBS) and incubated overnight at 4°C with primary antibodies. After washing, cells were incubated with fluorophore-conjugated secondary antibodies for 2 hours at room temperature. Cells were then washed twice with PBS, once with a solution of PBS and DAPI (2 μg/mL) and mounted with Fluoromount-G (Electron Microscopy Sciences). The PLA assay was performed using the Duolink Red Kit (O-link Bioscience, Sweden) in a humidity chamber according to the manufacturer's instructions. Briefly, cells were blocked with Duolink blocking buffer for 30 m and incubated overnight at 4°C in the presence of primary antibodies diluted in Duolink antibody diluent (1:100). After washing with Duolink washing buffer A, cells were incubated with plus and minus PLA probes for 1 hour at 37°C followed by ligase addition and incubation for 30 minutes. After washing with Duolink washing buffer A, samples were incubated with the polymerase for 100 minutes at 37°C. Finally, covers were washed twice with Duolink washing buffer B, once with a solution of PBS and DAPI (2 μg/mL) and mounted with Fluoromount-G (Electron Microscopy Sciences). Primary Antibodies used in these experiments were the following: anti-XPO1 (sc-5595 or sc-74454, Santa Cruz Biotechnology), anti-THOC4 (12655, Cell Signaling Technology), anti-EIF4E (9742, Cell Signaling Technology).
Image acquisition, processing, and quantification
Cells were imaged using a Zeiss LSM 880 microscope equipped with Airyscan detectors, a plan-Apochromat 63×1.4-NA oil objective, and Zeiss ZEN software. Depending on cell volume, 20 to 30 images at optimum step size were taken per field of view (FOV). Microscopy images were processed with Fiji using custom macros. Briefly, in the case of THOC4 or EIF4E mean fluorescence intensity, each image of a stack was split in its component channels. DAPI signal was used to determine the nuclear area in which the MFI of THOC4 and eIF4E were quantified. Finally, an average MFI was calculated for each cell. In the cases of the PLA, a Z projection was obtained for each FOV (using Maximum intensity) and the DAPI signal was used to determine the nuclear area in which the number of PLA spots were quantified. The data presented in all figures are from three technical replicates from at least two biological replicates.
RNA-seq
RNA was isolated from residual PDTX tissue by using the RNeasy Plus Mini kit (Qiagen) according to the manufacturers’ protocol. RNA integrity was verified by using the Agilent 2100 Bioanalyzer (Agilent Biotechnology). Library preparation for RNA-seq, sequencing, and post-processing of the raw data was performed at the Epigenomics Core at Weill Cornell Medicine. Sequencing libraries were generated with poly A+ RNA by using the TruSeq RNA Sample Prep Kit (Illumina). Sequencing was performed on an Illumina HiSeq 2500 to obtain ∼50M reads per sample. Primary processing of sequencing images was done using Illumina's Real Time Analysis software. Illumina's CASAVA 1.8.2 software was used to perform image capture, base calling, and demultiplexing. Raw fastq files were initially scanned for contaminants and genome distribution with fastQ screen and filtered using fastp with default settings. Samples were deconvoluted to split the human and mouse reads with a pipeline that consists of STAR (v2.6.1a) alignment followed by XenofilteR (v0.99 under R3.5.1) combined with samtools (v1.7). Transcript level quantification was performed with Salmon (v1.3.0). Genome versions used were hg38 and mm10. Gene-level count matrices were created using tximport. Human reads were analyzed with DESeq2.
Data representation and statistical analysis of preclinical research
Data represent the mean of three independent experiments, each performed in triplicate, and error bars represent standard error mean (unless otherwise indicated). Unless otherwise specified, we reported the mean, SEM and P values associated with a student t test with two-tailed distribution of equal variance (or nonparametric equivalent test when appropriate) for experimental data. For experiments including more than two treatment arms, statistical analysis was performed by one-way ANOVA followed by post-hoc multiple comparison test. P values < 0.05 and multiple comparison adjusted P values < 0.05 for multidimensional data were considered statistically significant.
Data availability
Microscopy images were deposited on figshare (10.6084/m9.figshare.20616009) and the codes used to analyze the images were deposited on GitHub (github.com/nahuelzamponi/xpo1_dlbcl_2022). The Gene Expression Omnibus accession number for the RNA-seq data: GSE211684. All other raw data generated in this study are available upon request from the corresponding author.
Results
XPO1 is amplified and overexpressed in a subset of chemoresistant GCB-DLBCL
XPO1 is targeted by genetic gains in DLBCL. XPO1 gains may also result from focal amplifications as well as from broad chromosomal 2p arm gains, more specifically those involving the 2p15–16 and/or 2p16.1 amplicons, that constitute one of the most significantly amplified regions in DLBCL (Supplementary Fig. S1A; refs. 19–21, 31). To determine the impact of XPO1 gains on XPO1 overexpression, we analyzed XPO1 copy-number gains in relation to its transcript and protein expression. In a compiled cohort of 750 primary DLBCLs (31), XPO1 was amplified (XPO1amp) in 16% of cases and associated with a higher level of transcript expression (P < 0.001, vs. XPO1 diploid DLBCLs; Fig. 1A). From 59 DLBCL cases for which XPO1 protein expression was available, 57 expressed levels ranging from low (n = 16%) to moderate (n = 33%) to high (n = 51%), according to an ad hoc IHC scoring system. Like XPO1 transcripts, XPO1amp DLBCLs expressed significantly higher levels of XPO1 protein (vs. XPO1-diploid, 75% vs. 40%, respectively; P < 0.04; Fig. 1B). Additional mechanisms seemed to increase XPO1 expression because a subgroup of DLBCLs with high XPO1 expression were diploid for XPO1 (Fig. 1B). Overall, these data suggest a positive advantage for increased expression of XPO1 in DLBCLs. We next determined the association between XPO1 protein expression at diagnosis and patient response to first-line R-CHOP (rituximab, cyclophosphamide, vincristine, prednisone, doxorubicin) chemoimmunotherapy using our dataset containing 50% responsive and 50% non-responsive patients. The proportion of XPO1 overexpression (XPO1high) in refractory or relapsed patients was 70% whereas in patients who achieved complete responses was 33% (P = 0.005; Fig. 1C). This indicates that patients with XPO1high DLBCLs have a lower likelihood of responding to first-line treatment with a HR of 2.28 (1.32–3.93; P = 0.0029).
Figure 1.

XPO1 is overexpressed in resistant and relapsed DLBCL. A, XPO1 copy number (x-axis) and transcript expression (y-axis) in a cohort of 750 treatment-naïve primary DLBCLs. B and C, Frequency of treatment-naïve primary DLBCL cases with low, medium, and high XPO1 protein expression (determined by IHC) in relation to XPO1 copy number (diploid vs. gains, n = 42 pts, WES and WGS; B) and response to R-CHOP (n = 57 pts; C). Statistical analysis was performed by Chi-square test. D, Frequency of XPO1 copy number in 750 treatment-naïve primary DLBCL cases classified as GCB-DLBCL versus ABC-DLBCL by RNA-seq profiling. E and F, IFNγ response (E) and cell proliferation and DNA repair (F) pathway analysis (GSEA) in 4,655 primary DLBCLs stratified by XPO1 increasing gene expression quartiles (Q) by RNA-seq.
