

Abstract
Structure-based design, synthesis, X-ray structural studies, and biological evaluation of a new series of potent HIV-1 protease inhibitors are described. These inhibitors contain various pyridyl-pyrimidine, aryl thiazole or alkylthiazole derivatives as the P2 ligands in combination with darunavir-like hydroxyethylamine sulfonamide isosteres. These heterocyclic ligands are inherent to kinase inhibitor drugs, such as nilotinib and imatinib. These ligands are designed to make hydrogen bonding interactions with the backbone atoms in the S2 subsite of HIV-1 protease. Various benzoic acid derivatives have been synthesized and incorporation of these ligands provided potent inhibitors that exhibited subnanomolar level protease inhibitory activity and low nanomolar level antiviral activity. Two high resolution X-ray structures of inhibitor-bound HIV-1 protease were determined. These structures provided important ligand-binding site interactions for further optimization of this class of protease inhibitors.
Keywords: HIV-1 protease inhibitors, Antiviral, Imatinib, Nilotinib, Kinase inhibitors, X-ray crystal structure, Backbone binding, Pyridylpyrimidine
1. Introduction
Therapeutic inhibition of virally encoded HIV-1 protease is one of the most important strategies for the treatment of HIV-1 infection and AIDS [1–3]. The development of HIV-1 protease inhibitor drugs using protein X-ray structure-based design is regarded as a major achievement in medicinal chemistry [4,5]. Protease inhibitors have been an integral part of current antiretroviral therapies (cART) with reverse transcriptase inhibitors which emerged in the mid-1990s [6,7]. The cART treatment regimens have dramatically transformed HIV/AIDS into a manageable chronic ailment [8–10]. As a result, the HIV-related mortality rates continued to decline due to improved treatment regimens [11,12]. Despite these major improvements, the growing emergence of drug-resistant strains, high pill burden, and drug side effects are becoming major concerns regarding the long-term prospects of HIV/AIDS management [13–15]. In particular, the first generation of HIV-1 protease inhibitors were associated with peptide-like features which contributed to poor oral bioavailability, high metabolic degradation, and other debilitating side effects [13,16]. The development of second generation protease inhibitor drugs alleviated some of these major issues, however, current treatments are far from ideal due to the emergence of drug resistance, cardiovascular, and CNS (central nervous system) problems associated with current treatment regimens [17,18]. In our continuing efforts towards the development of improved treatment options, our protease inhibitor design strategy has been aimed at maximizing hydrogen bonding interactions with the active site protease backbone atoms [19,20]. These strategies have resulted in a range of highly potent inhibitors including FDA approved inhibitor drug, darunavir (1, Fig. 1) and related compound TMC-126 (2) [21–24]. Darunavir, exhibits a dual mechanism of action in which it inhibits the catalytically active dimeric HIV-1 protease and also inhibits dimerization of the individual protease monomers, preventing the formation of catalytically active protease enzyme [25,26].
Fig. 1.

Structure of HIV-1 protease inhibitors 1–5.
Darunavir utilizes a unique design strategy that involves promoting a network of hydrogen-bonding interactions with protease backbone S2 to S2’ sites of the active site [19,27,28]. These properties most likely contributed to darunavir’s high genetic barrier to the development of drug-resistant variants compared to other approved protease inhibitor drugs [29,30]. While darunavir is used widely as a first line therapy for rescue treatment, darunavir resistant HIV-1 variants have emerged [31–33]. Therefore, further development of a new class of protease inhibitors with structurally novel features are important for future treatment options.
One of the important features of darunavir is the bicyclic polyether-like P2 ligand, the bis-tetrahydrofuranyl heterocycle [34,35]. Both oxygens of this polyether scaffold form tight hydrogen bonds with backbone amide NHs of Asp29 and Asp30 in the S2 subsite [27,28]. In an effort to design new functionalities that can accommodate similar hydrogen bonding interactions with the backbone atoms, we speculated that the pyridyl-pyrimidine scaffold of imatinib 3 or nilotinib 4 can be incorporated with substituted 3-aminobenzoic acid to mimic hydrogen bonding interactions in the S2 subsite similar to the bis-THF ligand of darunavir [36,37]. Nilotinib is a selective tyrosine kinase receptor inhibitor used for the treatment of chronic myelogenous leukemia. Nilotinib binds to and stabilizes the inactive conformation of the kinase domain of the Abl protein of the Bcr-Abl fusion protein. This results in the inhibition of the Bcr-Abl-mediated proliferation of Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) cells [38,39]. Nilotinib is orally bioavailable, and incorporation of its structural features may improve pharmacological properties of the resulting protease inhibitors.
The X-ray structural studies of imatinib- and nilotinib-bound Abl-Bcr kinase revealed donor-acceptor abilities of pyridyl-pyrimidine hetereocycles [40,41]. The major advantage is that such heterocyclic ligands would not have stereochemical complexities related to bis-THF ligand. We describe here our effort towards the design of a new class of HIV-1 protease inhibitors by incorporating a variety of hinge-binding heterocycles that would maintain the critical hydrogen bonding interactions in the S2 subsite of HIV-1 protease. The structural class is represented in compound 5. These heterocyclic scaffolds are key templates for kinase inhibitor drugs, imatinib and dasatinib. For our preliminary work, we investigated the new ligand scaffolds in combination with the hydroxyethylamine sulfonamide isosteres inherent to darunavir and TMC-126 [19,22].
2. Results and discussion
We recently reported a series of HIV-1 protease inhibitors that incorporated substituted isophthalamide derivatives as the P2-ligands [42,43]. A representative example, inhibitor 6 is shown in Fig. 2. These inhibitors were designed by taking advantage of the large hydrophobic pocket in the HIV-1 protease S1–S2 subsites [43]. While the P2-ligands resemble 3-hydroxy-2-methyl benzamide inherent to nelfinavir 7, an FDA approved drug, X-ray structural studies of inhibitor-bound HIV-1 protease revealed that the 3-hydroxy benzylamide carbonyl involved in hydrogen bonding interaction with the backbone amide NH of Asp29 [44,45]. However, the oxazolylmethyl segment is mostly solvent exposed and does not participate in any polar interactions in the actve site. Based upon this structural precedence, we planned to investigate pyridyl-pyrimidine and various alkyl, arylthiazolyl hetereocycles such as 8 and 9 in combination with hydroxyethylamine sulfonamide isosteres. Our preliminary models of pyridyl-pyrimidine derivatives, based upon the X-ray structure of nelfinavir-bound HIV-1 protease, revealed that pyridyl-pyrimidine functionalities can form hydrogen bonds with backbone amide NHs in the S2 subsite [45]. Also, we speculated that appropriately functionalized alkyl or aryl substituents can effectively fill in the hydrophobic pocket in the S2–S3 subsites. There is no previous report of the use of pyridyl-pyrimidine derived P2 ligands in the design of HIV-1 protease inhibitors.
Fig. 2.

Structure of HIV-1 protease inhibitors and ligands.
2.1. Synthesis of inhibitors
The synthesis of various nilotinib-based HIV-1 protease inhibitors 5a-d is shown in Scheme 1. Commercially available 4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]benzoic acid 10 was reacted with previously reported hydroxyethylene isosteres 11–14 in the presence of HATU and Et3N in DMF at 23 °C for 24 h to afford the desired coupling products 5a-d in good yield (60–76%). The syntheses of various methyl-3-[[4-(3-pyridinyl)-2-thiazolyl]amino]benzoic acids and syntheses of inhibitors 5e-i are shown in Scheme 2. Hantzsch thiazole synthesis [46] was utilized in order to obtain these desired pyridinyl-thiazolylamino benzoic acid derivatives. As shown, various methyl substituted amino-methylbenzoates 15 were reacted with potassium thiocyante and acetyl chloride in acetone at 23 °C to 60 °C to provide thiourea derivatives 16. Exposure of 16 to potassium carbonate in methanol, followed by addition of α-bromo ketone 17 furnished pyridinylthiazole derivatives 18. Saponification of methyl esters 18 with 1 M LiOH in THF at 23 °C afforded ligand carboxylic acids 19a-e. Coupling of these ligand acids with hydroxyethylsulfonamide isosteric amine 11 [22,34] provided inhibitors 5e-i in good yields.
Scheme 1.
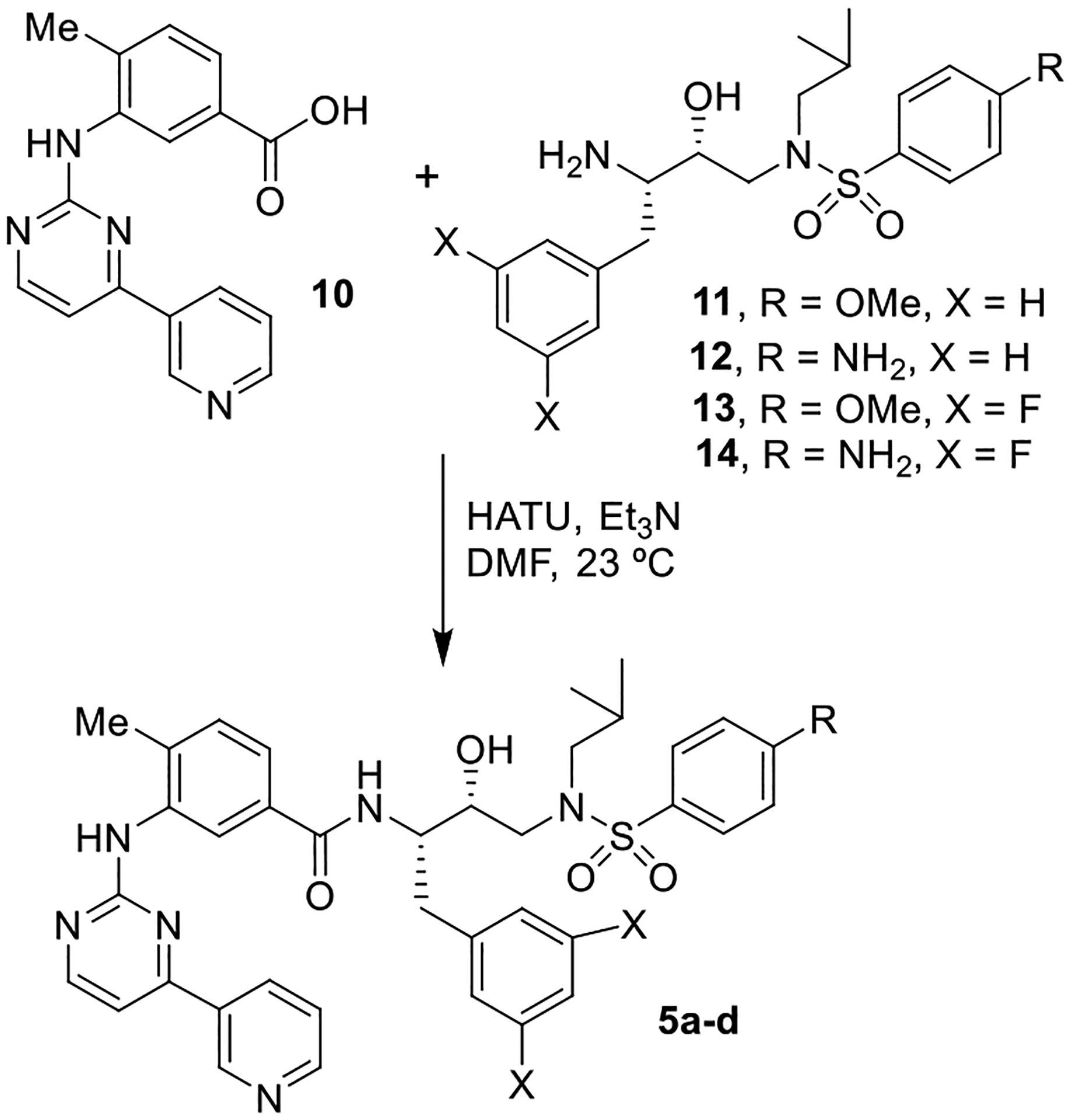
Synthesis of protease inhibitors 5a-d.
Scheme 2.

Synthesis of protease inhibitors 5e-i.
The synthesis of various methyl substituted thiazole ligands and their conversion to inhibitors 5j-m are shown in Scheme 3. Methyl thiazole derivatives 20a-d were synthesized utilizing an efficient one step procedure reported by Lagoja and Schantl [47]. This procedure involves reaction of potassium thiocyanate with chloroacetone at 23 °C followed by addition of amino methyl esters 15a-d and heating to 60 °C in methanol for 6 h to provide 20a-d. The procedure resulted in good yields of these thiazole derivatives. However, yields are lower if the reactions are carried out longer than 6 h. Saponification of methyl esters 20 with 1 M LiOH in THF at 23 °C provided carboxylic acids 21a-d. Coupling these ligand acids with amine 11 as described above provided inhibitors 5j-m in good yields.
Scheme 3.

