

Abstract
Satellite DNA are long tandemly repeating sequences in a genome and may be organized as high-order repeats (HORs). They are enriched in centromeres and are challenging to assemble. Existing algorithms for identifying satellite repeats either require the complete assembly of satellites or only work for simple repeat structures without HORs. Here we describe Satellite Repeat Finder (SRF), a new algorithm for reconstructing satellite repeat units and HORs from accurate reads or assemblies without prior knowledge on repeat structures. Applying SRF to real sequence data, we show that SRF could reconstruct known satellites in human and well-studied model organisms. We also find satellite repeats are pervasive in various other species, accounting for up to 12% of their genome contents but are often underrepresented in assemblies. With the rapid progress in genome sequencing, SRF will help the annotation of new genomes and the study of satellite DNA evolution even if such repeats are not fully assembled.
Satellite DNA (SatDNA) are long tandemly repeating sequences that look like “BBBBBB”, where each symbol “B” represents a repeat unit, also known as a monomer. A monomer “B” could range from a few base pairs (bp) to thousands of bp in length and an entire SatDNA could span megabases in large genomes. Several percent of the human genome, or a couple of hundred megabases in total, is composed of SatDNA (Altemose et al. 2022). Monomers in a SatDNA array are similar in sequence but often not identical because of random mutations and rearrangements between satellite arrays.
In some species, SatDNA may be organized as high-order repeats (HORs; Miga 2019). For example, the centromere of human Chromosome 2 has a pattern like “ABCDABCDABCD”. Letters “A”–“D” correspond to four diverged alpha repeat monomers of ∼171 bp each, respectively, and the “ABCD” unit is repeated many times in the centromere with all copies being similar to each other. Researchers who study centromere repeats usually say “ABCD” is a 4-mer HOR unit. Because we will often mention short nucleotide sequences in this article, we will call “ABCD” in this example as a 4-monomer HOR unit to avoid confusion.
In the human genome, the active centromeric regions that centromeric proteins bind to are primarily composed of alpha HORs (αHORs; Altemose et al. 2022). Conversely, though, not all αHORs are present in the active regions. These inactive αHORs tend to be shorter than active ones. Alpha repeat monomers are also present in pericentromeric regions without clear HOR structures. There are often not clear boundaries between αHOR and nonHOR monomers. In addition to alpha repeats, the human genome is also enriched with three types of human satellites (HSat1–3), contributing to a few percent of human genome (Altemose 2022). Almost all these satellites are located around centromeres or on the long arm of the Y Chromosome.
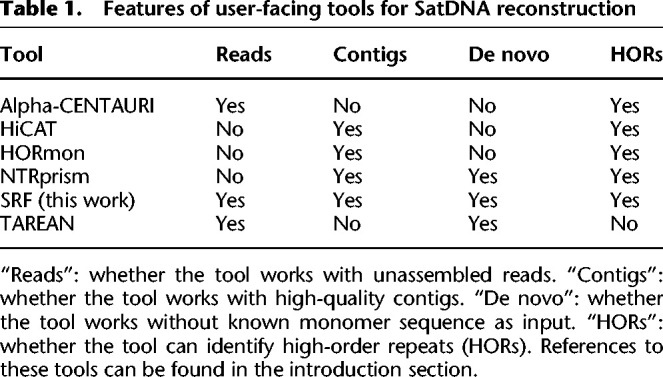
SatDNA is often not assembled in long contigs because of its repetitiveness. For example, in the human reference genome GRCh38 (Schneider et al. 2017), αHORs were computationally generated from a Hidden Markov model (Miga et al. 2014); HSats are underrepresented. As a result, we would have to reconstruct SatDNA from raw sequence reads in this case (Lower et al. 2018). Wei et al. (2014) developed k-Seek to study SatDNA consisting of 2–10 bp repeat units. Melters et al. (2013) applied tandem repeat finder (TRF; Benson 1999) to Sanger reads and fragmented short-read contigs to find the most common monomer in each species. TAREAN (Novák et al. 2017) does all-versus-all comparison between short reads, clusters the reads, and then identifies circular structures from the cluster graphs. These methods can reconstruct unknown monomers but they are unable to reveal HOR structures. On the contrary, Alpha-CENTAURI (Sevim et al. 2016) reconstructs HORs from long reads but it requires known monomer sequences.
With improved sequencing technologies, it is now possible to assemble through human centromeres (Nurk et al. 2022). More recent methods, including NTRprism (Altemose et al. 2022), HORmon (Kunyavskaya et al. 2022), and HiCAT (Gao et al. 2023), can identify detailed chromosome-specific HOR patterns from complete SatDNA sequences in human. These methods demand high-quality assembly and rich prior knowledge on SatDNA in the studied species. However, the finished human genome, CHM13, was derived from a near homozygous molar cell line that is easier to assemble. For a normal diploid human individual, we could only assemble through a fraction of SatDNA even with the best possible data and algorithm (Rautiainen et al. 2023). The complete assembly of other species is even rarer. This has limited the application of such assembly-based SatDNA reconstruction algorithms.
In this article, we will describe a new algorithm, Satellite Repeat Finder (SRF), for assembling SatDNA repeat units. SRF overcomes the limitation of previous methods. It is applicable to both accurate reads and high-quality assembly and is able to automatically reconstruct HORs with no prior knowledge on monomer sequences.
