

Introduction
Allergic asthma is characterized by Th2-type lung inflammation, increased mucus production, and changes to the airways, including remodeling, obstruction, and airway hyperresponsiveness (AHR). The evidence for mast cell involvement in asthma includes pulmonary mast cell hyperplasia and the abundance of leukotrienes, tryptase, chymase, and histamine in the sputum and bronchoalveolar lavage fluid of asthmatic patients (1–4). Furthermore, anti-IgE treatment is effective in ~60% of asthmatic pateitnts, which supports the involvement of IgE and mast cells in human asthma (5). Mouse models of allergic lung inflammation have shown that the involvement of mast cells is dependent on the type of antigen, the potency of sensitization, genetic background, and sex (6–12). Although these models do not fully recapitulate human disease, they have greatly expanded our knowledge of allergic asthma. We know that mast cells exacerbate AHR and lung inflammation and play a critical role in promoting pulmonary eosinophilia in mice (6, 7, 12). For example, mast cell activation causes AHR in mice (6) and the protective role of prostaglandin E2 (PGE2) in reducing AHR requires is likely due to PGE2-mediated suppression of mast cells (8, 10). Overall, the clinical and experimental evidence suggest that mast cells are an important target for treating allergic asthma.
Alterations in allergic inflammation include increased extracellular adenosine triphosphate ATP in the asthmatic lung (13). By binding to purinergic receptors, ATP functions as a damage-associated molecular pattern (DAMP) that amplifies inflammation (14–16). Human mast cells express mRNA for the ATP receptors P2X1, 4 and 7 (17, 18). Mast cell P2X receptors stimulate Ca2+ influx and cytokine/chemokine release upon ligation by ATP (18). Furthermore, ATP signaling through mast cell P2X7 promotes IL-33 release and intestinal inflammation in a murine colitis model and in human Crohn’s disease (19). ATP-driven P2X7 activation induces mast cell degranulation, while P2X1 and 4 enhance IgE/FcεRI-induced degranulation (20). The role of P2X receptors in mast cell cytokine secretion is poorly understood. These data suggest that ATP receptors are a possible therapeutic target in allergic disease and may act directly on mast cells.
Selective serotonin reuptake inhibitors (SSRIs) are antidepressants which may have off-target anti-inflammatory effects. For example, a meta-analysis of studies examining serum cytokine levels before and after treatment of patients diagnosed with major depressive disorder found that SSRI treatment reduced serum TNF and IL-6 (21). Patients with the co-morbid diagnoses of major depression and multiple sclerosis (MS) undergoing SSRI treatment had decreased IFNɣ production, results corroborated by work showing SSRIs reduced the onset of experimental MS in mice (22, 23). Relevant to our study, a trial studying the safety of administering SSRIs to asthmatics showed that the SSRI group had decreased corticosteroid use, an important measure of asthma severity (24). Collectively, these publications suggest that SSRIs may be a therapeutic option for multiple inflammatory diseases.
Although these studies show that SSRIs have anti-inflammatory activities, their effects in allergic disease are unclear. SSRIs inhibit the serotonin reuptake transporter (SERT) with high affinity (25), a feature that has proven predicative validity for antidepressant potential. Nonetheless, recent studies have suggested that these drugs may have therapeutic activity through other targets (26), though this contention remains a topic of significant debate (27). In this regard, SSRIs have been shown to inhibit P2X4, P2X7, and the inflammasome component NLRP3 (28–30), suggesting that the anti-inflammatory actions for these drugs and targets should be rigorously tested.
Using primary mouse and human mast cells, we showed that the SSRI fluoxetine suppresses IgE-mediated Ca2+ flux, degranulation, cytokine production, and inflammatory lipid secretion. Several other SSRIs showed similar effects. Reduced mast cell function was matched by decreased FcεRI signaling in the presence of fluoxetine. We found that FcεRI stimulation elicited rapid ATP release that augmented cytokine production. Fluoxetine suppressed ATP-mediated Ca2+ flux, degranulation, lipid secretion, cytokine production, and NFκB function. Fluoxetine effects were consistent in models of passive systemic anaphylaxis (PSA) and house dust mite (HDM)-induced AHR and lung inflammation. Collectively, our data supported the premise that fluoxetine might be re-purposed for use in allergic disease and that its mechanism of action involves blocking an ATP-mediated amplification of mast cell function.
Results
SSRIs inhibit IgE-mediated mast cell function
We first tested a panel of SSRIs for effects on mast cell cytokine production induced by antigen-mediated IgE crosslinkage (IgE XL) (Fig. 1A). The drug concentrations used were the highest that did not affect the viability of C57BL/6J bone marrow-derived mast cells (BMMCs). All 5 SSRIs suppressed cytokine production to varying degrees, with citalopram being the least inhibitory and paroxetine the most inhibitory.
Fig. 1. Fluoxetine suppresses mast cell function in vitro.
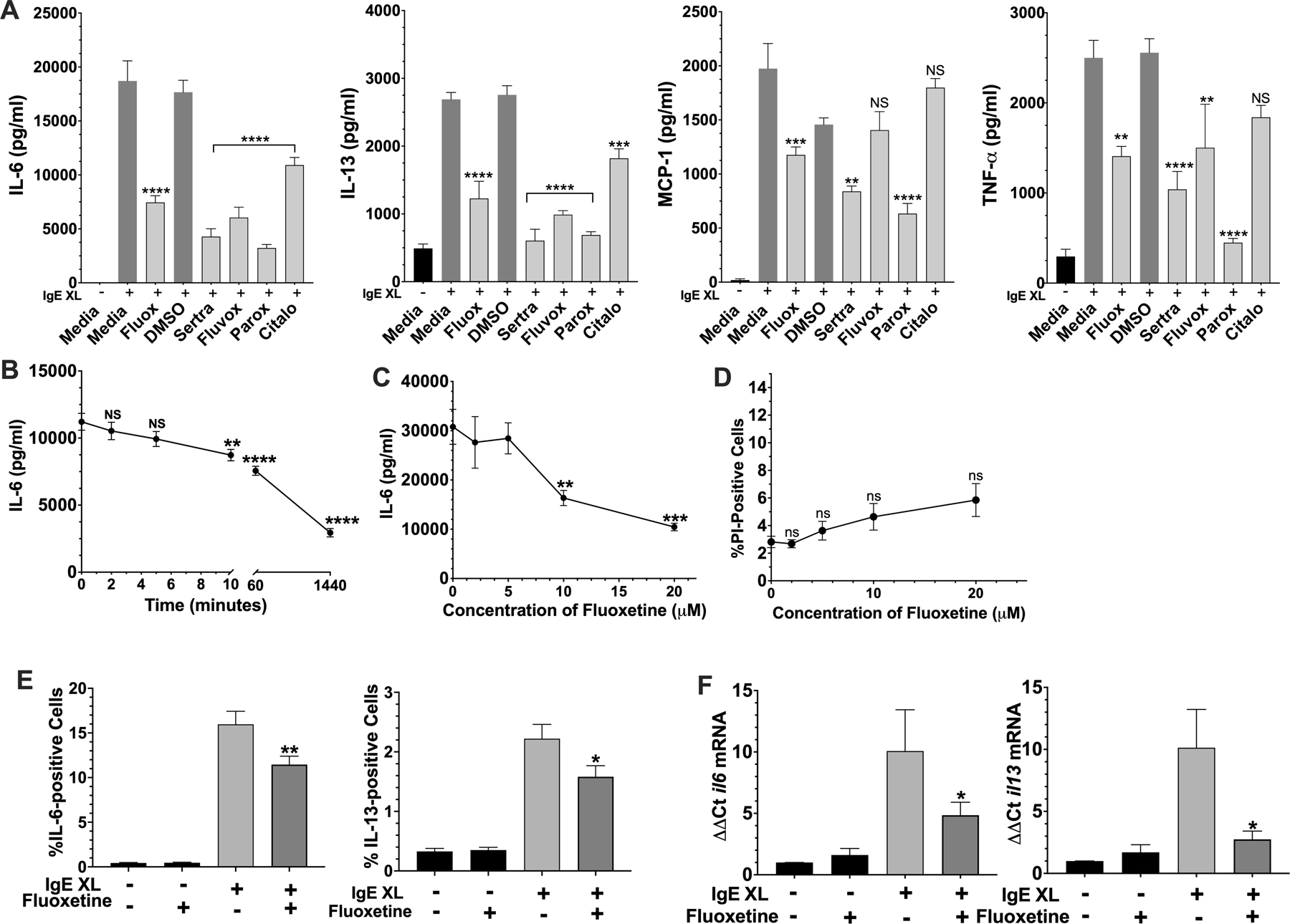
(A) BMMC pre-sensitized with dinitrophenyl (DNP)-specific IgE were cultured with fluoxetine, sertraline, fluvoxamine, paroxetine, citalopram or vehicles, for 24 hours. DNP-HSA was added for 16 hours and cytokine concentrations in culture supernatants were determined by ELISA. N=6/group (B) BMMCs were treated with fluoxetine for the indicated time before IgE XL and DNP-HSA was added to crosslink for 16 hours. IL-6 levels in culture supernatant were measured by ELISA. N=4–6/group (C) BMMCs were treated as in (A) with the indicated concentrations of fluoxetine, and IL-6 levels were measured by ELISA. N=6/group (D) BMMCs were treated as in (A) with the indicated concentrations of fluoxetine, and viability as measured by flow cytometry after staining with propidium iodide. N=6/group. (E) BMMCs were treated as in (A), except that IgE XL was performed for 2 hours prior to the addition of monensin for 5 hours, then analyzed by flow cytometry. N= 16–19/group. (F) BMMCs cultured as in (A) were activated with IgE XL for 4 hours and RNA was collected using Trizol. Cytokine mRNA levels were determined by RT-qPCR. N=9/group. Data in A-D are representative of three independent experiments done in biological triplicates or quadruplicates. In (E-F), data are pooled from at least three independent experiments done in biological triplicates. All p values were determined by 1-way ANOVA (Tukeys test). *p<.05; **p<.001; ****p<.0001.
Inbred laboratory mice are genetically variable across strains (31). To test for genotypic effects on SSRI-induced suppression, the experiment was repeated with BMMCs derived from BALB/cJ and 129S1/SvImJ strains (Suppl. Fig. 1A,B). Fluoxetine and paroxetine suppressed cytokine production regardless of mouse strain background (Figure 1A, Suppl. Figure 1A,B). Due to its consistent activity in these assays and its water solubility, we focused on fluoxetine.
