

Summary
Valvular heart disease presents a significant health burden, yet advancements in valve biology and therapeutics have been hindered by the lack of accessibility to human valve cells. In this study, we have developed a scalable and feeder-free method to differentiate human induced pluripotent stem cells (iPSCs) into endocardial cells, which are transcriptionally and phenotypically distinct from vascular endothelial cells. These endocardial cells can be challenged to undergo endothelial-to-mesenchymal transition (EndMT), after which two distinct populations emerge—one population undergoes EndMT to become valvular interstitial cells (VICs), while the other population reinforces their endothelial identity to become valvular endothelial cells (VECs). We then characterized these populations through bulk RNA-seq transcriptome analyses and compared our VIC and VEC populations to pseudobulk data generated from normal valve tissue of a 15-week-old human fetus. By increasing the accessibility to these cell populations, we aim to accelerate discoveries for cardiac valve biology and disease.
Subject areas: Biological sciences, Developmental biology, Cardiology, Stem cell research
Graphical abstract
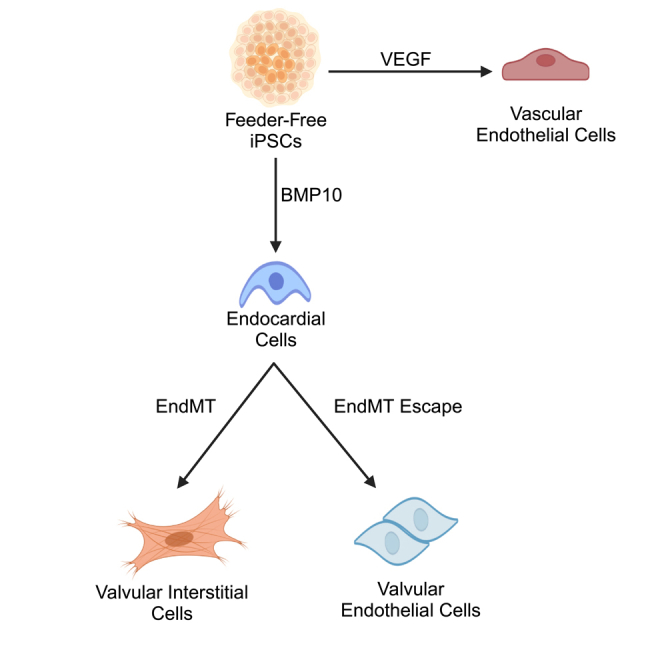
Highlights
-
•
Human iPSCs can be differentiated into endocardial cells with BMP10
-
•
Endocardial cells are distinct from VEGF-derived vascular endothelial cells
-
•
Endocardial cells undergo EndMT to form valvular interstitial cells
-
•
Some endocardial cells escape EndMT to become valvular endothelial cells
Biological sciences; Developmental biology; Cardiology; Stem cell research
Introduction
Valvular heart disease represents a significant health burden, with an estimated total U.S. prevalence of 2.5%.1 Additionally, aberrant development of cardiac valves is a frequent occurrence in congenital heart disease, representing 20%–30% of cases.2 Despite this high prevalence, valvular disease is primarily managed through surgical intervention, with limited pharmacological therapies available.3,4 A major barrier toward the development of targeted therapeutics for valvular disease is the lack of appropriate in vitro systems to model both developmental and pathological processes with human cells. One method of circumventing this barrier would be to develop a robust and scalable differentiation strategy by directing pluripotent stem cells (PSCs) toward normal cardiac valve developmental pathways.
Cardiac valvulogenesis is a complex process that involves numerous cell types and signaling pathways (reviewed extensively elsewhere, see2,5,6). Briefly, cardiac valvulogenesis begins after looping of the primitive heart tube, during which BMP signaling from the myocardium leads to increased extracellular matrix (ECM) deposition and swelling of the cardiac jelly at the atrioventricular canal and outflow tract.5,6,7,8 A subpopulation of the endocardial cells overlaying the cardiac jelly then undergoes endothelial-to-mesenchymal transition (EndMT) to become the putative valvular interstitial cells (VICs), migrating into the cardiac jelly to populate the developing endocardial cushion.8 Multiple signaling pathways have been implicated in promoting EndMT, including BMP2/4,7,9 TGFβ2,10 NOTCH,11,12 WNT/β-CATENIN,13 and HIPPO/YAP.14 Following EndMT, the endocardial cushions begin to elongate into valve leaflets—a process resulting from proliferation, apoptosis, and the secretion and remodeling of ECM proteins by the VICs.15,16,17 Notably, a subpopulation of endocardial cells escapes EndMT and continues to overlay the endocardial cushion to become valvular endothelial cells (VECs).6,18 Although other lineages, such as neural crest derivatives,19,20 epicardial cells,21,22,23 and hemogenic populations,24,25 also contribute to valve development, these are not the focus of the present study.
Given the central role of endocardium in valve development, generating an endocardial population from PSCs is of intense interest. Some previous studies have utilized VEGF to direct PSCs into an endothelial population, transitioning through a cardiac mesoderm stage26,27,28,29 or epicardial intermediate.30 Intriguingly, these endothelial populations express some endocardial markers and, in some cases, have the potential to undergo EndMT.26,29 However, recent work by Mikryukov et al. demonstrated that endocardial cells could be differentiated without the use of VEGF but rather via a combination of BMP10 and bFGF.31 In this strategy, PSCs were first maintained on mouse embryonic fibroblast feeder cells and then formed into embryoid bodies, prior to endocardial specification. Critically, this group demonstrated that this BMP10-induced endocardial population is transcriptionally and functionally distinct from VEGF-induced endothelial populations, has robust expression of endocardial markers, and maintains EndMT potential.
Feeder cells, such as mouse embryonic fibroblasts, are routinely used in PSC cultures to help support their pluripotency32,33 and have also been utilized to support a VEGF-directed strategy of differentiating endocardial-like cells.26 However, there are specific disadvantages to the use of feeder cells. In addition to increasing the technical difficulty of culturing PSCs, feeder cells are known to secrete various growth factors and cytokines, hindering the ability to precisely control the levels of signaling molecules that PSCs encounter. Incomplete removal and presence of residual feeder cells has also been shown to influence transcriptomic analysis of PSCs and can limit the suitability of differentiated cells for clinical purposes.34
To circumvent these limitations, we sought to develop a feeder-free, monolayer differentiation strategy incorporating GSK3 inhibition with CHIR-99021 (CHIR) that would be compatible with the BMP10-directed endocardial specification strategy. In this study, we utilized two wild-type human induced pluripotent stem cell (iPSC) lines to demonstrate that endocardial cells can be generated via CHIR in a feeder-free monolayer system. We confirmed that BMP10-induced endocardial cells are distinct from VEGF-induced endothelial cells and that these endocardial cells maintain their ability to undergo EndMT. Interestingly, we found that following an EndMT challenge, two distinct populations emerge and are representative of VICs and VECs. We then performed bulk RNA sequencing to further characterize our iPSC-derived populations and identified putative markers to distinguish them. The ability to generate these populations in a scalable fashion will facilitate future studies into human valvulogenesis and valvular disease.
Results
Monolayer differentiation of feeder-free iPSCs into endocardium
Since the identification of the utility of GSK3 inhibition in generating cardiomyocytes,35,36 the use of CHIR has been widely adapted in differentiating various mesoderm derivatives. To determine if this strategy would be compatible with producing endocardium, we utilized two wild-type human iPSC lines: WTC11 and MSN02-4. These iPSCs were grown on Matrigel without the use of feeder cells and subsequently directed to a mesoderm fate with 48 h of CHIR (8 μM). Afterward, we introduced BMP10 (10 ng/mL) and bFGF (50 ng/mL) over the next 12 days to generate a CD31+ (PECAM1) endocardial population. As a control, we also differentiated iPSCs toward a vascular endothelial fate with VEGF-A (100 ng/mL) and bFGF (50 ng/mL) (Figure 1A).
Figure 1.

Feeder-free generation of endocardial and endothelial populations from iPSCs
(A) Schematic of differentiation timeline.
(B) Flow cytometry plots assessing mesoderm generation on day 4 of their respective differentiation strategies.
(C) Quantification of CD13+/CD56+ mesoderm generation (n = 4, ∗p < 0.05, two-way ANOVA with Sidak’s multiple comparisons test, error bars ±SEM).
(D) Flow cytometry plots assessing endocardial/endothelial differentiation efficiency on day 14 (also see Figure S1).
(E) Timeline of CD31+ endocardial differentiation on days 10, 12, and 14 (n = 4, ∗p < 0.05 for WTC11, ##p < 0.01 for MSN02-4, two-way ANOVA with Dunnett’s multiple comparisons test, error bars ±SEM).
(F) Quantification of CD31+ endocardial efficiency on day 14 (n = 5, ∗p < 0.05, two-way ANOVA with Sidak’s multiple comparisons test, error bars ±SEM).
To determine the efficiency of mesoderm generation for each of the lines, we then dissociated differentiating iPSCs on day 4 and evaluated them for co-expression of CD13 and CD56 (Figures 1B and 1C). As CD56 has been identified as a marker of early mesoderm-committed populations37 and CD13 as an early pre-cardiac mesoderm marker,38 CD13+/CD56+ double-positive cells represent a mesoderm population capable of undergoing further cardiac cell-type specification. We found that both the endocardial and control endothelial differentiation conditions generated a robust CD13+/CD56+ population in the MSN02-4 line (∼60% yield). However, the WTC11 line had diminished CD13+/CD56+ specification under the endocardial protocol (∼40% yield), while the endothelial protocol performed similarly between both iPSC lines (∼60% yield).
