

Abstract
Aims
HNF1B syndrome is caused by defects in the hepatocyte nuclear factor 1B (HNF1B) gene, which leads to maturity‐onset diabetes of the young type 5 and congenital organ malformations. This study aimed to identify a gene defect in a patient presenting with diabetes and severe diarrhea, while also analyzing the prevalence of hypomagnesemia and its correlation with the HNF1B genotype.
Materials and Methods
Whole exome sequencing was used to identify responsible point mutations and small indels in the proband and their family members. Multiplex ligation‐dependent probe amplification was carried out to identify HNF1B deletions. Furthermore, an analysis of published data on 539 cumulative HNF1B cases, from 29 literature sources, was carried out to determine the correlation between the HNF1B genotype and the phenotype of serum magnesium status.
Results
Using multiplex ligation‐dependent probe amplification, we identified a de novo heterozygous HNF1B deletion in the patient, who showed dorsal pancreas agenesis and multiple kidney cysts, as detected by magnetic resonance imaging. Magnesium supplementation effectively alleviated the symptoms of diarrhea. Hypomagnesemia was highly prevalent in 192 out of 354 (54.2%) patients with HNF1B syndrome. Compared with patients with intragenic mutations, those with HNF1B deletions were more likely to suffer from hypomagnesemia, with an odds ratio of 3.1 (95% confidence interval 1.8–5.4).
Conclusions
Hypomagnesemia is highly prevalent in individuals with HNF1B syndrome, and those with HNF1B deletion are more susceptible to developing hypomagnesemia compared with those with intragenic mutations. The genotype–phenotype associations in HNF1B syndrome have significant implications for endocrinologists in terms of genotype detection, treatment decisions and prognosis assessment.
Keywords: Hepatocyte nuclear factor 1B, Hypomagnesemia, Maturity‐onset diabetes of the young
In this study, we identified the gene defect in a patient manifested as diabetes and severe diarrhea. Then we carried out an analysis to investigate the prevalence of hypomagnesemia and its correlation with the hepatocyte nuclear factor 1B (HNF1B) genotype. Our findings indicate that hypomagnesemia is very common in HNF1B syndrome, especially in patients with whole HNF1B deletion, and HNF1B deletion, but not intragenic mutation, was more susceptible to hypomagnesemia with a large sample size.
INTRODUCTION
Hepatocyte nuclear factor 1B (HNF1B), also known as transcription factor 2 (TCF2), plays an important role in organ specification from endoderm and mesoderm during embryogenesis 1 , 2 . Molecular defects of HNF1B are associated with a variety of congenital organ abnormalities, including pancreatic agenesis, kidney malformation, liver abnormalities, genital tract anomalies and maturity‐onset diabetes of the young 5 (MODY5) 3 . Additionally, affected patients might show abnormal biological variables, such as impaired renal function 3 , 4 , 5 , electrolyte disturbance 6 , hyperuricemia early‐onset gout 6 , deranged liver function tests 3 , 4 and exocrine pancreatic dysfunction 7 . The disease caused by the molecular defects in HNF1B was called HNF1B syndrome 3 .
To date, a total of 377 different HNF1B molecular defects have been identified, consisting of gross deletions (21%), missense or nonsense mutations (50%), small deletions (15%) and splice‐site mutations (6%). Patients with HNF1B deletions are usually found to have a 17q12 deletion spanning 15 genes 8 .
The phenotypes of HNF1B syndrome are remarkably heterogeneous 3 . In patients with HNF1B syndrome, diabetes is often the initial clinical manifestation, which is actually a form of monogenic diabetes known as MODY5. MODY (monogenic diabetes) is a rare autosomal dominant subtype of diabetes and accounts for 1–3% of diabetes diagnoses 9 , 10 . The most common MODY subtype is HNF1A‐MODY3 (58%), whereas MODY5 only accounts for 2–6% of MODY diagnoses 10 , 11 . A study showed that the frequency of Japanese MODY1‐3, 5 and 6 were 7.6%, 36.3%, 39.4%, 13.6% and 3.0%, respectively 12 . However, the prevalence of MODY5 might be underestimated due to its wide phenotypical variability. Along with the increasing awareness of endocrinologists 13 , progressive genetic testing methods and the emergence of the MODY clinical risk calculator 14 , many more monogenic diabetes cases would be efficiently diagnosed. Nowadays, the clinical characteristics, treatment and complications of MODY are better understood, including both the common (MODY1‐3, 5) and rarer (MODY4, 6–14) subtypes of MODY 15 .
Here, we reported a case with a heterozygous whole HNF1B gene deletion, which rarely manifested as diabetes combined with severe diarrhea. The diarrhea in this patient significantly eased when the hypomagnesemia was rectified. Furthermore, we carried out an analysis to investigate the prevalence of hypomagnesemia in individuals with HNF1B syndrome, and assessed genotype–phenotype correlations between hypomagnesemia and HNF1B genotype.
MATERIALS AND METHODS
Case presentation
A 33‐year‐old male patient presented to the Department of Metabolism & Endocrinology at the Second Xiangya Hospital, Central South University, Changsha, Hunan, China, with symptoms of diabetes, weight loss and severe diarrhea. He had a normal body stature (body mass index 23.1 kg/m2) before becoming sick, and had been showing symptoms of polyuria, polydipsia and polyphagia for over a year before seeking treatment. Due to worsening symptoms, he began to experience lower limb pain and numbness and was subsequently diagnosed with type 2 diabetes and diabetic neuropathy, for which he received insulin therapy. Then, he developed severe diarrhea, resulting in weight loss of 15 kg (body mass index 17.4 kg/m2) and the onset of a cachectic state (Figure 1a). Negative stool and blood cultures helped to exclude infectious enteritis and disease. Laboratory examinations showed impaired islet function, amylase deficiency, abnormal liver function, decreased renal function and hypomagnesemia (Table 1). The patient tested negative for islet autoantibodies (glutamic acid decarboxylase autoantibodies, insulinoma‐2 associated autoantibodies and zinc transporter‐8 autoantibodies) and had no family history of diabetes in the first degree. Magnetic resonance showed pancreatic hypoplasia and small cysts in bilateral kidneys, with a paucity of the distal major pancreatic duct shown by magnetic resonance cholangiopancreatography, as shown in Figure 1b–d.