Fluoxetine addition for at least 10 minutes before IgE XL suppressed IL-6 production (Fig. 1B). Because we noted maximal effects after 24 hours, we continued to use this timepoint for our studies. At least 10μM fluoxetine was needed to inhibit FcεRI-mediated cytokine secretion (Fig. 1C), conditions that did not affect BMMC viability (Fig. 1D). This is a clinically relevant dose because tissue concentrations of fluoxetine up to 60μM have been reported in adults, although plasma concentrations are much lower, typically 1–2μM (32, 33). Since mast cells are tissue resident cells, tissue concentrations are more relevant to our results.
To begin determining how fluoxetine suppresses cytokine secretion, we measured intracellular IL-6 and IL-13. As with secretion, IgE XL-mediated IL-6 and IL-13 accumulation were also decreased by fluoxetine (Fig. 1E), albeit to a lesser extent. We then measured cytokine mRNA levels using RT-qPCR for il6 and il13 mRNAs (Fig.1F). Since the induction of both cytokine mRNAs was diminished, we hypothesized that fluoxetine-mediated cytokine suppression by affecting mRNA stability or transcription.
Mouse peritoneal mast cells and human skin mast cells are suppressed by fluoxetine
Peritoneal mast cells (PMCs) are differentiated in vivo and thus provide a tool to test whether fluoxetine effects on BMMCs are due to in vitro maturation. PMCs were treated with fluoxetine and activated by IgE XL as described in Figure 1. Fluoxetine suppressed both cytokine production and degranulation in PMC cultures (Suppl. Fig. 2 A-B), suggesting that these effects are not linked to in vitro maturation.
We corroborated our mouse studies by testing fluoxetine effects on human skin-derived mast cells (HuMC; Fig. 2A). These cells matured in vivo and were expanded ex vivo, similar to the PMCs. IgE-induced cytokine production by HuMC from 5 donors was significantly inhibited by fluoxetine. Collectively, these data show that fluoxetine effects are consistent on distinct mast cell populations from mouse or human origin.
Fig. 2. Fluoxetine inhibits human mast cell responses to IgE XL.
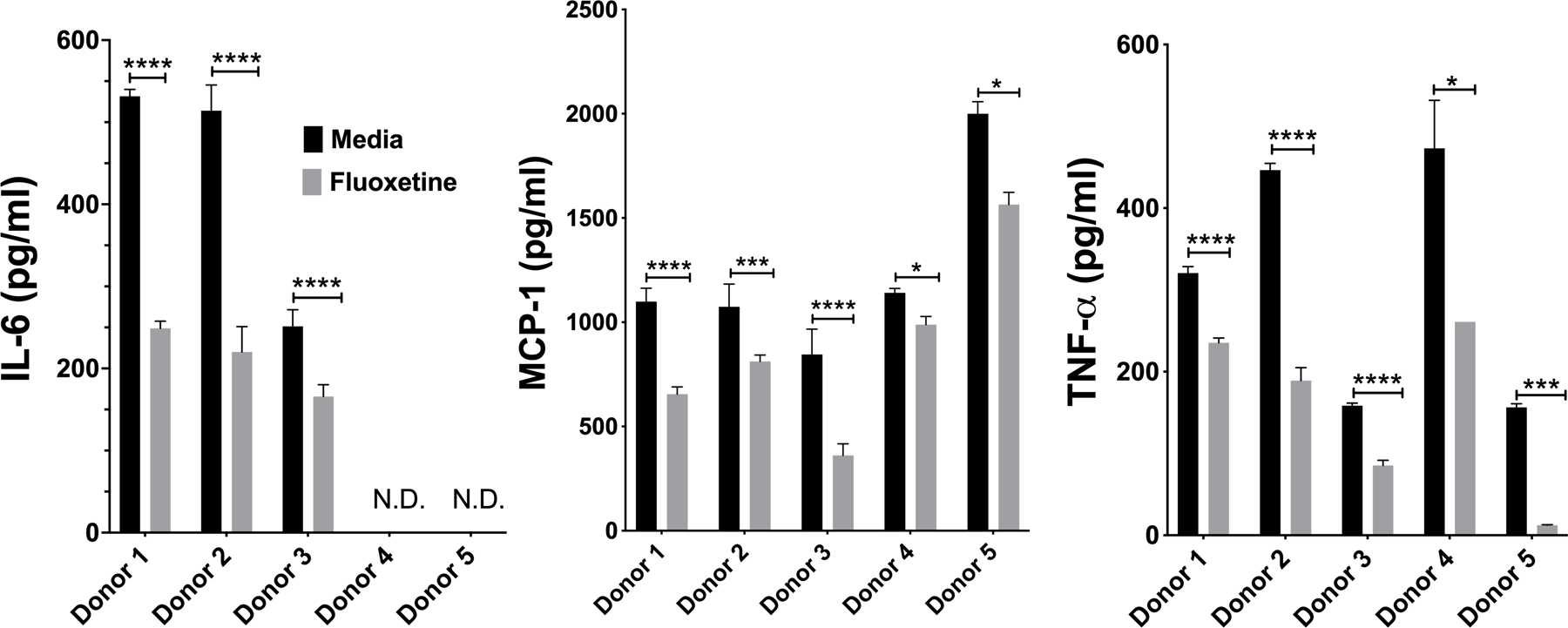
(A) HuMC were treated with anti-DNP IgE and fluoxetine, crosslinked with DNP-HSA and cytokines were measured by ELISA. Each donor represents an individual subject and were analyzed as technical triplicates, (biological N= 5/group). Statistical values were determined by paired Student’s t-test. *p<.05; **p<.001; ****p<.0001. N.D.= Not detected.
Fluoxetine inhibits FcεRI-mediated degranulation and eicosanoid secretion
Mast cell activation is multi-phasic. The early phase response involves the release of pre-formed mediators, most of which are stored in granules, and this process ofdegranulation occurs within minutes. We tested whether fluoxetine suppressed IgE-mediated degranulation. BMMCs were treated with fluoxetine, then activated by IgE XL. The cells were stained with anti-CD107a, a lysosomal marker whose surface expression corresponds to degranulation (34) and analyzed by flow cytometry. Degranulation was also measured by the release of histamine, a preformed mediator stored in mast cell granules. IgE-mediated CD107a expression and histamine release were both reduced by fluoxetine (Fig. 3A).
Fig. 3. Fluoxetine Inhibits the early phase of mast cell activation.
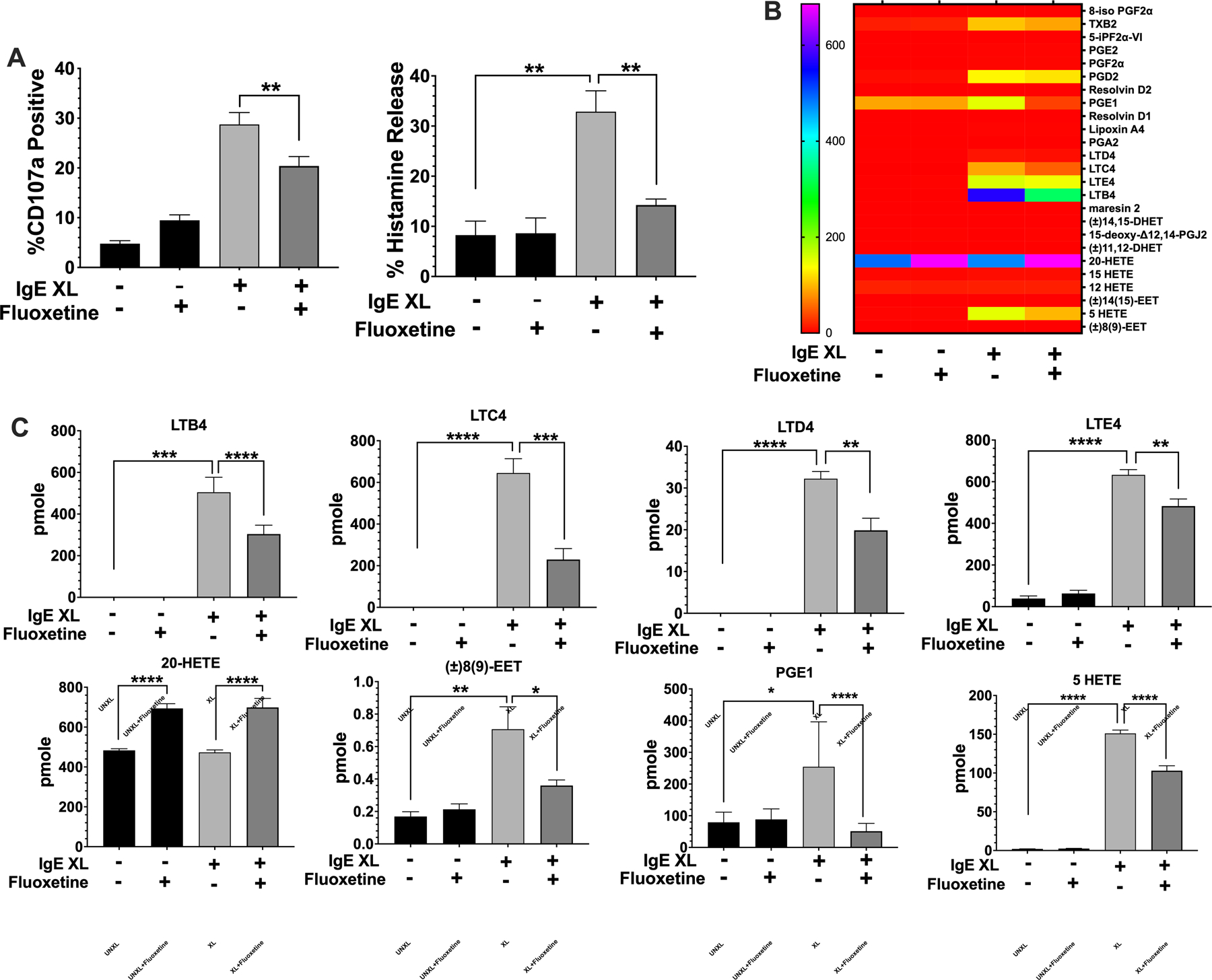
(A) BMMCs were treated with fluoxetine and activated by IgE XL for 30 minutes. Cells were either analyzed by flow cytometry for CD107a surface expression or cell pellets and supernatant were collected for histamine ELISA. N=12/group. (B,C) BMMCs were treated with fluoxetine and activated by IgE XL for 1 hour. Supernatant was analyzed for eicosanoid production by mass spectrometry. N=3/group. Data are pooled from at least three independent experiments with biological triplicates in A. B and C is representative of two independent experiments with biological triplicates, per experiment. Data are pmole/4 million cells from biological triplicate samples analyzed by 1-way ANOVA (Tukey’s test). *p<.05; **p<.001; ****p<.0001. NS, not statistically significant.