DLBCL comprises two major subgroups that have distinct clinical behavior and molecular features: germinal center B cell–like (GCB-DLBCL) and activated B cell–like (ABC-DLBCL; ref. 35). Patients with GCB-DLBCLs have better outcome following R-CHOP chemoimmunotherapy compared with ABC-DLBCLs (36, 37). Intriguingly, we found that XPO1amp occurs more frequently in GCB-DLBCLs (Fig. 1D; 33% vs. 14%, respectively), affecting all genetic subtypes but more commonly in EZB-MYCneg, EZB-MYCpos, ST2 and complex (Supplementary Fig. S1A; ref. 38). Consistently, XPO1 is overexpressed in GCB-DLBCL cell lines at levels similar or higher than in normal GCB cells (Supplementary Fig. S1B). We thus determined the effect of XPO1 overexpression on the clinical outcome using a real-world cohort of 1,296 primary GCB-DLBCL patients intended to be treated with chemoimmunotherapy R-CHOP or equivalent regimen (31). To uncover any dose effect, patients were clustered in quartiles based on XPO1 transcript levels. Patients expressing XPO1 at the highest (Q4) versus lowest (Q1) quartiles have levels of XPO1 transcript consistent with XPO1 gains vs. XPO1 neutral/losses patients, respectively (as in Fig. 1A). In univariate analysis, GCB-DLBCL patients in the highest quartile of XPO1 expression displayed significantly worse progression-free survival compared with patients in the lower quartiles (P = 0.03; Supplementary Fig. S1C). Taken together these data suggest a potential oncogenic role for XPO1 in DLBCL as increased XPO1 expression might confer a survival advantage to lymphoma cells in response to chemotherapy. To elucidate biological functions associated with XPO1 expression, we conducted gene expression pathway analysis in our cohort of 39 DLBCLs comparing XPO1high versus XPO1low cases (Supplementary Table S1). We then selected biological pathways that could indicate potential mechanistic associations with therapeutic response to chemoimmunotherapy and determine their activation in the cohort of 4,655 DLBCL patients (31) segregated into quartiles of XPO1 expression. Among the most significantly associated with XPO1 expression we identified a negative correlation with “IFNγ immune signaling” pathway activity (Fig. 1E), that is consistent with a statistically significant overrepresentation of the immune depleted lymphoma microenvironment category (i.e., DP-LME) among the GCB-DLBCLs harboring XPO1 gains (31). A DP-LME associates with the highest likelihood of chemoimmunotherapy resistance (31). Meanwhile, XPO1 expression positively correlated with higher activity of the “cell proliferation” and “DNA damage repair” pathways (Fig. 1F), overall suggesting mechanistic links between these cellular functions and XPO1 expression.
XPO1 enables genotoxic stress tolerance by facilitating DNA damage repair
The previous analysis of XPO1high DLBCLs led us to hypothesize that XPO1 may be involved in genotoxic stress tolerance. Cancer cells are continuously exposed to DNA damage due to replication stress that originates in the dysregulated S-phase progression driven by aberrant expression of specific oncogenes, among which is MYC (39, 40). Dysregulated MYC expression induces genotoxic stress in the form of S-phase DNA damage (41), of which, cells need to mitigate to prevent senescence and/or apoptosis (42, 43). Because MYC is a known driver of DLBCL, we speculated that XPO1 may promote tolerance toward MYC-induced replication stress. Supporting this hypothesis, we found that DLBCLs harboring MYC translocation express higher levels of XPO1 compared with DLBCL without MYC translocation (Fig. 2A). To determine whether XPO1 is required to tolerate MYC-induced replication stress, we used a B-lymphoblastoid cell line (P-493–6) harboring a conditionally expressed MYC allele, and the small molecule selinexor that blocks XPO1 by covalently binding to cysteine 528 in its cargo-binding pocket (44). Upon MYC induction, cells were exposed to vehicle or selinexor for 6 hours (Fig. 2B), and DNA damage level (γH2AX) and cell-cycle distribution were assessed by flow cytometry. The inhibition of XPO1 resulted in increased DNA damage level, specifically in the cells transitioning through S-phase (vs. vehicle; P < 0.0001; Fig. 2C) as well as in cell-cycle arrest in the G2–M phase (vs. vehicle; P = 0.003; Fig. 2D), overall indicating that XPO1 is required to mitigate MYC-induced replication stress. Increased γH2AX in selinexor-treated cells was not due to apoptosis, because no significant difference was observed in the percentage of sub-G1 cells between groups (Supplementary Fig. S2A).
Figure 2.

XPO1 promotes DNA damage repair enabling genotoxic stress tolerance. A, XPO1 expression in treatment-naïve primary GCB-DLBCLs harboring or not MYC translocation. B, Immunoblot for MYC expression in P493–6 B cells representing baseline (pretreatment), doxycycline treatment, doxycycline removal (30 min) plus vehicle and doxycycline removal (30 min) plus selinexor conditions. C and D, DNA damage levels (by H2AX staining; C) and cell-cycle distribution (by flow cytometry analysis; D) of P493–6 B cells upon MYC induction (doxycycline removal) and exposure to vehicle or selinexor (1 μmol/L) for 6 hours. E, Baseline DNA damage levels in a panel of 16 DLBCL cell lines assessed by flow cytometry analysis of γH2AX levels. Inset, average level of DNA damage in selinexor sensitive versus resistant DLBCL cell lines. F and G, Comet assay showing the amount of residual DNA damage after 4 hours of exposure (induction phase) and 4 hours withdrawal (repair phase) from 3 μmol/L etoposide (with and without 1 μmol/L selinexor) in OCI-Ly1 (F) and Toledo (G) cells. H and I, Cell-cycle analysis of OCI-Ly1 (H) and Toledo (I) cells exposed to vehicle, selinexor (1 μmol/L), etoposide (3 μmol/L) or their combination for 24 hours. J, Apoptosis analysis by caspase-7/3 activity in DoHH2 (selinexor sensitive) and Karpas422 (selinexor resistant) cell lines upon exposure to vehicle, etoposide, selinexor, and their combination for 72 hours. Data normalized to vehicle-treated and selinexor-treated cells. **, P < 0.005; ***, P < 0.0005.
If XPO1 is required to tolerate genotoxic stress in cancer cells, then lymphoma cells with higher levels of endogenous DNA damage should be more critically dependent on XPO1 for survival. To explore this notion, we first classified a panel of 16 DLBCL cell lines as “sensitive” and “resistant” to XPO1 inhibition accordingly to their GI50 after treatment with selinexor. Selinexor impaired proliferation in 62% (10/16) of cells at clinically relevant doses (selinexor-sensitive; GI50 = < 1 μmol/L) whereas 38% (6/16) of cells exhibited a GI50 > 10 μmol/L (selinexor-resistant; Supplementary Fig. S2B). We next assessed the amount of genotoxic stress in these cell lines by γH2AX and found that the “selinexor-sensitive” exhibited higher levels of baseline endogenous DNA damage than the “selinexor-resistant” cell lines (Fig. 2E). Exposing DLBCL cells to selinexor 1 μmol/L induced apoptosis (by caspase-7/3 activity) to a level that was in general agreement with their baseline degree of endogenous DNA damage (Supplementary Fig. S2C). Notably, there was no association between XPO1 expression levels and sensitivity to selinexor (Supplementary Fig. S2D). A mechanism by which XPO1 may promote genotoxic stress tolerance is by modulating DNA damage repair, which was suggested above from the pathway analysis in XPO1high DLBCLs. We experimentally tested this by exposing two MYC-driven GCB-DLBCL cell lines (OCI-Ly1 and Toledo) to the DNA damaging agent etoposide, alone or in combination with selinexor, and assessed DNA damage repair by single cell gel electrophoresis assay (Fig. 2F and G). While inhibition of XPO1 did not affect the amount of DNA damage induced by etoposide, we observed a significantly higher amount of unrepaired DNA damage in cells exposed to the drug combination compared with either drug alone (P < 0.001 for both cell lines; Fig. 2F and G). This effect was independent of the type of DNA damaging agent used as inhibition of XPO1 impaired DNA damage repair following exposure to doxorubicin (not shown). The impairing of the DNA damage repair capacity induced by XPO1 inhibition is biologically relevant because OCI-Ly1 and Toledo cells exposed to the combination of etoposide and selinexor showed increased S–G2 cell-cycle arrest (vs. etoposide alone; Fig. 2H and I). Moreover, we exposed the DoHH2 (selinexor sensitive) and Karpas422 (selinexor resistant) cell lines that showed, respectively, the highest and lowest levels of baseline DNA damage (Fig. 2E) to vehicle, etoposide, selinexor, and their combination for 72 hours and measured apoptosis by caspase-7/3 activity and senescence by β-galactosidase activity. Although there was no induction of senescence in any cell line in any condition tested (not shown); we found an increase of apoptosis upon etoposide treatment (vs. vehicle) that was higher for DoHH2 than for Karpas422 cells (Fig. 2J). The block of apoptosis in the selinexor resistant cell line Karpas422 was rescued by selinexor (Fig. 2J), whereas in the selinexor sensitive cell line DoHH2 selinexor increased the apoptosis level induced by etoposide (Fig. 2J). This data agrees with the notion that XPO1 overexpression allows DLBCL to overcome and survive a genotoxic stress likely through promoting DNA damage repair.
