

Abstract
Myeloid cells, including macrophages, play important roles as first responders to cardiac injury and stress. Epidermal growth factor receptor (EGFR) has been identified as a mediator of macrophage responsiveness to select diseases, though its impact on cardiac function or remodeling following acute ischemic injury is unknown. We aimed to define the role of myeloid cell-specific EGFR in the regulation of cardiac function and remodeling following acute myocardial infarction (MI)-induced injury. Floxed EGFR mice were bred with homozygous LysM-Cre (LMC) transgenic mice to yield myeloid-specific EGFR knockout (mKO) mice. Via echocardiography, immunohistochemistry, RNA sequencing and flow cytometry, the impact of myeloid cell-specific EGFR deletion on cardiac structure and function was assessed at baseline and following injury. Compared with LMC controls, myeloid cell-specific EGFR deletion led to an increase in cardiomyocyte hypertrophy at baseline. Bulk RNASeq analysis of isolated cardiac Cd11b+ myeloid cells revealed substantial changes in mKO cell transcripts at baseline, particularly in relation to predicted decreases in neovascularization. In response to myocardial infarction, mKO mice experienced a hastened decline in cardiac function with isolated cardiac Cd11b+ myeloid cells expressing decreased levels of the pro-reparative mediators Vegfa and Il10, which coincided with enhanced cardiac hypertrophy and decreased capillary density. Overall, loss of EGFR qualitatively alters cardiac resident macrophages that promotes a low level of basal stress and a more rapid decrease in cardiac function along with worsened repair following acute ischemic injury.
Introduction
In recent years, there has been an intense focus on understanding the origin, phenotype and response of various immune cell populations to cardiac stress and injury [1–6]. Myeloid cells, which predominantly encompass neutrophils, monocytes and macrophages (mfs), have been of particular interest as first responders to cardiac injury and stress. Hematopoiesis-derived myeloid cells such as neutrophils and monocytes are known to rapidly accumulate in the heart, within hours, and reside at the site of injury for several days; however non-hematopoiesis-derived resident cardiac mf have also been shown to respond to acute injury and help coordinate the subsequent inflammatory processes [5,7,8]. In addition to their contributions toward post-injury remodeling, cardiac resident mf have been demonstrated to regulate several developmental or homeostatic processes, with notable roles in capillary and vascular development, lymphatic vessel growth and maturation, as well as maintenance of cardiac cell turnover and electrical conduction [1,6,9–12].
Mf exist within a broad spectrum of phenotypes driven by environmental cues [13]. While resident mf can contribute to cardiac homeostasis and coordination of early inflammatory responses to acute cardiac injury [14], they are replaced over time by pro-inflammatory monocyte-derived mf that express lymphocyte antigen 6 complex locus c1 (LY6C) and c-c motif chemokine receptor 2 (CCR2) and secrete cytokines such as interleukin-6 (IL-6), enabling them to promote extracellular matrix (ECM) breakdown and debris clearance [8,15]. As time progresses post-injury, mf subtypes continue to change in response to their environment and begin to exhibit repair-promoting phenotypes, expressing higher levels of anti-inflammatory cytokines, scavenger receptors, and pro-angiogenic mediators, including IL-10, cluster of differentiation 206 (CD206) and vascular endothelial growth factor (VEGF), which act to dampen inflammation, facilitate efficient clearance of dying inflammatory cells and debris and promote neovascularization, respectively [5,16].
With an expanding understanding of the plasticity and impact of myeloid cell subpopulations on cardiac inflammation and repair, their potential as therapeutic targets to promote improved remodeling outcomes following acute injury continues to grow. Myeloid cells express an extensive repertoire of cell surface receptors that influence their biology across various disease etiologies and offer potentially useful targets for manipulating their behavior and responsiveness to injury [17,18]. Epidermal growth factor receptor (EGFR), a member of the erythroblastic leukemia viral oncogene homolog (ErbB) family of receptor tyrosine kinases, has historically been investigated for its role in cancer development due to its impact on downstream signaling pathways that promote tumor growth. However, EGFR has also been shown to be important in cardiac pathophysiology [19,20], where its cell-specific roles in the heart have largely focused on cardiomyocytes [21–24]. The impact of EGFR on myeloid cell function in the injured heart has not been specifically explored, though a handful of studies have demonstrated a context-dependent role for myeloid cell-specific EGFR in regulating adaptive immune responses to infection, including colitis, colitis-associated tumorigenesis and bacterial clearance during pneumonia [25–30]. Altogether, these led us to hypothesize that EGFR influences myeloid cell-dependent repair processes following acute injury.
Here, using mice with myeloid cell-specific EGFR deletion, we have demonstrated for the first time that loss of EGFR qualitatively alters cardiac myeloid cells with a predominant decrease in the expression of pro-angiogenic transcripts. Congruently, mice with myeloid cell-specific EGFR deletion experienced a worsened response to acute cardiac injury, with a more rapid drop in cardiac function, enhanced accumulation of inflammatory mf over time and decreased neovascularization.
Materials and methods
Animals
Experiments using mice were conducted in the Lewis Katz School of Medicine under the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and also approved by the Institutional Animal Care and Use Committee (IACUC) at Temple University with Animal Protocols 4891 and 4902. The B6.129s6-Egfrtm1Dwt/Mmnc mouse strain, ID 031765-UNC, was obtained from the Mutant Mouse Regional Resource Center (MMRRC), an NIH-funded strain repository, and was donated to the MMRRC by David Threadgill, Ph.D., North Carolina State University. These mice harbor a conditional allele of EGFR with exon 3 flanked by two loxP sites (EGFRf/f), where deletion results in early termination of translation [31]. EGFRf/f mice were bred with homozygous LysM-Cre transgenic mice to yield myeloid-specific EGFR knockout (EGFRmKO) [32]. LysM-Cre mice were obtained from Jackson Laboratory (B6.129P2-Lyz2tm1(cre)Ifo/J, Stock No: 004781). Both male and female EGFRmKO, LysM-Cre, and EGFRf/f controls were used throughout the study, with numbers of mice used in each experiment are represented individually in bar graphs, and/or indicated in figure legends. Surgical procedures were performed on mice anesthetized via continuous isoflurane inhalation with post-operative subcutaneous administration of buprenorphine for analgesia. At study endpoint, mice were euthanized via isoflurane overdose and removal of heart.
Mouse genotyping
Mouse genotypes were confirmed via agarose gel separation of polymerase chain reaction (PCR) products from tail tissue. At around 3 weeks of age, no more than 0.5 cm of tail tissue was clipped and digested in 50 μl of DirectPCR Lysis Reagent (Viagen Biotech, Los Angeles, CA, U.S.A.) at 95°C for 20 min. Tails then received an additional 50 μl of DirectPCR Lysis Reagent, containing 0.6 mg/ml of proteinase K (VWR, Solon, OH, U.S.A.), and were kept at 55°C overnight (O/N). Tail solutions were finally heated at 95°C for 10 min, and after thorough vortex/centrifugation, tail DNA was subject to PCR. PCR primers used to detect floxed Egfr and Cre are provided in Supplementary Table S1. PCR for both primer sets required 30 cycles of 94°C for 30 s, 56°C for 25 s, and 72°C for 60 s. Following PCR, products were evaluated on agarose gel (2% in 1X Tris-acetate-EDTA, Thermo Scientific, Wilmington, DE) based on size: homozygous Egfrf/f (348 bp), WT Egfr (253 bp), Cre (565–585 bp).
Quantitative Real-Time PCR (RT-qPCR)
Total RNA was isolated from frozen, quarter left ventricular (LV) or cell samples with the PureLink RNA mini kit (Invitrogen, Carlsbad, CA, U.S.A.), according to the manufacturer’s instructions. RNA yield was tested for quality and concentration using a Nanodrop 2000 (Thermo Scientific, Wilmington, DE). Following quantification, 500 ng (cells) or 1000 ng (tissue) of RNA was converted to complementary DNA (cDNA) with the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Waltham, MA, U.S.A.). Normalized quantities of cDNA (25 ng) were then subject to RT-qPCR in triplicate using the Powerup Sybr Select Master Mix (Applied Biosystems, Waltham, MA, U.S.A.). All gene expression was normalized to an internal control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and analyzed using the Applied Biosystems Comparative CT Method (2−ΔΔCT). All primers used in RT-qPCR are listed in Supplementary Table S1.
Immunofluorescence (IF) and Immunohistochemistry (IHC) of cardiac sections
Mouse hearts were saline-perfused, excised, and fixed (in whole) in 4% paraformaldehyde in phosphate-buffered saline (PBS). After 72 h, fixed hearts were washed in PBS, embedded in paraffin, and sectioned onto charged microscope slides (Globe Scientific, Mahwah, NJ, U.S.A.) at 5-micron thickness. Adhered heart sections were then deparaffinized in xylene (15 min) and ethanol (100%, 90%, 80% and 70% sequentially, 5 min each) prior to staining. To analyze cell cross-sectional areas, hearts were stained overnight at 4°C with Wheat Germ Agglutinin (WGA, 1:250, Vector Labs, San Francisco, CA, U.S.A.), washed (3× PBS), and stained with NucBlue Fixed Cell ReadyProbes (ThermoFisher, Fair Lawn, NJ, U.S.A.) for 10 min at room temperature (RT). Similarly, to assess capillary density, hearts were stained overnight at 4°C with isolectin B4 (Lectin, 1:250, Vector Labs, San Francisco, CA, U.S.A.), washed, and counterstained with NucBlue. To assess cardiac cell death, sections were subjected to TdT-mediated dUTP-X nick end labeling (TUNEL) via an In Situ Cell Death Detection Kit, TMR Red (Sigma-Aldrich, St. Louis, MO, U.S.A.). After proteinase K treatment, sections were incubated with TMR-dUTP for 1 h at 37°C, and counterstained with troponin I (TNNI, 1:100, Cell Signaling, Danvers, MA, U.S.A.) overnight at 4°C. Donkey anti-Rabbit ReadyProbes Secondary Antibody Alexa Fluor 488 (1:250, ThermoFisher, Fair Lawn, NJ, U.S.A.) was used to visualize TNNI. Nuclei were identified after staining with NucBlue Fixed Cell ReadyProbes as described above. All IF slides were preserved with 1–2 drops of ProLong Gold Antifade Mountant (ThermoFisher, Fair Lawn, NJ, U.S.A.). Heart sections were also stained with Masson’s trichrome (Sigma-Aldrich, St. Louis, MO, U.S.A.) to analyze infarct length and fibrosis. Here, sections were placed in Bouins fluid overnight at room temperature. After washing, sections were incubated in Hematoxylin-Eosin, and Biebrich Scarlet-Acid Fucshin for 5 min, with rinsing in between. Sections were then placed in phosphotungstic-phosphomolybdic acid, and finally stained with Aniline Blue. IHC slides were preserved with 1–2 drops of Permount Mounting Media (ThermoFisher, Fair Lawn, NJ, U.S.A.).
Stained hearts were visualized with the Nikon Eclipse NiE or TiE microscope at magnification (×) 0.8, 1, 20 or 40 where appropriate. The Nikon NIS-Elements software was used to record and analyze images. Cell size was determined after outlining and averaging the area of 80–100 cardiomyocytes over 8–10 individual images from each left ventricle (LV). Capillaries were identified as lectin positive capillaries over the tissue area in 8–10 individual images from each LV. Cell death was analyzed as number of TUNEL, NucBlue, TNNI positive cells over the total number of NucBlue, TNNI-positive cells in 8–10 images in the infarct zone and remaining area of each LV. The infarct length was quantified as the average percent of collagen scar length over total LV length in three images of each LV.