Synthesis of protease inhibitors 5j-m.
The synthesis of trifluoromethyl and methoxymethyl substituted thiazole ligand acids and their conversion to inhibitors 5n-p are shown in Scheme 4. For the synthesis of trifluoromethyl substituted ligand acids, thiourea derivatives 16a and 16b were reacted with trifluoromethyl ketone 22 to provide thiazole ester derivatives 23a and 23b. Saponification of the methyl esters furnished ligand acids 24a and 24b. Coupling of these acids with amine 11 resulted inhibitors 5m and 5n, respectively. Methoxymethyl substituted thiazole ligand acid was synthesized from thiourea derivative 16d and the known α-bromo ketone 25, which was prepared using the reported procedure [48]. This bromo ketone derivative is known to undergo Favorskii rearrangement upon heating, standing, or in the presence of base to provide the un-desired secondary α-bromide [48]. We therefore employed freshly prepared bromo ketone. The reaction of 25 with thiourea 16d was carried out with lower equivalents of potassium carbonate. The thiazole derivative 26 was obtained in low yield possibly due the extreme base sensitivity. Saponification of ester 26 followed by coupling of the resulting acid 27 with amine 11 afforded inhibitor 5p.
Scheme 4.

Synthesis of protease inhibitors 5m-p.
2.2. Biological evaluation and structure-activity relationship
We first investigated the ability of the nilotinib subunit, pyridyl-pyrimidinyl amino benzamides to serve as P2 ligands in the S2 subsite of HIV-1 protease. The structure and activity of these inhibitors are shown in Table 1. The HIV-1 protease inhibitory activity of these compounds was evaluated in an enzyme-inhibitory assay reported by Toth and Marshall [49].
Table 1.
Structure and activity of pyrimidine- and thiozole-derived protease inhibitors.
| Entry | Inhibitor | Ki (nM)a | IC50 (nM)b |
|---|---|---|---|
| 1. |

|
0.028 | 154 |
| 2. |
|
0.17 | 66.8 |
| 3. |

|
0.58 | 124 |
| 4. |

|
1.92 | 212 |
| 5. |

|
38.3 | 390 |
| 6. |

|
19.3 | >1,000 |
| 7. |

|
185.2 | >1,000 |
| 8. |

|
8.7 | 580 |
| 9. |

|
122 | 510 |
Ki values represent at least 5 data points. Standard error in all cases was less than 7%. Darunavir exhibited Ki = 16 pM.
Values are means of at least three experiments. Standard error in all cases was less than 5%. Darunavir exhibited IC50 = 2.1 nM.
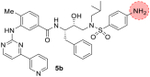
As shown, compound 5a with a nilotinib pyridyl-pyrimidinyl heterocycle as the P2 ligand and 4-methoxy sulfonamide as the P2′-ligand exhibited very potent HIV-1 protease inhibitory activity with a Ki of 0.028 nM. The corresponding 4-aminosulfonamide derivative 5b displayed a nearly 5-fold reduction in inhibitory Ki value (entry 2). We also determined the antiviral activity of these inhibitors in MT-2 human-T-lymphoid cells exposed to HIVLAI [50]. In this antiviral assay, compound 5a showed a potent antiviral IC50 value of 154 nM. Interestingly, compound 5b showed a further improvement in antiviral activity compared to its 4-methoxy derivative, 5a. Compound 5c with 3, 5-difluorophenylmethyl as the P1 ligand did not improve inhibitory activity and showed only a slight improvement in antiviral activity over compound 5a (entry 3). The corresponding 4-amino derivative 5d did not improve activity over compound 5b. Interestingly, compound 5d exhibited a 3-fold reduction in antiviral activity over compound 5c (entries 3 and 4). We then examined a pyridyl-thiazole heterocycle in place of the pyridyl-pyrimidinyl scaffold in compounds 5a-d. Compound 5e, with a 4-methylbenzamide scaffold showed reduced inhibitory activity compared to its pyridyl-pyrimidine derivative 5a. This compound showed an antiviral IC50 value of 390 nM. The 2-methyl-3-aminothiozolyl derivative 5f displayed an enzyme inhibitory Ki of 19 nM, but did not exhibit any appreciable antiviral activity (entry 6). Compound 5g with a 3-aminothiazoloyl-5-methyl heterocycle did not improve any activity (entry 7). Such discrepancies between activity in enzyme and antiviral assays are common possibly due to poor cell membrane permeability of compounds 5f and 5g. Interestingly, 3-aminothiazoloyl-6-methyl derivative 5h showed an improvement in Ki value (8.7 nM). Also, this compound showed an antiviral IC50 value of 580 nM (entry 8). Compound 5i with a 2,4-dimethyl-3-aminothiazolyl derivative showed reduction in Ki value, but maintained an antiviral IC50 of 510 nM, comparable to derivative 5h (entries 8, 9).
We then investigated the effect of an alkyl substitution on the 3-thiazolyl group as well as methyl substitution on the benzamide scaffold. The structure and activity of various derivatives are shown in Table 2. Compound 5j with a 3-methyl-thiazolylamino heterocycle showed a slight improvement in HIV-1 inhibitory activity (Ki value 26 nM) over the corresponding 3-pyridine derivative 5e. However, it did not show any appreciable antiviral activity (entry 1). We have examined the effect of methyl substitution on the benzamide ring. Compounds 5k-m with 2-methyl, 5-methyl, and 6-methyl substitutions resulted in no improvement of activity over 4-methyl derivative 5j (entries 1–4). Also, substitution of a 2-trifluoromethyl group in compounds 5n and 5o did not improve any activity over their methyl derivatives (entries 5 and 6). Incorporation of a methoxy group on the 2-methyl derivative in compound 5p showed a slight improvement in inhibitory activity. This compound also exhibited an antiviral activity IC50 value of 630 nM (entry 7). Our X-ray crystallographic studies of inhibitor 5b-bound HIV-1 protease provided molecular insight into the ligand-binding site interactions responsible for its inhibitory properties.
Table 2.
Structure and activity of substituted thiazolyl-derived protease inhibitors.
| Entry | Inhibitor | Ki (nM)a | IC50 (nM)b |
|---|---|---|---|
| 1. |

|
26.5 | >1,000 |
| 2. |

|
71.9 | >1,000 |
| 3. |

|
80.0 | >1,000 |
| 4. |

|
75.9 | >1,000 |
| 5. |

|
60.3 | >1,000 |
| 6. |

|
72 | >1,000 |
| 7. |

|
18.0 | 630 |
Ki values represent at least 5 data points. Standard error in all cases was less than 7%. Darunavir exhibited Ki = 16 pM.
Values are means of at least three experiments. Standard error in all cases was less than 5%. Darunavir exhibited IC50 = 2.1 nM.
2.3. X-ray structural studies
The X-ray structures of inhibitors 5b and 5c were determined in complex with wild-type HIV-1 protease at 1.25 Å and 1.12 Å, respectively [51]. Both complexes crystallized in the orthorhombic space group P21212 with one protease homodimer per asymmetric unit. The inhibitors were observed in the active site in two alternate conformations related by 180° rotation with relative occupancy of 0.65/0.35 and 0.55/0.45, respectively. These conformations are depicted in Fig. 3 and the 2Fo-Fc electron density maps showing the alternative conformations are in the Supplemental Material. Crystal structures of HIV protease with inhibitors frequently have alternate conformations for inhibitors bound in two orientations due to the pseudo-symmetrical arrangement of the two subunits in the protease dimer. In most cases, the two conformations are almost identical and show very similar interactions with the protease. However, unlike most of the reported X-ray structures of HIV protease, the two conformations of inhibitors 5b and 5c formed different interactions with HIV protease and nearby solvent molecules. In particular, inhibitor 5b had an unusually large difference in the two conformations for the P2–P3 rings. Conformation A has higher relative occupancy of 0.65 which suggests the A conformation is preferred over the B conformation. The protease dimers in 5b- and 5c-HIV protease complexes share similar backbone structures with HIV-1 PR/darunavir (DRV) complex [27,28] with a low RMSD of 0.13 Å and 0.17 Å, respectively, for superposition of 198 equivalent Ca atoms. The largest deviation of 0.5 Å occurred at residue Gly49’ in 5b-bound HIV-1 protease structure, and 0.7 Å at Ala71’ in 5c-bound HIV-1 protease structure. Both inhibitors formed similar hydrogen bonding interactions to those observed between DRV and the main chain atoms of HIV-1 protease. The key interactions of inhibitor 5b with HIV-1 protease and inhibitor 5c with HIV-1 protease are highlighted in stereoviews of the active sites in Figs. 4 and 5, respectively. As in many HIV protease structures, both inhibitors also formed a tetracoordinated water-mediated hydrogen bond interaction with one of the sulfonamide oxygens, the carbonyl oxygen and the amide nitrogen of flap residues Ile50 and Ile50’ [4,5].
Fig. 3.

Superimposed conformations of inhibitors 5b and 5c in the HIV-1 protease active site. 5b: conformation A in turquoise and conformation B in magenta with relative occupancies of 0.65 and 0.35 (PDB code, 5b: 8FUI); 5c: conformation A in orange and conformation B in green with relative occupancies of 0.55 and 0.45 (PDB code, 5c: 8FUJ).
Fig. 4.

Stereoview of the X-ray crystal structure of inhibitor 5b (turquoise) conformation A into the active site of HIV-1 protease (PDB code: 8FUI). The pyridyl-pyrimidine P2 ligand makes several hydrogen bonds in the S2 subsite. Hydrogen bond interactions were defined by standard geometric criteria. All key hydrogen bonds are shown as black dotted lines.
Fig. 5.

