

Abstract
Transcription and replication both require large macromolecular complexes to act on a DNA template, yet these machineries cannot simultaneously act on the same DNA sequence. Conflicts between the replication and transcription machineries (transcription-replication conflicts, or TRCs) are widespread in both prokaryotes and eukaryotes, and have the capacity both to cause DNA damage and to compromise complete, faithful replication of the genome. This review will highlight recent studies investigating the genomic locations of TRCs and the mechanisms by which they may be prevented, mitigated or resolved. We address work from both model organisms and mammalian systems, but predominantly focus on multicellular eukaryotes due to the additional complexities inherent in the coordination of replication and transcription in the context of cell-type-specific gene expression and higher-order chromatin organization.
Keywords: Transcription-replication conflicts, TRCs, genomic instability, replisome, RNA polymerase, DNA repair, origin firing, replication stress, transcription
1. Introduction
Transcription-replication conflicts (TRCs) can occur in two orientations: co-directional (CD) or head-on (HO) (Figure 1A). CD-TRCs represent encounters between the DNA replication machinery (known as the replisome) and RNA polymerase (RNAP) moving in the same direction on DNA, while HO-TRCs occur when the replisome and RNAP are moving towards one another. Most models assume that CD-TRCs are normally the result of replisomes ‘rear-ending’ transcription: this is likely to be almost invariably true in bacteria, since prokaryotic replisomes move many times faster than their RNA polymerase and CD-TRCs are widespread (14). In eukaryotes, RNA polymerase 2 (RNAP2) and the replisome move at approximately equal bulk rates with stochastic variability (20, 90). Therefore, both permutations for CD-TRCs are likely possible in eukaryotic genomes (Figure 1A). Early studies of TRCs in bacteria show that HO-TRCs compromise replication-fork progression more severely than CD-TRCs (68). Similarly, yeast studies show that HO-TRCs impede replication fork progression, leading to elevated DNA breaks, transcription-associated recombination (TAR), and co-transcriptionally formed RNA:DNA hybrids (R-loops) and related genomic instability (39)
Figure 1.
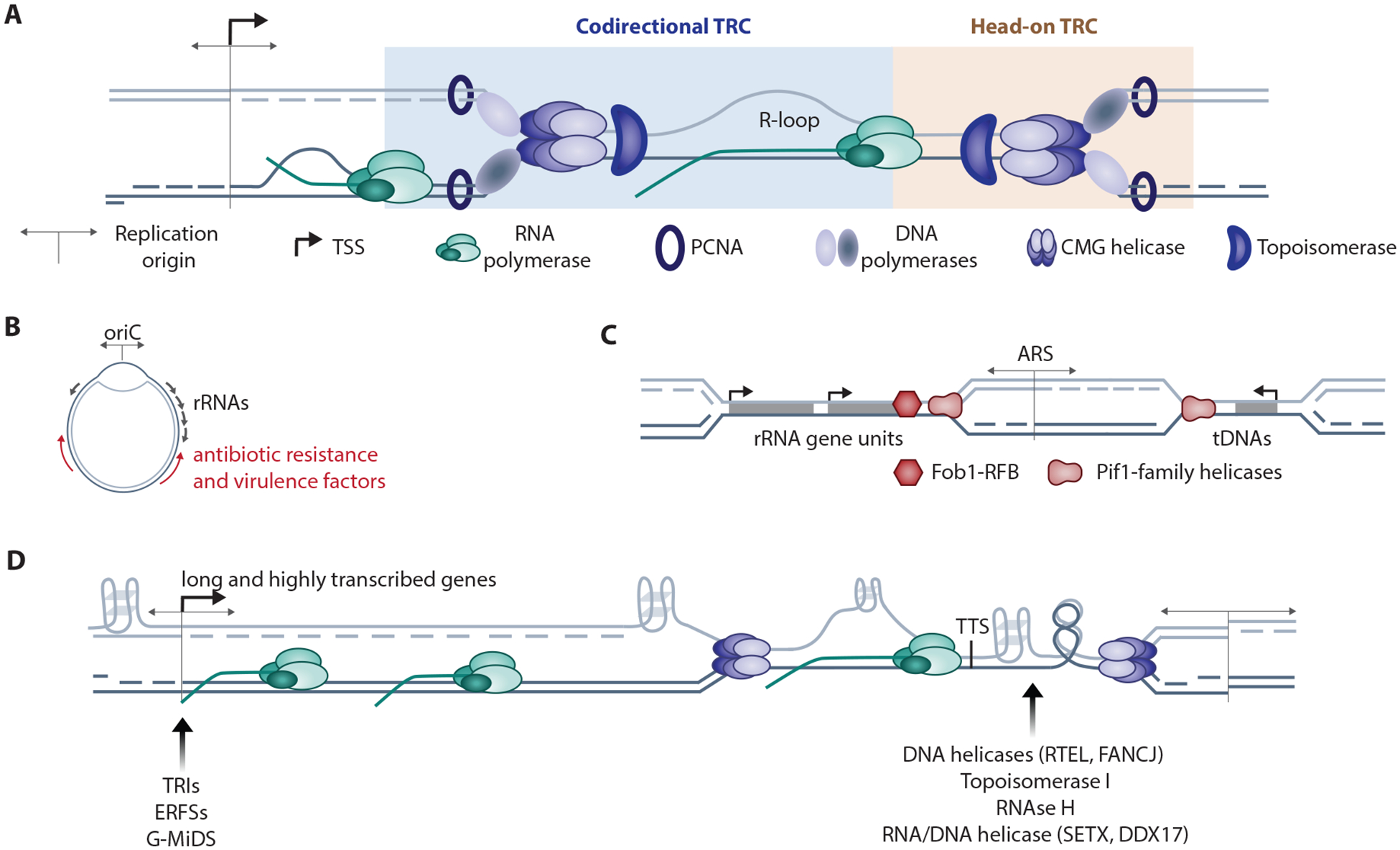
Genome organization and transcription-replication collisions (TRCs) across species.
A. Codirectional (CD) TRCs occur between the replisome and RNA polymerase (RNAP) oriented in the same direction on leading-strand DNA. CD-TRCs can occur in to permutations: RNAP following the replisome on newly-synthesized DNA (Codirectional TRC – 1), or the replisome following RNAP (Codirectional TRC – 2). Head-on (HO) TRCs occur between the replisome and RNAP oppositely oriented on lagging-strand DNA. Co-transcriptional R-loops, RNA:DNA hybrids, are linked to TRCs.
B. Bacterial genomes contain a single replication origin (oriC), and the most highly transcribed and essential genes are oriented codirectionally with oriC (ref). HO-oriented genes accumulate mutations, but are essential for bacterial survival. Specifically, lagging-strand genes are enriched antibiotic resistance and virulence factors.
C. Yeast origins occur at sequence-specific ARS sequences, and forks are blocked at the 3’ end of rRNA gene units by Fob1-replication fork barriers (RFBs). Pif1-family helicases are required for progression through programmed RFBs in yeast, and more generally the termini of highly-transcribed gene units such as tRNA genes.
D. Replication origins occur preferentially at transcription start sites (TSSs) of long and transcriptionally-active genes in mammalian cells, facilitating codirectional movement through gene bodies. CD-TRCs at TSSs could be endogenous sources of DNA damage and replication stress in murine lymphocyte models (TRIs as in REF, ERFSs as in REF), or regions of mitotic replication (G-MiDS as in REF). G4 quadruplex structures are enriched at TSSs, on lagging strand DNA, and on persistent R-loops. G4s, R-loops, and topological stress are barriers to replication fork progression. Helicases (DNA and RNA:DNA), topoisomerases, and RNases promote fork progression through unprogrammed barriers.
Both the propensity for TRCs and the diversity of possible conflicts differs greatly between prokaryotic and eukaryotic genomes. In E. coli, the single circular chromosome has one replication origin (oriC), and both replication and transcription are regulated primarily in response to external stimuli (e.g. nutrient availability). Mammalian genomes (with more than 200 different cell types in the human body) have complex replication and transcriptional programs that complicate the temporal and spatial regulation of when, where, and how TRCs occur. Tens of thousands of flexible replication origins are licensed in excess to ensure complete duplication of the entire genome prior to cell division. Additionally, the mammalian transcriptome is regulated by a dynamic and complex network of transcription factors and cis-regulatory elements that influence expression of genes that vary greatly in transcription efficiency and gene length. Temporal separation alone of replication and transcription does not dramatically limit TRCs. Although replication is confined to S-phase, transcription occurs throughout the entirety of the cell cycle (6). Transcription is high during G1-S due to CDK phosphorylation of Rb, which releases E2F transcriptional factors that promote transcription of cyclins/CDKs, housekeeping genes, and periodically transcribed products(6). Histone gene expression levels peak during S-phase to accommodate newly synthesized DNA. Additionally, some long genes take more than one cell cycle to transcribe (49). Therefore, RNAPs persist on DNA throughout replication, but how the coordination of these processes prevents detrimental collisions and genomic instability remains unclear and is an active area of research.
Within S-phase, the spatiotemporal separation of replication and transcription could represent a simple mechanism to limit TRCs via mutually exclusive chromatin occupancy. Euchromatin replicates early and heterochromatin replicates late (reviewed in (83), and the DNA replication and transcriptional machineries are spatially anticorrelated in both early and late S-phase (141). Additional recent high-resolution live-cell imaging shows a global anti-correlation of transcriptionally elongating RNAP2 with PCNA – a core replisome component (136). Sequencing of nascent transcripts in S-phase demonstrated that periodically transcribed genes in early-replicating regions are transcribed late, and vice versa (88), providing further support for a global separation of replication and transcription in mammalian cells as a general mechanism for limiting TRCs in dividing cells. However, spatiotemporal separation of replication and transcription outlined above is almost certainly non-binary. Early genome-wide studies predicting replication fork dynamics from nucleotide skew proposed that mammalian replication origins occur preferentially at transcriptional units in an expression-dependent manner (53, 132). More recent genome-wide replication-origin mapping has consistently shown that early-firing origins occur at or near transcription start sites (TSS) of genes with high transcriptional activity (106). Endogenous replication origins in human cells are associated with RNAP2 occupancy, G4-quadruplex structures, GC-skew, CpGs, and open chromatin, all of which are linked to increased transcriptional activity (7, 70, 106, 149). The coordination of tens of thousands of origins around tens of thousands of potentially active transcriptional units throughout gigabases of DNA in higher eukaryotes suggests that TRCs likely occur frequently in the course of a normal cell cycle. Despite recent progress, the specific mechanisms of how TRCs are negotiated and mitigated, and the mechanisms by which aberrant TRCs contribute to genomic instability, remain unresolved.
Many studies investigating TRCs have done so via the direct analysis of replication-fork dynamics: 2D-gel electrophoresis of reporter genes reveals locus-specific replisome pausing (105), while DNA fiber analysis shows non-specific, global changes in fork speed, origin density, and fork asymmetry (115). The development of several novel sequencing techniques such as Ok-seq (21, 100), INI-seq (44, 70), SNS-seq (7), Repli-seq (149), and Pu-seq (62) has permitted genome-wide mapping of replication origin usage, fork directionality, and replication termination, greatly increasing our understanding of the global dynamics of DNA replication in normal and perturbed conditions. Additionally, a large body of work has inferred the existence of TRCs by assaying genomic instability in response to dysregulated replication and/or transcription (39, 46). A genuinely direct analysis of TRCs has proven to be more challenging. Proximity ligation assays between the replication and transcription machineries (45) are sensitive but lack DNA sequence information for genome site-specific TRCs. Recent works aligning genome-wide ChIP-seq of DNA damage markers alongside replication-fork mapping suggested possible locations where deleterious TRCs might occur (107), but few studies have probed for direct transcription-replication colocalization or established direct and irrefutable mechanisms linking specific TRCs to genomic instability. The work here will highlight recent studies elucidating how endogenous TRCs are prevented through genome organization, and how both CD- and HO-TRCs are thought to contribute to genomic instability. We will also outline key studies exploring how replication and transcription dysregulation exacerbate TRCs. We note that several comprehensive reviews have been written on TRCs in the past few years; please refer to these excellent reviews that cover additional critical work not discussed here due to space constraints (39, 46, 64, 143).
