

Abstract
The molecular mechanisms of luminal cell differentiation are not understood well enough to determine how differentiation goes awry during oncogenesis. Using RNA-Seq analysis, we discovered that CREB1 plays a central role in maintaining new luminal cell survival and that oncogenesis dramatically changes the CREB1-induced transcriptome. CREB1 is active in luminal cells, but not basal cells. We identified ING4 and its E3 ligase, JFK, as CREB1 transcriptional targets in luminal cells. During luminal cell differentiation, transient induction of ING4 expression is followed by a peak in CREB1 activity, while JFK increases concomitantly with CREB1 activation. Transient expression of ING4 is required for luminal cell induction; however, failure to properly down-regulate ING4 leads to luminal cell death. Consequently, blocking CREB1 increased ING4 expression, suppressed JFK, and led to luminal cell death. Thus, CREB1 is responsible for the suppression of ING4 required for luminal cell survival and maintenance. Oncogenic transformation by suppressing PTEN resulted in constitutive activation of CREB1. However, the tumor cells could no longer fully differentiate into luminal cells, failed to express ING4, and displayed a unique CREB1 transcriptome. Blocking CREB1 in tumorigenic cells suppressed tumor growth in vivo, rescued ING4 expression, and restored luminal cell formation, but ultimately induced luminal cell death. IHC of primary prostate tumors demonstrated a strong correlation between loss of ING4 and loss of PTEN. This is the first study to define a molecular mechanism whereby oncogenic loss of PTEN, leading to aberrant CREB1 activation, suppresses ING4 expression causing disruption of luminal cell differentiation.
Keywords: prostate cancer, luminal differentiation, PTEN, ING4, CREB, JFK
INTRODUCTION
The normal prostate gland consists of a series of branched ducts of stratified basal epithelial and suprabasal secretory post-mitotic luminal cells. Well-defined markers distinguish luminal cells from basal cells [1, 2]. It is thought that bi-potential progenitor cells differentiate and give rise to both basal and luminal cells [3, 4]. A hallmark of prostate cancer is the loss of basal cells and retention of some, but not all, basal markers in luminal-like proliferative cancer cells [5, 6]. These histological anomalies predict that genetic dysregulation of basal and luminal cell differentiation results in prostate cancer development.
Genetic studies in mice have linked prostate cancer development to a defect in differentiation. Knock out of the prostate-specific differentiation gene, Nkx3.1, in mice is a tumor initiating event [7]. About 50% of human primary patient samples contain the TMPRSS2-ERG fusion, which leads to ERG overexpression, a member of the ETS family of transcription factors with known roles in differentiation [8]. Loss of Nkx3.1 cooperates with TMPRSS2-ERG to promote prostate tumor progression in mice [9]. MYC overexpression and PTEN loss are also common genetic abnormalities in human prostate cancer with roles in differentiation [10, 11]. Loss of PTEN in mouse prostates is reportedly accompanied by a decrease in tissue differentiation [12]. Loss of PTEN or overexpression of MYC downregulates Nkx3.1 expression, which is sufficient to induce prostate cancer in mice [13]. Although these studies identified specific oncogenic events that drive prostate cancer development and likely dysregulate prostate epithelial cell differentiation, the underlying mechanisms to explain how dysregulated differentiation leads to cancer development is not understood. Moreover, these genetic studies have largely been limited to mouse models and defining of the molecular mechanisms that dysregulate differentiation in human cancer has yet to be achieved.
In an in vitro human prostate epithelial cell differentiation model, transient induction of ING4, via MYC, was required for human prostate luminal cell differentiation and its loss was required for MYC-driven prostate tumorigenesis [14]. Furthermore, ING4 expression is lost in a subset of primary prostate tumors, and that genetic loss of PTEN in the context of MYC overexpression causes loss of ING4 expression and suppresses differentiation. These findings further support the view that prostate cancer arises from defective differentiation [14, 15]. Functionally ING4 is a chromatin reader that binds to tri-methylated histone H3, and recruits HBO1 acetyltransferase to increase histone acetylation [15]. However, the mechanisms regulating the transient expression of ING4 during normal differentiation and how loss of PTEN leads to loss of ING4 are unknown.
CREB1 is primarily known for its role in neurons, but is reportedly involved in the differentiation of several tissue types [16]. CREB1 also reportedly plays a role in some cancers, where it is either overexpressed [17], involved in gene fusions [18], or aberrantly activated [19]. In prostate cancer, CREB1 activation via cAMP signaling is implicated in the switch from adenocarcinoma to neuroendocrine prostate cancer [20]. While no role for CREB1 has been reported for normal prostate differentiation or development, a recent report suggests aberrant CREB activation may be associated with prostate cancer recurrence [21].
CREB1 is a bZIP transcription factor that homo- or hetero-dimerizes to activate target genes through cAMP response elements (CRE). CREB1 dimerization with different partners allows selective activation of numerous downstream target genes [16, 22]. CREB1 binds constitutively to CREs in open chromatin and is activated through phosphorylation by numerous kinases, including PKA, AMPK, MAPK, and AKT [16]. CREB1 can also be directly inactivated via PTEN’s protein phosphatase activity [23].
We unexpectedly identified CREB1 as a major regulator of human prostate luminal cell differentiation while comparing differentially expressed genes during prostate luminal cell differentiation [24]. In this study, we establish a novel link between CREB1 activation and transient expression of ING4 during normal human prostate luminal cell differentiation and demonstrate that oncogenic loss of PTEN disrupts the induction of ING4 expression via aberrant CREB1 activation.
RESULTS
CREB1 gene signature in differentiating luminal cells.
When grown to confluency and treated with KGF plus androgen, basal prostate epithelial cells (PrEC) undergo differentiation such that a suprabasal secretory luminal cell layer forms on top of the basal layer in about 2 weeks [1, 5, 14, 24]. To identify genetic programs associated with luminal cell differentiation, PrECs differentiated for 0-17 days were subjected to RNA sequencing. Over 600 genes were significantly (FDR adjusted p<0.05) differentially expressed over the time course (GSE77460) (Fig. 1A). Pathway analysis indicates enrichment of genes involved in mitosis, epithelial differentiation, translation, cytoskeleton, and cell adhesion (Supplementary Fig. S1, Table S1, S2). Transcription factors predicted to regulate expression of these differentially expressed genes were identified. Majority of the differentially expressed genes at days 8 and 12 are known targets of MYC, p53, AR, SP1, and E2F1 (Fig. 1B). Analysis of isolated suprabasal cells at days 14 (14L) and 17 (17L) identified CREB1 as a key transcriptional regulator of approximately 25% of the differentially expressed genes (Fig. 1B; Supplementary Table S3).
Figure 1: CREB1 gene signature in differentiating luminal cells.

