

ABSTRACT
Secondary immunoglobulin diversification by somatic hypermutation and class switch recombination in B cells is instrumental for an adequate adaptive humoral immune response. These genetic events may, however, also introduce aberrations into other cellular genes and thereby cause B cell malignancies. While the basic mechanism of somatic hypermutation and class switch recombination is now well understood, their regulation and in particular the mechanism of their specific targeting to immunoglobulin genes is still rather mysterious. In this review, we summarize the current knowledge on the mechanism and regulation of secondary immunoglobulin diversification and discuss known mechanisms of physiological targeting to immunoglobulin genes and mistargeting to other cellular genes. We summarize open questions in the field and provide an outlook on future research.
Keywords: Somatic hypermutation, germinal center, lymphomagenesis
Our immune system deals daily with dangerous events, such as infection or tissue damage. While innate immunity provides a quick and effective response, the adaptive immune system responds more slowly, but generates a memory of the adverse events which may be kept for a lifetime [1]. T and B cells are the effector cells of adaptive immunity in jawed vertebrates. They carry specific antigen receptors formed by V(D)J recombination during development of their precursor cells (Figure 1) [2], which may specifically recognize a wide variety of pathogens and other foreign agents. While T cells do not further change their antigen receptors when they are activated during an immune response, B cells do respond not only by cellular activation but also by further diversifying the genes coding for their B cell receptor/antibody, i.e. the immunoglobulin (Ig) genes. Diversification via somatic hypermutation alters the variable portion of Ig genes for affinity maturation, and class switch recombination alters the constant region for a change in antibody effector functions (Figure 1) [3].
Figure 1.
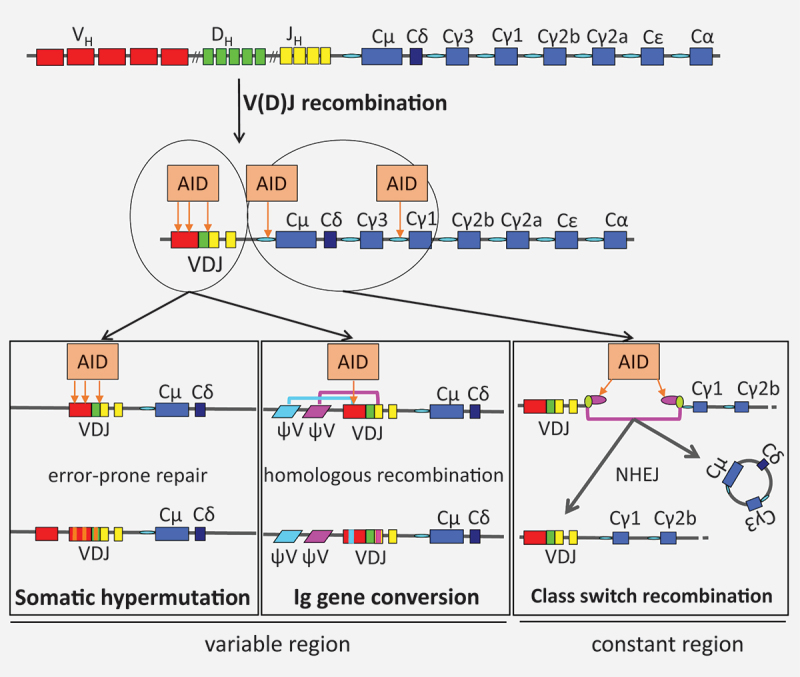
Ig diversification processes V(D)J recombination recombines one V, one D, and one J segment of the Ig genes to a V(D)J joint that codes for the variable region of the antibody. Somatic hypermutation is based on the introduction of AID-induced lesions into the V(D)J joint, followed by error-prone repair for mutagenesis. Ig gene conversion is based on the processing of AID-induced lesion in the V(D)J joint by homologous recombination with upstream pseudogenes. Class switch recombination relies on the processing of AID-induced lesions in the switch regions to DNA double-strand breaks, which are then joined by non-homologous end joining to replace the constant region of the Ig gene
While class switch recombination may occur upon B cell activation even outside of follicles [4], affinity maturation via somatic hypermutation is generally thought to require a follicular germinal center reaction (Figure 2) [5–7], although pre-germinal center diversification of marginal zone B cells has been suggested [8]. For germinal center formation, activated B cells proliferate massively in secondary lymphoid organs, and form a finally bipartite morphological structure within the follicle consisting of a dark and a light zone [9]. In the dark zone of germinal centers, proliferation of the B cells and somatic hypermutation of the Ig variable genes occurs, while in the light zone, B cells with variant antigen receptors are selected based on antigen-binding affinity. By taking up an antigen presented by follicular dendritic cells and processing it for presentation to antigen-specific T cells, B cells that recognize the cognate antigen with relatively higher affinity are positively selected as they receive survival signals from the T cells via CD40 ligation [10]. Class switch recombination was long thought to also occur in the germinal center light zone, but recent evidence suggests that it is a rare event in these structures [11] and rather occurs before B cells enter the germinal center reaction. B cells that have successfully undergone somatic hypermutation and selection in the germinal center may differentiate into either long-lived plasma cells to produce large amounts of high affinity antibodies, or memory B cells which carry the memory of the respective pathogenic insult throughout lifetime [12].
Figure 2.
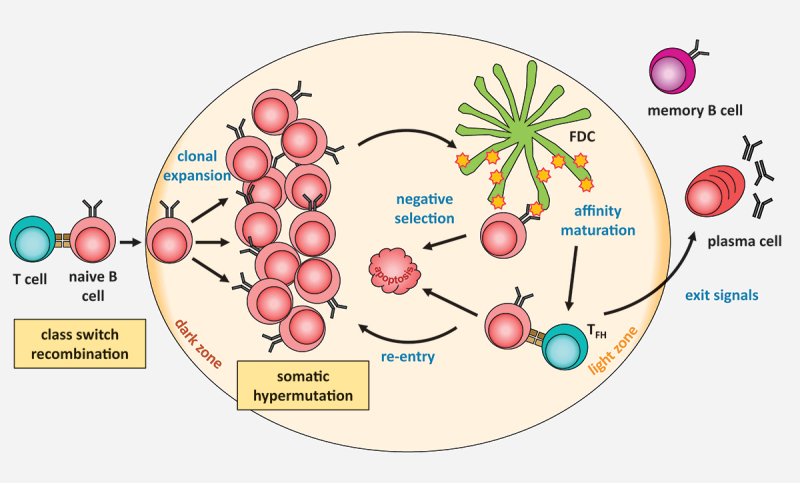
The germinal center reaction Naïve B cells are activated by contact with antigen-specific T cells and may undergo class switch recombination. They proliferate heavily within the dark zone of the germinal center, where they also undergo somatic hypermutation. For selection, they move to the light zone of the germinal center, take up the antigen presented by follicular dendritic cells, and present it to T cells to receive survival signals. They may then differentiate into either memory or plasma cells.