Cell Isolation and staining for flow cytometry analysis
Cells were isolated from heart and spleen for flow cytometry analysis or bone marrow for cell culture. In the heart, non-myocyte cells were obtained after manual and enzymatic digestion. After PBS perfusion, hearts were chopped into roughly 1 mm3 pieces and transferred to Roswell Park Memorial Institute 1640 Media (RPMI) (Cellgro Corning, Corning, NY) supplemented with 450 U collagenase II (Worthington Biochemical Lakewood, NJ, U.S.A.), and 60 U hyaluronidase(Sigma-Aldrich,St.Louis,MO,U.S.A.)foreachml.Roughly7mlsofdigestionbufferwasusedforevery 120 mg of tissue. After 1 h in a shaking 37°C water bath, cell suspensions were collected and strained through a PBS primed 40-μm strainer (Greiner Bio-One, Monroe, NC). Red blood cells (RBCs) were lysed at 37°C for 5 min with Ammonium Chloride Potassium (ACK) Lysing Buffer (ThermoFisher, Fair Lawn, NJ, U.S.A.), washed, and separated at 1000 rpm for 5 min at 4°C. Cells were then counted with Trypan Blue Solution (ThermoFisher, Fair Lawn, NJ, U.S.A.) to check viability. Spleens were carefully teased out, rinsed in PBS, and digested manually by mashing with syringes and rinsing over 40 μm strainers. Splenocytes were then pelleted at 1000 rpm for 5 min at 4°C. RBCs were lysed as described above, and the remaining cell pellet was separated and counted as described above. Femur and tibia bones were cleared of skin and muscle and flushed with Hanks Buffered Salt Solution (HBSS) using a 27 G needle (BD, Franklin Lakes, NJ, U.S.A.). After resuspension into single cell suspension (18 G needle, BD, Franklin Lakes, NJ, U.S.A.), leukocytes were spun at 1000 rpm for 5 min at 4°C. RBCs were lysed as described above, and the remaining cell pellet was separated and counted as described above.
Equal volumes of enzymatically digested cardiac single cell suspensions were taken for cell surface marker staining. Briefly, samples were spun at 300 × g for 5 min at 4°C and cells were incubated with fluorophore-labelled antibodies at concentrations described in Supplementary Table S2. After 45 min of incubation on ice, cells were washed in FACS buffer (1X PBS without Ca2+/Mg2+ + 10% fetal bovine serum (FBS, Gemini Bio-Products)). Cells were fixed in 4% paraformaldehyde for 10 min on ice and subsequently resuspended in 400 μl of FACS buffer to be acquired using an LSR-II flow cytometer (BD Biosciences, U.S.A.). Prior to antibody incubation, cells were treated with FcR blocking reagent (Miltenyi Biotec, Germany) for 15 min on ice. Fluorophore compatible isotype matched IgG were taken as control during staining (Supplementary Table S2; Supplementary Figures S1 and 2). Each experiment included an unstained control and single-color compensations using UltraComp eBeads™ (Thermo Fisher Scientific, U.S.A.). Exported FCS files were analyzed by FlowJo software (BD Biosciences, U.S.A.). First, cells were sorted by FSC and SSC, singlets were selected by FSC-H/FSC-A and then CD45+ cells were gated for further analyses, as indicated in figure legends. All positive gated cells obtained per individual heart were normalized to mg tissue weight.
Isolation and concentration of mouse cardiac cluster of differentiation (CD)11b cells
Non-myocyte cells were isolated from a total of 500–700 mg of combined heart tissue (3–6 hearts were pooled for each replicate) as described above. After counting with trypan blue, cells were resuspended in chilled, sterile PBS, and purified with Debris Removal Solution (Miltenyi Biotec, Auburn, CA, U.S.A.), according to the manufacturer’s instructions. Debris was discarded, and the remaining cells were subject to RBC lysis as described above. After washing with PBS, the cells were placed in a degassed, chilled, solution of PBS containing 2% bovine serum albumin (BSA) and 0.5 mM EDTA (isolation buffer). CD11b Microbead Cocktail (Miltenyi Biotec, Auburn, CA, U.S.A.) was then added to cell suspensions, paying attention to manufacture protocols. After 15 min at 4°C, cells were washed with isolation buffer and applied to an MS column in a magnetic separator (Miltenyi Biotec, Auburn, CA, U.S.A.). CD11b cells were retained in the column, and then plunged. Cells were counted with trypan blue and used for downstream applications. Debris removal and CD11b concentration were verified with flow cytometry using CD11b-BV650 (1:100, BD, Franklin Lakes, NJ, U.S.A.), Cd45-BUV395 (1:100, BD, Franklin Lakes, NJ, U.S.A.), and LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (1:40, Invitrogen, Carlsbad, CA, U.S.A.).
Whole transcriptome analysis
Freshly isolated cardiac CD11b+ cells from 12-week-old EgfrmKO vs. Egfrf/f control mice were immediately subject to RNA isolation and quantification as described above. Total RNA samples were then submitted for transcriptomic analysis at Fox Chase Cancer Center (Philadelphia, PA, U.S.A.) using the NextSeq2000 platform (Illumina, San Diego, CA, U.S.A.). Bioanalyzer (Agilent Technologies, Santa Clara, CA, U.S.A.), and Qubit (Invitrogen, Carlsbad, CA, U.S.A.) were used to determine RNA quality, and confirm concentrations. About 200 ng of samples with RNA integrity number (RIN) over 8 were used to prepare the mRNA library using the NEBNext ® Ultra™ II Directional RNA Library Prep Kit, Nextseq 2000 R2 100 cycle reagent (Illumina, San Diego, CA, U.S.A.). Here, mRNAs were enriched twice using poly-T based RNA purification beads and were then fragmented at 94°C (15 min) by the divalent cation method. 1st strand cDNA was synthesized at 42°C (15 min), followed by 2nd strand synthesis at 16°C (60 min). After End preparation, adapters with illuminaP5, P7 sequences, along with indices were ligated to the cDNA fragment at 20°C (15 min). Samples were then subjected to AMPure bead purification (Beckman Coulter, Pasadena, CA, U.S.A.), and subjected to PCR, at 9 cycles of 98°C (10 s) and 65°C (75 s). After additional purification using AMPure beads, libraries were checked for quality and quantity with the bioanalyzer and Qubit. Libraries were then pooled and sequenced with read length of 65 bp. Reads were aligned to mouse genome (GRCm38) using STAR [33]. The number of raw counts in each known gene from the RefSeq database was enumerated using htseq-count from the HTSeq package [34]. Differential expression between samples and across different conditions was assessed for statistical significance using the R/Bioconductor package DESeq2 [35]. Genes with a false discovery rate (FDR) ≥ 0.05 and a fold-change ≥ 2 by were considered significant. Functional annotation of data sets, networks, and upstream regulator analyses were performed in the Ingenuity Pathway Analysis software (Ingenuity Systems, Redwood City, CA, U.S.A.). RNA-seq data files were uploaded in GEO under accession number GSE207686.
Isolation and culture of mouse bone marrow macrophages
Macrophages (mf) were derived using isolated leukocytes from femur/tibias, yielding bone marrow-derived mf (BMDM). Bone leukocytes were isolated as described above, using sterile tools and conditions. Isolated, and strained cells were then resuspended in RPMI supplemented with 10% FBS, 1% penicillin/streptomycin/amphotericin B (PSF, Gemini Bio-Products), and 20% conditioned media from L929 fibroblast culture (BMDM media) [36]. Cells were plated, and mononuclear phagocytes were allowed to adhere in a humidified atmosphere of 5% CO2 at 37°C. Monocytes were then cultured for 5 days, and on day 3, BMDM media was replenished. Approximately 1 ml of media was added for every 1 × 106 cells. For mf derivation, leukocytes were initially seeded at a density of 3 × 106 in 6 well dishes, 6 × 106 in 60 mm dishes, or 10 × 106 in 100 mm dishes. Mf were confirmed by flow cytometric analysis of CD11b and F4/80 co-expression.
L929 fibroblast culture
L929 fibroblast cells were cultured in Minimum Essential Medium (MEM, Cellgro Corning, Corning NY, U.S.A.) supplemented with 10% FBS and 1% PSF. Cells were grown in a humidified atmosphere of 5% CO2 at 37°C. L929 cells were seeded at a density of 5 × 106 cells in a T175 flask and allowed to grow till 75% confluency (20 × 106). At confluency, media was taken off the cells and sterile filtered to be used for mf derivation.
Protein extraction and immunoblot analysis
Samples were lysed in RIPA buffer (1 M Tris pH 7.4, 10% NP-40, 10% sodium deoxycholate, 1 M sodium chloride, 0.5 M EDTA pH 8, 0.5 M sodium fluoride) containing 1X HALT protease inhibitor cocktail (ThermoFisher, Fair Lawn, NJ, U.S.A.) and phosphatase inhibitor cocktail set IV (Calbiochem, San Diego, CA, U.S.A.). Equal concentrations of lysates were subjected to SDS-polyacrylamide gel electrophoresis (8–10% gels), and then transferred onto Immobilon-PSQ polyvinylidene fluoride 0.2 μm pore membranes (Millipore, Burlington, MA, U.S.A.). To prevent non-specific binding, membranes were incubated in blocking buffer (Rockland, Pottstown, PA, U.S.A.), and then subjected to antibodies of interest. Primary antibodies used are provided in Supplementary Table S2. After washing membranes with TBS-T, they were subjected to secondary antibody at room temperature for 1 hr in the dark (1:15000, IRDye680 Donkey anti-rabbit IgG, or IRDye800 Goat anti-mouse IgG (LI-COR Biosciences, Lincoln, NE, U.S.A.). Membranes were then visualized on the LI-COR Biosciences Odyssey System.
Mouse model of non-reperfused myocardial infarction
Myocardial infarction was surgically induced after mice were anesthetized with 2% isoflurane inhalation [37]. A small skin incision was made over the left chest, and a purse suture was made, allowing the pectoral muscles to be dissected and retracted. The 4th intercostal space was then exposed, and a small hole was made to gently pop the heart out. Once the left main descending coronary artery (LCA) was located, it was sutured and ligated approximately 3 mm from its origin with a 6–0 silk suture. After ligation, the heart was placed back into the intra-thoracic space, and the chest was closed. Sham operated mice were treated similarly, except the LCA was not occluded. Injury level from MI surgery was standardized by a predefined parameter as having an ejection fraction of less than 45% as measured by echocardiography 7 days after MI, whereas animals having an ejection fraction of greater than 45% were considered as having an unsuccessful surgery and would be excluded from the study.