Figure 1.
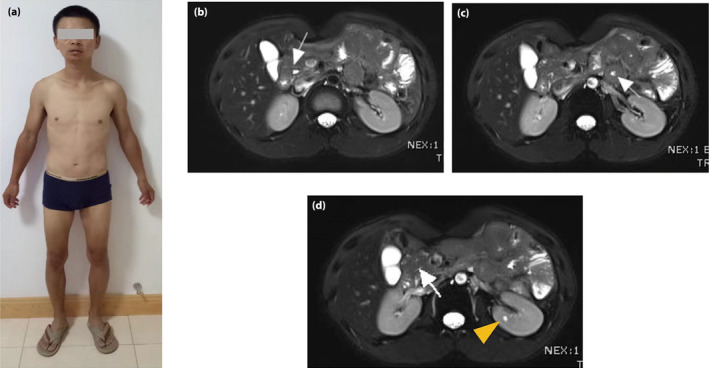
Images of the hepatocyte nuclear factor 1B deletion patient, and his magnetic resonance showing typical malformation of the visceral organs. (a) The patient showed a thin figure, but without facial and trunk dysmorphic features. (b) The pancreatic head (white arrow) was present, whereas the pancreatic body and tail was absent in the scan. (c) The pancreatic body and tail were not visible on serial slices, and the intestine tissue (white arrow) was present in the expected location of the distal pancreas. (d) Magnetic resonance cholangiopancreatography showed the short main pancreatic duct (white arrow) and small cysts in the kidney (yellow triangle).
Table 1.
Laboratory examinations of the patient with HNF1B syndrome
| Parameter | First admission | Follow up At 4 months later | Reference interval |
|---|---|---|---|
| 0 min glycemia (mmol/L) | 5.53 | 7.77 | 3.9–6.1 |
| 120 min glycemia (mmol/L) | 7.82 | 9.12 | <7.8 |
| 0 min C‐peptide (pmol/L) | 279.7 | 291.3 | 223–746 |
| 120 min C‐peptide (pmol/L) | 465.7 | 777.1 | ‐ |
| HbA1c (%) | 5.1 | 6.0 | 3.9–6.1 |
| Amylopsin (U/L) | 15.6 ↓ | 14.6 ↓ | 17–115 |
| ALT (U/L) | 116.3 ↑ | 41.4 | 9–55 |
| AST (U/L) | 56.2 ↑ | 37.6 | 15–40 |
| ALP (U/L) | 153.2 ↑ | 105.6 | 45–125 |
| Potassium (mmol/L) | 4.14 | 3.44 ↓ | 3.5–5.5 |
| Calcium (mmol/L) | 2.42 | 2.22 | 2.11–2.52 |
| Phosphate (mmol/L) | 1.12 | 1.1 | 0.85–1.51 |
| Cr (μmol/L) | 102.3 ↑ | 89.2 | 44–92 |
| Magnesium (mmol/L) | 0.59 ↓ | 0.75 ↓ | 0.85–1.51 |
ALP, alkaline phosphatase; ALT, glutamic pyruvic transaminase; AST, glutamic oxaloacetic transaminase; Cr, creatinine; HbA1c, hemoglobin A1c.
Given the multiorgan dysfunction and genetic factors, genetic testing was recommended for the patient and his family members, with written informed consent. All procedures were in accordance with the principles of the Declaration of Helsinki, and were approved by the Human Ethics Committee of the Second Xiangya Hospital of Central South University (No. 2019‐Research‐40).
Molecular genetic testing
Peripheral venous blood was collected from the proband, his younger sister and his parents. Deoxyribonucleic acid was extracted using Qiagen FlexiGene DNA Kit (Cat. 512206; Qiagen, Hilden, Germany) according to the protocol. Whole exome sequencing was used to identify responsible point mutations and small indels. The genomic deoxyribonucleic acid was randomly sheared into 180–280 bp fragments, which were amplified by polymerase chain reaction and sequenced by the Illumina HiSeq X Ten platform. Meanwhile, multiplex ligation‐dependent probe amplification was undertaken to identify the exon deletion of the MODY gene. The multiplex ligation‐dependent probe amplification kit (Cat. P241; MRC‐Holland, Amsterdam, the Netherlands) was designed to detect copy number variations in the HNF1B, GCK, HNF1A and HNF4A genes.
Literature search and data extraction
Literature search, study selection and data extraction were carried out independently by two investigators (Wang and Lin). A systematic search was carried out in PubMed for the published literature up to March 2022, with the terms HNF1B, TCF2, diabetes mellitus type MODY5 and 17q12 deletion. In total, 539 published cases with genetically verified HNF1B defects were assessed, and 354 of them were with serum magnesium status (Table 2). Hypomagnesemia is defined as serum magnesium <0.7 mmol/L or lower than the lower limit of reference value in each publication 6 , 16 . Finally, data from 227 cases with both serum magnesium and HNF1B genotype were extracted and pooled for phenotype–genotype analysis.
Table 2.