Prostate luminal cell differentiation was initiated by treating basal epithelial cells (PrECs) with 2ng/mL KGF and 5nM R1881 for 0-17 days. A) Isolated poly-A RNA was sequenced from biological triplicates of 0, 4 (no differentiation), 8, 11 (mixed basal and luminal), 14, and 17 (luminal only(L)) day cultures. Differentially expressed genes with p<0.05, relative to basal cells, for each time point were generated. B) Metacore analysis of transcription factors (Txf) likely involved in regulating the differentially expressed genes during differentiation. Top 6 Txf at each time point and the number of differentially expressed genes regulated by that Txf are indicated. C) Transcript counts (log2 CPM) for CREB1-specific target genes Blimp1, CLDN1, PLK2, and CHEK1 during differentiation. D) qRT-PCR validation of CREB1-specific target mRNAs. Normalized to 18s rRNA and shown as fold change relative to basal cells. *p<0.05; **p<0.01; ***p<0.001; n=3; error bars SD. E) BLIMP-1, PLK2, and CLDN1 protein levels were measured by immunoblotting. Tubulin (Tub) and GAPDH (GDH) served as loading controls. F) BLIMP1 immunostaining of PrECs differentiated for 8 days. Cells were imaged by phase or fluorescence microscopy.
Differential expression of predicted CREB1 targets, PRDM1, PLK2, CLDN1, and CHEK1 (Fig. 1C), known for their roles in differentiation and cell cycle suppression [25-28], were validated by qRT-PCR (Fig. 1D). BLIMP1 (PRDM1), PLK2, and CLDN1 proteins were also upregulated during differentiation, with BLIMP1 transiently peaking between days 8 and 12, while PLK2 and CLDN1 peaked in suprabasal cells at day 14 (Fig. 1E). Nuclear expression of BLIMP1 in suprabasal cells was evident at day 10 (Fig. 1F).
CREB1 is required for luminal cell survival.
CREB1 activity, as measured by immunoblotting for Ser133 phosphorylation, was induced after 12 days of differentiation and peaked in the suprabasal cells at day 14, while total CREB1 protein and mRNA were not significantly changed throughout differentiation (Fig. 2A,B). Immunofluorescent staining revealed that CREB1 activity was undetectable until day 12, where it was specifically localized in the suprabasal cells (Fig. 2C). To test the necessity of CREB1 for luminal differentiation, we generated cells expressing Tet-inducible shRNA targeting CREB1 (Fig. 2D,E). Knockdown of CREB1 with Dox did not prevent the induction of suprabasal cells at day 12 as visualized by the presence of AR-positive (luminal marker) integrin α6-negative (basal marker) suprabasal cells (Fig. 2F). These suprabasal cells express luminal-specific K18 and PSA and lack nuclear p63 (Supplementary Fig. S2). However, the luminal cells displayed increased activation of Caspase 3 and appeared unhealthy (Fig. 2G). Thus, CREB1 is not required for luminal differentiation, but is crucial for survival of the new luminal cells.
Figure 2: CREB1 is required for luminal cell survival.

Confluent PrECs were induced to differentiate with 2ng/ml KGF plus 5nM R1881 for 3-14 days. ‘L’ indicates only the luminal cells were analyzed. A) Total CREB1, ATF1, and activated levels, i.e., phosphorylation at Ser 133 (pCREB1, pATF1), measured by immunoblotting. Tubulin (Tub) served as loading control. B) Levels of CREB1 mRNA measured by qRT-PCR. C) Differentiated cultures immunostained with anti-pCREB (Ser133) antibody and DNA counterstained with Hoescht (Merge) and imaged by phase or fluorescence microscopy. D,E) Dox concentration required to suppress CREB1 protein and mRNA expression in PrECs expressing Tet-inducible shRNA (PrEC-TetON-shCREB1) assessed by D) immunoblotting at Day 12 and E) qRT-PCR at Day 3 and 12, **p<0.01; n=3; error bars SD. F,G) PrEC-TetON-shCREB1 cells induced to differentiate for 5 or 12 days in the absence (-Dox) or presence (+Dox) of 25ng/ml doxycycline. Cultures immunostained for integrin α6 (ITGα6, basal marker), AR (luminal marker), cleaved caspase 3 (Casp 3) and DNA counterstained with Hoescht (Merge) imaged by phase contrast and fluorescence microscopy.
To test the specificity of CREB1, we monitored activation of a related family member, ATF1, and found it was activated with different kinetics (Fig. 2A), being transiently activated at day 12 and back down by day 14. In contrast to CREB1, knockdown of ATF1 (Supplementary Figure S3A) blocked suprabasal induction (Supplementary Figure S3B). Strikingly, the levels of AR were significantly elevated in the shATF1 cells suggesting they may be converting to luminal-like cells in the absence of suprabasal induction. Thus, different CREB family members are likely involved in different aspects of luminal cell differentiation.
CREB1 limits ING4 expression during differentiation.
Transient induction of the chromatin remodeling protein ING4 (day 8-10) and its subsequent drop (after day 12), is required for proper luminal cell differentiation [14]. Constitutive ING4 expression initially accelerates suprabasal cell formation, but ultimately induces luminal cell death. Given that loss of CREB1 phenocopies ING4 overexpression (see Fig. 2G), we investigated whether CREB1 is responsible for the normal downregulation of ING4. ING4 protein is transiently elevated between days 8 and 12 of differentiation, and CREB1 activity peaks at the time ING4 declines (Fig. 3A). Knockdown of CREB1 in the PrECs leads to increased levels of ING4 protein (Fig. 3B-C) and mRNA expression (Fig. 3D). Thus, CREB1 is required to limit the duration of ING4 expression to prevent induction of luminal cell death.
Figure 3. CREB1 limits ING4 expression during differentiation.

Confluent PrECs induced to differentiate with 2ng/mL KGF and 5nM R1881 for 3-14 days. ‘L’ indicates only the luminal cells were analyzed. A) Levels of pCREB1, CREB, ING4, and PTEN during differentiation measured by immunoblotting. GAPDH served as loading control. B,C) PrEC-TetON-shCREB1 cells differentiated 5-12 days with (+Dox) or without (-Dox) 25ng/ml doxycycline. B) CREB1, ATF1, and ING4 expression measured by immunoblotting. C) Differentiated cultures immunostained for ING4, nuclei counterstained with Hoescht (Merge), and imaged by phase and fluorescence microscopy. D) Log2-fold expression of ING4 mRNA in PrEC-TetON-shCREB1 cells treated with (Dox) or without (Veh) doxycycline measured by qRT-PCR and normalized to day 0. ***p<0.005; ****p<0.001; n=4; error bars SD. E) ChIP of CREB1 at day 3 or 10 of differentiation versus IgG control. Enrichment of CREB1 binding at two CRE elements in the ING4 promoter. F) ChIP of CREB1 at day 3 or 10 of differentiation or day 3 of differentiation with cells overexpressing ING4 (I). G) Immunostaining of PrEC and PrEC-shPTEN for pCREB1.
