

Summary
CRISPR/Cas9 technology is revolutionizing the field of gene editing. While this technology enables the targeting of any gene, it may also target unplanned loci, termed off-target sites (OTS), which are a few mismatches, insertions, and deletions from the target. While existing methods for finding OTS up to a given mismatch threshold are efficient, other methods considering insertions and deletions are limited by long runtimes, incomplete OTS lists, and partial support of versatile thresholds. Here, we developed SWOffinder, an efficient method based on Smith-Waterman alignment to find all OTS up to some edit distance. We implemented an original trace-back approach to find OTS under versatile criteria, such as separate limits on the number of insertions, deletions, and mismatches. Compared to state-of-the-art methods, only SWOffinder finds all OTS in the genome in just a few minutes. SWOffinder enables accurate and efficient genomic search of OTS, which will lead to safer gene editing.
Subject areas: Techniques in genetics, Bioinformatics, Biological constraints, Algorithms
Graphical abstract
Highlights
-
•
SWOffinder finds off-target sites efficiently and accurately by SW-local alignment
-
•
SWOffinder enables versatile search thresholds of the number of mismatches and bulges
-
•
SWOffinder outperforms other off-target site search methods in runtime and accuracy
-
•
SWOffinder will lead to enhancements in the safety of gene-editing applications
Techniques in genetics; Bioinformatics; Biological constraints; Algorithms
Introduction
CRISPR/Cas9 has revolutionized gene-editing research and applications.1 CRISPR/Cas9 enables the precise and efficient modification of DNA sequences in living cells by a single-guide RNA (sgRNA) targeting specific loci of 20nt followed by a protospacer adjacent motif (PAM) sequence, which is NGG for Cas9. CRISPR/Cas9 can be used in a wide range of organisms, from bacteria to humans, and it can be applied in a variety of different fields, including agriculture and medicine.
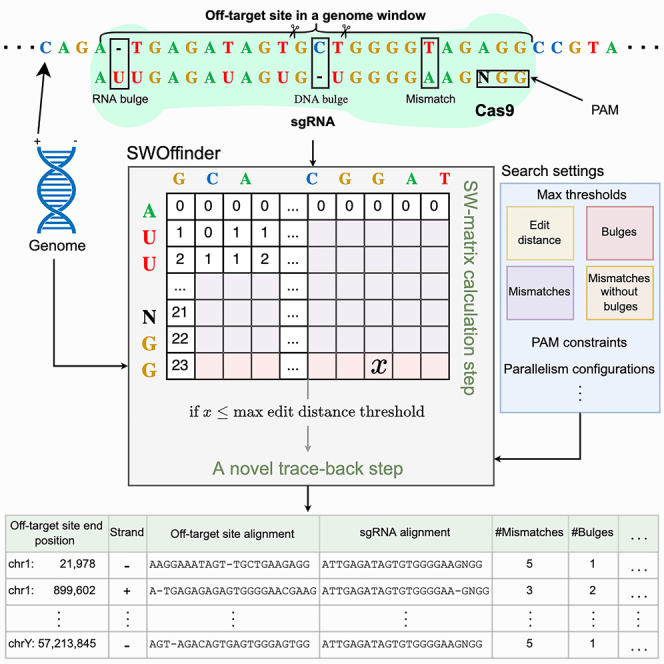
Unfortunately, there are concerns about the safety of using CRISPR/Cas9.2 One of the main risks associated with CRISPR/Cas9 is the possibility of off-target sites. Off-target sites occur when the CRISPR/Cas9 system edits unintended regions in the genome, which can lead to undesired consequences, such as the introduction of new mutations or the disruption of important genes. Off-target sites often occur in regions that are similar by sequence, i.e., in a small Hamming or edit distance, to the sgRNA (Figure 1).
Figure 1.

Off-target site alignment illustration
An off-target site alignment with 1 DNA bulge, 1 RNA bulge, and 1 mismatch of a sgRNA sequence AUUGAGAUAGUGUGGGGAAG with NGG PAM and a genome window of the forward strand.