Results
The SRF algorithm
In a SatDNA array “BBBBBB ”, suppose every monomer “B” is identical to each other. Under a long enough k, the k-mer de Bruijn graph of the SatDNA array will be a single cycle. When there are base pair differences between monomers, the de Bruijn graph will not be a simple cycle. If there are many different monomers, the de Bruijn graph can become very complex and cannot be resolved with classical graph cleaning algorithms (Zerbino and Birney 2008).
Our intuition is that if there are many copies of the monomer, we may still be able to find a cycle composed of highly abundant k-mers in the de Bruijn graph. We can start with the most abundant k-mer and at each bifurcation in the graph and greedily choose the k-mer of the highest occurrence. We repeat this process until we go back to the starting k-mer, which will reconstruct a repeat unit, or come to a dead end, which will be discarded. Algorithm 1 (Fig. 1) provides more details. Here, t → s indicates k-mer t and s are adjacent in the de Bruijn graph. For simplicity, this algorithm traverses a unidirected de Bruijn graph. In SRF, we implemented a bidirected de Bruijn graph such that we will not find a repeat unit on both strands.
Figure 1.

The SRF algorithm.
SRF works with Illumina short reads, Pacific Biosciences (PacBio) HiFi long reads, and high-quality assembly contigs and can identify HORs (Table 1). When assembling satellite repeats from PacBio HiFi reads in this article, we counted 151-mers with KMC (Kokot et al. 2017) and collected 151-mers occurring ≥10 times over the average read coverage. k-mer counting may take a few tens of minutes for a high-coverage human data set and is the performance bottleneck. SRF only takes seconds to reconstruct all repeat units after k-mer counting.
Table 1.
Features of user-facing tools for SatDNA reconstruction
SRF would not be able to distinguish two repeat units that share an identical k-mer. k needs to be large enough to separate human HORs. On the other hand, a large k may reconstruct highly similar but nonidentical repeat units. This is a relatively small problem as we can reduce redundancy later. When applying SRF to raw reads, we also need to consider the sequencing error rate . The probability of seeing an error-free k-mer is . If , only 28.4% of 251-mers are error free. We may miss high-occurrence k-mers with large k. Another practical consideration is that KMC by default only supports k ≤ 256. Counting long k-mers is also slow.
Estimating satellite abundance
The abundance of a repeat unit is the fraction of the genome size to which this repeat contributes. If we assume reads are evenly distributed along the genome, the abundance can also be estimated as the total repeat length in reads over the sum of all read lengths.
We may add up counts of k-mers in a repeat unit to estimate the abundance of the repeat. However, such an estimate does not count inexact repeats and would be an underestimate. To get a more accurate estimate, we mapped all input sequences against reconstructed repeat units to measure the total length of each repeat unit. For human CHM13 data, we observed many diverged hits between HORs and scattered monomers in pericentromeric regions. To get more accurate HOR length estimates, we filtered out hits of <90% identity to the repeat unit. The effect of the identity filter is determined by the repeat structure in a species. For example, switching off the filter would increase the total abundance estimate by 40% for human but only by 4% for Arabidopsis thaliana. We still applied this filter to all data sets even though this may lead to underestimates for some species.
As is explained in the previous section, SRF may reconstruct two repeat units similar to each other, up to (k − 1)/k in identity. Such repeats may be mapped to the same genomic loci. To remove redundancy, we only select the hit of the highest identity among hits overlapping on an input sequence. With this procedure, we map each base on an input sequence to at most one repeat unit.
Occasionally a small number of long terminal repeats (LTRs) may occur tandemly in a few regions. SRF may identify such LTRs even though they do not form long tandem arrays. When estimating abundance, we additionally filter out repeats with <2 tandem copies in the middle of a sequence or with <1.5 tandem copies when the repeat-to-read alignment reaches the end of a read. This filter is reliable when we apply SRF to assemblies but may miss long repeat units when applied to reads. We again opted for conservative estimates.
Annotating satellite repeats in human T2T-CHM13
We first ran SRF on each T2T-CHM13 chromosome separately and compared the results to existing annotations by HORmon (Kunyavskaya et al. 2022) and HiCAT (Gao et al. 2023). HORmon reports the same αHOR lengths as Altemose et al. (2022). In Table 2, column “SRF (k = 171) chromosome” shows the lengths of HORs identified by running SRF on individual chromosomes. SRF reported the same αHOR lengths as HORmon except for Chromosomes 5, 8, 9, 13, and 18. The Chromosome 8 of T2T-CHM13 has been well studied by Logsdon et al. (2021). Although the 11-monomer is the most abundant, it is interleaved with 4-, 7-, and 8-monomers that are derived from the 11-monomer. The 7-monomer is the second most abundant array and forms the longest αHOR array in the middle of the centromere. The greedy SRF algorithm chooses the 7-monomer over the 11-monomer possibly because the 7-monomer has a more conservative consensus. The SRF-HORmon differences in other chromosomes may have a similar cause.
Table 2.
Human chromosome-specific high-order alpha repeats (αHORs)

Unlike HORmon and HiCAT which require users to provide the monomer sequence and prepare centromeric sequences, SRF was directly applied to whole chromosome sequences with no prior knowledge. In addition to active αHORs, SRF identified shorter αHOR arrays outside the active regions. It also further found many other long satellite arrays including a repeat unit of 1814 bp on Chr 15, of 6112 bp on Chr 16, of 3569 bp on Chr Y, and of 2420 bp on Chr Y as well. These span over one megabase and have been reported previously (Altemose 2022).