PTEN controls CREB1 activation and differentiation.
Expression of PTEN, which suppresses CREB1 activity [10] and whose down regulation is required for AKT-dependent survival of the luminal cells [5], is elevated early in differentiation. On the other hand, PTEN drops at day 12 of differentiation, prior to ING4 loss and CREB1 activation, and is lowest in the suprabasal cells at Day 14, when active CREB1 is the highest (Fig. 3A), suggesting PTEN may control CREB1 activity. Indeed, knockdown of PTEN is sufficient to induce robust induction of nuclear-localized pCREB1 (Fig. 3G). Moreover, loss of PTEN suppressed PrEC differentiation (Supplementary Fig. S3C). Thus, the loss of PTEN in suprabasal cells may contribute to the timing of CREB1 activation and down regulation of ING4 required for nascent luminal cell survival.
Chromatin immunoprecipitation (ChIP) at day 3 (low ING4) and at day 10 (high ING4) detected CREB1 constitutively bound to a CRE half site (CRE1) in the ING4 promoter, while CREB1 was inducibly bound to the second full CRE site (CRE2) at day 10 (Fig. 3E). Since CREB1 binding is dictated by open chromatin [29] and ING4 influences chromatin state [15], we repeated the ChIP in ING4 overexpressing cells. Overexpression of ING4 is sufficient to enrich CREB1 binding to the full CRE at day 3 (Fig. 3F). CREB1 was not enriched at the Histone H3 gene. These data suggest ING4 is required for CREB1 binding to the CRE2 in the ING4 promoter and suppresses its transcription.
ING4 E3 Ligase JFK is a target of CREB1 and ING4.
ING4 protein levels are regulated by ubiquitination via the E3 ligase JFK [30]. We detected increased expression of JFK mRNA during differentiation (Fig. 4A). We found CREB1 to be constitutively associated with the CRE element in the JFK promoter, but not at a non-CRE site (Fig. 4B). In addition, low levels of ING4 were enriched at the JFK transcription initiation site at day 3, and more binding was observed at day 10 when ING4 levels increase. Overexpression of ING4 resulted in more binding at day 3 (Fig. 4C). Finally, knockdown of CREB1 leads to a reduction in JFK mRNA at the time when CREB1 activity peaks (Fig. 4D). Thus, ING4 and CREB1 appear to work cooperatively to increase JFK expression to coordinate the timing of ING4 destruction during differentiation.
Figure 4: ING4 E3 Ligase JFK is a target of CREB1 and ING4.
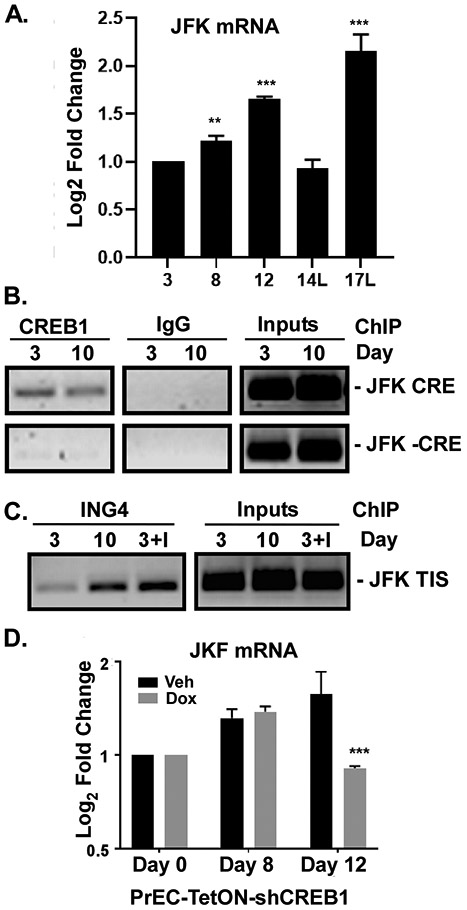
A) JFK mRNA levels during differentiation measured by qRT-PCR. B) ChIP of CREB1 on the CRE (+CRE) or non-CRE region (−CRE) of the JFK promoter. C) ChIP of ING4 at the transcriptional initiatin site (TIS) of the JFK promoter at day 3 or 10 post differentiation or at day 3 in cells overexpressing ING4 (I). D) Log2-fold expression of JFK mRNA in PrEC-TetON-shCREB1 cells treated with (Dox) or without (Veh) doxycycline measured by qRT-PCR and normalized to Day 0. **p<0.01; ***p<0.001; n=4; error bars SD
CREB1 targets different genes upon oncogenic transformation.
Overexpression of ERG (E), MYC (M), and shPTEN (P) in PrECs (EMP cells) is sufficient to generate tumors when cells are injected orthotopically into mice [14]. EMP cells fail to form a suprabasal layer, fail to fully differentiate into luminal cells, and co-express both basal and luminal markers [14]. EMP cells ‘differentiated’ for 0-17 days were subjected to the same RNA sequencing protocol as normal PrECs. Over 1700 genes were significantly (FDR adjusted p<0.05) differentially expressed compared to normal basal PrECs (GSE77460) (Fig. 5A). Pathway analysis indicates enrichment of genes involved in RNA metabolism, cell death, proliferation, transcription, motility/wounding, and morphogenesis (Supplementary Fig. S1, Table S4, S5). Surprisingly, a CREB1 signature was present in the tumor cells throughout the entire time course (Fig. 5B). CREB1 is predicted to regulate ~25% of the differentially expressed genes (Supplementary Table S6). The CREB1-regulated genes in PrEC suprabasal cells, when compared with the CREB1-regulated genes in EMP cells, are largely non-overlapping (Fig. 5C,D).
Figure 5. CREB1 targets different genes upon oncogenic transformation.

EMP cells (PrEC overexpressing ERG (E), MYC (M), and shPTEN (P)) subjected to the differentiation protocol for 0-17 days. RNA sequencing was performed on biological triplicates in parallel with PrECs (Fig.1). Differentially expressed genes with p<0.05 at each time point relative to normal basal cells were identified. A) Metacore analysis of transcription factors (Tfx) likely involved in regulating differentially expressed genes. B) Top 5 Tfx shown for each time point and the number of genes regulated by that Tfx are indicated. C) Heat map comparing CREB1-regulated genes in PrECs versus EMP cells. D) Comparison of CREB1-regulated genes from Day 14 luminal (L) PrECs vs. EMP cells. E,F) Levels of pCREB1 and pATF1 (Ser133), CREB1, and ATF1 measured by immunoblotting during differentiation in E) EMP or F) EMP versus PrEC. GAPDH served as loading control. G) IHC staining of pCREB1 in two EMP orthotopic xenograft tumors. H,I) Transcript counts from RNA-seq (log2 CPM) and qRT-PCR mRNA levels (log2 fold change) of EMP CREB1-specific target genes: H) GATA2 and Twist1, and I) NDN (Necdin) and PPM1F. J) Levels of Twist1 and GATA2 in differentiating PrEC versus EMP measured by immunoblotting. K) Levels of PMDR1 (BLIMP1) and CLDN1 mRNA measured by qRT-PCR from differentiating PrEC vs EMP. **p<0.01; ***p<0.001; n=4; error bars SD relative to PrEC.