2. Genome organization and TRCs
2.1. Evolutionary selective pressure for co-directional TRCs in highly transcribed genes
The organization of small genomes from rapidly replicating organisms shows an evolutionary selective pressure for CD-TRCs and against HO-TRCs in a gene expression-dependent manner. The circular genome of Phi X 174 bacteriophage is entirely codirectionally oriented with replication fork progression, and the linear bacteriophage T7 genome is almost entirely unidirectionally transcribed, with the origin of replication within the first 15% of its 5’ end: the handful of genes in the T7 phage genome that are upstream of the replication origin are early genes transcribed by host RNAP before viral replication occurs (14). In E. coli most highly transcribed bacterial operons, including rRNA gene arrays and nearly 90% of genes involved in DNA, RNA, and protein synthesis, all run in the same direction relative to replication forks emanating from oriC.(14) (Figure 1B). While this suggests that HO-TRCs are disfavored in order to minimize genomic instability, recent work suggests that HO-TRCs may represent a mechanism to potentiate rapid genomic evolution and enhance pathogen survival under stress. Evidence from E. coli and B. subtilis suggests that a gene’s essentiality, as opposed to solely its expression level, can drive evolutionary selection for CD- and HO-gene orientation in bacteria (111). Therefore, the majority of essential genes are oriented codirectionally with oriC, and the 17% of essential genes found on the lagging strand in B. subtilis have increased nonsynonymous point mutations in a transcription- and length-dependent manner(97). Moreover, across several bacterial species genes oriented head-on with respect to replication are enriched for virulence and antibiotic resistance factors, likely the result of randomly inverted codirectional operons (87). These data are consistent with a model in which genomic instability from HO-TRCs is tolerated to an extent in order to preserve mutagenic capacity and genetic flexibility in rapidly dividing and evolving bacteria.
Evidence for a gene orientation bias in budding yeast, a long-standing eukaryotic model system for TRCs (30) is more mixed than for prokaryotes and bacteriophage. S. cerevisiae tRNA genes, which are highly transcribed by RNAP3, lead to substantially more replication-fork stalling (93) and R-loop-dependent gross chromosomal rearrangements (133) in the HO orientation relative to the CD orientation. Consistent with HO-TRCs being more deleterious than CD, tRNA gene orientation is strongly biased such that CD-TRCs will predominate (93). However, a significant fraction of S. cerevisiae tRNA genes are found in the HO orientation, and no detectable orientation bias exists for protein-coding genes transcribed by RNAP2 in the yeast genome despite the association of very highly transcribed genes with replisome pausing (24) and mitotic recombination (105).
Replication origin firing in both bacteria and yeast occurs efficiently at fixed DNA sequences. A higher propensity for DNA breaks at HO-TRCs would, over time and in response to cellular adaptation, presumably reorient problematic HO genes to favor codirectional movement in an expression-dependent manner. However, such sequence-specific fixed replication origins would likely pose a challenge for multicellular eukaryotes with diverse cell-type-specific transcriptomes. Initiation of DNA replication immediately upstream of every active TSS would naturally co-orient replication with transcription, minimizing both HO- and CD-TRCs due to the matched speeds of replication and transcription. Indeed, a significant body of evidence is consistent with cell-type specific origin definition in metazoa occurring via the licensing of replication origins (by deposition of the MCM2–7 complex) upstream of active genes (79, 104, 118) to facilitate codirectional movement in highly transcribed regions (44). The most efficient replication origins in human cells appear to be conserved across cell types, although these origins differ in firing efficiency in a cell-type dependent manner (7) and are supplemented by stochastically firing cell-type specific origins (2). These conserved core origins contain a signature G-rich sequence(2) that could represent a vestige of ancestral sequence-specific origin definition. Thus, the genome-wide organization of replication and transcription across multiple species suggests a conserved evolution for CD movement of transcription and replication. However, recent single-molecule analysis of origin firing demonstrates that initiation events are largely stochastic in human cells (140); therefore, a replication program that on average minimizes the propensity for HO-TRCs may still generate a large number of such conflicts in any given cell in a population.
Replication origin firing does not occur immediately after origin licensing, allowing time for RNAPs to modulate the location of excess MCM2–7 complexes loaded in late G1. RNA polymerases can relocate topologically loaded MCM proteins both in vitro and in vivo, although extensive MCM2–7 repositioning may be associated with a reduction in origin firing efficiency (43, 104, 118). Consistent with the clearance of intragenic MCMs by RNAP2 in unperturbed human cells, replication origins are depleted within transcriptionally-active gene bodies (2, 82) but enriched downstream of genes (21, 62), either at a gene’s transcription termination site (TTS) or the nearest downstream TSS. Whether or not these downstream origins are indeed predominantly licensed by mobile MCMs, and whether they lead to HO-TRCs, remain as open questions; additional work will be needed to define both the distance over which MCMs can be moved in the context of a chromatinized genome, and the extent to which MCM repositioning impacts the ability of this complex to recruit origin firing factors. Intuitively, if MCM2–7 can be extensively loaded within gene bodies and subsequently repositioned by RNAP, a substantial fraction of licensed origins – perhaps even the majority – would lie downstream of the most highly transcribed genes. Given that replication predominantly initiates upstream of such genes, it is likely that additional factors regulate replication-origin firing. An understanding of whether dynamically redistributed origin licensing factors are linked to dormant origin firing or oncogene-induced origin firing could reveal new mechanisms of replication-stress tolerance to ensure local DNA replication (discussed in section 3).
2.2. How are endogenous HO-TRCs prevented and resolved?
HO-TRCs, even if deleterious to replication fork progression and genomic integrity, are inevitable in complex genomes due to stochastic origin firing. In human cells, there is no bias for origin firing at divergent, tandem, or convergent gene neighbors (100), with the exception of a preference for efficient origin firing in the context of closely spaced (<5kb) transcriptionally active divergent genes. Thus, replication-origin firing upstream of a transcriptionally active gene has the potential to induce a HO-TRC in one or more neighboring genes. Furthermore, genome-wide analysis replication-fork direction by both Ok-seq (21) and Pu-seq(62) analysis has shown that the use of replication origins downstream of transcribed genes – another potential cause of HO-TRCs – is widespread in unperturbed cells. Programmed replication-fork barriers (RFBs), for example the Tus-Ter system in E. coli (113), and the binding of Fob1 to the RFB sequence in yeast (51) (Figure 1C), represent a simple mechanism to prevent HO-TRCs in specific regions of the genome. RFBs are programmed protein-DNA complexes that unidirectionally block replication-fork progression and thereby allow only those forks moving codirectionally with transcription to complete replication of the downstream region. However, with the exception of an intriguing study suggesting that polar poly(AT) tracts may function as endogenous blocks to fork progression at rRNA termini and other sites in replicating murine B-cells (135), there is little evidence that programmed RFBs area common strategy to prevent HO-TRCs in higher eukaryotes.
Failure to progress through non-programmed blocks to DNA replication is associated with genomic instability: in yeast, the partially redundant Pif1 and Rrm3 helicases function genome-wide to promote fork progression through R-loops, G-quadruplex structures (G4s), and highly-transcribed gene units (28, 63, 117); loss of PIF1-helicases leads to severe fork arrest, R-loop-dependent gross chromosomal rearrangements, and increased point mutagenesis (93, 133, 150) (Figure 1C). RNAP, R-loops, and G4s appear to be able to serve as barriers to replication-fork progression in yeast and mammalian cells (39, 73, 147). G4s also stabilize GC-rich R-loops, which are enriched at gene termini (123). In human cells, the RTEL helicase is required for resolution of G4s on R-loops in order to protect against TRCs and replication stress (60); FANCJ also exhibits G4-unwinding activity necessary for replication fork progression, and numerous other Fanconi complex proteins have been extensively implicated in preventing R-loops, TRCs, and TRC-related genomic instability (12, 72, 115, 119, 144, 146) (Figure 1D). We anticipate that future work in mammalian systems will define both the extent to which, and the mechanisms by which, transcription-associated secondary structures compromise replication-fork progression in gene bodies and at gene termini.
Topoisomerases can prevent genomic instability associated with G4s, and additionally promote fork progression through protein-DNA barriers (122, 145) (Figure 1D). Genome-wide mapping of TRC-induced genomic instability in human cells demonstrated that R-loops and topological stress are linked to dysregulated replication fork progression at gene termini: ATR and topoisomerase 1 protect against R-loops, replication stress, and DNA-breaks at TTSs of highly transcribed genes (107). HO-TRCs are likely particularly prone to topological stress due to the additive downstream supercoiling in front of two processive polymerases, whereas negative supercoiling behind polymerases would create a net neutral torsional stress in CD-TRCs (Figure 1D). Indeed, in the model prokaryote B. subtilis, type II topoisomerases relieve R-loop and replication-associated torsional stress (69), and B. subtilis cells with engineered HO-TRCs are sensitive to topoisomerase inhibitors. Studies interrogating the impact of HO- vs CD-TRCs are more challenging to directly engineer in mammalian genomes, limiting the extent to which the implications of this work have so far been extended to mammalian genomes.
It is unclear whether replication-forks approaching the 3’ ends of genes pause at gene termini or enter gene bodies, and whether HO replication of these regions leads to direct contact between the replication and transcription machinery. Ok-seq suggests that transcription plays a role in orienting replication termination: replication termination is enriched at TTSs of highly transcribed genes, consistent with a possible role for RNAP in blocking progression of downstream forks into genes, thereby promoting genic replication by codirectional movement (21). However, hydroxyurea-induced dormant origin firing causes replication termination within gene bodies, so this transcriptional barrier might be permissible to an extent. Future work uncovering the specific mechanisms of transcription-dependent replication termination could shed light on how TRCs are endogenously prevented or resolved.
2.3. Endogenous CD-TRCs can compromise genomic instability
RNAP pausing and backtracking are inherent mechanisms of transcriptional regulation(92). RNAP pausing is positively correlated with R-loop formation, and various R-loop sequencing methods suggest that R-loops are most prevalent at TSSs and TTSs of highly transcribed genes (19, 116). Therefore, endogenous CD-TRCs are likely prevalent even during unperturbed DNA replication in mammalian cells, and dysregulation of either replication or transcription could further increase the frequency of such CD-TRCs as well as inducing ectopic HO-TRCs (discussed in section 3 and 4).
In an episomal system in human cells, CD-TRCs induced DNA breaks and ATM-checkpoint activation, distinct from ATR activation by an analogous HO-TRC in the same system (45). In a genomic context, a recent analysis in human cancer cells by Koyanagi and colleagues showed variation in replicative polymerase usage consistent with widespread leading- and lagging-strand polymerase uncoupling within gene bodies in a transcription- and length-dependent manner (62). Polymerase uncoupling was observed to be strongest at TSSs and TTSs and present in both conflict configurations (HO and CD). These results suggest that a basal level of fork uncoupling might be tolerated in cancer cell lines with dysregulated checkpoint function, and perhaps to a lesser extent in unperturbed cells.