Stereoview of the X-ray crystal structure of inhibitor 5c (orange) conformation A into the active site of HIV-1 protease (PDB code: 8FUJ). The pyridyl-pyrimidine P2 ligand makes interesting hydrogen bonds in the S2 subsite. Hydrogen bond interactions were defined by standard geometric criteria. All key hydrogen bonds are shown as black dotted lines.
Both inhibitors 5b and 5c are quite different from darunavir as the bis-THF, P2 ligand is replaced with a P2–P3 group which contains three linked aromatic rings, a 4-methyl-benzamide, a pyrimidine, and a pyridine moeity. The 4-methyl-benzamide group is closest to the P1 group of the inhibitor, the pyrimidine group is in the center, and the pyridine group is the outermost ring. Other modifications were introduced in inhibitor 5c which contains a methoxy group in place of the aniline NH2 in the P2′ ligand of DRV, and a 3,5-difluorophenylmethyl moiety replaces the P1 phenyl ligand in DRV. These altered P1 and P2′ groups show similar interactions with protease as described for the corresponding groups in other inhibitors [52,53]. For both inhibitors, the outer two rings occur in two distinct conformations for the two orientations of the inhibitor due to different rotations of the flexible connections combined with weaker interactions formed with the protease. In conformation A (Fig. 3) of inhibitor 5b-bound HIV-1 protease structure, the outer two rings (P3) are rotated toward Asp29 and Arg8′ and are stabilized by π-π stacking interactions. In conformation B, the outer rings are rotated toward the flap region and form weak C–H⋯O with Gly48 and the carbonyl oxygen of Met46. The 4-methyl-benzamide is a mimic of 4-aminobenzene at P2’, however, the methyl group of 4-methyl-benzamide in P2–P3 does not coincide with the position of the amide (N1) of the P2’ ligand on 180° rotation, but is shifted by 1.6 Å and 1.2 Å, respectively, in the two orientations of the inhibitor.
The hydrogen bond interactions of inhibitor 5b with HIV protease are shown in Fig. 4. The interactions between the P2–P3 group in inhibitor 5b and protease include standard hydrogen bonds and multiple weaker interactions, such as C–H⋯O and π-π stacking interactions. The geometrical criteria used to define the weaker interactions are stated in the Supplementary material. As shown in Fig. 3, the N62 atom of the pyrimidine group forms a hydrogen bond with the amine of Asp29 at 3.2 Å. The 4-methyl-benzamide group forms additional C–H … π interactions with Ala28. A chloride ion in the solvent interacts with atom N60 of the 4-methyl benzamide group and the amine of Gly48 at distances of 3.3 Å and 3.2 Å, respectively. The pyrimidine N66 of the pyrimidine group forms a hydrogen bond with a water molecule, while C76 forms a C–H⋯O interaction with a second water molecule. The first water molecule forms a direct hydrogen bond with the carbonyl oxygen atom of Gly48, and the second water molecule interacts via a network of other water molecules with the side chains of Glu21′ and Arg8’. The pyrimidine and pyridine rings form additional π-π stacking interactions with the carboxylic oxygen of Asp29 and the guanidine of Arg8’. The pyrimidine of P2–P3 also forms internal π-π stacking interactions with the P1 phenyl of the inhibitor. Additional C–H … π interactions also occur with Val82’. These favorable interactions were omitted from Fig. 3 for clarity.
In conformation B of inhibitor 5b and in both orientations of inhibitor 5c in Fig. 5, the N60 atom of the 4-methyl-benzamide group forms a hydrogen bond with the carbonyl oxygen of Gly48 at 2.8–3.0 Å. A water molecule mediates hydrogen bond interactions between the N62 atom of the pyrimidine group and the amine atom of Asp29. The 4-methyl-benzamide group is sandwiched between the carbonyl oxygen atoms of Asp30 and Gly48 via C–H⋯O interactions at distances of 3.5 Å and 3.2 Å, respectively in conformation B of inhibitor 5b. In both orientations of inhibitor 5c, similar interactions with Gly48 are maintained at distances of 3.0–3.1 Å. The pyrimidine group of the N66 atom forms a hydrogen bond with the amine of Gly48 at 3.0–3.3 Å distance, while the C63 atom of the pyrimidine group forms a C–H⋯O interaction with the side chain carboxylate of Asp29.
Several other weak interactions link the P2–P3 group of the inhibitor with the protease. C–H … π interactions connect the 4-methyl-benzamide group with Ala28 sidechain and the pyrimidine group with the Ile47 sidechain. Two water molecules form OH⋯π interactions with the pyrimidine group and the pyridine group, however, the second water interaction with the pyridine group is lost in one conformation of inhibitor 5c. The carbonyl oxygen atom of Met46 forms a lone-pair … π interaction with the pyridine group at 3.2–3.8 Å. One parallel-displaced π-π stacking interaction is formed between the pyrimidine group and the carboxylic group of Asp30. Therefore, both inhibitors can attain multiple interactions with protease due to the distinct conformations of the P2–P3 rings. These interactions are responsible for the high affinity of inhibitors 5b and 5c by the HIV-1 protease.
3. Conclusion
In summary, we have designed and synthesized a new class of HIV-1 protease inhibitors incorporating the pyridyl-pymiridine nilotinib subunit and aryl thiazole derivatives of dasatinib as the P2 ligands. We have examined the activity profiles of methyl thiazole derivatives. Also, we examined the effect of methyl substitution on the benzamide ring. Various alkyl and aryl thiazoles were conveniently prepared using the Hantzsch thiazole synthesis. Several compounds exhibited very potent enzyme inhibitory activity in low nanomolar to subnanomolar level. A number of compounds also displayed low nanomolar antiviral activity. In general, pyridyl-pymiridine benzamide derivatives provided the most promising results. Compound 5a with a 4-methoxysulfonamide as the P2′-ligand turned out be the most potent compound, which exhibited an enzyme inhibitory Ki of 28 pM and antiviral activity of 154 nM. Compound 5b with a 4-aminosulfonamide as the P2′-ligand displayed potent antiviral activity with an IC50 value of 66 nM. Incorporation of lipophilic 1,3-difluorophenyl P1-ligand did not improve antiviral activity. Among thiazole derived inhibitors, compound 5h with a pyridyl thiazole as the P2 ligand showed the best result exhibiting an HIV-1 protease inhibitory Ki of 8.7 nM and antiviral activity of 580 nM. Methyl thiazole-derived compounds showed low nanomolar HIV-1 protease activity but no appreciable antiviral activity. Inhibitors 5a and 5b differ only in the presence of OMe instead of NH2 in the P2′ ligand, although the conformations of the P2–P3 rings may differ to an unknown extent and influence the binding potency. Our X-ray structural studies of inhibitor 5b-bound HIV-1 protease provided intriguing molecular insight into their ligand-binding site interactions. As it turns out, one of the pyrimidine nitrogens forms a hydrogen bond with the backbone amide NH of Asp29. The pyrimidine group also forms additional C–H … π interactions with Ala28. Also, the other pyrimidine nitrogen forms a water mediated hydrogen bond with the amide NH of Gly48, located in the flap region of HIV-1 protease. Moreover, the pyrimidine and pyridine rings make favorable π-π and C–H … π interactions with the protease side chains. In contrast, inhibitor 5c cannot form these interactions likely due to steric hindrance from the fluorines on the P1 phenyl, which may explain its lower potency. The preliminary results of these pyridyl-pyrimidine-derived inhibitors are very encouraging and warrant further optimization of ligand-binding site interactions. We are utilizing these current results for further improvement of inhibitor properties.
4. Experimental section
All moisture-sensitive reactions were carried out in oven-dried glassware under an argon atmosphere unless otherwise stated. Anhydrous solvents were obtained as follows: Tetrahydrofuran was distilled from sodium metal/benzophenone under argon. Dichloromethane was distilled from calcium hydride under argon. All other solvents were reagent grade. Column chromatography was performed using Silicycle Silia Flash F60 230–400 mesh silica gel. Thin layer chromatography was carried out using EMD Millipore TLC silica gel 60 F254 plates. 1H NMR and 13C NMR spectra were recorded on a Bruker AV–III–400, Bruker DRX500, or Bruker AV-111–800. Low-resolution mass spectra were collected on an LCMS. High-resolution mass spectra were collected by the Purdue University Campus-Wide Mass Spectrometry Center. HPLC analysis and purification were done on an Agilent 1100 series instrument using a YMC-Pak ODS-A column of 4.6 mm i.d. for analysis and either 10 mm i.d. or 20 mm i.d. for purification. The purity of all test compounds was determined by HPLC analysis to be >90% pure.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino) benzamide (5a). To a stirring solution of commercially available carboxylic acid 10 (15 mg, 0.05 mmol) in DMF (1 mL) was added triethylamine (0.05 mL, 0.35 mmol), HATU (29 mg, 0.08 mmol), and amine 11 (0.06 mmol). The reaction was then stirred for 24 h at 23 °C. The reaction mixture was concentrated under reduced pressure and purified via silica gel column chromatography (70% EtOAc/hexanes) to afford 5a (22 mg, 76%): 1H NMR (500 MHz, CDCl3) δ 9.30 (s, 1H), 8.73 (d, J = 4.7 Hz, 1H), 8.62 (s, 1H), 8.51 (dd, J = 5.2, 0.8 Hz, 1H), 8.35 (dt, J = 7.9, 2.0 Hz, 1H), 7.72–7.66 (m, 2H), 7.45 (dd, J = 8.0, 4.8 Hz, 1H), 7.32–7.10 (m, 7H), 6.95–6.89 (m, 2H), 6.54 (d, J = 8.4 Hz, 1H), 4.42 (dt, J = 7.4, 3.7 Hz, 1H), 4.08 (dt, J = 8.0, 4.0 Hz, 1H), 3.82 (s, 3H), 3.22 (dd, J = 15.0, 4.2 Hz, 1H), 3.14–3.00 (m, 3H), 2.94 (dd, J = 13.5, 7.7 Hz, 1H), 2.84 (dd, J = 13.4, 7.3 Hz, 1H), 2.38 (s, 3H), 1.90 (dq, J = 13.7, 6.8 Hz, 1H), 0.85 (dd, J = 11.7, 6.6 Hz, 6H);13C NMR (125 MHz, CDCl3) δ 168.10, 163.01, 162.64, 160.45, 159.31, 151.56, 148.44, 138.23, 137.76, 134.87, 133.12, 131.45, 130.88, 130.42, 129.61, 129.59, 128.67, 126.63, 124.05, 121.93, 119.21, 114.38, 108.69, 72.65, 58.68, 55.69, 54.55, 53.69, 35.02, 29.84, 27.25, 20.23, 20.19, 18.30; LRMS-ESI (m/z): [M+H] 695.3. HRMS (ESI) m/z: [M+H] calcd C38H42N6O5SH 695.30157; found 695.30138.