Both somatic hypermutation and class switch recombination are initiated by the same enzyme, activation-induced cytidine deaminase (AID) [13,14], although low-level class switch recombination can occur in the absence of AID due to intrinsic properties of the switch regions [15]. AID deaminates cytosines leading to uracils in transcribed DNA [16,17]. The lesions are then processed by error-prone repair pathways, leading to either point mutation introduction during somatic hypermutation, or recombination of new constant regions during class switch recombination [18]. In some farm animals like chicken and rabbits, but not in humans and mice, a related process called Ig gene conversion is based on processing of AID-induced lesions by homologous recombination with upstream pseudogenes (Figure 1) [19,20]. All three processes are focused largely, but not entirely, on the Ig genes by multiple regulatory processes. Their deregulation may lead to mutations or recombination in other genes and hence to genetic instability and lymphoma development [21]. In fact, most human B cell lymphoma entities are derived from B cells that have undergone a germinal center reaction [22]. It is therefore imperative to understand mechanisms of normal targeting of somatic hypermutation and class switch recombination to the Ig genes, to better identify mechanisms of deregulation of these processes during lymphomagenesis. It has been shown that adequate targeting of somatic hypermutation is based, on the one hand, on targeting of AID, and, on the other hand, on targeting of locus-specific error-prone repair processes [23]. We will therefore discuss these two regulatory branches separately here.
Regulation of AID
AID is a dangerous mutator, and is thus regulated at multiple levels, including regulation of mRNA expression, regulation of protein localization and stability, and regulation of its targeting to Ig genes. Regulation of AID mRNA expression. AID expression is largely confined to activated and germinal center B cells, which is due to regulation of its mRNA expression by a multitude of transcription factors (Figure 3A). There are four conserved regulatory regions in the AID gene, on which binding sites for 19 transcription factors cluster [24]. Among them are B cell-specific transcription factors such as Pax-5 and E2A [25,26], as well as factors specific for B cell activation, such as NF-kappaB, HoxC4 and Stat6 [27–29]. Also, the PI3 kinase pathway and MAP kinase signaling impinge upon AID expression [30,31]. The key germinal center transcription factor Bcl-6 increases AID gene expression indirectly by repressing the plasma cell-specific transcription factor Blimp-1, which in turn represses the activating factor Pax-5 [32,33]. Also, AID mRNA is regulated by miRNAs, such as the inhibitory miRNA155 which is repressed by Bcl-6 [34–36].
Figure 3.
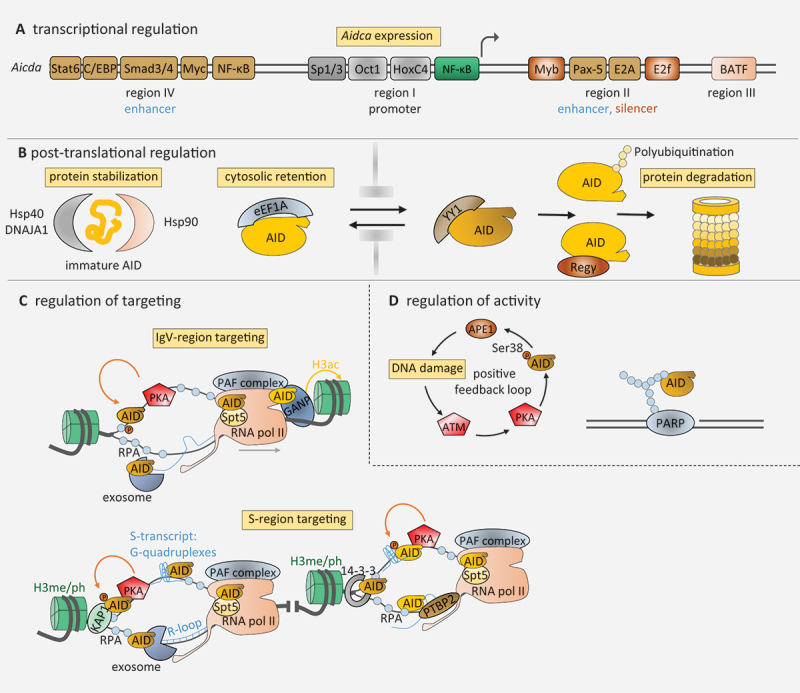
Regulation of AID A) transcriptional regulation of AID. Binding sites for transcription factors cluster in four sites of the Aicda locus and contribute to either induction or repression of AID expression. B) posttranslational regulation of AID. AID is stabilized in the cytoplasm by interaction with HSP 40 and 90. It is retained in the cytoplasm by interaction with eEF1A. Upon nuclear translocation, AID is quickly degraded by the proteasome, unless complexed with YY1. C) regulation of AID targeting to Ig loci. Multiple factors contribute to the specific targeting of AID to either the V region or switch regions. D) regulation of AID activity at the Ig locus. A positive feedback loop involving ATM and PKA regulates AID at switch regions. A negative feedback loop involving PARP regulates AID at the V region. For more details, see text.