Echocardiography
The VisualSonic Vevo 2100 Imaging System (FUJIFILM, Toronto, ON, U.S.A.) was used to assess cardiac function. Hair was removed from the mouse chest with Nair prior to echocardiography. All mice were sedated with 3% isoflurane (Patterson Veterinary, Greeley, CO) to maintain a heart rate (HR) of 450–550 beats per minute (bpm). Long and short axis orientation images of the left ventricle (LV) were taken in B and M mode using the MS400 (30-MHz centerline frequency) probe. Cardiac functional and structural parameters were measured using M mode images. These included LV diastolic (d) and systolic (s) internal diameters (ID), posterior and anterior wall thickness (PW, AW), and volumes (V). Stroke volume (SV) was calculated as the difference between diastolic and systolic end volumes. Percentage of fractional shortening (FS) was calculated using the equation ([LVIDd – LVIDs]/LVIDd) × 100 and the percentage of ejection fraction (EF) was calculated using the equation ([LVVd – LVVs]/LVVd) × 100. Cardiac output (CO) was calculated as the SV multiplied by HR.
Statistical analysis
All statistical analysis was performed using GraphPad Prism 9 software (GraphPad Software Inc., San Diego, CA, U.S.A.). All data graphs were generated by plotting the mean ± standard error of the mean (SEM) with number of individual samples per group indicated in the figure legends. Statistical comparisons were made using two-tailed unpaired t-tests for two groups, ordinary one-way ANOVA with Tukey’s multiple comparisons for three or more groups or two-way repeated measures ANOVA to determine the row (timepoint) × column (i.e. genotype/sex or surgical intervention) factor interaction with Tukey’s multiple comparisons test of column factors at individual timepoints, or Log-rank (Mantel-Cox) test for survival analysis. All differences were considered significant at P<0.05.
Results
Myeloid cell-specific EGFR deletion induces a small cardiomyocyte hypertrophic response
To determine how myeloid cell-expressed EGFR impacts cardiac physiology, we crossed mice harboring loxP sites flanking exon 3 of EGFR (EGFRf/f, CTL mice) with mice carrying a LysM-Cre transgene (LMC mice), producing mice with EGFR deletion in Lyz2-expressing myeloid cells (EGFRmKO, mKO mice) (Figure 1A) [32,38,39]. We derived mfs in vitro from bone marrow cells isolated from mKO versus the CTL and LMC control lines, where a majority were shown to be CD11b+F4/80+ and confirmed EGFR down-regulation at both the transcript and protein levels (Figure 1B and Supplementary Figure S3). Since we have previously shown that EGFR can modulate cardiac homeostasis [22], and cardiac mf are essential for cardiac development and homeostasis [6], we sought to understand whether constitutive myeloid cell-specific EGFR deletion resulted in any structural or functional consequences in mKO mice over time (Figure 1C). By and large, we did not observe significant alterations in cardiac structure or function of mKO versus CTL mice, including ejection fraction (%EF, Figure 1D), which also did not differ by sex (Supplementary Figure S3b), or other parameters (Supplementary Figure S4a–m), as assessed via echocardiography up to 18 months of age. Only stroke volume and diastolic LV volume differed between mKO and CTL mice at 15 months of age but was not sustained to 18 months of age (Supplementary Figure S4c,d). Notably, LysM-Cre control mice inconsistently displayed significant differences in some echocardiographic parameters versus either mKO or CTL mice at various timepoints (Figure 1D and Supplementary Figure 4a–m). Additionally, mKO mice did not display altered mortality over time, with similar dropout of mice over time as the control lines (Figure 1F). Despite a lack of alterations in mKO hearts as detected by echocardiography, we observed a small increase in cardiomyocyte cross-sectional area in the LV of mKO mice versus their control lines (Figure 2A) at 3–4 months of age, with a significant difference observed compared with the LMC controls which was accompanied by increases in LV gene expression of the hypertrophic markers β-myosin heavy chain (Myh7) and B-type natriuretic peptide (Nppb) (Figure 2B). Thus, myeloid-cell specific EGFR deletion induces a small increase in cardiomyocyte hypertrophy, but overall this effect does not lead to altered cardiac function or overall survival of the mice over time.
Figure 1. Myeloid cell-specific EGFR deletion does not negatively impact cardiac function over time.
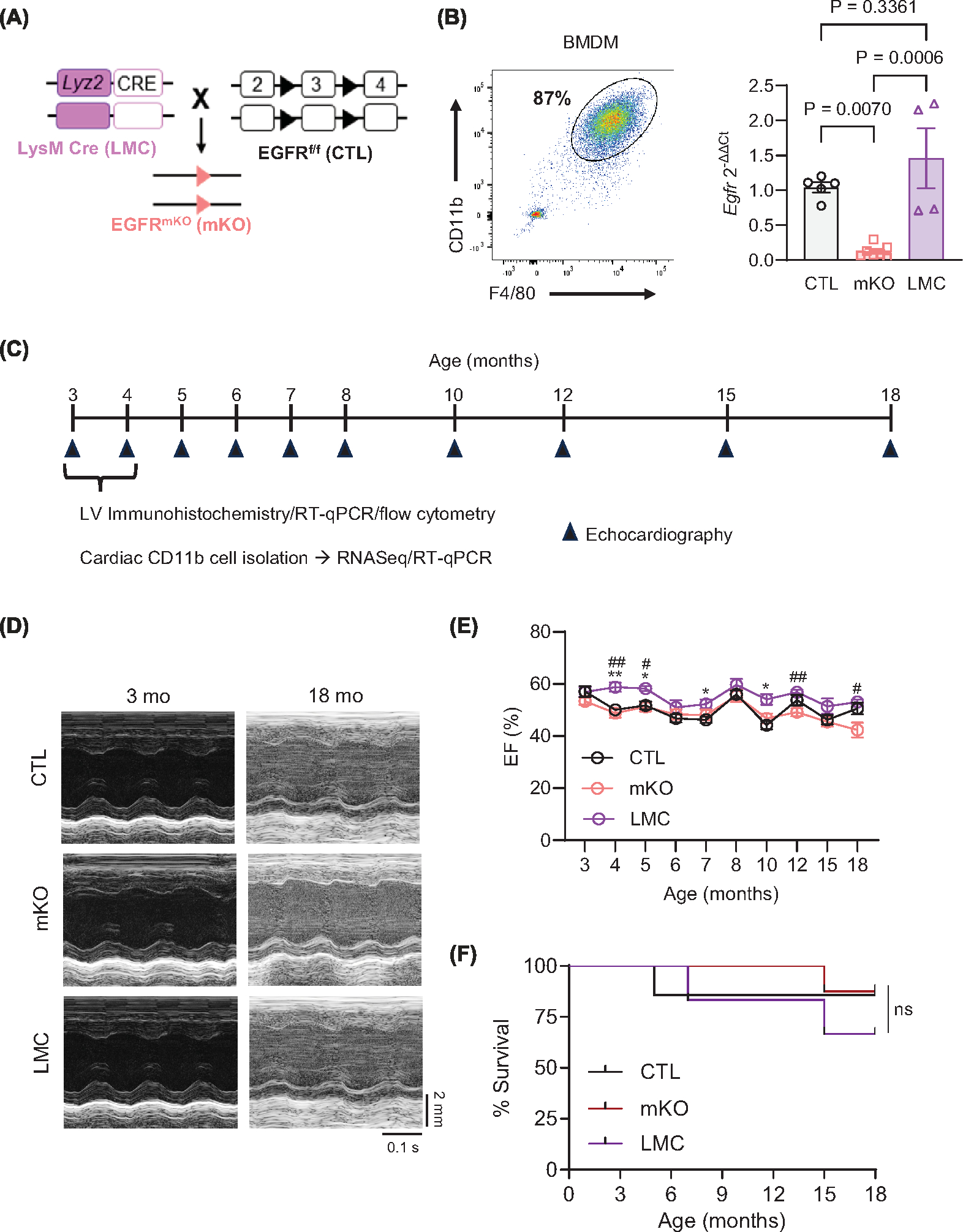
(A) Schematic showing generation of myeloid cell-specific EGFR knockout mice (EGFRmKO, mKO). Homozygous LysM-Cre transgenic mice (LMC) were crossed to mice carrying a conditional allele of Egfr with exon 3 flanked by two loxP sites (EGFRf/f, control (CTL)). (B) Bone marrow derived macrophages (BMDM) were subject to flow cytometry to verify macrophage (mf) derivation using CD11b, and F4/80. RT-qPCR was used to measure Egfr in mKO versus CTL and LMC BMDM, normalized to Gapdh. N = 5 (CTL), 7 (mKO) or 4 (LMC); data are mean ± SEM, one-way ANOVA with Tukey’s multiple comparisons test. (C) Timeline of comparative assessments of CTL, mKO and LMC mice via echocardiography, immunohistochemistry, RNASeq and RT-qPCR analyses. (D) Representative m-mode echocardiograms of CTL, mKO and LMC hearts at 3 and 18 mo. Upper scale depicts 2 mm. Bottom scale depicts 0.1 seconds. (E) Ejection fraction (EF, %) of CTL, mKO and LMC mouse hearts measured via echocardiography from 3 to 18 months of age. Data are mean ± SEM; *P<0.05 CTL vs. LMC, #P<0.05, ##P<0.01, two-way repeated measures ANOVA to determine the row (timepoint) × column (genotype/age) factor interaction (P=0.1445) with Tukey’s multiple comparisons post-hoc analyses at the individual timepoints. N = 8 (CTL), 7 (mKO) or 6 (LMC). (F) Survival analysis of CTM, mKO and LMC mice until 18 months of age; ns = not significant, Log-rank (Mantel-Cox) test. Initial N = 8 (CTL), 7 (mKO) and 6 (LMC).
Figure 2. Myeloid cell-specific EGFR deletion induces mild cardiomyocyte hypertrophy.