N-((2S,3R)-4-((4-amino-N-isobutylphenyl)sulfonamido)-3-hydroxy-1-phenylbutan-2-yl)-4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino) benzamide (5b). Inhibitor 5b was synthesized according to the same procedure as inhibitor 5a utilizing carboxylic acid 10 (12 mg, 0.04 mmol), DMF (1 mL), DIPEA (0.04 mL, 0.24 mmol), HATU (20 mg, 0.05 mmol), and amine 12 (0.05 mmol). The crude product was purified via silica gel column chromatography (5% methanolic ammonia/CH2Cl2) to afford 5b (20 mg, 74%): 1H NMR (500 MHz, CDCl3) δ 9.31 (s, 1H), 8.74 (d, J = 4.8 Hz, 1H), 8.62 (s, 1H), 8.53 (d, J = 5.1 Hz, 1H), 8.37 (dt, J = 8.1, 2.0 Hz, 1H), 7.54 (d, J = 8.4 Hz, 2H), 7.46 (dd, J = 8.0, 4.8 Hz, 1H), 7.36–7.16 (m, 8H), 7.13 (s, 1H), 6.63 (d, J = 8.4 Hz, 2H), 5.16 (d, J = 9.0 Hz, 1H), 4.45 (td, J = 7.3, 3.8 Hz, 1H), 4.18 (s, 2H), 4.10 (dt, J = 8.2, 4.2 Hz, 1H), 3.30–3.04 (m, 4H), 2.88 (ddd, J = 50.0, 13.4, 7.5 Hz, 2H), 2.40 (s, 3H), 1.95–1.89 (m, 1H), 0.88 (dd, J = 10.3, 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 168.07, 162.64, 160.49, 159.36, 151.64, 150.74, 148.51, 138.28, 137.75, 134.77, 133.12, 132.62, 131.59, 130.84, 129.61, 129.58, 128.62, 126.64, 126.57, 123.99, 121.91, 119.39, 114.17, 108.68, 72.74, 58.83, 54.49, 54.41, 53.79, 35.03, 27.28, 20.24, 18.28, 14.24; LRMS-ESI (m/z): [M+H] 680.3. HRMS (ESI) m/z: [M+H] calcd C37H41N7O4SH 680.30189; found 680.30245.
N-((2S,3R)-1-(3,5-difluorophenyl)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)butan-2-yl)-4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)benzamide (5c). Inhibitor 5c was synthesized according to the same procedure as inhibitor 5a utilizing carboxylic acid 10 (15 mg, 0.05 mmol), DMF (1 mL), DIPEA (0.05 mL, 0.29 mmol), HATU (25 mg, 0.06 mmol), and amine 13 (0.06 mmol). The crude product was purified via silica gel column chromatography (30% acetone/hexanes) to afford 5c (15 mg, 41%): 1H NMR (500 MHz, CDCl3) δ 9.34 (d, J = 2.3 Hz, 1H), 8.77–8.68 (m, 2H), 8.53 (d, J = 5.1 Hz, 1H), 8.33 (dt, J = 8.1, 1.9 Hz, 1H), 7.75–7.67 (m, 2H), 7.46 (dd, J = 8.0, 4.8 Hz, 1H), 7.29 (dd, J = 8.9, 7.1 Hz, 1H), 7.21 (dd, J = 5.2, 1.9 Hz, 1H), 7.10 (s, 1H), 6.97–6.92 (m, 2H), 6.90–6.81 (m, 2H), 6.66–6.56 (m, 1H), 5.52 (s, 1H), 4.41 (dt, J = 8.9, 4.7 Hz, 1H), 4.10 (s, 1H), 3.83 (s, 3H), 3.23–3.16 (m, 1H), 3.14–2.94 (m, 4H), 2.84 (dd, J = 13.4, 7.1 Hz, 1H), 2.40 (s, 3H), 1.89 (dt, J = 13.9, 6.9 Hz, 1H), 0.87 (dd, J = 18.6, 6.5 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 168.10, 164.12, 164.01, 163.11, 162.56, 160.40, 159.44, 151.64, 148.50, 142.63, 137.88, 134.77, 132.67, 131.02, 130.25, 129.59, 124.03, 121.89, 118.33, 114.44, 112.55, 112.36, 108.80, 102.14, 72.57, 58.86, 55.72, 53.99, 53.74, 34.72, 27.29, 20.22, 18.30; LRMS-ESI (m/z): [M+H] 731.2. HRMS (ESI) m/z: [M+H] calcd C38H40F2N6O5SH 731.28272; found 731.28315.
N-((2S,3R)-4-((4-amino-N-isobutylphenyl)sulfonamido)-1-(3,5-difluorophenyl)-3-hydroxybutan-2-yl)-4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)benzamide (5d). Inhibitor 5d was synthesized according to the same procedure as inhibitor 5a utilizing carboxylic acid 10 (15 mg, 0.05 mmol), DMF (1 mL), triethylamine (0.05 mL, 0.29 mmol), HATU (24 mg, 0.06 mmol), and amine 14 (0.06 mmol). The crude product was purified via silica gel column chromatography (50% acetone/hexanes) to afford 5d (32 mg, 89%): 1H NMR (500 MHz, CDCl3) δ 9.32 (d, J = 2.3 Hz, 1H), 8.74 (dd, J = 4.8, 1.6 Hz, 1H), 8.67 (d, J = 2.0 Hz, 1H), 8.52 (d, J = 5.1 Hz, 1H), 8.33 (dt, J = 8.1, 1.9 Hz, 1H), 7.57–7.51 (m, 2H), 7.45 (dd, J = 8.0, 4.8 Hz, 1H), 7.29 (dd, J = 10.4, 8.5 Hz, 1H), 7.21 (dd, J = 5.2, 1.9 Hz, 1H), 7.10 (s, 1H), 6.87–6.83 (m, 2H), 6.66–6.57 (m, 3H), 5.37 (s, 1H), 4.41 (dd, J = 7.3, 3.6 Hz, 1H), 4.13 (s, 2H), 4.08 (s, 1H), 3.18 (dd, J = 15.0, 4.5 Hz, 1H), 3.14–2.88 (m, 4H), 2.80 (dd, J = 13.4, 7.1 Hz, 1H), 2.39 (s, 3H), 1.94–1.82 (m, 1H), 0.87 (dd, J = 16.1, 6.6 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 168.07, 162.61, 160.44, 159.41, 151.67, 150.76, 148.50, 142.60, 137.87, 134.77, 133.01, 132.66, 131.26, 131.00, 129.61, 126.61, 124.02, 121.90, 118.67, 114.23, 112.55, 112.36, 108.78, 102.10, 72.70, 59.02, 54.01, 53.82, 34.76, 29.84, 27.34, 20.24, 18.31; LRMS-ESI (m/z): [M+H] 716.3. HRMS (ESI) m/z: [M+H] calcd C37H39F2N7O4SH 716.28306; found 716.28351.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-4-methyl-3-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzamide (5e). To a stirring solution of 19a (16.5 mg, 0.05 mmol) in CH2Cl2 (1.0 mL) was added triethylamine (0.04 mL, 0.32 mmol) and HATU (26.0 mg, 0.07 mmol) at 23 °C. To the reaction was then added 11 (0.058 mmol) and the reaction was stirred for 24h. The reaction mixture was washed with 1 M citric acid and then successively with H2O. The organic layer was then washed with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified via silica gel column chromatography (40–45% EtOAc/hexanes) to afford 5e (11 mg, 29.7%): 1H NMR (500 MHz, CDCl3) δ 9.12 (d, J = 1.8 Hz, 1H), 8.51 (d, J = 5.9 Hz, 1H), 8.24 (s, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.68 (d, J = 8.8 Hz, 2H), 7.46 (bs, 1H), 7.33–7.23 (m, 6H), 7.16 (t, J = 7.3 Hz, 1H), 6.95–6.87 (m, 3H), 6.72 (d, J = 8.4 Hz, 1H), 4.46–4.38 (m, 1H), 4.1–4.06 (m, 1H), 3.83 (s, 3H), 3.22–3.04 (m, 4H), 2.95–2.78 (m, 2H), 2.34 (s, 3H), 1.93–1.81 (m, 1H), 0.85 (t, J = 6.4 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 167.66, 165.70, 163.08, 148.60, 148.32, 147.55, 138.88, 138.16, 133.66, 133.45, 131.99, 131.37, 130.45, 130.22, 129.61, 129.58, 128.72, 126.69, 123.81, 122.70, 118.38, 114.42, 104.03, 72.89, 58.81, 55.72, 54.55, 53.78, 35.16, 27.30, 20.23, 18.01; LRMS-ESI (m/z): [M+H] 700.3. HRMS (ESI) m/z: [M+H] calcd C37H41N5O5S2H 700.262739; found 700.26288.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-2-methyl-3-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzamide (5f). Inhibitor 5f was synthesized according to the same procedure as inhibitor 5e utilizing 19b (14.0 mg, 0.05 mmol), CH2Cl2 (1.0 mL), triethylamine (0.04 mL, 0.27 mmol), HATU (22.0 mg, 0.06 mmol), and 11 (0.05 mmol). The crude product was purified via silica gel column chromatography (50–60% EtOAc/hexanes) to afford 5f (16.7 mg, 53.9%): 1H NMR (500 MHz, CD3OD) δ 9.01 (s, 1H), 8.44 (d, J = 4.3 Hz, 1H), 8.26 (dt, J = 8.0, 1.6 Hz, 1H), 7.88 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 8.9 Hz, 2H), 7.45 (dd, J = 8.0, 4.9 Hz, 1H), 7.33–7.23 (m, 4H), 7.24–7.16 (m, 3H), 7.06 (d, J = 8.9 Hz, 2H), 6.86 (d, J = 7.5 Hz, 1H), 4.34–4.29 (m, 1H), 3.94–3.86 (m, 1H), 3.84 (s, 3H), 3.60 (dd, J = 14.9, 2.9 Hz, 1H), 3.14–3.04 (m, 2H), 2.93 (dd, J = 13.6, 6.8 Hz, 1H), 2.82 (s, 1H), 2.65 (dd, J = 13.9, 11.8 Hz, 1H), 2.08 (dt, J = 14.2, 7.0 Hz, 1H), 1.90 (s, 3H), 0.92 (dd, J = 30.3, 6.6 Hz, 6H); 13C NMR (125 MHz, CD3OD) δ 172.75, 168.69, 164.55, 148.67, 148.64, 147.70, 141.13, 140.21, 139.89, 135.21, 132.73, 132.20, 130.64, 130.52, 129.38, 127.55, 127.36, 127.32, 125.28, 124.80, 123.94, 115.39, 105.60, 74.57, 58.93, 56.19, 55.63, 54.11, 36.88, 28.11, 20.56, 20.48, 14.72; LRMS-ESI (m/z): [M+H] 700.5. HRMS (ESI) m/z: [M+H] calcd C37H41N5O5S2H 700.262739; found 700.26285.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-methyl-5-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzamide (5g). Inhibitor 5g was synthesized according to the same procedure as inhibitor 5e utilizing 19c (23.0 mg, 0.07 mmol), CH2Cl2 (1.2 mL), triethylamine (0.06 mL, 0.1 mmol), HATU (36.0 mg, 0.1 mmol), and 11 (0.08 mmol). The crude product was purified via silica gel column chromatography (50–60% EtOAc/hexanes) to afford 5g (11.2 mg, 22%): 1H NMR (500 MHz, CD3OD) δ 9.09 (d, J = 1.8 Hz, 1H), 8.44 (dd, J = 4.9, 1.6 Hz, 1H), 8.34 (dt, J = 8.0, 1.9 Hz, 1H), 8.18 (d, J = 9.2 Hz, 1H), 7.91 (s, 1H), 7.67–7.59 (m, 3H), 7.46 (ddd, J = 8.0, 4.9, 0.9 Hz, 1H), 7.31–7.26 (m, 3H), 7.21 (t, J = 7.6 Hz, 2H), 7.14–7.10 (m, 2H), 6.89–6.83 (m, 2H), 4.22 (d, J = 9.6 Hz, 1H), 3.99 (td, J = 8.3, 2.7 Hz, 1H), 3.72 (s, 3H), 3.41 (dd, J = 15.2, 2.7 Hz, 1H), 3.10 (dd, J = 13.5, 8.7 Hz, 1H), 2.92 (dd, J = 15.2, 8.7 Hz, 1H), 2.88–2.74 (m, 3H), 2.40 (s, 3H), 2.09–2.02 (m, 1H), 0.90 (dd, J = 38.4, 6.6 Hz, 8H); 13C NMR (125 MHz, CD3OD) δ 170.39, 165.74, 164.42, 148.84, 148.55, 147.67, 142.72, 140.37, 140.26, 136.85, 135.45, 132.72, 131.49, 130.61, 130.37, 129.29, 127.23, 125.40, 121.94, 121.85, 115.24, 114.79, 105.68, 74.78, 61.53, 59.28, 56.05, 54.59, 36.94, 28.02, 21.69, 20.50, 14.45; LRMS-ESI (m/z): [M+H] 700.5. HRMS (ESI) m/z: [M+H] calcd C37H41N5O5S2H 700.262739; found 700.26397.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-2-methyl-5-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzamide (5h). Inhibitor 5h was synthesized according to the same procedure as inhibitor 5e utilizing 19d (33.1 mg, 0.11 mmol), CH2Cl2 (1.8 mL), triethylamine (0.09 mL, 0.64 mmol), HATU (52.0 mg, 0.14 mmol), and 11 (0.12 mmol). The crude product was purified via silica gel column chromatography (60% EtOAc/hexanes) to afford 5h (24.1 mg, 32%): 1H NMR (500 MHz, CDCl3) δ 9.12 (s, 1H), 8.47 (s, 1H), 8.01 (d, J = 7.8 Hz, 2H), 7.71 (d, J = 8.9 Hz, 2H), 7.61 (s, 1H), 7.27 (dt, J = 15.0, 7.4 Hz, 3H), 7.17 (dd, J = 15.3, 8.1 Hz, 2H), 7.04 (d, J = 8.3 Hz, 1H), 6.91 (d, J = 8.9 Hz, 2H), 6.84 (s, 1H), 6.51 (d, J = 9.0 Hz, 1H), 4.59–4.49 (m, 1H), 4.13 (td, J = 7.4, 3.7 Hz, 1H), 3.80 (s, 3H), 3.29–3.10 (m, 3H), 3.02–2.90 (m, 3H), 2.03 (s, 3H), 1.92 (tt, J = 14.9, 7.6 Hz, 1H), 0.89 (dd, J = 13.4, 6.6 Hz, 6H). 13C NMR (125 MHz, CDCl3) δ 169.94, 164.73, 163.05, 148.16, 147.88, 147.69, 138.20, 138.12, 136.87, 133.25, 131.83, 130.80, 130.48, 129.60, 128.66, 127.92, 126.64, 123.81, 119.87, 117.06, 114.43, 114.06, 103.61, 72.91, 58.61, 55.69, 53.72, 34.74, 29.84, 27.29, 20.25, 18.79; LRMS-ESI (m/z): [M+H] 700.5. HRMS (ESI) m/z: [M+H] calcd C37H41N5O5S2H 700.262739; found 700.26323.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-2,4-dimethyl-3-((4-(pyridin-3-yl)thiazol-2-yl)amino) benzamide (5i). Inhibitor 5i was synthesized according to the same procedure as inhibitor 5e utilizing 19e (17.8 mg, 0.06 mmol), CH2Cl2 (0.9 mL), triethylamine (0.05 mL, 0.33 mmol), HATU (27.0 mg, 0.07 mmol), and 11 (0.06 mmol). The crude product was purified via silica gel column chromatography (2% MeOH/CH2Cl2) to afford 5i (7.5 mg, 19%): 1H NMR (800 MHz, CDCl3) δ 8.80 (s, 1H), 8.37 (s, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.72 (d, J = 8.9 Hz, 2H), 7.29 (m, 4H), 7.21 (t, J = 7.1 Hz, 1H), 7.07 (d, J = 7.8 Hz, 1H), 6.96 (dd, J = 15.9, 8.3 Hz, 3H), 6.79 (d, J = 20.7 Hz, 1H), 6.74 (s, 1H), 4.46–4.36 (m, 1H), 4.08–4.00 (m, 1H), 3.86 (s, 3H), 3.26–3.12 (m, 3H), 3.02 (dd, J = 14.2, 10.3 Hz, 1H), 2.92 (ddd, J = 38.8, 13.4, 7.5 Hz, 2H), 2.28 (s, 3H), 2.16 (s, 3H), 1.91 (dt, J = 13.7, 6.9 Hz, 1H), 0.89 (dd, J = 27.0, 6.6 Hz, 6H); 13C NMR (200 MHz, CDCl3) δ 170.60, 170.40, 163.21, 148.06, 146.48, 139.16, 138.13, 137.70, 136.74, 135.02, 133.98, 130.77, 130.04, 129.61, 129.57, 128.86, 128.78, 127.94, 126.80, 126.61, 123.87, 114.54, 114.08, 103.67, 73.27, 59.07, 55.78, 54.56, 53.86, 35.06, 27.48, 20.28, 20.11, 18.57, 15.04; LRMS-ESI (m/z): [M+H] 714.3. HRMS (ESI) m/z: [M+H] calcd C38H43N5O5S2H 714.27839; found 714.27875.