Off-target search considering only mismatches, i.e., up to some Hamming distance, has been solved efficiently by a plethora of computational methods, with the most popular being Cas-OFFinder.3 But, the efficient search for potential off-target sites up to some edit distance, i.e., when considering mismatches, insertions, and deletions, is still an open problem. It has been shown that the problem of computing the edit distance between two strings cannot be computed in subquadratic time.4 Nonetheless, several methods have been developed for CRISPR off-target search considering insertions and deletions, termed DNA and RNA bulges, including CRISPRitz,5 CALITAS,6 and an unpublished variant of Cas-OFFinder.
Unfortunately, these methods fail to find all potential off-target sites in a feasible runtime, report incomplete lists of off-target sites, and/or do not support versatile threshold criteria. While CALITAS overcomes the most prominent limitations of Cas-OFFinder and CRISPRitz (the exponential dependence of runtime in the number of gaps, allowing only DNA or RNA insertions, and reporting all possible alignments6), its runtime is prohibitive, especially when running it on thousands of sgRNAs in parallel, which is required in most sgRNA-design scenarios.7 Moreover, CALITAS searches for off-target sites with specific scores for mismatches, insertions, and deletions, while common applications require searching for off-target sites with a separate threshold for each edit operation.8 As a result, there is currently no feasible solution to the CRISPR off-target search problem, when considering insertions and deletions and a separate threshold for each edit operation.
Here, we developed SWOffinder, a new highly efficient method to find all off-target sites up to some edit distance, which is based on the classic Smith-Waterman (SW) alignment (Figure 2). We implemented a novel trace-back method to find off-target sites under versatile criteria, such as separate limits on the number of bulges and mismatches. Our results compared to state-of-the-art methods show that SWOffinder finds all off-target sites in the human genome in only a couple of minutes. We expect our method to enable accurate and efficient genomic search of potential off-target sites, which will make gene-editing applications safer.
Figure 2.

Overview of SWOffinder for searching off-target sites across a given genome
First, in the SW-matrix calculation step, SWOffinder scans the genome for all end positions of sites with an edit distance that meets a user-specified threshold using a novel version of the Smith-Waterman alignment algorithm. Then, in the trace-back step, SWOffinder applies a novel recursive procedure on the subset of end positions obtained by the SW-matrix calculation step to filter sites, which do not meet the user-specified operation-specific thresholds, and find their alignment that meets those thresholds with minimum edit distance (Algorithm 1).
Results
To evaluate both off-target site identification and runtime performance of SWOffider and existing methods, we used as a test case the set of chromosomes derived from the hg38 reference genome and the sgRNA AUUGAGAUAGUGUGGGGAAG with NGG PAM which was also used in CALITAS becnmarks.6 We present results with the NGG PAM, which fits CRISPR/Cas9, the most popular nuclease, and note that SWOffider can run on sgRNAs of any length and any PAM. One of the key strengths of SWOffider is its versatile search options, which are particularly suited for common use cases. However, since existing methods do not support such versatile search options, we compared them to SWOffider only over their search options.
Comparison of the number of identified sites and runtime at an edit-distance threshold
We compared SWOffidner to CALITAS and CRISPRitz in the number of identified off-target sites with up to 4 edits and in a single-threaded runtime. The specific parameters of this run for the different methods were.
-
(1)
SWOffinder: without PAM edits allowed.
-
(2)
CALITAS: Up to 4 edits without PAM edits allowed.
-
(3)
CRISPRitz: Up to 4 mismatches and 4 bulges with an index file for NGG PAM. Note that CRISPRitz has the inability to search up to 4 edits directly, so we filtered the obtained sites to the desired edit threshold.
Regarding off-target site identification, SWOffinder found the greatest number of unique off-target sites (Figure 3A). While the total number of alignments produced by CRISPRitz is the greatest ( when filtering the results to up to 4 edits), this number is the greatest due to the redundancy in the number of alignments for the same off-target site. After removing redundant sites, we discovered that CRISPRitz found only unique sites compared to unique sites found by CALITAS and by SWOffinder.