We then assessed the induction of endocardial cells on days 10, 12, and 14 (Figure 1E). Over these time points, both the WTC11 and MSN02-4 lines demonstrated increased specification of an endocardial population, marked by cell surface expression of CD31. By day 14, both iPSC lines produced an endocardial population, with yield ranging from 15% to 30% (Figures 1D–1F). Additionally, we found that the endocardial population had heterogeneous expression of CD34, indicative of endocardial cells at varying levels of maturation. When we directed our iPSCs toward an endothelial population with VEGF-A, we generated a distinct CD31+/CD34+ population by day 14. Interestingly, the WTC11 line appeared to be less efficient at generating this endothelial population compared to the MSN02-4 line (∼12% vs. ∼25% yield).
BMP10-derived endocardial cells upregulate endocardial-specific genes
Following specification of both endocardial and endothelial cell populations on day 14, these populations were isolated using magnetic-activated cell sorting (MACS) for CD31 and CD34, respectively. This strategy allowed us to routinely isolate these populations to ∼95% or greater purity (Figures S1A and S1B). We then performed RT-qPCR on these day 14 populations to assess the expression of cardiac- and endocardial-specific genes (Figure 2A). Crucially, we found that for both the WTC11 and MSN02-4 lines, BMP10-derived endocardial cells upregulated expression of cardiac-specific transcription factors NKX2.5, GATA4, and GATA5 compared to the endothelial population. In addition, we found upregulation of endocardial genes ISL1, HAPLN1, NPR3, NRG1, MEIS2, and TMEM100 in our endocardial population. Interestingly, NFATc1 expression was detected at similar levels between both endocardial and endothelial populations.
Figure 2.

BMP10-derived endocardial cells are distinct from VEGF-derived endothelial cells
(A) RT-qPCR analysis of cardiac and endocardial gene expression on day 14 endocardial and endothelial cells (n = 4, ∗p < 0.05, ∗∗p < 0.01, two-way ANOVA, error bars ±SEM).
(B) Representative confocal microscopy of MSN02-4 endocardial and endothelial cells, stained for CD31 (GFP) and NKX2-5 (RFP). Scale bar, 50 μm.
(C) Representative confocal microscopy of MSN02-4 endocardial and endothelial cells, stained for NFATc1 (GFP) and CDH5 (RFP) (also see Figure S2). White arrowheads = diffuse NFATc1 in the cytoplasm of endothelial cells. Scale bar, 50 μm.
Next, we performed confocal microscopy on endocardial and endothelial cells. Both endocardial and endothelial populations had expression of CD31, localized to the plasma membrane. Notably, only endocardial cells had co-expression of CD31 and NKX2.5, while NKX2.5 was undetectable in the endothelial cells (Figure 2B). Although we detected NFATc1 in both the endocardial and endothelial cells, we found that NFATc1 was predominantly localized in the nucleus in endocardial cells while the NFATc1 staining pattern was more diffused in the endothelial cells (Figure 2C). Lastly, we showed that both the endocardial and endothelial populations co-localized CD31 and CDH5 (VE-CADHERIN) to the plasma membrane (Figure S2).
EndMT challenge of endocardial cells generates valvular interstitial cells and valvular endothelial cells
After sorting out the endocardial population on day 14 with MACS, we then plated the endocardial cells on fibronectin and induced EndMT with a combination of BMP2 (100 ng/mL), TGFβ2 (0.3 ng/mL), and bFGF (10 ng/mL) (Figure 1A). Following 6–7 days of this EndMT condition, we found that a subpopulation of the endocardial cells lost CD31 expression to become VICs and began to express the mesenchymal surface marker PDGFRβ. Additionally, we noticed a population that consistently escaped the EndMT process and maintained expression of CD31 (Figures 3A and 3B). We hypothesized that this population, having been challenged with EndMT conditions and subsequently failing to undergo EndMT, represents VECs—consistent with in vivo observations that only a subset of endocardial cells overlaying the endocardial cushion undergo EndMT.39,40 Notably, we found that our control endothelial population was unable to survive this EndMT process, with nearly 100% cell death observed by the end of the EndMT period (data not shown).
Figure 3.

EndMT of endocardial cells generates VIC and VEC populations
(A) Flow cytometry plots following EndMT (also see Figure S1).
(B) Quantification of CD31− VIC and CD31+ VEC populations following EndMT (n = 4, error bars ±SEM).
(C) RT-qPCR analysis of mesenchymal and endothelial gene expression on day 14 endocardial cells and day 20/21 VICs/VECs (n = 4, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, two-way ANOVA with Tukey’s multiple comparisons test, error bars ±SEM).
By utilizing our CD31 MACS strategy, we were then able to sort the corresponding CD31− VICs from CD31+ VECs (Figures S1C and S1D). RT-qPCR analysis on the starting day 14 endocardial cells and the post-EndMT day 20/21 VIC/VEC populations revealed that VICs strongly upregulated mesenchymal-specific genes, such as SOX9, N-CAD, CDH11, VIM, TGFβ2, COL1A1, COL3A1, and POSTN. Furthermore, the VICs lost expression of endothelial genes CD31 and CDH5 (Figure 3C). Interestingly, we found that, following the EndMT challenge, VECs reinforced their endothelial identity and upregulated endothelial genes PECAM1 and CDH5 compared to day 14 endocardial cells.
Next, we performed confocal microscopy on our sorted VICs and VECs. Notably, we found that VICs became morphologically distinct from their initial endocardial population; in addition to having larger cell sizes, these cells displayed a more elongated morphology (Figures 4A and 4B). Furthermore, we found that the VICs had robust staining of mesenchymal markers PERIOSTIN, VIMENTIN, αSMA, and SOX9. In contrast to the VIC morphology, VECs exhibited typical cuboidal endothelial morphology with localization of endothelial proteins CD31 and CDH5 at the plasma membrane (Figure 4C).
Figure 4.

Immunofluorescence analysis of VICs and VECs
(A) Representative confocal microscopy of WTC11 VICs stained for PERIOSTIN (GFP) and VIMENTIN (RFP).
(B) Representative confocal microscopy of MSN02-4 VICs stained for αSMA (GFP) and SOX9 (RFP).
(C) Representative confocal microscopy of WTC11 VECs stained for CDH5 (GFP) and CD31 (RFP).
Transcriptional characterization of iPSC-derived endocardial and endothelial cells
To further validate each of our respective cell types and identify potential markers, we conducted bulk RNA sequencing on day 14 endocardial and endothelial cells, as well as our D20/21 VICs and VECs. Results from principal component analysis (PCA) showed that the two iPSC lines for each cell type grouped together, with the first two principal components (PC1 and PC2) accounting for 58% and 17% of the variance, respectively (Figure S3A). Importantly, VICs were the most distinct from the control endothelial cells, separated primarily by PC1. We also conducted differential gene expression analysis and performed unsupervised hierarchical clustering on the upper quartile of genes ranked by variance. This similarly revealed that each cell type grouped together, with all endothelial cell types clustering together, and the two cardiac endothelial subtypes (endocardial and VECs) clustering closer than the control endothelial population (Figure S3B).
Next, we assessed the top differentially expressed genes (DEGs) between our endocardial and endothelial populations. Notably, we found that genes upregulated in the endocardial cells include the cardiac-specific transcription factors NKX2.5, GATA4, GATA5, HAND2, and TBX20 (Figures 5A and 5B). Many genes that have been recently proposed to be endocardial markers were also strongly upregulated in our endocardial cells, such as TK1,41 CYTL1,42 TMEM108,42 and SGK1.31 Cell cycle genes TOP2A, UBE2C, and CDC20 were also found to be upregulated, which have been reported to be specifically enriched in embryonic endocardium compared to postnatal and adult endocardium,42 consistent with the fetal-like phenotype of most iPSC-derived cell types. Gene ontology (GO) analysis of upregulated DEGs in the endocardial cells supported an increase in proliferation in our endocardial cells, with numerous top terms related to mitosis, such as “mitotic spindle assembly checkpoint signaling” and “microtubule cytoskeleton organization involved in mitosis.” Other intriguing terms involve extracellular matrix development, such as “collagen fibril organization” and “extracellular matrix organization,” indicating an early developmental role of endocardial cells in the production of matrix within the cardiac jelly and endocardial cushion (Figure 5C).
Figure 5.

Bulk RNA-seq characterization of iPSC-derived endocardial and endothelial cells (also see Figure S3 and File S1)
(A) Heatmap of the top 50 DEGs ranked by variance between endocardial and endothelial populations.
(B) Volcano plot of DEGs between endocardial vs. endothelial populations. Cutoff for log2 fold change was set to 1 and -1; padj < 10−3.
(C) Top 10 biological process GO terms for upregulated DEGs in endocardial cells, ranked by p value.
(D) Top 10 biological process GO terms for upregulated DEGs in endothelial cells, ranked by p value.
In contrast, analysis of endothelial cell DEGs revealed upregulation of known endothelial or blood vessel development genes, such as CXCR4,43 ESM1,44 ANGPT2,45 FLT1,46 and NOTCH447 (Figures 5A and 5B). Accordingly, GO term analysis of upregulated endothelial DEGs included “regulation of angiogenesis,” “endothelial cell development,” and “establishment of endothelial barrier,” confirming the vascular endothelial identity of our differentiated endothelial cells (Figure 5D).