Immunoblot analysis of CREB1 activity in EMPs subjected to the differentiation protocol showed sustained constitutive CREB1 activity throughout compared to PrECs differentiated for 3 or 10 days (Fig. 5E,F). Additionally, pCREB was highly elevated in EMP prostate tumor xenografts (Fig. 5G). Differential expression of predicted CREB1 targets GATA2, TWIST1, NND (Necdin, tumor suppressor), and PPM1F (CAMK Phosphatase) seen by RNA-Seq (Fig. 5H,I), were validated by qRT-PCR (Fig. 5H,I). Levels of TWIST1 and GATA2 protein were elevated in EMP cells relative to normal PrECs (Fig. 5J). In addition, CREB1 targets BLIMP1 and CLDN1 induced during differentiation in PrECs, were not induced in the EMP cells (Fig. 5K).
PTEN loss correlates with ING4 loss.
ING4 expression is reduced or lost in a subset of primary prostate tumors and loss of PTEN leads to loss of ING4 in vitro [14]. To assess the relationship between PTEN and ING4 loss and pCREB1, a tissue microarray, consisting of 81 primary tumors and 48 adjacent normal tissues, was scored for pCREB1, ING4, PTEN, and AR expression (Fig. 6A). We found that ~50% of the tumors had higher levels of pCREB1 relative to normal tissue. Over 35% of the tumors in this array had low ING4 expression and 47% had low PTEN. We found that 80% of the tumors with low ING4 also had low PTEN (Fig. 6B), suggesting loss of PTEN contributes to loss of ING4 in primary tumors. We were unable to detect a relationship between activated CREB1 and ING4 or PTEN. We attribute this in part to the high CREB1 activity present in normal tissues (Fig. 6C).
Figure 6: ING4 loss is associated with PTEN loss.

A TMA containing 81 tumors and 48 adjacent normal samples was interrogated by IHC for the expression of AR, pCREB1, ING4, and PTEN. A) Levels of expression of each indicated marker in the tumor samples. 0-1 scored as negative, 2-3 scored as positive. B) Fisher test demonstrating the association between low ING4 and low PTEN. ****p<0.0001. C) Representative IHC images.
Loss of CREB1 in tumorigenic cells rescues luminal cell differentiation and suppresses tumor growth.
To determine if loss of CREB1 in the tumor cells could rescue the defect in EMP differentiation, we induced knock-down of CREB1 in the EMP cells (Fig. 7A). Knock-down of CREB1 did not affect expression of oncogenic ERG, MYC, or shPTEN (Fig. 7B). Untreated EMP cells failed to properly differentiate as seen by the lack of a suprabasal layer compared to normal PrECs (Fig. 7C). Dox-induced suppression of CREB1 lead to re-induction of the suprabasal layer (Fig. 7C). However, as seen in normal PrECs, these suprabasal cells appeared stressed and had elevated caspase activity (Fig. 7D). The induced suprabasal cells are luminal, as indicated by exclusive expression of K18 and PSA and loss of p64, compared to EMP cells, which co-express both luminal and basal markers (Supplementary Fig. S4). Thus, inhibiting CREB1 in the tumor cells restores the differentiation phenotype, but since CREB1 is required for luminal cell survival, the luminal cells cannot persist. Inhibiting CREB1 expression in EMP cells also suppressed tumor growth in vivo (Fig. 7E). In contrast, suppression of ATF1 expression in EMP cells did not rescue suprabasal induction. As seen in normal PrECs (see Fig. S3B), loss of ATF1 in EMP cells also lead to increased AR expression in non-suprabasal cells (Supplementary Fig. S5A,B).
Figure 7: CREB1 suppresses luminal cell differentiation in tumorigenic EMP cells.

A) Doxycycline concentration required to knock-down CREB1 in EMP-TetON-shCREB1 cells. B) Validation of ERG, MYC, and PTEN expression in tumorigenic EMP cells relative to normal PrEC by immunoblotting. C,D) EMP-TetON-shCREB1 cells differentiated for 12 days with (+Dox) or without (−Dox) 100ng/ml doxycycline. Untreated 12-day differentiated PrECs were included as a control. Cultures were immunostained for C) integrin α6 (ITGα6, basal marker) and AR (luminal marker) or D) cleaved caspase 3 (Casp3) and counterstained with Hoescht (Merge), and imaged by phase or fluorescence microscopy. E) Growth of EMP-TetON-shCREB1 orthotopic tumors in SCID mice treated with (Dox) or without (Suc) doxycycline. *p<0.05; n=6; error bars SD. F) Models for how PTEN influences CREB1 activation and ING4 expression dynamics in normal (PrEC) versus tumor (EMP) cells. G) Pathways by which PTEN and CREB1 control ING4 expression.
MATERIALS AND METHODS
Cell lines:
Primary basal prostate epithelial cells (PrEC) isolated from clinical prostectomies, as previously described [31, 32], were immortalized as a pool with retroviruses expressing HPV E6/E7 and hTert [14]. Cultures were routinely tested for Mycoplasma and validated to be derived from prostate basal cells by STR. Oncogenic EMP cells were generated by stably overexpressing Erg (E), MYC (M), and shPTEN (P) as previously described and maintained in 2.0μg/ml puromycin [14]. Tet-inducible lentiviral shRNAs targeting CREB1 and ATF1 were used to generate PrEC-TetON-shCREB1, PrEC-TetON-shATF1, EMP-TetON-shCREB1, and EMP-TetON-shATF1 cells, which were selected and maintained in 2.0μg/ml puromycin (PrECs), 200μg/ml hygromycin (EMP-shCREB1), or 5μg/ml blasticidin (EMP-shATF1). Non-inducible shRNA targeting PTEN was used to generate PrEC-shPTEN cells, which were selected and maintained in 2.0μg/ml puromycin. All lines were maintained in Keratinocyte Serum-Free Media (1X)/(KSFM) (Gibco/Thermo Fisher Scientific) supplemented with bovine pituitary extract and epidermal growth factor as previously described [14, 31, 32].
Differentiation Protocol:
Each cell line, grown to confluency, were treated in complete growth medium containing 2-5ng/ml keratinocyte growth factor (KGF) (Cell Sciences) and 5nM R1881 (PerkinElmer) every other day for up to 17 days. For genetic and biochemical analysis, the differentiated suprabasal layer was separated from the basal layer at days 14 or 17 as previously described [5].