Regulation of AID protein localization and stability. The AID protein needs to act in the nucleus but is largely confined to the cytoplasm in normal cells by interactions with eEF1A (Figure 3B) [37–39]. Nuclear translocation of AID involves a N-terminal structural nuclear localization sequence recognized by importins, and CTNNBL1, SRSF1–3 and GANP aid AID nuclear translocation [40–44]. A C-terminal nuclear export sequence regulates export of AID by CRM-1 [45–48]. Also, the nuclear-to-cytoplasmic ratio of AID is determined by a differential regulation of its half-life within the two compartments. While cytoplasmic AID is relatively stable with a half-life of 18–20 hours, due to interactions with HSP40 and HSP90 [49,50], the nuclear half-life of AID is about 2.5 hours [51–53], showing that AID is subject to rapid proteasomal degradation mediated by the E3 ubiquitin ligase CUL7 or REG- when it enters the nucleus [52,53]. It has been shown that AID interacts with the transcription factor YY1 to increase its nuclear stability [54].
The cellular localization and stability of the AID protein is also regulated by the cell cycle. Even though the AID protein was found to be expressed throughout the cell cycle [51], the fluorescence of an AID-YFP fusion protein was detected in the nucleus of chicken B cells mainly in G1-phase cells [55]. A later study showed via time-lapse imaging of AID-EGFP-expressing primary mouse B cells that AID accumulates in the nucleus exclusively in the early G1 phase, and this has been shown to correlate with the activity of AID being restricted to this phase of the cell cycle [56]. In S/G2, nuclear AID has been proposed to undergo faster degradation than in G1 [57], and this observation, together with the yet unexplored possibility of increased active export to the cytoplasm, may explain the reduced AID nuclear levels in S/G2.
Regulation of AID targeting to Ig loci. A lot of effort has been made to clarify mechanisms of AID targeting to the Ig locus (Figure 3C). AID detects and deaminates single-stranded DNA by interacting with RPA [58]. A role of Ig transcription in opening up the double-stranded DNA was detected early by several studies, and a specific role for Ig enhancers has been shown convincingly [59,60]. In particular, the 3’ regulatory region enhancer (3’RR) has been shown to boost secondary Ig diversification by increasing accessibility of the region for AID as well as regulating locus contraction and loop extrusion during CSR [61–64].
A more focused role of transcription in directing AID targeting emerged when it was shown that AID localizes to promotor-proximal stalled transcription complexes via interaction with Spt-5 [65–68]. Transcriptional stalling shortly after initiation is a feature of paused genes [69] and affects less genes than transcription as such; this mechanism may also explain why only the 5’ portion of the gene rather than the entire transcription unit is targeted by AID. Also, a role of super-enhancer-induced convergent transcription and the resultant non-coding RNA in directing AID targeting to its on- (Ig genes) and off-targets has been observed [70–73]. A particular example is provided by the non-coding germline transcript produced in switch regions, which promotes the formation of R-loops and quadruplexes, thereby facilitating access of AID to the single-stranded DNA [74,75]. This may explain why processing of the switch transcript by splicing is required for class switch recombination to occur [76,77].
Also, AID interaction factors are involved in bringing AID to the correct locus. The splicing regulator PTBP2 is required for AID binding to switch regions, likely via binding of PTBP2 to switch transcripts [78]. GANP helps to bring AID to the Ig variable region by modulating chromatin assembly via its histone acetyltransferase domain [79]. The RNA PolII associated PAF complex apparently serves as a binding platform for AID at the switch and variable regions [80]. 14-3-3 adapter proteins bring AID to the switch regions by binding to their repetitive 5’-AGCT-3’ motifs and to the AID C-terminus [81]. The association of 14-3-3 with the switch region is also facilitated by the combinatorial H3K9acS10ph mark that selectively labels donor and acceptor switch regions [82]. AID interaction with KAP-1 and HP-1 tethers AID to the donor switch region by specific binding of HP-1 to H3K9me3 marks [83]. Other epigenetic marks are also associated with Ig diversification, but whether they affect AID recruitment or other processes is unclear [84]. Once bound to the locus, a licensing step activates AID for deamination by a thus far undefined mechanism [85]. AID’s access to both coding and non-coding strand of the transcribed DNA is mediated by the exosome that degrades prematurely terminated transcripts near enhancers and promoters [86]. Finally, certain sequence motifs within the Ig genes have evolved to focus AID activity to its AGCT hotspots [87,88].
An apparent locus-specific event is the regulation of AID by phosphorylation (Figure 3D). Early work indicated a role of protein kinase A (PKA), which phosphorylates serine 38 of AID and thereby contributes to efficient somatic hypermutation, class switch recombination, and Ig gene conversion [89–95]. In B cells, PKA has been shown to localize specifically to the Ig genes [96], but how this occurs and how PKA activity is induced was not clear. Apparently, AID-induced DNA breaks induce PKA-mediated AID phosphorylation via ATM activation [97], leading to a positive feedback loop for locus-specific AID activity during class switch recombination. During somatic hypermutation, a potential locus-specific regulation by a negative feedback loop occurs, involving the DNA break-associated activation of PARP-1 and inhibition of AID activity by binding of AID to the poly(ADP-ribose) chains synthesized by PARP1 [98]. While genome-wide induction of AID-PARP-1 binding may be mediated by treatment of cells with genotoxic agents [99], under physiological circumstances, the process is likely rather specific for certain genomic locations as PARP-1 activation requires either an (AID-induced) DNA break or PARP-1 targeting to certain genes by transcription factors [100]. Interestingly, the Bcl-6 locus has been described to be regulated by PARP-1 activity [101], so one of the most prominent non-Ig genes subject to hypermutation in the germinal center [102] may be regulated by such a mechanism.
In summary, multiple processes regulate specific AID activity at the target loci. It appears possible that most of them are now identified, and deregulation of either process has the potential to cause unrestrained mutagenesis [103]. Interestingly, though, AID has been shown to associate with thousands of genes in B cells [104] and to induce lesions in hundreds of them [23,72,73,105], but not all of these genes eventually hypermutate [23,105,106]. Thus, DNA repair processes must be (de)regulated in a locus-specific manner as well.