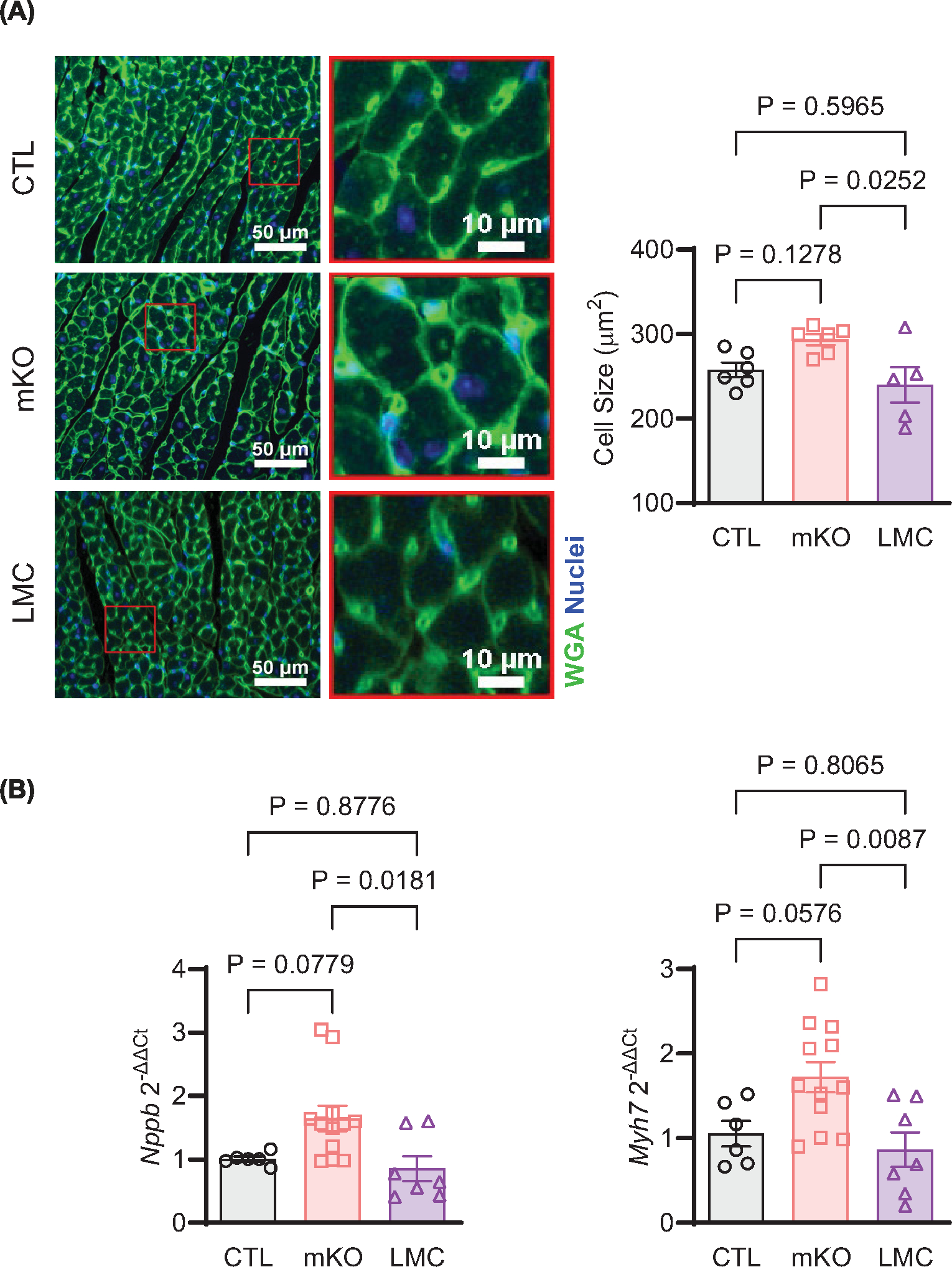
(A) Cardiomyocyte size was assessed in the LV of 12- to 16-week-old CTL, mKO and LMC mice after WGA staining. Panels shown are representative regions of interest (ROI) from 20× magnification of the LV from each group, scale bar denotes 50 (left) or 10 (right) μm. Quantification of cardiomyocyte size is shown in the histogram. N = 6 (CTL, mKO) or 5 (LMC); data are mean ± SEM, one-way ANOVA with Tukey’s multiple comparisons test. (B) RT-qPCR was used to measure the expression of Nppb (left) and Myh7 (right) in the LV of 12- to 16-week-old CTL, mKO or LMC mice, normalized to Gapdh. N = 6 (CTL), 12 (mKO) or 7 (LMC); data are mean ± SEM, one-way ANOVA with Tukey’s multiple comparisons test.
Myeloid cell-specific EGFR deletion alters the cardiac macrophage transcriptome
Since myeloid cell-specific EGFR deletion induced a modest cardiomyocyte hypertrophy response in the uninjured heart, we surmised that there could be an alteration in resident cardiac mf quantity or quality in mKO mouse hearts. However, via FACS analysis, cardiac mf did not quantitatively differ between mKO mice and their control lines (Figure 3A), suggesting that EGFR deletion in cardiac mf may rather lead to qualitative differences that promotes a low level of cardiomyocyte hypertrophy. Therefore, to assess qualitative alterations in cardiac mf, we isolated cardiac non-myocytes from the LV of adult mKO versus CTL mice, concentrated the CD11b+ myeloid cells via magnetic bead separation and subjected them to RNA sequencing. The RNASeq data confirmed a reduction of Egfr and Lyz2 expression in mKO cardiac CD11b+ cells and mf enrichment in both mKO and CTL CD11b+ cells, including several known plasma membrane-expressed mf markers (i.e. Adgre1 (F4/80), Adgre5 (Cd97), Cd14, Cd68, Fgcr3 (Cd16), H2-Aa (MHC-II), Mrc1 (Cd206), Timd4) and efferocytosis-related genes (i.e. Axl, Lrp1, Stab1) (Supplementary Table S3). Evaluation of gene expression changes between mKO and CTL CD11b+ cells revealed 763 differentially expressed transcripts, of which 612 were down-regulated and 151 were up-regulated in mKO cardiac myeloid cells (Figure 3B and Supplementary Tables S4 and 5).
Figure 3. Myeloid cell-specific EGFR deletion alters the cardiac myeloid cell transcriptome.
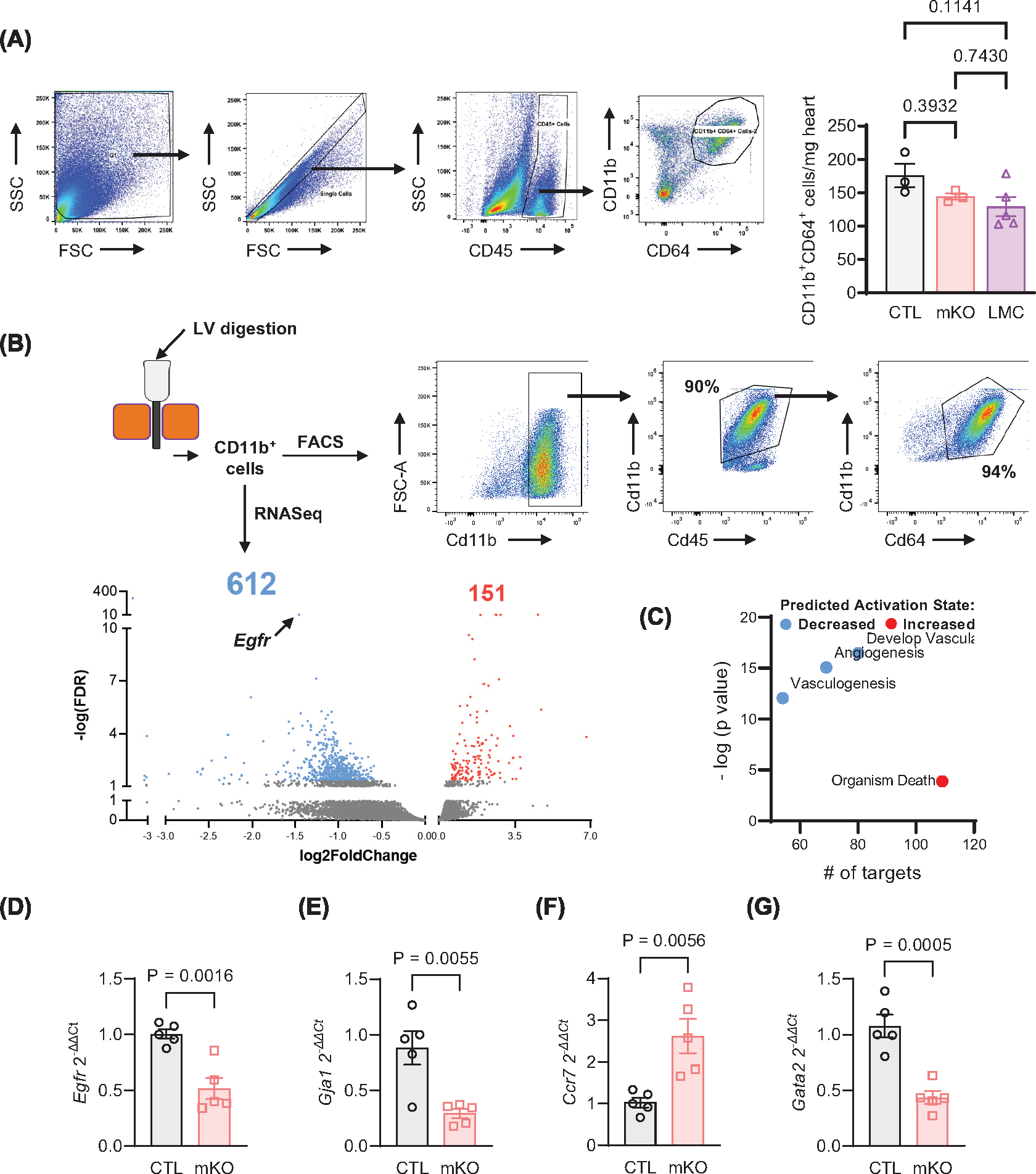
(A) Cardiac non-myocytes isolated from 12-week-old CTL, mKO or LMC hearts at baseline underwent flow cytometry analysis for CD11b+CD64+cardiac mf, as quantified in the accompanying histogram. N = 3 (CTL, mKO) and 5 (LMC); data are mean +− SEM, one-way ANOVA with Tukey’s multiple comparisons test. (B) CD11b+ cells were isolated from CTL or mKO hearts via magnetic bead separation, of which >90% were confirmed to be mf as shown via FACS, and then subjected to bulk RNAseq analyses. Isolated Cd11b+ cells from 3 to 6 CTL or mKO mouse hearts per replicate were combined. Volcano plot of differentially expressed RNA transcripts highlighting the number of genes increased and decreased in expression out of total transcripts analyzed. Transcripts were only considered significantly differentially expressed at cutoffs: fold change absolute value (>1.5) and false discovery rate (<0.05). Significant transcripts are coded in blue (down-regulated) or red (up-regulated). (C) IPA analysis grouped transcripts select decreased and increased biological functions predicted by IPA analysis that are altered in mKO versus CTL cardiac myeloid cells. The y axis indicates level of significance, and the × axis indicates number of targets assigned to biological functions. Blue dots indicate decreased activation, and red dot indicates increased activation. RT-qPCR was used to measure Egfr (D), Gja1 (E), Ccr7 (F) and Gata2 (G) expression in CTL and mKO cardiac myeloid cells, normalized to Gapdh. N = 5 (CTL, mKO); data are mean ± SEM, unpaired t-test.
Many of the transcripts up-regulated in mKO cardiac myeloid cells have predicted roles in antigen binding, activation of complement, activation of the immune response, and phagocytosis (Supplementary Table S5) [40,41], suggesting the inflammatory status of the cardiac myeloid cells could be altered toward a more reactive state. However, the majority of the differentially altered transcripts were down-regulated in mKO cardiac myeloid cells, with the top 3 significantly down-regulated biological functions being ‘development of vasculature’, ‘angiogenesis’, and ‘vasculogenesis’ and the topmost activated biological pathway being ‘organism death’ (Figure 3C). To validate targets identified in the RNASeq data, we performed RT-qPCR analysis of differentially expressed transcripts in cardiac CD11b+ cells isolated from mKO and CTL control mouse hearts, initially confirming down-regulation of EGFR in mKO cardiac myeloid cells (Figure 3D). Notably, recent reports indicate roles for cardiac mf in scavenging of cardiac cell debris and cardiac intercellular communication [2,10,12,42]. Thus, we analyzed targets reported to regulate mf in a manner that could alter cardiac homeostasis or play roles in altering cardiac remodeling responses following injury, including mf coupling with conducting cardiomyocytes (Gja1 (connexin 43, Cx43) [10]), regulation of inflammatory mf motility (chemokine receptor 7, Ccr7[43]) and regulation of phagocytic activity (GATA binding protein 2, Gata2[44]) (Figure 3E–G). The expression profile of these transcripts coincided with the RNASeq data, confirming their altered expression in mKO cardiac myeloid cells. Collectively, these findings indicate that EGFR deletion may prime myeloid cells for altered responses to injury.