Constructs:
pLKO-TetON-shCREB1 and shATF1 vectors were generated by subcloning validated sequences (Supplementary Table S7) from the TRC shRNA library (Broad Institute) into the lentiviral EZ-Tet-pLKO-Puro, EZ-Tet-pLKO-Hygro, or EZ-Tet-pLKO-Blast vectors [33]. pLKO-shPTEN was generated by subcloning sequences complementary to the 3’-UTR of PTEN into a lentiviral vector pLKO.1-puro as previously described [14, 33].
Virus Generation and Infection:
Lentiviral shRNAs were generated by co-transfecting HEK293FT cells with shRNA vector and packaging plasmids as previously described [33]. Virus was harvested 3 days later and immediately used to infect cells.
Antibodies:
Immunofluorescence and IHC: AR (C-19) and p63 (BC4A4) were purchased from Santa Cruz. ITGα6 (GoH3) was purchased from BD Pharmingen, ING4 polyclonal antibody was purchased from ProteinTech. K18 (CY-90) came from Sigma and K5/14 (HMW-34βE12/M0630) from DAKO. (pCREB/pATF1-Ser 133(87G3), CREB (D76D11), cleaved Caspase3 (Asp175)(5A1E), PSA (D11E1), and PTEN (138G6) were purchased from Cell Signaling Technology. Immunoblotting: ING4 (EP3804) and ATF1 (EPR1590) antibodies were purchased from Abcam. pCREB-Ser 133(87G3), CREB (D76D11) and PTEN (138G6) were purchased from Cell Signaling Technology. Tubulin antibody (DM1A) was purchased from Sigma and GAPDH (6CS) from Millipore. Chromatin Immunoprecipitations: ING4 (EP3804) was purchased from Abcam and CREB (D76D11) was purchased from Cell Signaling.
Immunofluorescent Microscopy:
Differentiated cultures were fixed in 4% paraformaldehyde for 30 minutes, neutralized with 1M glycine, and permeabilized with 0.2% Triton-X 100 for 5 minutes and immunostained as previously described [14]. Coverslips were mounted using Fluoromount-G (Southern Biotech). Images were acquired on a Nikon Eclipse TE300 fluorescence microscope using OpenLab v5.5.0 image analysis software (Improvision).
Immunohistochemistry:
Paraffin-embedded and formalin fixed tissues were processed for IHC staining using automated immunostaining (Ventana Discovery XT). Tissue Microarray (TMA-47) containing 81 tumors and 48 adjacent normal tissues were stained for AR, pCREB, ING4, and PTEN. TMA staining was scored manually by two blinded individuals with IHC assigned to each core as composite scores of 0, 1, 2, or 3 with 0 to 1 representing complete to major loss of protein, and 2 to 3 near normal to wild-type levels.
Immunoblotting:
Total cell lysates were prepared for immunoblotting as previously described after lysing in RIPA buffer [31]. PVDF membranes were blocked in 5% BSA in TBST overnight at 4°C then probed with primary antibody, and HRP-conjugated secondary antibodies (Bio-Rad). Signals were visualized by chemiluminescence reagent with a CCD camera in a Bio-Rad Chemi-Doc Imaging System using Quantity One software v4.5.2 (Bio-Rad).
RT-PCR:
Total RNA was isolated using Life Technology’s RNeasy PureLink Kit. RNA was purified with RNase-free DNase and RNeasy PureLink Mini Kits. For qRT-PCR, 0.5μg RNA was reversed transcribed using a reverse transcription system (Promega). Synthesized cDNA was amplified for qRT-PCR using SYBR green master mix (Roche) with gene-specific primers and an ABI 7500 RT-PCR system (Applied Biosystems). Gene expression was normalized to 18S rRNA by the 2-ΔΔCt method [34]. Primers for target genes are listed in Supplementary Table S8.
RNA-Sequencing:
Biological triplicates of PrECs were differentiated for 0, 4, 8, 11, 14, and 17 days. Suprabasal cells on days 14 and 17 were detached from basal cells prior to RNA extraction. Total RNA was isolated using Life Technologies RNeasy kit. RNA was purified using Life Technologies Purelink RNA mini kit. TruSeq mRNA libraries were prepared for sequencing using standard Illumina protocols from PolyA-enriched RNA. Samples were sequenced using Illumina RNAseq as single reads at 50bp and approximately 30M reads per sample. Sequenced reads were mapped to the hg19 whole genome using the Subread aligner (v1.4.3). Reads were assigned to genes using featureCounts. Raw read counts were voom transformed and differential expression performed using limma. Differentially expressed genes with an FDR adjusted p<0.05 (n=3) and their expression levels were uploaded into the web-based Metacore program. Since samples from Day 4, 8, and 11 are a mixture of basal and luminal cells, we did not set a fold change cut off, as some of these may not be false positives. Gene lists were analyzed using MetaCore’s Pathway Enrichment and Transcription Factors one-click analysis and the transcriptional regulation network algorithm. Access to data: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=utahcoigpdubpcj&acc=GSE77460
Chromatin Immunoprecipitation (ChIP):
Cells (3.0X106) were fixed in 1% formaldehyde (Thermo Scientific) for 1 min and washed 3 times with ice cold calcium-magnesium free PBS (CMF-PBS) supplemented with protease inhibitors: pepstatin, aprotinin, leupeptin, and phenylmethylsulfonyl (PMSF). Cells were scraped and pelleted at 2,000 rpm for 8 min at 4°C. Pellet was resuspended in swelling buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% IGEPAL), and incubated on ice for 30 min. Nuclei were dounce homogenized and then pelleted at 4,000 rpm for 10 min, 4°C. Nuclei were resuspended in sonication buffer (0.1% SDS, 10 mM EDTA, 50 mM Tris–HCl pH 8.1) and incubated on ice for 10 min prior to sonication. Chromatin was sheared at 4°C using the Covaris E220 Ultra Sonicator following manufacturer’s suggested settings of 2% Duty Cycle, 105-Watt Peak Intensity, 200 Cycles/Burst. Chromatin was sonicated for 10 min to achieve 300–500 bp fragments.
Chromatin immunoprecipitations (ChIP) were performed with 1 million cells/IP using magnetic beads (NEB). Chromatin was incubated with 6mg appropriate antibody overnight at 4°C with rotation. Following incubation, magnetic beads blocked with 1% BSA supplemented with 10 mg/ml salmon sperm, were added to samples and incubated at 4°C with rotation for 6 ‘hr. Following immunoprecipitations, beads were washed in the following buffers at 4°C for 10 min with rotation: Triton Wash Buffer (50 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% Triton X-100), followed by Lysis Buffer 500 (0.1% NaDOC, 1 mM EDTA, 50 mM HEPES pH 7.5, 500 mM NaCl, 1% Triton X-100), LiCl Detergent buffer (0.5% NaDOC, 1 mM EDTA, 250 Mm LiCl, 0.5% IGEPAL, 10 mM Tris–HCl pH 8.1), and Tris–EDTA pH 8.1. Chromatin was eluted from beads in Elution Buffer (10 mM EDTA, 1% SDS, Tris–HCl pH 8.0) for 30 min at 65°C. Samples were then treated with 20 mg proteinase K, and 10 mg RNase A, and NaCl (200 mM) was added and incubated at 65°- C overnight to reverse cross-links. DNA was purified using phenol/chloroform extraction followed by ethanol precipitation. Primers (Supplementary Table S9) were designed referencing the UCSC Genome Browser to determine the transcriptional start sites and locations of CRE sites within ING4 and JFK promoters.