Published studies reported HNF1B cases with serum magnesium status
| No. | Year published | First author | Hypomagnesemia (n) | Normomagnesemia (n) | Cases with magnesium data (n) | Frequency of hypomagnesemia (%) | Total HNF1B cases (n) | Country | Details of each study |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2009 | Adalat 20 | 8 | 10 | 18 | 44.4 | 21 | UK | Screened out 21 HNF1B cases from 91 patients with RM |
| 2 | 2011 | Faguer 25 | 10 | 6 | 16 | 62.5 | 27 | France | Focused on HNF1B nephropathy in 27 cases |
| 3 | 2012 | Roelandt 33 | 3 | 0 | 3 | 100.0 | 3 | Belgium | Liver biopsy of 3 HNF1B cases |
| 4 | 2012 | Faguer 34 | 4 | 7 | 11 | 36.4 | 11 | France | Studied the expression of renal cystic genes in 11 HNF1B cases |
| 5 | 2013 | Ferrè 35 | 8 | 3 | 11 | 72.7 | 11 | The Netherlands | Reported 11 HNF1B cases with early hyperparathyroidism |
| 6 | 2015 | van der Made 30 | 3 | 0 | 3 | 100.0 | 3 | France | Hypomagnesemia in 3 HNF1B cases |
| 7 | 2015 | Raaijmakers 36 | 5 | 15 | 20 | 25.0 | 20 | Belgium | Screened out 20 HNF1B cases from 205 patients with CAKUT |
| 8 | 2016 | Gondra 37 | 1 | 4 | 5 | 20.0 | 7 | France | Reported 7 HNF1B fetuses presenting with hyperechogenic kidneys |
| 9 | 2016 | Dotto 38 | 1 | 0 | 1 | 100.0 | 1 | Brazil | Reported an HNF1B case suffered DKA and died of renal failure and sepsis |
| 10 | 2016 | Clissold 16 | 18 | 16 | 34 | 52.9 | 38 | UK | Kidney disease and psychiatric disorder in 38 HNF1B cases |
| 11 | 2016 | Verhave 17 | 3 | 2 | 5 | 60.0 | 5 | The Netherlands | Reported 5 HNF1B cases |
| 12 | 2016 | Bech 39 | 5 | 0 | 5 | 100.0 | 5 | The Netherlands | Reported 5 HNF1B cases with hypomagnesemia |
| 13 | 2017 | Wang 40 | 0 | 2 | 2 | 0.0 | 2 | China | Reported 2 HNF1B cases |
| 14 | 2017 | Kettunen 41 | 10 | 2 | 12 | 83.3 | 14 | Finland | Enrolled 14 HNF1B cases to study biliary anomalies |
| 15 | 2017 | Dubois‐Laforgue 3 | 49 | 16 | 65 | 75.4 | 201 | France | Included 201 HNF1B cases for genotype–phenotype study |
| 16 | 2017 | Carrillo 42 | 1 | 0 | 1 | 100.0 | 1 | Spain | Reported a case with good response to oral therapy |
| 17 | 2017 | Horinouchi 19 | 1 | 0 | 1 | 100.0 | 1 | Japan | Reported 1 HNF1B case |
| 18 | 2018 | Raman 43 | 1 | 0 | 1 | 100.0 | 1 | America | Reported 1 HNF1B case |
| 19 | 2018 | Stiles 32 | 1 | 0 | 1 | 100.0 | 1 | UK | Reported 1 HNF1B case |
| 20 | 2019 | Madariaga 44 | 9 | 8 | 17 | 52.9 | 23 | Spain | Screened out 23 HNF1B cases from 60 suspected patients |
| 21 | 2019 | Omura 45 | 1 | 0 | 1 | 100.0 | 1 | Japan | Reported 1 HNF1B case |
| 22 | 2019 | Nagano 46 | 13 | 18 | 31 | 41.9 | 33 | Japan | Screened out 33 HNF1B cases from 596 suspected patients |
| 23 | 2019 | Dotto 47 | 3 | 2 | 5 | 60.0 | 5 | Brazil | Reported 2 HNF1B pedigrees |
| 24 | 2019 | Okorn 48 | 12 | 38 | 50 | 24.0 | 62 | Germany | Recruited 62 children with HNF1B nephropathy |
| 25 | 2019 | Pinon 49 | 1 | 3 | 4 | 25.0 | 4 | Italy | Reported 4 HNF1B cases presenting with neonatal cholestasis |
| 26 | 2019 | Al‐Khawaga 50 | 1 | 0 | 1 | 100.0 | 1 | Qatar | Reported 1 HNF1B case |
| 27 | 2019 | Adalat 22 | 23 | 19 | 42 | 54.8 | 52 | UK | Screened out 52 HNF1B cases from 199 pediatric patients with RM |
| 28 | 2019 | Li 51 | 1 | 0 | 1 | 100.0 | 1 | USA | Reported 1 HNF1B case |
| 29 | 2021 | Motyka 52 | 3 | 1 | 4 | 75.0 | 4 | Poland | Reported 4 HNF1B cases |
| 30 | 2021 | Patouni 53 | 1 | 0 | 1 | 100.0 | 1 | Greece | Reported 1 HNF1B case |
| In total | 192 | 162 | 354 | 54.2 | 539 |
CAKUT, congenital abnormalities of kidney and urinary tract; HNF1B, hepatocyte nuclear factor 1B; RM, renal malformations.
RESULTS
Genetic study result
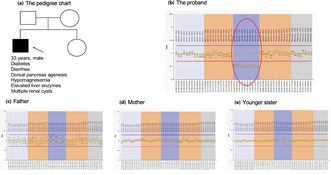
As the present patient was rather young and did not present with metabolic syndrome, the diagnosis of type 2 diabetes was doubtful. Therefore, the etiology of his diabetes remained unclear. Whole exome sequencing was used to search for gene mutations responsible for the patient's multiorgan dysfunction, but no such mutations were found. However, copy number variation analysis using CODEX and XHMM software showed a predicted segment deletion spanning nearly the entire HNF1B gene. Therefore, successive multiplex ligation‐dependent probe amplification was carried out to identify the MODY gene deletion in the proband and his family members, which confirmed a de novo heterozygous whole HNF1B deletion in the proband (Figure 2b–e).