SRF further found a satellite array on the long arm of Chromosome 1 between coordinate 227,746,662 and 228,024,151. The repeat unit is 2240 bp in length, composed of an AluY repeat, a 5S-RNA, and dinucleotide repeats. This is the only noncentromeric array in T2T-CHM13 longer than 100 kb.
The SRF inference on the whole T2T-CHM13 genome (column “SRF/171 assembly” in Table 2) is close to the inference on individual chromosomes. SRF missed the αHOR array on Chr 14 and Chr 21 because Chr 22 and Chr 13, respectively, have very similar arrays which are merged during the whole-genome inference. SRF found the 68 bp beta satellite in a 1906 bp contig. TRF estimated the contig consists of 28 copies of the beta repeat unit with an average sequence identity of 77% according to TRF. This contig occurs tandemly for 39 times on Chr 22: 1,531,778–1,606,098 or 31 times on Chr 15: 579,188–638,269 at ≥98% identity to the contig sequence, suggesting these two regions locally have a high-order structure. Nonetheless, such a structure is not obvious in many other loci in the genome. SRF failed to identify the 220 bp gamma repeat possibly because of its lower abundance or higher sequence divergence.
The T2T consortium annotated 70.3 Mb of αHOR sequences, including 61.9 Mb of active ones. From the whole genome, SRF identified 55.4 Mb of αHOR sequences, nearly all of which (99.98%) are annotated by T2T. 96.6% of SRF αHORs are annotated as active by T2T with the remaining being inactive αHORs. Conversely, 86.3% of active and 76.0% of all αHOR annotated by T2T are overlapping with SRF αHORs. The two numbers are increased to 96.0% and 84.6%, respectively, if we change the identity filter from 90% to 85%. At threshold 85%, SRF found 63.5 Mb of αHOR with 99.67% annotated by T2T.
SRF works on sequence reads which HORmon, HiCAT, and NTRprism are not applicable to. On PacBio High-Fidelity (HiFi) reads, SRF reconstructed αHORs similar to the whole-genome reconstruction (column “SRF/171 HiFi reads” in Table 2). It can also identify the majority of αHORs from Illumina short reads (column “SRF/101 Illumina” in Table 2), though the use of shorter 101-mer reduces the sensitivity to some arrays.
From Table 2 we can see that some αHOR arrays, such as those on Chr 3 and Chr 11, can be consistently reconstructed by various tools on different types of input data. However, some other arrays, such as those on Chr 8 and Chr 19, are intrinsically harder to reconstruct. These are probably because monomers in a HOR may be connected in different ways, as is shown by Kunyavskaya et al. (2022).
We used k = 171 for human because an alpha unit is about 171 bp in length. Changing k to 101, 151, 201, or 251 bp generally led to the same conclusion: There are 24–30 αHORs contributing to 1.65%–1.81% of the genome. However, with k = 51, SRF started to miss distinct αHORs that share 51-mers. It identified only 12 αHORs contributing to 0.52% of the genome.
Satellite repeats in multiple human assemblies
We applied SRF independently to each phased human assembly produced by the Human Pangenome Reference Consortium (HPRC). We identified αHORs with dna-brnn (Li 2019), aligned them to the T2T-CHM13 genome and HORmon consensus, and manually assigned the αHORs to chromosomes based on the similarity to existing annotations. The last column of Table 2 shows the αHOR lengths and their frequencies. We consider two αHORs are different if they have different lengths and the shorter αHOR cannot be aligned into the longer one at <2% sequence divergence.
There are 47 diploid samples and 94 haploid assemblies. We could find αHORs assigned to individual chromosomes in most cases. We sometimes see αHORs of different lengths assigned to the same chromosome but their sequence divergences are small. This again could be caused by the different ways HOR monomers are connected in individual samples (Logsdon et al. 2021; Kunyavskaya et al. 2022).
We also applied SRF to the pool of all haploid assemblies. This procedure may miss infrequent satellite arrays but it helps to simplify the study of shared arrays. In addition to active αHORs, we identified supposedly inactive αHORs that are at >10% divergence from existing annotated αHORs. Notably, there is a 20-monomer αHOR that is mapped to the Chromosome 15 of T2T-CHM13 and spans several hundred kilobases in most samples. There are other examples like this. SRF also found long HSat arrays and nonHOR satellites, including those found in CHM13, which are easy to identify as they do not have internal structures.
Satellite repeats in Arabidopsis thaliana
We obtained four HiFi data sets (Table 3) and downsampled them to about 40-fold coverage each. Col-0N and Ey15-2R were sequenced from a pool of multiple samples and Col-0R and Col-0W from a single sample.
Table 3.
A. thaliana PacBio HiFi data sets

The A. thaliana centromeres are composed of CEN180 satellites. Although Naish et al. (2021) have identified HORs in centromeres, these HORs are fragmented to blocks of 429 bp on average and each centromere consists of over 10,000 different HORs; monomers in a HOR may only differ by 2.8%. In contrast, each human centromere only consists of one major type of HOR and monomers in a HOR often differ by >10%. HORs in A. thaliana are local and each individual HOR has low abundance.