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-4-methyl-3-((4-methylthiazol-2-yl)amino)benzamide (5j). Inhibitor 5j was synthesized according to the same procedure as inhibitor 5e utilizing 21a (17.0 mg, 0.05 mmol), CH2Cl2 (0.9 mL), triethylamine (0.05 mL, 0.33 mmol), HATU (27.0 mg, 0.07 mmol), and 11 (0.06 mmol). The crude product was purified via silica gel column chromatography (35% EtOAc/hexanes) to afford 5j (13.2 mg, 38.8%): 1H NMR (500 MHz, CDCl3) δ 7.72–7.69 (m, 2H), 7.33–7.17 (m, 5H), 7.11 (d, J = 8.3 Hz, 1H), 7.01 (d, J = 2.5 Hz, 1H), 6.99–6.94 (m, 2H), 6.19 (s, 1H), 6.06 (d, J = 8.6 Hz, 1H), 4.41–4.35 (m, 1H), 4.20 (bs, 1H), 4.01–3.98 (m, 1H), 3.86 (s, 3H), 3.20–3.11 (m, 3H), 3.05–2.91 (m, 2H), 2.93–2.85 (m, 1H), 2.28 (s, 3H), 2.16 (s, 3H), 1.92–1.87 (m, 1H), 0.90 (dd, J = 17.6, 6.6 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 170.08, 164.33, 163.21, 148.96, 138.33, 137.90, 136.77, 132.17, 130.51, 130.01, 129.61, 129.55, 128.84, 126.84, 120.00, 116.83, 114.53, 102.42, 73.08, 59.07, 55.78, 54.38, 53.85, 35.15, 27.48, 20.27, 20.09, 18.97, 17.46; LRMS-ESI (m/z): [M+H] 637.2. HRMS (ESI) m/z: [M+H] calcd C33H40N4O5S2H 637.25184; found 637.25175.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-2-methyl-3-((4-methylthiazol-2-yl)amino)benzamide (5k). Inhibitor 5k was synthesized according to the same procedure as inhibitor 5e utilizing 21b (20.1 mg, 0.08 mmol), CH2Cl2 (1.35 mL), triethylamine (0.07 mL, 0.49 mmol), HATU (40.0 mg, 0.11 mmol), and 11 (0.09 mmol). The crude product was purified via silica gel column chromatography (35% EtOAc/hexanes) to afford 5k (13.2 mg, 25.3%): 1H NMR (500 MHz, CDCl3) δ 7.75–7.69 (m, 2H), 7.64 (d, J = 8.1 Hz, 1H), 7.32–7.27 (m, 3H), 7.25–7.22 (m, 1H), 7.15 (t, J = 7.9 Hz, 1H), 7.01–6.95 (m, 2H), 6.76 (dd, J = 7.6, 1.2 Hz, 1H), 6.17 (d, J = 1.3 Hz, 1H), 6.01 (d, J = 8.7 Hz, 1H), 4.39 (dd, J = 9.2, 4.8 Hz, 1H), 4.00 (dt, J = 7.5, 4.5 Hz, 1H), 3.87 (d, J = 1.6 Hz, 3H), 3.22–3.11 (m, 3H), 3.01–2.83 (m, 3H), 2.28 (d, J = 1.1 Hz, 3H), 2.04 (s, 3H), 1.95–1.87 (m, 1H), 0.96–0.86 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 170.37, 165.60, 163.24, 139.67, 138.16, 137.85, 129.97, 129.61, 129.50, 128.83, 127.03, 126.88, 126.79, 126.63, 122.29, 121.59, 114.55, 102.54, 73.14, 59.08, 55.78, 54.36, 53.85, 35.18, 27.48, 20.28, 20.08, 17.29, 14.09; LRMS-ESI (m/z): [M+H] 637.2. HRMS (ESI) m/z: [M+H] calcd C33H40N4O5S2H 637.25184; found 637.25154.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-3-methyl-5-((4-methylthiazol-2-yl)amino)benzamide (5l). Inhibitor 5l was synthesized according to the same procedure as inhibitor 5e utilizing 21c (28.4 mg, 0.09 mmol), CH2Cl2 (1.5 mL), triethylamine (0.07 mL, 0.52 mmol), HATU (43.0 mg, 0.11 mmol), and 11 (0.10 mmol). The crude product was purified via silica gel column chromatography (35% EtOAc/hexanes) to afford 5l (10.2 mg, 18.5%): 1H NMR (800 MHz, CDCl3) δ 7.68 (dq, J = 9.0, 2.4, 1.6 Hz, 2H), 7.42 (s, 1H), 7.32–7.24 (m, 4H), 7.22–7.17 (m, 1H), 7.02 (s, 1H), 6.95–6.91 (m, 2H), 6.49 (d, J = 8.3 Hz, 1H), 6.21 (s, 1H), 4.39 (dq, J = 8.2, 3.0 Hz, 1H), 4.00 (dt, J = 8.4, 4.3 Hz, 1H), 3.84 (s, 3H), 3.19 (dd, J = 15.1, 4.1 Hz, 1H), 3.12–3.08 (m, 3H), 2.88 (d, J = 7.5 Hz, 2H), 2.34 (s, 3H), 2.29 (s, 3H), 1.89–1.83 (m, 1H), 0.86 (dd, J = 9.7, 6.6 Hz, 6H); 13C NMR (200 MHz, CDCl3) δ 167.92, 164.05, 163.17, 147.93, 140.62, 140.18, 137.92, 135.50, 129.99, 129.59, 128.82, 126.82, 122.00, 121.67, 114.49, 113.83, 102.45, 72.94, 58.98, 55.75, 54.72, 53.68, 35.23, 27.37, 21.64, 20.24, 20.13, 17.14; LRMS-ESI (m/z): [M+H] 637.3. HRMS (ESI) m/z: [M+H] calcd C33H40N4O5S2H 637.25184; found 637.25228.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-2-methyl-5-((4-methylthiazol-2-yl)amino)benzamide (5m). Inhibitor 5m was synthesized according to the same procedure as inhibitor 5e utilizing 21d (25.0 mg, 0.1 mmol), CH2Cl2 (0.17 mL), triethylamine (0.084 mL, 0.6 mmol), HATU (49.0 mg, 0.13 mmol), and 11 (0.11 mmol). The crude product was purified via silica gel column chromatography (35–40% EtOAc/hexanes) to afford 5m (13.4 mg, 25.8%): 1H NMR (500 MHz, CDCl3) δ 7.74–7.66 (m, 2H), 7.33–7.17 (m, 5H), 7.11 (d, J = 8.2 Hz, 1H), 7.01 (d, J = 2.5 Hz, 1H), 6.99–6.93 (m, 2H), 6.19 (d, J = 1.2 Hz, 1H), 6.07 (d, J = 8.6 Hz, 1H), 4.37 (dq, J = 9.4, 4.6 Hz, 1H), 3.99 (dd, J = 7.4, 4.6 Hz, 1H), 3.86 (s, 3H), 3.22–3.09 (m, 3H), 3.06–2.82 (m, 3H), 2.28 (d, J = 1.1 Hz, 3H), 2.16 (s, 3H), 1.90 (dt, J = 13.8, 6.8 Hz, 1H), 0.90 (dd, J = 17.8, 6.6 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 170.07, 164.43, 163.21, 138.27, 137.91, 136.78, 132.18, 130.58, 130.01, 129.61, 129.55, 128.83, 126.84, 120.02, 116.84, 114.53, 102.35, 73.08, 59.07, 55.78, 54.38, 53.85, 35.15, 29.85, 27.48, 20.27, 20.09, 18.97, 17.39; LRMS-ESI (m/z): [M+H] 637.3. HRMS (ESI) m/z: [M+H] calcd C33H40N4O5S2H 637.25184; found 637.25186.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-4-methyl-3-((4-(trifluoromethyl)thiazol-2-yl)amino) benzamide (5n). Inhibitor 5n was synthesized according to the same procedure as inhibitor 5e utilizing 24a (24.9 mg, 0.079 mmol), CH2Cl2 (1.0 mL), triethylamine (0.066 mL, 0.472 mmol), HATU (39.0 mg, 0.102 mmol), and 11 (0.094 mmol). The crude product was purified via silica gel column chromatography (50% EtOAc/hexanes) to afford 5n (25.9 mg, 35%): 1H NMR (500 MHz, CDCl3) δ 7.89 (d, J = 1.8 Hz, 1H), 7.79–7.69 (m, 3H), 7.32 (dd, J = 9.9, 5.8 Hz, 7H), 7.12–6.96 (m, 3H), 6.57 (d, J = 8.3 Hz, 1H), 4.43 (dd, J = 7.9, 4.5 Hz, 1H), 4.09–4.03 (m, 1H), 3.90 (s, 3H), 3.30–3.10 (m, 4H), 2.92 (dd, J = 7.6, 3.0 Hz, 2H), 2.37 (s, 3H), 1.96–1.87 (m, 1H), 0.90 (s, 7H). 13C NMR (126 MHz, CDCl3) δ 167.96, 167.16, 163.07, 138.38, 137.82, 134.37, 133.41, 131.59, 129.90, 129.47, 129.43, 128.68, 126.68, 123.62, 121.39, 120.06, 119.24, 114.38, 109.74, 72.82, 58.82, 55.65, 54.67, 53.52, 45.95, 35.09, 30.98, 29.74, 27.25, 20.12, 20.01, 17.88, 8.64; LRMS-ESI (m/z): LRMS-ESI (m/z): [M+H] 691.2. HRMS (ESI) m/z: [M+H] calcd C33H37F3N4O5S2H 691.22357; found 691.22424.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-2-methyl-3-((4-(trifluoromethyl)thiazol-2-yl)amino) benzamide (5o). Inhibitor 5o was synthesized according to the same procedure as inhibitor 5e utilizing 24b (20.1 mg, 0.063 mmol), CH2Cl2 (1.0 mL), triethylamine (0.05 mL, 0.378 mmol), HATU (31.1 mg, 0.08 mmol), and 11 (0.08 mmol). The crude product was purified via silica gel column chromatography (50% EtOAc/hexanes) to afford 5o (15.4 mg, 35.4%): 1H NMR (500 MHz, CDCl3) δ 7.76–7.68 (m, 2H), 7.56–7.44 (m, 1H), 7.30 (d, J = 6.3 Hz, 4H), 7.25–7.16 (m, 2H), 7.03–6.94 (m, 3H), 6.89 (dd, J = 8.1, 2.0 Hz, 1H), 6.10 (dd, J = 18.5, 8.8 Hz, 1H), 4.42 (s, 1H), 3.87 (s, 3H), 3.16 (ddd, J = 14.6, 8.7, 5.8 Hz, 3H), 3.00–2.93 (m, 2H), 2.91–2.84 (m, 1H), 2.05 (d, J = 10.2 Hz, 3H), 1.91 (t, J = 7.0 Hz, 1H), 0.91 (dd, J = 14.3, 6.5 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ 172.15, 171.31, 163.53, 137.95, 137.60, 136.98, 131.81, 131.54, 129.59, 129.33, 129.18, 129.02, 127.85, 127.27, 126.56, 126.29, 126.00, 114.70, 73.04, 58.97, 55.81, 54.60, 53.57, 53.11, 34.51, 31.09, 29.85, 27.44, 26.92, 20.15, 19.89, 19.46, 14.23. LRMS-ESI (m/z): [M+H] 691.2. HRMS (ESI) m/z: [M+H] calcd C33H37F3N4O5S2H 691.22357; found 691.22400.
N-((2S,3R)-3-hydroxy-4-((N-isobutyl-4-methoxyphenyl)sulfonamido)-1-phenylbutan-2-yl)-5-((4-(methoxymethyl)thiazol-2-yl)amino)-2-methylbenzamide (5p). Inhibitor 5p was synthesized according to the same procedure as inhibitor 5e utilizing 27 (17.4 mg, 0.06 mmol), CH2Cl2 (1.1 mL), triethylamine (0.05 mL, 0.38 mmol), HATU (31.0 mg, 0.08 mmol), and 11 (0.07 mmol). The crude product was purified via silica gel column chromatography (35% EtOAc/hexanes) to afford 5p (10.6 mg, 25.2%): 1H NMR (800 MHz, CDCl3) δ 7.71 (d, J = 8.8 Hz, 2H), 7.31–7.27 (m, 3H), 7.24–7.19 (m, 2H), 7.12 (d, J = 8.1 Hz, 2H), 6.98–6.96 (m, 3H), 6.53 (s, 1H), 6.07 (d, J = 8.7 Hz, 1H), 4.40 (s, 2H), 4.18 (s, 1H), 4.01 (d, J = 6.1 Hz, 1H), 3.87 (s, 3H), 3.44 (s, 3H), 3.18–3.14 (m, 3H), 3.01 (dd, J = 14.3, 10.0 Hz, 1H), 2.96–2.93 (m, 1H), 2.88 (d, J = 6.4 Hz, 1H), 2.17 (s, 3H), 1.90 (dt, J = 14.0, 6.8 Hz, 1H), 0.90 (dd, J = 26.3, 6.6 Hz, 6H); 13C NMR (200 MHz, CDCl3) δ 169.96, 163.22, 138.07, 137.93, 136.87, 132.26, 130.04, 129.62, 129.55, 129.37, 128.85, 127.95, 126.85, 120.24, 117.16, 114.54, 114.08, 105.18, 73.09, 70.53, 59.06, 58.71, 55.79, 54.37, 53.83, 35.08, 27.48, 20.28, 20.10, 19.00; LRMS-ESI (m/z): [M+H] 667.2. HRMS (ESI) m/z: [M+H] calcd C34H42N4O6S2H 667.26241; found 667.26216.
Methyl 3-(3-acetylthioureido)-4-methylbenzoate (16a). To a solution of potassium thiocyanate (292 mg, 3.0 mmol) in acetone (7.5 mL) was added acetyl chloride (0.14 mL, 2.0 mmol) dropwise. The reaction mixture was stirred at 60 °C for 1.5 h. To the reaction was then added methyl 3-amino-4-methylbenzoate (165 mg, 1.0 mmol) and the reaction was stirred at 23 °C for 4 h. The reaction was then filtered and concentrated under reduced pressure. The crude product was purified via silica gel column chromatography (25% ethyl acetate/hexanes) to afford the desired thiourea in quantitative yield as a white solid: 1H NMR (500 MHz, CD3OD) δ 8.27 (d, J = 1.8 Hz, 1H), 7.85 (dd, J = 8.0, 1.8 Hz, 1H), 7.39 (d, J = 8.0 Hz, 1H), 3.90 (d, J = 0.8 Hz, 3H), 2.33 (s, 3H), 2.19 (d, J = 0.8 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 181.89, 173.93, 173.15, 168.03, 140.84, 138.52, 131.82, 129.59, 129.01, 52.64, 23.87, 18.30.
Methyl 3-(3-acetylthioureido)-2-methylbenzoate (16b). Thiourea 16b was synthesized according to the same procedure as thiourea 16a utilizing potassium thiocyanate (292 mg, 3.0 mmol), acetone (7.5 mL), acetyl chloride (0.14 mL, 2.0 mmol), methyl 3-amino-2-methylbenzoate (165 mg, 1.0 mmol). The crude mixture was purified via silica gel column chromatography (25% ethyl acetate/hexanes) to afford the desired thiourea in quantitative yield as a white solid: 1H NMR (500 MHz, CDCl3) δ 9.39 (s, 1H), 7.85 (d, J = 7.7 Hz, 1H), 7.69 (d, J = 7.8 Hz, 1H), 7.31 (t, J = 7.9 Hz, 1H), 3.90 (s, 3H), 2.50 (s, 3H), 2.22 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 180.24, 171.67, 167.91, 137.46, 135.87, 131.83, 130.97, 130.17, 126.04, 52.29, 24.56, 15.37.
Methyl 3-(3-acetylthioureido)-5-methylbenzoate (16c). Thiourea 16c was synthesized according to the same procedure as thiourea 16a utilizing potassium thiocyanate (292 mg, 3.0 mmol), acetone (7.5 mL), acetyl chloride (0.14 mL, 2.0 mmol), methyl 3-amino-2-methylbenzoate (165 mg, 1.0 mmol). The crude mixture was purified via silica gel column chromatography (25% ethyl acetate/hexanes) to afford methyl 3-(3-acetylthioureido)-5-methylbenzoate (138 mg, 42.5%): 1H NMR (500 MHz, CDCl3) δ 12.31 (s, 1H), 8.90 (s, 1H), 3.91 (s, 3H), 2.42 (2, 3H), 2.22 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.71, 171.20, 166.60, 139.36, 137.69, 131.01, 129.41, 128.89, 122.81, 52.42, 24.66, 21.44.