Regulation of DNA repair processes
Deamination of cytosine by AID leads to uracil in the DNA [16,17]. Multiple DNA repair pathways process these lesions in Ig genes (Figure 4). First and foremost, the base excision repair pathway is responsible for error-free processing of such lesions in all cells of our body. This pathway includes the removal of the uracil by Uracil-N-Glycosylase (UNG), followed by the nicking of the resultant abasic site by apurinic endonuclease (APE). The resultant break then serves as substrate for Polymerase ß (in concert with XRCC1) to fill the gap and insert the original nucleotide. Error-free base excision repair also processes AID-induced lesions in the Ig genes, as Ig diversification is increased in B cells haploinsufficient for XRCC1 or deficient for Pol [107,108]. However, base excision repair must be at least somewhat perturbed during Ig diversification, as the intermediates of this pathway serve as substrates for error-prone processing by other pathways. The uracil may be simply replicated over, leading to transition mutations at the respective C:G position (phase 1A of somatic hypermutation). Also, the abasic site formed upon uracil excision may be bypassed by specialized translesion polymerases such as Rev1, which insert random nucleotides opposite of the uninstructive lesion leading to transition and transversion mutations at C:G (phase 1B) [109]. As an alternative to base excision repair, mismatch repair factors may recognize the U:G mismatch and catalyze the removal of nucleotides from one strand of DNA in the attempt to repair the damage. The process generates a single-stranded gap which is subsequently refilled mainly by polymerase upon PCNA monoubiquitination by Rad6/Rad18 [110–115]. This results in mutations specifically at A:T base pairs (phase 2). Also, the strand breaks introduced by APE may combine to form DNA double strand breaks, which are a substrate for non-homologous end joining during class switch recombination [116]. Mismatch recognition and uracil removal have been shown to be complementary pathways for A:T mutagenesis and class switch recombination [117], so some residual switching occurs in UNG deficient cells, while some A:T mutagenesis can still be found in mismatch repair deficient cells. Finally, the single- or double-strand breaks may serve as substrates for homologous recombination, leading to Ig gene conversion or an alternative pathway of error-free repair of the AID-induced lesions [118,119].
Figure 4.
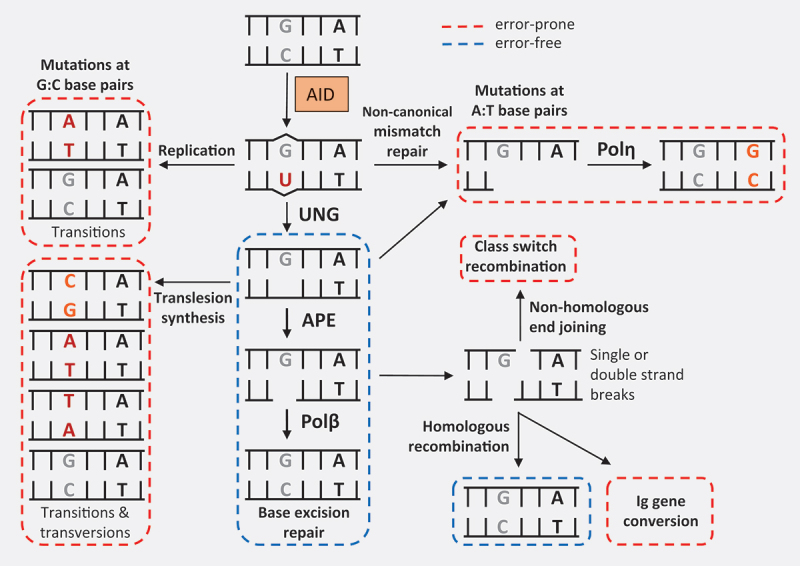
DNA repair during Ig diversification AID induces uracils, which may be removed by UNG, followed by strand cleavage by APE and repair by Pol/XRCC1 for classical base excision repair. Uracils may also be replicated over to yield transition mutations at C:G base pairs. Alternatively, the abasic site resulting from uracil excision may be replicated over by specialized translesion polymerases such as Rev1 to yield transition and transversion mutations at C:G base pairs. Also, the U:G mismatch may be recognized by mismatch repair enzymes, leading to cleavage of a strand fragment and DNA synthesis by Pol, which requires PCNA ubiquitination for its action, resulting in mutations at A:T residues. The breaks caused by APE may lead to double-strand breaks, which are processed by non-homologous end joining during class switch recombination. Uracil excision and mismatch repair both contribute to A:T mutagenesis and class switch recombination. Alternatively, single- or double-strand breaks may be processed by homologous recombination, leading to Ig gene conversion or error-free repair of the DNA lesion. Error-free pathways are marked in blue, while error-prone pathways are marked in red.
There is an interesting relationship between somatic hypermutation, class switch recombination, and homologous recombination. When homologous recombination is inactivated in the chicken DT40 cell line, the cells stop diversifying their Ig locus via Ig gene conversion and instead switch to human/murine-like somatic hypermutation [120]. This indicates that homologous recombination could counteract the error-prone processing of AID-induced lesions via translesion polymerases, suggesting that homologous recombination potentially needs to be downregulated in the human/murine Ig locus to enable somatic hypermutation in the first place. However, mouse studies have later demonstrated that homologous recombination in S/G2 repairs AID-induced DNA double-strand breaks in off-target genes but also in the switch regions if class switch recombination was not accomplished, thereby protecting the B cell genome from chromosomal aberrations [119,121]. By analyzing the hypermutating variable region, our lab has then shown that homologous recombination is indeed active in the Ig locus but prevents only a fraction of C:G transversion mutations and is rather needed to support survival of highly mutating B cells [122]. During class switch recombination, the decision of repair of the resultant DNA double strand breaks via classical non-homologous end joining rather than alternative repair pathways is mediated by the p53bp1/Rif1 module with the help of other factors such as Shieldin and hnRNPU [123–125].
Accordingly, several mutagenic DNA repair pathways contribute to somatic hypermutation, while error-prone recombination forms the basis of class switch recombination and Ig gene conversion. As mentioned before, this error-prone processing of DNA lesions in the Ig genes is locus-specific, as AID-induced lesions in other cellular genes are largely, but not entirely, repaired in an error-free fashion by the same repair pathways mediating Ig diversification [23]. In principle, one may envision four potential ways of how this may occur: 1) each repair pathway is separately deregulated (globally in B cells undergoing Ig diversification, or in a locus-specific manner) by a pathway-specific process; 2) AID itself affects DNA repair in the genes it targets; 3) an upstream regulator of all associated repair pathways, such as checkpoint signaling, is deregulated in a locus-specific fashion or 4) repair is error-prone in the Ig genes as it occurs in the wrong cell cycle phase. The truth likely lies within a combination of these possibilities, as explained next.