Myeloid cell-specific EGFR deletion promotes adverse outcomes following myocardial infarction
Myeloid cells are key components of the post-injury immune response and have been shown to influence cardiac repair outcomes [7]. As mf-expressed EGFR has been implicated in altering disease outcomes [25–30,45], we investigated how mKO mice would respond to overt cardiac pathological stress. To this end, mKO and their control lines were subjected to permanent coronary artery ligation to induce MI, after which we assessed initial injury response, cardiac function over time via weekly echocardiography and chronic remodeling parameters after 4 weeks (Figure 4A). Quantification of TUNEL+ cardiomyocytes in the infarct areas of mKO versus control line mouse hearts at 24 h post-MI revealed an equivalent acute injury response across groups (Figure 4B), indicating that the initial response to acute ischemic injury is not altered in mKO mice. However, starting at 1-week post-MI, mKO hearts showed a hastened decline in function, as indicated by a rapid drop in %EF (Figure 4C), again not differing by sex (Supplementary Figure S5a) or leading to enhanced mortality versus the control lines (Supplementary Figure S5b), as well as other morphometric parameters (Supplementary Figure S5c–o). We subsequently assessed chronic remodeling outcomes at 4 weeks post-MI, observing equivalent infarct lengths and LV border zone fibrosis (Supplementary Figure S6a,b) between mKO mice and their control lines. However, cardiac hypertrophy continued to be impacted by myeloid cell-specific EGFR deletion as post-MI mKO mouse heart weights were elevated in comparison to their control lines (Figure 5A,B) and WGA staining indicated more of an increase in post-MI cardiomyocyte size in mKO mouse hearts than their control lines (Figure 5C). Additionally, injured mKO mouse hearts exhibited lower capillary density relative to the hearts of the control lines (Figure 5D), indicating an impairment in reparative neovascularization, despite similar levels of capillary density in sham-operated hearts. These data are consistent with the decrease in vasculogenesis/angiogenesis pathways predicted from the RNASeq data (Figure 3) and demonstrate that in response to MI, myeloid cell-specific EGFR deletion leads to a rapid worsening of cardiac function and long-term deleterious remodeling.
Figure 4. Myeloid cell-specific EGFR deletion hastens cardiac dysfunction following myocardial infarction.
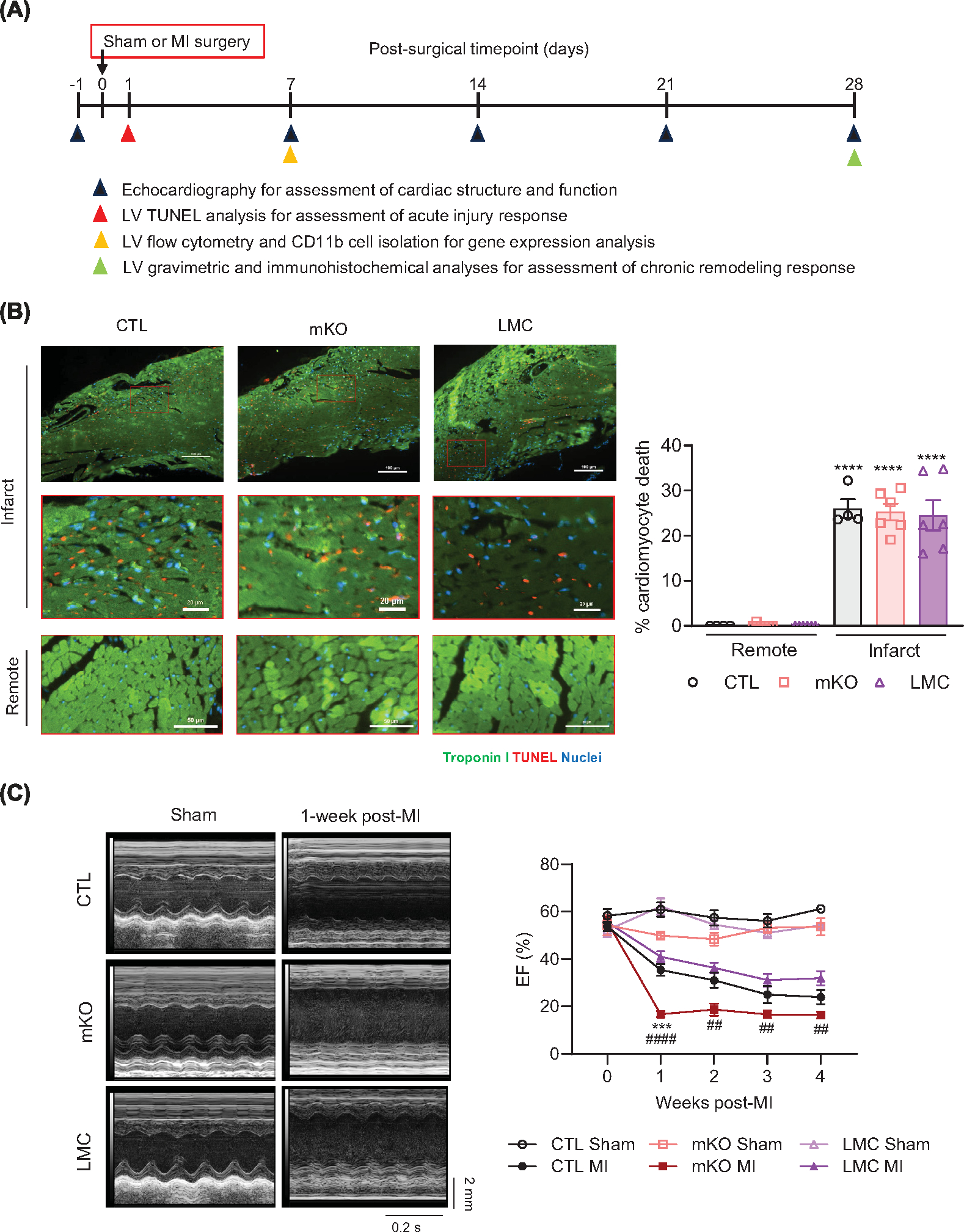
(A) Timeline of comparative assessments of CTL, mKO and LMC mice following sham or MI surgery via echocardiography, immunohistochemistry, flow cytometry and RT-qPCR analyses. (B) Representative panels and insets of TUNEL (red)-, and troponin I (Green) and NucBlue (blue)-stained LVs at 24 post-MI in CTL, mKO and LMC hearts. Scale bar denotes 100 μm (infarct), 20 μm (infarct insets) and 50 μm (remote). Accompanying histogram details quantification of cardiomyocyte death at 24 h post-MI in the infarcts versus remote zones. N = 4 (CTL) or 6 (mKO, LMC); data are mean ± SEM; ****P<0.0001 versus remote zone of same genotype, one-way ANOVA with Tukey’s multiple comparisons test. (C) Representative m-mode echocardiograms of CTL, mKO and LMC hearts 1 week following sham or MI surgery. Upper scale depicts 2 mm. Bottom scale depicts 0.2 s. Histogram depicts ejection Fraction (EF, %) of CTL, mKO and LMC mouse hearts prior to (week 0), and 1–4 weeks post-MI. Two-way repeated measures ANOVA was used to determine the row (timepoint) × column (genotype/surgical condition) factor interaction P<0.001. Tukey’s multiple comparisons test was subsequently used to determine statistical significance between genotype/surgical conditions at the individual timepoints; ns, not significant, ***P<0.001 mKO MI versus CTL MI, ##P<0.01, ####P<0.0001 mKO versus LMC. N = 5 (CTL Sham, mKO Sham), 6 (LMC Sham), 8 (CTL MI, LMC MI) or 7 (mKO MI).
Figure 5. Myeloid cell-specific EGFR deletion promotes cardiomyocyte hypertrophy and reduces capillary density following myocardial infarction.
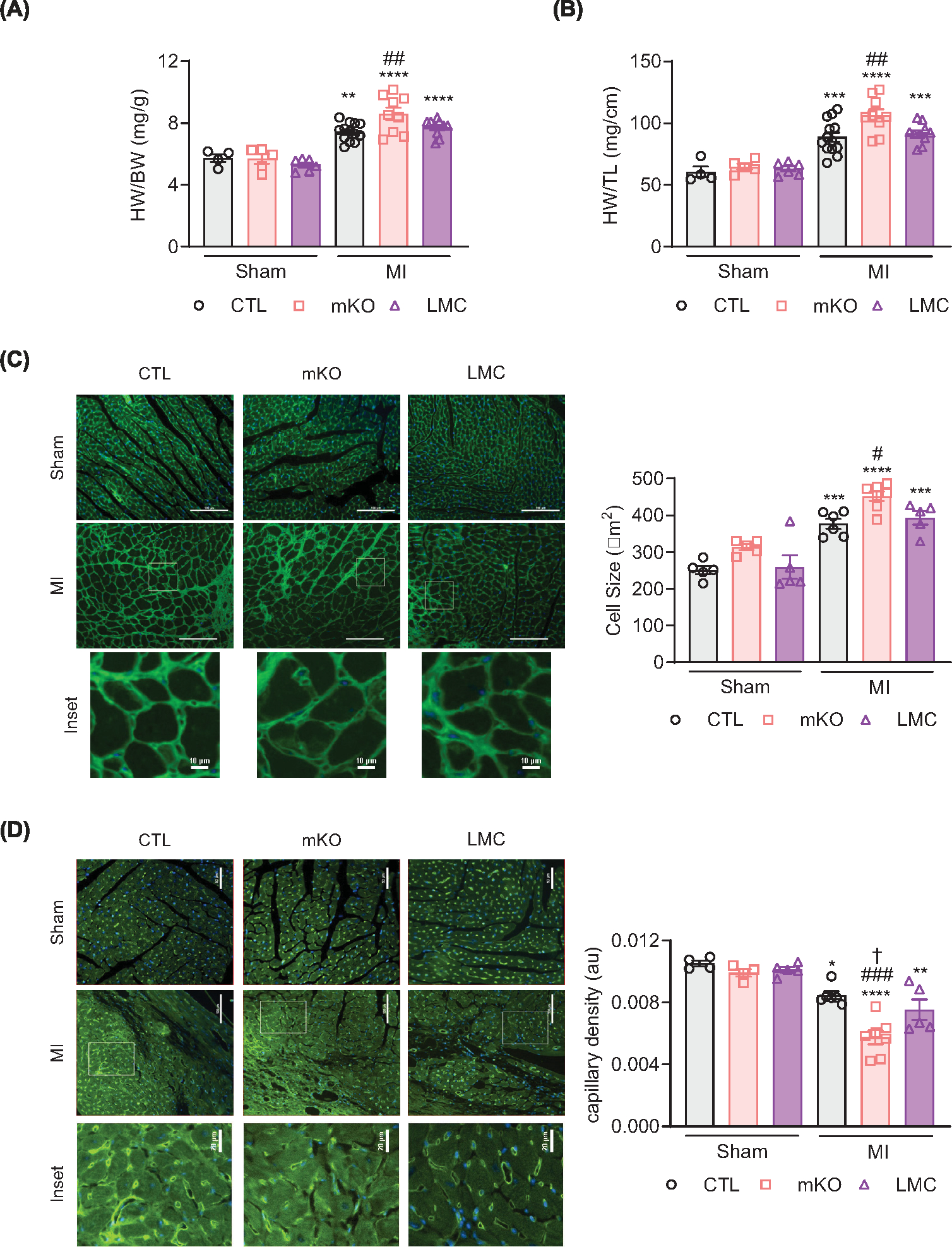
Heart weights from CTL, mKO and LMC mice were normalized to body weight (A) or tibia lengths (B) 4 weeks post-MI. Data are mean ± SEM; **P<0.01, ***P<0.001, ****P<0.0001 versus sham of same genotype, ##P<0.01 versus CTL MI, one-way ANOVA with Tukey’s multiple comparisons test. N = 4 (CTL sham), 5 (mKO sham), 6 (LMC sham), 13 (CTL MI), 9 (mKO MI) or 10 (LMC MI). (C) Representative 20× panels and insets (scale bar: 100 μm, upper panels, and 10 μm, lower insets) of WGA (green) and NucBlue (blue)-stained hearts 4 weeks post-surgery in CTL, mKO and LMC hearts. Histogram shows quantification of cardiomyocyte cell size. Data are mean ± SEM; ***P<0.001, ****P<0.0001 versus sham of same genotype, #P<0.05 versus CTL MI, one-way ANOVA with Tukey’s multiple comparisons test. N = 5 (all shams), 6 (CTL), 7 (mKO) or 5 (LMC). (D) Representative 20× images and insets (scale bar: 100 μm, upper panels, and 20 μm insets) of isolectin B4 (ILB4, green)- and NucBlue (blue)-stained hearts 4 weeks post-surgery in CTL, mKO or LMC hearts. Histogram shows quantification of ILB4-stained capillary density. Data are mean ± SEM; *P<0.05, **P<0.01, ****P<0.0001 versus sham of same genotype, ###P<0.001 versus CTL MI, †P<0.05 versus LMC MI, one-way ANOVA with Tukey’s multiple comparisons test. N = 4 (CTL sham, mKO sham), 5 (LMC sham, LMC MI), 6 (CTL MI) and 7 (mKO MI).