Inhibition of XPO1 is sufficient to enhance the effect of cytotoxic chemotherapy
Data from above suggests that XPO1 inhibition could be sufficient to sensitize DLBCL cells to DNA-damaging agents. We tested this in a panel of five MYC-driven GCB-DLBCL and double-hit lymphoma cell lines with different intrinsic chemosensitivity (45). Cells were exposed to etoposide, doxorubicin, mechlorethamine (as in vitro equivalent to cyclophosphamide and ifosfamide), vincristine, gemcitabine, or carboplatin alone and in combination with selinexor. These DNA damaging agents were selected as they are used to treat DLBCL patients. We observed a favorable dose-reduction index for each cytotoxic agent in almost each cell line across a range of different doses (Fig. 3A), indicating that XPO1 inhibition broadly sensitizes MYC-driven DLBCL cells in an agnostic fashion to the chemotherapy agent and regardless of their intrinsic chemosensitivity. This chemosensitizing effect was observed when chemotherapy (i.e., doxorubicin or etoposide) was administered either before or after selinexor but not with concomitant administration, which resulted mostly in an additive effect (Supplementary Fig. S3A).
To determine whether the chemosensitizing effect of selinexor can be achieved in vivo, we developed a PDTX model of chemoresistant MYC-driven XPO1high GCB-DLBCL. The specimen was isolated from a treatment-naïve patient with stage IVb DLBCL harboring amplifications of MYC, BCL2, and XPO1. Mice with established PDTXs were randomized to receive vehicle, CHOP, selinexor (7.5 mg/kg), or their combination (Fig. 3B). While either selinexor or CHOP showed an anti-lymphoma effect (Fig. 3C; Supplementary Fig. S2B), the PDTX growth was more potently suppressed by the combination (P = 0.03 vs. CHOP; and P < 0.001 vs. selinexor; Fig. 3C; Supplementary Fig. S3B). The higher effect of the combination was associated with significant increase in apoptotic cells as assessed by the TUNEL assay (P = 0.006 vs. CHOP; and P < 0.001 vs. selinexor; Fig. 3D; Supplementary Fig. S3C). The combination of selinexor and CHOP was well tolerated as there was no evidence of toxicity based on mice weight monitoring (Supplementary Fig. S3D) or blood chemistry.
To determine the molecular effects of selinexor addition to cytotoxic chemotherapy, we conducted gene expression analysis by RNA-seq in PDTX tissues harvested at the end of the treatments. Gene set enrichment analysis was performed on human transcripts differentially expressed between PDTX exposed to selinexor plus CHOP vs. CHOP alone (all vs. vehicle). The top enriched pathways in PDTX exposed to the combination were “oxidative phosphorylation” and “DNA damage repair” (Fig. 3E); likely representing compensatory mechanisms due to the impairment of DNA damage repair induced by selinexor in vivo. Another top enriched pathway was “MYC target genes” (Fig. 3E), that included transcripts associated with XPO1 function like RAN and RANBP1, as well as others associated with mRNA processing and protein biosynthesis like EIF4E, EIF3B, HNRNPR, EXOSC7, and RPL14, suggesting that XPO1 inhibition may affect these cellular functions in response to genotoxic stress. Conversely, the top pathway negatively enriched in the combination treated PDTX was “mitotic spindle” (Fig. 3E), compatible with higher proliferation arrest in these tumors.
Selinexor in combination with chemoimmunotherapy is tolerable in patients with aggressive B-cell lymphomas
On the basis of these preclinical findings, we conducted a phase I trial of selinexor plus a standard second-line chemotherapy regimen, R-ICE (rituximab, ifosfamide, carboplatin, and etoposide), in patients with R/R aggressive B-cell lymphomas (Fig. 4A). Primary endpoint was to determine the MTD of selinexor in combination with R-ICE; secondary endpoints were to assess the efficacy of selinexor plus R-ICE and to evaluate the feasibility of stem cell collection after treatment with selinexor plus R-ICE. The study comprised a dose-finding cohort with a 3+3 dose escalation design and a planned enrollment of a maximum of 18 subjects. An expansion cohort of a maximum of 10 subjects was to be treated at the MTD (including those previously treated at that dose) to better define the toxicity profile. Eligible subjects included patients with DLBCL, DH-HGBL, and indolent lymphomas transformed to DLBCL who had received one prior regimen administered with curative intent. A separate cohort enrolled patients with RT from CLL; there was no requirement regarding prior therapies in this cohort. Patients could undergo cellular therapy after two or more cycles at the discretion of the treating physician. Selinexor was initially dosed before R-ICE according to the following schedule: selinexor on days -5, -3, 1, 3, and 5 and R-ICE on days 1–3 of a 21-day cycle. However, due to CNS toxicity thought to be primarily related to ifosfamide, the dosing schedule was amended so that R-ICE was given on days 1 to 3 and, following completion of ifosfamide, selinexor was given on days 3, 5, and 7 of each cycle (Fig. 4A). Our preclinical data (Supplementary Fig. S3A) supported either dosing schedule as potentially similarly effective in improving chemosensitivity. Dose levels were 40 mg (dose level, DL-1), 60 mg (DL1), and 80 mg (DL2) and the DLT period was cycle 1.
Figure 4.

Selinexor in combination with R-ICE in patients with aggressive B-cell lymphomas. A, Flowchart of clinical trial design with allocation results and number of DLT events. Twenty-two subjects were enrolled (19 in standard aggressive B-cell lymphoma cohort and three in RT cohort). DLBCL-NOS, DLBCL not otherwise specified; PMBL, primary mediastinal B-cell lymphoma. B, Dosing schedule with patient allocation and DLT for the 19 subjects enrolled in the DLBCL cohort. From the three RT subjects, one subject was taken off the trial prior to receiving selinexor because of neurotoxicity due to ifosfamide and the other two subjects did not experience a DLT. In the initial dosing schedule (n = 3) at 60 mg there were no DLTs. Two DLTs were observed when dose subsequently increased to 80 mg, prompting de-escalation to 60 mg and enrolling three additional subjects at that dose level. One of the second group of three subjects experienced grade 3 AMS. At that point, the protocol was modified so that the selinexor was dosed after completion of ifosfamide. C, Waterfall plot of best relative percent change from baseline in tumor size for 18 patients. Of 20 subjects evaluable for response, one progressed prior to imaging response assessment and one with complete response on PET had no measurable disease, and thus, is not included in the waterfall plot. Solid columns, modified schedule; dashed columns, initial schedule.