Transcriptional characterization of iPSC-derived valve cells
We then analyzed DEGs between our VICs and endocardial cells, which revealed upregulation of extracellular matrix genes, including COL1A1, COL3A1, COL8A1, COL12A1, FN1, FBN1, MMP10, ITGA4, and ADAMTS16, consistent with the primary function of VICs to secrete and remodel the extracellular matrix of the valve leaflet. Additionally, we found upregulation of genes known to be involved in valve development and VIC function, such as SOX9,41,48,49 CXCL12,50,51 INHBA,41 LOXL1,52,53 PTX3,54 BMP6,55 ERBB2,56 and ACTA257,58 (Figures 6A and 6B). GO analysis on upregulated VIC DEGs revealed numerous terms involving the extracellular matrix, including “extracellular matrix organization,” and “collagen fibril organization” (Figure 6C). Although not in the top 10, other notable GO terms included processes known to be involved in valve development, such as “positive regulation of apoptotic process” (p = 4.9 x 10−6), “cell-matrix adhesion” (p = 7.8 x 10−6), “regulation of epithelial to mesenchymal transition” (p = 1x10−6), and “glycosaminoglycan biosynthetic process” (p = 1.2 x 10−6).
Figure 6.

Bulk RNA-seq characterization of iPSC-derived VIC and VECs (also see Figure S4 and File S1)
(A) Heatmap of top 50 DEGs ranked by variance between VIC and endocardial populations.
(B) Volcano plot of DEGs between VIC vs. endocardial populations. Cutoffs for log2 fold change was set to 1 and -1; padj < 10−3.
(C) Top 10 biological process GO terms for upregulated DEGs in VICs, ranked by p value.
(D) Heatmap of top 50 DEGs ranked by variance between VEC, endocardial, and endothelial populations.
(E) Volcano plot of DEGs between VECs vs. endocardial populations. Cutoff for log2 fold change was set to 1 and -1; padj < 10−3.
(F) Top 10 biological process GO terms for upregulated DEGs in VECs, ranked by p value.
Next, we characterized our VECs by comparing its gene expression pattern with the other endothelial cell types present in our dataset—endocardial and vascular endothelial cells. By looking at the most variable genes between these three populations, we found a subset of genes that were uniquely upregulated in our VECs: PALMD, EFEMP1, GJA5, IL33, and SULF1 (Figure 6D). In addition, DEG analysis between VECs and endocardial cells identified numerous upregulated genes that have been recently proposed to be VEC-specific markers, such as PALMD,41 AQP1,41 CD44,29 CD55,53 KFL9,29 VCAM1,29 and DKK2/329,53 (Figures 6D and 6E). Other genes that were upregulated included CXCL12,50,51 BMP6,55 PRND,59 and ENFA1,59 which have specific roles in valve development and VEC function (Figure 6E). GO term analysis of upregulated VEC DEGs identified endothelial-specific terms, such as “regulation of angiogenesis” and “positive regulation of vasculature development.” Notably, other terms such as “positive regulation of fibroblast proliferation” and “positive regulation of cell motility” suggest that our VECs may support or regulate VIC function through paracrine signals.
We then performed DEG analysis between VECs and VICs to increase the contrast between comparisons. This further confirmed the endothelial identity of VECs with upregulation of endothelial markers SOX17, CDH5, TIE1, FLT4, CD34, NOTCH4, and CXCR4 (Figure S4A). With these DEGs, we queried BioPlanet GO terms to dissect differential signaling pathways associated with VECs and VICs. We found that the most significant terms associated with VECs involve the VEGF, insulin, NGF, EGF, PDGFB, and MAPK signaling pathways (Figure S4B). In contrast, signaling pathways in VICs included TGFb, BDNF, and integrin signaling pathways (Figure S4C). Lastly, we analyzed CXCL12, INHBA, and BMP6 expression between VICs and VECs, as these were found to be upregulated in both populations relative to endocardial cells. We found that CXCL12 and INHBA were much more upregulated in the VICs, while VECs had high levels of CXCR4 expression. Interestingly, BMP6 was not differentially expressed between VICs and VECs (Figure S4D).
Comparison of iPSC-derived valve cells to 15-week human fetal valve cells
In order to confirm the cell identity of our iPSC-derived VICs and VECs, we sought to compare our cells to ones from human fetal cardiac valves. To do so, we leveraged a recently curated single-cell RNA-seq dataset that sequenced micro-dissected valves from a normal 15-week male human fetus.53 In order to compare our bulk RNA-seq dataset with this single-cell RNA-seq dataset, we created a pseudobulk expression profile that averages the raw counts across the VICs and VECs. After performing batch correction, normalization, and filtering, we analyzed the degree of concordance between our iPSC-derived populations with these primary cells. We found that PCA separated VICs and VECs by PC1, comprising 85% of the variance (Figure 7A). Pearson correlation analysis with unsupervised clustering revealed that our iPSC-derived VICs clustered closely with the 15-week fetal VICs, while our iPSC-derived VECs clustered with 15-week fetal VECs. Notably, the degree of correlation between our differentiated cells and the primary cells within each cell type was >90% (Figure 7B). Lastly, we performed unsupervised clustering on the top quartile of genes ranked by variance. This further confirmed a high degree of similarity between our iPSC-derived VICs and VECs with those from a 15-week human fetus, with iPSC-derived populations clustering with their respective primary cell population (Figure 7C).
Figure 7.

Comparison of iPSC-derived VICs and VECs to human fetal heart valve cells
(A) PCA plot of iPSC-derived VICs, VECs, and human fetal heart valve cells from a normal 15-week-old fetus (labeled Liu_W15).
(B) Pearson correlation matrix comparing iPSC-derived VICs and VECs to their respective human fetal valve populations.
(C) Heatmap with unsupervised clustering on the top quartile of genes sorted by variance, showing human fetal valve VICs and VECs clustering with their respective iPSC-derived populations.
Collectively, these data support the differentiation of iPSCs into an endocardial population with BMP10 that are distinct from VEGF-derived endothelial cells, and that these endocardial cells can then undergo an EndMT challenge to generate both VICs and VECs.
Discussion
The ability for iPSCs to differentiate into various cell types has allowed researchers to further their understanding of development and the biological mechanisms that govern healthy and disease states. Expanding the repertoire of cell types that can be differentiated from iPSCs will only further these goals. In the present study, we developed a protocol that allowed us to differentiate iPSCs into endocardial cells as a monolayer, without requiring the presence of feeder cells or the formation of embryoid bodies in suspension. We then demonstrate that these endocardial cells could be challenged to undergo EndMT, resulting in the segregation of two distinct populations—VICs and VECs. We also provide robust transcriptional characterization of these iPSC-derived populations and provide evidence that these differentiated VICs and VECs closely resemble their respective populations in a normal human fetal heart.
Our protocol was validated using two wild-type iPSC lines. We found that CHIR alone was sufficient to induce mesoderm specification in iPSCs cultured as a monolayer and that the efficiency of our mesoderm specification under the endocardial condition differed between our two wild-type iPSC lines (Figure 1C). Notably, we have found that this efficiency can be improved by titrating the CHIR concentration and that optimizing this step can increase the efficiency of endocardial generation by day 14 (data not shown). These differences suggest that the genetic background can influence iPSC response to CHIR and that optimal CHIR concentrations should be determined for each potential cell line.
Following specification of mesoderm, we promoted endocardial differentiation with BMP10 and bFGF and found that key cardiac transcription factors, such as NKX2.5, GATA4, and GATA5, were upregulated in our endocardial population compared to VEGF-derived endothelial cells (Figure 2A). These results are in keeping with the findings from Mikryukov et al. that BMP10-derived endocardial cells are transcriptionally and phenotypically distinct from VEGF-derived endothelial cells.31 Interestingly, we found that NFATc1 was expressed in both of our endocardial and endothelial populations and that it was predominantly localized in the nucleus within the endocardial population, while endothelial cells had more diffuse localization of NFATc1 throughout the cell (Figure 2C). This is consistent with prior work identifying NFATc1 as one of the earliest markers of the endocardium, as its expression in E8.5 mice is restricted to the heart tube endocardium and becomes specific to the cushion endocardium between E9.5 to E11.5.60,61 In this context, NFATc1 is found in the nucleus and functions to regulate EndMT.39 In endothelial cells, however, NFATc1 expression has also been described, where it functions to regulate angiogenesis genes62 and is also involved in the development and patterning of lymphatic endothelial cells.63 This suggests that NFATc1 specificity to the endocardium may be temporally restricted and that NFATc1 levels may be reduced or absent at earlier time points for our endothelial cells.
After EndMT of our endocardial cells, we identified and characterized two distinct populations that represent VICs and VECs. Multiple lines of evidence support the establishment of these cell types. First, our transcriptomic data confirmed the divergence of endocardial cells from endothelial cells, with upregulation of numerous cardiac and endocardial-specific markers in our endocardial population (Figure 5). Given the normal developmental trajectory for valvulogenesis, the populations derived from EndMT of the endocardial cells should represent cardiac valve populations. Indeed, our transcriptional and immunofluorescence analyses confirmed that VICs are of a mesenchymal identity and are involved in extracellular matrix production and organization, including production of both collagen and glycosaminoglycans (Figures 3C, 4A, 4B, and 6A–6C). In addition to upregulation of ECM genes, we also identify numerous genes thought to be markers of VICs and function during valve development (Figures 6A–6C).
Furthermore, transcriptional and immunofluorescence analyses revealed that VECs reinforced their endothelial identity after being challenged with EndMT, with upregulation of endothelial markers compared to their endocardial populations (Figures 3C, 4C, and S4). By comparing our transcriptomic data for VECs with endocardial and endothelial populations, we also identified PALMD, EFEMP1, GJA5, IL33, and SULF1 as genes uniquely upregulated in VECs, which may serve as VEC-specific markers (Figure 6D). To the best of our knowledge, the function of these genes within VECs has yet to be explored. Our analysis also revealed a number of upregulated genes that have been recently proposed to be VEC markers, further supporting the VEC phenotype of our differentiated cells. Notably, we found that KLF9, PRND, and ENFA1 to be upregulated in our VECs—genes regulated by shear stress. For example, KLF9 has been shown to be upregulated specifically in VECs from the aortic side of the aortic valve29—the side of the valve that experiences turbulent rather than laminar flow. In addition, laminar shear stress on aortic VECs has been shown to upregulate miR-486-5p, resulting in inhibition of PRND and ENFA1 transcription.59 As our cell cultures were maintained in static fluid conditions and did not include shear stress or fluid flow, the upregulation of these genes in our VECs is consistent with known VEC biology.