Myeloid-cell specific EGFR deletion reduces pro-reparative mediators following MI
Since mKO mice experience an accelerated decline in function in the first week post-MI, coinciding with the period in which mfs play an important role in the initiation of reparative processes, we sought to determine whether this may be due to quantitative or qualitative differences in the mfs themselves. By FACS analysis, we found that the amount of CD64+CD11b+ mfs in the LV of 7-day post-MI mKO hearts were not significantly different from those of the control lines, and although there was a slight decrease in both inflammatory Ly6CHi and reparative Ly6CLo mfs in mKO versus CTL mouse hearts, this phenomenon appeared to be driven non-specifically by LysM-Cre (Supplementary Figure S7), thus unrelated to EGFR deletion and unable to explain the worsened phenotype at this timepoint. During the repair phase of injury resolution, mf gene expression and phenotypes are focused on facilitating scar stabilization, and angiogenesis [46,47]. Since we observed a decrease in capillary density in the post-MI mKO hearts, we investigated the expression of related genes in the mfs during this phase. We isolated CD11b+ cells from 7-day post-MI hearts, wherein the relative percentage of CD11b+CD64+ mf was lower than that observed in basal hearts (~65% vs. >90%), due to the presence of additional recruited myeloid cells such as monocytes and neutrophils but retained a similar level of EGFR down-regulation (Figure 6A). Seven-day post-MI mKO cardiac CD11b+ cells expressed increased expression of the inflammatory cytokine Il6, as well as decreased expression of transcripts involved in cardiac repair processes, including Cd206, Il10, and Vegfa relative to CTL cardiac CD11b+ cells (Figure 6B). In particular, reduced expression of the pro-repair and pro-angiogenic transcripts Il10 and Vegfa correspond with the decreased cardiac function observed by 7d post-MI (Figure 4C) and the reduced capillary density by 4 weeks post-MI in the mKO mice (Figure 5D). In all, these data are consistent with mKO myeloid cells exhibiting an impaired ability to promote repair and neovascularization in the first week post-injury, leading to a more rapid decline in cardiac function and decreased capillary density over time (Figure 6C).
Figure 6. EGFR-deficient cardiac myeloid cells have reduced expression of reparative transcripts following MI.
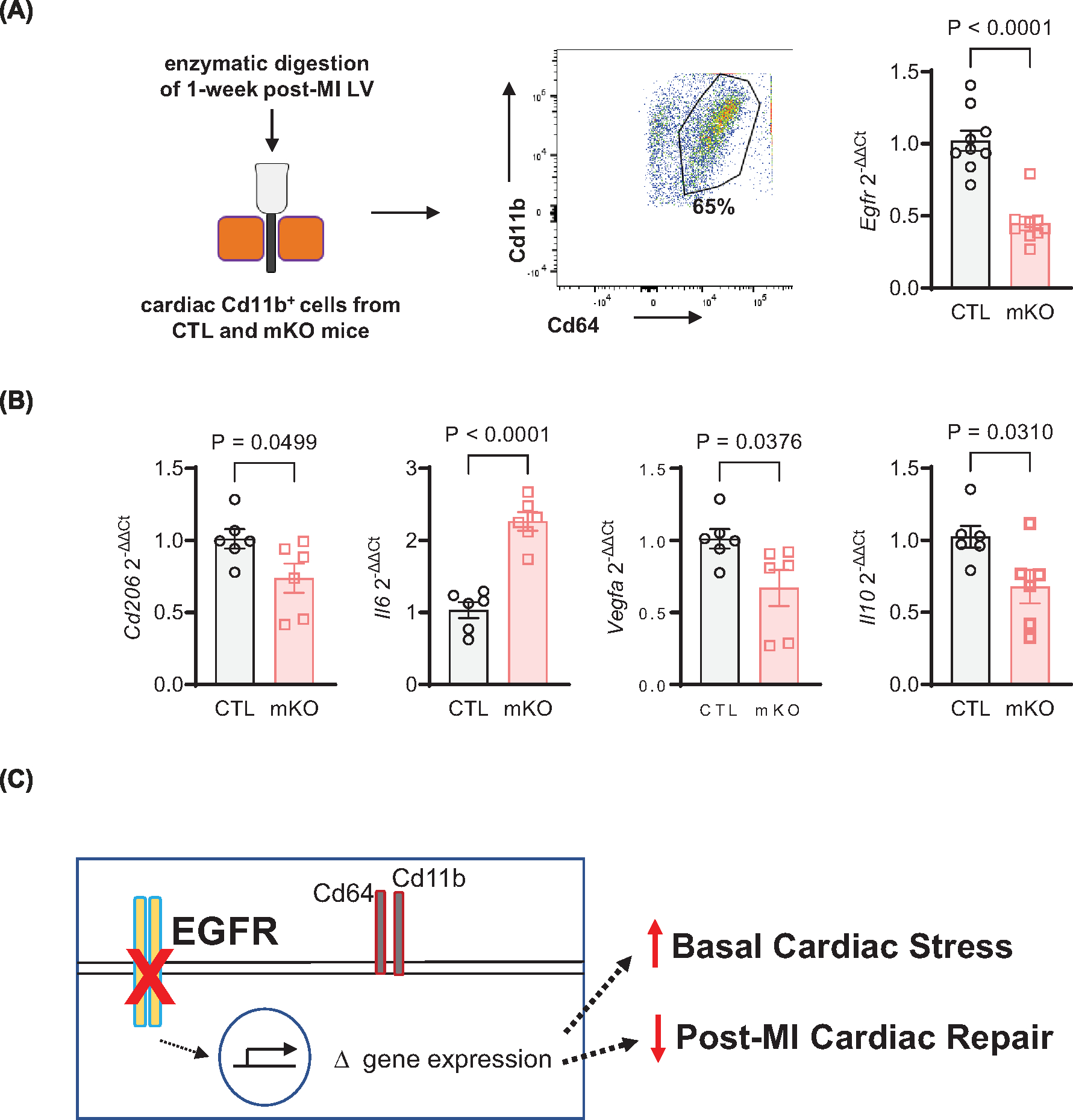
(A) CD11b magnetic beads were applied to cardiac non-myocytes to separate CD11b+ myeloid cells at 7d post-MI using a magnetic column, then subjected to flow cytometry to gate for CD45, CD11b, and CD64 mf. RT-qPCR was used to measure Egfr expression in cardiac CD11b+ myeloid cells from CTL and mKO mice at 7d post-MI, normalized to Gapdh. N = 9 (CTL, mKO); data are mean ± SEM, unpaired t-test. (B) RT-qPCR was used to measure Cd206, Il6, Vegfa and Il10, with Gapdh for normalization, in cardiac CD11b+ cells from CTL and mKO mice at 7d post-MI. N = 6 (CTL, mKO); data are mean ± SEM, unpaired t-test. (C) Myeloid cell-specific deletion of EGFR leads to altered regulation of mf gene expression that contributes to mild basal cardiac stress and a decrease in reparative genes following MI-induced injury, ultimately contributing to an early decrease in cardiac function and delaying neovascularization.
Discussion
The healthy mammalian heart maintains a complex cellular landscape to support its homeostatic functions [48,49], including resident mf, the most abundant immune cell type under normal conditions [3,5,50]. While studies continue to define mf sub-populations, as well as other myeloid cell subtypes, both at baseline and following cardiac injury [1,2,11,42,51], understanding how they may be regulated to impart changes in cardiac homeostasis or reparative processes at different phases post-injury is an ongoing challenge. Receptor tyrosine kinases, including those in the ErbB family, have been shown to regulate myeloid cell biology [52], however the impact of EGFR specifically on response to injury has not been studied. Here, using a mouse model with myeloid cell-specific EGFR deletion (EGFRmKO or mKO), we have demonstrated that loss of myeloid cell-expressed EGFR results in substantial changes to resident cardiac mf transcript expression, but aside from a slight increase in baseline cardiomyocyte hypertrophy, does not lead to a substantial reduction in cardiac function over time in the absence of injury. However, pathway analysis of the RNASeq data did suggest that EGFR-deficient myeloid cells may be more prone to promoting inflammation with a substantially diminished capacity for supporting neovascularization. Indeed, by 7 days post-MI, mKO mice harbored CD11b+ myeloid cells with altered expression of reparative factors, ultimately leading to a hastened decline in cardiac function and diminished LV neovascularization.
Myeloid cell-specific EGFR has been shown previously to regulate mf biology primarily in the context of bacterial infection and cancer [25–30]. Briefly, myeloid cell-specific EGFR deletion has been shown in mice to promote recovery from dextran sulfate sodium (DSS)-induced colitis, as well as bacterial infection-induced gastritis, wherein colonic or gastric mf or tissues isolated from these mouse models expressed less inflammatory cytokines, including IL-6, but more of the anti-inflammatory cytokine IL-10 [26,28]. Another study of DSS-induced colitis also reported a decrease in IL-6 expression in colonic CD11b+ cells isolated from myeloid cell-specific EGFR knockout mice, however in this case the mice were observed to experience worsened colitis due to decreased IL-6-mediated proliferation of intestinal epithelial cells [29]. These studies contrast with our own, in which cardiac myeloid cells isolated 7 d post-MI expressed more IL-6 and less IL-10, suggesting that EGFR deletion does not induce the same effects universally across all mf and all pathologies. Indeed, the impact of mf-expressed EGFR on its local environment has also been explored in gastric cancer studies, wherein myeloid cell-specific deletion resulted in reduced azoxymethane (AOM)-DSS-induced colorectal tumor formation [25,29], which was associated with decreased reparative mf transcript expression, and ultimately less angiogenesis within the tumors of mice with myeloid cell-specific deletion of EGFR [25], results more akin to our own in which mKO cardiac myeloid cells expressed less markers of repair and mKO mice exhibited decreased post-MI cardiac neovascularization.