Mast cells also produce eicosanoids that contribute to chemotaxis and vasodilation. Thus, we cultured BMMC for 24 hours with or without fluoxetine and measured lipids by mass spectrometry 1 hour after IgE XL. Thirteen inflammatory lipids were induced by IgE XL (Fig. 3B). Fluoxetine significantly reduced production of LTB4 and the cysteinyl leukotrienes LTC4, LTD4, and LTE4. Two additional eicosinoids, PGE1 and 8(9)-EET were also inhibited (Fig. 3C). Fluoxetine induced 20-HETE (Fig. 3C) and had no suppressive effect on FcεRI-mediated induction of five eicosanoids, including several prostaglandins (Suppl. Fig. 3).
Fluoxetine inhibits early FcεRI signaling events
One explanation for reduced IgE-mediated function is decreased expression of either FcεRI or c-Kit, the SCF receptor that augments FcεRI signaling and is stimulated by SCF in the culture media (35–37). Therefore, we measured surface FcεRI and c-Kit with or without fluoxetine treatment (Suppl. Fig. 4). Fluoxetine did not alter c-Kit and unexpectedly increased FcεRI expression. Therefore, we concluded that reduced receptor expression is not a mechanism of action for the observed suppression of FcεRI-mediated mast cell function.
We next assessed fluoxetine effects on the FcεRI signaling cascade. This revealed that fluoxetine suppressed IgE XL-mediated Ca2+ flux and the phosphorylation of Syk, Btk, Akt, Erk and STAT5 (Fig. 4 A,B). These data suggested an inhibitory mechanism acting early in FcεRI signaling that has broad downstream effects.
Fig. 4. Fluoxetine inhibits early mast cell signaling events.
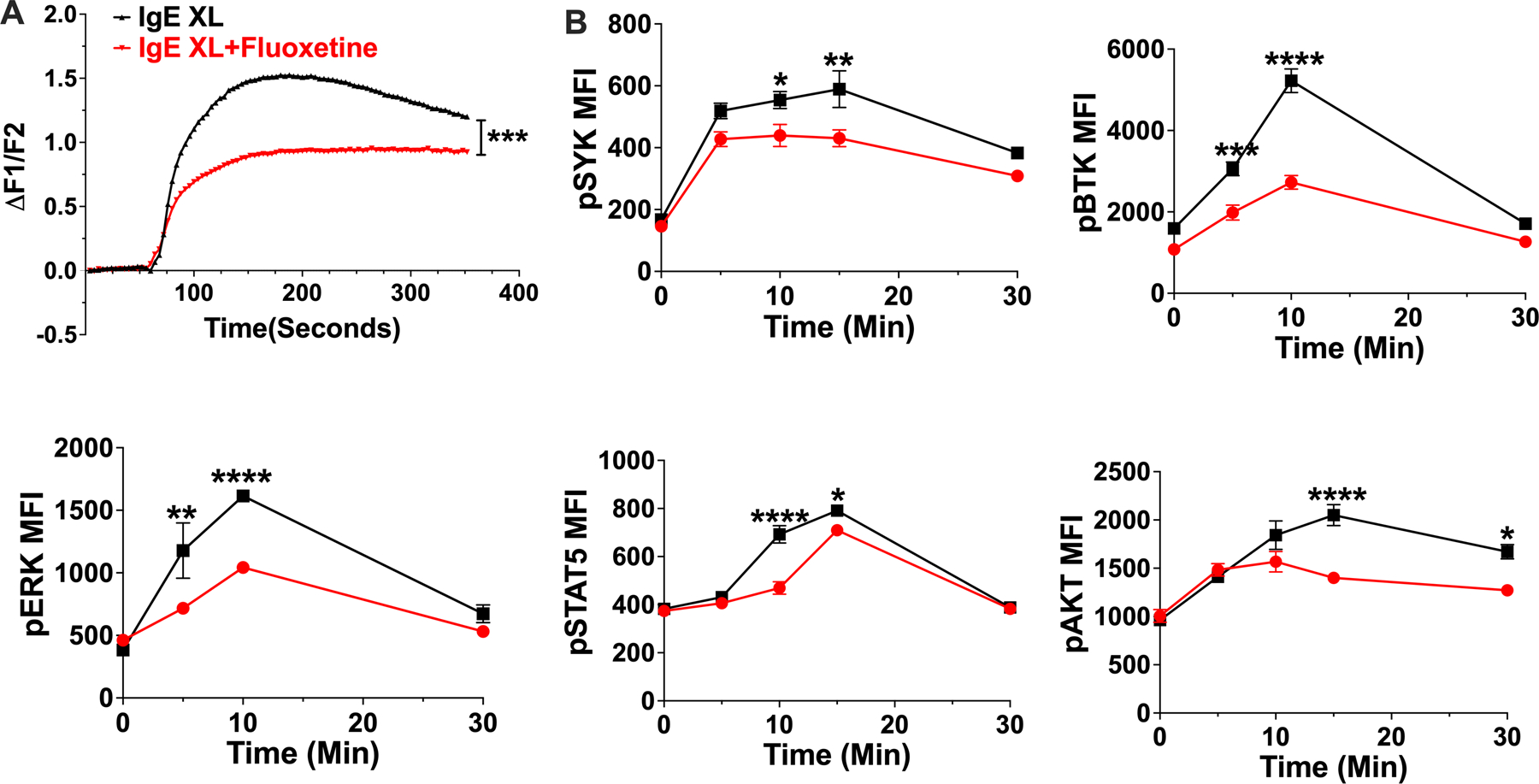
(A) BMMCs were treated with fluoxetine and activated by IgE XL. Ca2+ flux was measured by FURA-2 staining. N=11–12/group. (B) BMMCs treated with fluoxetine, then crosslinked for the indicated times and flow cytometry was used to measure phosphoprotein expression. N= 3/group. Data are representative of two (A) or (B) three independent experiments performed in biological triplicates (A) or quadruplicates (B). Statistical values were determined by t-Test for (A) and 1-way ANOVA (Tukey’s or Sidak’s test). *p<.05; **p<.001; ****p<.0001.
SERT expression or increased extracellular serotonin do not explain fluoxetine effects
Mast cells can produce serotonin, and express several serotonin receptors and the serotonin reuptake transporter (SERT) (38). Since fluoxetine targets SERT, whose inhibition might increase extracellular serotonin availability, we determined whether serotonin or SERT is involved in suppressing mast cell function. BMMCs were treated with 50–400 ng/ml of mouse serotonin prior to IgE XL, butno effect on cytokine production was noted (Suppl. Fig. 5A). We then tested the involvement of SERT by performing a fluoxetine dose response on SERT knock down (KD) BMMCs (Suppl. Fig. 5B) and on SERT deficient (KO) BMMCs (Suppl. Fig. 5C). Both the KO and KD responded to fluoxetine similarly to control BMMCs. We confirmed that SERT KO BMMC lacked SERT protein and possessed the targeted genomic allele (Suppl. Fig. 5D). These cells expressed the mast cell surface markers c-Kit, ST2, and FcεRI at levels comparable to WT BMMC (Suppl. Fig. 5E). These data showed that SERT expression is not required for fluoxetine-mediated suppression, suggesting an off-target drug effect.
Fluoxetine suppresses ATP-induced mast cell activation
It has been previously reported that fluoxetine may mimic the structure of ATP and that paroxetine can suppress ATP-mediated signaling by P2X4 and P2X7 receptors (28–30). Since extracellular ATP can activate mast cells through P2X receptors (17, 18), we initially tested if fluoxetine inhibits ATP-mediated activation. Fluoxetine suppressed all aspects of ATP-induced signaling measured, including Ca2+ flux, degranulation, inflammatory lipid production, NFκB function, and IL-6 secretion, and (Figure 5 A-F).
Fig. 5. Fluoxetine inhibits ATP-mediated mast cell activation.
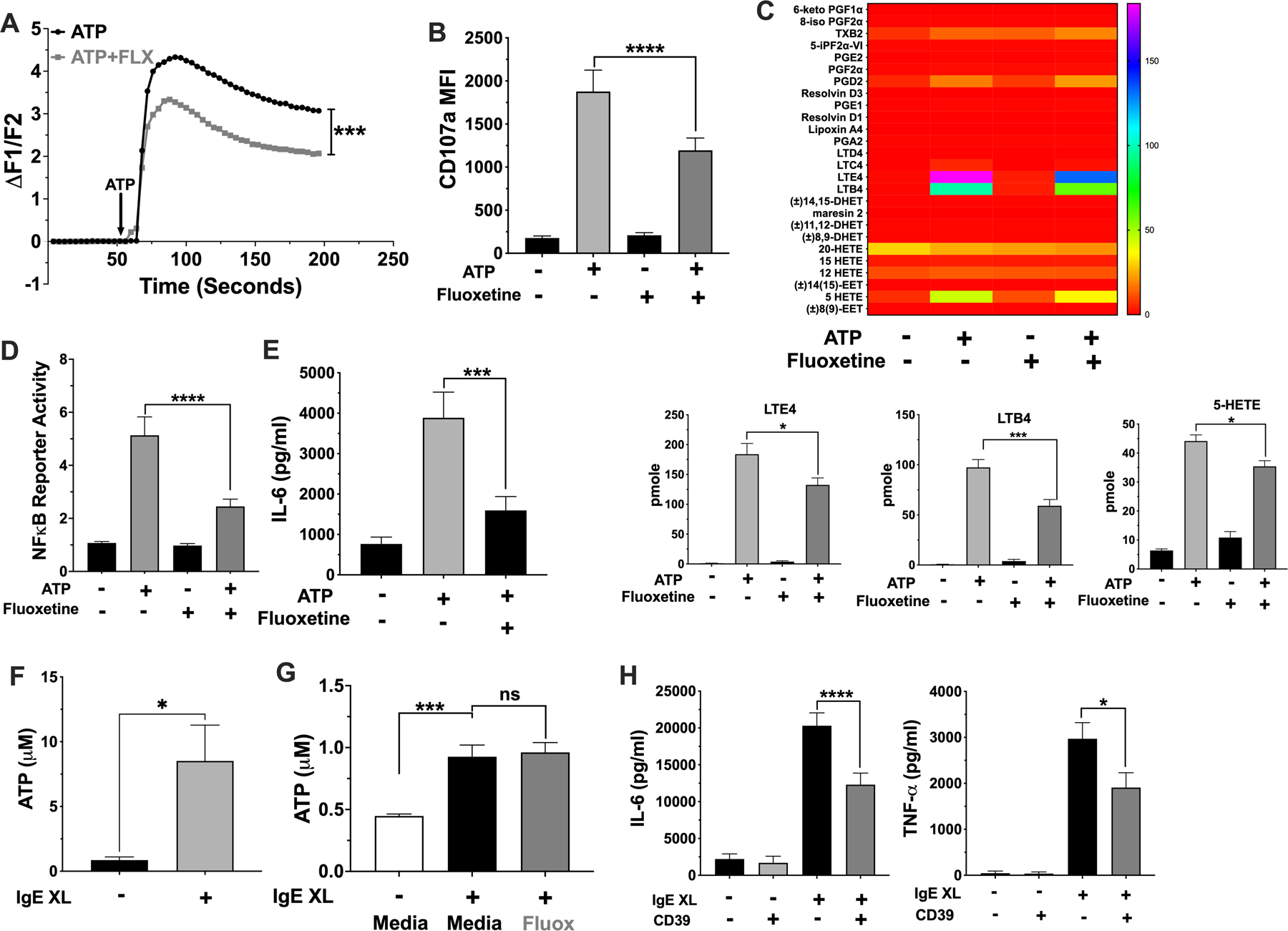
(A-E) BMMCs were treated with fluoxetine and activated with ATP (A) prior to measuring Ca2+ influx. N= 6/group. (B) BMMCs surface CD107a expression was measured by flow cytometry. N=12/group. (C) BMMC supernatant lipids were measured by mass spectroscopy. Lipids that were significantly reduced by fluoxetine are shown in the bar charts; all others are shown in the heatmap. Data are pmole/4 million cells and analyzed by ANOVA (Tukey’s test). N=4/group. (D) BMMCs NFκB reporter activity was measured by luciferase assay. N=9/group. (E) BMMC IL-6 was measured by ELISA. N=12/group. (F) BMMCs were activated by IgE XL for 60 seconds and supernatant ATP was measured using a luminescent ATP assay. N=6/group from 2 independent experiments. (G) IgE-sensitized BMMCs were treated with fluoxetine, then crosslinked with DNP-HSA for 1 minute and supernatant was used to detect ATP by luminescence. N=9/group. (H) BMMCs were activated by IgE XL in the presence or absence of recombinant mouse CD39. Supernatant cytokines were measured by ELISA. N=6–12/group. All data are pooled from at least 2 independent experiments or a representative of two independent experiments done in quadruplicates. Statistical values were determined by 1-way ANOVA (Tukey’s test) or Student’s t-Test (A, using area under curve values). *p<.05; **p<.001; ****p<.0001.