Global downmodulation of base excision repair in cells undergoing Ig diversification. As mentioned above, base excision repair must be at least somewhat perturbed in cells undergoing Ig diversification, as its intermediates (uracils, abasic sites and strand breaks) serve as starting points for other mutagenic repair pathways in these cells. Within the last years, four studies have revealed that base excision repair is apparently globally impaired by several mechanisms in cells undergoing Ig diversification. Concerning the uracil, two groups reported the upregulation of Fam72A in activated and germinal center B cells, which binds to UNG2 and triggers its degradation [126,127]. Thereby, uracils may remain in the DNA and serve as templates for error-prone replication or recognition by the mismatch repair pathway for A:T mutagenesis. Concerning the abasic site, the downregulation of APE1 in germinal centers and replacement by the less proficient APE2 has been shown [128]. This likely results in abasic sites lingering in the DNA until translesion synthesis converts them to mutations. Finally, concerning the strand break, Pol has been shown to be downregulated in the germinal center, likely leading to unprocessed strand breaks that may serve as starting points for A:T mutagenesis or class switch recombination [129]. It will be interesting to investigate the molecular mechanism of up- and downmodulation of these proteins in the germinal center.
Modulation of DNA repair by AID. The C-terminus of AID is required for class switch recombination but not for somatic hypermutation [130]. It has been shown that this C-terminus recruits repair factors to the switch region, thereby likely affecting repair capacity [131,132]. Also, the above-described positive feedback loop leads to phosphorylation-dependent recruitment of APE1 to the switch regions by AID [97]. However, this likely helps rather than prevents error-free repair. Therefore, any potential contribution of AID to error-prone repair in Ig genes still needs to be investigated.
Regulation of Ig diversification by checkpoint signaling. Checkpoint responses involve the kinases ATM, which recognizes DNA double-strand breaks, and ATR, which recognizes exposed single-stranded regions (Figure 5) [133]. ATM phosphorylates Chk2 and p53, which is additionally phosphorylated by Chk2 itself. ATR phosphorylates Chk1 and p53, which is also a substrate for Chk1 kinase activity. p53 stabilization upon phosphorylation as well as phosphorylation of Cdc25 proteins lead to a checkpoint response mediating a blockade of the cell cycle [134].
Figure 5.
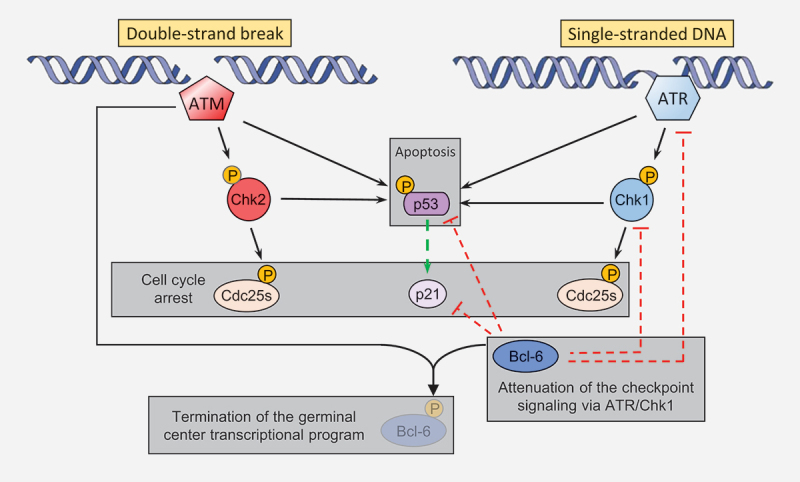
Regulation of checkpoint signaling in the germinal center DNA double-strand breaks lead to ATM activation, which phosphorylates Chk2, which in turn phosphorylates p53 for stabilization and induction of checkpoint responses. Single-stranded regions exposed upon replication stress or occurring during DNA repair activate ATR, which phosphorylates Chk1, which in turn phosphorylates p53 for induction of checkpoint responses. Both Chk1 and Chk2 also phosphorylate Cdc25 proteins for contributions to the checkpoint response. Activation of ATM upon DNA damage leads to phosphorylation and degradation of the master regulator of the germinal center program Bcl-6, which in turns attenuates the DNA damage response via ATR/Chk1/p53/p21 in germinal center B cells. Continuous lines indicate phosphorylation events, dashed lines indicate transcriptional regulation.
Early work on checkpoint signaling in the regulation of secondary Ig diversification has employed memory B cells from human patients with defects in ATM and ATR. It was found that ATM-deficient patients showed no defect in somatic hypermutation frequency or pattern [135], while ATR-deficient B cells showed an altered pattern of hypermutation [136]. While these results implied that the ATR axis is involved in the regulation of hypermutation, studies on the absolute efficacy of hypermutation could not be performed in this system, as selection had already occurred for these memory B cells.
Interestingly, other studies revealed a striking regulation of checkpoint signaling in the germinal center reaction (Figure 5). Recognition of DNA damage by ATM leads to phosphorylation and degradation of Bcl-6, thus terminating the germinal center program [137]. Also, ATM activation contributes to termination of the germinal center program through LKB1 and CRTC2 [138]. Conversely, members of the ATR axis, namely ATR itself, Chk1, p53 and its target gene p21 are all downregulated in the germinal center by Bcl-6 activity [139–142]. Moreover, the generation of chromosomal translocations during class switch recombination is repressed by checkpoint signaling [143,144]. Accordingly, it was interesting to investigate the impact of the ATM and ATR signaling axis on secondary Ig diversification.