Figure 2.
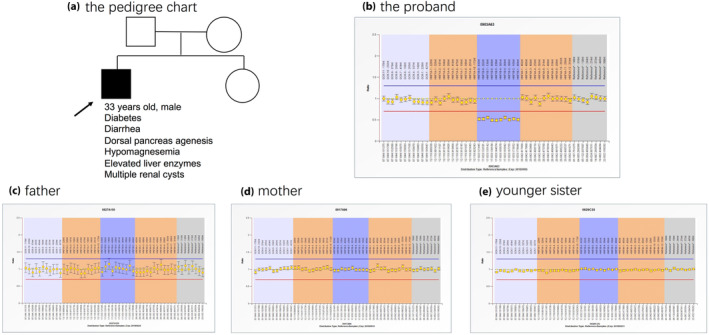
Multiplex ligation‐dependent probe amplification showed a de novo heterozygous whole hepatocyte nuclear factor 1B (HNF1B) deletion in the proband. (a) The pedigree chart of the family. (b–e) The multiplex ligation‐dependent probe amplification results of the proband and family members. The multiplex ligation‐dependent probe amplification kit contained probes detecting the maturity‐onset diabetes of the young 5 gene, GCK, HNF1A, HNF1B and HNF4A. The fluorescence signal between 0.7 and 1.33 (from the red horizontal line to blue horizontal line) was considered to be normal. A heterozygous whole HNF1B deletion was found in the proband (the ellipse in b).
Diagnosis and treatment
As a result, the diagnosis of this patient was HNF1B syndrome, and the diabetes type was MODY5, not type 2 diabetes. The patient's severe and refractory diarrhea was likely attributed to a combination of digestive enzyme deficiency, diabetic autonomic neuropathy and hypomagnesemia that increased intestinal peristalsis. The treatment plan involved insulin aspart 30, digestive enzyme capsules, neurotrophic drugs and oral magnesium supplements. At the 4‐month follow up, hypomagnesemia persisted; however, there was a remarkable improvement in serum magnesium levels (Table 1). Furthermore, diarrhea was significantly alleviated, with well‐formed bowel movements and a reduced frequency of two or three times per day. The patient was well managed in terms of blood glucose control (hemoglobin A1c 6.0%) and experienced a weight gain of 6 kg. After being discharged for 1 year, the patient ceased taking the digestive enzyme capsules and neurotrophic drugs. However, diarrhea remained well controlled with the use of magnesium supplements.
Genotype–phenotype correlations
Persistent hypomagnesemia and refractory diarrhea were observed in the HNF1B case. However, no previous systematic study has focused on hypomagnesemia in the HNF1B genotype. Therefore, our aim was to determine the genotype–phenotype correlations related to hypomagnesemia in HNF1B syndrome.
After carrying out a PubMed search and independent review by two investigators, a total of 30 publications were included in the present study. Finally, a total of 354 cases with serum magnesium status were extracted from 539 cases in 29 publications. The frequency of hypomagnesemia was 54.2% (192/354 cases). Among the 227 cases with both HNF1B genotype and serum magnesium status individually (Table S1), the χ2‐test showed that hypomagnesemia was more frequently present in patients with HNF1B deletion than in those with intragenic mutations (61.8 vs 34.1%, P < 0.001; Table 3). More specifically, patients with HNF1B deletions were more prone to suffering from hypomagnesemia than those with intragenic mutations, with an odds ratio of 3.1 (95% confidence interval 1.8–5.4). Next, we aimed to classify the genotypes into HNF1B deletion, missense mutation, nonsense mutation and splice‐site mutation for subsequent analysis. Ultimately, the corresponding genotypes were extracted from 202 cases (Table S2). The χ2‐test showed statistical differences in magnesium ion status among HNF1B deletion, missense mutation, nonsense mutation and splice‐site mutation (Table S3). Furthermore, patients with HNF1B deletions were more likely to have hypomagnesemia than those with HNF1B missense mutations, with an odds ratio of 5.2 (95% confidence interval 2.3–11.4). Subsequently, we also extracted the corresponding magnesium ion concentrations from 65 patients and found that diagnosing HNF1B deletion or intragenic mutation based on serum magnesium concentration yielded an area under the curve of 0.74 (Youden index 0.4, cut‐off value 1.6 mg/dL, sensitivity 61.1%, specificity 83.0%; Figure 3).
Table 3.
Genotype–phenotype correlation of hypomagnesemia in HNF1B syndrome
| Hypomagnesemia | Normomagnesemia | Total (n) | Frequency (%) † | OR (95% CI) | P‐value | |
|---|---|---|---|---|---|---|
| Deletion | 84 | 52 | 136 | 61.8 | 3.1 (1.8, 5.4) | <0.001 |
| Intragenic mutation | 31 | 60 | 91 | 34.1 | ||
| Total (n) | 115 | 112 | 227 | 50.7 |
Frequency of hypomagnesemia. CI, confidence interval; OR, odds ratio.
Figure 3.