We pooled the HiFi reads of the three Col-0 data sets, applied SRF, and reconstructed two 1-monomers, three 2-monomers, one 3-monomer, and two 7-monomer HORs with monomers in each HOR being 90%–96% identical. They are hard to be distinguished from unstructured monomers. We thus ignored the HOR structures and focused on the abundance of CEN180 only.
All three data sets have assemblies that go through all centromeres. We used CEN180 repeats reconstructed from reads to estimate the total CEN180 length in each assembly. We identified 9.9, 10.0, and 9.9 Mb of CEN180 repeats in Col-0R, Col-0N, and Col-0W assemblies, respectively. All assemblies have similar CEN180 content.
SRF inferred that 7.5% of Col-0R read bases, 5.2% of Col-0N, and 12.5% of Col-0W are comprised of CEN180 repeats. Col-0W contains more CEN180 than the other two samples. To check whether this large difference is caused by an SRF artifact, we directly extracted high-occurrence 179-mers from raw reads, without running SRF, and compared their counts in Col-0N and Col-0W (Fig. 2A). It seemed that Col-0W did have more CEN180 content. We further aligned Col-0N and Col-0W reads against the Col-0R assembly (AC:GCA_946499705.1). Consistent with the SRF result, the read depth of the Chr 1 centromere is noticeably higher in Col-0W than in Col-0R and Col-0N (Fig. 2C). We saw a similar pattern on other chromosomes and on all raw reads without downsampling. It is not impossible that CEN180 regions in Col-0W were duplicated after its divergence from other laboratory samples. Such recent duplications would not be separated by assemblers. We would need original samples to test this hypothesis.
Figure 2.

Normalized counts of 179-mers in three A. thaliana read data sets. Raw 179-mer counts in reads are normalized by coverage. A 179-mer is selected in the plot if it matches the CEN180 satellite and if its normalized count is at least 50 in one of the data sets. (A) Counts between two different samples from the same strain. (B) Counts between two different strains. (C) Read depths averaged in 100 kb windows across Chromosome 1. The shaded area indicates the centromere enriched with the CEN180 satellite repeat.
SRF estimated that 11.5% of Ey15-2R is composed of CEN180 satellites, higher than both Col-0R and Col-0N but comparable to Col-0W. Furthermore, whereas Col-0W and Col-0N share similar high-occurrence 179-mers that match CEN180 (Fig. 2A), Col-0 and Ey15-2 share few common 179-mers (Fig. 2B). The centromere sequences between strains are distinct both in sequence and in length.
In addition to the CEN180 satellite, SRF also reconstructed a 10,201 bp rDNA unit from the three Col-0 data sets. It has 3.1% abundance in Col-0R, 1.7% in Col-0N, and 2.5% in Col-0W. If we assume the A. thaliana genome is 132 Mb in length according to the nuclear assembly, Col-0R has ∼400 copies of this rDNA unit whereas Col-0N has ∼220 copies and Col-0W has ∼320 copies. The Col-0N assembly (Naish et al. 2021) only has seven copies, located towards the telomeric ends of Chr 2 or Chr 4 short arms. SRF did not reconstruct an rDNA unit from Ey15-2R. We mapped the Col-0 rDNA unit to Ey15-2R reads and estimated that Ey15-2R has ∼200 copies. SRF also identified the (TTTAGGG)n telomere repeat but missed the less frequent CEN160 repeat (Round et al. 1997).
Satellite repeats in other model organisms
We applied SRF to the HiFi reads of three model organisms (Hon et al. 2020): the reference C57BL/6J strain of Mus musculus (mouse; AC:SRR11606870), the F1 generation of the reference ISO1 strain and the A4 strain of Drosophila melanogaster (AC:SRR10238607), and the B73 strain of Zea mays (maize; AC:SRR11606869).
In mouse, SRF identified two satellite units. The second most abundant repeat is the 234 bp major satellite around centromeres (Arora et al. 2021; Thakur et al. 2021). The first is 1199 bp in length, composed of 10 copies of the 120 bp minor satellite unit. This confirms the high-order organization of minor satellites observed by Pertile et al. (2009). The full-length hits of this repeat in the mouse reference genome mostly come from the sex chromosomes and are all below 75% in identity. Nonetheless, this repeat is abundant in reads with the majority of alignments at 95% identity or higher. To further investigate this repeat, we assembled the HiFi reads with hifiasm (Cheng et al. 2021). We can find long tandem arrays of this repeat on multiple contigs, all shorter than 1.1 Mb. Hifiasm keeps the repeat content but is unable to assemble this satellite.
In Drosophila, the most abundant satellite SRF identified is a 358 bp repeat unit hitting 0.90% of read bases. It belongs to the 1.688 family (Khost et al. 2017). The abundance of the 358 bp repeat is lower in the BDGP6 reference genome, at 0.26% only. SRF assembled the 240 bp intergenic spacer (IGS; Shatskikh et al. 2020) into two sequences, at 240 bp and 239 bp, respectively. The edit distance between the two IGS sequences is 5. They hit to 0.43% of read bases in total but are depleted in the reference at <0.01% only. SRF also found other known satellite repeats such as (AAGAC)n, (AACAC)n, (AATAG)n, (GGTCCCGTACT)n, and (AATAACATAG)n (Shatskikh et al. 2020; Thakur et al. 2021). There are more copies of these repeat units but because they are short, they contribute less to the genome in comparison to the 1.688 and IGS satellites.