We have therefore studied the role of Chk1 as well as Chk2 in the regulation of somatic hypermutation, class switch recombination, and Ig gene conversion in vitro. Interestingly, we found an inverse behavior upon inactivation of each of these two proteins. Chk1 downregulation led to increased hypermutation and decreased Ig gene conversion, likely due to reduced Chk1-dependent facilitation of homologous recombination, while class switch recombination was found to be barely affected [145]. Chk2-deficient cells showed lower rates of somatic hypermutation and class switch recombination but higher Ig gene conversion, likely upon higher activity of Chk1 in Chk2-deficient cells [146]. Investigation of the role of Chk1 in vivo also revealed an interesting phenotype: a decreased activity of Chk1 influences the mutational pattern during somatic hypermutation by facilitating A:T over C:G mutagenesis. We suggest that the reduced activation of Chk1 signaling in response to the single-stranded DNA stretches generated in the course of repair via mismatch repair facilitates gap refilling via Polη [147]. Moreover, we found that p53 regulates translesion synthesis-mediated mutagenesis at the switch regions during class switch recombination, while it does not affect somatic hypermutation of the Ig variable region in vivo [148]. The question still stands of whether the attenuation of the checkpoint signaling pathways in B cells undergoing Ig diversification is a global rather than a local phenomenon induced specifically at the Ig locus, as it may be presumed that the same outcome would be achieved in terms of Ig diversification. Interestingly, though, AID-induced DNA double-strand breaks, unlike radiation-induced breaks, do not strongly activate the G1/S checkpoint [121]. Also, PARP-1 activity regulates Chk1 at stalled replication forks [149], so local perturbations in PARP-1 activity might not only allow for higher AID function but also trigger more error-prone repair.
Regulation of DNA repair by the cell cycle. An increasing amount of evidence has shed light on the role of the cell cycle in regulating not only AID activity, but also the downstream mechanisms of error-prone repair at the Ig loci (Figure 6). Double-strand breaks formed during class switch recombination are generated in the G1 phase [150]. Accordingly, our recent study shows that AID activity in G1 is indispensable for class switch recombination to occur [151]. Interestingly, however, breaks generated at off-target genes were shown to preferentially persist till the S phase and to be repaired in an error-free fashion by homologous recombination [121,152]. The detection of uracils at the switch regions of in vitro-cultured primary mouse B cells revealed that uracils are processed quickly after generation in G1 at the Ig loci, as a scarce amount of those was detected in S/G2 [56]. UNG is known to be active mainly in the S phase where it removes uracils which have been misincorporated during replication. Nevertheless, the inhibition of UNG activity in G1 but not in S/G2 was shown to impair Ig diversification in vitro, indicating that UNG is highly active in G1 in B cells [153]. The G1 phase is also the predominant window for the activity of the A:T mutator during somatic hypermutation, as previously suggested and recently proved [151,154]. Concerning the pathways involved in C/G mutagenesis, we and others have shown that they are active throughout the cell cycle, even though the findings of our recent study point to an increased activity of UNG/translesion synthesis pathways in G1 [151,155].
Figure 6.
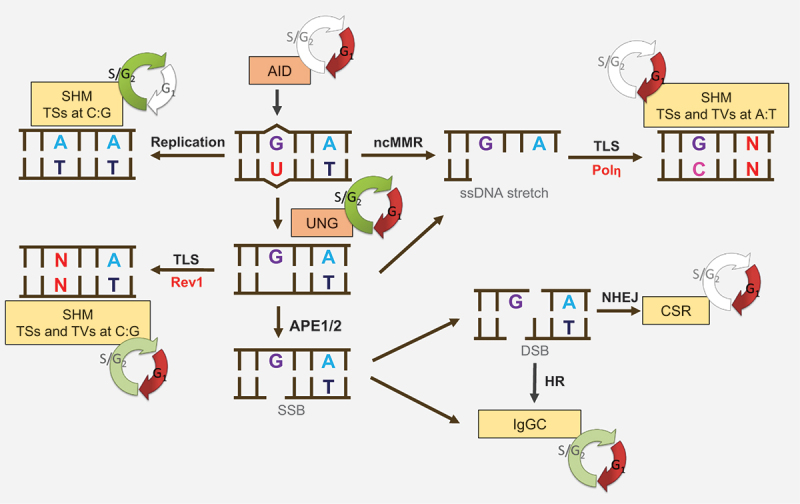
Role of cell cycle during secondary Ig diversification In physiological contexts AID acts exclusively in G1 phase. During SHM, the DNA repair is regulated by the cell cycle. A/T mutagenesis is accomplished in G1, while UNG acts throughout the cell cycle. TLS via Rev1 is not restricted to a certain cell cycle phase, but is more efficient in G1. The process of replication is associated with TSs at C:G base pairs and acts in S/G2. Class switch recombination (CSR) is initiated in G1 and completed immediately after the G1/S transition. Ig gene conversion (IgGC) can occur throughout the cell cycle but is facilitated when initiated in G1. SSB single-strand break, DSB double-strand break.
Therefore, in at least two cases DNA repair at the Ig genes is indeed mainly error-prone because of occurring in the wrong cell cycle phase. In case of A:T mutagenesis, PCNA monoubiquitination and translesion synthesis by Pol is performed in an error-prone manner in G1, representing non-canonical mismatch repair which has been shown to occur also in G1 cells [156]. In this instance, the error-free alternative via PCNA polyubiquitination and a template switch to the sister chromatid [157] would only be available in S/G2, and thus the pathway responsible for the A:T mutator is error-prone in G1. For the C:G mutator, a less pronounced but similar effect may be seen: Uracils persisting in S/G2 are more likely to generate transition mutations while transversions are generated more efficiently in the G1 phase by translesion synthesis via Rev1, which may act in a PCNA ubiquitination-independent manner [158]. Likewise, during class switch recombination, the ligation of the AID-induced breaks by error-prone non-homologous end joining occurs only at lesions induced in G1, while the alternative error-free process of homologous recombination takes care of the breaks in non-Ig genes during S/G2 [121]. While these notions are highly intriguing, they of course now pose the question of why lesions in Ig genes would be repaired in G1 to achieve class switch recombination and A:T (+ some C:G) mutagenesis, while lesions in other genes are preferentially processed in S/G2. Also, it remains to be identified why the C:G mutator is error-prone both in G1 as well as in S/G2. Potentially, though, mutagenesis needs to occur mainly in G1 as it would be too deleterious in replicating cells. All these topics will need to be investigated in future studies.