Figure 3.

Off-target sites identification and runtime comparison of SWOffinder, CALITAS, and CRISPRitz
(A) Comparison of the number of sites found by each method. We ran SWOffinder and CALITAS to find off-target sites up to edit distance 4, and CRISPRitz with up to 4 mismatches and 4 bugles (as it does not support an edit-distance threshold).
(B) The overlap between sites that were identified by SWOffinder and sites that were found by CALITAS and CRISPRitz in a window. SWOffinder found all sites that were found by CALITAS and CRISPRitz.
(C) Runtime comparison of SWOffinder, CALITAS, and CRISPRitz in searching off-target sites up to edit distance 4. CRISPRitz was run with up to 4 mismatches and 4 bulges (as it does not support an edit-distance threshold).
While SWOffinder enables the user to choose whether to output all possible off-target sites or optimal sites within a user-defined genome window, CALITAS is limited to the second option due to its use of semi-global alignment, where the default window size is 1000, which may lead to missing proximal off-target sites. Therefore, we compared the number of non-consecutive unique sites and performed an identification in-a-window test (for definitions, see Subsection Benchmarking off-target sites identification). SWOffinder found the greatest number of non-consecutive unique off-target sites with sites compared to and by CALITAS and CRISPRitz, respectively (Figure 3A). In the identification in-a-window test, only out of the unique sites found by SWOffinder could be associated with CRISPRitz (Figure 3B). When comparing CALITAS and SWOffinder in this test, we found that 2 of the sites found by SWOffinder could not be associated with CALITAS. The results show the supremacy of SWOffidner in identifying all potential off-target sites in various settings.
Regarding the runtime in a single-threaded run, SWOffinder’s runtime is 626 seconds, which is significantly faster than CALITAS with a runtime of seconds (Figure 3C). In this experiment, SWOffinder was also faster than CRISPRitz, which ran in seconds. We note that the last is not a direct comparison since CRISPRitz cannot search up to 4 edits directly. In this experiment, the maximum memory usage of SWOffinder and CALITAS were very similar, at 2.3GB and 2.1GB, respectively. CRISPRitz had maximum memory usage of 5GB, probably due to the extended search, unique data structure, and output of multiple alignments.
Comparison of the number of identified sites and runtime over various combinations of bulges and mismatches thresholds
We compared SWOffidner to CRISPRitz in a multi-threaded run over various combinations of bulges and mismatch thresholds. We excluded CALITAS from this comparison as it does not support a search with separate thresholds for bulges and mismatches, which is the common usage of CRISPR off-target search.9,10 In this comparison, we examined the number of identified off-target sites and the runtime for searches over thresholds combinations of x bulges and y mismatches where and .
The results show that for all comparisons SWOffider finds a greater number of unique off-target sites compared to CRISPRitz (Figure 4A). For example, for the combination of up to 1 bulge and 2 mismatches, SWOffinder found 90 sites while CRISPRitz found only 86. In the greater thresholds, the nominal difference in the number of unique sites is greater. For example, for the combination of up to 2 bulges and 5 mismatches, SWOffider found non-consecutive unique sites compared to only found by CRISPRitz.
Figure 4.

A comparison of the number of unique off-target sites found and the runtime between SWOffinder and CRISPRitz over various combinations of bulges and mismatches thresholds
We did not compare to CALITAS in this benchmark as it does not support a search with a combination of separate thresholds.
(A) A comparison of the unique off-target sites that were found. Note that the x-axis is on a logarithmic scale.
(B) A comparison of the runtime. Note that the runtime of SWOffinder trace-back step was calculated by reducing the runtime of the SWOffinder SW-matrix calculation step from the total runtime.