ATP is a well-recognized DAMP (13, 14, 16) that can amplify inflammation (14–16). For example, mice deficient in ecto-nuclease expression have exacerbated allergic lung inflammation and allergic dermatitis (19, 39, 40). IgE-induced ATP production and ATP effects on allergic inflammation have been partly ascribed mast cells and basophils (40). To test for the possibility of an ATP autocrine loop in mast cells, ATP release was measured after IgE XL. We detected extracellular ATP in the first minute after IgE XL (Fig. 5F), which was unchanged by fluoxetine treatment (Fig. 5G). Furthermore, the addition of recombinant CD39 ATPase at the time of stimulation reduced IgE-mediated IL-6 secretion (Fig. 5H). Collectively, these data suggested that IgE XL elicits rapid ATP release that amplifies mast cell function, and that fluoxetine reduces ATP signaling rather than ATP release.
Fluoxetine suppresses mast cell-dependent anaphylaxis in vivo
We next examined fluoxetine effects on FcεRI-induced mast cell function in vivo. Female C57BL/6J mice were passively sensitized with anti-DNP IgE and treated with saline (PBS) or fluoxetine by intraperitoneal (IP) injection. A second injection of PBS or fluoxetine was given 16 hours later, followed by antigen. Hypothermia (Figure 6A), plasma histamine (Figure 6B), and plasma cytokines (Figure 6C) were measured. Fluoxetine significantly reduced hypothermia, which correlated with reduced plasma histamine and cytokines. We also subjected mice to histamine-induced anaphylaxis to test whether fluoxetine affected the vascular response to histamine rather than acting on mast cells directly. Mice treated with PBS or fluoxetine showed similar hypothermic responses to histamine, indicating that fluoxetine does not prevent the vascular histamine response (Figure 6D). Thus, our data showed that multiple parameters of mast cell function are suppressed by fluoxetine, in vitro and in vivo.
Fig. 6. Fluoxetine inhibits passive systemic anaphylaxis.
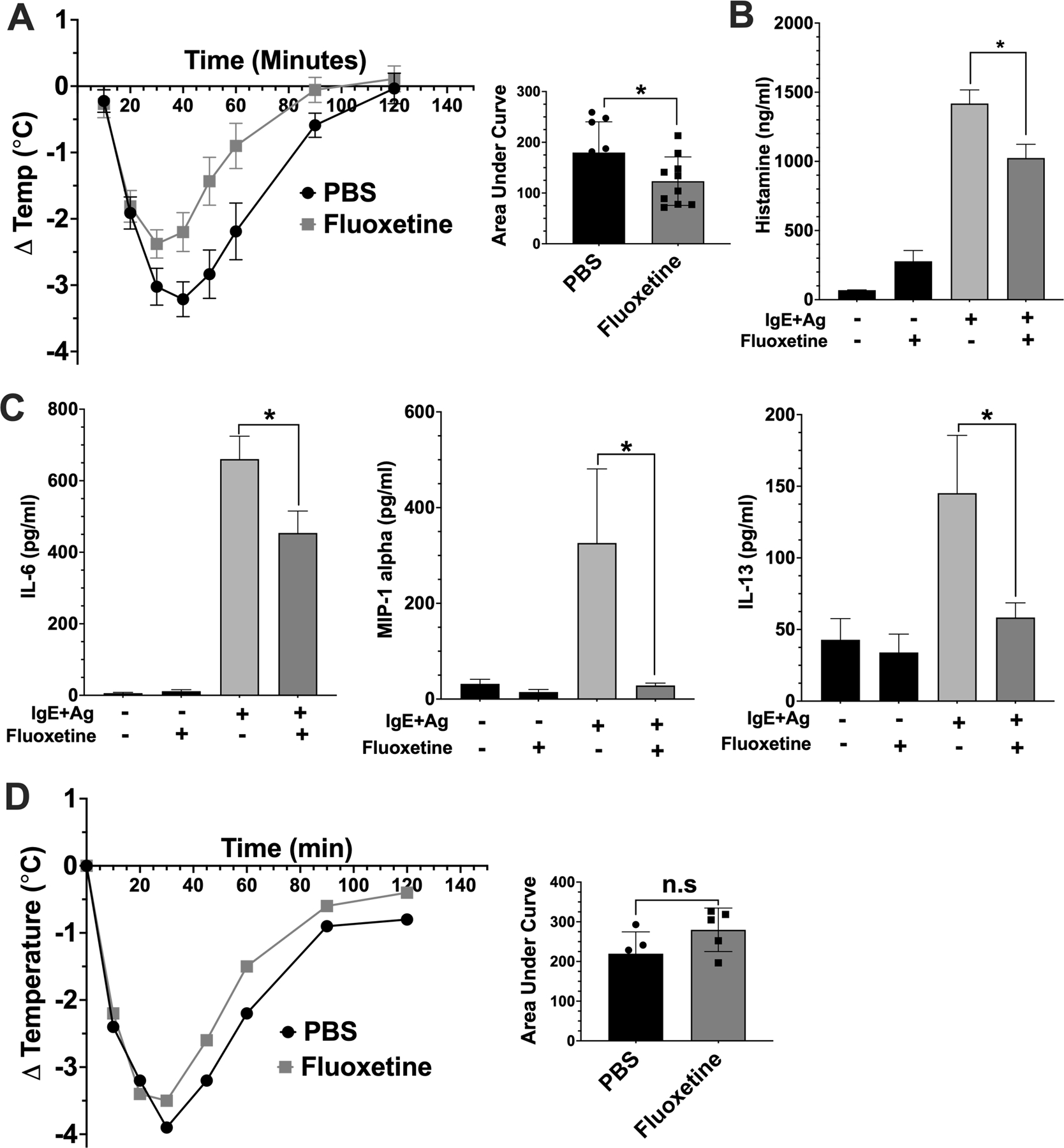
(A) Mice were sensitized with anti-DNP IgE and treated with PBS or fluoxetine. The next day, anaphylaxis was induced with DNP-has and core body temperature was measured (A, N=9/group). (B) Mice were euthanized 30 minutes after DNP-HSA injection to assess plasma histamine levels by ELISA (N=10/group). (C) Mice were euthanized after 2 hours, and plasma was collected for cytokine analysis by ELISA (N=10/group). (D) Mice (N=5/group) were treated with PBS or fluoxetine as in (A). Histamine was administered and Core body temperature was measured. Data are pooled from two independent repeats. Statistical values were determined by 1-way ANOVA (Tukey’s test) and area under the curve and Student’s t-test in (A). *p<.05; **p<.001; ****p<.0001. n.s.=not statistically significant.
Fluoxetine reduces HDM-induced AHR and lung inflammation
Next, we determined fluoxetine effects in a house dust mite (HDM)-induced allergic lung inflammation model that mimics some aspects of human asthma. Mice were sensitized to HDM in alum intraperitoneally and exposed to HDM intranasally as shown in Figure 7A. On days 11–22, mice received daily intranasal PBS or fluoxetine (Figure 7A). Fluoxetine significantly decreased methacholine-induced airway responsiveness as measured by resistance (Rrs) and elastance (Ers) (Figure 7B), indicating that the drug improved lung function.
Fig. 7. Fluoxetine diminishes HDM-induced lung inflammation and airway hyperresponsiveness.