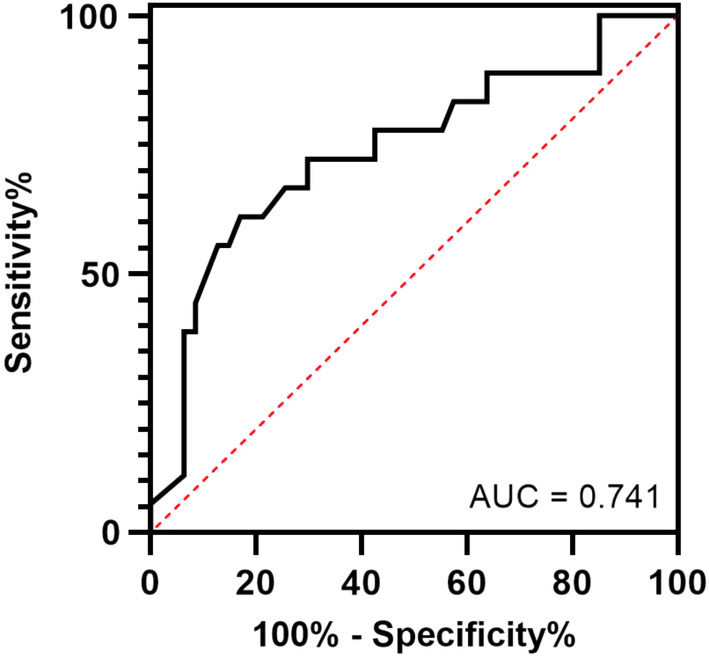
The receiver operating characteristic curve showed that serum magnesium concentration had a high predictive value in diagnosing hepatocyte nuclear factor 1B deletion or intragenic mutation. AUC, area under the curve.
DISCUSSION
The results of the present study showed that hypomagnesemia is very common in HNF1B syndrome, especially in patients with a whole HNF1B deletion. The various manifestations in HNF1B syndrome can be attributable to the fact that HNF1B acts as a promiscuous transcription factor, regulating over 25 genes in multiple systems 17 . In the present study, a case with a heterozygous whole HNF1B deletion was genetically diagnosed with manifestations including HNF1B‐MODY (MODY5), exocrine pancreatic dysfunction, dorsal pancreatic agenesis and extrapancreas phenotypes consisting of abnormal liver test, hypomagnesemia, reduced estimated glomerular filtration rate and renal cysts. The accurate diagnosis is of great benefit for the treatment, and would be great help for antenatal genetic counseling for this patient and others with genetic diabetes.
There have been nearly 20 genes associated with inherited hypomagnesemia, which can be classified into four groups: hypercalciuric hypomagnesemia, Gitelman‐like hypomagnesemia, mitochondrial hypomagnesemia and other hypomagnesemia 18 . Combined with clinical manifestations, a differential diagnosis strategy in a flowchart has been established for inherited hypomagnesemia 19 . Hypomagnesemia was first reported to be associated with HNF1B syndrome in 2009 20 . It was found that 44% (8/18) of HNF1B patients had hypomagnesemia versus 2% (1/48) of patients with renal malformations, but without HNF1B defects. Analogously, but in a larger group, we showed that hypomagnesemia was present in 54.2% (192/354) of patients with HNF1B syndrome. Given the high prevalence of hypomagnesemia in HNF1B defects, hypomagnesemia is one of the sensitive features that show HNF1B defects. Consequently, hypomagnesemia, along with diabetes and morphological abnormalities of the kidney and other organs, has been weighted to create the HNF1B score for selecting patients for HNF1B genetic screening 6 , 21 .
Previous studies have shown that serum magnesium levels were lower in the deletion group 16 . Nevertheless, they found no difference in magnesium status between patients with HNF1B intragenic mutations and deletions 16 , 20 , 22 . In the present study, we showed that patients with HNF1B deletion were more likely to suffer from hypomagnesemia than those with intragenic mutation by a large sample size. The contradiction might be due to the fact that the frequency of hypomagnesemia increased with age in pediatric patients 22 , and became obviously discriminatory in the adult population, but not in the pediatric cohort 21 . Therefore, previous studies with only samples of children or small sample sizes have drawn divergent conclusions. Therefore, a genotype–phenotype correlation exists between HNF1B and hypomagnesemia, to the endocrinologists. Hypomagnesemia has a high positive predictive value for HNF1B deletion, especially in adult patients.
HNF1B defects lead to the transcriptional inactivation of FXYD2 23 , a gene that encodes the γ subunit of the Na+/K+–ATPase, which mediates the reabsorption of magnesium in the distal convoluted tubule 20 , 24 . Therefore, HNF1B mutation results in hypomagnesemia due to renal wasting 3 , 20 , 25 . HNF‐1α and HNF‐1β are homeoproteins, and have been found to form heterodimers both in vitro and in vivo over the past 30 years 26 , 27 , 28 . HNF‐1β and HNF‐1α are sequentially expressed and committed to the renal tubular differentiation during embryogenesis 26 , 28 . Meanwhile, patients with MODY3 (HNF1A mutation) have also been observed with kidney malformations 29 . A previous study showed that the epithelia of distal convoluted tubule expressed HNF‐1α, as well as HNF‐1β 20 . Hence, HNF‐1β probably binds to HNF‐1α to form heterodimers in the distal convoluted tubule to regulate the magnesium absorption, which indicates that haploinsufficiency is the preponderant mechanism for hypomagnesemia in patients with HNF1B deletion. Thus, our hypothesis about this genotype–phenotype correlation in hypomagnesemia might be that haploinsufficiency of HNF1B allele deletion insufficiently activates the FXYD2 transcription and leads to decreased absorption of magnesium in the renal. In contrast, there might be some relationships between the other genes on chromosome 17q12 and hypomagnesemia.