In terms of runtime, we see that both CRISPRitz and SWOffinder have comparable runtime performances of minutes over the human genome for small mismatch and bulge thresholds (Figure 4B). For combinations of up to 1 bulge and combinations of up to 2 bulges with up to 3 mismatches, CRISPRitz is outperforming SWOffinder, which achieves an almost constant runtime of 2 minutes due to the operation of step 1—the SW dynamic programming (DP) matrix filling that is the same for all thresholds. Strikingly, when we tested the combinations of up to 2 bulges and 4 or 5 mismatches, the trend changed. For example, for the combination of up to 2 bulges and 5 mismatches, SWOffinder was much faster than CRISPRitz with a runtime of 161.6 seconds compared to 552.62. This change in trends shows the advantage of using the two-step procedure in SWOffidner, where the SW-matrix calculation step is constant for all thresholds, and the trace-back step is negligible for most parameter combinations, including the more challenging ones. Another advantage of SWOffinder compared to CRISPRitz lies in its memory utilization. While SWOffinder is robust to the number of threads, CRISPRitz’s memory utilization increases with the number of threads. For example, for the combination of up to 2 bulges and 5 mismatches, the maximum memory usage of SWOffinder was slightly more than 2GB for both 1- and 8-threaded instances, while the usage of CRISPRitz was 3.3GB and 16.7GB, respectively. As a result, CRISPRitz’s memory utilization may become prohibitive with more threads.
Discussion
In this work, we developed SWOffinder to enable a fast and accurate search of off-target sites defined by an edit-distance threshold, and separate thresholds for mismatches, and bulges. We modified the SW algorithm for local alignment and developed a novel post-processing step to find off-target sites with separate user-specified operation-specific thresholds. Our results show that SWOffinder finds more sites than existing off-target search methods. In addition, in our benchmarks, we show that even without genome indexing or unique data structures, SWOffinder achieves state-of-the-art performance in terms of runtime and memory utilization, as it runs across the human genome in only a few minutes with low memory usage.
The novelty in our approach lies in our novel modification of SW alignment and our novel post-processing approach. Our new post-processing algorithm has efficient runtime due to its DP implementation, and the fact that it runs on a limited set of off-target sites—only those that passed the edit-distance threshold as found by SW alignment. The new features we developed for searching under versatile criteria are much more relevant than the search enabled by current off-target search methods, as prior biological knowledge demonstrated that at most two bulges, or even just one bulge, can occur in an off-target site as opposed to tolerance to multiple mismatches.11
Our study may be extended in the future in several promising directions. First, SWOffinder can be extended to consider genetic variants to enable off-target search across individual genomes. This feature can be easily incorporated in both SW alignment and our novel post-processing approach, as it only affects the match and mismatch operations, which can be supported by encoding multiple nucleotides instead of just one. Second, we hope to speed up SWOffinder by utilizing dedicated hardware. Previous studies have shown that SW alignment can run much faster by implementing and running it on GPUs or even FPGAs,12 so similar improvements are expected for SWOffinder. Third, a possible solution to obtain all possible alignments in one pass of filling the DP matrix is to extend the recursive formula to include the number of bulges, and the number of mismatches. In this way, the matrix encodes whether an alignment of the number of bulges and mismatches exists. This comes at the cost of increasing the asymptotic runtime complexity by a factor of the number of allowed bulges and the number of allowed mismatches. Fourth, we plan to extend SWOffinder to run efficiently on multiple sgRNAs by reading the genome and traversing it only once for all sgRNAs.
To conclude, by addressing one of the major concerns associated with CRISPR/Cas9 technology, the possibility of unintended genetic modifications, SWOffinder promises to enhance the safety and accuracy of gene-editing processes. SWOffinder enables researchers to comprehensively identify potential off-target sites across the genome, allowing for thorough assessment and mitigation of off-target risks. Ultimately, the introduction of SWOffinder unlocks new avenues for precise genetic manipulation and paves the way for future advancements in the fields of medicine, agriculture, and biotechnology.