Beyond colitis and cancer, few studies have investigated the impact of myeloid cell-specific EGFR in a cardiovascular context. Low-density lipoprotein receptor knockout (Ldlr−/−) mice, having undergone transplantation with bone marrow from donor mice with myeloid cell-specific EGFR deletion and subsequent feeding with a high fat diet (HFD), were shown to have reduced size and necrosis of atherosclerotic lesions [45]. These observations were associated with decreased mf accumulation within the lesions themselves and decreased oxidized lipid uptake in EGFR-deficient macrophages. Similarly, another group recently reported that CD11b-Cre-mediated EGFR deletion resulted in decreased HFD-induced obesity and insulin resistance with decreased pro-inflammatory mf accumulation in peripheral adipose tissue [53]. Finally, one group reported that mice with myeloid cell-specific EGFR deletion with prior MI experienced improved survival outcomes upon a subsequent bacterial pneumonia challenge [27]. While the authors of this study did not characterize the impact of myeloid cell-specific EGFR deletion on post-MI cardiac function and remodeling, their results were notable since they demonstrated a selective enhancement of alveolar versus interstitial mf activation, which was associated with decreased expression of inflammatory cytokines and improved survival.
Collectively, these studies suggest that EGFR-dependent regulation of inflammation in myeloid subsets is not uniform and can result in distinct cell phenotypes and disease outcomes, the mechanistic underpinnings of which may be due to factors related to differences in EGFR expression, activation and signaling in these distinct tissue and disease environments. For instance, EGFR has been well-described as a signaling hub that can integrate inputs from several sources including a variety of extracellular ligands, many of which have been shown to differentially impact tissue development and disease [54,55]. EGFR ligands themselves can be differentially regulated via other signaling systems, for instance numerous G protein-coupled receptors (GPCRs) have been shown to regulate the extracellular cleavage of various EGFR pro-ligands to induce EGFR transactivation [56], thus the impact of EGFR on cardiac myeloid cell biology may differ from those in other tissues by virtue of their GPCR expression patterns. Additionally, EGFR can heterodimerize with other members of the ErbB receptor tyrosine kinase family, including ErbB2 that lacks a ligand and requires heterodimerization with other ligand-activated ErbB members to relay signals, and ErbB3, which lacks kinase activity, thus also requires heterodimerization to relay the effects of its own ligands, such as neuregulins, which have been shown to regulate myeloid cells [52,57]. The complex interplay between potential EGFR ligands, GPCR transactivators and ErbB heterodimerization partners within myeloid cell subtypes at various stages of injury and disease progression may therefore dictate the ultimate impact of EGFR on molecular outcomes in distinct tissue environments, such as regulation of neovascularization in the context of cardiac injury.
Indeed, the aforementioned studies had investigated the role of myeloid cell-specific EGFR primarily in the context of bacterial challenge and tumorigenesis, whereas the role of EGFR on baseline tissue resident mf function was not assessed. Via FACS analysis, we did not detect changes in mf quantities between control and mKO hearts, either at baseline or following injury. These findings indicate that myeloid cell-specific EGFR deletion does not lead to quantitative changes in cardiac mf populations at these timepoints, however our RNASeq data suggests qualitative changes in the properties of the cells that could contribute to enhanced stress at baseline. For instance, a majority of the up-regulated transcripts in mKO cardiac myeloid cells were associated with immune cell reactivity or inflammation, as evidenced by increased expression of inflammatory mf marker Ccr7(43). A potentially important transcript that was shown to be down-regulated in mKO cardiac myeloid cells by RNASeq, and confirmed via RT-qPCR, was Gja1 (Cx43), a gap junction protein involved in intercellular communication through the transfer of ions and other signaling molecules [58]. Mf-expressed Cx43 has been shown to allow their coupling with conducting cardiomyocytes within the AV node, deletion of which altered AV conduction [10]. While Cx43 expression was confirmed to be down-regulated in mKO cardiac myeloid cells, whether this relayed any changes in the electrophysiologic properties of the heart remains to be determined. We did not observe enhanced mortality of mKO mice over time, suggesting that altered Cx43 expression in these mice does not relay detrimental effects, at least under normal conditions. However, mKO mice exhibited slight cardiomyocyte hypertrophy and increased expression of fetal gene transcripts at baseline. The fact that these effects were not associated with overt pathological cardiac remodeling over time suggests this may be indicative of the promotion of an adaptive growth pathway, which would be of interest to follow up on, but wouldn’t explain the worsened cardiac function acutely following MI.
Both resident and infiltrating monocyte-derived mfs play important roles in the regulation of cardiac repair after injury [1–3,5–8], and in the days following acute ischemic injury, resident mf are largely cleared out [2]. Thus, circulating monocytes infiltrate injured hearts, providing an additional source of phagocytes to clear dead cells and debris, as well as differentiate into mf that over time adopt a reparative phenotype [5,8,59]. Notably, it was identified that improved systolic function in HF patients receiving an LVAD, as measured by EF, was associated with having a lower percentage of CCR2+ mf in the myocardium, both pre- and post-LVAD implantation [14]. In our study, mKO hearts experienced worse cardiac function following injury with a greater reduction in systolic function in comparison with either control group. Cardiac myeloid cells isolated from 7-day post-MI mKO mice exhibited higher expression of Il6, while transcripts associated with repair and neovascularization, including Il10 and Vegfa, were decreased. Ultimately, this coincided with a more rapid decrease in cardiac function and decreased revascularization post-MI in mice with myeloid cell-specific EGFR deletion.
While our study is the first to describe the impact of myeloid cell-specific EGFR deletion on cardiac parameters, there are two key limitations. First, we employed a model of constitutive LysM-Cre-mediated myeloid cell-specific EGFR deletion, which results in EGFR deletion in all cells that highly express Lyz2 [39]. As such, use of inducible Cre mouse models with promotors that are more selective for neutrophils, peripheral monocytes or mf would provide additional clarity in the myeloid cell subtypes that are primarily influenced by EGFR, and that could be controlled at different timepoints pre- and post-injury. For instance, because myeloid cell-specific EGFR deletion resulted in elevated stress in hearts before injury, it is important to understand how EGFR deletion only after injury may impact cardiac function and remodeling outcomes. Second, we concentrated cardiac CD11b+ cells before and after injury to perform RNAseq and/or RT-qPCR analysis specifically on myeloid cell populations, which at baseline was comprised of >90% mfs. While our FACS data were consistent with a lack of quantitative changes in mfs at baseline and following injury in mKO hearts, our RNASeq data revealed qualitative changes that could reflect subtle differences in cardiac resident mf subpopulations that promote cardiomyocyte hypertrophy or may be primed for promoting worse outcomes following injury. After injury, the cellular composition of cardiac myeloid cells is more diverse and although mf still made up a majority of the cardiac myeloid cells isolated 7 days post-MI, whether the alteration in reparative transcripts detected were exclusive to mfs is unclear. Thus, future work would benefit from single-cell RNASeq analysis to define the cellular composition of mKO hearts before and after injury for enhanced resolution of beneficial or detrimental changes in their populations.
In summary, using mice with myeloid cell-specific EGFR deletion, we have demonstrated that loss of EGFR qualitatively alters cardiac mfs, resulting in a small cardiomyocyte hypertrophy response and a predominant decrease in the expression of transcripts involved in neovascularization. Following acute cardiac injury, mice with myeloid cell-specific EGFR deletion accumulate mfs with decreased expression of reparative mediators, concurrent with a rapid decline in cardiac function and decreased reparative remodeling. These data support the concept that myeloid cell-specific EGFR-dependent signaling may be targeted early following injury to improve long-term cardiac remodeling outcomes.
Supplementary Material
Clinical perspectives.
Myeloid cells express an extensive repertoire of cell surface receptors that offer potential targets for manipulating their responsiveness to injury. EGFR can regulate myeloid cell behavior, but its influence on the response to cardiac injury is unknown.
We found that myeloid cell-specific EGFR deletion alters the cardiac macrophage transcriptome and, following acute myocardial infarction, myeloid cell-specific EGFR deletion enhances the accumulation of macrophages with less reparative markers, hastening cardiac dysfunction and diminishing neovascularization.
Targeting EGFR may offer a strategy to alter myeloid cell behavior to impart better outcomes following acute cardiac injury.
Funding
This work was supported by National Institutes of Health [grant numbers R01 HL139522 and P01 HL147841 (to D.G.T.), and F31 HL154814 (to A.D.O.)].
Abbreviations
- CCR2
c-c motif chemokine receptor 2
- CD
cluster of differentiation
- EF
ejection fraction
- EGFR
epidermal growth factor receptor
- IL
interleukin
- LV
left ventricle
- LysM
lysozyme 2
- Mf
macrophage
- MI
myocardial infarction
- VEGF
vascular endothelial growth factor
Footnotes
Competing Interests
All authors have read and approved the manuscript and have no conflicts of interest or relationships with industry to declare, except for current employment at Merck (EM), which is not relevant to this study.
Ethics Approval
This study was approved by the Institutional Animal Care and Use Committee (IACUC) at Temple University with Animal Protocols 4891 and 4902.
CRediT Author Contribution
Ama D. Okyere: Conceptualization, Formal analysis, Visualization, Methodology, Writing—original draft, Project administration. Tapas K. Nayak: Investigation. Viren Patwa: Investigation. David Teplitsky: Validation, Investigation. Erin McEachern: Validation. Rhonda L. Carter: Validation. Heli Xu: Investigation. Erhe Gao: Investigation, Methodology. Yan Zhou: Data curation, Formal analysis. Douglas G. Tilley: Conceptualization, Formal analysis, Supervision, Funding acquisition, Writing—review & editing.
Data Availability
RNA-seq data files were uploaded in GEO under accession number GSE207686. All other relevant data of the study are available from the corresponding author upon reasonable request.