Methyl 5-(3-acetylthioureido)-2-methylbenzoate (16d). Thiourea 16d was synthesized according to the same procedure as thiourea 16a utilizing potassium thiocyanate (353 mg, 3.6 mmol), acetone (9 mL), acetyl chloride (0.17 mL, 2.4 mmol), 5-amino-2-methylbenzoate (200 mg, 1.2 mmol). The crude mixture was purified via silica gel column chromatography (25% ethyl acetate/hexanes) to afford methyl 5-(3-acetylthioureido)-2-methylbenzoate (181 mg, 56%): 1H NMR (500 MHz, CDCl3) δ 8.87 (s, 1H), 8.08 (d, J = 2.2 Hz, 1H), 7.76 (dd, J = 8.3, 2.3 Hz, 1H), 7.28 (d, J = 8.3 Hz, 1H), 3.90 (s, 3H), 2.60 (s, 3H), 2.22 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 178.74, 171.18, 167.24, 139.40, 135.31, 132.35, 130.04, 128.05, 126.67, 52.18, 24.66, 21.57.
Methyl 3-(3-acetylthioureido)-2,4-dimethylbenzoate (16e). Thiourea 16e was synthesized according to the same procedure as thiourea 16a utilizing potassium thiocyanate (33 mg, 0.34 mmol), acetone (0.84 mL), acetyl chloride (0.02 mL, 0.22 mmol), and methyl 3-amino-2,4-dimethylbenzoate (0.02 mg, 0.11 mmol). The crude product was purified via silica gel column chromatography (30% ethyl acetate/hexanes) to afford methyl 3-(3-acetylthioureido)-2,4-methylbenzoate (26.7 mg, 85%): 1H NMR (500 MHz, CDCl3) δ 9.91 (s, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.17 (d, J = 8.1 Hz, 1H), 3.88 (s, 3H), 2.48 (s, 3H), 2.31 (s, 3H), 2.22 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 180.55, 172.07, 167.86, 140.28, 137.77, 136.42, 130.64, 129.18, 127.95, 52.09, 24.43, 18.90, 15.67; LRMS-ESI (m/z): [M+H]+ 281.0.
Methyl 4-methyl-3-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoate (18a). To a stirring solution of 16a (58 mg, 0.22 mmol) in dry methanol (1.3 mL) was added K2CO3 (182 mg, 1.32 mmol) and stirred for 5 min. To the reaction was then added 3-(bromoacetyl)pyridine • HBr (62 mg, 0.22 mmol) and the reaction was stirred for 6 h. The reaction mixture was then concentrated under reduced pressure and diluted with ethyl acetate and water. The aqueous layer was extracted with ethyl acetate (3x) and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified via silica gel column chromatography (50% ethyl acetate/hexanes) to afford 18a as a white solid (45 mg, 60%): 1H NMR (500 MHz, CDCl3) δ 9.14 (d, J = 2.3 Hz, 1H), 8.55–8.47 (m, 2H), 8.11 (dt, J = 7.9, 2.0 Hz, 1H), 7.76 (dd, J = 7.9, 1.7 Hz, 1H), 7.59–7.51 (bs, 1H), 7.32–7.27 (m, 2H), 6.91 (s, 1H), 3.93 (s, 3H), 2.39 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 166.82, 166.18, 148.83, 148.60, 147.68, 138.90, 134.34, 133.43, 131.26, 130.53, 129.51, 125.63, 123.62, 121.52, 103.72, 52.36, 18.24; LRMS-ESI (m/z): [M+H]+ 326.1.
Methyl 4-methyl-3-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoate (18b). Thiazole 18b was synthesized according to the same procedure as thiazole 18a utilizing 16b (77.7 mg, 0.29 mmol), methanol (1 mL), K2CO3 (242 mg, 1.75 mmol), and 3-(bromoacetyl)pyridine • HBr (82 mg, 0.29 mmol). The crude product was then purified via silica gel column chromatography (50% ethyl acetate/hexanes) to afford 18b as a white solid (58 mg, 61%): 1H NMR (500 MHz, CDCl3) δ 9.29 (s, 1H), 8.64 (bs, 1H), 8.52 (d, J = 4.0 Hz, 1H), 8.07 (dt, J = 7.9, 1.7 Hz, 1H), 7.87 (d, J = 7.4 Hz, 1H), 7.73 (d, J = 7.7 Hz, 1H), 7.33 (t, J = 7.9 Hz, 1H), 7.28 (dd, J = 7.8, 4.8 Hz, 1H), 6.87 (s, 1H), 3.95 (s, 3H), 2.57 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 168.27, 168.11, 148.53, 148.40, 147.64, 140.12, 133.29, 132.76, 132.29, 130.59, 127.34, 126.66, 126.62, 123.44, 103.29, 52.22, 15.01; LRMS-ESI (m/z): [M+H]+ 326.1.
Methyl 3-methyl-5-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoate (18c). Thiazole 18c was synthesized according to the same procedure as thiazole 18a utilizing 16c (57 mg, 0.215 mmol), methanol (0.77 mL), K2CO3 (178 mg, 1.29 mmol), and 3-(bromoacetyl)pyridine • HBr (60 mg, 0.215 mmol). The crude product was purified via silica gel column chromatography (50% EtOAc/hexanes) to afford 17c (60 mg, 86%): 1H NMR (500 MHz, CDCl3) δ 9.21–9.15 (m, 1H), 8.56 (d, J = 4.9 Hz, 1H), 8.17 (dt, J = 8.1, 1.9 Hz, 1H), 7.96 (s, 1H), 7.57 (s, 1H), 7.51 (s, 1H), 7.37 (dd, J = 8.0, 4.8 Hz, 1H), 6.96 (s, 1H), 3.93 (s, 3H), 2.42 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.01, 164.66, 148.28, 147.27, 140.51, 139.89, 133.87, 131.38, 130.72, 124.95, 123.86, 123.13, 116.37, 113.27, 103.79, 52.44, 21.68; LRMS-ESI (m/z): [M+H]+ 326.1.
Methyl 2-methyl-5-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoate (18d). Thiazole 18d was synthesized according to the same procedure as thiazole 18a utilizing 16d (24 mg, 0.09 mmol), methanol (0.32 mL), K2CO3 (75 mg, 0.54 mmol), 3-(bromoacetyl)pyridine • HBr (25 mg, 0.09 mmol). The crude product was purified via silica gel column chromatography (50% ethyl acetate/hexanes) to afford 17d (65 mg, 66%): 1H NMR (500 MHz, CDCl3) δ 9.14 (d, J = 1.6 Hz, 1H), 8.56 (d, J = 6.3 Hz, 1H), 8.21 (dt, J = 7.9, 1.7 Hz, 1H), 8.06 (d, J = 2.5 Hz, 1H), 7.56 (dd, J = 8.2, 2.6 Hz, 1H), 7.41 (dd, J = 7.8, 5.0 Hz, 1H), 7.26 (d, J = 8.2 Hz, 3H), 6.95 (s, 1H), 3.93 (s, 3H), 2.58 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.66, 165.14, 147.50, 146.52, 138.05, 135.32, 134.49, 132.96, 130.42, 128.73, 127.39, 124.10, 122.24, 120.78, 103.94, 52.21, 21.28; LRMS-ESI (m/z): [M+H]+ 326.2.
Methyl 2,4-dimethyl-3-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoate (18e). Thiazole 18e was synthesized according to the same procedure as thiazole 18a utilizing 16e (12.7 mg, 0.05 mmol), methanol (0.28 mL), K2CO3 (0.04 mg, 0.27 mmol), 3-(bromoacetyl)pyridine • HBr (0.01 mg, 0.05 mmol. The crude product was purified via silica gel column chromatography (50% ethyl acetate/hexanes) to afford 17e (12 mg, 78.4%): 1H NMR (500 MHz, CDCl3) δ 9.07 (d, J = 2.0 Hz, 1H), 8.50 (dd, J = 4.8, 1.4 Hz, 1H), 8.05 (dt, J = 7.9, 1.9 Hz, 1H), 7.82 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 8.1 Hz, 1H), 6.77 (s, 1H), 3.90 (s, 3H), 2.58 (s, 3H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 170.52, 167.98, 149.03, 148.44, 148.00, 147.03, 141.81, 139.24, 138.27, 133.86, 130.37, 130.02, 128.60, 123.76, 103.41, 52.20, 19.01, 15.76; LRMS-ESI (m/z): [M+H]+ 340.1.
Methyl 4-methyl-3-((4-methylthiazol-2-yl)amino)benzoate (20a). To a stirring solution of chloroacetone (0.1 mL, 1.2 mmol) and KSCN (175 mg, 1.8 mmol) at 23 °C in dry methanol (0.8 mL) was stirred for 1 h. To the reaction was then added methyl 3-amino-4methylbenzoate (200 mg, 1.2 mmol) and stirred at 60 °C for 5 h. The reaction was then cooled to 23 °C, filtered and concentrated under reduced pressure. The crude product was purified via silica gel column chromatography (25% ethyl acetate/hexanes) to afford methyl 20a (55 mg, 17.5%): 1H NMR (500 MHz, CDCl3) δ 8.24 (s, 1H), 7.74 (d, J = 6.4 Hz, 1H), 7.30 (d, J = 7.9 Hz, 1H), 6.20 (s, 1H), 3.91 (s, 3H), 2.37 (s, 3H), 2.31 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 166.61, 165.91, 146.99, 138.49, 134.55, 131.26, 129.45, 125.71, 120.77, 102.19, 52.24, 18.16, 16.76; LRMS-ESI (m/z): [M+H]+ 263.0.
Methyl 2-methyl-3-((4-methylthiazol-2-yl)amino)benzoate (20b). Thiazole 20b was synthesized according to the same procedure as thiazole 20a utilizing chloroacetone (0.1 mL, 1.2 mmol), KSCN (175 mg, 1.8 mmol), methanol (0.8 mL), and methyl 3-amino-2-methylbenzoate (200 mg, 1.2 mmol). The crude product was purified via silica gel column chromatography (25% EtOAc/hexanes) to afford methyl 20b (100 mg, 32%): 1H NMR (500 MHz, CDCl3) δ 7.75 (dd, J = 8.0, 1.8 Hz, 1H), 7.63 (dd, J = 7.8, 1.4 Hz, 1H), 7.33–7.17 (m, 1H), 6.14 (dt, J = 2.8, 1.1 Hz, 1H), 3.90 (d, J = 0.9 Hz, 3H), 2.50 (s, 3H), 2.25 (dd, J = 3.1, 1.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 168.45, 166.98, 148.90, 140.13, 132.37, 131.70, 126.81, 126.65, 125.38, 102.30, 52.28, 17.38, 14.89; LRMS-ESI (m/z): [M+H]+ 263.2.
Methyl 3-methyl-5-((4-methylthiazol-2-yl)amino)benzoate (20c). Thiazole 20c was synthesized according to the same procedure as thiazole 20a utilizing chloroacetone (0.05 mL, 0.61 mmol), KSCN (90 mg, 0.92 mmol), methanol (0.41 mL), methyl 3-amino-5-methylbenzoate (100 mg, 0.61 mmol). The crude product was purified via silica gel column chromatography (25% EtOAc/hexanes) to afford 20c (110 mg, 69%): 1H NMR (500 MHz, CDCl3) δ 7.80 (s, 1H), 7.53 (s, 1H), 7.40 (s, 1H), 6.19 (s, 1H), 3.90 (s, 3H), 2.37 (s, 3H), 2.30 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.04, 165.14, 148.55, 141.04, 139.67, 131.22, 124.62, 123.37, 116.68, 102.11, 52.28, 21.50, 17.41; LRMS-ESI (m/z): [M+H] 263.1.
Methyl 2-methyl-5-((4-methylthiazol-2-yl)amino)benzoate (20d). Thiazole 20d was synthesized according to the same procedure as thiazole 20a utilizing chloroacetone (0.05 mL, 0.61 mmol), KSCN (90 mg, 0.92 mmol), methanol (0.41 mL), and methyl 3-amino-5-methylbenzoate (100 mg, 0.61 mmol). The crude product was purified via silica gel column chromatography (25% EtOAc/hexanes) to afford 20d (94 mg, 59%): 1H NMR (500 MHz, CDCl3) δ 7.87 (d, J = 2.6 Hz, 1H), 7.44 (dd, J = 8.2, 2.6 Hz, 1H), 7.20 (d, J = 8.3 Hz, 1H), 6.14 (s, 1H), 3.88 (s, 3H), 2.55 (s, 3H), 2.26 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.68, 166.06, 148.49, 138.75, 134.93, 132.77, 130.29, 122.78, 121.44, 101.60, 52.01, 21.17, 17.42; LRMS-ESI (m/z): [M+H] 263.1.
Methyl 4-methyl-3-((4-(trifluoromethyl)thiazol-2-yl)amino)benzoate (23a). Thiazole 23a was synthesized according to the same procedure as thiazole 18a utilizing 16a (150 mg, 0.636 mmol), methanol (3.25 mL), K2CO3 (528 mg, 3.816 mmol), 3-Bromo-1,1,1-trifluoroacetone (134 mg, 0.699 mmol). The crude product was purified via silica gel column chromatography (50% ethyl acetate/hexanes) to afford 23a (141 mg, 60% yield): 1H NMR (500 MHz, CDCl3) δ 8.27 (s, 1H), 8.17 (d, J = 1.7 Hz, 1H), 7.85 (dd, J = 7.9, 1.7 Hz, 1H), 7.36 (d, J = 7.9 Hz, 1H), 6.97 (s, 1H), 3.91 (s, 3H), 2.37 (s, 3H); 13C NMR (126 MHz, CDCl3) δ 169.53, 166.52, 141.01, 140.72, 138.41, 137.62, 131.62, 129.75, 127.51, 124.33, 121.42, 119.27, 109.39, 109.35, 52.38, 18.08. [M+H]+ = 317.2.
Methyl 2-methyl-3-((4-(trifluoromethyl)thiazol-2-yl)amino)benzoate (23b). Thiazole 23b was synthesized according to the same procedure as thiazole 18a utilizing 16b (90 mg, 0.338 mmol), methanol (1.20 mL), K2CO3 (280 mg, 2.025 mmol), 3-Bromo-1,1,1-trifluoroacetone (71 mg, 0.372 mmol). The crude product was purified via silica gel column chromatography (50% ethyl acetate/hexanes) to afford 23b (75.9 mg, 71% yield): 1H NMR (400 MHz, CDCl3) δ 7.79 (dd, J = 7.9, 1.3 Hz, 1H), 7.64 (dd, J = 7.9, 1.4 Hz, 1H), 7.33 (td, J = 7.9, 0.7 Hz, 1H), 6.96 (d, J = 1.2 Hz, 1H), 3.93–3.90 (m, 3H), 2.53 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 170.33, 167.98, 138.95, 134.64, 132.73, 129.18, 127.93, 127.06, 121.35, 118.67, 109.25, 52.39, 15.09. LRMS-ESI (m/z): [M+H]+ 317.1.