Limitations of the study
The main limitation of SWOffinder is its worst-case asymptotic runtime complexity. The SW-matrix calculation step runs in , where the length of the sgRNA sequence is m, and the length of the genome is n. This runtime is independent of the user-specified thresholds. On the other hand, the trace-back step runs in time that depends linearly on the number of sites found in the SW-matrix calculation step, and exponentially on the number of bulges allowed. While the dependence on parameter values, and especially exponential dependence, is a disadvantage, as well as the dependence on the genomic content, in practice due to the number of sites found in the SW-matrix calculation step and our novel approach, which stops searching past the maximum effective edit distance, the runtime of the trace-back step is negligible compared to the SW-matrix calculation step. So, while SWOffinder does not benefit from a guaranteed polynomial runtime in the worst case, in practice, it runs in only a few minutes over the human genome for various combinations of thresholds and performs on par or outperforms existing methods depending on the thresholds.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Human reference genome NCBI build 38, GRCh38 | The Genome Sequencing Consortium | https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/ |
| Software and algorithms | ||
| CRISRitz | Cancellieri et al.5 | https://github.com/pinellolab/CRISPRitz |
| CALITAS | Fennell et al.6 | https://github.com/editasmedicine/calitas |
| SWOffinder | This paper | https://doi.org/10.5281/zenodo.10115908 |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Prof. Yaron Orenstein (yaron.orenstein@biu.ac.il).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The publicly available data that we used in this study is listed in the key resources table.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
An overview of SWOffinder
SWOffinder utilizes the SW algorithm for local alignment, which is based on dynamic programming (DP).13 SWOffinder comprises of two steps:
-
(1)
Using a novel version of SW alignment (Figure 2), we find all positions of sites under a user-specified edit-distance threshold.
-
(2)
We apply a novel recursive procedure, which, given an end position of a potential off-target site obtained in the first step, finds the start position and the alignment of a site that satisfies user-specified separate thresholds with minimum edit distance (Algorithm 1).
Algorithm 1. The trace-back step of SWOffinder to post-process off-target sites.
Input DP matrix (M), sgRNA (a), Genome window (b), sgRNA pointer index (i), Genome-window
pointer index (j), Mismatch count (), Bulge count (), Partial alignment - sgRNA (), Partial
alignment - DNA ()
Output Alignment that follows the user-specified thresholds with minimum edit distance
Initialization sgRNA length, matching end position, , ϵ ϵ (empty string),
edits, mismatches, and bulges thresholds () are pre-defined
1: procedure Post()
2: if or are greater than the mismatches or bulges thresholds, respectively then
3: return Could not find alignment - returns
4: end if
5: if then
6: return Found alignment - returns alignment object
7: end if
8: if then
9: return Could not find alignment - returns
10: end if
11: Calculate effective max edit distance
12: if then
13: return Could not find alignment - returns
14: end if
15: Mismatch penalty
16: Alignment without bulge at position
17: Alignment with sgRNA bulge
18: Alignment with DNA bulge
19: if then
20: return Could not find alignment - returns
21: end if
22: return The alignment with minimum bulges and mismatches among
, , and
23: end procedure
SW-matrix calculation step details
Given a sgRNA sequence a of length m, and a DNA window b of length n, the first step of SWOffinder finds all end positions of sites in the DNA window that satisfy the edit-distance threshold, which we denote as . There are some key differences in how we fill and use the DP matrix M of size (Figure 2) compared to the original SW alignment.
-
•
While SW alignment zeros the first row and column cells, we zero only the first row ( for ). We do not zero the first column since it is impossible to have DNA bulges at the beginning of a sgRNA and DNA site alignment by definition.
-
•
Since it is impossible to achieve separate thresholds for mismatches, insertions, and deletions with SW alignment, we use it for filtering sites that do not meet the edit-distance threshold. Therefore, we set unit penalty costs for all edit operations in the penalty matrix function (Equation 1) resulting in an asymptotic runtime complexity of .
| (Equation 1) |
-
•
The main difference from SW alignment is how we use the DP matrix. While in general, the last column of the matrix is used to find an optimal local alignment end position, we use the last column to find end positions of all local alignments that have a total penalty cost smaller or equal to the edit-distance threshold, enabling us to find all off-target sites in a single fill of the DP matrix. Other methods, such as CALITAS,6 cannot find all off-target sites as they partition the genome into windows and find a global alignment in each window, which might miss multiple alignments in the same window.