Tumor Studies:
1x106 Tet-inducible EMP-shCREB1 cells were injected orthotopically into male SCID mice at 8 weeks of age. Two days after tumor injection 6 mice each were randomly placed into 2 cohorts; controls were fed 5% glucose in their drinking water, and induced mice were given 2mg/ml doxycycline in 5% glucose. Tumors were growth was blindly measured by palpitation and caliper weekly for 12 weeks. All mouse studies were conducted according to an IACUC approved protocol.
Statistical Analyses:
TMA analysis: Two-tailed Fishers exact test was used to determine co-occurrence between high pCREB, low PTEN, and low ING4. Power was based on previously published results for ING4 IHC [14]. Tumor studies: Tumor volumes for each cohort, n=6, was averaged and 2-way Nova used to calculate statistical significance, which was set at p<0.05. Power was based on previously published results [14]. qRT-PCR: Biological triplicates were averaged and statistical significance determined by unpaired student T-test.
DISCUSSION
Previous studies demonstrated the importance of ING4, MYC, Miz1, and p38 MAPK in normal human prostate luminal cell differentiation [1, 14, 24]. Transient induction of ING4 is required for luminal cell differentiation and its loss leads to MYC-driven prostate tumorigenesis [14]. One objective of the current study was to uncover the molecular mechanisms that regulate transient ING4 expression during luminal cell differentiation. Using RNA-Seq analysis, we identified CREB1 as a major transcriptional driver of gene expression in suprabasal luminal cells. CREB1 is activated via phosphorylation late in differentiation. Knock-down of CREB1 does not suppress luminal cell formation, but the new luminal cells cannot survive, indicating CREB1 is required for luminal cell survival and maintenance. Generation of these short-lived luminal cells by CREB1 loss, phenocopies what is seen upon overexpression of ING4 [14]. Correspondingly, we observe an increase in both ING4 protein and mRNA in differentiating cells in which CREB1 is missing, and these cells ultimately die. Thus, the role of activated CREB1 is to limit the duration of ING4 expression (Fig. 7F). CREB1 does this by two mechanisms, through repression of ING4 mRNA transcription and transcriptional induction of the ING4 E3 ligase, JFK (Fig. 7G).
In addition to controlling ING4 expression, we identified a set of genes whose expression is regulated during differentiation and potentially controlled by CREB1. We validated that the CREB1 targets BLIMP1, PLK2, and CLDN1 mRNA and proteins are highly upregulated in suprabasl cells. BLIMP-1 is known to play a fundamental role in embryonic development in many organisms [35]. Epidermal terminal differentiation in the mouse depends on BLIMP1 protein expression in the granular layer of keratinocytes, the most differentiated corneocyte precursors. [36]. PLK2 belongs to the polo family of serine/ threonine protein kinases involved in normal cell division. In contrast to the role of PLK1 in promoting proliferation, PLK2 activity is associated with loss of proliferation. Recently a study identified PLK2 as a coordinator of early lineage commitment of cardiac progenitor cells [37].
Claudin1, a protein well known for its role in promoting cell-cell adhesion through tight junctions, is expressed in numerous epithelia [38]. Changes in claudin expression are part of a larger program of epithelial differentiation in the gut [38, 39]. There is a 3-fold increase in E-cadherin expression, another cell-cell adhesion protein, during luminal cell differentiation [5]. Increased cell-cell adhesion is required for survival of the new luminal cells, mediated by E-cadherin-dependent induction of PI3K [5]. The RNA-Seq data also identified cell adhesion molecules as a major set of enriched genes during differentiation. These data suggest that CREB1 not only promotes survival of the luminal cells by suppressing ING4, but may also enhance expression of cell-cell adhesion proteins required for survival.
CREB1 is crucial for neuronal survival and morphological maturation [40-42]. CREB has also been found to be involved in odontogenic differentiation of dental pulp stem cells [43], osteoclast differentiation [44], and mesoderm-derived embryonic stem cells [45]. In the above reports, a p38-dependent CREB signaling pathway is the most common axis involved. p38-MAPK is required for luminal cell differentiation, where in cooperation with MYC, it induces Notch3 [1]. Further investigation is required to establish whether p38 is also involved in setting up the CREB1 and ING4 interaction.
The most unexpected finding from these studies, is that CREB target genes are also induced upon oncogenic conversion of prostate epithelial cells. However, the CREB signature in tumor cells is highly distinct from the differentiation signature, and includes genes such as GATA2 and TWIST1, which are associated with prostate cancer progression. While aberrant CREB1 expression has been demonstrated for several cancers, including hematopoietic malignancies [46], non-small cell lung cancer, glioblastoma [47], breast cancer [48], and melanoma [49], very little has been investigated in prostate cancer. A previous report indicated that activated CREB1 is elevated in prostate cancer cell lines [50], and a more recent study identified a CREB1/FoxA1 signature in patient samples associated with pro-survival, cell cycle, and metabolic transcription programs in prostate cancer [21].
Here we demonstrate that the loss of PTEN is sufficient to activate CREB1, a known target of AKT, which is known to phosphorylate and activate CREB1. We found that PTEN is normally elevated in basal cells, which limits AKT activation, but drops during luminal cell differentiation, coincident with the peak in ING4 expression, but prior to CREB1 activation (Fig. 7F). PTEN appears to be responsible for setting the timing for when ING4 expression is suppressed. Once PTEN levels drop, then CREB1 is activated, which in turn suppresses ING4 (Fig. 7G). However, during oncogenic transformation via PTEN loss, CREB1 is activated prematurely, which prevents ING4 induction, because CREB1’s normal role is to suppress ING4 expression. Thus, ING4-dependent luminal cell differentiation cannot occur when CREB1 is activated too early in differentiation by high Akt levels due to PTEN loss (Fig. 7F).