SRF reconstructed a 5045 bp repeat unit at 0.34% abundance in reads and 0.08% in the reference genome. It harbors histone genes and is located in a small region on Chromosome 2L. To investigate further, we assembled the HiFi reads using the hifiasm trio-binning mode with ISO1 and A4 short reads from SRR6702604, SRR457665, SRR457666, and SRR457707. When aligning the ISO1 haplotype assembly to the reference genome, we see a clean 242 kb insertion entirely composed of the 5045 bp histone repeat. The insertion has 48 tandem copies at >99% identity between the copies. The BDGP6 reference genome might have misassembled this region.
In maize, SRF reconstructed a 741 bp repeat unit at 0.25% abundance. It matches the SAT1_ZM record in Repbase. This SRF unit includes four copies of a 180 bp knob-associated repeat (Ananiev et al. 1998b). In the NAM-5.0 reference genome or the hifiasm assembly, this repeat tends to be present in short contigs and towards ends of long contigs. It is not assembled well. SRF also identified many potential repeat units at <0.09% abundance in reads. Nonetheless, none of them form long tandem arrays. Meanwhile, under the 151-mer setting, SRF failed to identify the 156 bp CentC repeat (Ananiev et al. 1998a). SRF could find this repeat if we counted 101-mers. Only 0.045% of read bases were mapped to CentC. Low-abundance SatDNA is harder to assemble correctly.
Comparison to TAREAN
TAREAN (Novák et al. 2017) can identify novel satellite repeats from sequence reads. Its developers recommend to use reads at up to 0.5-fold coverage to avoid redundancy between reads sequenced from the same loci. We ran TAREAN on simulated short reads at 0.2-fold from the Drosophila HiFi data set described above, without introducing additional sequencing errors. TAREAN found six high-confidence satellite repeats, including the 1.688 family and the histone cluster, (GGTCCCGTACT)n and (AATAACATAG)n. The other two TAREAN repeats also hit to SRF contigs. SRF assembled eight more SatDNA repeat units at >0.05% abundance. Manually inspecting the alignment of SRF contigs to raw HiFi reads, we observed a tandem pattern for all of them, suggesting they were real SatDNA.
To evaluate whether TAREAN can reconstruct HORs, we ran TAREAN on 0.2-fold CHM13 reads randomly sampled from SRR2088062. TAREAN took 5 h and found four high-confidence satellite repeats, including a 2-monomer alpha repeat at 1.0% abundance, an HSat2 repeat at 0.9%, a SAR satellite, and a beta satellite. TAREAN did not identify other HORs.
Satellite repeats in other species
We randomly selected 14 species from the Darwin Tree of Life project and collected two species from Hon et al. (2020) (Table 4). We assembled SatDNA in these and several other species described in earlier sections. SRF may reconstruct mitochondria or chloroplast from sequence reads. We manually removed them based on NCBI BLAST against the nt database. We then estimated the abundance of SatDNA in each of these species (Fig. 3).
Table 4.
HiFi data sets for nonmodel organisms
Figure 3.

Abundance of satellite DNA in 21 species.
Red deer (C. elaphus) has the highest abundance at 11.9%. A single 796 bp repeat unit accounts for 10.3% of satellite DNA. Killer whale (O. orca) in the same order is also enriched with satellite DNA. Yellow-legged frog (R. muscosa) is next to killer whale. SRF reconstructed many distant variants of a 131 bp repeat unit. On the other extreme, apples (M. domestica and M. sylvestris) barely have satellite repeats partly because they have transposon-rich centromeres (Zhang et al. 2019).
It is worth noting that our abundance estimate may be an underestimate because of the additional filters we used. For example, chicken mushroom (L. sulphureus) had a repeat unit of 9659 bp at 0.9% abundance. As we discarded alignments shorter than 1.5 times 9659 bp, we filtered out many HiFi reads shorter than this threshold even if entire reads were aligned to the repeat. The abundance estimate would be doubled without this filter. Such long repeat units are infrequent in the species we studied.
To investigate what satellites are organized as HOR, we ran TRF (Benson 1999) on SRF-assembled repeat motif. A repeat motif is considered to have a high-order structure if TRF identifies a tandem repeat repeating at least three times and covering 90% of the motif. With this criterion, 98.5% of human satellites are HORs with a variety of number of monomers. 8.6% of satellites in Eurasian badger (M. meles) are HORs of a 138 bp monomer, contributing to 0.14% of the genome. The other species in our survey either do not have multiple HORs composed of similar monomers or only have HORs at <0.1% abundance. Consistent with our observation, Melters et al. (2013) rarely identified HORs consisting of ≥3 monomers. The authors attributed this to the limited Sanger read length. Based on longer reads and a different algorithm, our result suggests that most species do not show rich HOR structures.
Discussion
SRF is a de novo assembler for reconstructing SatDNA repeat units and can identify most known HORs and SatDNA in well-studied species without prior knowledge on monomer sequences or repeat structures. It is the only de novo algorithm for reconstructing HORs from sequence reads as well as high-quality assemblies. SRF only depends on a third-party k-mer counter. It is easy to run and fast to execute.