As HNF1B syndrome is a multiorgan disease, individualized treatment should be developed for patients according to the organs affected. The clinical features of hypomagnesemia caused by HNF1B syndrome range from asymptomatic to fatigue, tetany, seizures, cardiac arrhythmias and even death 18 , 30 . Diarrhea was rarely reported in HNF1B syndrome. Physiologically, magnesium inhibits the sensitivity of acetylcholine receptors on the endplate membrane. During hypomagnesemia, the repressive effect on the smooth muscle of digestive tract is relieved, which enhances intestinal motility and causes diarrhea in patients 31 . As previously mentioned, the lack of digestive enzyme due to exocrine pancreatic dysfunction was another factor for diarrhea in HNF1B patients.
The glycemia in the present patient was well controlled with insulin administration. However, the refractory diarrhea was not cured after receiving neurotrophic drugs and loperamide. With the accurate genetic diagnosis, it has raised our attention that besides diabetic autonomic neuropathy, deficiencies in digestive enzymes and hypomagnesemia were also major causes of the severe diarrhea. Therefore, compound digestive enzymes (dietary advanced glycation end‐products) and magnesium salts were added to this patient's treatment. Other treatments for HNF1B syndrome include protecting the liver, relieving jaundice, lowering uric acid levels and maintaining electrolyte balance, depending on the systems affected.
The present study had several limitations. First, the sample size included in the study was relatively small, and future studies with larger prospective designs are needed to establish causal relationships. Second, only English‐language studies were included, which might introduce bias due to the lack of literature in other languages. Third, many of the collected samples lacked specific subtyping of mutation types and serum magnesium concentrations, potentially limiting the clinical significance. Finally, the present study focused on investigating the impact of HNF1B mutations on hypomagnesemia, without extensively analyzing the effects of other genes within the deleted segment. Further research is needed to explore the potential impact of these genes in more detail.
In summary, the present study identified a case of MODY5 with a heterozygous HNF1B deletion. Furthermore, we showed for the first time that HNF1B deletion, rather than intragenic mutation, is associated with a higher susceptibility to hypomagnesemia, supported by a large sample size. When diabetes is concomitant with multisystem phenotypes, HNF1B syndrome should be taken into consideration. The utilization of the HNF1B score is valuable for screening potential HNF1B candidates, and the understanding of the genotype–phenotype association provides significant assistance to endocrinologists in selecting appropriate methods for detecting HNF1B deletion or intragenic mutation, devising treatment plans and evaluating prognosis.
DISCLOSURE
The authors declare no conflict of interest.
Approval of the research protocol: This study was approved by the ethics committee of The Second Xiangya Hospital of Central South University (No. 2019‐Research‐40), and carried out according to the principles of the Declaration of Helsinki in 1995 (as revised in Fortaleza, Brazil, October 2013).
Informed consent: The patient provided his written informed consent to participate in this study.
Registry and the registration no. of the study/trial: N/A.
Animal studies: N/A.
Supporting information
Table S1 | The case number with both HNF1B genotype and serum magnesium status in each publication.
Table S2 | The number of cases with both HNF1B genotype (categorized as Deletion, Missense mutation, Nonsense mutation, Splice‐site mutation) and serum magnesium. status in each publication.
Table S3 | Genotypes (categorized as Deletion, Missense mutation, Nonsense mutation, Splice‐site mutation)‐Phenotypes of hypomagnesemia in HNF1B syndrome.
ACKNOWLEDGMENTS
The work was supported by the Basic and Applied Basic Research Foundation of Guangdong Province (2021A1515110995 to YW), Innovation project of Foshan Science and Technology Bureau (2020001005311 to YW), and the National Natural Science Foundation of China (82200933 to JH). We appreciate the cooperation from Mr Pan, the patient reported in the study, and his family, and congratulate Mr Pan for getting married after discharge and having two healthy daughters.
Contributor Information
Yaoming Xue, Email: xueyaoming999@126.com.
Jingyi Hu, Email: hujingyi0066@csu.edu.cn.
REFERENCES
- 1. Duval H , Michel‐Calemard L, Gonzales M, et al. Fetal anomalies associated with HNF1B mutations: Report of 20 autopsy cases. Prenat Diagn 2016; 36: 744–751. [DOI] [PubMed] [Google Scholar]
- 2. Haumaitre C, Reber M, Cereghini S. Functions of HNF1 family members in differentiation of the visceral endoderm cell lineage. J Biol Chem 2003; 278: 40933–40942. [DOI] [PubMed] [Google Scholar]
- 3. Dubois‐Laforgue D, Cornu E, Saint‐Martin C, et al. Diabetes, associated clinical spectrum, long‐term prognosis, and genotype/phenotype correlations in 201 adult patients with hepatocyte nuclear factor 1B (HNF1B) molecular defects. Diabetes Care 2017; 40: 1436–1443. [DOI] [PubMed] [Google Scholar]
- 4. Bellanne‐Chantelot C, Chauveau D, Gautier JF, et al. Clinical spectrum associated with hepatocyte nuclear factor‐1beta mutations. Ann Intern Med 2004; 140: 510–517. [DOI] [PubMed] [Google Scholar]
- 5. Alvelos MI, Rodrigues M, Lobo L, et al. A novel mutation of the HNF1B gene associated with hypoplastic glomerulocystic kidney disease and neonatal renal failure: A case report and mutation update. Medicine (Baltimore) 2015; 94: e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Faguer S, Chassaing N, Bandin F, et al. The HNF1B score is a simple tool to select patients for HNF1B gene analysis. Kidney Int 2014; 86: 1007–1015. [DOI] [PubMed] [Google Scholar]
- 7. Tjora E, Wathle G, Erchinger F, et al. Exocrine pancreatic function in hepatocyte nuclear factor 1beta‐maturity‐onset diabetes of the young (HNF1B‐MODY) is only moderately reduced: Compensatory hypersecretion from a hypoplastic pancreas. Diabet Med 2013; 30: 946–955. [DOI] [PubMed] [Google Scholar]
- 8. Laffargue F, Bourthoumieu S, Llanas B, et al. Towards a new point of view on the phenotype of patients with a 17q12 microdeletion syndrome. Arch Dis Child 2015; 100: 259–264. [DOI] [PubMed] [Google Scholar]
- 9. Shepherd M, Shields B, Hammersley S, et al. Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. pediatric diabetes population with monogenic diabetes. Diabetes Care 2016; 39: 1879–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fajans SS, Bell GI. MODY: History, genetics, pathophysiology, and clinical decision making. Diabetes Care 2011; 34: 1878–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ivanoshchuk DE, Shakhtshneider EV, Rymar OD, et al. The mutation spectrum of maturity onset diabetes of the young (MODY)‐associated genes among Western Siberia patients. J Pers Med 2021; 11: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Horikawa Y. Maturity‐onset diabetes of the young as a model for elucidating the multifactorial origin of type 2 diabetes mellitus. J Diabetes Investig 2018; 9: 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Broome DT, Pantalone KM, Kashyap SR, et al. Approach to the patient with MODY‐monogenic diabetes. J Clin Endocrinol Metab 2021; 106: 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Exeter Diabetes App. Available from: https://www.diabetesgenes.org/exeter‐diabetes‐app/ Accessed May 16, 2022.