Trace-back step details
In the second step of SWOffinder, given user-specified operation-specific thresholds, e.g. separate limits on the number of bulges and mismatches, SWOffinder returns an alignment that satisfies the separate thresholds with minimum edit distance, if there exists one, for any end position obtained in the first step. In SWOffinder, in addition to , the edit-distance threshold which is used in the SW-matrix calculation step, the user may provide 3 additional thresholds on the number of mismatches and bulges:
-
(1)
: The number of bulges allowed in the alignment.
-
(2)
: The number of mismatches allowed in the alignment when no bulges exist in the alignment.
-
(3)
: The number of mismatches allowed in the alignment when bulges exist in the alignment.
These versatile criteria enable a user to set diverse thresholds for mismatches and bulges, including distinct mismatch thresholds based on bulge presence or absence, which is common when processing and analyzing experimental off-target sites data.14
In addition to using the SW DP matrix in the SW-matrix calculation step to filter non-potential sites based on , SWOffinder utilizes the matrix to terminate the alignment search when the matrix values indicate that it is impossible to achieve such alignment. To do so, we first calculate an effective max edit, , that considers the separate operation-specific thresholds. is calculated in each instance of the function to be either when there are already bulges in the alignment or when there are no bulges. Then, as described in lines of Algorithm 1, the alignment search is terminated when the SW DP matrix value is greater than the allowed number of edits.
The runtime of the trace-back step is negligible compared to the SW-matrix calculation step in the most common cases for two main reasons: (i) it is applied to a small subset of the potential off-target sites that were obtained in the SW-matrix calculation step; and (ii) although the complexity of the trace-back step increases exponentially with the number of allowed bulges, this number is usually only 1 or 2 as prior CRISPR off-target studies showed that observing more than one DNA or RNA bulge is unlikely, even at the cost of multiple mismatches.15
Technical implementation details and features
Parallelism
Since the problem of searching off-target sites across a genome is easily parallelizable, to speed up the runtime of SWOffinder, we partition the given genome into multiple overlapping windows and then apply SWOffinder to each genome window in parallel. By the overlap, we ensure that SWOffinder does not miss any site in the border between the windows.
PAM search
SWOffinder supports off-target search with any PAM sequence and gives the user the flexibility to allow edits in the PAM sequence. If the user decides to prohibit mismatches or bulges in the PAM sequence, this can be easily achieved by modifying the SW recursive function in the relevant positions or by modifying the trace-back step of SWOffinder when the sgRNA pointer index (i in Algorithm 1) is in the PAM region. In the current implementation of SWOffinder, we implemented the second option. For clarity, we did not include this part in the pseudo-code of Algorithm 1.
Double-stranded search
As in other off-target search methods, SWOffinder supports off-target search on both the forward and reverse strands. To reduce I/O operations and avoid computing the reverse complement of the given genome when searching over the reverse strand, SWOffinder reads the genome only once and computes the reverse-complement of the sgRNA sequence instead of the genome. As a result, SWOffinder reverses the orientation of the PAM, which is taken into account when aligning the PAM sequence.
Versatile search options
There are multiple options for user-defined thresholds when searching for off-target sites by SWOffinder. Any combination of thresholds is allowed. In the most simple form, the user can search for all off-target sites up to a given edit-distance threshold. If additional thresholds are provided, i.e. for mismatches, mismatches with bulges, and/or bulges, the sites found under the edit-distance threshold are further filtered to meet the user-specified separate thresholds.