ING4 is a tumor suppressor and forms a four-subunit histone acetylation complex containing ING4, HBO1, hEaf6, JADE1/2/3 paralogs [51]. When ING4 associates with HBO1 it prefers acetylation of H4 but also targets H3 to a lesser extent [51, 52]. The transient induction of ING4 during basal to luminal differentiation hints towards a possible role for ING4 in loosening of chromatin structure by acetylation of H4/H3 residues to allow transcription of genes required for the commitment to terminal differentiation [53, 54]. Interestingly, CREB1 is not activated until after ING4 is induced. CREB1 only binds open chromatin, suggesting that chromatin rearrangements, due in part to ING4, could open new binding sites for CREB1. This is supported by our findings that overexpression of ING4 results in induced CREB1 binding to the CRE site in ING4. This could explain the distinct difference in CREB1 targets activated late in differentiation, versus those that are activated early under oncogenic conditions. The failure to induce ING4 in the context of PTEN loss might prevent chromatin rearrangement and deny CREB1 access to terminal-differentiation genes. This would prevent permanent exit from mitosis and allow for oncogenesis.
We are the first to demonstrate an association between loss of ING4 and loss of PTEN in primary human prostate cancer. We did not see a correlation with high levels of pCREB1 and low PTEN or low ING4. This is likely due to the high levels of pCREB1 present in the luminal cells of the normal prostate, making it difficult to reliably distinguish normal from cancer tissues based solely on this marker. We would expect to see a better correlation if we were to interrogate some of the cancer-specific CREB1 targets rather than pCREB1 itself. Our ongoing studies are aimed at determining the role of the CREB1 targets in prostate cancer development.
Supplementary Material
ACKNOWLEDGEMENTS
We wish to thank the bioinformatics and pathology cores at the Van Andel Institute and University of Arizona (BBSR, TACMSR), and the experimental mouse core (EMSR) at University of Arizona for all their help with this study. These studies were supported by funding from the Department of Defense, W81XWH-14-1-0479 (MJW, PLB, CKM) and W81XWH-17-1-0570 (SBF, KB, LT, CKM) and funding from the Van Andel Research Institute. Cores at the University of Arizona were supported by funds from NIH/NCI P30 CA023074.
Funding:
MJW, PLB, KB, SBF, TL, SSG CKM – 2 grants from DOD; GH, MW – VAI
Footnotes
CONFLICTS
The authors declare no conflicts.
REFERENCES
- 1.Frank SB, Berger PL, Ljungman M, Miranti CK. Human prostate luminal cell differentiation requires NOTCH3 induction by p38-MAPK and MYC. J Cell Sci. 2017;130:1952–1964. [DOI] [PubMed] [Google Scholar]
- 2.Henry GH, Malewska A, Joseph DB, Malladi VS, Lee J, Torrealba J et al. A cellular anatomy of the normal adult human prostate and prostatic urethra. Cell Rep. 2018;25:3530–3542.e3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burger PE, Xiong X, Coetzee S, Salm SN, Moscatelli D, Goto K et al. Sca-1 expression identifies stem cells in the proximal region of prostatic ducts with high capacity to reconstitute prostatic tissue. Proc Natl Acad Sci U S A. 2005;102:7180–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwon OJ, Zhang L, Xin L. Stem Cell Antigen-1 identifies a distinct androgen-independent murine prostatic luminal cell lineage with bipotent potential. Stem Cells. 2016;34:191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamb LE, Knudsen BS, Miranti CK. E-cadherin-mediated survival of androgen-receptor-expressing secretory prostate epithelial cells derived from a stratified in vitro differentiation model. J Cell Sci. 2010;123:266–276. [DOI] [PubMed] [Google Scholar]
- 6.Xiao GQ, Golestani R, Pham H, Sherrod AE. Stratification of atypical intraepithelial prostatic lesions based on basal cell and architectural patterns. Am J Clin Pathol. 2020;153:407–416. [DOI] [PubMed] [Google Scholar]
- 7.Abate-Shen C, Shen MM, Gelmann E. Integrating differentiation and cancer: the Nkx3.1 homeobox gene in prostate organogenesis and carcinogenesis. Differentiation. 2008;76:717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandi F, Grupp K, Hube-Magg C, Kluth M, Lang D, Minner S et al. High concordance of TMPRSS-ERG fusion between primary prostate cancer and its lymph node metastases. Oncology Lett. 2018;16:6238–6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thangapazham R, Saenz F, Katta S, Mohamed AA, Tan SH, Petrovics G et al. Loss of the NKX3.1 tumorsuppressor promotes the TMPRSS2-ERG fusion gene expression in prostate cancer. BMC Cancer. 2014;14:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu W, Xie CC, Thomas CY, Kim ST, Lindberg J, Egevad L et al. Genetic markers associated with early cancer-specific mortality following prostatectomy. Cancer. 2013;119:2405–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang H, Jung WY, Kang Y, Lee H, Kim A, Kim BH. Expression of ROR1, pAkt, and pCREB in gastric adenocarcinoma. Ann Diagn Pathol. 2015;19:330–334. [DOI] [PubMed] [Google Scholar]
- 12.Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U S A. 2006;103:1480–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koh CM, Bieberich CJ, Dang CV, Nelson WG, Yegnasubramanian S, De Marzo AM. MYC and prostate cancer. Genes Cancer. 2010;1:617–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger PL, Frank SB, Schulz VV, Nollet EA, Edick MJ, Holly B et al. Transient induction of ING4 by Myc drives prostate epithelial cell differentiation and its disruption drives prostate tumorigenesis. Cancer Res. 2014;74:3357–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hung T, Binda O, Champagne KS, Kuo AJ, Johnson K, Chang HY et al. ING4 mediates crosstalk between histone H3 K4 trimethylation and H3 acetylation to attenuate cellular transformation. Mol Cell. 2009;33:248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. [DOI] [PubMed] [Google Scholar]
- 17.Shankar DB, Cheng JC, Kinjo K, Federman N, Moore TB, Gill A et al. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell. 2005;7:351–362. [DOI] [PubMed] [Google Scholar]
- 18.Thway K, Fisher C. Tumors with EWSR1-CREB1 and EWSR1-ATF1 fusions: the current status. The Am J Surg Pathol. 2012;36:e1–e11. [DOI] [PubMed] [Google Scholar]
- 19.Fang Z, Lin A, Chen J, Zhang X, Liu H, Li H et al. CREB1 directly activates the transcription of ribonucleotide reductase small subunit M2 and promotes the aggressiveness of human colorectal cancer. Oncotarget. 2016;7:78055–78068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Zheng D, Zhou T, Song H, Hulsurkar M, Su N et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat Comm. 2018;9:4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sunkel B, Wu D, Chen Z, Wang CM, Liu X, Ye Z et al. Integrative analysis identifies targetable CREB1/FoxA1 transcriptional co-regulation as a predictor of prostate cancer recurrence. Nucleic Acids Res. 2016;44:4105–4122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rehfuss RP, Walton KM, Loriaux MM, Goodman RH. The cAMP-regulated enhancer-binding protein ATF-1 activates transcription in response to cAMP-dependent protein kinase A. J Biol Chem. 1991;266:18431–18434. [PubMed] [Google Scholar]