We also sought to validate our iPSC-derived VICs and VECs by comparing their transcriptional profile with that of VICs and VECs obtained from a normal 15-week-old human fetus. Through PCA, Pearson’s correlation analysis, and unsupervised clustering on the top quartile of genes sorted by variance, we find that our VICs and VECs cluster with their respective primary cell population. Some of the variance from this analysis can be explained by differences in sequencing depth (bulk RNA-seq vs. single-cell RNA-seq-derived pseudobulk) and the fact that iPSC-derived populations are thought to be fetal-like, perhaps even earlier than week 15 of human development. Despite these confounders, we find >90% similarity between our iPSC-derived valve cells to their primary cells, consistent with our interpretation that a subset of endocardial cells undergoes EndMT to become VICs, while the others escape EndMT and reinforce their endothelial identity to become VECs. How endocardial cells decide to enter EndMT or not remains an open question in the field, but is thought to be regulated through NFATc1 expression and its subcellular localization.18,39,40 Our in vitro iPSC model could be particularly well suited to explore such questions regarding EndMT mechanisms during heart development.
Our transcriptomic analysis also identified genes that appeared to be upregulated in both VICs and VECs, relative to endocardial cells, such as CXCL12, VGLL3, BMP6, and INHBA (Figures 6 and S4D). These genes may serve as transcriptional markers that distinguish cardiac valve cells from other endothelial and mesenchymal populations. CXCL12 is known to be important in valve development, as Cxcl12-null mice develop dysplastic and misaligned semilunar valves.50 CXCL12 signaling has also been shown to influence positioning and orientation of CXCR4+ VECs undergoing EndMT, along with regulation of VIC proliferation during remodeling and maturation of the valve leaflet.50 Intriguingly, we find that CXCL12 is expressed at higher levels within VICs compared to VECs, while VECs have much higher levels of CXCR4 (Figure S4D). This suggests that while VECs contribute some CXCL12 ligand, VICs actually express much more of the ligand within the endocardial cushion to regulate the progression of EndMT (Figure S4D). Additionally, upregulation of VGLL3 is particularly intriguing as its function in valve development, to the best of our knowledge, is unexplored. However, in other contexts, it is thought to activate HIPPO signaling and regulate YAP activity by competing for TEAD binding.64 Notably, the function of Vgll4 has recently been explored in mice, where knockout of Vgll4 led to semilunar valve thickening—a phenotype that was rescued with partial knockout of Yap.65 As YAP also responds differentially to changes in mechanical force and regulates valve growth and remodeling,66 it would be worth investigating the influence of VGLL3 on these processes and how its function in this context differs from VGLL4.
In summary, our study presents a simplified protocol for differentiating endocardial and valve cell populations from iPSCs grown in a feeder-free monolayer format. This allows for an easily scalable approach that decreases the technical challenges of other protocols, while simultaneously providing an unlimited number of cells to investigate endocardial and valve biology. As iPSC-derived cell populations are generally considered to be fetal-like, our protocol would be particularly advantageous for modeling congenital valve disease. Furthermore, the transcriptomic dataset we have generated is vast, with many differentially expressed genes that have yet to be thoroughly investigated within the context of valve biology and disease (see File S1). With the development of this protocol, we hope to expand access to these human cell populations and promote the advancement of discoveries in cardiac valve biology and disease.
Limitations of the study
Our study was limited to bulk RNA-seq analysis, precluding the identification of subpopulations within each of cell types. To address this, future directions include performing single-cell RNA sequencing to explore the heterogeneity of our populations. Additionally, we only cultured our cells in 2D without the influence of mechanical forces. Future work can include the generation of tissues with these cells and examining the effects of mechanical stimuli on signaling between cell populations. Lastly, iPSC-derived cells are fetal-like, and methods to mature the valve cells generated here may need to be developed in order to recapitulate adult forms of valve biology and disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal to CD31 (clone WM59), APC | ThermoFisher | Cat. #17-0319-42; RRID: AB_10852842 |
| Mouse monoclonal to CD31 (clone JC/70A) | ThermoFisher | Cat. #MA5-13188; RRID: AB_10982120 |
| Mouse monoclonal to NFATC1 (clone 7A6) | ThermoFisher | Cat. #MA3-024; RRID: AB_2236037 |
| Rabbit monoclonal to VIMENTIN (clone SP20) | ThermoFisher | Cat. #MA5-16409; RRID: AB_2537928 |
| Mouse monoclonal to CD34 (clone 561), PE-Cy7 | BioLegend | Cat. #343616; RRID: AB_2629726 |
| Mouse monoclonal to CD56 (clone 5.1H11), APC | BioLegend | Cat. #362504; RRID: AB_2563913 |
| Mouse monoclonal to CD13 (clone WM15), PE | BioLegend | Cat. #301704; RRID: AB_314180 |
| Rabbit monoclonal to NKX2.5 (clone E1Y8H) | Cell Signaling Technology | Cat. #8792; RRID: AB_2797667 |
| Rabbit polyclonal to VE-CADHERIN | Cell Signaling Technology | Cat. #2158; RRID: AB_2077970 |
| Mouse monoclonal to PERIOSTIN (clone F-10) | Santa Cruz Biotechnology | Cat. #sc-398631; RRID: AB_2934053 |
| Rabbit polyclonal to SOX9 | MilliporeSigma | Cat. #AB5535; RRID: AB_2239761 |
| Mouse monoclonal to αSMA (Clone 1A4) | Abcam | Cat. #ab7817; RRID: AB_262054 |
| Mouse monoclonal to PDGFRβ (clone 28D4), BV786 | BD Biosciences | Cat. #743038; RRID: AB_2741234 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher | Cat. #A-1100; RRID: AB_2534069 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher | Cat. #A-11008; RRID: AB_143165 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | ThermoFisher | Cat. #A-11004; RRID: AB_2534072 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | ThermoFisher | Cat. #A-11011; RRID: AB_143157 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human BMP10 | R&D | Cat. #2926-BP |
| Recombinant Human bFGF | R&D | Cat. #233-FB |
| Recombinant Human VEGF-165 | R&D | Cat. #293-VE |
| Recombinant Human/Mouse/Rat BMP2 | R&D | Cat. #355-BM |
| Recombinant Human TGF-beta 2 | R&D | Cat. #302-B2 |
| Human Apo-Transferrin | R&D | Cat. #3188-AT |
| Penicillin/streptomycin | ThermoFisher | Cat. #15140122 |
| GlutaMAX | ThermoFisher | Cat. #35050061 |
| Bovine Serum Albumin Fraction V (BSA) | ThermoFisher | Cat. #15260037 |
| Ascorbic Acid | MilliporeSigma | Cat. #A4544 |
| 2-Mercaptoethanol | MilliporeSigma | Cat. #M3148 |
| DAPI | MilliporeSigma | Cat. #9564 |
| Fetal Bovine Serum (FBS) | MilliporeSigma | Cat. #F4135 |
| Fibronectin Human Plasma | MilliporeSigma | Cat. #F0895 |
| Triton X-100 | MilliporeSigma | Cat. #93418 |
| Sodium Azide | MilliporeSigma | Cat. #S2002 |
| Tween-20 | MilliporeSigma | Cat. #P1379 |
| DNase I | MilliporeSigma | Cat. # 260913 |
| DMSO | MilliporeSigma | Cat. #D2650 |
| 4% Paraformaldehyde | ThermoFisher | Cat. # J19943K2 |
| CHIR 99021 | Tocris | Cat. #4423 |
| Collagenase Type II | Worthington | Cat. #LS004176 |
| Thiazovivin | Selleckchem | Cat. #S1459 |
| Matrigel, Growth Factor Reduced | Corning | Cat. #354230 |
| HEPES | Research Products International | Cat. #H75050 |
| Glycine | Bio-Rad | Cat. #1610718 |
| Proteinase K | Qiagen | Cat. #19131 |
| Accutase | Accutase | Cat. #AT104 |
| Critical commercial assays | ||
| Superscript IV VILO | ThermoFisher | Cat. #11756050 |
| PowerTrack SYBR Green | ThermoFisher | Cat. #A46012 |
| RNA 6000 Nano Kit | Agilent | Cat. # 5067-1511 |
| CD31 MicroBead Kit | Miltenyi Biotec | Cat. #130-091-935 |
| CD34 MicroBead Kit | Miltenyi Biotec | Cat. #130-046-702 |
| Deposited data | ||
| Raw & processed data from iPSC-derived populations | This study | GEO: GSE226476 |
| 15-week-old fetal human valve single-cell RNAseq data | Liu et al., 202353 | GEO: GSE228638 |
| Experimental models: Cell lines | ||
| WTC11 (ACTN2-eGFP tagged, monoallelic) | Coriell Institute | Cat. #AICS-0075-085 |
| MSN02-452,53 | Gift from Dr. Dubois | N/A |
| Oligonucleotides | ||
| See Table S1 | This study | Table S1 |
| Software and algorithms | ||
| FCS Express 7 | De Novo Software | https://denovosoftware.com/ |
| GraphPad Prism | GraphPad | https://www.graphpad.com/ |
| R CRAN, Version 4.2.0 | https://cran.r-project.org/ | |