Finding an optimal site within a user-defined genome window
Most of the current state-of-the-art experimental techniques to detect off-target sites in the genome cannot identify the exact position of an off-target site within a sequencing read.14,16 Therefore, given a genome window (i.e., sequencing read) in which the edit occurred, a post-processing step of the experimental data aims to find an alignment to the sgRNA achieving minimum edit distance. In analogue, SWOffinder provides an option to find sites achieving the minimum edit distance within a genome window of a user-specified length. To enable this option, SWOffinder keeps two consecutive potential sites that were found in the trace-back step. If two sites are found in the same window, the site with the minimum edit distance is selected. In case of a tie, SWOffinder returns the first site.
Evaluation and comparison to existing off-target search methods
To evaluate the performance of SWOffinder and compare it to the state-of-the-art off-target search methods, CALITAS6 and CRISPRitz,5 we performed two types of evaluations: off-target site identification and runtime. We did not include Cas-OFFinder3 in the benchmarks and comparisons as its developers stated that their unpublished bulge-supported version is unstable.17 Indeed, previous studies included Cas-OFFinder in their benchmarks and comparisons and showed its inferior performance.5,6
For both CALITAS and CRISPRitz, we had to create a reference genome index prior to running the methods. In CRISPRitz, this is required for each combination of a PAM sequence and bulge size. We excluded the creation of the index from the runtime measurement.
We ran CALITAS with the following command line: calitas SearchReference -i <sgRNA with PAM> -r <genome reference and index path> -max-total-diffs <integer> -max-pam-mismatches <integer> -max-gaps-between-guide-and-pam <integer> -threads <integer>.
We ran CRISPRitz with the following command line: crispritz.py search <genome index path> <PAM file path> <sgRNAs list file path> <output path> -index -mm <integer> -bDNA <integer> -bRNA <integer> -r -th <integer>.
Benchmarking off-target sites identification
Although SWOffider is guaranteed to find all potential off-targets in a given genome thanks to the correctness of the SW algorithm, it is still necessary to validate the implementation of SWOffinder compared to other methods. To compare the methods in terms of off-target sites identification, we defined three tests:
-
(1)
The number of unique sites, where a site is defined as the (chromosome, strand, end position) triplet.
-
(2)
The number of non-consecutive unique sites, where sites with consecutive end positions are considered in the count as one site. This allows for avoiding scenarios where different sites are counted as unique only due to the permitted bulge size.
-
(3)
The number of sites identified in a window of a site found by SWOffinder. We examined for each off-target site identified by SWOffinder whether other sites were found by existing methods in the genome window of the SWOffinder off-target site. The genome window is defined as the SWOffinder off-target site plus additional flanks of the size of the maximum bulge size allowed.
Runtime comparison
To compare the runtime performance of SWOffinder to state-of-the-art methods for CRISPR off-target search, we tested the runtime of the methods over various edit-distance thresholds, and combinations of mismatch and bulge thresholds, in both single- and multi-threaded runs. To conduct a fair comparison, we used the same AWS machine with 1st generation Intel Xeon Platinum 8000 eight 3.1GHz cores, and 32GB RAM for all runtime tests, and reported the Elapsed (wall clock) time and Maximum Resident Set Size as measured by the Unix time -v command for runtime and maximum memory usage, respectively.
Acknowledgments
This study was partially supported by the Israel Science Foundation (grant no. 358/21). We gratefully acknowledge the support of the CRISPR-IL consortium for supporting our research and for giving access to the AWS platform used for part of this work. Ofir acknowledges the support of the Azrieli PhD fellowship, the Kretiman PhD fellowship at Ben-Gurion University, Nehemia Levtzion Ph.D. fellowship from the Israeli Council for Higher Education, and the cloud-compute credit by the Planning and Budgeting Committee through IUCC.
Author contributions
OY conducted the analyses and developed all the code used in this study. AM and EC assisted with the analyses. YO supervised this study. OY and YO wrote the manuscript.
Declaration of interests
The authors declare that they have no competing interests.
Published: November 23, 2023
Contributor Information
Ofir Yaish, Email: yaishof@post.bgu.ac.il.