- 23.Gu T, Zhang Z, Wang J, Guo J, Shen WH, Yin Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011;71:2821–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berger PL, Winn ME, Miranti CK. Miz1, a novel target of ING4, can drive prostate luminal epithelial cell differentiation. The Prostate. 2017;77:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan J, Jiang J, Lim CA, Wu Q, Ng HH, Chin KC. BLIMP1 regulates cell growth through repression of p53 transcription. Proc Natl Acad Sci U S A. 2007;104:1841–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cizmecioglu O, Warnke S, Arnold M, Duensing S, Hoffmann I. Plk2 regulated centriole duplication is dependent on its localization to the centrioles and a functional polo-box domain. Cell Cycle. 2008;7:3548–3555. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Han X, Zhang Y. Autoregulatory mechanisms of phosphorylation of checkpoint kinase 1. Cancer Res. 2012;72:3786–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirschner N, Rosenthal R, Furuse M, Moll I, Fromm M, Brandner JM. Contribution of tight junction proteins to ion, macromolecule, and water barrier in keratinocytes. J Invest Dermatol. 2013;133:1161–1169. [DOI] [PubMed] [Google Scholar]
- 29.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–13508. [DOI] [PubMed] [Google Scholar]
- 30.Yan R, He L, Li Z, Han X, Liang J, Si W et al. SCF(JFK) is a bona fide E3 ligase for ING4 and a potent promoter of the angiogenesis and metastasis of breast cancer. Genes Dev. 2015;29:672–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Edick MJ, Tesfay L, Lamb LE, Knudsen BS, Miranti CK. Inhibition of integrin-mediated crosstalk with epidermal growth factor receptor/Erk or Src signaling pathways in autophagic prostate epithelial cells induces caspase-independent death. Mol Biol Cell. 2007;18:2481–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gmyrek GA, Walburg M, Webb CP, Yu HM, You X, Vaughan ED et al. Normal and malignant prostate epithelial cells differ in their response to hepatocyte growth factor/scatter factor. Am J Pathol. 2001;159:579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frank SB, Schulz VV, Miranti CK. A streamlined method for the design and cloning of shRNAs into an optimized Dox-inducible lentiviral vector. BMC Biotechnol. 2017;17:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 35.Martins G, Calame K. Regulation and functions of Blimp-1 in T and B lymphocytes. Ann Rev Immunol. 2008;26:133–169. [DOI] [PubMed] [Google Scholar]
- 36.Magnusdottir E, Kalachikov S, Mizukoshi K, Savitsky D, Ishida-Yamamoto A, Panteleyev AA et al. Epidermal terminal differentiation depends on B lymphocyte-induced maturation protein-1. Proc Natl Acad Sci U S A 2007;104:14988–14993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mochizuki M, Lorenz V, Ivanek R, Della Verde G, Gaudiello E, Marsano A et al. Polo-like kinase 2 is dynamically regulated to coordinate proliferation and early lineage specification downstream of Yes-associated protein 1 in cardiac progenitor cells. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Capaldo CT, Nusrat A. Claudin switching: physiological ppasticity of the tight junction. Sem Cell Dev Biol. 2015;42:22–29. [DOI] [PubMed] [Google Scholar]
- 39.Garcia-Hernandez V, Quiros M, Nusrat A. Intestinal epithelial claudins: expression and regulation in homeostasis and inflammation. Ann NY Acad Sci. 2017;1397:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landeira BS, Santana T, Araujo JAM, Tabet EI, Tannous BA, Schroeder T et al. Activity-independent effects of CREB on neuronal survival and differentiation during mouse cerebral cortex development. Cereb Cortex. 2018;28:538–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhat NR, Zhang P, Mohanty SB. p38 MAP kinase regulation of oligodendrocyte differentiation with CREB as a potential target. Neurochem Res. 2007;32:293–302. [DOI] [PubMed] [Google Scholar]
- 42.Jeong SG, Cho GW. The tubulin deacetylase sirtuin-2 regulates neuronal differentiation through the ERK/CREB signaling pathway. Biochem Biophys Res Comm. 2017;482:182–187. [DOI] [PubMed] [Google Scholar]
- 43.Zhu Q, Gao J, Tian G, Tang Z, Tan Y. Adrenomedullin promotes the odontogenic differentiation of dental pulp stem cells through CREB/BMP2 signaling pathway. Acta Biochim Biophys Sin. 2017;49:609–616. [DOI] [PubMed] [Google Scholar]
- 44.Kim JH, Kim K, Kim I, Seong S, Lee KB, Kim N. BCAP promotes osteoclast differentiation through regulation of the p38-dependent CREB signaling pathway. Bone. 2018;107:188–195. [DOI] [PubMed] [Google Scholar]
- 45.Rahman F, Bordignon B, Culerrier R, Peiretti F, Spicuglia S, Djabali M et al. Ascorbic acid drives the differentiation of mesoderm-derived embryonic stem cells. Involvement of p38 MAPK/CREB and SVCT2 transporter. Mol Nutrit Food Res. 2017;61. [DOI] [PubMed] [Google Scholar]
- 46.Cheng JC, Kinjo K, Judelson DR, Chang J, Wu WS, Schmid I et al. CREB is a critical regulator of normal hematopoiesis and leukemogenesis. Blood. 2008;111:1182–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steven A, Seliger B. Control of CREB expression in tumors: from molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget. 2016;7:35454–35465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chhabra A, Fernando H, Watkins G, Mansel RE, Jiang WG. Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol Rep. 2007;18:953–958. [PubMed] [Google Scholar]
- 49.Melnikova VO, Dobroff AS, Zigler M, Villares GJ, Braeuer RR, Wang H et al. CREB inhibits AP-2α expression to regulate the malignant phenotype of melanoma. PloS One. 2010;5:e12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garcia GE, Nicole A, Bhaskaran S, Gupta A, Kyprianou N, Kumar AP. Akt-and CREB-mediated prostate cancer cell proliferation inhibition by Nexrutine, a Phellodendron amurense extract. Neoplasia. 2006;8:523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell. 2006;21:51–64. [DOI] [PubMed] [Google Scholar]
- 52.Lalonde ME, Avvakumov N, Glass KC, Joncas FH, Saksouk N, Holliday M et al. Exchange of associated factors directs a switch in HBO1 acetyltransferase histone tail specificity. Genes Dev. 2013;27:2009–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruan K, Yamamoto TG, Asakawa H, Chikashige Y, Kimura H, Masukata H et al. Histone H4 acetylation required for chromatin decompaction during DNA replication. Sci Rep. 2015;5:12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palacios A, Moreno A, Oliveira BL, Rivera T, Prieto J, Garcia P et al. The dimeric structure and the bivalent recognition of H3K4me3 by the tumor suppressor ING4 suggests a mechanism for enhanced targeting of the HBO1 complex to chromatin. J Mol Biol. 2010;396:1117–1127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.