Recently, St Germain and colleagues developed a novel technique for genome-wide mapping of TRCs. TRIPn-seq (Transcription–Replication ImmunoPrecipitation on Nascent DNA Sequencing) combines immunoprecipitation of nascent BrdU-labeled DNA with RNAP2-pSer5 ChIP, followed by sequencing. This technique was used to identify regions co-occupied by Ser5-phosphorylated RNAP2 and nascent DNA in mouse primary B-cells (38). The Ser5 phospho-form of RNAP2 is most prevalent at TSS of highly transcribed genes, and consistently, the majority of TRIs (transcription-replication interactions) detected by this technique were found to be TSS-proximal and in early-replicating regions of the genome (Figure 1D). While at least some of the TRIs identified may represent transcription of newly synthesized DNA, it is likely that many reflect bona fide TRCs. Because origin firing is suppressed within gene bodies (81), TRCs identified by TRIPn-seq are most likely co-directional. TRIs are enriched for divergent gene pairs, consistent with previous reports of RNAPs at divergent genes contributing to genomic instability through excessive negative supercoiling and topological stress behind two opposed replisomes (94). TRIs are sites of spontaneous DSBs, high GC-skew, increased G4-quadruplex formation, and increased RPA association with DNA under replication stress. These results provocatively suggest that a subset of TSS’s are associated with replication origins from which at least one resulting replication-fork that commonly engages in a CD-TRC, leading to DNA damage.
TRIs mapped by TRIPn-seq are reminiscent of early-replicating fragile sites (ERFSs), which were also characterized in a mouse B-lymphocyte models (4). Like TRIs, ERFSs undergo spontaneous DNA breaks in a transcription- and replication-dependent manner (Figure 1D). Genomic instability at ERFSs is exacerbated by inhibition of ATR, suggesting that ATR signaling mitigates a basal level of DNA-damage associated with TRCs in unperturbed cells. It’s important to note that B-lymphocytes have endogenously high levels of DNA damage because they rely on R-loops and DNA breaks for AID-dependent recombination and generation of diverse immunoglobulins, but AID can generate double-strand breaks at sites other than immunoglobulin genes (40).
Recent work from Wang and colleagues suggests that TRCs at TSSs can also be associated with under-replication of short genomic regions in human cell lines, but that such under-replication is well tolerated (139) (Figure 1D). BrdU-seq and Repli-seq in unperturbed human fibroblasts show that a narrow window of DNA (< 5kb) at TSSs of highly-transcribed and long genes remains under-replicated throughout the majority of S-phase, with replication completed only in late G2/M. Strand-specific Ok-seq analysis in human retinal epithelial cells (RPE-1) was consistent with replication fork uncoupling at TSSs of long genes, and under-replication of the lagging strand. The G-MiDS sites (G2/M DNA synthesis) identified in this study are distinct from mitotic DNA synthesis (MiDAS) and TRIs in that they do not appear to be associated with genomic instability.
It is unclear what distinguishes a benign CD-TRC from one that leads to DNA damage. Future work across several other mammalian systems will hopefully shed light on this important unresolved question. It is possible that the presence or absence of multiple RNAP2 molecules at a conflict site may play a role in determining the outcome of a CD-TRC – i.e. that the number of RNAPs ahead of (or behind) a replisome would correlate with the impediment to replication fork progression and/or DNA breakage. Indeed, CD-TRCs in an E. coli model show increased nascent DNA gaps in an RNAP/R-loop array-dependent manner (16). The precise disposition of RNA polymerase may also be important. For example TRC-dependent DNA double-strand breaks in E. coli were found to be specifically associated with backtracked RNA polymerase in the context of CD-, but not HO-TRCs (33): to our knowledge, there is as yet no evidence for an analogous effect of RNAP backtracking on TRC outcome in eukaryotic genomes.
2.4. TRCs occur at common fragile sites
Common fragile sites (CFSs) are highly-conserved large genomic regions (kb to Mb) interspersed throughout mammalian genomes and characterized by increased DNA breaks and recombination, which can, in turn, lead to chromosomal rearrangements and copy-number variation (32). CFSs are highly sensitive to replication stress (32), and are enriched for long genes (>300 kb) that take more than one cell cycle to transcribe (49). Consistent with a direct role for TRCs in CFS instability, DNA breakage at these sites depends on the presence of active transcription and R-loops, and is mitigated by endogenous RNaseH1 (49, 115). CFSs are also late-replicating and reside in origin-poor regions of the genome. These regions rely on long-range moving replication forks for faithful DNA replication, and are at risk of under-replication, MiDAS, and/or DNA breakage (9). Origin density within CFSs and CFS instability is not conserved between cell types or even between cell generations of the same identity (74, 75); this is likely due to transcriptome-specific orientation of replication origins. CFS instability is not linearly correlated to transcriptional activity of long CFS genes: lowly transcribed long genes with weak promoters are more prone to destabilization than highly transcribed long genes with strong promoters (10). This instability is driven by under-replication of large genes whose origins fire too late in S-phase and cannot complete replication (15). Therefore, it is possible that even low levels of transcription or subtle changes to transcription programming can alter replication origin usage and cause genomic instability. Replication of CFSs could also be compromised by RPA exhaustion in settings of elevated levels of replication stress (129).
In addition to low origin density, CFSs are sensitive to replication stress due to AT-rich sequences that increase the propensity for non-B-form DNA structures in these regions. Artificial insertion of an AT-dinucleotide repeat sequence into a stable region of the genome causes increase non-B-form DNA structures that exacerbate replication stress and DNA breaks (54). Conversely, long, homopolymeric poly(dA:dT) tracts are sites of collapsed DNA replication forks that flank CFS genes (135). In this model, replication fork impairment and collapse are driven by secondary structure formation of poly(dA), evidenced by a lack of RPA-coated ssDNA in these regions. In yeast, long poly(dA:dT) tracts are also R-loop prone, suggesting that transcription and TRCs could contribute to AT-rich secondary structure instability (138)
3. TRCs caused by DNA replication stress
Given that DNA replication and transcription must be coordinated, it stands to reason that dysregulation of either process has the potential to disrupt this coordination – and therefore to compromise genomic integrity via mechanisms that include, but are not necessarily limited to, increased TRCs. Replication stress leads to aberrant origin firing and stalled replication forks, both of which can increase the propensity for TRCs (section 3.1). Additionally, recent studies have revealed multiple mechanisms of replication recovery following fork stalling with transcription machinery (section 3.2), including direct RNAP removal (section 3.3).
3.1. TRCs are caused by aberrant origin firing
Replication origins are licensed in excess in late G1, but only a subset of euchromatic origins will be activated in early S to generate productive replication forks. Excess origin licensing safeguards against replication stress (37) by ensuring that nearby dormant origins can be activated to ensure complete replication after fork stalling, facilitated by ATR (23). Dormant replication origins do not appear to be distinct from constitutive origins, but rather are distinguished by their passive usage when the forks coming from constitutive origins stall or fail to fire (20). Fork progression and origin density are tightly linked: defects in fork progression (fork slowing or stalling) lead to concomitant increase in dormant origin firing. The pool of MCM2–7 complexes loaded onto DNA and available for dormant origin firing may be at least partly positioned by RNAP2 (43, 118) or pushed ahead of the replication fork consistent with ‘trains’ of MCM complexes observed in reconstituted systems (50).
Depletion of nucleotide pools by low-dose hydroxyurea treatment in human cells causes compensatory origin firing immediately downstream of TTSs of highly-transcribed genes, although it is unclear whether this TTS-proximal origin firing would potentiate HO-TRCs if occurring solely due to replication-fork slowing (20, 21). In contrast to HU-induced fork stalling, Cyclin E overexpression causes replication stress through premature S-phase entry and aberrant replication initiation; such unscheduled origin firing has the potential to increase TRCs, and to deplete or exhaust nucleotide pools and RPA, resulting in slow and/or stalled replication-forks (55). Cyclin E-induced replication stress is transcription- and R-loop dependent (55). Surprisingly, EdU-seq analysis demonstrated that rapid induction of Cyclin E overexpression in human cells induces aberrant origin firing in the middle of protein-coding genes (82). Intragenic origin firing is associated with DSBs and break-induced translocations in human cells, consistent with Cyclin E1 overexpression as a driver for oncogenesis in multiple cancers, including p53 wild-type models that tolerate Cyclin E1-induced whole-genome duplication via a mechanism unrelated to TRCs (148). Intragenic origins induced by Cyclin E1 overexpression compared to intergenic origins downstream of TTSs induced by HU-induced dormant origin firing is likely dependent on the time available for RNAP2 to redistribute MCMs in low-dose HU, orienting them towards gene termini (Figure 2A); this time window is shortened in Cyclin E1 overexpression, and MCM loading inversely proportional to G1 length is uncoupled in Cyclin E1 overexpression (85). However, recent work in yeast suggests premature S-phase entry alone does not cause new origins to fire and does not cause TRCs (110). In an engineered yeast model with induced CDK/DDK regulation-bypass mechanisms allows for premature activation and replication in G1. Unscheduled G1 replication initiation increased canonical origin usage and causes genomic instability through over-replication and head-to-tail replication-fork collisions, suggesting that uncoordinated firing of new origins may be required for TRCs (Figure 2A). Future work continuing to elucidate how premature S-phase entry, Cyclin E1 overexpression, and dormant origin firing distinctly cause genomic instability could reveal novel mechanisms of TRCs.
Figure 2.