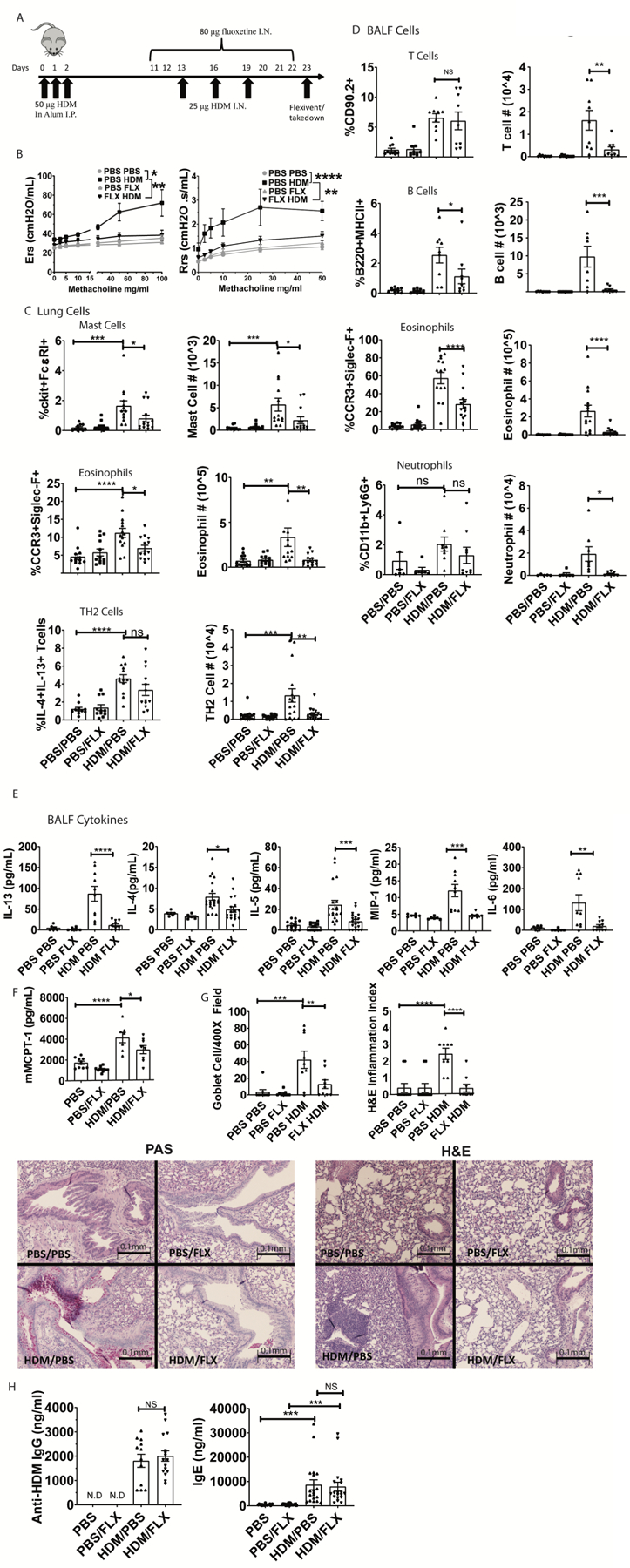
(A) Mice were sensitized and challenged with HDM while receiving fluoxetine or PBS treatments. Animals and tissue were analyzed on day 23. (B) Methacholine-induced lung resistance was measured in anesthetized mice using a Scrieq flexivent instrument. Data are pooled from 2 independent experiments with N=8/group total. (C) Digested lung tissue single cell suspension was analyzed by flow cytometry for mast cells (FcεRI+cKit+), eosinophils (siglecF+CCR3hi) and Th2 cells (CD3+CD4+IL13+IL4+). N=11–14/group. (D) BALF cells were analyzed for T cells (CD90.2+/MHC II-neg), B cells (B220+MHCII+), eosinophils (SiglecF+CCR3+) and neutrophils (CD11b+Ly6G+). Data are pooled from at least 2 independent repeats with each point representing an individual mouse. N=9/group. (E) BALF cytokines were measured by ELISA. Data are pooled from 3 independent experiments. N=10–18/group. (F) Plasma MCPT-1 levels were measured by ELISA from two independent experiments. N=8/group. (G) Lung sections were stained for mucus production with PAS (left) or H&E (right) and blinded samples were scored as described in Materials and Methods. Data are pooled from 2 independent experiments. N=9/group. (H) Anti-HDM IgG and total IgE in plasma were determined by ELISA from two independent experiments. N=11–15/group. All replicates are biological. Statistical values were determined by 1-way ANOVA (Tukey’s test) *p<.05; **p<.001; ****p<.0001 N.D.=not detected.
These data correlated with changes in lung tissue and BALF cells. HDM-sensitized mice had a greater abundance of pulmonary mast cells, an effect that was reduced by fluoxetine (Figure 7C). Mast cell presence in the lungs was confirmed by histology (Suppl. Fig. 6). We also measured eosinophils in the lung tissue and found that fluoxetine reduced HDM-mediated pulmonary eosinophilia (Figure 7C, Suppl. Fig. 7). We tested for direct effects on eosinophils in vitro and found that fluoxetine inhibited ATP-induced degranulation (Suppl. Fig. 8A) and reduced CCR3 surface expression on bone marrow derived mouse eosinophils (Suppl. Fig. 8B). Fluoxetine also decreased the number of IL-4/IL-13-producing T cells elicited by HDM (Figure 7C, Suppl. Fig. 7). Like lung tissue, BALF analysis showed that fluoxetine inhibited HDM-mediated recruitment of eosinophils, neutrophils, T and B cells into the bronchoalveolar space (Figure 7D, Suppl. Fig. 7). Matching the decline in cell recruitment, BALF cytokine analysis also showed reduced IL-5, MIP-1α, IL-13, IL-4, and IL-6 in fluoxetine-treated versus PBS-treated mice (Figure 7E). In addition to changes in the BALF, we noted that the HDM challenge induced plasma mast cell protease 1 (MCPT-1), which was indicative of mast cell activation. Similar to cytokine levels, MCPT-1 was significantly reduced by fluoxetine treatment (Fig. 7F).
Histological analysis of lung sections by PAS and H&E staining showed that HDM induced goblet cell hyperplasia and cell infiltration, respectively. These effects were significantly diminished by fluoxetine treatment (Figure 7G). Finally, total IgE and anti-HDM IgG were increased in HDM-sensitized mice and unaffected by fluoxetine (Fig. 7H). This was expected, since fluoxetine treatment was begun after HDM sensitization and demonstrated that even without suppressing the B cell response, fluoxetine can be beneficial in ongoing allergic disease.
Discussion
The many connections between depression and inflammation include observations that SSRIs have anti-inflammatory effects (21–23, 41–43). In searching for FDA-approved drugs that might be repurposed for allergic disease, these findings drew our attention. SSRIs are widely prescribed with an excellent safety profile (44). Fluoxetine has fewer side effects than other antidepressants and a long half-life of 3–4 days, with a 7–10 day half-life for its active metabolite, norfluoxetine. These characteristics could be useful for repurposing, so our initial data showing fluoxetine suppresses mast cell function prompted further study. Our interest was furthered by recent evidence that several antidepressants, including fluoxetine, have positive clinical outcomes in COVID-19 patients (45, 46). Because mast cells may contribute to COVID-19 disease severity and lung damage, understanding how fluoxetine affects mast cell function may be beneficial (47–49).
We show that fluoxetine inhibits the early and late phases of IgE-induced mast cell function, reducing very rapid Ca2+ flux and degranulation and the delayed secretion of eicosanoids and cytokines. These data were matched by a broad reduction in FcεRI signaling events that collectively suggest fluoxetine acts at receptor-proximal stages of signal propagation. The signaling disruptions yielded some incongruities, including a minor effect on intracellular cytokine staining that contrasted with a 50–70% inhibition of cytokine mRNA and protein secretion. These differences may be attributed to assay sensitivity, with intracellular staining for cytokines being less quantifiable. Alternatively, the data could suggest that fluoxetine has more potent effects on portions of the signaling cascades. Fluoxetine’s inhibitory effects were consistent using in vivo models of anaphylaxis and airway inflammation and AHR.
Previous data showing that fluoxetine may mimic the structure of ATP and that the related drug paroxetine can suppress P2X7 prompted our interest in how FcεRI and ATP signaling may be connected (28–30, 50). We found that activated mast cells rapidly secrete ATP and that this is necessary for full cytokine production. Fluoxetine reduced all stages of ATP-induced mast cell function measured. We therefore postulate that IgE-induced ATP secretion amplifies the mast cell response in a feedforward loop that can be antagonized by fluoxetine (Figure 8).
Figure 8: Model for fluoxetine-mediated suppression of mast cell function.
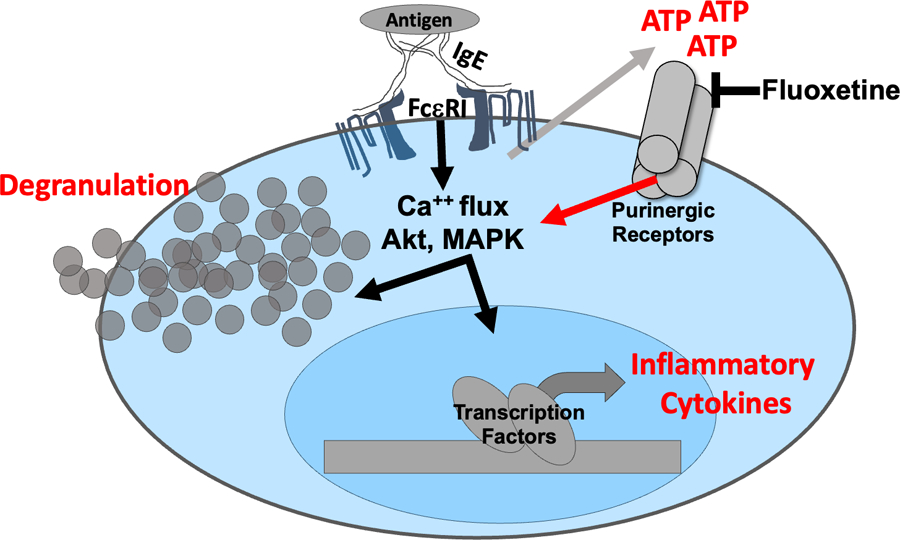
The data collectively support a model in which IgE crosslinkage with multivalent antigen elicits rapid ATP release that subsequently activates purinergic receptors and amplifies FcεRI-mediated signaling. Fluoxetine is proposed to inhibit purinergic receptor activation, as the drug does not alter FcεRI-induced ATP secretion but inhibits ATP-mediated mast cell function.
These amplifying, DAMP-like functions of ATP have important clinical implications. Extracellular ATP activates mast cells and accumulates in the airways of human asthmatics and in mouse models of asthma (13, 18, 19). Purinergic signaling involves two subclasses of trimeric receptors, P2X and P2Y. P2X receptors are ligand-gated ionotropic channels, while P2Y are G-protein coupled metabotropic receptors. P2X1–7 bind ATP, as do P2Y2 and P2Y11. While our data support an FcεRI-ATP-P2X/Y amplification loop that is disrupted by fluoxetine, the drug target remains unclear. Conversely, we found that SERT blockade is an unlikely answer, since SERT deficiency did not alter fluoxetine effects. Additionally, citalopram, a highly selective SERT antagonist, and paroxetine, a highly effective SERT inhibitor (25), did not consistently mimic fluoxetine effects, which further suggests that SERT inhibition is not the fluoxetine mechanism of action. Similarly, increasing the concentration of serotonin during FcεRI signaling had no effect. Therefore, we propose that fluoxetine has an off-target effect on purinergic receptors. This fits our data, since purinergic receptors induce calcium flux, an event that is critical for FcεRI function and reduced by fluoxetine. In support of fluoxetine disrupting ATP receptor interactions, Ambati et al. showed that fluoxetine antagonizes NLRP3 by mimicking ATP (28). Our ongoing work is focused on determining how fluoxetine inhibits the FcεRI-ATP feedback loop and which of the nine purinergic ATP receptors may be suppressed by fluoxetine. Although mice lacking high affinity fluoxetine interactions with SERT also display losses in antidepressant activity (51), others have concluded that non-serotoninergic mechanisms drive antidepressant actions in humans (26), though counter-arguments exist (27). Purinergic receptors are widely expressed in the nervous system and have been implicated in depression (52–54). Given the association between inflammation and depression, and our findings of non-SERT-mediated fluoxetine activity, it is possible that fluoxetine may exert multiple effects, including antagonizing purinergic receptors.