Deregulation of secondary Ig diversification during lymphomagenesis
Human lymphomas often resemble certain stages of normal B cells in marker expression and behavior, a finding which has led to the concept that B cell lymphomas are trapped at the state of their cell of origin [22]. Several molecules or signaling pathways that are constitutively active in lymphomas have been identified, such as c-Myc overexpression in Burkitt’s lymphoma, Bcl-2 overexpression in follicular lymphoma, or Bcl-6 deregulation in diffuse large B cell lymphoma (DLBCL) [21]. In these cases, chromosomal translocations of the respective genes into the Ig locus occur, which may be due to aberrant processing of AID-induced lesions [159]. Alternatively, somatic hypermutation-like mutagenesis in the 5’ region of certain genes may impinge on their expression [102,160]. Accordingly, it has been shown in mouse models that AID is required for germinal center derived lymphomagenesis [161,162].
While Bcl-6 is a master transcription factor of the germinal center program [163,164], and thus a germinal center appearance of DLBCL is not unexpected, the contribution of Bcl-2 and c-Myc to the germinal center phenotype of the respective lymphoma cells is less clear. We have shown that c-Myc deregulation leads to a germinal center phenotype and constitutive somatic hypermutation in Burkitt’s lymphoma, but other pathways must contribute as well, as mere c-Myc overexpression in B cells does not trigger constitutive hypermutation [165]. Genetic mouse models are being used to clarify such remaining questions [166].
In general, though, Burkitt’s lymphoma is characterized by ongoing somatic hypermutation [167,168], as is follicular lymphoma [169,170]. DLBCL is characterized by aberrant hypermutation [171], i.e. the appearance of mutations in genes that are not hypermutation targets in normal germinal center B cells. The molecular pathways leading to such constitutive or aberrant hypermutation still need to be identified, as mere AID overexpression is not always sufficient to trigger somatic hypermutation [172,173].
Future perspectives
Intensive research in the last two decades since the discovery of AID has provided us with a thorough understanding of the basic mechanism of secondary immunoglobulin diversification. At first glance, one may even get the impression that the key questions of the field are answered, but in fact several completely mysterious topics remain.
Concerning the basic mechanism of Ig diversification, the pathways of deregulation of DNA repair at the Ig locus deserve further study. This includes the G1 restriction of the A:T mutator and class switch recombination, as well as the error-prone processing of AID-induced lesions by the C:G mutator at Ig loci, while other loci in the cell are repaired by the same repair pathways in an error-free manner. This understanding is crucial to solve the main puzzle of secondary Ig diversification: specific targeting to Ig loci.
Concerning deregulated Ig diversification, the main pathways leading to constitutive hypermutation as well as aberrant hypermutation in B cell lymphoma still need to be identified. While for Burkitt lymphoma adequate cell lines to study this do exist [165,167,174], clarification of the molecular mechanism of constitutive or aberrant hypermutation in follicular lymphoma as well as DLBCL will require the establishment of adequate cellular models.
Accordingly, research in the field will still prosper and make major contributions to our understanding of major pathways of normal and deregulated immunity.
Acknowledgements
We apologize to all colleagues whose important work could not be cited due to space and reference number limitations. We thank Liane Giebeler and Vanessa Gölling for contributions to the figures of this review, and Katherine Dickinson for critical reading of the manuscript.
Funding Statement
The work was supported by the Deutsche Forschungsgemeinschaft [JU2690/1-1 and 1-2, JU2690/4-1 and 4-2]; Deutsche Krebshilfe [70112155].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- [1].Boehm T, Swann JB.. Origin and evolution of adaptive immunity. Ann Rev Anim Biosci. 2014;2(1):259–283. doi: 10.1146/annurev-animal-022513-114201 [DOI] [PubMed] [Google Scholar]
- [2].Roth DB. V(D)J recombination: mechanism, errors, and fidelity. Microbiol Spectr. 2014;2(6). doi: 10.1128/microbiolspec.MDNA3-0041-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381(6585):751–758. doi: 10.1038/381751a0 [DOI] [PubMed] [Google Scholar]
- [4].MacLennan IC, Toellner KM, Cunningham AF, et al. Extrafollicular antibody responses. Immunol Rev. 2003;194(1):8–18. doi: 10.1034/j.1600-065X.2003.00058.x [DOI] [PubMed] [Google Scholar]
- [5].Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67(6):1121–1129. doi: 10.1016/0092-8674(91)90289-B [DOI] [PubMed] [Google Scholar]
- [6].Jacob J, Kelsoe G, Rajewsky K, et al. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354(6352):389–392. doi: 10.1038/354389a0 [DOI] [PubMed] [Google Scholar]
- [7].Elsner RA, Shlomchik MJ. Germinal center and extrafollicular B cell responses in vaccination, immunity, and autoimmunity. Immunity. 2020;53(6):1136–1150. doi: 10.1016/j.immuni.2020.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Weller S, Faili A, Garcia C, et al. CD40-CD40L independent Ig gene hypermutation suggests a second B cell diversification pathway in humans. Proc Natl Acad Sci U S A. 2001;98(3):1166–1170. doi: 10.1073/pnas.98.3.1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. 2015;15(3):137–148. doi: 10.1038/nri3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shlomchik MJ, Luo W, Weisel F. Linking signaling and selection in the germinal center. Immunol Rev. 2019;288(1):49–63. doi: 10.1111/imr.12744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Roco JA, Mesin L, Binder SC, et al. Class-switch recombination occurs infrequently in germinal centers. Immunity. 2019;51(2):337–50 e7. doi: 10.1016/j.immuni.2019.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lau AW, Brink R. Selection in the germinal center. Curr Opin Immunol. 2020;63:29–34. doi: 10.1016/j.coi.2019.11.001 [DOI] [PubMed] [Google Scholar]