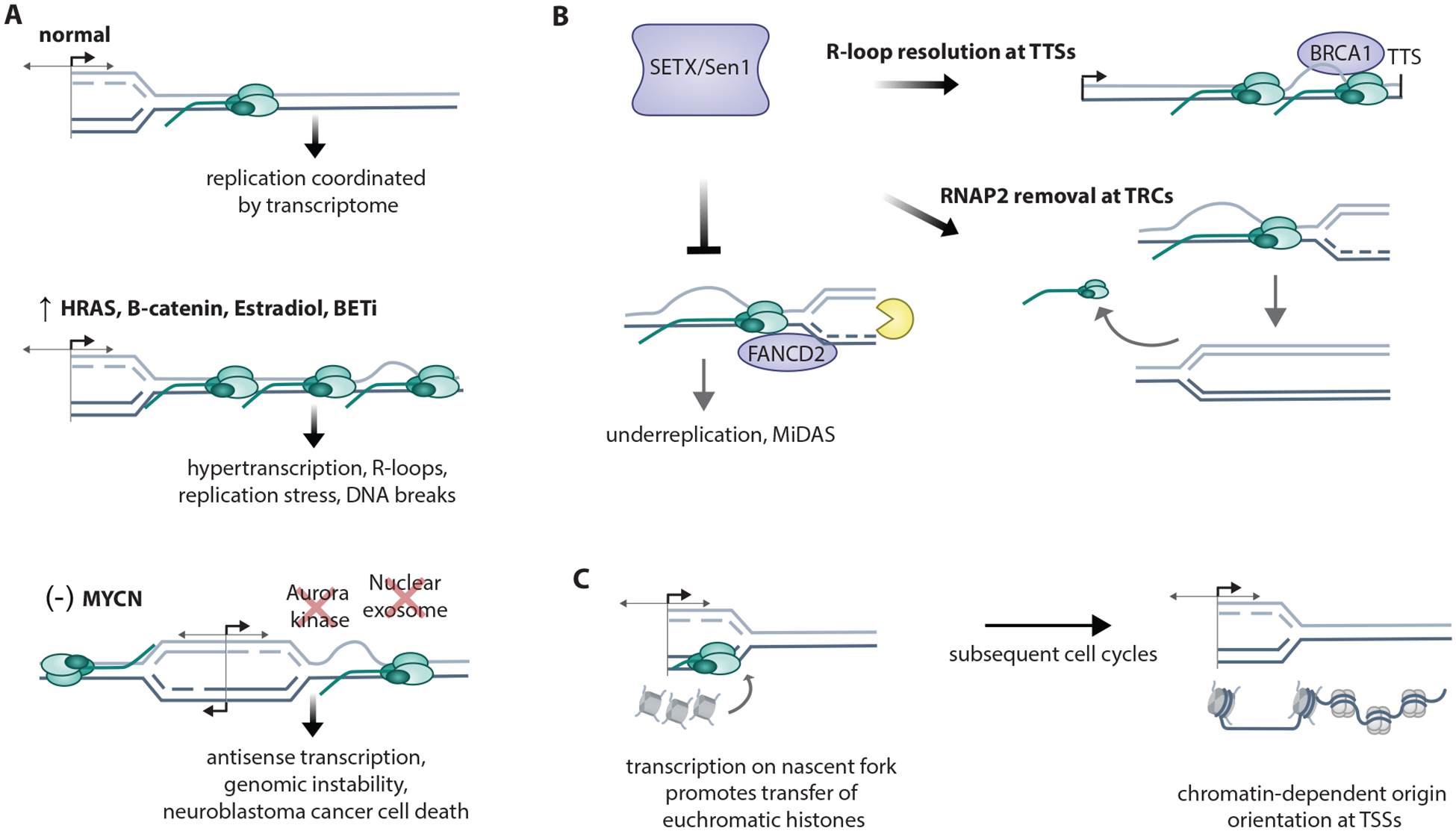
Replication dysregulation causes TRCs
A. Short G1 and premature S-phase entry causes increased usage of canonical origins and over-replication leading to double strand breaks (DSBs) resulting from head-to-tail-fork collisions from mixed G1- and S-phase forks. (Pfander ref) Cyclin E1 overexpression causes intragenic origin firing and TRCs linked to fork collapse and genomic rearrangements (Halazonetis ref), as the result of incomplete MCM redistribution by transcription machinery. In contrast, low-dose HU causes dormant origin firing at transcription termination sites of highly-transcribed genes due to MCM clearance of gene bodies by transcription.
B. Several pathways of replication fork recovery follow TRCs. Replication fork restart by MUS81 fork cleavage leads to RAD52/LIG4/POLD3-dependent fork religation. Replication fork reversal is triggered by MUS81 cleavage, which activates ATR, which limits excessive MUS81 cleavage. Replication fork repriming past R-loop and G4 secondary structures requires PRIMPOL (primase polymerase). Local RNAP removal by ATAD5, RECQ5, or PNUTS resolves TRCs.
3.2. Replication stress response mitigates TRCs
TRCs, particularly HO-TRCs, stall replication forks (39, 46, 68), and both fork reversal and bypass mechanisms have been shown to resolve TRCs (discussed herein). Both of these fork recovery processes are mediated by replication stress response and checkpoint activation pathways (23). Replication fork recovery relies on chromatin remodelers for fork reversal, the HR machinery for fork protection against nucleases, and translesion synthesis polymerases for fork restart (31, 108, 130). Lesions impeding replication forks can be bypassed by translesion synthesis polymerases and Fanconi proteins (109). In yeast, fork reversal and repriming pathways are mutually exclusive: however, pathway choice is not as well understood in mammalian models and might be influenced by the availability of specific DNA repair and DNA polymerase factors, and the type of lesion encountered by the replisome (35, 109).
Replication fork reversal facilitates resolution of R-loop-induced TRCs by promoting fork recovery past RNAP and R-loops: MUS81 endonuclease-mediated ATR activation responds to excessive R-loop accumulation, replication stress, and TRCs (84). In this model, ATR coordinates replication stress response to negotiate R-loop-induced TRCs by orchestrating replication recovery through limiting excessive MUS81 nuclease activity and upregulating the G2/M checkpoint to prevent further TRCs (84) (Figure 2B). Further work has shown that a unique MUS81-SLX4-RAD52-POLD3 axis is responsible for fork cleavage and re-ligation after fork stalling, this is mediated by RECQ5 helicase (17) (Figure 2B). In the latter model, recovered replication forks might be able to progress past RNA polymerase without compromising transcription, reminiscent of foundational work demonstrating that the replisome is able to bypass ternary transcription complexes without compromising transcription in a bacteriophage model system. (78)
Tumor suppressor proteins involved in homologous recombination (HR) also play an important role in replication fork reversal and protection. BRCA2 binds reversed replication forks to enable Rad51 loading to prevent MRE11 nucleolytic degradation of nascent DNA (108). In addition to facilitating fork protection, Rad51 is required for fork reversal in a BRCA2-independent manner (108). HR-deficiency and replication stress have also been previously associated with MiDAS (mitotic DNA synthesis), which results from defective S-phase DNA synthesis and requires Rad52 for resolution (9). In recent work, HR-deficient cells were found to have increased TRCs and MiDAS due to replication timing dysregulation(8, 42). In both BRCA2- and Rad51-deficient human cells, MiDAS sites have been identified using MiDAS-seq, and correspond to regions of early S-phase replication that also exhibit early S-phase transcription: these data demonstrate a model wherein TRCs delay replication of these regions (8, 42), possibly consistent with previously described TRIs (section 2.3). Specifically, CD-TRCs trigger BRCA2-deficient MiDAS associated with regions that align with R-loops, consistent with a previously reported role for BRCA2 in promoting transcription elongation and preventing R-loops (121). In several models, BRCA2-deficient cells exhibit slowed replication forks, the speed of which can be rescued by PARP inhibition at the expense of ssDNA gaps (25, 86). Future work exploring whether fork stalling instigates TRCs that can be rescued by modulating fork speed or compensatory dormant origin firing will shed light on the role of TRCs in replication regulation. Other mechanisms of BRCA2-mediated transcription regulation could contribute to preventing TRCs: damage-induced long-noncoding RNAs (dilncRNAs), and subsequent RNA-DNA hybrid formation, accumulate around DSBs specifically in S-phase, and require BRCA2 for repair (29). Whether these breaks are sites of collapse replication forks that use local RNAP/R-loops for dilnc synthesis is unknown but could explain a mechanism of RNA-mediated DNA repair specifically at TRCs (29).
PRIMPOL (primase polymerase)-mediated leading strand repriming and synthesis has also been shown to have the potential to resolve TRCs (discussed in this paragraph). The specific mechanisms of PRIMPOL-mediated lesion bypass are unclear, but initial work in mammalian models has characterized that PRIMPOL activity is required for replication fork bypass of UV-adducts, inter-strand crosslinks, and G4 quadruplex structures in unperturbed cells, at the expense of generating replication gaps (31). Endogenous PRIMPOL activity has been shown to be required for replication through expansive (GAA) repeats in DT40 cells in an RNA:DNA hybrid-dependent manner (Figure 2B), and defective PRIMPOL activity leads to S-phase specific accumulation of R-loops associated with G4 motifs (128). Although traditionally thought to be primarily involved in bypass of DNA adducts and nucleotide-level lesions, translesion polymerases have recently been implicated in other fork bypass mechanisms that might make them candidates for resolving TRCs. TLS polymerase eta has RNA primase and extension activity, and TLS polymerase kappa is required for fork recovery (36, 130). Continued work to explore pathway choice of replication forks stalled at TRCs in various cellular and genetic contexts will be needed to understand mechanisms of TRC-prevention and resolution.
In prokaryotes, the mRNA associated with the RNAP in a TRC can be used as a primer for lesion bypass in the co-directional orientation. RNAPs and R-loops associated with mRNA transcripts can by bypassed in vitro by mRNA takeover or downstream repriming using the mRNA as a primer for continued leading-strand synthesis, resulting in nascent ssDNA gaps (102). Intriguingly, in a reconstituted yeast replication system, analogous mechanisms to PRIMPOL repriming and translesion synthesis have recently been described as mechanisms to bypass unique TRCs: leading strand synthesis hindered by G4s or R-loops are able to be reprimed downstream of these lesions using the mRNA as a template, resulting in fork uncoupling and nascent DNA gaps (63). Whether mammalian replisomes have an analogous capacity for leading-strand repriming downstream of transcription complexes, particularly RNAP arrays, is yet to be demonstrated.
3.3. RNAP removal by replication machinery and signaling
One straightforward means of resolving TRCs is direct removal of RNAP or R-loops by the DNA replication machinery. Replication is high-stakes, as evidenced by several checkpoint pathways promoting tight cell cycle regulation and DNA replication fidelity. Conversely, transcription of any given mRNA is likely dispensable, with the possible exception of extremely long genes that require more than one cell cycle to transcribe. Therefore, local transcription inhibition directly associated with the replisome could prevent unanticipated TRCs that arise during DNA replication. This would facilitate quick TRC resolution without globally compromising cell cycle progression or genomic integrity with prolonged cell cycle checkpoint arrest. Although there is limited evidence for such transcriptional inhibition, recent live-cell imaging of transcription and replication machinery at an inducible-transcribed gene in yeast suggests that transcription is transiently inhibited ahead of replication forks, which remain unaffected except in the setting of excessive RNAP accumulation (134). Transient transcription inhibition might be orientation-specific, as only CD-TRCs in an engineered episomal system are associated with R-loop resolution(45). Immediate R-loop resolution could also impact local RNAP chromatin occupancy and resolve TRCs. ATAD5 regulates PCNA and prevents genomic instability by unloading it from DNA after replication completion and promoting deubiquitination of PCNA in response to DNA damage along with UAF1 (71). ATAD5 resolves R-loops by recruiting DEAD/DDX RNA helicases directly to replication forks (58). Therefore, ATAD5 depletion might lead to increased TRCs through dual mechanisms of defective R-loop resolution and persistent PCNA on DNA. (Figure 2B).
Accessory replicative helicases in both prokaryotes and eukaryotes remove protein-DNA complexes ahead of oncoming replication forks: in bacteria, Rep and UvrD remove RNAP without resulting ssDNA gaps (48); RecQL4 helicase tethered to pre-replicative helicases induces unscheduled origin firing and replication stress that is rescued by transcription inhibition (120). RECQ5 helicase and TRIM28 E3 ligase facilitate SUMO2-PCNA conjugation to promote RNAP2 removal from chromatin to prevent TRCs and TRC-induced breaks (76). Similarly, protein phosphatase PNUTS prevents replication stress checkpoint through direct dephosphorylation of phospho-Ser5 residues on RNAP to facilitate its removal at TRCs (66). (Figure 2B).
Collectively, these results show that several helicases associated with replisomes might resolve or prevent TRCs through transcription inhibition. Future work exploring the impact of this mechanism on gene expression will be needed: although transient inhibition of transcription could be favored to replication inhibition, whether inhibition of transcription impacts mRNA transcript maturity and gene expression that drives disease is unknown.
4. TRCs caused by transcriptional dysregulation
Transcription dysregulation leads to deleterious TRCs through multiple mechanisms including the formation of R-loops, RNAP stalling, and backtracking. Oncogene-related hyper-transcription, defective mRNA processing, and dysregulated chromatin all can lead to TRCs (sections 4.1, 4.2, 4.3). Failure to resolve TRCs also cause dysregulated gene expression, although the mechanisms underlying this are unclear (67, 134). Continued work exploring how TRCs impact proximal gene expression could elucidate novel genomic instability pathways that drive disease.