Twenty-two patients were enrolled with a median age of 67 years (Supplementary Table S2). Diagnoses were DLBCL (n = 12), DH-HGBL (n = 4), transformed indolent lymphoma (n = 2), primary mediastinal large B-cell lymphoma (n = 1) and RT from CLL (n = 3, Supplementary Table S2; Fig. 4A). Seven patients were treated on the initial dosing schedule and 12 patients with DLBCL were treated on the modified dosing schedule (Fig. 4A). At the initial 60 mg dosing, 2 of 3 patients had a DLT (Fig. 4B) whereas at the downscaled 40 mg dosing only 1 of 6 patients had a DLT (Fig. 4B), therefore 40 mg was declared the RP2D, and three additional subjects were enrolled in the expansion cohort. Three subjects with RT were treated on the modified dosing schedule; two received selinexor and neither had a DLT. The median number of cycles administered was 2 and the most common grade 3 and 4 toxicities were cytopenias followed by fatigue, hyponatremia, and transaminitis (Supplementary Table S3). Neutropenic fever occurred in four subjects; all were grade 3 except for one grade 5 episode in a subject who had undergone allogenic stem cell transplantation four months prior to study enrollement (Supplementary Table S3). As mentioned above, 3 patients treated on the initial schedule with selinexor administered before R-ICE experienced grade 3 altered mental status (AMS) thought to be primarily due to ifosfamide. In these 3 patients, AMS began either during or immediately after completion of the ifosfamide infusion. They were treated with methylene blue and thiamine and underwent neurologic imaging, which was negative for acute findings. The symptoms improved in all patients, and completely resolved in two after 3 and 6 days. The third patient underwent a lumbar puncture concerning for CNS involvement of the lymphoma, which was also thought to be a contributor to the AMS. He was taken off the study as CNS involvement was an exclusion criterion. Neurologic toxicity was not observed in the modified dosing cohort with selinexor dosed after R-ICE (Supplementary Table S3).
In the 21 patients who received selinexor plus R-ICE, the objective response rate (ORR) was 71% with seven CR, eight partial response (PR), and three SD (Fig. 4C). Of the 7 patients achieving CR, three underwent autologous stem cell transplant (ASCT), one was treated with CAR T-cell therapy, and three were monitored without further treatment. Of the 8 patients achieving PR, one underwent ASCT, one underwent allogeneic SCT after bendamustine bridging protocol (both of whom remain in CR at last follow-up; ref. 46), four were treated with CAR T-cells, and two died without further therapy. Of the 3 patients achieving SD, one underwent ASCT after bendamustine protocol, one was treated with CAR T-cells, and the other died without further therapy. Of note, at the RP2D in the DLBCL cohort (n = 9), ORR was 78% with 4 CR, 3 PR, and 2 SD.
Stem cell collection was successful in four of 8 patients, while the remaining four were unable to be collected. Details regarding stem cell collection are included in Supplementary Table S4. To our knowledge, all four patients who were unable to have stem cells collected remain alive. Three have not required additional therapy since receiving selinexor plus R-ICE with follow-up of over four (n = 1) and five (n = 2) years. The fourth was found to have relapsed CNS disease at the time of attempted collection, was treated with CNS-directed systemic chemotherapy, underwent allogeneic SCT, and remains in CR for over 2 years. Further studies should investigate the potential of selinexor for CNS toxicity, which may in part be related to the crossing of the blood-brain barrier, potentially advantageous in DLBCL, as well as the impact on stem cells. The long remissions of patients who were unable to have stem cells collected suggest that selinexor's impact on the bone marrow may provide clinical benefit. Overall, this clinical trial indicates that the combination of selinexor with chemotherapy is tolerable and potentially effective in aggressive B-cell lymphomas.
XPO1 sustains the turnover of proteins involved in DNA damage repair
To elucidate the molecular mechanism by which XPO1 inhibition affects DNA damage repair in DLBCL, we first determined the effect of selinexor on the expression of key proteins involved in DNA damage response and repair (CHEK1, RPA1, RAD51, WEE1, and KU70, or XRCC6) in MYC-driven OCI-Ly1 cells (Fig. 5A). We found that selinexor impaired the expression of CHEK1, RAD51, and WEE1 but not RPA1 and KU70. While etoposide increased the expression of some of these proteins, selinexor blunted this effect (Fig. 5A). Similarly, while MYC induction in P-493–6 cells was associated with upregulation of CHEK1, RAD51, and WEE1 (Fig. 5B), a likely consequence of increased replication stress (47, 48), this effect was almost completely blunted by XPO1 inhibition (Fig. 5B). Remarkably, MYC induction did not increase XPO1 levels, indicating that XPO1 is not a direct MYC target gene (Fig. 5B).
Figure 5.

XPO1 sustains the turnover of proteins modulating DNA damage repair. A, Representative immunoblot of indicated DNA damage repair proteins in OCI-Ly1 cells exposed to vehicle, selinexor (1 μmol/L), etoposide (3 μmol/L), or their combination for 24 hours. B, Representative immunoblot of indicated DNA damage repair proteins in P493–6 B-cells after induction of MYC (30 minutes after doxycycline withdrawal) and exposure to vehicle or selinexor (1 μmol/L) for 24 hours. C, Expression levels of transcripts encoding for DNA damage repair protein in OCI-Ly1 cells exposed to vehicle, selinexor (1 μmol/L), etoposide (3 μmol/L), or their combination for 6 hours. D and E, Representative immunoblots (D) of cycloheximide (CHX) chase assay of CHEK1, RAD51, and WEE1 (and actin as control) in OCI-Ly1 cells exposed to vehicle or selinexor (1 μmol/L) for the indicated times. The relative amount of each protein compared with β-actin was quantified by densitometry and plotted with respect to time (E). The protein to β-actin level at baseline was defined as 100%. F, Immunoblots showing CHEK1, RAD51, and WEE1 (and actin as control) protein levels in the newly synthesized fraction (AHA-pulldown) over total protein abundance (input) in OCI-Ly1 cells exposed to vehicle, selinexor (1 μmol/L), etoposide (3 μmol/L), or their combination for 6 hours.
To determine whether decreased CHEK1, RPA1, RAD51, and WEE1 protein expression was due to decreased template availability, we analyzed their transcripts expression in the conditions and cell lines described before. Both etoposide administration and MYC induction upregulated these transcripts (Fig. 5C; Supplementary Fig. S4), likely accounting for the observed increased protein expression. Transcript upregulation was also observed for KU70 (Fig. 5C), a protein whose expression did not significantly change, suggesting that etoposide may increase KU70 turnover (49, 50). On the contrary, selinexor did not affect the expression level of these transcripts (Fig. 5C; Supplementary Fig. S4). Furthermore, selinexor did not affect the stability of already synthesized CHEK1, RAD51, and WEE1 proteins as demonstrated by measuring their half-life (Fig. 5D and E). Likewise, the effect of selinexor on protein expression was not a global phenomenon because expression of RPA1 and KU70 was not affected. We thus assessed newly synthesized CHEK1, RAD51, and WEE1 proteins in OCI-Ly1 cells treated with etoposide, selinexor, and their combination. We found that selinexor impaired new synthesis of CHEK1, RAD51, and WEE1 in response to genotoxic stress (but not of actin, Fig. 5F), indicating that XPO1 activity is necessary to sustain the turnover of proteins involved the repair of DNA damage.