References
- 1.Alvarez-Argote S and O’Meara CC (2021) The evolving roles of cardiac macrophages in homeostasis, regeneration, and repair. Int. J. Mol. Sci. 22 (15), 7923, 10.3390/ijms22157923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG et al. (2019) Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 20, 29–39, 10.1038/s41590-018-0272-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, Godwin J et al. (2018) The macrophage in cardiac homeostasis and disease: JACC Macrophage in CVD Series (Part 4). J. Am. Coll. Cardiol. 72, 2213–2230, 10.1016/j.jacc.2018.08.2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K et al. (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90, 10.1126/science.1219179 [DOI] [PubMed] [Google Scholar]
- 5.Swirski FK and Nahrendorf M (2018) Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 18, 733–744, 10.1038/s41577-018-0065-8 [DOI] [PubMed] [Google Scholar]
- 6.Williams JW, Giannarelli C, Rahman A, Randolph GJ and Kovacic JC (2018) Macrophage Biology, Classification, and Phenotype in Cardiovascular Disease: JACC Macrophage in CVD Series (Part 1). J. Am. Coll. Cardiol. 72, 2166–2180, 10.1016/j.jacc.2018.08.2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Y, Mouton AJ and Lindsey ML (2018) Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 191, 15–28, 10.1016/j.trsl.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prabhu SD and Frangogiannis NG (2016) The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ. Res. 119, 91–112, 10.1161/CIRCRESAHA.116.303577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cahill TJ, Sun X, Ravaud C, Villa Del Campo C, Klaourakis K, Lupu IE et al. (2021) Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development 148 (3), dev194563, 10.1242/dev.194563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A et al. (2017) Macrophages facilitate electrical conduction in the heart. Cell 169, 510e20–522e20, 10.1016/j.cell.2017.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE and Lavine KJ (2016) Primitive embryonic macrophages are required for coronary development and maturation. Circ. Res. 118, 1498–1511, 10.1161/CIRCRESAHA.115.308270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ et al. (2020) A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183, 94e23–109e23, 10.1016/j.cell.2020.08.031 [DOI] [PubMed] [Google Scholar]
- 13.Okyere AD and Tilley DG (2020) Leukocyte-dependent regulation of cardiac fibrosis. Front Physiol. 11, 301, 10.3389/fphys.2020.00301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I et al. (2019) Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ. Res. 124, 263–278, 10.1161/CIRCRESAHA.118.314028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Witherel CE, Abebayehu D, Barker TH and Spiller KL (2019) Macrophage and fibroblast interactions in biomaterial-mediated fibrosis. Adv. Healthc. Mater. 8, e1801451, 10.1002/adhm.201801451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peet C, Ivetic A, Bromage DI and Shah AM (2020) Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc. Res. 116, 1101–1112, 10.1093/cvr/cvz336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mosser DM and Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969, 10.1038/nri2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nayak TK and Tilley DG (2021) Recent advances in GPCR-regulated leukocyte responses during acute cardiac injury. Curr. Opin. Physiol. 19, 55–61, 10.1016/j.cophys.2020.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makki N, Thiel KW and Miller, Jr, F.J. (2013) The epidermal growth factor receptor and its ligands in cardiovascular disease. Int. J. Mol. Sci. 14, 20597–20613, 10.3390/ijms141020597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holbro T and Hynes NE (2004) ErbB receptors: directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 44, 195–217, 10.1146/annurev.pharmtox.44.101802.121440 [DOI] [PubMed] [Google Scholar]
- 21.Barrick CJ, Roberts RB, Rojas M, Rajamannan NM, Suitt CB, O’Brien KD et al. (2009) Reduced EGFR causes abnormal valvular differentiation leading to calcific aortic stenosis and left ventricular hypertrophy in C57BL/6J but not 129S1/SvImJ mice. Am. J. Physiol. Heart Circ. Physiol. 297, H65–H75, 10.1152/ajpheart.00866.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo S, Okyere AD, McEachern E, Strong JL, Carter RL, Patwa VC et al. (2022) Epidermal growth factor receptor-dependent maintenance of cardiac contractility. Cardiovasc. Res. 118, 1276–1288, 10.1093/cvr/cvab149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreier B, Rabe S, Schneider B, Bretschneider M, Rupp S, Ruhs S et al. (2013) Loss of epidermal growth factor receptor in vascular smooth muscle cells and cardiomyocytes causes arterial hypotension and cardiac hypertrophy. Hypertension 61, 333–340, 10.1161/HYPERTENSIONAHA.112.196543 [DOI] [PubMed] [Google Scholar]
- 24.Schreier B, Stern C, Dubourg V, Nolze A, Rabe S, Mildenberger S et al. (2021) Endothelial epidermal growth factor receptor is of minor importance for vascular and renal function and obesity-induced dysfunction in mice. Sci. Rep. 11, 7269, 10.1038/s41598-021-86587-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardbower DM, Coburn LA, Asim M, Singh K, Sierra JC, Barry DP et al. (2017) EGFR-mediated macrophage activation promotes colitis-associated tumorigenesis. Oncogene 36, 3807–3819, 10.1038/onc.2017.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardbower DM, Singh K, Asim M, Verriere TG, Olivares-Villagomez D, Barry DP et al. (2016) EGFR regulates macrophage activation and function in bacterial infection. J. Clin. Invest. 126, 3296–3312, 10.1172/JCI83585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoyer FF, Naxerova K, Schloss MJ, Hulsmans M, Nair AV, Dutta P et al. (2019) Tissue-specific macrophage responses to remote injury impact the outcome of subsequent local immune challenge. Immunity 51, 899e7–914e7, 10.1016/j.immuni.2019.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu N, Wang L, Cao H, Liu L, Van Kaer L, Washington MK et al. (2014) Activation of the epidermal growth factor receptor in macrophages regulates cytokine production and experimental colitis. J. Immunol. 192, 1013–1023, 10.4049/jimmunol.1300133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srivatsa S, Paul MC, Cardone C, Holcmann M, Amberg N, Pathria P et al. (2017) EGFR in tumor-associated myeloid cells promotes development of colorectal cancer in mice and associates with outcomes of patients. Gastroenterology 153, 178e10–190e10, 10.1053/j.gastro.2017.03.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao G, Liu L, Peek RM Jr., Hao X., Polk DB., Li H. et al. (2016) Activation of epidermal growth factor receptor in macrophages mediates feedback inhibition of M2 polarization and gastrointestinal tumor cell growth. J. Biol. Chem. 291, 20462–20472, 10.1074/jbc.M116.750182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee TC and Threadgill DW (2009) Generation and validation of mice carrying a conditional allele of the epidermal growth factor receptor. Genesis 47, 85–92, 10.1002/dvg.20464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clausen BE, Burkhardt C, Reith W, Renkawitz R and Forster I (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 8, 265–277, 10.1023/A:1008942828960 [DOI] [PubMed] [Google Scholar]
- 33.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, 10.1093/bioinformatics/bts635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anders S, Pyl PT and Huber W (2015) HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169, 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anders S and Huber W (2010) Differential expression analysis for sequence count data. Genome Biol. 11, R106, 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weischenfeldt J and Porse B (2008) Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc. 2008, pdb.prot5080, 10.1101/pdb.prot5080 [DOI] [PubMed] [Google Scholar]
- 37.Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M et al. (2010) A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 107, 1445–1453, 10.1161/CIRCRESAHA.110.223925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abram CL, Roberge GL, Hu Y and Lowell CA (2014) Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J. Immunol. Methods 408, 89–100, 10.1016/j.jim.2014.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi J, Hua L, Harmer D, Li P and Ren G (2018) Cre driver mice targeting macrophages. Methods Mol. Biol. 1784, 263–275, 10.1007/978-1-4939-7837-324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuchs T, Hahn M, Ries L, Giesler S, Busch S, Wang C et al. (2018) Expression of combinatorial immunoglobulins in macrophages in the tumor microenvironment. PLoS ONE 13, e0204108, 10.1371/journal.pone.0204108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S et al. (2019) Gut-associated IgA(+) immune cells regulate obesity-related insulin resistance. Nat. Commun. 10, 3650, 10.1038/s41467-019-11370-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B et al. (2014) Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104, 10.1016/j.immuni.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mueller PA, Zhu L, Tavori H, Huynh K, Giunzioni I, Stafford JM et al. (2018) Deletion of macrophage low-density lipoprotein receptor-related protein 1 (LRP1) accelerates atherosclerosis regression and increases C-C chemokine receptor type 7 (CCR7) expression in plaque macrophages. Circulation 138, 1850–1863, 10.1161/CIRCULATIONAHA.117.031702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yin C, Vrieze AM, Rosoga M, Akingbasote J, Pawlak EN, Jacob RA et al. (2020) Efferocytic defects in early atherosclerosis are driven by GATA2 overexpression in macrophages. Front Immunol. 11, 594136, 10.3389/fimmu.2020.594136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeboudj L, Giraud A, Guyonnet L, Zhang Y, Laurans L, Esposito B et al. (2018) Selective EGFR (epidermal growth factor receptor) deletion in myeloid cells limits atherosclerosis-brief report. Arterioscler. Thromb. Vasc. Biol. 38, 114–119, 10.1161/ATVBAHA.117.309927 [DOI] [PubMed] [Google Scholar]
- 46.Ferraro B, Leoni G, Hinkel R, Ormanns S, Paulin N, Ortega-Gomez A et al. (2019) Pro-Angiogenic macrophage phenotype to promote myocardial repair. J. Am. Coll. Cardiol. 73, 2990–3002, 10.1016/j.jacc.2019.03.503 [DOI] [PubMed] [Google Scholar]
- 47.Gombozhapova A, Rogovskaya Y, Shurupov V, Rebenkova M, Kzhyshkowska J, Popov SV et al. (2017) Macrophage activation and polarization in post-infarction cardiac remodeling. J. Biomed. Sci. 24, 13, 10.1186/s12929-017-0322-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Litvinukova M, Talavera-Lopez C, Maatz H, Reichart D, Worth CL, Lindberg EL et al. (2020) Cells of the adult human heart. Nature 588, 466–472, 10.1038/s41586-020-2797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi, Jr., K.C. et al. (2020) Transcriptional and cellular diversity of the human heart. Circulation 142, 466–482, 10.1161/CIRCULATIONAHA.119.045401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lafuse WP, Wozniak DJ and Rajaram MVS (2020) Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells 10 (1), 51, 10.3390/cells10010051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dick SA, Wong A, Hamidzada H, Nejat S, Nechanitzky R, Vohra S et al. (2022) Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci Immunol. 7, eabf7777, 10.1126/sciimmunol.abf7777 [DOI] [PubMed] [Google Scholar]
- 52.Yin H, Favreau-Lessard AJ, deKay JT, Herrmann YR, Robich MP, Koza RA et al. (2021) Protective role of ErbB3 signaling in myeloid cells during adaptation to cardiac pressure overload. J. Mol. Cell Cardiol. 152, 1–16, 10.1016/j.yjmcc.2020.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cao S, Pan Y, Tang J, Terker AS, Arroyo Ornelas JP, Jin GN et al. (2022) EGFR-mediated activation of adipose tissue macrophages promotes obesity and insulin resistance. Nat. Commun. 13, 4684, 10.1038/s41467-022-32348-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneider MR and Wolf E (2009) The epidermal growth factor receptor ligands at a glance. J. Cell. Physiol. 218, 460–466, 10.1002/jcp.21635 [DOI] [PubMed] [Google Scholar]
- 55.Harris RC, Chung E and Coffey RJ (2003) EGF receptor ligands. Exp. Cell. Res. 284, 2–13, 10.1016/S0014-4827(02)00105-2 [DOI] [PubMed] [Google Scholar]
- 56.Grisanti LA, Guo S and Tilley DG (2017) Cardiac GPCR-mediated EGFR transactivation: impact and therapeutic implications. J. Cardiovasc. Pharmacol. 70, 3–9, 10.1097/FJC.0000000000000462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ryzhov S, Matafonov A, Galindo CL, Zhang Q, Tran TL, Lenihan DJ et al. (2017) ERBB signaling attenuates proinflammatory activation of nonclassical monocytes. Am. J. Physiol. Heart Circ. Physiol. 312, H907–H918, 10.1152/ajpheart.00486.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rodjakovic D, Salm L and Beldi G (2021) Function of connexin-43 in macrophages. Int. J. Mol. Sci. 22 (3), 1412, 10.3390/ijms22031412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dehn S and Thorp EB (2018) Myeloid receptor CD36 is required for early phagocytosis of myocardial infarcts and induction of Nr4a1-dependent mechanisms of cardiac repair. FASEB J. 32, 254–264, 10.1096/fj.201700450r [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data files were uploaded in GEO under accession number GSE207686. All other relevant data of the study are available from the corresponding author upon reasonable request.