Methyl 5-((4-(methoxymethyl)thiazol-2-yl)amino)-2-methylbenzoate (26). Thiazole 26 was synthesized according to the same procedure as thiazole 18a utilizing 16d (12 mg, 0.05 mmol), methanol (0.45 mL), K2CO3 (6 mg mg, 0.05 mmol), and 1-bromo-3-methoxypropan-2-one (9 mg, 0.05 mmol). The crude product was purified via silica gel column chromatography (20% EtOAc/hexanes) to afford 26 (6 mg, 42%): 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 2.6 Hz, 1H), 7.44 (dd, J = 8.3, 2.6 Hz, 1H), 7.22 (d, J = 8.3 Hz, 1H), 6.52 (s, 1H), 4.39 (s, 2H), 3.89 (s, 3H), 3.43 (s, 3H), 2.56 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 167.66, 165.85, 149.51, 138.24, 135.28, 132.93, 130.46, 122.65, 121.04, 104.85, 70.51, 58.67, 52.14, 21.23; LRMS-ESI (m/z): [M+H]+ 293.1, [M+Na]+ 315.0.
4-Methyl-3-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoic acid (19a). To a stirring solution of 18a (0.028g, 0.086 mmol) in 1 M LiOH (0.15 mL) was added a few drops of methanol and THF. The reaction was stirred until completion, and then the organic solvent was evaporated under reduced pressure. The aqueous layer was acidified to pH 3–5 utilizing 10% citric acid. The precipitate was collected via filtration and used without further purification.
2-Methyl-3-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoic acid (19b). Carboxylic acid 18b was synthesized according to the same procedure as carboxylic acid 19a utilizing 18b (53 mg, 0.16 mmol) and 1 M LiOH (0.5 mL).
3-Methyl-5-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoic acid (19c). Carboxylic acid 19c was synthesized according to the same procedure as carboxylic acid 19a utilizing 18c (0.02 g, 0.062 mmol) and 1 M LiOH (0.15 mL).
2-Methyl-5-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoic acid (19d). Carboxylic acid 19d was synthesized according to the same procedure as carboxylic acid 19a utilizing 18d (0.03 g, 0.09 mmol) and 1 M LiOH (0.28).
2,4-Dimethyl-3-((4-(pyridin-3-yl)thiazol-2-yl)amino)benzoic acid (19e). Carboxylic acid 19e was synthesized according to the same procedure as carboxylic acid 19a utilizing 18e (13 mg, 0.04 mmol) and 1 M LiOH (0.08).
4-Methyl-3-((4-methylthiazol-2-yl)amino)benzoic acid (21a). Carboxylic acid 21a was synthesized according to the same procedure as carboxylic acid 19a utilizing 20a (0.05 mg, 0.18 mmol) and 1 M LiOH (0.55 mL).
3-Methyl-5-((4-methylthiazol-2-yl)amino)benzoic acid (21b). Carboxylic acid 21b was synthesized according to the same procedure as carboxylic acid 19a utilizing 20b (51.0 mg, 0.19 mmol) and 1 M LiOH (0.38 mL).
2-Methyl-3-((4-methylthiazol-2-yl)amino)benzoic acid (21c). Carboxylic acid 21c was synthesized according to the same procedure as carboxylic acid 19a utilizing 20c (19.4 mg, 0.074 mmol) and 1 M LiOH (0.15 mL).
2-Methyl-5-((4-methylthiazol-2-yl)amino)benzoic acid (21d). Carboxylic acid 21d was synthesized according to the same procedure as carboxylic acid 19a utilizing 20d (28.1 mg, 0.11 mmol) and 1 M LiOH (0.2 mL).
4-Methyl-3-((4-(trifluoromethyl)thiazol-2-yl)amino)benzoic acid (24a). Carboxylic acid 24a was synthesized according to the same procedure as carboxylic acid 19a utilizing 23a (0.280 g, 0.885 mmol) and 1 M LiOH (1.95 mL).
2-Methyl-3-((4-(trifluoromethyl)thiazol-2-yl)amino)benzoic acid (24b). Carboxylic acid 24b was synthesized according to the same procedure as carboxylic acid 19a utilizing 23b (0.40 g, 0.127 mmol) and 1 M LiOH (1.55 mL).
5-((4-(Methoxymethyl)thiazol-2-yl)amino)-2-methylbenzoic acid (27). Carboxylic acid 27 was synthesized according to the same procedure as carboxylic acid 19a utilizing 26 (22.0 mg, 0.08 mmol) and 1 M LiOH (0.23 mL).
4.1. Determination of X-ray structure of HIV-1 protease-inhibitor complex
HIV-1 protease was expressed and purified as described previously [54]. Both protease complexes were crystallized by the hanging drop vapor diffusion method with well solution of 1.1 M NaCl, 0.1 M Sodium Acetate, pH 5.4 for PR/5b and 0.57 M NaCl, 0.1 M Sodium Acetate, pH 6.0 for PR/5c, respectively. Diffraction data were collected on a single crystal cooled to 90 K at SER-CAT (22-ID beamline), Advanced Photon Source, Argonne National Lab (Chicago, USA) with X-ray wavelength of 1.0 Å. The diffraction data were processed by HKL-2000 [55], to Rmerge of 10.4% in PR/5b and 6.2% in PR/5c, respectively. Both structures were solved by PHASER [56] in CCP4i Suite [57–59], using the previously reported isomorphous structure with PDB code 3NU3 [60] as the initial model. Both complexes were refined with REFMAC5 [61]. Jligand [62] was used to construct the restraints for refinement. COOT [63,64] was used for model building. Anisotropic atomic displacement parameters (B factors) were applied for all atoms including solvent molecules. The final refined solvent structures comprised two Na+ ions, four Cl− ions, one acetate ion, one glycerol, one formic acid and 250 water molecules for PR/5b and one Na+ ion, four Cl− ions, one glycerol, two formic acid and 312 water molecules for PR/5c, respectively. The crystallographic statistics are listed in Table S1 (Please see supporting information). The coordinates and structure factors of both PR/5b, GRL-03419A and PR/5c, GRL-02519A were deposited in the Protein Data Bank [65] with code 8FUI and 8FUJ, respectively.
Supplementary Material
Acknowledgements
This research was supported by the National Institutes of Health (Grant AI150466, AKG and Grant AI150461, ITW). X-ray data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22BM at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31-109-Eng-38. This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (HM), and in part by grants for the promotion of AIDS research from the Ministry of Health; grants from Welfare and Labor of Japan (HM); grants for the Research Program on HIV/AIDS from the Japan Agency for Medical Research and Development (AMED) under grant numbers JP15fk0410001 and JP18fk041001 (HM); a grant from the National Center for Global Health and Medicine (NCGM) (HM); and grants from JSPS KAKENHI under grants numbers 17K19577, 17H04222, 20K21609, 20H03727 (HM). The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
Abbreviations used
- ART
antiretroviral therapies
- bis-THF
bis-tetrahydrofuran
- CML
chronic myeloid leukemia
- CNS
central nervous system
- DRV
darunavir
- PR
protease
- PDB
Protein Data Bank
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2023.115385.
Data availability
All data within the article is available.
References
- [1].Ogden RC, Flexner CW (Eds.), Protease Inhibitors in AIDS Therapy, Marcel Dekker, New York, 2011, pp. 1–300. [Google Scholar]
- [2].Ghosh AK, Osswald HL, Prato G, Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS, J. Med. Chem 59 (2016) 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Esté JA, Cihlar TC, Current status and challenges of antiretroviral research and therapy, Antivir. Res 85 (2010) 25–33. [DOI] [PubMed] [Google Scholar]
- [4].Wlodawer A, Vondrasek J, Inhibitors of HIV-1 protease: a major success of structure-assisted drug design, Annu. Rev. Biophys. Biomol. Struct 27 (1998) 249–284. [DOI] [PubMed] [Google Scholar]
- [5].Appelt K, Crystal structures of HIV-1 protease-inhibitor complexes, Perspect. Drug Discov. Des 1 (1993) 23–48. [Google Scholar]
- [6].Maenza J, Flexner C, Combination antiretroviral therapy for HIV infection, Am. Fam. Physician 57 (1998) 2789–2798. [PubMed] [Google Scholar]
- [7].Vitoria M, Rangaraj A, Ford N, Doherty M, Current and future priorities for the development of optimal HIV drugs, Curr. Opin. HIV AIDS 14 (2019) 143–149. [DOI] [PubMed] [Google Scholar]
- [8].Cohen MS, Chen YQ, McCauley M, Prevention of HIV-1 infection with early antiretroviral therapy, N. Engl. J. Med 365 (2011) 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Diffenbach CW, Fauci AS, Thirty years of HIV and AIDS: future challenges and opportunities, Ann. Intern. Med 154 (2011) 766–771. [DOI] [PubMed] [Google Scholar]
- [10].Braitstein P, Brinkhof MWG, Dabis F, Schechter M, Boulle A, Miotti P, Wood R, Laurent C, Sprinz E, Seyler C, Bangsberg DR, Balestre E, Sterne JAC, May M, Egger M, Mortality of HIV-1-infected patients in the first year of antiretroviral therapy: comparison between low-income and high-income countries, Lancet 367 (2006) 817–824. [DOI] [PubMed] [Google Scholar]
- [11].Bosh KA, Johnson AS, Hernandez AL, Prejean J, Taylor J, Wingard R, Valleroy LA, Hall HI, Vital signs: deaths among persons with diagnosed HIV infection, United States, 2010–2018, MMWR Morb. Mortal. Wkly. Rep 69 (2020) 1717–1724, 10.15585/mmwr.mm6946a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gheibi Z, Shayan Z, Joulaei H, Fararouei M, Beheshti S, Shokoohi M, Determinants of AIDS and non-AIDS related mortality among people living with HIV in Shiraz, southern Iran: a 20-year retrospective follow-up study, BMC Infect. Dis 19 (2019) 1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].(a) Lv Z, Chu Y, Wang Y, HIV protease inhibitors: a review of molecular selectivity and toxicity, HIV AIDS (Auckl) 7 (2015) 95–107; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Waters L, Nelson M, Why do patients fail HIV therapy? Int. J. Clin. Pract 61 (2007) 983–990. [DOI] [PubMed] [Google Scholar]
- [14].Chawla A, Wang C, Patton C, Murray M, Punekar Y, de Ruiter A, Steinhart C, A review of long-term toxicity of antiretroviral treatment regimens and implications for an aging population, Infect. Dis. Ther (2018) 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reust CE, Common adverse effects of antiretroviral therapy for HIV disease, Am. Fam. Physician 83 (2011) 1443–1451. [PubMed] [Google Scholar]
- [16].Barbaro G, Long-term effects of protease-inhibitor-based combination therapy, Lancet 363 (2004) 900–901. [DOI] [PubMed] [Google Scholar]
- [17].Ghosh AK, Chapsal BD, Second-generation approved HIV protease inhibitors for the treatment of HIV/AIDS, in: Ghosh AK(Ed.), Aspartic Acid Proteases as Therapeutic Targets, Wiley-VCH, Weinheim, Germany, 2010, pp. 169–204. [Google Scholar]
- [18].Lu Z, Second generation HIV protease inhibitors against resistant virus, Expet Opin. Drug Discov 3 (2008) 775–786. [DOI] [PubMed] [Google Scholar]
- [19].Ghosh AK, Anderson DD, Weber IT, Mitsuya H, Enhancing protein backbone binding–a fruitful concept for combating drug-resistant HIV, Angew. Chem. Int. Ed 51 (2012) 1778–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ghosh AK, Chapsal B, Weber IT, Mitsuya H, Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance, Acc. Chem. Res 41 (2008) 78–86. [DOI] [PubMed] [Google Scholar]