- 15. Aarthy R, Aston‐Mourney K, Mikocka‐Walus A, et al. Clinical features, complications and treatment of rarer forms of maturity‐onset diabetes of the young (MODY) ‐ A review. J Diabetes Complications 2021; 35: 107640. [DOI] [PubMed] [Google Scholar]
- 16. Clissold RL, Shaw‐Smith C, Turnpenny P, et al. Chromosome 17q12 microdeletions but not intragenic HNF1B mutations link developmental kidney disease and psychiatric disorder. Kidney Int 2016; 90: 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verhave JC, Bech AP, Wetzels JF, et al. Hepatocyte nuclear factor 1beta‐associated kidney disease: More than renal cysts and diabetes. J Am Soc Nephrol 2016; 27: 345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Viering D, de Baaij JHF, Walsh SB, et al. Genetic causes of hypomagnesemia, a clinical overview. Pediatr Nephrol 2017; 32: 1123–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horinouchi T, Nozu K, Kamiyoshi N, et al. Diagnostic strategy for inherited hypomagnesemia. Clin Exp Nephrol 2017; 21: 1003–1010. [DOI] [PubMed] [Google Scholar]
- 20. Adalat S, Woolf AS, Johnstone KA, et al. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 2009; 20: 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Clissold R, Shields B, Ellard S, et al. Assessment of the HNF1B score as a tool to select patients for HNF1B genetic testing. Nephron 2015; 130: 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adalat S, Hayes WN, Bryant WA, et al. HNF1B mutations are associated with a Gitelman‐like tubulopathy that develops during childhood. Kidney Int Rep 2019; 4: 1304–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferre S, Veenstra GJ, Bouwmeester R, et al. HNF‐1B specifically regulates the transcription of the gammaa‐subunit of the Na+/K+‐ATPase. Biochem Biophys Res Commun 2011; 404: 284–290. [DOI] [PubMed] [Google Scholar]
- 24. de Baaij J, Dorresteijn E, Hennekam E, et al. Recurrent FXYD2 p.Gly41Arg mutation in patients with isolated dominant hypomagnesaemia. Nephrol Dial Transplant 2015; 30: 952–957. [DOI] [PubMed] [Google Scholar]
- 25. Faguer S, Decramer S, Chassaing N, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int 2011; 80: 768–776. [DOI] [PubMed] [Google Scholar]
- 26. Rey‐Campos J, Chouard T, Yaniv M, et al. vHNF1 is a homeoprotein that activates transcription and forms heterodimers with HNF1. EMBO J 1991; 10: 1445–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mendel DB, Hansen LP, Graves MK, et al. HNF‐1 alpha and HNF‐1 beta (vHNF‐1) share dimerization and homeo domains, but not activation domains, and form heterodimers in vitro. Genes Dev 1991; 5: 1042–1056. [DOI] [PubMed] [Google Scholar]
- 28. Lazzaro D, De Simone V, De Magistris L, et al. LFB1 and LFB3 homeoproteins are sequentially expressed during kidney development. Development 1992; 114: 469–479. [DOI] [PubMed] [Google Scholar]
- 29. Matsukura H, Nagamori M, Miya K, et al. MODY3, renal cysts, and Dandy‐Walker variants with a microdeletion spanning the HNF1A gene. Clin Nephrol 2017; 88: 162–166. [DOI] [PubMed] [Google Scholar]
- 30. van der Made CI, Hoorn EJ, de la Faille R, et al. Hypomagnesemia as first clinical manifestation of ADTKD‐HNF1B: A case series and literature review. Am J Nephrol 2015; 42: 85–90. [DOI] [PubMed] [Google Scholar]
- 31. Lewis TV, Neely S, Turman MA. Efficacy and tolerability of magnesium plus protein for managing hypomagnesemia in pediatric kidney transplant patients. Pediatr Transplant 2018; 22: e13170. [DOI] [PubMed] [Google Scholar]
- 32. Stiles CE, Thuraisingham R, Bockenhauer D, et al. De novo HNF1 homeobox B mutation as a cause for chronic, treatment‐resistant hypomagnesaemia. Endocrinol Diabetes Metab Case Rep 2018; 2018: 17‐0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Roelandt P, Antoniou A, Libbrecht L, et al. HNF1B deficiency causes ciliary defects in human cholangiocytes. Hepatology 2012; 56: 1178–1181. [DOI] [PubMed] [Google Scholar]
- 34. Faguer S, Decramer S, Devuyst O, et al. Expression of renal cystic genes in patients with HNF1B mutations. Nephron Clin Pract 2012; 120: c71–c78. [DOI] [PubMed] [Google Scholar]
- 35. Ferrè S, Bongers EM, Sonneveld R, et al. Early development of hyperparathyroidism due to loss of PTH transcriptional repression in patients with HNF1β mutations? J Clin Endocrinol Metab 2013; 98: 4089–4096. [DOI] [PubMed] [Google Scholar]
- 36. Raaijmakers A, Corveleyn A, Devriendt K, et al. Criteria for HNF1B analysis in patients with congenital abnormalities of kidney and urinary tract. Nephrol Dial Transplant 2015; 30: 835–842. [DOI] [PubMed] [Google Scholar]