Yaron Orenstein, Email: yaron.orenstein@biu.ac.il.
References
- 1.Jiang F., Doudna J.A. CRISPR–Cas9 structures and mechanisms. Annu. Rev. Biophys. 2017;46:505–529. doi: 10.1146/annurev-biophys-062215-010822. [DOI] [PubMed] [Google Scholar]
- 2.Pineda M., Lear A., Collins J.P., Kiani S. Safe CRISPR: challenges and possible solutions. Trends Biotechnol. 2019;37:389–401. doi: 10.1016/j.tibtech.2018.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Bae S., Park J., Kim J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Backurs A., Indyk P. Proceedings of the forty-seventh annual ACM symposium on Theory of computing. 2015. Edit distance cannot be computed in strongly subquadratic time (unless SETH is false) pp. 51–58. [Google Scholar]
- 5.Cancellieri S., Canver M.C., Bombieri N., Giugno R., Pinello L. CRISPRitz: rapid, high-throughput and variant-aware in silico off-target site identification for CRISPR genome editing. Bioinformatics. 2020;36:2001–2008. doi: 10.1093/bioinformatics/btz867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fennell T., Zhang D., Isik M., Wang T., Gotta G., Wilson C.J., Marco E. CALITAS: a CRISPR-cas-aware ALigner for in silico off-TArget Search. CRISPR J. 2021;4:264–274. doi: 10.1089/crispr.2020.0036. [DOI] [PubMed] [Google Scholar]
- 7.Bao X.R., Pan Y., Lee C.M., Davis T.H., Bao G. Tools for experimental and computational analyses of off-target editing by programmable nucleases. Nat. Protoc. 2021;16:10–26. doi: 10.1038/s41596-020-00431-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones S.K., Jr., Hawkins J.A., Johnson N.V., Jung C., Hu K., Rybarski J.R., Chen J.S., Doudna J.A., Press W.H., Finkelstein I.J. Massively parallel kinetic profiling of natural and engineered CRISPR nucleases. Nat. Biotechnol. 2021;39:84–93. doi: 10.1038/s41587-020-0646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin J., Zhang Z., Zhang S., Chen J., Wong K.-C. Crispr-net: A recurrent convolutional network quantifies crispr off-target activities with mismatches and indels. Adv. Sci. 2020;7 [Google Scholar]
- 10.Zhang Z.-R., Jiang Z.-R. Effective use of sequence information to predict crispr-cas9 off-target. Comput. Struct. Biotechnol. J. 2022;20:650–661. doi: 10.1016/j.csbj.2022.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: a web-based tool for identifying and validating CRISPR/Cas off-target sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y., Huang W., Johnson J., Vaidya S. Computational Science–ICCS 2006: 6th International Conference, Reading, UK, May 28-31, 2006, Proceedings, Part IV. Vol. 6. 2006. GPU accelerated smith-waterman; pp. 188–195. [Google Scholar]
- 13.Smith T.F., Waterman M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 14.Lazzarotto C.R., Malinin N.L., Li Y., Zhang R., Yang Y., Lee G., Cowley E., He Y., Lan X., Jividen K., et al. Change-seq reveals genetic and epigenetic effects on crispr–cas9 genome-wide activity. Nat. Biotechnol. 2020;38:1317–1327. doi: 10.1038/s41587-020-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pacesa M., Lin C.-H., Cléry A., Saha A., Arantes P.R., Bargsten K., Irby M.J., Allain F.H.-T., Palermo G., Cameron P., et al. Structural basis for cas9 off-target activity. Cell. 2022;185:4067–4081.e21. doi: 10.1016/j.cell.2022.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P., et al. Guide-seq enables genome-wide profiling of off-target cleavage by crispr-cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeongbin P., Sangsu B. GitHub repository; 2022. Cas-offinder.https://github.com/snugel/cas-offinder [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
-
•
The publicly available data that we used in this study is listed in the key resources table.
-
•
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.