We found that fluoxetine effects are not mast cell-restricted, since the drug also suppressed ATP-mediated eosinophil degranulation. Eosinophils are effector cells with potent granular mediators that elicit damage in allergic inflammation (55). Elevated ATP in inflamed tissues can potentiate eosinophil activation and recruitment (56, 57). Eosinophils can be activated by extracellular ATP in the asthmatic airways through P2X1 signaling, which induces eosinophil adhesion and migration (58). Human eosinophils were also shown to migrate towards ATP in vitro (48). Thus, fluoxetine effects on eosinophils have clinical relevance.
We found evidence that fluoxetine inhibits allergic inflammation in vivo. The PSA and airway inflammation models showed suppressive effects that can be partly attributed to inhibiting mast cell function. PSA relies heavily on IgE-mediated histamine release from mast cells (59). Since fluoxetine had no effect on histamine-induced anaphylaxis, mast cell suppression is the most likely explanation. In contrast, the HDM airway inflammation model is more complex than PSA, likely requiring fluoxetine to antagonize multiple immune cell lineages. Fluoxetine reduced multiple parameters of HDM-induced airway hyperresponsiveness and inflammation. It also suppressed allergic inflammation in sensitized mice after the humoral response had begun. Allergic rhinitis and allergic asthma affect 15% and 11% of the US population, respectively (60, 61). Current treatments have limited efficacy (e.g., leukotriene receptor antagonists), problematic side effects (e.g., steroids) or high costs (e.g., monoclonal antibodies) that limit clinical use or patient access. Given the results of this study and fluoxetine’s capacity to concentrate in tissues where mast cells reside (32, 33) and many allergens are encountered, fluoxetine seems to be a viable option for clinical evaluation in allergy therapy, perhaps in combination with other treatments. Furthermore, because fluoxetine improved both AHR and inflammation, it could circumvent issues with treatments that address only one of these pathologies.
A limitation of our study is that we did not test fluoxetine effects in humans in vivo. The literature reports unclear data regarding fluoxetine’s ability to suppress allergy skin prick tests in humans. For example, these studies show that fluoxetine treatment had no effect on histamine- induced skin reaction but did not report allergy-induced skin reaction results before and after fluoxetine treatment (62, 63). It is noteworthy that leukotriene receptor inhibitors have also been reported to have no effect on skin prick tests but are effective at treating allergic diseases (63–65). Additionally, self-reported allergic rhinitis was significantly decreased after fluoxetine treatment in one study, encouraging further evaluation (62). Overall, our work supports further investigation in allergic patients.
In summary, our study indicates that FcεRI-induced ATP release amplifies IgE-mediated mast cell function. Fluoxetine suppressed IgE- and ATP-mediated signals, suggesting the drug can interact with ATP receptors that have myriad functions in the immune and nervous systems. This hypothesis warrants further study due to the potential of repurposing fluoxetine in allergic disease.
Materials and Methods
Mouse mast cell culture:
Bone marrow derived mast cells (BMMCs) were derived from marrow collected from mice femurs. Red blood cells were lysed with ACK lysis buffer. Cells were plated in RPMI (Invitrogen life technology) with 10% FBS (Sigma), 2mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 5μg/ml ciprofloxacin, 1 mM sodium pyruvate and 1mM HEPES buffer. WEHI supernatant containing 3ng/mL IL-3, and BHK-MKL supernatant containing 15 ng/mL SCF was added to the growth medium. Cells were cultured for 21–28 days, at which point cells were approximately 90% cKit/FcεRI-positive. Peritoneal mast cells (PMCs) were cultured the cRPMI with 10 ng/ml IL-3 and SCF for 7–10 days.
Human skin mast cells:
All protocols involving human tissues were approved by the Institutional Review Board at the University of South Carolina or Virginia Commonwealth University. Cells were cultured as previously described (66). Human skin mast cells were cultured with 10μM fluoxetine, 1 μg/ml mouse IgE, and human recombinant SCF for 24 hours. Cells were washed and re-plated in SCF ± 10µM fluoxetine for 24 hours and 50 ng/ml DNP-HSA for 16 hours. Supernatant was collected for cytokine analysis by ELISA.
Animals:
C57BL/6J, 129/SvImJ, BALB/cJ, and B6.129(Cg)-Slc6a4tm1Kpl/J (SERT KO) female mice were purchased from Jackson Laboratory, bred and maintained at the VCU animal facilities. Experiments using mice employed animals housed in the same room and compared age- and sex-matched mice. All animal use was approved by the Virginia Commonwealth University Institutional Animal Care and Use Committee (IACUC).
Mouse bone marrow derived eosinophil culture:
Mouse bone marrow-derived eosinophils were grown in RPMI with Glutamax (Invitrogen), 20% FBS, 25mM HEPES, 100 U/mL penicillin, 100ug/mL streptomycin, 1X NEAA, 55 µM βME, supplemented with 100ng/ml SCF and 100ng/mL FLT-3 (Shenandoah Inc) for 4 days and then 10ng/mL IL-5 (Shenandoah Inc). On day 12 of culture, cells were approximately 90% SiglecF-positive and ready for experimentation (67). Bone marrow derived eosinophils were treated with the indicated concentrations of fluoxetine for 24 hours prior to activation with 300μM ATP for 15 minutes. Cells were stained for CD63 as a marker for degranulation and analyzed by flow cytometry. Some eosinophils were treated with fluoxetine (10μM), stained for surface expression of CCR3, and analyzed by flow cytometry.
Cell Culture Supernatant Cytokine Measurement:
BMMCs were treated with fluoxetine (10 μM), sertraline (2 μM), fluvoxamine (8 μM), paroxetine (5 μM), citalopram (30 μM) or vehicle at 1x106 cells/mL in cRPMI with 10ng/mL IL-3 and SCF with 0.5μg/mL anti-DNP IgE for 24 hours. Some experiments were done with pre-sensitized BMMCs as indicated in figure legends. Cells were then washed and re-plated with fluoxetine and 50ng/mL DNP-HSA for 16 hours. Other BMMCs treated with fluoxetine for 24 hours were activated with 150μM ATP for 4 hours. Some experiments included pre-treatment with recombinant serotonin (R&D) at the indicated concentrations for 24 hours. Supernatant was collected and analyzed for IL-13, MIP-1α (CCL-3) (Peprotech), IL-6, MCP-1 and TNF (Biolegend) by ELISA as instructed by the manufacturer.
Degranulation:
IgE-sensitized BMMCs were treated with 10 µM fluoxetine for 24 hours, washed, and stimulated with 50 ng/ml DNP-HSA with fluoxetine present for 30 minutes. In some experiments, BMMCs treated with fluoxetine for 24 hours were activated with 300 μM ATP for 15 minutes. Cells were placed on ice and washed with ice cold PBS. Cells were stained with anti-CD107a and anti-CD63, then analyzed using a FACSCelesta. Histamine was measured in the supernatant and cell lysates after +/− 30 minutes of activation by ELISA (Neogen).
RT-qPCR:
Cells treated +/− fluoxetine were stimulated with IgE crosslinkage as described above for 4 hours. Trizol (Life Sciences) was used to extract total RNA. Nucleic acid purity was measured using a Nanodrop 1000 UV-Vis Spectrophotometer (Thermo Scientific). To measure mRNA expression, cDNA was synthesized using the qScript cDNA Synthesis Kit (Quanta Biosciences) following the manufacturer’s protocol. qPCR analysis was performed with Bio Rad CFX96 Touch™ Real-Time PCR Detection System and SYBR® Green detection using a relative Livak Method normalized to β-actin. Reactions consisted of a heat-activation step at 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds, 55°C for 30 seconds and 60°C for 1 minute. Fluorescence data were collected during the extension step of the reaction. Primers for IL-6 (forward: 5′TCCAGTTGCCTTCTTGGGAC3′, reverse: TCCAGTTGCCTTCTTGGGAC3′), β-actin (forward: 5′GATGACGATATCGCTGCGC3′, Reverse: 5′CTCGTCACCCACATAGGAGTC3′) and IL-13 (forward: ATGGCGTCTGGGTGACTGCAGTCC’, Reverse: GAAGGGGCGTGGCGAAACAGTTGC) were purchased from Eurofins.
Drugs and Inhibitors:
Fluoxetine, paroxetine, fluvoxamine, citalopram and sertraline were purchased from Sigma Aldrich. Serotonin was purchased from Tocris. Propidium Iodine exclusion staining of mast cells and flow cytometry was used to determine toxicity of all drugs before use.
Intracellular Cytokine Staining:
BMMCs were treated with IgE and fluoxetine (10 μM, 24 hours) as previously described, crosslinked with DNP-HSA (50 ng/ml) for 2 hours, then treated with 5 μM monensin for 5 hours. Cells were fixed in 4% paraformaldehyde for 20 minutes at room temperature and stored in PBS overnight at 4°C. The next day, fixed cells were permeabilized in saponin buffer for 20 minutes at room temperature and then stained in saponin buffer at 4°C for 40–60 minutes with fluorochrome-labeled anti-IL6 (BD Bioscience clones MP5–20F3), or anti-IL13 (BD Bioscience clone JES10–5A2).
Mass Spectrometry for Lipid Detection:
BMMCs were treated with IgE and fluoxetine as previously described and activated with 50 ng/mL DNP-HSA or 150 μM ATP for 1 hour. Supernatants were stored at −80 °C until analysis. Eicosanoids were extracted using a process previously described (68). Eicosanoids were analyzed via mass spectrometry using an AB Sciex Triple Quad 5500 Mass Spectrometer (68).
Passive Systemic Anaphylaxis:
Female mice were sensitized with 50 μg IgE and treated with PBS or fluoxetine at 1 mg/kg intraperitoneally. The next day, mice were given a second injection of PBS or fluoxetine. Baseline core body temperature was measured using a rectal thermometer (Braintree Scientific Inc). Two hours later, mice received 100μg DNP-HSA by IP injection. Core body temperature was recorded every 10 minutes for 1 hour and then every 30 minutes for 1 hour for a total of 2 hours. Mice were sacrificed using CO2 asphyxiation as approved by the VCU IACUC. Plasma cytokine levels were determined using IL-13 and MIP-1 ELISA kits from Peptrotech, and IL-6 ELISA kit from Biolegend. Plasma histamine levels were determined using a Neogen Histamine ELISA Kit. Histamine-induced anaphylaxis was elicited by IP injection of 2mg histamine.