4.1. TRCs caused by hypertranscription
Oncogene induction can drive cancer through transcriptome reprogramming that promotes tumorigenic pathways. Hyper-transcription from oncogenes has been consistently linked to R-loop-induced replication stress (13). Activating mutations in H-Ras cause increased global RNA synthesis, R-loops, impaired replication fork progression and DNA breaks through the general transcription factor TBP (61) (Figure 3A). Beta-catenin activation causes increased global transcription, elevated R-loops, and transcriptional reprogramming, all of which are further exacerbated in BRCA2-deficient cells (27) (Figure 3A). In this study hypertranscription causes TRCs through downregulation of CDKN1A gene, which encodes the CDK inhibitor p21, leading to aberrant origin firing, fork collapse, and DNA breaks. Estradiol-induced transcriptional activity resembles oncogene-induced hypertranscription (Figure 3A): in human mammary epithelial cells, estrogen drives genomic instability in both a transcription- and replication-dependent manner and rapidly increases R-loop formation at estradiol-responsive genes, which correspond to regions of translocations in breast cancer, elucidating a potential mechanism by which estradiol drives luminal breast cancer (127).
Figure 3.
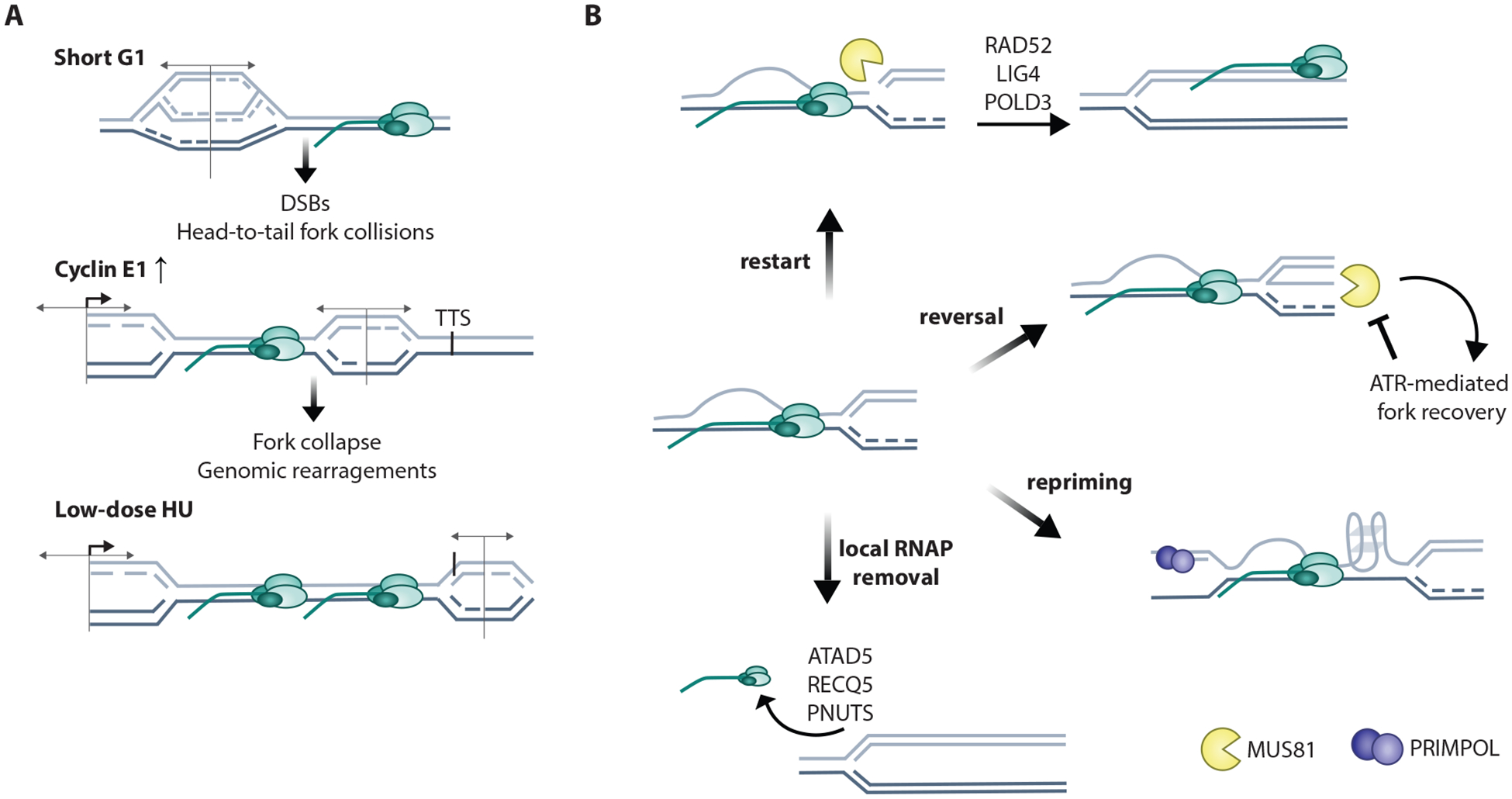
Transcription dysregulation causes TRCs
A. Replication and transcription are normally coordinated through several mechanisms (discussed in section 2). Hypertranscription by oncogenes (HRAS, B-catenin), estradiol, or BET inhibitors leads to hypertranscription that causes replication stress and TRCs. MYCN-low neuroblastoma cancer cells are prone to TRCs through defective recruitment of Aurora kinase and the nuclear exosome to transcribed gene units, resulting in antisense transcription, genomic instability, and cancer cell death.
B. Senataxin (SETX), yeast homolog Sen1, guards against TRCs through multiple mechanisms: SETX and BRCA1 resolve R-loops at transcription termination sites (TTSs); Sen1 removes RNAP at TRCs in yeast; SETX prevents against FANCD2- and MUS81-mediated fork cleavage and mitotic DNA synthesis (MiDAS). C. Transcription on nascent DNA promotes transfer of epigenetic histone marks, which encourages site-specific origin firing in subsequent cell cycles.
MYC oncoproteins are transcription factors that globally bind the majority of RNAPs to facilitate release from promoter-proximal pause sites to stimulate transcription elongation. However, the impact of MYC-family genes on actual gene expression of MYC-regulated gene targets is minimal, suggesting that MYC plays other roles in promoting oncogenesis, particularly in preventing TRCs: MYCN recruits the nuclear exosome to its promoter proximal pause sites for facilitation of transcription elongation and prevention of TRCs (95); MYCN multimers form in response to transcriptional stress to shield stalled replication forks and block anti-sense transcription at promoters (125) (Figure 3A). Intriguingly, these studies were carried out in MYCN-amplified neuroblastoma models. Therefore, MYCN’s role in preventing TRCs might be a consequence of mitigating catastrophic levels of replication stress, and this work highlights how TRCs could be used to induce genomic instability for cancer cell survival. Indeed, Aurora-A kinase inhibition induces TRCs, and combined Aurora-A kinase inhibition and ATR inhibition both led to increased DNA damage in in vivo MYCN-high neuroblastoma mouse models (112).
4.2. TRCs caused by defective mRNA processing
Defective RNA processing or persistent RNAP on chromatin can lead to R-loops and TRCs. R-loops have been comprehensively reviewed (26, 99), and recent reports continue to identify specific helicases involved in R-loop resolution to prevent or mitigate TRCs (11, 89). Impaired mRNA export can also impact TRCs: mutations in the THO/TREX (transcription export) complex that facilitate mRNP biogenesis and nuclear export cause TRC-induced genomic instability through TAR (142). Nuclear exosome-mediated mRNA processing also prevents TRCs: MYCN multimers inhibits transcriptional pausing by facilitating exosome recruitment (95). Defective transcription termination could promote TRCs at TTSs. HO-TRCs caused by origin firing downstream of genes may already make these sites TRC-prone, and the persistence of the transcription machinery and/or defective RNAP/R-loop removal could inhibit fork progression, or vice versa (i.e. origin firing at TTSs can interfere with proper transcription termination). SETX, acting in concert with BRCA1, plays a role in resolving R-loops at TTSs, promoting efficient transcription termination and thereby potentially preventing TRCs downstream of genes (47, 124) (Figure 3B). Recent work has shown that Sen1, yeast homolog of SETX, interacts with both the replisome and transcription machinery to resolve TRCs (1) (Figure 3B). Excessive R-loops that stall forks in SETX-depleted mammalian cells require FANCD2/FANCI for MUS81-mediated MiDAS (114); these events might be more frequent at long, CFS genes (Figure 3B).
Pre-mRNA splicing can also impact genomic instability, potentially through TRCs (98). Mutations in splicing factors cause R-loops to accumulate in pre-leukemia models, resulting in ATR activation and replication stress (18). The role of splicing on RNAP turnover and TRCs is unclear. TRCs occur at long genes due to their need of >1 cell cycle to transcribe (49). Long genes are also rich in both alternatively-spliced variants and premature stop codons (41). Proper splicing is mutually exclusive to premature transcription termination, which is a driver of transcriptome regulation and diversification (57). Therefore, long, transcriptionally-active genes might be susceptible to defective transcriptional processing caused by replication stress. Under-replication through gene bodies, or origin firing downstream of a long gene’s TTS could impact transcription elongation and termination (cleavage and polyadenylation), thereby leading to dysfunctional gene expression.
Improper transcription termination could also result in dysregulation of chromatin structure and function. Interestingly, R-loops induced by defective RNAi processing machinery (Dicer, Argonaute) induce antisense transcription at TTSs and histone repressive marks mediated by HP1g (123). Similarly, paused RNAP encourages local heterochromatin formation in yeast (96). Transcription is increasingly linked to modulating epigenetic marks (91), and histones are post-translationally modified in response to replication stress (52). Much of the work exploring impact of TRCs on genomic instability has focused on DNA breaks coming from collapsed forks, R-loops, or HR repair at TRCs. Future work exploring how TRCs directly impact gene expression could reveal novel mechanisms of epigenetically driven replication and transcription reprogramming.
4.3. Chromatin conformation
Active chromatin remodeling represents yet another mechanism by which cells can resolve TRCs. In yeast and murine models, the INO80 complex is directly bound to replication origins to locally inhibit transcription (131). This finding extends earlier work demonstrating that INO80 interacts with PAF1 complex to facilitate RNAP2 removal from early firing origins in response to replication stress (101). Additionally, BRG1, a SWI/SNF chromatin remodeling complex factor, cooperates with FANCD2 to resolve R-loops associated with TRCs at stalled forks (5).
Local and global chromatin status is tightly linked to coordination of replication and transcription. H3K4 activating histone modifications are strongly associated with replication-origin firing (100), and both H2A.Z and H4K20me2 play a role in the definition of early replication origins (80). Intriguingly, H3K4 methylation gradients have been shown to slow replication fork speed of oncoming replication forks as they approach genes, which may represent a mechanism to prevent HO-TRCs(22). In large chromatin domains, early origins are enriched at the borders of cell-type-specific topologically associating domains (TADs) (103), which are themselves high in transcriptional activity, R-loops, and cis-regulatory units that facilitate transcription throughout the TAD loops extruded through CTCF-cohesin complexes. Super-resolution microscopy has provided additional evidence that transcription and CTCF chromatin structure sets replication origins near TAD borders in G1/S (77). The mechanism(s) by which chromatin impacts replication-transcription coordination, specifically at TSSs, remain to be fully elucidated. Evidence suggests that transcription drives epigenetic memory that specifies origins throughout each cell division. Immediate reloading of transcription factors and RNAP onto the leading strand of newly replicated DNA promotes the efficient transfer of chromatin-accessible marks (3, 126), which may propagate the epigenetic landscape and indirectly impact transcription and replication over generations (59) (Figure 3C). Additionally, Ok-seq analysis shows replication origins occur preferentially at nucleosome-poor enhancers in a transcription-independent manner (21), suggesting that open chromatin structure may be sufficient for origin licensing and firing in some cases.