SRF uses a greedy algorithm to assemble SatDNA repeat units. When two repeat units share long similar sequences, the one of lower abundance and higher diversity may be missed. We plan to improve the current algorithm by reporting multiple overlapping cycles. This may be able to find a more complete collection of HORs in the human genome.
Meanwhile, although SRF can reconstruct known HORs in human, it may report incidental HORs in species, such as mouse and A. thaliana, that only have weak high-order patterns. We need to run TRF (Benson 1999) on SRF contigs to obtain minimal repeat units. SRF may also assemble the same class of repeat into multiple similar but nonidentical copies. We can align assembled repeat units to identify such redundancy. In general, SRF only provides an initial list of repeat units but does not attempt to classify repeats into families. We recommend manual curation for a deeper insight into the SatDNA structure of a new species.
Estimating the abundance of SatDNA is challenging. Sometimes ancient SatDNA repeats may be too diverged from the assembled repeat consensus to be aligned confidently. In human, whether to count scattered monomers in pericentromeric regions as long SatDNA arrays would affect the estimate as well. In addition, occasionally SatDNA units can be >5 kb in length. We may not observe clear tandem patterns in ∼10 kb HiFi reads, which would lead to an underestimate. We do not have an automated algorithm to provide accurate abundance estimates in corner cases. The lack of ground truth in nonhuman species further complicates the evaluation of abundance estimation.
SatDNA is pervasive in many species. It is, however, often underrepresented in current reference genomes such as the human GRCh38 genome and the Drosophila BDGP6 genome. Even with improved sequencing technologies and assembly algorithms, the assembly of SatDNA is often fragmented. With thousands of species sequenced recently (Challis et al. 2020; Rhie et al. 2021) and more to come in the future, SRF may become an important tool to identify and annotate SatDNA in these species. It may also supplement RepeatModeler (Flynn et al. 2020) to provide a more comprehensive repeat library for masking SatDNA in assembled genomes.
Methods
Running SRF for human assemblies
We counted 171-mers occurring 20 times or more with KMC, using command line kmc -fm -k171 -ci20 -cs100000 and extracted the 171-mer counts with kmc_dump. SRF is directly applied to the output of kmc_dump output in the default setting.
Running SRF on sequence reads
We estimated the approximate read depth by dividing the total number of read bases by the number of bases in the reference genome or the corresponding read assembly. We counted 151-mers with kmc -fq -k151 -ciXX -cs1000000, where XX is 10 times the average read depth of each sample.
Estimating the abundance of SatDNA
We aligned reconstructed repeat units to HiFi reads or contigs with minimap2 (Li 2018), using command line minimap2 -c -N1000000 -f1000 -r100,100 <(srfutils.js enlong srf.fa), where srfutils.js is a companion script along with the SRF tool. Option -N1000000 asks minimap2 to report up to a million hits per query sequence; -f1000 considers high-occurrence seeds; -r100,100 enables a small bandwidth of 100 bp during alignment.
After the alignment, we used srfutils.js paf2bed to filter poor alignments and to merge adjacent alignments. By default, this step filters out alignments of <90% identity to the reconstructed repeat units. Users may adjust this threshold with option -d. We then used srfutils.js bed2abun to calculate the abundance of each repeat unit.
Running TAREAN
For human CHM13, we used real short reads. We ran TAREAN with singularity exec --bind ${PWD}:/data/ shub://repeatexplorer/repex_tarean seqclust -p -c 32 -r 50000000. For Drosophila, we simulated 125 bp paired-end reads from HiFi reads with dwgsim -N 146000 -1 125 -2 125 -y0 -e0 -E0 -r0 -F0 -R0. This command line did not add additional sequencing errors; the simulated reads only carried real sequencing errors on the original HiFi reads.
Data access
The SRF implementation and associated analysis scripts are provided at GitHub (https://github.com/lh3/srf) and a modified TRF with an alternative command-line interface at GitHub (https://github.com/lh3/TRF-mod). The source code is also available as Supplemental Code. Assembled repeat units and their abundance estimates can be found as Supplemental Material and at Zenodo (https://doi.org/10.5281/zenodo.8412861).
Supplementary Material
Acknowledgments
This work is supported by U.S. National Human Genome Research Institute (NHGRI) grants R01HG010040 and U01HG010961 to H.L.
Author contributions: H.L. developed the SRF algorithm. H.L. and Y.Z. validated the algorithm on public data and analyzed the result. J.C. and H.C. provided technical help and discussed the result interpretations. H.L. and Y.Z. prepared and revised the manuscript. All authors read and approved the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.278005.123.
Competing interest statement
The authors declare no competing interests.