- [21].Ghosh AK, Kincaid JF, Cho W, Walters DE, Krishnan K, Hussain KA, Koo Y, Cho H, Rudall C, Holland L, Buthod J. Bioorg. Med. Chem. Lett 8 (1998) 687–690. [DOI] [PubMed] [Google Scholar]
- [22].Ghosh AK, Dawson ZL, Mitsuya Darunavir H, A conceptually new HIV-1 protease inhibitor for the treatment of drug-resistant HIV, Bioorg. Med. Chem 15 (2007) 7576–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang Y-F, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H, Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro, Antimicrob. Agents Chemother 47 (2003) 3123–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Béthune M-P, TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates, Antimicrob. Agents Chemother 49 (2005) 2314–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hayashi H, Takamune N, Nirasawa T, Aoki M, Morishita Y, Das D, Koh Y, Ghosh AK, Misumi S, Mitsuya H, Dimerization of HIV-1 protease occurs through two steps relating to the mechanism of protease dimerization inhibition by darunavir, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 12234–12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Koh Y, Matsumi S, Das D, Amano M, Davis DA, Li J, Leschenko S, Baldridge sA., Shioda T, Yarchoan R, Ghosh AK, Mitsuya H, Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization, J. Biol. Chem 282 (2007) 28709–28720. [DOI] [PubMed] [Google Scholar]
- [27].Kovalevsky AY, Liu F, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT, Ultra-high resolution crystal structure of HIV-1 protease mutant reveals two binding sites for clinical inhibitor TMC114, J. Mol. Biol 363 (2006) 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tie Y, Boross PI, Wang Y-F, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT, High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multidrug-resistant clinical strains, J. Mol. Biol 338 (2004) 341–352. [DOI] [PubMed] [Google Scholar]
- [29].Ghosh AK, Chapsal BD, Design of the anti-HIV protease inhibitor darunavir, in: Ganellin CR, Roberts SM(Eds.), From Introduction to Biological and Small Molecule Drug Research and Development, R. Jefferis., 2013, pp. 355–384. [Google Scholar]
- [30].Aoki M, Das D, Hayashi H, Aoki-Ogata H, Takamatsu Y, Ghosh AK, Mitsuya H, Mechanism of darunavir (DRV)’s high genetic barrier to HIV-1 resistance: a key V32I substitution in protease rarely occurs, but once it occurs, it predisposes HIV-1 to develop DRV resistance, mBio 9 (2018) e02425–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mallolas J, Darunavir stands up as preferred HIV protease inhibitor, AIDS Rev. 19 (2017) 105–112. [PubMed] [Google Scholar]
- [32].Curran A, Pascuet ER, [Darunavir as first-line therapy. The TITAN study], Enferm. Infecc. Microbiol. Clín 10 (2008) 14–22. [DOI] [PubMed] [Google Scholar]
- [33].De Meyer S, Lathouwers E, Dierynck I, De Paepe E, Van Baelen B, Vangeneugden T, Spinosa-Guzman S, Lefebvre E, Picchio G, de Béthune MP, Characterization of virologic failure patients on darunavir/ritonavir in treatment-experienced patients, AIDS 23 (2009) 1829–1840. [DOI] [PubMed] [Google Scholar]
- [34].Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H, Bis-tetrahydrofuran: a privileged ligand for darunavir and a new generation of HIV protease inhibitors that combat drug resistance, ChemMedChem 1 (2006) 939–950. [DOI] [PubMed] [Google Scholar]
- [35].Ghosh AK, Chapsal B, Mitsuya H, Darunavir, a new PI with dual mechanism: from a novel drug design concept to new hope against drug-resistant HIV, in: Ghosh AK(Ed.), Aspartic Acid Proteases as Therapeutic Targets, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, pp. 205–243. [Google Scholar]
- [36].Deininger M, Buchdunger E, Druker BJ, The development of imatinib as a therapeutic agent for chronic myeloid leukemia, Blood 105 (2005) 2640–2653. [DOI] [PubMed] [Google Scholar]
- [37].Dohse M, Scharenberg C, Shukla S, Robey RW, Volkmann T, Deeken JF, Brendel C, Ambudkar SV, Neubauer A, Bates SE, Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib, Drug Metab. Dispos 38 (2010) 1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].DeRemer DL, Ustun C, Natarajan Nilotinib K, A second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia, Clin. Therapeut 30 (2008) 1956–1975. [DOI] [PubMed] [Google Scholar]
- [39].Blay JY, von Mehren Nilotinib M, A novel, selective tyrosine kinase inhibitor, Semin. Oncol 38 (2011) S3–S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J, Structural mechanism for STI-571 inhibition of abelson tyrosine kinase, Science 289 (2000) 1938–1942. [DOI] [PubMed] [Google Scholar]
- [41].Tokarski JS, Newitt JA, Chang CYJ, Cheng JD, Wittekind M, Kiefer SE, Kish K, Lee FYF, Borzillerri R, Lombardo LJ, Xie D, Zhang Y, Klei HE, The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants, Cancer Res. 66 (2006) 5790–5797. [DOI] [PubMed] [Google Scholar]
- [42].Ghosh AK, Takayama J, Kassekart L, Ella-Menye J-R, Yashchuk S, Agniswamy J, Wang Y-F, Aoki M, Amano M, Weber IT, Mitsuya H, Structure-based design, synthesis, X-ray studies, and biological evaluation of novel HIV-1 protease inhibitors containing isophthalamide-derived P2-ligands, Bioorg. Med. Chem. Lett 25 (2015) 4903–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ghosh AK, Brindisi M, Nyalapatla PR, Takayama J, Ella-Menye J-R, Yashchuk S, Agniswamy J, Wang Y-F, Aoki M, Amano M, Weber IT, Mitsuya H, Design of novel HIV-1 protease inhibitors incorporating isophthalamide-derived P2–P3 ligands: synthesis, biological evaluation and X-ray structural studies of inhibitor-HIV-1 protease complex, Bioorg. Med. Chem 25 (2017) 5114–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kozisek M, Bray J, Rezacova P, Saskova K, Brynda J, Pokorna J, Mammano F, Rulisek L, Konvalinka J, J. Mol. Biol 374 (2007) 1005–1016. [DOI] [PubMed] [Google Scholar]
- [45].Kaldor SW, Kalish VJ, Davies JF, Shetty BV, Fritz JE, Appelt K, Burgess JA, Campanale KM, Chirgadze NY, Clawson DK, Dressman BA, Hatch SD, Khalil DA, Kosa MB, Lubbehusen PP, Muesing MA, Patick AK, Reich SH, Su KS, Tatlock JH, Viracept (nelfinavir mesylate, AG1343): a potent, orally bioavailable inhibitor of HIV-1 protease, J. Med. Chem 40 (1997) 3979–3985. [DOI] [PubMed] [Google Scholar]
- [46].Wang Z, Hantzsche Thiazole Synthesis. Chapter 296 in Comprehensive Organic Name Reactions and Reagents, John Wiley & Sons Inc, 2010, https://doi.org/0.1002/9780470638859.conrr296. [Google Scholar]
- [47].Schantl JG, Lagoja IM, Expedient synthesis of N-substituted 2-aminothiazoles, Synth. Commun 28 (8) (1998) 1451–1462. [Google Scholar]
- [48].Bell TW, Sondheimer F, Synthesis and wittig reaction of 1-(triphenylphosphoranylidene)-3-methoxy-2-propanone, J. Org. Chem 46 (1) (1981) 217–219. [Google Scholar]
- [49].Toth MV, Marshall GR, A simple, continuous fluorometric assay for HIV protease. Int, J. Pept. Protein Res 36 (1990) 544–550. [DOI] [PubMed] [Google Scholar]
- [50].Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H, Vitro selection of highly darunavir-resistant and replication-competent HIV-1 variants by using a mixture of clinical HIV-1 isolates resistant to multiple conventional protease inhibitors, J. Virol 84 (2010) 11961–11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].For Details of X-Ray Studies, please see Supplementary Information.
- [52].Ghosh AK, Rao KV, Nyalapatla PR, Kovela S, Brindisi M, Osswald HL, Reddy BS, Agniswamy J, Wang Y-F, Aoki M, Hattori S.-i., Weber IT, Mitsuya H, Design of highly potent, dual acting and central nervous system penetrating HIV-1 protease inhibitors with excellent potency against multidrug-resistant HIV-1 variants, ChemMedChem 13 (2018) 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ghosh AK, Kovela S, Osswald HL, Amano M, Aoki M, Agniswamy J, Wang Y-F, Weber IT, Mitsuya H, Structure-based Design of highly potent HIV1 protease inhibitors containing new tricyclic ring P2-ligands: design synthesis, biological, and xray structural studies, J. Med. Chem 63 (2020) 4867–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Agniswamy J, Kneller DW, Brothers R, Wang Y-F, Harrison RW, Weber IT, Highly drug-resistant HIV-1 protease mutant PRS17 shows enhanced binding to substrate analogues, ACS Omega 4 (2019) 8707–8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Otwinowski Z, Minor W, Processing of X-ray diffraction data collected in oscillation mode, in: Carter CW, Sweet RM (Eds.), Methods in Enzymology, 276: Macromolecular Crystallography, Part A, Academic Press, New York, 1997, pp. 307–326. [DOI] [PubMed] [Google Scholar]
- [56].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, R. J.; read. Phaser crystallographic software, J. Appl. Crystallogr 40 (2007) 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Winn MD, et al. , Overview of the CCP4 suite and current developments, Acta Crystallogr., Sect. D: Biol. Crystallogr 67 (2011) 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Collaborative computational project, number 4, the CCP4 suite: programs for protein crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr 50 (1994) 760–763. [DOI] [PubMed] [Google Scholar]
- [59].Potterton E, Briggs P, Turkenburg M, Dodson E, A graphical user interface to the CCP4 Program suite, Acta Crystallogr., Sect. D: Biol. Crystallogr 59 (2003) 1131–1137. [DOI] [PubMed] [Google Scholar]
- [60].Shen C-H, Wang Y-F, Kovalevsky AY, Harrison RW, Weber IT, Amprenavir complexes with HIV-1 protease and its drug-resistant mutants altering hydrophobic clusters, FEBS J. 277 (2010) 3699–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Murshudov GN, Vagin AA, Dodson EJ, Refinement of macromolecular structures by the maximum-likelihood method, Acta. Crystallogr. D Biol. Crystallogr Part III 53 (1997) 240–255. [DOI] [PubMed] [Google Scholar]
- [62].Lebedev AA, Young P, Isupov MN, Moroz OV, Vagin AA, Murshudov GN Jligand, A graphical tool for the CCP4 template-restraint library, Acta Crystallogr D Biol Crystallogr., Part IV 68 (2012) 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of coot, Acta Crystallogr., Sect. D: Biol. Crystallogr 66 (2010) 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Emsley P, Cowtan K, Coot: model-building tools for molecular graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr 60 (2004) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [65].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, The protein Data Bank, Nucleic Acids Res. 28 (2000) 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data within the article is available.