Alterations in chromatin conformation that impact transcriptional processivity could also elucidate the impact of RNAP status on TRCs. BET bromodomain family proteins, including BRD2/4, bind acetylated histones to facilitate positive transcriptional elongation. Paradoxically, inhibition of BET family proteins causes increased nascent transcription of certain genes linked to replication stress, increased origin firing, and TRCs (13) (Figure 3A). BRD4 loss increases R-loops through inhibition of transcription elongation in S-phase cells, and DNA damage (34, 65). Similarly, HDAC inhibitors open chromatin, increase transcription of certain highly-expressed genes, and cause replication stress (13). Whether HDACi causes TRCs is unknown. Both BETi and HDACi have been implicated in oncogenic transcriptional programming, and several of these inhibitors are promising anti-cancer targets.
Chromatin conformation impacting transcriptional processivity might also alter the propensity of TRCs to occur endogenously within gene bodies. It is unclear whether hyper-transcription that is associated with modulations in transcriptional speed impacts TRCs. Genes of varying length could be a model to explore the impact of transcriptional speed on TRCs, as long genes contain more introns and cotranscriptional splicing events, which both have been linked to increased transcriptional speed over these regions (56, 137). Ok-seq and Pu-seq analysis shows increased efficiency of replication origins upstream long genes (21, 62). Whether or not this is linked to increased transcriptional processivity along these genes is unknown, but open chromatin linked to transcriptional efficiency may help to ensure efficient fork progression over long gene bodies. Overall, and with the exception of R-loops, the transcriptional aspect of TRCs has been less extensively studied than replication:future studies exploring the impact of transcription dynamics on TRCs could reveal new contributions to genomic integrity.
6. Future directions and conclusions
Several techniques employing genome-wide mapping of replication fork dynamics have revealed critical discoveries surrounding the coordination of replication and transcription, but TRIPn-seq is one of few methods to directly detect transcription and replication co-occupancy. More nuanced genome-wide analysis of transcription dynamics will be required to extend our understanding of RNAP occupancy, processivity, and backtracking status affect TRCs. Novel methods that utilize long-read sequencing will also bring new information – for example Replicon-seq, which allows single-molecule genome-wide mapping of fork dynamics and could presumably be extended to analyze DNA mutations associated with transcription (24). We anticipate that future methods will directly interrogate TRCs, for example single-molecule super-resolution microscopy or biotin proximity labeling of transcription and replication factors.
Despite an immense body of work studying replication-transcription coordination, the impact of TRCs in specific contexts on genomic integrity in humans remains unclear. Continued efforts to map regions of transcription and replication co-occupancy could reveal specific sites of DNA damage, mutagenesis, and alterations in gene expression that drive disease progression, particularly in cancer. Cancer is characterized by dysregulation of transcription and replication: recent work in TRCs increasingly suggest the transcriptome drives coordination of these two processes, and TRCs cause genomic instability. Future work exploring specific mechanisms of unique TRCs and their impact on genomic instability could elucidate whether inter- or intra-tumor transcriptome heterogeneity facilitates transcription-replication coordination to evade catastrophic levels of DNA damage, and reveal novel therapeutic candidates.
Okazaki fragment sequencing (Ok-seq): Isolation of nucleotide-analog labeled Okazaki fragments for replication fork and origin dynamics without temporal resolution
Short nascent strand sequencing (SNS-seq): Isolation of short, nascent replication initiation intermediates for high-sensitivity replication origin mapping
Bubble sequencing (bubble-seq): DNA of replicating cells is fragmented and run on an agarose gel for isolation of non-linear DNAs and replication bubbles
Initiation-site sequencing (Ini-seq): Isolated nuclei are pulsed with replication factors, and sequencing of nascent DNA allows for origin analysis
Repli-seq: Cells divide with BrdU analog and are sorted by FACS for cell-cycle specific temporal resolution of fork dynamics
Polymerase usage seq (Pu-seq): Cells expressing mutant RNaseH and mutant DNA polymerases promote incorporation of ribonucleotides, allowing alkali extraction and sequencing of short DNAs
Hydroxyurea: ribonucleotide reductase inhibitor that depletes nucleotide pools and causes dose-dependent replication stress
Acknowledgments
The authors would like to acknowledge the numerous scientists whose research has contributed to our understanding of transcription-replication conflicts in prokaryotes, model eukaryotes, and mammalian systems. Due to space limitations, we were unable to directly reference all contributions to the field and apologize to those whose work we could not cite directly. The authors of this review are supported by funding from the National Institutes of Health (GM139610 and ES031658 to T.T.H.; GM134918 to D.J.S), and NYU MSTP Scholar Award to L.G.
Footnotes
Disclosure statement
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
References
- 1.Aiello U, Challal D, Wentzinger G, Lengronne A, Appanah R, et al. 2022. Sen1 is a key regulator of transcription-driven conflicts. Mol Cell. 82(16):2952–2966.e6 [DOI] [PubMed] [Google Scholar]
- 2.Akerman I, Kasaai B, Bazarova A, Sang PB, Peiffer I, et al. 2020. A predictable conserved DNA base composition signature defines human core DNA replication origins. Nat Commun. 11(1):4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez V, Bandau S, Jiang H, Rios-Szwed D, Hukelmann J, et al. 2023. Proteomic profiling reveals distinct phases to the restoration of chromatin following DNA replication. Cell Reports. 42(1):111996. [DOI] [PubMed] [Google Scholar]
- 4.Barlow JH, Faryabi RB, Callén E, Wong N, Malhowski A, et al. 2013. Identification of Early Replicating Fragile Sites that Contribute to Genome Instability. Cell. 152(3):620–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bayona-Feliu A, Barroso S, Muñoz S, Aguilera A. 2021. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription–replication conflicts. Nat Genet, pp. 1–14 [DOI] [PubMed] [Google Scholar]
- 6.Bertoli C, Skotheim JM, M de Bruin RA. 2013. Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Bio. 14(8):518–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, et al. 2012. Unraveling cell type–specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol. 19(8):837–44 [DOI] [PubMed] [Google Scholar]
- 8.Bhowmick R, Lerdrup M, Gadi SA, Rossetti GG, Singh MI, et al. 2022. RAD51 protects human cells from transcription-replication conflicts. Mol Cell. 82(18):3366–3381.e9 [DOI] [PubMed] [Google Scholar]
- 9.Bhowmick R, Minocherhomji S, Hickson ID. 2016. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol Cell. 64(6):1117–26 [DOI] [PubMed] [Google Scholar]
- 10.Blin M, Tallec BL, Nähse V, Schmidt M, Brossas C, et al. 2019. Transcription-dependent regulation of replication dynamics modulates genome stability. Nat Struct Mol Biol. 26(1):58–66 [DOI] [PubMed] [Google Scholar]
- 11.Boleslavska B, Oravetzova A, Shukla K, Nascakova Z, Ibini ON, et al. 2022. DDX17 helicase promotes resolution of R-loop-mediated transcription–replication conflicts in human cells. Nucleic Acids Res. 50(21):gkac1116- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bosch PC, Segura-Bayona S, Koole W, Heteren JT, Dewar JM, et al. 2014. FANCJ promotes DNA synthesis through G-quadruplex structures. Embo J. 33(21):2521–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowry A, Kelly RDW, Petermann E. 2021. Hypertranscription and replication stress in cancer. Trends Cancer [DOI] [PubMed] [Google Scholar]
- 14.Brewer BJ. 1988. When polymerases collide: Replication and the transcriptional organization of the E. coli chromosome. Cell. 53(5):679–86 [DOI] [PubMed] [Google Scholar]
- 15.Brison O, El-Hilali S, Azar D, Koundrioukoff S, Schmidt M, et al. 2019. Transcription-mediated organization of the replication initiation program across large genes sets common fragile sites genome-wide. Nat Commun. 10(1):5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brüning J-G, Marians KJ. 2020. Replisome bypass of transcription complexes and R-loops. Nucleic Acids Res. 48(18):gkaa741- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chappidi N, Nascakova Z, Boleslavska B, Zellweger R, Isik E, et al. 2020. Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-transcriptional R-Loops. Mol Cell. 77(3):528–541.e8 [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Chen J-Y, Huang Y-J, Gu Y, Qiu J, et al. 2018. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol Cell. 69(3):412–425.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen L, Chen J-Y, Zhang X, Gu Y, Xiao R, et al. 2017. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol Cell. 68(4):745–757.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y-H, Jones MJK, Yin Y, Crist SB, Colnaghi L, et al. 2015. ATR-Mediated Phosphorylation of FANCI Regulates Dormant Origin Firing in Response to Replication Stress. Mol Cell. 58(2):323–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y-H, Keegan S, Kahli M, Tonzi P, Fenyö D, et al. 2019. Transcription shapes DNA replication initiation and termination in human cells. Nat Struct Mol Biol. 26(1):67–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chong SY, Cutler S, Lin J-J, Tsai C-H, Tsai H-K, et al. 2020. H3K4 methylation at active genes mitigates transcription-replication conflicts during replication stress. Nat Commun. 11(1):809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cimprich KA, Cortez D. 2008. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Bio. 9(8):616–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Claussin C, Vazquez J, Whitehouse I. 2022. Single-molecule mapping of replisome progression. Mol Cell. 82(7):1372–1382.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, et al. 2021. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crossley MP, Bocek M, Cimprich KA. 2019. R-Loops as Cellular Regulators and Genomic Threats. Mol Cell. 73(3):398–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dagg RA, Zonderland G, Lombardi EP, Rossetti GG, Groelly FJ, et al. 2021. A transcription-based mechanism for oncogenic β-catenin-induced lethality in BRCA1/2-deficient cells. Nat Commun. 12(1):4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dahan D, Tsirkas I, Dovrat D, Sparks MA, Singh SP, et al. 2018. Pif1 is essential for efficient replisome progression through lagging strand G-quadruplex DNA secondary structures. Nucleic Acids Res. 46(22):gky1065- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.D’Alessandro G, Whelan DR, Howard SM, Vitelli V, Renaudin X, et al. 2018. BRCA2 controls DNA:RNA hybrid level at DSBs by mediating RNase H2 recruitment. Nat Commun. 9(1):5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deshpande AM, Newlon CS. 1996. DNA Replication Fork Pause Sites Dependent on Transcription. Science. 272(5264):1030–33 [DOI] [PubMed] [Google Scholar]
- 31.Díaz-Talavera A, Montero-Conde C, Leandro-García LJ, Robledo M. 2022. PrimPol: A Breakthrough among DNA Replication Enzymes and a Potential New Target for Cancer Therapy. Biomol. 12(2):248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durkin SG, Glover TW. 2007. Chromosome Fragile Sites. Genetics. 41(1):169–92 [DOI] [PubMed] [Google Scholar]