XPO1 prioritizes the nuclear export of nucleoproteins carrying genotoxic stress transcripts
Because XPO1 inhibition impaired the production of a subset of DNA damage repair proteins without affecting their transcript levels, we speculated that this could be the consequence of decreased nuclear export of their transcripts. We thus determined whether XPO1 inhibition alters the intracellular distribution of CHEK1, RPA1, RAD51, WEE1, and KU70 transcripts. B-actin transcript (BACT) whose protein level did not change upon selinexor treatment was used as a control. In agreement with the selective effect observed in protein expression, XPO1 inhibition in MYC-driven Toledo and OCI-Ly1 cells resulted in nuclear retention of CHEK1, WEE1, and RAD51, but not of RPA1, KU70, or BACT (control) transcripts (Fig. 6A; Supplementary Fig. S5A). Because XPO1 is unable to directly bind these transcripts (not shown), we hypothesized that XPO1 inhibition may affect the export of ribonucleoproteins carrying them. As potential candidates, we investigated THOC4, FUS, and EIF4E, which have been independently reported by us and others to bind transcripts coding for proteins regulating DNA damage repair in cancer cells (34, 51). Specifically, re-analysis of our reported (34) EIF4E RIP-sequencing conducted in OCI-Ly1 cells at baseline conditions demonstrated a significant enrichment of DNA repair transcripts (vs. IgG RIP-seq; P < 0.001, Supplementary Fig. S5B) as EIF4E binders. To determine whether the nuclear export of these proteins is XPO1-dependent, we first determined their nuclear expression in Toledo and OCI-Ly1 cells at baseline and following exposure to selinexor by immunofluorescence. In accordance with what was previously reported for other cell types (13, 52), we found that THOC4 localization is prevalently nuclear while EIF4E localization is prevalently cytoplasmic in lymphoma cells at baseline. Inhibition of XPO1 in Toledo and OCI-Ly1 cells resulted in increased nuclear expression of THOC4 and EIF4E (P < 0.001 vs. vehicle for both proteins in Toledo and for THOC4 in OCI-Ly1 cells; Fig. 6B and Supplementary Fig. S5C) in absence of an increase in total THOC4 and EIF4E protein levels (Supplementary Fig. S5D), indicating that the export of these proteins is at least partially XPO1-dependent. We did not observe nuclear retention of FUS in OCI-Ly1 or Toledo cells exposed to selinexor (not shown).
Figure 6.

XPO1 prioritizes the nuclear export of nucleoproteins carrying genotoxic stress transcripts. A, Nuclear/cytoplasmic ratio of selected DNA damage repair transcripts (i.e., CHEK1, RAD51, WEE1, RPA1, and KU70) in Toledo cells exposed to vehicle or selinexor (1 μmol/L) for 6 hours. B, Nuclear levels of THOC4 and EIF4E in Toledo cells exposed to vehicle or selinexor (1 μmol/L) for 6 hours. Left, representative images; right, quantification. The bar represents pixel intensity. C, Proximity ligation assays (PLA) of XPO1-THOC4 and XPO1-EIF4E complexes in Toledo cells exposed to vehicle or etoposide for 6 hours (top) and in P493–6 B-cells with or without MYC expression (bottom). D, Nuclear level of THOC4 in P493–6 B-cells after MYC induction, followed by vehicle or selinexor (1 μmol/L) for 6 hours. The bar represents pixel intensity. E, Ribonucleoprotein immunoprecipitation assays of DNA damage repair transcripts (i.e., CHEK1, RAD51, WEE1, RPA1, and KU70) bound by THOC4 (left) or EIF4E (right) in the nuclear fraction of Toledo cells. Data are presented as fold enrichment over input. F, Change in the amount of DNA damage repair transcripts CHEK1, RAD51, WEE1, RPA1, and KU70 (and actin as control) bound by THOC4 (left) and EIF4E (right) in Toledo lymphoma cells exposed to vehicle or etoposide for 6 hours. Data are presented as fold enrichment over vehicle (normalized by their respective inputs). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
This data suggested that THOC4 and EIF4E are exported through XPO1. Thus, we analyzed THOC4-XPO1 and EIF4E-XPO1 interactions at baseline and in response to etoposide-induced DNA damage by conducting proximity ligation assays in Toledo cells. We found that XPO1 binds both THOC4 and EIF4E in Toledo cells at baseline, and such interactions significantly increase in response to genotoxic stress (P < 0.001, vs. baseline, for both proteins, Fig. 6C, top). Similarly, we observed increased THOC4-XPO1 and eIF4E-XPO1 interactions upon acute MYC expression in P-493–6 lymphoblastoid B cells (P < 0.001, vs. MYC negative condition, for both proteins, Fig. 6C, bottom). Accordingly, XPO1 inhibition in MYC expressing P-493–6 cells resulted in a significant increase in the nuclear retention of THOC4 (P < 0.001, vs. vehicle, Fig. 6D). Overall, these data indicate that XPO1 preferentially binds and exports THOC4 and EIF4E in response to endogenous (i.e., MYC) and exogenous (i.e., chemotherapy) genotoxic stresses.
To further refine our model, we determined whether THOC4 and EIF4E carried DNA damage transcripts by performing quantitative ribonucleoprotein immunoprecipitation (RIP) in OCI-Ly1 and Toledo cells. We found that THOC4 binds to CHEK1, RAD51, WEE1, and KU70 transcripts, while EIF4E binds to CHEK1 and WEE1 transcripts in both cell lines (Fig. 6E; Supplementary Fig. S5E). There was no binding of BACT transcripts used as a control (Fig. 6E; Supplementary Fig. S5E). We then determined whether an increase in the transcription of DNA damage repair genes associates with their preferential XPO1-dependent nuclear export. We thus conducted quantitative THOC4-RIPs and EIF4E-RIPs in nuclear lysates from OCI-Ly1 and Toledo cells treated with etoposide. We found that upon etoposide-induced DNA damage, THOC4 and EIF4E bound more CHEK1, RAD51, WEE1, and KU70 transcripts but not BACT transcripts (Fig. 6F; Supplementary Fig. S5F). Moreover, while EIF4E did not bind RAD51 transcripts at baseline, it did after genotoxic stress in both cell lines (Fig. 6F; Supplementary Fig. S5F). These data indicate that, in response to genotoxic stress, transcripts coding for proteins required for DNA damage repair are preferentially bound by THOC4 and EIF4E and exported in an XPO1-dependent manner.
Taken together our data support a model in which XPO1 enables the adaptive regulation of mRNA export required for genotoxic stress tolerance in lymphoma cells. Preferential nuclear export of selected transcripts is achieved at two levels of regulation, the increased binding of these transcripts by THOC4 and EIF4E followed by the increased export of the corresponding ribonucleoprotein complexes by XPO1. This mechanism potentially confers XPO1 overexpressing cancer cells a competitive advantage by allowing them to sustain higher levels of endogenous and exogenous genotoxic stresses as ultimately evidenced in R/R DLBCLs and/or in MYC-driven B-cell lymphomas. However, this creates a therapeutic vulnerability to XPO1 inhibition particularly in conditions of a sudden increase in DNA damage such as upon cytotoxic chemotherapy exposure that can be exploited for therapeutic gain in combinatorial scheduling.
Discussion
Our data demonstrate a novel oncogenic mechanism of XPO1 characterized by the hierarchization of ribonucleoprotein complexes export to enable endogenous and exogenous genotoxic stress tolerance. Specific post-translational reprogramming of the proteome is a general mechanism by which cells respond to different types of stress. To maximize survival, cells induce a general shutdown of global protein production while promoting instantaneous and coordinated expression of the proteins required to overcome the stressful condition. Such mechanism becomes critical in response to genotoxic stress in cancer cells as DNA damage needs to be rapidly repaired to prevent cell death. At the same time, DNA damage repair proteins belonging to the same repair pathway needs to be coordinately expressed as an imbalance in their expression can negatively impact cellular homeostasis (53–55). While previous reports supported a role for the NP complex as a hub for gene expression regulation in response to stress, the role of XPO1 in this process has not been fully explored.