- 37. Gondra L, Decramer S, Chalouhi GE, et al. Hyperechogenic kidneys and polyhydramnios associated with HNF1B gene mutation. Pediatr Nephrol 2016; 31: 1705–1708. [DOI] [PubMed] [Google Scholar]
- 38. Dotto RP, Giuffrida FM, Franco L, et al. Unexpected finding of a whole HNF1B gene deletion during the screening of rare MODY types in a series of Brazilian patients negative for GCK and HNF1A mutations. Diabetes Res Clin Pract 2016; 116: 100–104. [DOI] [PubMed] [Google Scholar]
- 39. Bech AP, Wetzels JF, Bongers EM, et al. Thiazide responsiveness testing in patients with renal magnesium wasting and correlation with genetic analysis: A diagnostic test study. Am J Kidney Dis 2016; 68: 168–170. [DOI] [PubMed] [Google Scholar]
- 40. Wang Y, Zhao Y, Zhang J, et al. A case of a novel mutation in HNF1β‐related maturity‐onset diabetes of the young type 5 with diabetic kidney disease complication in a Chinese family. J Diabetes Complications 2017; 31: 1243–1246. [DOI] [PubMed] [Google Scholar]
- 41. Kettunen JLT, Parviainen H, Miettinen PJ, et al. Biliary anomalies in patients with HNF1B diabetes. J Clin Endocrinol Metab 2017; 102: 2075–2082. [DOI] [PubMed] [Google Scholar]
- 42. Carrillo E, Lomas A, Pines PJ, et al. Long‐lasting response to oral therapy in a young male with monogenic diabetes as part of HNF1B‐related disease. Endocrinol Diabetes Metab Case Rep 2017; 2017: 17‐0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raman V, Cohen RA. Hypomagnesemia in a patient with an eating disorder. Am J Kidney Dis 2018; 71: A12–A14. [DOI] [PubMed] [Google Scholar]
- 44. Madariaga L, Garcia‐Castano A, Ariceta G, et al. Variable phenotype in HNF1B mutations: Extrarenal manifestations distinguish affected individuals from the population with congenital anomalies of the kidney and urinary tract. Clin Kidney J 2019; 12: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Omura Y, Yagi K, Honoki H, et al. Clinical manifestations of a sporadic maturity‐onset diabetes of the young (MODY) 5 with a whole deletion of HNF1B based on 17q12 microdeletion. Endocr J 2019; 66: 1113–1116. [DOI] [PubMed] [Google Scholar]
- 46. Nagano C, Morisada N, Nozu K, et al. Clinical characteristics of HNF1B‐related disorders in a Japanese population. Clin Exp Nephrol 2019; 23: 1119–1129. [DOI] [PubMed] [Google Scholar]
- 47. Dotto RP, Santana LS, Lindsey SC, et al. Searching for mutations in the HNF1B gene in a Brazilian cohort with renal cysts and hyperglycemia. Arch Endocrinol Metab 2019; 63: 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Okorn C, Goertz A, Vester U, et al. HNF1B nephropathy has a slow‐progressive phenotype in childhood‐with the exception of very early onset cases: Results of the German Multicenter HNF1B Childhood Registry. Pediatr Nephrol 2019; 34: 1065–1075. [DOI] [PubMed] [Google Scholar]
- 49. Pinon M, Carboni M, Colavito D, et al. Not only Alagille syndrome. Syndromic paucity of interlobular bile ducts secondary to HNF1β deficiency: A case report and literature review. Ital. J Pediatr 2019; 45: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Al‐Khawaga S, Mohammed I, Saraswathi S, et al. The clinical and genetic characteristics of permanent neonatal diabetes (PNDM) in the state of Qatar. Mol Genet Genomic Med 2019; 7: e00753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li HJ, Groden C, Hoenig MP, et al. Case report: Extreme coronary calcifications and hypomagnesemia in a patient with a 17q12 deletion involving HNF1B. BMC Nephrol 2019; 20: 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Motyka R, Kołbuc M, Wierzchołowski W, et al. Four cases of maturity onset diabetes of the young (MODY) type 5 associated with mutations in the hepatocyte nuclear factor 1 beta (HNF1B) gene presenting in a 13‐year‐old boy and in adult men aged 33, 34, and 35 years in Poland. Am J Case Rep 2021; 22: e928994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Patouni K, Cinek O, Pruhova S, et al. A case of digenic maturity onset diabetes of the young with heterozygous variants in both HNF1A and HNF1B genes. Eur J Med Genet 2021; 64: 104264. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 | The case number with both HNF1B genotype and serum magnesium status in each publication.
Table S2 | The number of cases with both HNF1B genotype (categorized as Deletion, Missense mutation, Nonsense mutation, Splice‐site mutation) and serum magnesium. status in each publication.
Table S3 | Genotypes (categorized as Deletion, Missense mutation, Nonsense mutation, Splice‐site mutation)‐Phenotypes of hypomagnesemia in HNF1B syndrome.