Flow Cytometry:
For direct surface markers single cell suspension was stained with live/dead indicator (aqua, Biolegend), Fcγ receptors were blocked with anti-CD16/CD32 clone 2.4G2, and cells were stained with fluorescently conjugated antibodies for 20–30 minutes at 4°C. All flow data were acquired using FACSCelesta and analyzed by FACS Diva or FlowJo software. Flow cytometry with BMMCs was gated on high FSC and SSC and singlets. Further gates were determined using isotype and fluorescence-minus-one controls.
Intracellular Ca2+ Flux Assay:
BMMCs were treated −/+ fluoxetine and IgE for 24 hours. Cells were washed and loaded with Fura 2-AM (Thermofisher) at 37°C for 40 minutes. Cells were washed and plated in Tyrode’s buffer with 1% FBS, 10ng/mL IL-3 and SCF −/+ fluoxetine. Cells were allowed to rest at room temperature for 30 minutes and 37°C for 10 minutes. Cells were activated with 150 μM ATP, 50ng/mL DNP-HSA or 1 μM ionomycin. Some samples received DNP-HSA and fluoxetine together. Cells were analyzed using a Flexstation plate reader (Molecular Devices). Data were collected every 4 seconds and reported as a ratio of 340/380 nm excitation detected at 510 nm emission.
NFκB reporter activity:
B10.Cg-H2k Tg(NFkB/Fos-luc)26Rinc/J mice (Catalog number 006100 from Jackson Laboratory) were used to generate NFκB reporter BMMCs as described above. BMMCs were treated with fluoxetine and activated with 150 µM ATP for 2 hours. Cells were lysed and luciferase activity was measured with the Promega Luciferase Assay Substrate and Glomax 20/20 Luminometer (Promega). Luciferase expression is normalized to the unactivated control.
DNA extraction and PCR
Qiagen DNeasy Blood and Tissue Kit (Cat. 69504) was used for DNA extraction of cells following manufacturer’s instructions. To determine SERT allele, oligonucleotide primers used for PCR-based genotyping were as follows: 5’-CCT AGA TAC CAG GCC CAC AA-3’ (sense, wildtype SERT), 5’-GCC AGA GGC CAC TTG TGT AG-3’ (sense, knockout SERT), and 5’-AAT GGT GAG GAG TGG TGG AG-3’ (antisense, common primer). Genotyping was performed on a MJ Research PTC-200 Thermal Cycler using RedTaq ReadyMix PCR Reaction Mix (Millipore Sigma, Cat. R2523) under the following conditions: 95⁰C for 5 minutes, then cycling 39 times through three steps including 95⁰C for 30 seconds, 64⁰C for 1 minute (then −0.3⁰C per cycle), and 72⁰C for 30 seconds, followed by a final 72⁰C for 10 minutes. All samples were then resolved on a 1.5% agarose gel to determine genotype, with wildtype SERT producing a 318 bp amplicon and the knockout SERT producing a 210 bp amplicon.
Protein extraction and Western blotting
Cells were pelleted and washed with ice cold PBS and resuspended in RIPA lysis buffer (10 mM Tris-HCl pH7.5, 300 mM NaCl, 1mM EDTA, 1% Triton X-100, 1% SDS, and 0.1% sodium deoxycholate) containing protease inhibitor cocktail (Millipore Sigma, Cat. P8340, 1:100 dilution). Samples rotated at 4⁰C for 30 minutes. Lysates were centrifuged at 20,000 g for 30 minutes at 4⁰C, supernatants were collected and a Pierce BCA Protein Assay (Thermo Fisher, Cat. 23225) was performed. Mouse brain tissue (midbrain) was collected and rapidly frozen on dry ice for storage. Brain tissue was thawed, homogenized using a handheld sonicator in RIPA lysis buffer containing protease inhibitor cocktail, rotated at 4C for 60 minutes, then centrifuged at 17,000 g for 10 minutes. Supernatants were collected and protein concentration was determined using a Pierce BCA Protein Assay. 90μg of protein from cells and 40μg from brain tissue was taken and added to 4x Laemmli sample buffer (plus beta-mercaptoethanol) (Bio-Rad, Cat. 1610747) and left to denature at room temperature for 45 minutes. Samples were loaded onto a NuPAGE 4–12% Bis-Tris Gel (Invitrogen, Cat. NP0335BOX), transferred to a PVDF membrane (20V for 18 hours at 4⁰C) and probed for SERT using a Guinea Pig anti-SERT antibody (Frontier Institute co., ltd., Cat. HTT-GP-Af1400, 1:1000 dilution). HRP-conjugated secondary antibody (Goat anti-Guinea Pig HRP, Jackson Immunoresearch, Cat. 106–085-003, 1:10,000 dilution) was used in conjunction with chemiluminescence (SuperSignal West Pico PLUS Chemiluminescent Substrate, Thermo Scientific, Cat. 34577) to visualize bands on the Li-COR Odyssey FC (OFC-0978). Subsequently, the membrane was stripped and reprobed with mouse anti-beta actin (Millipore Sigma, A3854, HRP conjugated, 1:10,000 dilution).
siRNA Knock Down Experiments:
BMMCs were starved of growth factors for 3 hours, then10ng/mL IL-3 and SCF was added 16 hours prior to transfection. For transfection, 3 million cells were resuspended in 100μL transfection media (DMEM, 20% FBS, 50 mM HEPES) with 2 μM siRNA (scramble control, 4 sequences pooled SERT specific siRNA purchased from Qiagen), placed in a transfection cuvette and electroporated using program T-05 in the Amaxa Nucleoporation instrument. Cells were plated at 0.5x106 cells/mL and allowed to rest for 48 hours in cRMPI+10 ng/mL IL-3 and SCF. After 48 hours, cells were collected to determine knock down efficiency by staining via flow cytometry using anti-SERT antibody (Alomone Labs catalog # AMT-004). Cells were resuspended in fresh media, sensitized with IgE, treated with fluoxetine at various doses, and crosslinked with DNP-HSA as previously described.
HDM Sensitization, Airway Hypersensitivity and Lung Inflammation Model:
Female C57BL/6J mice were sensitized with 50μg HDM (Stallergenes Greer Cat# XPB70D3A2.5, Lot 360924) in 4mg Imject Alum (Thermofisher) daily on days 0–2. Starting on day 13, every 3 days, mice were given 25μg HDM intranasally, under isoflurane anesthesia. Mice were given PBS or fluoxetine dissolved in PBS on days 11–23 intranasally. On day 24, mice were anesthetized, and a cannula was inserted in the trachea. Mice were then subjected to forced oscillation on a computer-controlled ventilator (Flexivent system, Scireq Inc.) during increasing nebulized methacholine exposure. BALF was collected for cytokine ELISA and cells were analyzed by flow cytometry. Lung lobes were either (1) flash frozen for RNA collection, (2) digested and single cell suspension was stained by flow cytometry, or (3) fixed in 10% formalin for H&E and PAS staining. For flow cytometry analysis, all cells were stained with Zombie aqua dye, blocked with 2.4G2 antibody, and stained with anti-CD45, anti-FcεRI, and anti-cKit for mast cells; anti-CD90.2 (Biolegend clone 30-H12) for T cells; anti-B220 (BD Bioscience clone RA3–6B2) and anti-MHC II (BD Bioscience clone M5/114) for B cells; anti-SiglecF and anti-CCR3 (Biolegend J073E5) for eosinophils; anti-CD3 (BD Bioscience clone 17A2), anti-CD4 (Biolegend clone GK1.5), anti-IL4, and anti-IL13 for Th2 cells; or anti-CD11b (Biolegend clone M1/70) and anti-Ly6G (BD Bioscience clone 1A8) for neutrophils. Cell number was determined using Precision Counting Beads as directed by the manufacturer (BD). For re-stimulation, lung cells were plated in cRPMI with 1μM ionomycin, 25 ng/mL PMA and 5 μM monensin at 37°C for 3 hours prior to staining. All gating was determined by isotype and fluorescence minus one controls. Blood plasma was collected for total IgE (Biolegend) and anti-HDM-IgG (Condrex) by ELISA.
The scoring system for PAS and H&E stainned slides was as follows. PAS: 0=no GBC present, 1=<25% of epithelial lining are GBC, 2=25–50% of epithelial lining are GBC, 3=51–75% of epithelial lining are GBC, 4=>75% of epithelial lining are GBC. H&E: 0=no cell infiltration in alveoli/bronchi space, 1=<25% cell infiltration alveoli and bronchi space, 2=25–50% cell infiltration alveoli and bronchi space, 3=51–75% cell infiltration alveoli and bronchi space, 4=>75% cell infiltration alveoli and bronchi space.
Supplementary Material
Acknowledgments:
The authors thank Dr. D’Arcy Mays for statistical assistance.
Funding:
This work was funded by NIH grants R01AI164710 and R01AI138495 awarded to JJR, F31AI140609–01A1 awarded to TTH, partly by NIH/NIAID R01AI095494 and NIH/NIAMS R21AR067996 to CAO, NIH grant MH120966 to P.A.G-K., Veteran’s Administration [VA Merit Review, I BX001792 to CEC and a Research Career Scientist Award, BX004603to CEC) and from the NIH via DK126444, HL125353, and AI139072 to CEC. The contents of this manuscript do not represent the views of the Department of Veterans Affairs or the U.S. government.
Footnotes
Competing interests: The authors have no competing interests to report.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. To access raw data obtained from mass spectroscopy analysis, see our public Open Science Framework account: https://osf.io/4grx5/, DOI 10.17605/OSF.IO/4GRX5.