We demonstrated that in cancer cells by binding EIF4E and THOC4, XPO1 is positioned to modulate and amplify specific messages by facilitating the export of groups of mRNAs acting in the same biochemical pathway and thereby eliciting potent biological outcomes. Although not completely elucidated, mRNA containing specific recognition elements (or USER codes) permit the engagement of selected export factors to facilitate their transit through the NP complex (56). In this model, multiple copies of each mRNA can use their multiple USER codes to join groups of functionally related mRNAs and ribonucleoproteins in time and space to coordinate complex functions (56). We and others demonstrated that mRNA containing the USER code 4ESE in their 3′UTR (e.g., transcripts supporting lymphoma cell proliferation such as MYC, BCL2, and BCL6) bind to EIF4E and LRPPRC to be exported via XPO1 (34, 57–60). Although THOC4 is usually recruited by NXF1/NXT1 to the NP complex for transit to the cytoplasm, we showed here that XPO1 can also host THOC4-bound mRNAs regulating DNA damage repair, suggesting the presence of a yet to-be-identified USER code for this RNA regulon. Another level of functional coherence to RNA regulons is established by different expression and functional states of their constituent ribonucleoproteins (56). Because we did not observe a change in the global expression of EIF4E or THOC4 in response to genotoxic stress, it is possible that post-translational modifications may regulate the binding of these ribonucleoproteins to XPO1 as reported for others XPO1 cargoes (61).
Analysis of genomic and transcriptomic data from primary DLBCLs as well as our functional assays suggest that XPO1 overexpression is selected for in DLBCL. Several data support a dose–effect for XPO1 activity because XPO1-mediated transport is saturable (62), XPO1 haploinsufficiency is associated with congenital development defects (63, 64), and overexpression of XPO1 results in increased export of its cargoes (65, 66). These observations are compatible with the notion that XPO1 overexpression results in a more effective hijacking of the NP complex. Accordingly, increased XPO1 expression, frequently associated with poor prognosis, has been observed in multiple cancer types. Nevertheless, efforts to implement XPO1 expression as a patient selection biomarker for XPO1 inhibitors has been unsuccessful. This is in part due to XPO1 expression in normal cells as well as the dynamic XPO1 expression in heterogenous cancer tissues. The mechanistic data we provided justifies the exploration of a potential more informative biomarker that relies on XPO1 overexpression in association with elevated levels of genotoxic stress. In this regard, patients with MYC-driven tumors including DH-HGBL, and/or harboring mutations in DNA damage repair genes and/or exhibiting an immune depleted microenvironment may likely benefit from XPO1 inhibitors. Moreover, a shared observation across tumor types is that TP53 mutations are associated with lower sensitivity to XPO1 inhibition; and because TP53 is a major effector of DNA damage-induced apoptosis our model may contribute to explaining this empirical observation.
The described mechanism also contributes to explain the chemosensitizing effect of XPO1 inhibitors as XPO1 activity may become rate limiting under conditions in which the abundance of DNA repair proteins becomes critical, for example upon administration of DNA damaging agents that exert sublethal damage in a fraction of cancer cells. This notion is compatible with the observation that sequential administration of chemotherapy and selinexor killed cells in a synergistic rather than additive manner as seen with their concomitant administration. Our clinical trial demonstrated that specifically at lower selinexor doses (i.e., 40 mg) a relevant therapeutic window does exist for combinatorial administration after chemotherapy agents. Noteworthy, limited data suggest that a potential immune effect could be elicited upon selinexor and could be eventually exploited in combination with immunotherapeutic strategies. Here, we described that DLBCL patients with high XPO1 expression associated with low interferon-dependent immune response, a notion compatible with the presence of an immune depleted LME in GCB-DLBCL carrying XPO1 amplification. In solid tumors, other groups have reported a synergistic effect of selinexor with DNA damaging agents (67), PARP inhibitors (68), and immune checkpoint inhibitors (69), suggesting a conserved function of XPO1 in broadly modulating DNA damage repair and, secondarily, the immune response associated with DNA damage pathways.
In summary, our findings described that mRNA export can be modified as part of the genotoxic stress response. Within this mechanism, XPO1 overexpression in cancer cells plays a critical role in their increased tolerance to DNA damage while providing new insights to optimize the clinical development of XPO1 inhibitors.
Supplementary Material
Supplementary Figures and Tables
Supplementary Table 1
Acknowledgments
ShaoNing Yang provided technical assistance. Dr. Kristy Richards (deceased) provided patient care. Silvia Senese, Trisha Ali-Shaw, Riyaad Rahim, Amelyn Rodriguez, Arcania Garcia, and Jennifer Santamala provided clinical trial coordination and patient care. R. Marullo was supported by NIH training grant T32 CA203702. Funding for this work was provided by the Leukemia and Lymphoma Society (LLS TRP R6510–19), the NIH-NCI (R01CA242069 and R01CA249843), and a research grant from Karyopharm, Inc.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
S.C. Rutherford reports grants from Karyopharm during the conduct of the study; personal fees from ADC, BMS, Genmab, Karyopharm, Kite; other support from Constellation, Genentech; and personal fees from Seagen outside the submitted work. N. Kotlov reports other support from Bostongene Corp. during the conduct of the study and other support from Bostongene Corp. outside the submitted work; in addition, N. Kotlov has a patent for Tumor Microenvironment-Based Methods for Assessing CAR-T and Other Immunotherapies issued to Bostongene Corp. J. Lara-Garcia reports ownership of stock in Johnson & Johnson and Pfizer. J.N. Allan reports personal fees from AbbVie, Adaptive Therapeutics, ADC Therapeutics, AstraZeneca; grants and personal fees from BeiGene, Genentech, Janssen; personal fees from Epizyme, Lava Therapeutics, Lilly, Pharmacyclics; and grants and personal fees from TG Therapeutics outside the submitted work. J. Ruan reports grants and personal fees from AstraZeneca; grants from BMS, Genentech, Daiichi Sankyo; personal fees from Janssen, SecuraBio; and personal fees from Kite Pharma outside the submitted work. S.A. Mayer reports grants from Karyopharm during the conduct of the study. J.P. Leonard reports personal fees from AbbVie, Astellas, AstraZeneca, Bayer, Beigene, BMS, Calithera, Constellation, Caribou Biosciences, Eisai, Lilly, Epizyme, Genmab, Grail, Incyte, Janssen, MEI Pharma, Merck, Mustang Bio, Novartis, Pfizer, Seagen, Second Genome; grants and personal fees from Roche/Genentech; and personal fees from Sutro outside the submitted work. P. Martin reports other support from Karyopharm during the conduct of the study and personal fees from AstraZeneca, Beigene, Genentech, Janssen, Merck, and BMS outside the submitted work. L. Cerchietti reports grants from NIH, NCI, Leukemia and Lymphoma Society and Karyopharm, Inc. during the conduct of the study and grants from Celgene outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
R. Marullo: Conceptualization, formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. S.C. Rutherford: Conceptualization, formal analysis, investigation, visualization, methodology, writing–original draft, writing–review and editing. M.V. Revuelta: Data curation, formal analysis, investigation. N. Zamponi: Formal analysis, investigation. B. Culjkovic-Kraljacic: Formal analysis, investigation. N. Kotlov: Data curation, formal analysis, visualization. N. Di Siervi: Formal analysis, investigation. J. Lara-Garcia: Formal analysis, investigation. J.N. Allan: Formal analysis, investigation. J. Ruan: Formal analysis, investigation. R.R. Furman: Formal analysis, investigation. Z. Chen: Formal analysis, investigation. T.B. Shore: Formal analysis, investigation. A.A. Phillips: Formal analysis, investigation. S. Mayer: Formal analysis, investigation. J. Hsu: Formal analysis, investigation. K. van Besien: Formal analysis, investigation. J.P. Leonard: Supervision. K.L.B. Borden: Methodology. G. Inghirami: Methodology. P. Martin: Conceptualization, supervision, methodology. L. Cerchietti: Conceptualization, resources, formal analysis, supervision, funding acquisition, investigation, writing–original draft, project administration, writing–review and editing.