References
- Altemose N. 2022. A classical revival: Human satellite DNAs enter the genomics era. Semin Cell Dev Biol 128: 2–14. 10.1016/j.semcdb.2022.04.012 [DOI] [PubMed] [Google Scholar]
- Altemose N, Logsdon GA, Bzikadze AV, Sidhwani P, Langley SA, Caldas GV, Hoyt SJ, Uralsky L, Ryabov FD, Shew CJ, et al. 2022. Complete genomic and epigenetic maps of human centromeres. Science 376: eabl4178. 10.1126/science.abl4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananiev EV, Phillips RL, Rines HW. 1998a. Chromosome-specific molecular organization of maize (zea mays l.) centromeric regions. Proc Natl Acad Sci 95: 13073–13078. 10.1073/pnas.95.22.13073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananiev EV, Phillips RL, Rines HW. 1998b. Complex structure of knob DNA on maize chromosome 9: retrotransposon invasion into heterochromatin. Genetics 149: 2025–2037. 10.1093/genetics/149.4.2025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora UP, Charlebois C, Lawal RA, Dumont BL. 2021. Population and subspecies diversity at mouse centromere satellites. BMC Genomics 22: 279. 10.1186/s12864-021-07591-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. 1999. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580. 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis R, Richards E, Rajan J, Cochrane G, Blaxter M. 2020. BlobToolKit - interactive quality assessment of genome assemblies. G3 (Bethesda) 10: 1361–1374. 10.1534/g3.119.400908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Concepcion GT, Feng X, Zhang H, Li H. 2021. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18: 170–175. 10.1038/s41592-020-01056-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. 2020. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci 117: 9451–9457. 10.1073/pnas.1921046117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote A, Bunskoek P, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Darwin Tree of Life Consortium. 2022. The genome sequence of the killer whale, Orcinus orca (Linnaeus, 1758). Wellcome Open Res 7: 250. 10.12688/wellcomeopenres.18278.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Yang X, Zhao X, Wang B, Ye K. 2023. HiCAT: a tool for automatic annotation of centromere structure. Genome Biol 24: 58. 10.1186/s13059-023-02900-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon T, Mars K, Young G, Tsai Y-C, Karalius JW, Landolin JM, Maurer N, Kudrna D, Hardigan MA, Steiner CC, et al. 2020. Highly accurate long-read HiFi sequencing data for five complex genomes. Sci Data 7: 399. 10.1038/s41597-020-00743-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khost DE, Eickbush DG, Larracuente AM. 2017. Single-molecule sequencing resolves the detailed structure of complex satellite DNA loci in Drosophila melanogaster. Genome Res 27: 709–721. 10.1101/gr.213512.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokot M, Długosz M, Deorowicz S. 2017. KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33: 2759–2761. 10.1093/bioinformatics/btx304 [DOI] [PubMed] [Google Scholar]
- Könyves K, Mian S, Johns J, Royal Botanic Garden Edinburgh Genome Acquisition Lab, Royal Botanic Gardens Kew Genome Acquisition Lab, Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Ruhsam M, et al. 2022. The genome sequence of the apple, Malus domestica (Suckow) Borkh., 1803. Wellcome Open Res 7: 297. 10.12688/wellcomeopenres.18646.1 [DOI] [Google Scholar]
- Kunyavskaya O, Dvorkina T, Bzikadze AV, Alexandrov IA, Pevzner PA. 2022. Automated annotation of human centromeres with HORmon. Genome Res 32: 1137–1151. 10.1101/gr.276362.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawniczak MK, Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective and Darwin Tree of Life Consortium. 2021. The genome sequence of the spiny starfish, Marthasterias glacialis (Linnaeus, 1758). Wellcome Open Res 6: 295. 10.12688/wellcomeopenres.17344.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawniczak MK, Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Darwin Tree of Life Consortium. 2022. The genome sequence of the blue-rayed limpet, Patella pellucida Linnaeus, 1758. Wellcome Open Res 7: 126. 10.12688/wellcomeopenres.17825.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2018. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34: 3094–3100. 10.1093/bioinformatics/bty191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2019. Identifying centromeric satellites with dna-brnn. Bioinformatics 35: 4408–4410. 10.1093/bioinformatics/btz264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon GA, Vollger MR, Hsieh P, Mao Y, Liskovykh MA, Koren S, Nurk S, Mercuri L, Dishuck PC, Rhie A, et al. 2021. The structure, function and evolution of a complete human chromosome 8. Nature 593: 101–107. 10.1038/s41586-021-03420-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse K, García-Berro A, Talavera G, Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Darwin Tree of Life Consortium. 2021. The genome sequence of the red admiral, Vanessa atalanta (Linnaeus, 1758). Wellcome Open Res 6: 356. 10.12688/wellcomeopenres.17524.1 [DOI] [Google Scholar]
- Lower SS, McGurk MP, Clark AG, Barbash DA. 2018. Satellite DNA evolution: old ideas, new approaches. Curr Opin Genet Dev 49: 70–78. 10.1016/j.gde.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melters DP, Bradnam KR, Young HA, Telis N, May MR, Ruby JG, Sebra R, Peluso P, Eid J, Rank D, et al. 2013. Comparative analysis of tandem repeats from hundreds of species reveals unique insights into centromere evolution. Genome Biol 14: R10. 10.1186/gb-2013-14-1-r10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga KH. 2019. Centromeric satellite DNAs: hidden sequence variation in the human population. Genes (Basel) 10: 352. 10.3390/genes10050352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga KH, Newton Y, Jain M, Altemose N, Willard HF, Kent WJ. 2014. Centromere reference models for human chromosomes x and y satellite arrays. Genome Res 24: 697–707. 10.1101/gr.159624.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naish M, Alonge M, Wlodzimierz P, Tock AJ, Abramson BW, Schmücker A, Mandáková T, Jamge B, Lambing C, Kuo P, et al. 2021. The genetic and epigenetic landscape of the Arabidopsis centromeres. Science 374: eabi7489. 10.1126/science.abi7489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman C, Tsai M-S, Buesching CD, Holland PWH, Macdonald DW, Darwin Tree of Life Consortium, University of Oxford and Wytham Woods Genome Acquisition Lab, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective. 2022. The genome sequence of the European badger, Meles meles (Linnaeus, 1758). Wellcome Open Res 7: 239. 10.12688/wellcomeopenres.18230.1 [DOI] [Google Scholar]