| STAR v.2.7.10a54 | https://github.com/alexdobin/STAR | |
| Picard v2.26.10 | http://broadinstitute.github.io/picard/ | |
| HTSeq v2.0.255 | https://htseq.readthedocs.io/en/master/ | |
| DESeq256 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | |
| Enrichr58 | https://maayanlab.cloud/Enrichr/ | |
| Seurat v467 | https://satijalab.org/seurat/ | |
| FIJI | https://imagej.net/software/fiji/ | |
| Other | ||
| RPMI 1640 | ThermoFisher | Cat. #11875093 |
| B27 Supplement | ThermoFisher | Cat. #17504044 |
| B27 Supplement, Minus Insulinf | ThermoFisher | Cat. #A1895601 |
| DMEM, high glucose | ThermoFisher | Cat. #11965092 |
| PBS, no calcium, no magnesium | ThermoFisher | Cat. #14190144 |
| 6-well clear flat bottom TC-treated culture plates | Fisher Scientific | Cat. #FB012927 |
| 24-well clear flat bottom TC-treated culture plates | Fisher Scientific | Cat. #FB012929 |
| 40um cell strainer | Fisher Scientific | Cat. #22363547 |
| Fibroblast Growth Medium, Kit | PromoCell | Cat. #C-23110 |
| Endothelial Cell Growth Medium MV2, Kit | PromoCell | Cat. #C-22121 |
| HBSS | Corning | Cat. #21022CV |
| EasySep Buffer | StemCell Technologies | Cat. #20144 |
| 12-well chamber slide | Ibidi | Cat. #81201 |
| Coverslips | Ibidi | Cat. #10811 |
| VECTASHIELD PLUS Antifade Mounting Medium | Vector Laboratories | Cat. #H-1900 |
| LS Column | Miltenyi Biotec | Cat. #130-042-401 |
| Cell Counting Slides | Bio-Rad | Cat. #1450015 |
| Trypan Blue | Bio-Rad | Cat. #1450021 |
| Human Reference cDNA | Takara | Cat. #636693 |
Resource availability
Lead contact
Additional information or requests for reagents should be directed to the lead contact, Dr. Bruce Gelb (bruce.gelb@mssm.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Data generated from the bulk RNA-sequencing were uploaded to GEO and can be accessed with accession number GEO: GSE226476.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Culture and maintenance of human iPSCs
Two wild-type human iPSC lines were utilized: WTC11 (XY, mono-allelic ACTN2-mEGFP, obtained from Coriell Institute #AICS-0075-085) and MSN02-4 (XX, generated from skin biopsy of a healthy 36-year-old at Icahn School of Medicine at Mount Sinai).68,69 The iPSC lines were maintained in 6-well plates coated with a thin layer of Matrigel (6.6% v/v, Corning), diluted in DMEM (Gibco) and supplemented with penicillin/streptomycin (5%, ThermoFisher). iPSCs were maintained and fed daily with iPSC culture medium consisting of mTesR Plus (Stem Cell Technologies) supplemented with penicillin/streptomycin (1%, ThermoFisher). iPSCs were passaged every 3 to 4 days by first dissociating to single cells with Accutase (Innovative Cell Technologies, Inc) and then replating at a 1:10 ratio with iPSC culture medium, supplemented with Thiazovivin (2 μM, Selleckchem) for the first 24 h. iPSC cultures were incubated at 37°C in a normoxic environment (5% CO2). iPSCs were visually authenticated daily to ensure proper stem cell morphology without presence of spontaneous differentiation. Mycoplasma contamination was checked quarterly via PCR (ATCC).
Method details
Directed monolayer differentiation of iPSCs into endocardial & endothelial cells
Prior to the start of differentiation, iPSCs were passaged onto Matrigel coated 24-well plates at a ratio of 1:12. Day 0 (start of differentiation) was begun when iPSCs had reached ∼80-90% confluency. Each well was washed with DMEM (Gibco), followed by a media change into phase 1 media consisting of RPMI 1640 (Gibco) supplemented with B27 minus insulin supplement (0.5x, Gibco), penicillin/streptomycin (1%, ThermoFisher), GlutaMAX (2 mM, Gibco), ascorbic acid (50 μg/mL, Sigma), apo-transferrin (150 μg/mL, R&D Systems), and monothioglycerol (50 μg/mL, Sigma). CHIR-99021 (8 μM, Tocris) was added into phase 1 media for Day 0 of differentiation. For endocardial differentiation, media was changed after 48 h to phase 1 media supplemented with BMP10 (10 ng/mL, R&D Systems) and bFGF (50 ng/mL, R&D Systems). On Day 6, media was changed into phase 2 media consisting of RPMI 1640 (Gibco) supplemented with B27 supplement (0.5x, Gibco), penicillin/streptomycin (1%, ThermoFisher), Glutamax (2 mM, Gibco), ascorbic acid (50 μg/mL, Sigma), apo-transferrin (150 μg/mL, R&D Systems), and monothioglycerol (50 μg/mL, Sigma). Phase 2 media was further supplemented with BMP10 (10 ng/mL, R&D Systems) and bFGF (50 ng/mL, R&D Systems) for media changes between Days 6-14.
For endothelial differentiation, media change was performed after 48 h of CHIR-99021 (8 μM, Tocris) to phase 1 media supplemented with VEGF (100 ng/mL, R&D Systems) and bFGF (50 ng/ml, R&D Systems). On Day 6, media was changed to phase 2 media supplemented with VEGF (100 ng/mL, R&D Systems) and bFGF (50 ng/ml, R&D Systems) and maintained until Day 14. For both endocardial and endothelial conditions, media change was performed every other day and incubated at 37°C in a hypoxic incubator (5% CO2, 5% O2, 90% N2).
Cell sorting and flow cytometry
For mesoderm analysis, cells undergoing either endocardial or endothelial differentiation were dissociated on Day 4 with Accutase (Innovative Cell Technologies, Inc) to a single-cell suspension. Cells were then resuspended in FACS staining buffer consisting of PBS-/- (Gibco) supplemented with FBS (10%, Gibco), DNase (10 μg/mL, Millipore), and Thiazovivin (2 μM, Selleckchem) and labeled with anti-CD56 APC (1:100, BioLegend) and anti-CD13 PE (1:100, BioLegend) on ice for 1 h. Following washes with PBS-/- (Gibco), cells were resuspended in FACS running buffer consisting of PBS-/- (Gibco) supplemented with FBS (1%, Gibco), penicillin/streptomycin (2%, ThermoFisher), DNase (10 μg/mL, Millipore), thiazovivin (2 μM, Selleckchem), and DAPI (0.1 μg/mL, Sigma).
For endocardial and endothelial analysis, cells were dissociated with collagenase type II (0.6 mg/ml, Worthington) in HBSS (Corning) supplemented with HEPES (5 mM, Sigma) and incubated at 37°C for 1 h. Dissociated cells were collected on ice with wash media consisting of DMEM (Gibco), BSA (0.05%, Gibco), DNAse (10 μg/mL, Millipore) and thiazovivin (2 μM, Selleckchem) and passed through a 40-μm cell strainer (Fisher Scientific). Enrichment of CD31+ endocardial cells or CD34+ endothelial cells to a purity greater than 95% was then performed by magnetic activated cell sorting (MACS, Miltenyi). Cells from the endocardial differentiation were incubated with human anti-CD31 microbeads (Miltenyi #130-091-935) according to manufacturer instructions in EasySep Buffer (Stem Cell Technologies) for 15 min on ice, followed by purification on an LS column. Similarly, cells from the endothelial differentiation were incubated with human anti-CD34 microbeads (Miltenyi #130-046-702) and purified on an LS column. Both endocardial and endothelial populations were stained with anti-CD31 APC (1:100, Invitrogen) and anti-CD34 PE-Cy7 (1:100, BioLegend) and analyzed via flow cytometry as described above.
Following EndMT, cells were dissociated with collagenase type II (0.6 mg/ml, Worthington) and collected as described above. VIC and VEC populations were separated with human CD31 MACS kit (Miltenyi #130-091-935) and purified on an LS column. Negative selection was performed to isolate CD31- VICs, which are not magnetically bound to the LS column. Conversely, the cells bound to the LS column were then eluted to isolate CD31+ VECs. These populations were stained with anti-CD31 APC (1:100, Invitrogen) and anti-PDGFRβ BV786 (1:100, BD Biosciences) and analyzed via flow cytometry as described above. All samples were analyzed on either the BD FACSCelesta, BD LSRFortessa, or BD LSR II flow cytometers.
EndMT induction of endocardial cells to generate VICs and VECs
On Day 14 of endocardial differentiation, endocardial cells were dissociated with collagenase type II (0.6 mg/ml, Worthington) for 1 h and enriched for a CD31+ endocardial population with CD31 MACS, as described above. Following purification, cell counts were determined with an automated cell counter (TC20, BioRad) and replated 3x105 cells/well on fibronectin-coated 24-well plates in the EndMT condition consisting of phase 2 media supplemented with BMP2 (100 ng/ml, R&D Systems), TGFβ2 (0.3 ng/mL, R&D Systems), and bFGF (10 ng/mL, R&D Systems). Thiazovivin (2 μM, Selleckchem) was also included for the first 16 h following plating. Fibronectin coating was performed with human fibronectin plasma (1:100, Sigma) diluted in PBS-/- (Gibco) and incubated for at least 1 h at 37°C. Plated cells were maintained in the EndMT condition for 6-7 days with media changes every other day and incubated at 37°C in a hypoxic incubator (5% CO2, 5% O2, 90% N2).