References
- 1. Cautain B, Hill R, de Pedro N, Link W. Components and regulation of nuclear transport processes. FEBS J 2015;282:445–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wente SR, Rout MP. The nuclear pore complex and nuclear transport. Cold Spring Harb Perspect Biol 2010;2:a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kosar M, Giannattasio M, Piccini D, Maya-Mendoza A, García-Benítez F, Bartkova J, et al. The human nucleoporin Tpr protects cells from RNA-mediated replication stress. Nat Commun 2021;12:3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zimmerli CE, Allegretti M, Rantos V, Goetz SK, Obarska-Kosinska A, Zagoriy I, et al. Nuclear pores dilate and constrict in cellulo. Science 2021;374:eabd9776. [DOI] [PubMed] [Google Scholar]
- 5. Strambio-De-Castillia C, Niepel M, Rout MP. The nuclear pore complex: bridging nuclear transport and gene regulation. Nat Rev Mol Cell Biol 2010;11:490–501. [DOI] [PubMed] [Google Scholar]
- 6. Sloan KE, Gleizes PE, Bohnsack MT. Nucleocytoplasmic transport of RNAs and RNA-protein complexes. J Mol Biol 2016;428:2040–59. [DOI] [PubMed] [Google Scholar]
- 7. Regot S, de Nadal E, Rodríguez-Navarro S, González-Novo A, Pérez-Fernandez J, Gadal O, et al. The Hog1 stress-activated protein kinase targets nucleoporins to control mRNA export upon stress. J Biol Chem 2013;288:17384–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zander G, Hackmann A, Bender L, Becker D, Lingner T, Salinas G, et al. mRNA quality control is bypassed for immediate export of stress-responsive transcripts. Nature 2016;540:593–6. [DOI] [PubMed] [Google Scholar]
- 9. Saavedra C, Tung KS, Amberg DC, Hopper AK, Cole CN. Regulation of mRNA export in response to stress in Saccharomyces cerevisiae. Genes Dev 1996;10:1608–20. [DOI] [PubMed] [Google Scholar]
- 10. Ramachandran S, Tran DD, Klebba-Faerber S, Kardinal C, Whetton AD, Tamura T. An ataxia-telangiectasia-mutated (ATM) kinase mediated response to DNA damage downregulates the mRNA-binding potential of THOC5. RNA 2011;17:1957–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amin R, Shukla A, Zhu JJ, Kim S, Wang P, Tian SZ, et al. Nuclear pore protein NUP210 depletion suppresses metastasis through heterochromatin-mediated disruption of tumor cell mechanical response. Nat Commun 2021;12:7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. The nucleoporin POM121 enhances prostate cancer aggressiveness. Cancer Discov 2018;8:OF7–OF. [Google Scholar]
- 13. Culjkovic-Kraljacic B, Baguet A, Volpon L, Amri A, Borden KL. The oncogene eIF4E reprograms the nuclear pore complex to promote mRNA export and oncogenic transformation. Cell Rep 2012;2:207–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wen W, Meinkoth JL, Tsien RY, Taylor SS. Identification of a signal for rapid export of proteins from the nucleus. Cell 1995;82:463–73. [DOI] [PubMed] [Google Scholar]
- 15. Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, et al. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 1997;390:308–11. [DOI] [PubMed] [Google Scholar]
- 16. Stade K, Ford CS, Guthrie C, Weis K. Exportin-1 (Crm1p) is an essential nuclear export factor. Cell 1997;90:1041–50. [DOI] [PubMed] [Google Scholar]
- 17. Hutten S, Kehlenbach RH. CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol 2007;17:193–201. [DOI] [PubMed] [Google Scholar]
- 18. Xu D, Farmer A, Collett G, Grishin NV, Chook YM. Sequence and structural analyses of nuclear export signals in the NESdb database. Mol Biol Cell 2012;23:3677–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fukuhara N, Tagawa H, Kameoka Y, Kasugai Y, Karnan S, Kameoka J, et al. Characterization of target genes at the 2p15–16 amplicon in diffuse large B-cell lymphoma. Cancer Sci 2006;97:499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bea S, Zettl A, Wright G, Salaverria I, Jehn P, Moreno V, et al. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene expression–based survival prediction. Blood 2005;106:3183–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shaffer AL 3rd, Young RM, Staudt LM. Pathogenesis of human B-cell lymphomas. Annu Rev Immunol 2012;30:565–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coiffier B, Lepage E, Brière J, Herbrecht R, Tilly H, Bouabdallah R, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large B-cell lymphoma. N Engl J Med 2002;346:235–42. [DOI] [PubMed] [Google Scholar]
- 23. Pfreundschuh M, Trümper L, Österborg A, Pettengell R, Trneny M, Imrie K, et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomized controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol 2006;7:379–91. [DOI] [PubMed] [Google Scholar]
- 24. Elstrom RL, Martin P, Ostrow K, Barrientos J, Chadburn A, Furman R, et al. Response to second-line therapy defines the potential for cure in patients with recurrent diffuse large B-cell lymphoma: implications for the development of novel therapeutic strategies. Clin Lymphoma Myeloma Leuk 2010;10:192–6. [DOI] [PubMed] [Google Scholar]
- 25. Van Den Neste E, Schmitz N, Mounier N, Gill D, Linch D, Trneny M, et al. Outcome of patients with relapsed diffuse large B-cell lymphoma who fail second-line salvage regimens in the International CORAL study. Bone Marrow Transplant 2016;51:51–7. [DOI] [PubMed] [Google Scholar]
- 26. Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 2017;130:1800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo B, Huang L, Gu Y, Li C, Lu H, Chen G, et al. Expression of exportin-1 in diffuse large B-cell lymphoma: immunohistochemistry and TCGA analyses. Int J Clin Exp Pathol 2018;11:5547–60. [PMC free article] [PubMed] [Google Scholar]
- 28. Kalakonda N, Maerevoet M, Cavallo F, Follows G, Goy A, Vermaat JSP, et al. Selinexor in patients with relapsed or refractory diffuse large B-cell lymphoma (SADAL): a single-arm, multinational, multicenter, open-label, phase II trial. Lancet Haematol 2020;7:e511–e22. [DOI] [PubMed] [Google Scholar]
- 29. Kırlı K, Karaca S, Dehne HJ, Samwer M, Pan KT, Lenz C, et al. A deep proteomics perspective on CRM1-mediated nuclear export and nucleocytoplasmic partitioning. eLife 2015;4:e11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fung HYJ, Fu S-C, Brautigam CA, Chook YM. Structural determinants of nuclear export signal orientation in binding to exportin CRM1. eLife 2015;4:e10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kotlov N, Bagaev A, Revuelta MV, Phillip JM, Cacciapuoti MT, Antysheva Z, et al. Clinical and biological subtypes of B-cell lymphoma revealed by microenvironmental signatures. Cancer Discov 2021;11:1468–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marullo R, Castro M, Yomtoubian S, Calvo-Vidal MN, Revuelta MV, Krumsiek J, et al. The metabolic adaptation evoked by arginine enhances the effect of radiation in brain metastases. Sci Adv 2021;7:eabg1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fernando TM, Marullo R, Pera Gresely B, Phillip JM, Yang SN, Lundell-Smith G, et al. BCL6 evolved to enable stress tolerance in vertebrates and is broadly required by cancer cells to adapt to stress. Cancer Discov 2019;9:662–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Culjkovic-Kraljacic B, Fernando TM, Marullo R, Calvo-Vidal N, Verma A, Yang S, et al. Combinatorial targeting of nuclear export and translation of RNA inhibits aggressive B-cell lymphomas. Blood 2016;127:858–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000;403:503–11. [DOI] [PubMed] [Google Scholar]