- 33.Dutta D, Shatalin K, Epshtein V, Gottesman ME, Nudler E. 2011. Linking RNA Polymerase Backtracking to Genome Instability in E. coli. Cell. 146(4):533–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edwards DS, Maganti R, Tanksley JP, Luo J, Park JJH, et al. 2019. BRD4 Prevents R-Loop Formation and Transcription-Replication Conflicts by Ensuring Efficient Transcription Elongation. Cell Reports. 32(12):108166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D. 2015. Error-Free DNA Damage Tolerance and Sister Chromatid Proximity during DNA Replication Rely on the Polα/Primase/Ctf4 Complex. Mol Cell. 57(5):812–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gali VK, Balint E, Serbyn N, Frittmann O, Stutz F, Unk I. 2017. Translesion synthesis DNA polymerase η exhibits a specific RNA extension activity and a transcription-associated function. Sci Rep-uk. 7(1):13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ge XQ, Jackson DA, Blow JJ. 2007. Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Gene Dev. 21(24):3331–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Germain CPS, Zhao H, Sinha V, Sanz LA, Chédin F, Barlow JH. 2022. Genomic patterns of transcription–replication interactions in mouse primary B cells. Nucleic Acids Res. 50(4):2051–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gómez-González B, Aguilera A. 2019. Transcription-mediated replication hindrance: a major driver of genome instability. Gene Dev. 33(15–16):1008–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gostissa M, Alt FW, Chiarle R. 2011. Mechanisms that Promote and Suppress Chromosomal Translocations in Lymphocytes. Immunology. 29(1):319–50 [DOI] [PubMed] [Google Scholar]
- 41.Grishkevich V, Yanai I. 2014. Gene length and expression level shape genomic novelties. Genome Res. 24(9):1497–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Groelly FJ, Dagg RA, Petropoulos M, Rossetti GG, Prasad B, et al. 2022. Mitotic DNA synthesis is caused by transcription-replication conflicts in BRCA2-deficient cells. Mol Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gros J, Kumar C, Lynch G, Yadav T, Whitehouse I, Remus D. 2015. Post-licensing Specification of Eukaryotic Replication Origins by Facilitated Mcm2–7 Sliding along DNA. Mol Cell. 60(5):797–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guilbaud G, Murat P, Wilkes HS, Lerner LK, Sale JE, Krude T. 2022. Determination of human DNA replication origin position and efficiency reveals principles of initiation zone organisation. Nucleic Acids Res. 50(13):7436–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA. 2017. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell. 170(4):774–786.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamperl S, Cimprich KA. 2016. Conflict Resolution in the Genome: How Transcription and Replication Make It Work. Cell. 167(6):1455–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hatchi E, Skourti-Stathaki K, Ventz S, Pinello L, Yen A, et al. 2015. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol Cell. 57(4):636–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hawkins M, Dimude JU, Howard JAL, Smith AJ, Dillingham MS, et al. 2019. Direct removal of RNA polymerase barriers to replication by accessory replicative helicases. Nucleic Acids Res. 47(10):gkz170- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helmrich A, Ballarino M, Tora L. 2011. Collisions between Replication and Transcription Complexes Cause Common Fragile Site Instability at the Longest Human Genes. Mol Cell. 44(6):966–77 [DOI] [PubMed] [Google Scholar]
- 50.Hill J, Eickhoff P, Drury LS, Costa A, Diffley JFX. 2020. The eukaryotic replisome requires an additional helicase to disarm dormant replication origins. Biorxiv. 2020.09.17.301366 [Google Scholar]
- 51.Hori Y, Engel C, Kobayashi T. 2023. Regulation of ribosomal RNA gene copy number, transcription and nucleolus organization in eukaryotes. Nat Rev Mol Cell Bio, pp. 1–16 [DOI] [PubMed] [Google Scholar]
- 52.Hsu C-L, Chong SY, Lin C-Y, Kao C-F. 2021. Histone dynamics during DNA replication stress. J Biomed Sci. 28(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huvet M, Nicolay S, Touchon M, Audit B, d’Aubenton-Carafa Y, et al. 2007. Human gene organization driven by the coordination of replication and transcription. Genome Res. 17(9):1278–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Irony-Tur Sinai M, Salamon A, Stanleigh N, Goldberg T, Weiss A, et al. 2019. AT-dinucleotide rich sequences drive fragile site formation. Nucleic Acids Res. 47(18):9685–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones RM, Mortusewicz O, Afzal I, Lorvellec M, García P, et al. 2013. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene. 32(32):3744–53 [DOI] [PubMed] [Google Scholar]
- 56.Jonkers I, Kwak H, Lis JT. 2014. Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife. 3:e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kamieniarz-Gdula K, Proudfoot NJ. 2019. Transcriptional Control by Premature Termination: A Forgotten Mechanism. Trends Genet. 35(8):553–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim S, Kang N, Park SH, Wells J, Hwang T, et al. 2020. ATAD5 restricts R-loop formation through PCNA unloading and RNA helicase maintenance at the replication fork. Nucleic Acids Res. 48(13):gkaa501- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klein KN, Zhao PA, Lyu X, Sasaki T, Bartlett DA, et al. 2021. Replication timing maintains the global epigenetic state in human cells. Science. 372(6540):371–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kotsantis P, Segura-Bayona S, Margalef P, Marzec P, Ruis P, et al. 2020. RTEL1 Regulates G4/R-Loops to Avert Replication-Transcription Collisions. Cell Reports. 33(12):108546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kotsantis P, Silva LM, Irmscher S, Jones RM, Folkes L, et al. 2016. Increased global transcription activity as a mechanism of replication stress in cancer. Nat Commun. 7(1):13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koyanagi E, Kakimoto Y, Minamisawa T, Yoshifuji F, Natsume T, et al. 2022. Global landscape of replicative DNA polymerase usage in the human genome. Nat Commun. 13(1):7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar C, Batra S, Griffith JD, Remus D. 2021. The interplay of RNA:DNA hybrid structure and G-quadruplexes determines the outcome of R-loop-replisome collisions. Elife. 10:e72286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lalonde M, Trauner M, Werner M, Hamperl S. 2021. Consequences and Resolution of Transcription–Replication Conflicts. Life. 11(7):637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lam FC, Kong YW, Huang Q, Han T-LV, Maffa AD, et al. 2020. BRD4 prevents the accumulation of R-loops and protects against transcription–replication collision events and DNA damage. Nat Commun. 11(1):4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Landsverk HB, Sandquist LE, Bay LTE, Steurer B, Campsteijn C, et al. 2020. WDR82/PNUTS-PP1 Prevents Transcription-Replication Conflicts by Promoting RNA Polymerase II Degradation on Chromatin. Cell Reports. 33(9):108469. [DOI] [PubMed] [Google Scholar]
- 67.Lang KS, Hall AN, Merrikh CN, Ragheb M, Tabakh H, et al. 2017. Replication-Transcription Conflicts Generate R-Loops that Orchestrate Bacterial Stress Survival and Pathogenesis. Cell. 170(4):787–799.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lang KS, Merrikh H. 2018. The Clash of Macromolecular Titans: Replication-Transcription Conflicts in Bacteria. Annu Rev Microbiol. 72(1):1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lang KS, Merrikh H. 2021. Topological stress is responsible for the detrimental outcomes of head-on replication-transcription conflicts. Cell Reports. 34(9):108797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langley AR, Gräf S, Smith JC, Krude T. 2016. Genome-wide identification and characterisation of human DNA replication origins by initiation site sequencing (ini-seq). Nucleic Acids Res. 44(21):10230–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee K, Fu H, Aladjem MI, Myung K. 2013. ATAD5 regulates the lifespan of DNA replication factories by modulating PCNA level on the chromatin. J Cell Biol. 200(1):31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee WTC, Yin Y, Morten MJ, Tonzi P, Gwo PP, et al. 2021. Single-molecule imaging reveals replication fork coupled formation of G-quadruplex structures hinders local replication stress signaling. Nat Commun. 12(1):2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lerner LK, Sale JE. 2019. Replication of G Quadruplex DNA. Genes-basel. 10(2):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, Debatisse M. 2013. Common Fragile Site Profiling in Epithelial and Erythroid Cells Reveals that Most Recurrent Cancer Deletions Lie in Fragile Sites Hosting Large Genes. Cell Reports. 4(3):420–28 [DOI] [PubMed] [Google Scholar]
- 75.Letessier A, Millot GA, Koundrioukoff S, Lachagès A-M, Vogt N, et al. 2011. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 470(7332):120–23 [DOI] [PubMed] [Google Scholar]
- 76.Li M, Xu X, Chang C-W, Liu Y. 2020. TRIM28 functions as the SUMO E3 ligase for PCNA in prevention of transcription induced DNA breaks. Proc National Acad Sci. 117(38):23588–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Y, Xue B, Zhang M, Zhang L, Hou Y, et al. 2021. Transcription-coupled structural dynamics of topologically associating domains regulate replication origin efficiency. Genome Biol. 22(1):206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu B, Wong ML, Alberts B. 1994. A transcribing RNA polymerase molecule survives DNA replication without aborting its growing RNA chain. Proc National Acad Sci. 91(22):10660–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y, Ai C, Gan T, Wu J, Jiang Y, et al. 2021. Transcription shapes DNA replication initiation to preserve genome integrity. Genome Biol. 22(1):176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Long H, Zhang L, Lv M, Wen Z, Zhang W, et al. 2020. H2A.Z facilitates licensing and activation of early replication origins. Nature. 577(7791):576–81 [DOI] [PubMed] [Google Scholar]
- 81.Macheret M, Bhowmick R, Sobkowiak K, Padayachy L, Mailler J, et al. 2020. High-resolution mapping of mitotic DNA synthesis regions and common fragile sites in the human genome through direct sequencing. Cell Res. 30(11):997–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Macheret M, Halazonetis TD. 2018. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature. 555(7694):112–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marchal C, Sima J, Gilbert DM. 2019. Control of DNA replication timing in the 3D genome. Nat Rev Mol Cell Bio. 20(12):721–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matos DA, Zhang J-M, Ouyang J, Nguyen HD, Genois M-M, Zou L. 2020. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol Cell. 77(3):514–527.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matson JP, Dumitru R, Coryell P, Baxley RM, Chen W, et al. 2017. Rapid DNA replication origin licensing protects stem cell pluripotency. Elife. 6:e30473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. 2018. High speed of fork progression induces DNA replication stress and genomic instability. Nature. 559(7713):279–84 [DOI] [PubMed] [Google Scholar]
- 87.Merrikh CN, Merrikh H. 2018. Gene inversion potentiates bacterial evolvability and virulence. Nat Commun. 9(1):4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Meryet-Figuiere M, Alaei-Mahabadi B, Ali MM, Mitra S, Subhash S, et al. 2014. Temporal separation of replication and transcription during S-phase progression. Cell Cycle. 13(20):3241–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mosler T, Conte F, Longo GMC, Mikicic I, Kreim N, et al. 2021. R-loop proximity proteomics identifies a role of DDX41 in transcription-associated genomic instability. Nat Commun. 12(1):7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Muniz L, Nicolas E, Trouche D. 2021. RNA polymerase II speed: a key player in controlling and adapting transcriptome composition. Embo J. 40(15):e105740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Niehrs C, Luke B. 2020. Regulatory R-loops as facilitators of gene expression and genome stability. Nat Rev Mol Cell Biology. 21(3):167–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nudler E 2012. RNA Polymerase Backtracking in Gene Regulation and Genome Instability. Cell. 149(7):1438–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Osmundson JS, Kumar J, Yeung R, Smith DJ. 2017. Pif1-family helicases cooperatively suppress widespread replication-fork arrest at tRNA genes. Nat Struct Mol Biol. 24(2):162–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pannunzio NR, Lieber MR. 2016. Dissecting the Roles of Divergent and Convergent Transcription in Chromosome Instability. Cell Reports. 14(5):1025–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Papadopoulos D, Solvie D, Baluapuri A, Endres T, Ha SA, et al. 2021. MYCN recruits the nuclear exosome complex to RNA polymerase II to prevent transcription-replication conflicts. Mol Cell [DOI] [PubMed] [Google Scholar]