Immunohistochemistry
Day 14 endocardial and endothelial cells were dissociated with collagenase type II (0.6 mg/ml, Worthington) and sorted with CD31-MACS and CD34-MACS, respectively, as described above. Endocardial and endothelial cells were plated on 12-chamber microscope slides (Ibidi) coated with Matrigel (6.6% v/v, Corning). Endocardial cells (1x105) were plated with phase 2 media supplemented with BMP10 (10 ng/mL, R&D Systems) and bFGF (50 ng/mL, R&D Systems). Endothelial cells (1x105) were plated with phase 2 media supplemented with VEGF (100 ng/mL, R&D Systems) and bFGF (50 ng/ml, R&D Systems).
Following EndMT, VICs and VECs were dissociated with collagenase type II (0.6 mg/ml, Worthington) and sorted with CD31-MACS for positive and negative selection, as described above. Isolated VICs and VECs were plated on 12-chamber microscopy slides (Ibidi) coated with human fibronectin plasma (1:100, Sigma). VICs were plated in fibroblast growth media (PromoCell) supplemented with penicillin/streptomycin (1%, ThermoFisher) and FBS (10%, Gibco). VECs were plated in MV2 endothelial media (PromoCell) supplemented with penicillin/streptomycin (1%, ThermoFisher).
All cells were incubated at 37°C overnight in a hypoxic incubator (5% CO2, 5% O2, 90% N2). Cells were fixed with 4% PFA in PBS (ThermoFisher) for 10 min at room temperature, followed by permeabilization with 0.1% Triton X-100 in PBS for 10 min. Cells were washed with PBS and blocked for 1 h at room temperature with PBS containing BSA (1%, Sigma), glycine (22.52 mg/mL, BioRad), Tween-20 (0.1%, Sigma), and sodium azide (0.02%, Sigma). Incubation with primary antibodies was performed overnight at 4°C in PBS containing BSA (1%, Sigma), Tween-20 (0.1%, Sigma), and sodium azide (0.02%, Sigma). Stained cells were then washed 3x with PBS for 10 min each and stained with secondary antibodies for 1 h at room temperature, followed by another wash step. Cell nuclei were stained with DAPI (0.25 μg/mL, Sigma) in PBS for 10 min at room temperature. Coverslips were then mounted onto the samples with Vectashield Plus Antifade Mounting Medium (Vector Laboratories). The following primary antibodies were used: anti-NKX2.5 (1:100, Cell Signaling Technology), anti-CD31 (1:100, Invitrogen), anti-NFATc1 (1:50, Invitrogen), anti-CDH5 (1:100, Cell Signaling Technology), anti-VIMENTIN (1:100, Invitrogen), anti-PERIOSTIN (1:100, Santa Cruz Biotechnology), anti-SOX9 (1:100, Sigma), and anti-αSMA (1:100, Abcam). The following secondary antibodies were used for detection: goat anti-rabbit IgG Alexa Fluor 488 (1:400, Invitrogen), goat anti-mouse IgG Alexa Flour 488 (1:400, Invitrogen), goat anti-rabbit IgG Alexa Flour 568 (1:400, Invitrogen), and goat anti-mouse IgG Alexa Flour 568 (1:400, Invitrogen). Detailed antibody information is described in the key resources table. Stained cells were imaged on a Zeiss LSM780 confocal microscope.
Quantitative reverse transcriptase PCR
Total RNA from iPSC-derived populations was isolated using the Qiagen RNeasy Micro Kit. Briefly, samples were lysed in Buffer RLT containing β-mercaptoethanol (Sigma) according to manufacturer’s instructions. Samples were incubated with proteinase K (Qiagen) at 55°C for 10 min prior to RNA isolation on the RNeasy column. On-column DNase treatment was performed according to manufacturer instructions and total RNA was eluted in RNase-free H2O. RNA quality was determined with the Agilent 2100 Bioanalyzer and the RNA 6000 Nano chip. RIN numbers were confirmed to be >8.0 for all samples. cDNA was generated using the SuperScript IV VILO Master Mix (Invitrogen). qRT-PCR was performed on a ViiA 7 Real-Time PCR System (Applied Biosystems) using the PowerTrack SYBR Green Master Mix (ThermoFisher). All experiments were run in triplicates using the ΔΔCt method relative to a human reference cDNA (Takara) and the housekeeping gene TBP. Primer sequences are listed in Table S1.
Bulk RNA sequencing analysis
Total RNA from sorted iPSC-derived populations was isolated as described above and confirmed to have a RIN > 8.0. Library preparation and sequencing were performed by Novogene. Library preparation was performed using polyA selection without strand-specificity and sequenced with an Illumina NovaSeq instrument using paired-end 150bp reads at a depth of ∼20 million reads per sample. Raw sequencing data in FASTQ format were mapped to the human reference genome (NCBI build38/UCSC hg38) with 2-pass mapping using STAR v2.7.10a.70 Duplicate reads were marked and removed with Picard v2.26.10 (http://broadinstitute.github.io/picard/). Gene counts were then obtained with HTSeq v2.0.2.71
Count matrices were then analyzed in R with the DESeq2 package.72 To reduce variability in our dataset stemming from sex differences between the WTC11 and MSN02-4 iPSC lines, genes on the X and Y chromosomes were filtered out prior to differential analysis with the biomaRt package. Batch correction was performed using limma and routine filtering for low counts was performed. Subsequent differential analysis was performed with DESeq2 using the likelihood ratio test. PCA was performed with the ‘plotPCA’ function in DESeq2. Heatmaps were generated using the R package ‘pheatmap’ on count data normalized with variance stabilizing transformation. Volcano plots were generated using the R package ‘Enhanced Volcano’ and performed on count data following adaptive shrinkage estimation with ‘ashr’.73 Gene ontology analysis was performed with Enrichr using either the GO Biological Processes 2021 or BioPlanet 2019 annotations.74
For comparison with normal human valve cells, single-cell RNAseq data generated from a normal 15-week-old male fetus’s heart valves53 was obtained (GSE228638) and analyzed using Seurat.67 The AverageExpression function in Seurat was used to generate average raw counts for each of the cell types. To account for differences in read depth between bulk RNAseq and pseudobulk data, analysis was restricted to genes that were detected in both sequencing formats. X and Y chromosome genes were also removed from this analysis as described above. Following batch correction with limma, scaling to balance read depth between samples, filtering out low reads, and normalization with variance stabilizing transformation, 12,563 genes were analyzed with DESeq2. PCA and heatmaps were generated as described above. Pearson correlation analysis was performed with the ‘cor’ function.
Quantification and statistical analysis
All data are represented as mean ± standard error of mean (SEM). Biological replicates of each differentiation experiment are represented by the sample size (n). Sample sizes were not predetermined and due to the nature of the experiments, samples were not randomized or blinded from investigators. Statistical significance was determined using 2-way ANOVA ± multiple comparison analysis with Sidak, Tukey, or Dunnet’s post hoc test in GraphPad Prism 9 software. Results were marked as significant at the following cutoffs p < 0.05 (∗), p < 0.01 (∗∗), p < 0.001 (∗∗∗), p < 0.0001 (∗∗∗∗). All statistical tests and parameters are reported in the respective figures and figure legends.
Acknowledgments
We extend our gratitude to Dr. Nicole Dubois for providing the MSN02-4 iPSC line and experimental advice as well as to the lab of Dr. Gordon Keller for fruitful discussions on our results. We also acknowledge the Flow Cytometry Core, Microscopy Core, and Scientific Computing and Data at the Icahn School of Medicine at Mount Sinai for their valuable support. This project was made possible by the generous funding from the National Institutes of Health/National Heart, Lung, and Blood Institute (R35 HL135742), the Additional Ventures (Single Ventricle Research Fund 1019125), the American Heart Association/The Children’s Heart Foundation Predoctoral Fellowship (23PRECHF1025586, https://doi.org/10.58275/AHA.23PRECHF1025586.pc.gr.161749), and the NIDCR Interdisciplinary Training in Systems and Developmental Biology and Birth Defects (T32HD075735). Lastly, the differentiation schematic shown here was created using BioRender.com.
Author contributions
Conceptualization, C.Z.L. and B.D.G.; methodology, C.Z.L. and B.D.G.; validation: C.Z.L. and B.D.G.; formal analysis, C.Z.L. and B.D.G.; investigation, C.Z.L., A.P., and B.J.; resources, B.D.G. and A.J.S.; data curation: C.Z.L., B.J., Y.L., and M.G.; writing – original draft: C.Z.L. and B.D.G.; visualization: C.Z.L.; supervision, B.D.G. and A.J.S.; project administration: B.D.G. and A.J.S.; funding acquisition, C.Z.L. and B.D.G.
Declaration of interests
The authors declare no competing interests.
Published: November 30, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108599.