- 36. Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA 2003;100:9991–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large B-cell lymphoma. N Engl J Med 2002;346:1937–47. [DOI] [PubMed] [Google Scholar]
- 38. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A probabilistic classification tool for genetic subtypes of diffuse large B-cell lymphoma with therapeutic implications. Cancer Cell 2020;37:551–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science 2008;319:1352–5. [DOI] [PubMed] [Google Scholar]
- 40. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006;444:638–42. [DOI] [PubMed] [Google Scholar]
- 41. Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, et al. Non-transcriptional control of DNA replication by c-Myc. Nature 2007;448:445–51. [DOI] [PubMed] [Google Scholar]
- 42. Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, et al. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS One 2008;3:e2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Robinson K, Asawachaicharn N, Galloway DA, Grandori C. c-Myc accelerates S-phase and requires WRN to avoid replication stress. PLoS One 2009;4:e5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Neggers Jasper E, Vercruysse T, Jacquemyn M, Vanstreels E, Baloglu E, Shacham S, et al. Identifying drug-target selectivity of small-molecule CRM1/XPO1 inhibitors by CRISPR/Cas9 genome editing. Chem Biol 2015;22:107–16. [DOI] [PubMed] [Google Scholar]
- 45. Clozel T, Yang S, Elstrom RL, Tam W, Martin P, Kormaksson M, et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov 2013;3:1002–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Orfali N, Jhanwar Y, Koo C, Pasciolla M, Baldo M, Cuvilly E, et al. Sequential intensive chemotherapy followed by autologous or allogeneic transplantation for refractory lymphoma. Leuk Lymphoma 2021;62:1629–38. [DOI] [PubMed] [Google Scholar]
- 47. Juncheng H, Tianci W, Jin X, Sanyun W, Liyuan W, Hexiu S, et al. WEE1 inhibition induces glutamine addiction in T-cell acute lymphoblastic leukemia. Haematologica 2020;106:1816–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luoto KR, Meng AX, Wasylishen AR, Zhao H, Coackley CL, Penn LZ, et al. Tumor cell kill by c-MYC depletion: role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res 2010;70:8748–59. [DOI] [PubMed] [Google Scholar]
- 49. Huh J, Piwnica-Worms H. CRL4CDT2Targets CHK1 for PCNA-independent destruction. Mol Cell Biol 2013;33:213–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Selvarajah J, Elia A, Carroll VA, Moumen A. DNA damage-induced S and G2–M cell-cycle arrest requires mTORC2-dependent regulation of Chk1. Oncotarget 2014;6:427–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wickramasinghe VO, Savill JM, Chavali S, Jonsdottir AB, Rajendra E, Gruner T, et al. Human inositol polyphosphate multikinase regulates transcript-selective nuclear mRNA export to preserve genome integrity. Mol Cell 2013;51:737–50. [DOI] [PubMed] [Google Scholar]
- 52. Yang X, Yang Y, Sun BF, Chen YS, Xu JW, Lai WY, et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res 2017;27:606–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 2012;12:104–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Calvo JA, Moroski-Erkul CA, Lake A, Eichinger LW, Shah D, Jhun I, et al. Aag DNA glycosylase promotes alkylation-induced tissue damage mediated by Parp1. PLos Genet 2013;9:e1003413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sobol RW, Kartalou M, Almeida KH, Joyce DF, Engelward BP, Horton JK, et al. Base excision repair intermediates induce p53-independent cytotoxic and genotoxic responses *. J Biol Chem 2003;278:39951–9. [DOI] [PubMed] [Google Scholar]
- 56. Blackinton JG, Keene JD. Post-transcriptional RNA regulons affecting cell cycle and proliferation. Semin Cell Dev Biol 2014;34:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Volpon L, Culjkovic-Kraljacic B, Sohn HS, Blanchet-Cohen A, Osborne MJ, Borden KLB. A biochemical framework for eIF4E-dependent mRNA export and nuclear recycling of the export machinery. RNA 2017;23:927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Topisirovic I, Siddiqui N, Lapointe VL, Trost M, Thibault P, Bangeranye C, et al. Molecular dissection of the eukaryotic initiation factor 4E (eIF4E) export-competent RNP. EMBO J 2009;28:1087–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Culjkovic B, Topisirovic I, Skrabanek L, Ruiz-Gutierrez M, Borden KLB. eIF4E is a central node of an RNA regulon that governs cellular proliferation. J Cell Biol 2006;175:415–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Monecke T, Güttler T, Neumann P, Dickmanns A, Görlich D, Ficner R. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science 2009;324:1087–91. [DOI] [PubMed] [Google Scholar]
- 61. Santiago A, Li D, Zhao LY, Godsey A, Liao D. p53 SUMOylation promotes its nuclear export by facilitating its release from the nuclear export receptor CRM1. Mol Biol Cell 2013;24:2739–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Askjaer P, Bachi A, Wilm M, Bischoff FR, Weeks DL, Ogniewski V, et al. RanGTP-regulated interactions of CRM1 with nucleoporins and a shuttling DEAD-box helicase. Mol Cell Biol 1999;19:6276–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fannemel M, Barøy T, Holmgren A, Rødningen OK, Haugsand TM, Hansen B, et al. Haploinsufficiency of XPO1 and USP34 by a de novo 230 kb deletion in 2p15, in a patient with mild intellectual disability and cranio-facial dysmorphisms. Eur J Med Genet 2014;57:513–9. [DOI] [PubMed] [Google Scholar]
- 64. Hucthagowder V, Liu T-C, Paciorkowski AR, Thio LL, Keller MS, Anderson CD, et al. Chromosome 2p15p16.1 microdeletion syndrome: 2.5 Mb deletion in a patient with renal anomalies, intractable seizures and a choledochal cyst. Eur J Med Genet 2012;55:485–9. [DOI] [PubMed] [Google Scholar]
- 65. Callanan M, Kudo N, Gout S, Brocard M, Yoshida M, Dimitrov S, et al. Developmentally regulated activity of CRM1/XPO1 during early Xenopus embryogenesis. J Cell Sci 2000;113:451–9. [DOI] [PubMed] [Google Scholar]
- 66. Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 1997;90:1051–60. [DOI] [PubMed] [Google Scholar]
- 67. Kashyap T, Argueta C, Unger T, Klebanov B, Debler S, Senapedis W, et al. Selinexor reduces the expression of DNA damage repair proteins and sensitizes cancer cells to DNA damaging agents. Oncotarget 2018;9:30773–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Marijon H, Gery S, Chang H, Landesman Y, Shacham S, Lee DH, et al. Selinexor, a selective inhibitor of nuclear export, enhances the antitumor activity of olaparib in triple negative breast cancer regardless of BRCA1 mutation status. Oncotarget 2021;12:1749–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Farren MR, Hennessey RC, Shakya R, Elnaggar O, Young G, Kendra K, et al. The exportin-1 inhibitor selinexor exerts superior antitumor activity when combined with T-cell checkpoint inhibitors. Mol Cancer Ther 2017;16:417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Tables
Supplementary Table 1
Data Availability Statement
Microscopy images were deposited on figshare (10.6084/m9.figshare.20616009) and the codes used to analyze the images were deposited on GitHub (github.com/nahuelzamponi/xpo1_dlbcl_2022). The Gene Expression Omnibus accession number for the RNA-seq data: GSE211684. All other raw data generated in this study are available upon request from the corresponding author.