- 96.Parsa J-Y, Boudoukha S, Burke J, Homer C, Madhani HD. 2018. Polymerase pausing induced by sequence-specific RNA-binding protein drives heterochromatin assembly. Gene Dev. 32(13–14):953–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Paul S, Million-Weaver S, Chattopadhyay S, Sokurenko E, Merrikh H. 2013. Accelerated gene evolution through replication–transcription conflicts. Nature. 495(7442):512–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee M-C, et al. 2009. A Genome-wide siRNA Screen Reveals Diverse Cellular Processes and Pathways that Mediate Genome Stability. Mol Cell. 35(2):228–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Petermann E, Lan L, Zou L. 2022. Sources, resolution and physiological relevance of R-loops and RNA–DNA hybrids. Nat Rev Mol Cell Bio, pp. 1–20 [DOI] [PubMed] [Google Scholar]
- 100.Petryk N, Kahli M, d’Aubenton-Carafa Y, Jaszczyszyn Y, Shen Y, et al. 2016. Replication landscape of the human genome. Nat Commun. 7(1):10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Poli J, Gerhold C-B, Tosi A, Hustedt N, Seeber A, et al. 2016. Mec1, INO80, and the PAF1 complex cooperate to limit transcription replication conflicts through RNAPII removal during replication stress. Gene Dev. 30(3):337–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pomerantz RT, O’Donnell M. 2008. The replisome uses mRNA as a primer after colliding with RNA polymerase. Nature. 456(7223):762–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pope BD, Ryba T, Dileep V, Yue F, Wu W, et al. 2014. Topologically associating domains are stable units of replication-timing regulation. Nature. 515(7527):402–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Powell SK, MacAlpine HK, Prinz JA, Li Y, Belsky JA, MacAlpine DM. 2015. Dynamic loading and redistribution of the Mcm2–7 helicase complex through the cell cycle. Embo J. 34(4):531–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Prado F, Aguilera A. 2005. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. Embo J. 24(6):1267–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Prioleau M-N, MacAlpine DM. 2016. DNA replication origins—where do we begin? Gene Dev. 30(15):1683–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Promonet A, Padioleau I, Liu Y, Sanz L, Biernacka A, et al. 2020. Topoisomerase 1 prevents replication stress at R-loop-enriched transcription termination sites. Nat Commun. 11(1):3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Quinet A, Lemaçon D, Vindigni A. 2017. Replication Fork Reversal: Players and Guardians. Mol Cell. 68(5):830–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Quinet A, Tirman S, Cybulla E, Meroni A, Vindigni A. 2021. To skip or not to skip: choosing repriming to tolerate DNA damage. Mol Cell. 81(4):649–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Reusswig K-U, Bittmann J, Peritore M, Courtes M, Pardo B, et al. 2022. Unscheduled DNA replication in G1 causes genome instability and damage signatures indicative of replication collisions. Nat Commun. 13(1):7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rocha EPC, Danchin A. 2003. Essentiality, not expressiveness, drives gene-strand bias in bacteria. Nat Genet. 34(4):377–78 [DOI] [PubMed] [Google Scholar]
- 112.Roeschert I, Poon E, Henssen AG, Garcia HD, Gatti M, et al. 2021. Combined inhibition of Aurora-A and ATR kinases results in regression of MYCN-amplified neuroblastoma. Nat Cancer. 2(3):312–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rothstein R, Michel B, Gangloff S. 2000. Replication fork pausing and recombination or “gimme a break.” Gene Dev. 14(1):1–10 [PubMed] [Google Scholar]
- 114.Said M, Barra V, Balzano E, Talhaoui I, Pelliccia F, et al. 2022. FANCD2 promotes mitotic rescue from transcription-mediated replication stress in SETX-deficient cancer cells. Commun Biology. 5(1):1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sanchez A, de Vivo A, Tonzi P, Kim J, Huang TT, Kee Y. 2020. Transcription-replication conflicts as a source of common fragile site instability caused by BMI1-RNF2 deficiency. Plos Genet. 16(3):e1008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sanz LA, Chédin F. 2019. High-resolution, strand-specific R-loop mapping via S9.6-based DNA–RNA immunoprecipitation and high-throughput sequencing. Nat Protoc. 14(6):1734–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schauer GD, Spenkelink LM, Lewis JS, Yurieva O, Mueller SH, et al. 2020. Replisome bypass of a protein-based R-loop block by Pif1. Proc National Acad Sci. 117(48):30354–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Scherr MJ, Wahab SA, Remus D, Duderstadt KE. 2022. Mobile origin-licensing factors confer resistance to conflicts with RNA polymerase. Cell Reports. 38(12):110531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, et al. 2015. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol Cell. 60(3):351–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shin G, Jeong D, Kim H, Im J-S, Lee J-K. 2019. RecQL4 tethering on the pre-replicative complex induces unscheduled origin activation and replication stress in human cells. J Biol Chem. 294(44):16255–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shivji MKK, Renaudin X, Williams ÇH, Venkitaraman AR. 2018. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Reports. 22(4):1031–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shyian M, Albert B, Zupan AM, Ivanitsa V, Charbonnet G, et al. 2020. Fork pausing complex engages topoisomerases at the replisome. Gene Dev. 34(1–2):87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Skourti-Stathaki K, Kamieniarz-Gdula K, Proudfoot NJ. 2014. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature. 516(7531):436–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Skourti-Stathaki K, Proudfoot NJ, Gromak N. 2011. Human Senataxin Resolves RNA/DNA Hybrids Formed at Transcriptional Pause Sites to Promote Xrn2-Dependent Termination. Mol Cell. 42(6):794–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Solvie D, Baluapuri A, Uhl L, Fleischhauer D, Endres T, et al. 2022. MYC multimers shield stalled replication forks from RNA polymerase. Nature. 612(7938):148–55 [DOI] [PubMed] [Google Scholar]
- 126.Stewart-Morgan KR, Reverón-Gómez N, Groth A. 2019. Transcription Restart Establishes Chromatin Accessibility after DNA Replication. Mol Cell. 75(2):284–297.e6 [DOI] [PubMed] [Google Scholar]
- 127.Stork CT, Bocek M, Crossley MP, Sollier J, Sanz LA, et al. 2016. Co-transcriptional R-loops are the main cause of estrogen-induced DNA damage. Elife. 5:e17548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Šviković S, Crisp A, Tan-Wong SM, Guilliam TA, Doherty AJ, et al. 2019. R-loop formation during S phase is restricted by PrimPol-mediated repriming. Embo J. 38(3):e99793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Toledo LI, Altmeyer M, Rask M-B, Lukas C, Larsen DH, et al. 2013. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell. 155(5):1088–1103 [DOI] [PubMed] [Google Scholar]
- 130.Tonzi P, Yin Y, Lee CWT, Rothenberg E, Huang TT. 2018. Translesion polymerase kappa-dependent DNA synthesis underlies replication fork recovery. Elife. 7:e41426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Topal S, Van C, Xue Y, Carey MF, Peterson CL. 2020. INO80C Remodeler Maintains Genomic Stability by Preventing Promiscuous Transcription at Replication Origins. Cell Reports. 32(10):108106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Touchon M, Nicolay S, Audit B, Brodie of B E-B, d’Aubenton-Carafa Y, et al. 2005. Replication-associated strand asymmetries in mammalian genomes: Toward detection of replication origins. Proc National Acad Sci. 102(28):9836–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tran PLT, Pohl TJ, Chen C-F, Chan A, Pott S, Zakian VA. 2017. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat Commun. 8(1):15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tsirkas I, Dovrat D, Thangaraj M, Brouwer I, Cohen A, et al. 2022. Transcription-replication coordination revealed in single live cells. Nucleic Acids Res. 50(4):2143–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tubbs A, Sridharan S, van Wietmarschen N, Maman Y, Callen E, et al. 2018. Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell. 174(5):1127–1142.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Uchino S, Ito Y, Sato Y, Handa T, Ohkawa Y, et al. 2021. Live imaging of transcription sites using an elongating RNA polymerase II–specific probe. J Cell Biology. 221(2):e202104134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Veloso A, Kirkconnell KS, Magnuson B, Biewen B, Paulsen MT, et al. 2014. Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res. 24(6):896–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wahba L, Costantino L, Tan FJ, Zimmer A, Koshland D. 2016. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Gene Dev. 30(11):1327–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang J, Rojas P, Mao J, Sadurnì MM, Garnier O, et al. 2021. Persistence of RNA transcription during DNA replication delays duplication of transcription start sites until G2/M. Cell Reports. 34(7):108759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang W, Klein KN, Proesmans K, Yang H, Marchal C, et al. 2021. Genome-wide mapping of human DNA replication by optical replication mapping supports a stochastic model of eukaryotic replication. Mol Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wansink DG, Manders EE, van der Kraan I, Aten JA, van Driel R, de Jong L. 1994. RNA polymerase II transcription is concentrated outside replication domains throughout S-phase. J Cell Sci. 107 (Pt 6)(6):1449–56 [DOI] [PubMed] [Google Scholar]
- 142.Wellinger RE, Prado F, Aguilera A. 2006. Replication Fork Progression Is Impaired by Transcription in Hyperrecombinant Yeast Cells Lacking a Functional THO Complex. Mol Cell Biol. 26(8):3327–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wu W, Hickson ID, Liu Y. 2020. The prevention and resolution of DNA replication–transcription conflicts in eukaryotic cells. Genome Instab Dis. 1(3):114–28 [Google Scholar]
- 144.Wu Y, Shin-ya K, Brosh RM. 2008. FANCJ Helicase Defective in Fanconia Anemia and Breast Cancer Unwinds G-Quadruplex DNA To Defend Genomic Stability. Mol Cell Biol. 28(12):4116–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yadav P, Harcy V, Argueso JL, Dominska M, Jinks-Robertson S, Kim N. 2014. Topoisomerase I Plays a Critical Role in Suppressing Genome Instability at a Highly Transcribed G-Quadruplex-Forming Sequence. Plos Genet. 10(12):e1004839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yang Y, Xu W, Gao F, Wen C, Zhao S, et al. 2022. Transcription–replication conflicts in primordial germ cells necessitate the Fanconi anemia pathway to safeguard genome stability. Proc National Acad Sci. 119(34):e2203208119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yeung R, Smith DJ. 2020. Determinants of Replication-Fork Pausing at tRNA Genes in Saccharomyces cerevisiae. Genetics. 214(4):825–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zeng J, Hills SA, Ozono E, Diffley JFX. 2023. Cyclin E-induced replicative stress drives p53-dependent whole-genome duplication. Cell. 186(3):528–542.e14 [DOI] [PubMed] [Google Scholar]
- 149.Zhao PA, Sasaki T, Gilbert DM. 2020. High-resolution Repli-Seq defines the temporal choreography of initiation, elongation and termination of replication in mammalian cells. Genome Biol. 21(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhou Z-X, Follonier C, Lujan SA, Burkholder AB, Zakian VA, Kunkel TA. 2022. Pif1 family helicases promote mutation avoidance during DNA replication. Nucleic Acids Res. 50(22):12844–55 [DOI] [PMC free article] [PubMed] [Google Scholar]