Supplemental information
References
- 1.Nkomo V.T., Gardin J.M., Skelton T.N., Gottdiener J.S., Scott C.G., Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368:1005–1011. doi: 10.1016/S0140-6736(06)69208-8. [DOI] [PubMed] [Google Scholar]
- 2.Combs M.D., Yutzey K.E. Heart Valve Development: Regulatory Networks in Development and Disease. Circ. Res. 2009;105:408–421. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saef J.M., Ghobrial J. Valvular heart disease in congenital heart disease: a narrative review. Cardiovasc. Diagn. Ther. 2021;11:818–839. doi: 10.21037/cdt-19-693-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borer J.S., Sharma A. Drug Therapy for Heart Valve Diseases. Circulation. 2015;132:1038–1045. doi: 10.1161/CIRCULATIONAHA.115.016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Donnell A., Yutzey K.E. Mechanisms of heart valve development and disease. Development. 2020;147:dev183020. doi: 10.1242/dev.183020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tao G., Kotick J.D., Lincoln J. Heart Valve Development, Maintenance, and Disease The Role of Endothelial Cells. Curr. Top. Dev. Biol. 2012;100:203–232. doi: 10.1016/B978-0-12-387786-4.00006-3. [DOI] [PubMed] [Google Scholar]
- 7.Ma L., Lu M.-F., Schwartz R.J., Martin J.F. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601–5611. doi: 10.1242/dev.02156. [DOI] [PubMed] [Google Scholar]
- 8.Eisenberg L.M., Markwald R.R. Molecular Regulation of Atrioventricular Valvuloseptal Morphogenesis. Circ. Res. 1995;77:1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- 9.McCulley D.J., Kang J.O., Martin J.F., Black B.L. BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev. Dynam. 2008;237:3200–3209. doi: 10.1002/dvdy.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romano L.A., Runyan R.B. Slug is an Essential Target of TGFβ2 Signaling in the Developing Chicken Heart. Dev. Biol. 2000;223:91–102. doi: 10.1006/dbio.2000.9750. [DOI] [PubMed] [Google Scholar]
- 11.Timmerman L.A., Grego-Bessa J., Raya A., Bertrán E., Pérez-Pomares J.M., Díez J., Aranda S., Palomo S., McCormick F., Izpisúa-Belmonte J.C., de la Pompa J.L. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Gene Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacGrogan D., D’Amato G., Travisano S., Martinez-Poveda B., Luxán G., Del Monte-Nieto G., Papoutsi T., Sbroggio M., Bou V., Gomez-Del Arco P., et al. Sequential Ligand-Dependent Notch Signaling Activation Regulates Valve Primordium Formation and Morphogenesis. Circ. Res. 2016;118:1480–1497. doi: 10.1161/CIRCRESAHA.115.308077. [DOI] [PubMed] [Google Scholar]
- 13.Liebner S., Cattelino A., Gallini R., Rudini N., Iurlaro M., Piccolo S., Dejana E. Beta-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J. Cell Biol. 2004;166:359–367. doi: 10.1083/jcb.200403050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H., von Gise A., Cai C.L., Liu Q., Hu T., Tian X., He L., Pu W., Huang X., et al. Yap1 Is Required for Endothelial to Mesenchymal Transition of the Atrioventricular Cushion. J. Biol. Chem. 2014;289:18681–18692. doi: 10.1074/jbc.M114.554584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinton R.B., Yutzey K.E. Heart Valve Structure and Function in Development and Disease. Physiology. 2011;73:29–46. doi: 10.1146/annurev-physiol-012110-142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kodigepalli K.M., Thatcher K., West T., Howsmon D.P., Schoen F.J., Sacks M.S., Breuer C.K., Lincoln J. Biology and Biomechanics of the Heart Valve Extracellular Matrix. J. Cardiovasc. Dev. Dis. 2020;7:57. doi: 10.3390/jcdd7040057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu A.C., Joag V.R., Gotlieb A.I. The Emerging Role of Valve Interstitial Cell Phenotypes in Regulating Heart Valve Pathobiology. Am. J. Pathol. 2007;171:1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu B., Baldwin H.S., Zhou B. Nfatc1 directs the endocardial progenitor cells to make heart valve primordium. Trends Cardiovasc. Med. 2013;23:294–300. doi: 10.1016/j.tcm.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura T., Colbert M.C., Robbins J. Neural Crest Cells Retain Multipotential Characteristics in the Developing Valves and Label the Cardiac Conduction System. Circ. Res. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- 20.Jain R., Engleka K.A., Rentschler S.L., Manderfield L.J., Li L., Yuan L., Epstein J.A. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J. Clin. Invest. 2011;121:422–430. doi: 10.1172/JCI44244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wessels A., van den Hoff M.J.B., Adamo R.F., Phelps A.L., Lockhart M.M., Sauls K., Briggs L.E., Norris R.A., van Wijk B., Perez-Pomares J.M., et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012;366:111–124. doi: 10.1016/j.ydbio.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lockhart M.M., Boukens B.J.D., Phelps A.L., Brown C.-L.M., Toomer K.A., Burns T.A., Mukherjee R.D., Norris R.A., Trusk T.C., van den Hoff M.J.B., Wessels A. Alk3 mediated Bmp signaling controls the contribution of epicardially derived cells to the tissues of the atrioventricular junction. Dev. Biol. 2014;396:8–18. doi: 10.1016/j.ydbio.2014.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu K., Yu W., Tang M., Tang J., Liu X., Liu Q., Li Y., He L., Zhang L., Evans S.M., et al. A dual genetic tracing system identifies diverse and dynamic origins of cardiac valve mesenchyme. Development. 2018;145:dev167775. doi: 10.1242/dev.167775. [DOI] [PubMed] [Google Scholar]
- 24.Kim A.J., Xu N., Yutzey K.E. Macrophage lineages in heart valve development and disease. Cardiovasc. Res. 2021;117:663–673. doi: 10.1093/cvr/cvaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shigeta A., Huang V., Zuo J., Besada R., Nakashima Y., Lu Y., Ding Y., Pellegrini M., Kulkarni R.P., Hsiai T., et al. Endocardially Derived Macrophages Are Essential for Valvular Remodeling. Dev. Cell. 2019;48:617–630.e3. doi: 10.1016/j.devcel.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neri T., Hiriart E., van Vliet P.P., Faure E., Norris R.A., Farhat B., Jagla B., Lefrancois J., Sugi Y., Moore-Morris T., et al. Human pre-valvular endocardial cells derived from pluripotent stem cells recapitulate cardiac pathophysiological valvulogenesis. Nat. Commun. 2019;10:1929. doi: 10.1038/s41467-019-09459-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palpant N.J., Pabon L., Friedman C.E., Roberts M., Hadland B., Zaunbrecher R.J., Bernstein I., Zheng Y., Murry C.E. Generating high-purity cardiac and endothelial derivatives from patterned mesoderm using human pluripotent stem cells. Nat. Protoc. 2017;12:15–31. doi: 10.1038/nprot.2016.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palpant N.J., Pabon L., Roberts M., Hadland B., Jones D., Jones C., Moon R.T., Ruzzo W.L., Bernstein I., Zheng Y., Murry C.E. Inhibition of β-catenin signaling respecifies anterior-like endothelium into beating human cardiomyocytes. Development. 2015;142:3198–3209. doi: 10.1242/dev.117010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng L., Xie M., Qiao W., Song Y., Zhang Y., Geng Y., Xu W., Wang L., Wang Z., Huang K., et al. Generation and characterization of cardiac valve endothelial-like cells from human pluripotent stem cells. Commun. Biol. 2021;4:1039. doi: 10.1038/s42003-021-02571-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bao X., Bhute V.J., Han T., Qian T., Lian X., Palecek S.P. Human pluripotent stem cell-derived epicardial progenitors can differentiate to endocardial-like endothelial cells. Bioeng. Transl. Med. 2017;2:191–201. doi: 10.1002/btm2.10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mikryukov A.A., Mazine A., Wei B., Yang D., Miao Y., Gu M., Keller G.M. BMP10 Signaling Promotes the Development of Endocardial Cells from Human Pluripotent Stem Cell-Derived Cardiovascular Progenitors. Cell Stem Cell. 2021;28:96–111.e7. doi: 10.1016/j.stem.2020.10.003. [DOI] [PubMed] [Google Scholar]
- 32.Theunissen T.W., Powell B.E., Wang H., Mitalipova M., Faddah D.A., Reddy J., Fan Z.P., Maetzel D., Ganz K., Shi L., et al. Systematic Identification of Culture Conditions for Induction and Maintenance of Naive Human Pluripotency. Cell Stem Cell. 2014;15:471–487. doi: 10.1016/j.stem.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yue X.-S., Fujishiro M., Nishioka C., Arai T., Takahashi E., Gong J.-S., Akaike T., Ito Y. Feeder Cells Support the Culture of Induced Pluripotent Stem Cells Even after Chemical Fixation. PLoS One. 2012;7 doi: 10.1371/journal.pone.0032707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wan H., Fu R., Tong M., Wang Y., Wang L., Wang S., Zhang Y., Li W., Wang X.J., Feng G. Influence of feeder cells on transcriptomic analysis of pluripotent stem cells. Cell Prolif. 2022;55 doi: 10.1111/cpr.13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lian X., Hsiao C., Wilson G., Zhu K., Hazeltine L.B., Azarin S.M., Raval K.K., Zhang J., Kamp T.J., Palecek S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA. 2012;109:E1848–E1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lian X., Zhang J., Azarin S.M., Zhu K., Hazeltine L.B., Bao X., Hsiao C., Kamp T.J., Palecek S.P. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat. Protoc. 2013;8:162–175. doi: 10.1038/nprot.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evseenko D., Zhu Y., Schenke-Layland K., Kuo J., Latour B., Ge S., Scholes J., Dravid G., Li X., MacLellan W.R., Crooks G.M. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc. Natl. Acad. Sci. USA. 2010;107:13742–13747. doi: 10.1073/pnas.1002077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Skelton R.J.P., Brady B., Khoja S., Sahoo D., Engel J., Arasaratnam D., Saleh K.K., Abilez O.J., Zhao P., Stanley E.G., et al. CD13 and ROR2 Permit Isolation of Highly Enriched Cardiac Mesoderm from Differentiating Human Embryonic Stem Cells. Stem Cell Rep. 2016;6:95–108. doi: 10.1016/j.stemcr.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monaghan M.G., Linneweh M., Liebscher S., Van Handel B., Layland S.L., Schenke-Layland K. Endocardial-to-mesenchymal transformation and mesenchymal cell colonization at the onset of human cardiac valve development. Development. 2016;143:473–482. doi: 10.1242/dev.133843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu B., Wang Y., Lui W., Langworthy M., Tompkins K.L., Hatzopoulos A.K., Baldwin H.S., Zhou B. Nfatc1 Coordinates Valve Endocardial Cell Lineage Development Required for Heart Valve Formation. Circ. Res. 2011;109:183–192. doi: 10.1161/CIRCRESAHA.111.245035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lotto J., Cullum R., Drissler S., Arostegui M., Garside V.C., Fuglerud B.M., Clement-Ranney M., Thakur A., Underhill T.M., Hoodless P.A. Cell diversity and plasticity during atrioventricular heart valve EMTs. Nat. Commun. 2023;14:5567. doi: 10.1038/s41467-023-41279-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng W., Chen L., Nguyen P.K., Wu S.M., Li G. Single Cell Analysis of Endothelial Cells Identified Organ-Specific Molecular Signatures and Heart-Specific Cell Populations and Molecular Features. Front. Cardiovasc. Med. 2019;6:165. doi: 10.3389/fcvm.2019.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rathjen T., Kunkemoeller B., Cederquist C.T., Wang X., Lockhart S.M., Patti J.C., Willenbrock H., Olsen G.S., Povlsen G.K., Beck H.C., et al. Endothelial Cell Insulin Signaling Regulates CXCR4 (C-X-C Motif Chemokine Receptor 4) and Limits Leukocyte Adhesion to Endothelium. Arterioscler. Thromb. Vasc. Biol. 2022;42:e217–e227. doi: 10.1161/ATVBAHA.122.317476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rocha S.F., Schiller M., Jing D., Li H., Butz S., Vestweber D., Biljes D., Drexler H.C.A., Nieminen-Kelhä M., Vajkoczy P., et al. Esm1 Modulates Endothelial Tip Cell Behavior and Vascular Permeability by Enhancing VEGF Bioavailability. Circ. Res. 2014;115:581–590. doi: 10.1161/CIRCRESAHA.115.304718. [DOI] [PubMed] [Google Scholar]
- 45.Hegen A., Koidl S., Weindel K., Marmé D., Augustin H.G., Fiedler U. Expression of Angiopoietin-2 in Endothelial Cells Is Controlled by Positive and Negative Regulatory Promoter Elements. Arterioscler. Thromb. Vasc. Biol. 2004;24:1803–1809. doi: 10.1161/01.ATV.0000140819.81839.0e. [DOI] [PubMed] [Google Scholar]
- 46.Kearney J.B., Ambler C.A., Monaco K.-A., Johnson N., Rapoport R.G., Bautch V.L. Vascular endothelial growth factor receptor Flt-1 negatively regulates developmental blood vessel formation by modulating endothelial cell division. Blood. 2002;99:2397–2407. doi: 10.1182/blood.v99.7.2397. [DOI] [PubMed] [Google Scholar]
- 47.Shirayoshi Y., Yuasa Y., Suzuki T., Sugaya K., Kawase E., Ikemura T., Nakatsuji N. Proto-oncogene of int-3, a mouse Notch homologue, is expressed in endothelial cells during early embryogenesis. Gene Cell. 1997;2:213–224. doi: 10.1046/j.1365-2443.1997.d01-310.x. [DOI] [PubMed] [Google Scholar]
- 48.Akiyama H., Chaboissier M.-C., Behringer R.R., Rowitch D.H., Schedl A., Epstein J.A., de Crombrugghe B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA. 2004;101:6502–6507. doi: 10.1073/pnas.0401711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garside V.C., Cullum R., Alder O., Lu D.Y., Vander Werff R., Bilenky M., Zhao Y., Jones S.J.M., Marra M.A., Underhill T.M., Hoodless P.A. SOX9 modulates the expression of key transcription factors required for heart valve development. Development. 2015;142:4340–4350. doi: 10.1242/dev.125252. [DOI] [PubMed] [Google Scholar]
- 50.Ridge L.A., Kewbank D., Schütz D., Stumm R., Scambler P.J., Ivins S. Dual role for CXCL12 signaling in semilunar valve development. Cell Rep. 2021;36 doi: 10.1016/j.celrep.2021.109610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sierro F., Biben C., Martínez-Muñoz L., Mellado M., Ransohoff R.M., Li M., Woehl B., Leung H., Groom J., Batten M., et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA. 2007;104:14759–14764. doi: 10.1073/pnas.0702229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu S., Kumar V., Xiao P., Kuss M., Lim J.Y., Guda C., Butcher J., Duan B. Age related extracellular matrix and interstitial cell phenotype in pulmonary valves. Sci. Rep. 2020;10 doi: 10.1038/s41598-020-78507-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Z., Liu Y., Yu Z., Pek N., O’Donnell A., Glass I., Winlaw D.S., Guo M., Chen Y.-W., Wu J.C., et al. APOE-NOTCH Axis Governs Elastogenesis During Human Cardiac Valve Remodeling. bioRxiv. 2023 doi: 10.1101/2023.04.26.538443. Preprint at. [DOI] [PubMed] [Google Scholar]
- 54.Torregrosa-Carrión R., Luna-Zurita L., García-Marqués F., D’Amato G., Piñeiro-Sabarís R., Bonzón-Kulichenko E., Vázquez J., de la Pompa J.L. NOTCH Activation Promotes Valve Formation by Regulating the Endocardial Secretome. Mol. Cell. Proteomics. 2019;18:1782–1795. doi: 10.1074/mcp.RA119.001492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim R.Y., Robertson E.J., Solloway M.J. Bmp6 and Bmp7 Are Required for Cushion Formation and Septation in the Developing Mouse Heart. Dev. Biol. 2001;235:449–466. doi: 10.1006/dbio.2001.0284. [DOI] [PubMed] [Google Scholar]
- 56.Camenisch T.D., Schroeder J.A., Bradley J., Klewer S.E., McDonald J.A. Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2–ErbB3 receptors. Nat. Med. 2002;8:850–855. doi: 10.1038/nm742. [DOI] [PubMed] [Google Scholar]
- 57.Bischoff J., Aikawa E. Progenitor Cells Confer Plasticity to Cardiac Valve Endothelium. J. Cardiovasc. Transl. 2011;4:710–719. doi: 10.1007/s12265-011-9312-0. [DOI] [PubMed] [Google Scholar]
- 58.Kovacic J.C., Dimmeler S., Harvey R.P., Finkel T., Aikawa E., Krenning G., Baker A.H. Endothelial to Mesenchymal Transition in Cardiovascular Disease. J. Am. Coll. Cardiol. 2019;73:190–209. doi: 10.1016/j.jacc.2018.09.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernández Esmerats J., Heath J., Jo H. Shear-Sensitive Genes in Aortic Valve Endothelium. Antioxidants Redox Signal. 2016;25:401–414. doi: 10.1089/ars.2015.6554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de la Pompa J.L., Timmerman L.A., Takimoto H., Yoshida H., Elia A.J., Samper E., Potter J., Wakeham A., Marengere L., Langille B.L., et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- 61.Ranger A.M., Grusby M.J., Hodge M.R., Gravallese E.M., de la Brousse F.C., Hoey T., Mickanin C., Baldwin H.S., Glimcher L.H. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 62.Suehiro J.i., Kanki Y., Makihara C., Schadler K., Miura M., Manabe Y., Aburatani H., Kodama T., Minami T. Genome-wide Approaches Reveal Functional Vascular Endothelial Growth Factor (VEGF)-inducible Nuclear Factor of Activated T Cells (NFAT) c1 Binding to Angiogenesis-related Genes in the Endothelium. J. Biol. Chem. 2014;289:29044–29059. doi: 10.1074/jbc.M114.555235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kulkarni R.M., Greenberg J.M., Akeson A.L. NFATc1 regulates lymphatic endothelial development. Mech. Dev. 2009;126:350–365. doi: 10.1016/j.mod.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hori N., Okada K., Takakura Y., Takano H., Yamaguchi N., Yamaguchi N. Vestigial-like family member 3 (VGLL3), a cofactor for TEAD transcription factors, promotes cancer cell proliferation by activating the Hippo pathway. J. Biol. Chem. 2020;295:8798–8807. doi: 10.1074/jbc.RA120.012781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu W., Ma X., Xu J., Heumüller A.W., Fei Z., Feng X., Wang X., Liu K., Li J., Cui G., et al. VGLL4 plays a critical role in heart valve development and homeostasis. PLoS Genet. 2019;15 doi: 10.1371/journal.pgen.1007977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang M., Lin B.Y., Sun S., Dai C., Long F., Butcher J.T. Shear and hydrostatic stress regulate fetal heart valve remodeling through YAP-mediated mechanotransduction. Elife. 2023;12 doi: 10.7554/eLife.83209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaniel C., Dhanan P., Hu B., Xiong Y., Raghunandan T., Gonzalez D.M., Dariolli R., D’Souza S.L., Yadaw A.S., Hansen J., et al. A library of induced pluripotent stem cells from clinically well-characterized, diverse healthy human individuals. Stem Cell Rep. 2021;16:3036–3049. doi: 10.1016/j.stemcr.2021.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wickramasinghe N.M., Sachs D., Shewale B., Gonzalez D.M., Dhanan-Krishnan P., Torre D., LaMarca E., Raimo S., Dariolli R., Serasinghe M.N., et al. PPARdelta activation induces metabolic and contractile maturation of human pluripotent stem cell-derived cardiomyocytes. Cell Stem Cell. 2022;29:559–576.e7. doi: 10.1016/j.stem.2022.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Putri G.H., Anders S., Pyl P.T., Pimanda J.E., Zanini F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics. 2022;38:2943–2945. doi: 10.1093/bioinformatics/btac166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stephens M. False discovery rates: a new deal. Biostatistics. 2017;18:275–294. doi: 10.1093/biostatistics/kxw041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V., Clark N.R., Ma’ayan A. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data generated from the bulk RNA-sequencing were uploaded to GEO and can be accessed with accession number GEO: GSE226476.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.