References Cited
- 1.Brightling CE et al. , Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med 346, 1699–1705 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Broide DH et al. , Evidence of ongoing mast cell and eosinophil degranulation in symptomatic asthma airway. J Allergy Clin Immunol 88, 637–648 (1991). [DOI] [PubMed] [Google Scholar]
- 3.Dougherty RH et al. , Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J Allergy Clin Immunol 125, 1046–1053 e1048 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang G et al. , Sputum mast cell subtypes relate to eosinophilia and corticosteroid response in asthma. Eur Respir J 47, 1123–1133 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Bousquet J et al. , Predicting response to omalizumab, an anti-IgE antibody, in patients with allergic asthma. Chest 125, 1378–1386 (2004). [DOI] [PubMed] [Google Scholar]
- 6.de Boer JD et al. , Mast cell-deficient kit mice develop house dust mite-induced lung inflammation despite impaired eosinophil recruitment. J Innate Immun 6, 219–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li S et al. , Antigen-induced mast cell expansion and bronchoconstriction in a mouse model of asthma. Am J Physiol Lung Cell Mol Physiol 306, L196–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin TR et al. , Mast cell activation enhances airway responsiveness to methacholine in the mouse. J Clin Invest 91, 1176–1182 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKnight CG, Jude JA, Zhu Z, Panettieri RA Jr., Finkelman FD, House Dust Mite-Induced Allergic Airway Disease Is Independent of IgE and FcepsilonRIalpha. Am J Respir Cell Mol Biol 57, 674–682 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serra-Pages M et al. , Activation of the Prostaglandin E2 receptor EP2 prevents house dust mite-induced airway hyperresponsiveness and inflammation by restraining mast cells' activity. Clin Exp Allergy 45, 1590–1600 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Takeda M et al. , Gender difference in allergic airway remodelling and immunoglobulin production in mouse model of asthma. Respirology 18, 797–806 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Williams CM, Galli SJ, Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med 192, 455–462 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Idzko M et al. , Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med 13, 913–919 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Gazzerro E et al. , The Danger Signal Extracellular ATP Is Involved in the Immunomediated Damage of alpha-Sarcoglycan-Deficient Muscular Dystrophy. Am J Pathol 189, 354–369 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Patel S, Danger-Associated Molecular Patterns (DAMPs): the Derivatives and Triggers of Inflammation. Curr Allergy Asthma Rep 18, 63 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Tanaka K, Choi J, Cao Y, Stacey G, Extracellular ATP acts as a damage-associated molecular pattern (DAMP) signal in plants. Front Plant Sci 5, 446 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradding P, Okayama Y, Kambe N, Saito H, Ion channel gene expression in human lung, skin, and cord blood-derived mast cells. J Leukoc Biol 73, 614–620 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Wareham K, Vial C, Wykes RC, Bradding P, Seward EP, Functional evidence for the expression of P2X1, P2X4 and P2X7 receptors in human lung mast cells. Br J Pharmacol 157, 1215–1224 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurashima Y et al. , Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat Commun 3, 1034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida K, Ito M, Matsuoka I, Divergent regulatory roles of extracellular ATP in the degranulation response of mouse bone marrow-derived mast cells. Int Immunopharmacol 43, 99–107 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Hannestad J, DellaGioia N, Bloch M, The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: a meta-analysis. Neuropsychopharmacology 36, 2452–2459 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhat R, Mahapatra S, Axtell RC, Steinman L, Amelioration of ongoing experimental autoimmune encephalomyelitis with fluoxetine. J Neuroimmunol 313, 77–81 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Mohr DC, Goodkin DE, Islar J, Hauser SL, Genain CP, Treatment of depression is associated with suppression of nonspecific and antigen-specific T(H)1 responses in multiple sclerosis. Arch Neurol 58, 1081–1086 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Brown ES et al. , A randomized trial of citalopram versus placebo in outpatients with asthma and major depressive disorder: a proof of concept study. Biol Psychiatry 58, 865–870 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Tatsumi M, Groshan K, Blakely RD, Richelson E, Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 340, 249–258 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Moncrieff J et al. , The serotonin theory of depression: a systematic umbrella review of the evidence. Mol Psychiatry, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jauhar S et al. , A leaky umbrella has little value: evidence clearly indicates the serotonin system is implicated in depression. Mol Psychiatry, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ambati M et al. , Identification of fluoxetine as a direct NLRP3 inhibitor to treat atrophic macular degeneration. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dao-Ung P, Skarratt KK, Fuller SJ, Stokes L, Paroxetine suppresses recombinant human P2X7 responses. Purinergic Signal 11, 481–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagata K et al. , Antidepressants inhibit P2X4 receptor function: a possible involvement in neuropathic pain relief. Mol Pain 5, 20 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang H, Bell TA, Churchill GA, Pardo-Manuel de Villena F, On the subspecific origin of the laboratory mouse. Nat Genet 39, 1100–1107 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Bolo NR et al. , Brain pharmacokinetics and tissue distribution in vivo of fluvoxamine and fluoxetine by fluorine magnetic resonance spectroscopy. Neuropsychopharmacology 23, 428–438 (2000). [DOI] [PubMed] [Google Scholar]
- 33.Johnson RD, Lewis RJ, Angier MK, The distribution of fluoxetine in human fluids and tissues. J Anal Toxicol 31, 409–414 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Betts MR et al. , Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 281, 65–78 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Iwaki S et al. , Btk plays a crucial role in the amplification of Fc epsilonRI-mediated mast cell activation by kit. J Biol Chem 280, 40261–40270 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Yamaguchi M et al. , IgE enhances mouse mast cell Fc(epsilon)RI expression in vitro and in vivo: evidence for a novel amplification mechanism in IgE-dependent reactions. J Exp Med 185, 663–672 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamaguchi M et al. , IgE enhances Fc epsilon receptor I expression and IgE-dependent release of histamine and lipid mediators from human umbilical cord blood-derived mast cells: synergistic effect of IL-4 and IgE on human mast cell Fc epsilon receptor I expression and mediator release. J Immunol 162, 5455–5465 (1999). [PubMed] [Google Scholar]
- 38.Conti P, Shaik-Dasthagirisaheb YB, Mast Cell Serotonin Immunoregulatory Effects Impacting on Neuronal Function: Implications for Neurodegenerative and Psychiatric Disorders. Neurotox Res 28, 147–153 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi T, Kouzaki H, Kita H, Human eosinophils recognize endogenous danger signal crystalline uric acid and produce proinflammatory cytokines mediated by autocrine ATP. J Immunol 184, 6350–6358 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsai SH et al. , The ectoenzyme E-NPP3 negatively regulates ATP-dependent chronic allergic responses by basophils and mast cells. Immunity 42, 279–293 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Kao CY et al. , Fluoxetine treatment prevents the inflammatory response in a mouse model of posttraumatic stress disorder. J Psychiatr Res 76, 74–83 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Nazimek K et al. , Repeatedly administered antidepressant drugs modulate humoral and cellular immune response in mice through action on macrophages. Exp Biol Med (Maywood) 241, 1540–1550 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Talmon M et al. , Vortioxetine exerts anti-inflammatory and immunomodulatory effects on human monocytes/macrophages. Br J Pharmacol 175, 113–124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossi A, Barraco A, Donda P, Fluoxetine: a review on evidence based medicine. Ann Gen Hosp Psychiatry 3, 2 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonnet U, Juckel G, COVID-19 Outcomes: Does the Use of Psychotropic Drugs Make a Difference? Accumulating Evidence of a Beneficial Effect of Antidepressants-A Scoping Review. J Clin Psychopharmacol 42, 284–292 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mas M et al. , Antidepressant Drugs and COVID-19: A Review of Basic and Clinical Evidence. J Clin Med 11, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gebremeskel S et al. , Mast Cell and Eosinophil Activation Are Associated With COVID-19 and TLR-Mediated Viral Inflammation: Implications for an Anti-Siglec-8 Antibody. Front Immunol 12, 650331 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schaller T et al. , Mast cells in lung damage of COVID-19 autopsies: A descriptive study. Allergy 77, 2237–2239 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wechsler JB, Butuci M, Wong A, Kamboj AP, Youngblood BA, Mast cell activation is associated with post-acute COVID-19 syndrome. Allergy 77, 1288–1291 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang W, Xiang ZH, Jiang CL, Liu WZ, Shang ZL, Effects of antidepressants on P2X7 receptors. Psychiatry Res 242, 281–287 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Nackenoff AG, Moussa-Tooks AB, McMeekin AM, Veenstra-VanderWeele J, Blakely RD, Essential Contributions of Serotonin Transporter Inhibition to the Acute and Chronic Actions of Fluoxetine and Citalopram in the SERT Met172 Mouse. Neuropsychopharmacology 41, 1733–1741 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sikka P et al. , Scrutinizing the Therapeutic Promise of Purinergic Receptors Targeting Depression. Neurotox Res 40, 1570–1585 (2022). [DOI] [PubMed] [Google Scholar]
- 53.Szopa A et al. , Purinergic transmission in depressive disorders. Pharmacol Ther 224, 107821 (2021). [DOI] [PubMed] [Google Scholar]
- 54.Zou Y, Yang R, Li L, Xu X, Liang S, Purinergic signaling: a potential therapeutic target for depression and chronic pain. Purinergic Signal 19, 163–172 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Acharya KR, Ackerman SJ, Eosinophil granule proteins: form and function. J Biol Chem 289, 17406–17415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alberto AV et al. , Role of P2 Receptors as Modulators of Rat Eosinophil Recruitment in Allergic Inflammation. PLoS One 11, e0145392 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi T et al. , ATP drives eosinophil effector responses through P2 purinergic receptors. Allergol Int 64 Suppl, S30–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wright A et al. , Impaired P2X1 Receptor-Mediated Adhesion in Eosinophils from Asthmatic Patients. J Immunol 196, 4877–4884 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Kanjarawi R et al. , Regulatory CD4+Foxp3+ T cells control the severity of anaphylaxis. PLoS One 8, e69183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Backman H et al. , Increased prevalence of allergic asthma from 1996 to 2006 and further to 2016-results from three population surveys. Clin Exp Allergy 47, 1426–1435 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Meltzer EO et al. , Burden of allergic rhinitis: results from the Pediatric Allergies in America survey. J Allergy Clin Immunol 124, S43–70 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Isik SR, Celikel S, Karakaya G, Ulug B, Kalyoncu AF, The effects of antidepressants on the results of skin prick tests used in the diagnosis of allergic diseases. Int Arch Allergy Immunol 154, 63–68 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Shah KM, Rank MA, Dave SA, Oslie CL, Butterfield JH, Predicting which medication classes interfere with allergy skin testing. Allergy Asthma Proc 31, 477–482 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Cuhadaroglu C, Erelel M, Kiyan E, Ece T, Erkan F, Role of Zafirlukast on skin prick test. Allergol Immunopathol (Madr) 29, 66–68 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Hill SL 3rd, Krouse JH, The effects of montelukast on intradermal wheal and flare. Otolaryngol Head Neck Surg 129, 199–203 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Kambe N, Kambe M, Kochan JP, Schwartz LB, Human skin-derived mast cells can proliferate while retaining their characteristic functional and protease phenotypes. Blood 97, 2045–2052 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Dyer KD et al. , Functionally competent eosinophils differentiated ex vivo in high purity from normal mouse bone marrow. J Immunol 181, 4004–4009 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.MacKnight HP et al. , The interaction of ceramide 1-phosphate with group IVA cytosolic phospholipase A(2) coordinates acute wound healing and repair. Sci Signal 12, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.