- Novák P, Ávila Robledillo L, Koblížková A, Vrbová I, Neumann P, Macas J. 2017. TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res 45: e111. 10.1093/nar/gkx257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, Vollger MR, Altemose N, Uralsky L, Gershman A, et al. 2022. The complete sequence of a human genome. Science 376: 44–53. 10.1126/science.abj6987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton J, Johnston SE, Fletcher TJ, Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Darwin Tree of Life Consortium. 2021. The genome sequence of the red deer, Cervus elaphus Linnaeus 1758. Wellcome Open Res 6: 336. 10.12688/wellcomeopenres.17493.1 [DOI] [Google Scholar]
- Pertile MD, Graham AN, Choo KHA, Kalitsis P. 2009. Rapid evolution of mouse Y centromere repeat DNA belies recent sequence stability. Genome Res 19: 2202–2213. 10.1101/gr.092080.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabanal FA, Gräff M, Lanz C, Fritschi K, Llaca V, Lang M, Carbonell-Bejerano P, Henderson I, Weigel D. 2022. Pushing the limits of HiFi assemblies reveals centromere diversity between two Arabidopsis thaliana genomes. Nucleic Acids Res 50: 12309–12327. 10.1093/nar/gkac1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautiainen M, Nurk S, Walenz BP, Logsdon GA, Porubsky D, Rhie A, Eichler EE, Phillippy AM, Koren S. 2023. Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat Biotechnol 41: 1474–1482. 10.1038/s41587-023-01662-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, Uliano-Silva M, Chow W, Fungtammasan A, Kim J, et al. 2021. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592: 737–746. 10.1038/s41586-021-03451-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round EK, Flowers SK, Richards EJ. 1997. Arabidopsis thaliana centromere regions: genetic map positions and repetitive DNA structure. Genome Res 7: 1045–1053. 10.1101/gr.7.11.1045 [DOI] [PubMed] [Google Scholar]
- Ruhsam M, Bell D, Hart M, Hollingsworth P, Royal Botanic Garden Edinburgh Genome Acquisition Lab, Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Darwin Tree of Life Consortium. 2022. The genome sequence of the European crab apple, Malus sylvestris (L.) Mill., 1768. Wellcome Open Res 7: 296. 10.12688/wellcomeopenres.18645.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen H-C, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. 2017. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27: 849–864. 10.1101/gr.213611.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevim V, Bashir A, Chin C-S, Miga KH. 2016. Alpha-CENTAURI: assessing novel centromeric repeat sequence variation with long read sequencing. Bioinformatics 32: 1921–1924. 10.1093/bioinformatics/btw101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatskikh AS, Kotov AA, Adashev VE, Bazylev SS, Olenina LV. 2020. Functional significance of satellite DNAs: Insights from Drosophila. Front Cell Dev Biol 8: 312. 10.3389/fcell.2020.00312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinding M-HS, Gopalakrishnan S, Raundrup K, Dalén L, Threlfall J, Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Darwin Tree of Life Consortium, et al. 2021. The genome sequence of the grey wolf, Canis lupus Linnaeus 1758. Wellcome Open Res 6: 310. 10.12688/wellcomeopenres.17332.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur J, Packiaraj J, Henikoff S. 2021. Sequence, chromatin and evolution of satellite DNA. Int J Mol Sci 22: 4309. 10.3390/ijms22094309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Yang X, Jia Y, Xu Y, Jia P, Dang N, Wang S, Xu T, Zhao X, Gao S, et al. 2022. High-quality arabidopsis thaliana genome assembly with nanopore and HiFi long reads. Genomics Proteomics Bioinformatics 20: 4–13. 10.1016/j.gpb.2021.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei KH-C, Grenier JK, Barbash DA, Clark AG. 2014. Correlated variation and population differentiation in satellite DNA abundance among lines of Drosophila melanogaster. Proc Natl Acad Sci 111: 18793–18798. 10.1073/pnas.1421951112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood C, Bishop J, Harley J, Mrowicki R, Marine Biological Association Genome Acquisition Lab, Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Darwin Tree of Life Consortium. 2022. The genome sequence of the orange-striped anemone, Diadumene lineata (Verrill, 1869). Wellcome Open Res 7: 93. 10.12688/wellcomeopenres.17763.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright R, Woof K, Douglas B, Gaya E, Royal Botanic Gardens Kew Genome Acquisition Lab, Darwin Tree of Life Barcoding collective, Wellcome Sanger Institute Tree of Life programme, Wellcome Sanger Institute Scientific Operations: DNA Pipelines collective, Tree of Life Core Informatics collective, Darwin Tree of Life Consortium. 2022. The genome sequence of the chicken of the woods fungus, Laetiporus sulphureus (Bull.) Murrill, 1920. Wellcome Open Res 7: 83. 10.12688/wellcomeopenres.17750.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. 10.1101/gr.074492.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Hu J, Han X, Li J, Gao Y, Richards CM, Zhang C, Tian Y, Liu G, Gul H, et al. 2019. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat Commun 10: 1494. 10.1038/s41467-019-09518-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.