- [13].Muramatsu M, Kinoshita K, Fagarasan S, et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–563. doi: 10.1016/S0092-8674(00)00078-7 [DOI] [PubMed] [Google Scholar]
- [14].Revy P, Muto T, Levy Y, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell. 2000;102(5):565–575. doi: 10.1016/S0092-8674(00)00079-9 [DOI] [PubMed] [Google Scholar]
- [15].Dalloul I, Laffleur B, Dalloul Z, et al. UnAIDed class switching in activated B-cells reveals intrinsic features of a self-cleaving IgH locus. Front Immunol. 2021;12:737427. doi: 10.3389/fimmu.2021.737427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419(6902):43–48. doi: 10.1038/nature00981 [DOI] [PubMed] [Google Scholar]
- [17].Maul RW, Saribasak H, Martomo SA, et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nat Immunol. 2011;12(1):70–76. doi: 10.1038/ni.1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Methot SP, Di Noia JM. Molecular mechanisms of somatic hypermutation and class switch recombination. Adv Immunol. 2017;133:37–87. [DOI] [PubMed] [Google Scholar]
- [19].Arakawa H, Buerstedde JM. Immunoglobulin gene conversion: insights from bursal B cells and the DT40 cell line. Dev Dyn. 2004;229(3):458–464. doi: 10.1002/dvdy.10495 [DOI] [PubMed] [Google Scholar]
- [20].Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295(5558):1301–1306. doi: 10.1126/science.1067308 [DOI] [PubMed] [Google Scholar]
- [21].Pasqualucci L. Molecular pathogenesis of germinal center-derived B cell lymphomas. Immunol Rev. 2019;288(1):240–261. doi: 10.1111/imr.12745 [DOI] [PubMed] [Google Scholar]
- [22].Kuppers R, Klein U, Hansmann ML, et al. Cellular origin of human B-cell lymphomas. N Engl J Med. 1999;341(20):1520–1529. doi: 10.1056/NEJM199911113412007 [DOI] [PubMed] [Google Scholar]
- [23].Liu M, Duke JL, Richter DJ, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451(7180):841–845. doi: 10.1038/nature06547 [DOI] [PubMed] [Google Scholar]
- [24].Tran TH, Nakata M, Suzuki K, et al. B cell-specific and stimulation-responsive enhancers derepress aicda by overcoming the effects of silencers. Nat Immunol. 2010;11(2):148–154. doi: 10.1038/ni.1829 [DOI] [PubMed] [Google Scholar]
- [25].Gonda H, Sugai M, Nambu Y, et al. The balance between Pax5 and Id2 activities is the key to AID gene expression. J Exp Med. 2003;198(9):1427–1437. doi: 10.1084/jem.20030802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sayegh CE, Quong MW, Agata Y, et al. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. 2003;4(6):586–593. doi: 10.1038/ni923 [DOI] [PubMed] [Google Scholar]
- [27].Park SR, Zan H, Pal Z, et al. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nat Immunol. 2009;10(5):540–550. doi: 10.1038/ni.1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yadav A, Olaru A, Saltis M, et al. Identification of a ubiquitously active promoter of the murine activation-induced cytidine deaminase (AICDA) gene. Mol Immunol. 2006;43(6):529–541. doi: 10.1016/j.molimm.2005.05.007 [DOI] [PubMed] [Google Scholar]
- [29].Dedeoglu F, Horwitz B, Chaudhuri J, et al. Induction of activation-induced cytidine deaminase gene expression by IL-4 and CD40 ligation is dependent on STAT6 and NFkappaB. Int Immunol. 2004;16(3):395–404. doi: 10.1093/intimm/dxh042 [DOI] [PubMed] [Google Scholar]
- [30].Gehringer F, Weissinger SE, Swier LJ, et al. FOXO1 confers maintenance of the dark zone proliferation and survival program and can be pharmacologically targeted in Burkitt lymphoma. Cancers (Basel). 2019;11(10):1427. doi: 10.3390/cancers11101427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gallagher E, Enzler T, Matsuzawa A, et al. Kinase MEKK1 is required for CD40-dependent activation of the kinases jnk and p38, germinal center formation, B cell proliferation and antibody production. Nat Immunol. 2007;8(1):57–63. doi: 10.1038/ni1421 [DOI] [PubMed] [Google Scholar]
- [32].Tunyaplin C, Shaffer AL, Angelin-Duclos CD, et al. Direct repression of prdm1 by Bcl-6 inhibits plasmacytic differentiation. J Immunol. 2004;173(2):1158–1165. doi: 10.4049/jimmunol.173.2.1158 [DOI] [PubMed] [Google Scholar]
- [33].Alinikula J, Nera KP, Junttila S, et al. Alternate pathways for Bcl6-mediated regulation of B cell to plasma cell differentiation. Eur J Immunol. 2011;41(8):2404–2413. doi: 10.1002/eji.201141553 [DOI] [PubMed] [Google Scholar]
- [34].Basso K, Schneider C, Shen Q, et al. BCL6 positively regulates AID and germinal center gene expression via repression of miR-155. J Exp Med. 2012;209(13):2455–2465. doi: 10.1084/jem.20121387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Teng G, Hakimpour P, Landgraf P, et al. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity. 2008;28(5):621–629. doi: 10.1016/j.immuni.2008.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Dorsett Y, McBride KM, Jankovic M, et al. MicroRNA-155 suppresses activation-induced cytidine deaminase-mediated Myc-igh translocation. Immunity. 2008;28(5):630–638. doi: 10.1016/j.immuni.2008.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pasqualucci L, Guglielmino R, Houldsworth J, et al. Expression of the AID protein in normal and neoplastic B cells. Blood. 2004;104(10):3318–3325. doi: 10.1182/blood-2004-04-1558 [DOI] [PubMed] [Google Scholar]
- [38].Hasler J, Rada C, Neuberger MS. Cytoplasmic activation-induced cytidine deaminase (AID) exists in stoichiometric complex with translation elongation factor 1alpha (eEF1A). Proc Natl Acad Sci U S A. 2011;108(45):18366–18371. doi: 10.1073/pnas.1106729108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Methot SP, Litzler LC, Trajtenberg F, et al. Consecutive interactions with HSP90 and eEF1A underlie a functional maturation and storage pathway of AID in the cytoplasm. J Exp Med. 2015;212(4):581–596. doi: 10.1084/jem.20141157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Patenaude AM, Orthwein A, Hu Y, et al. Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nat Struct Mol Biol. 2009;16(5):517–527. doi: 10.1038/nsmb.1598 [DOI] [PubMed] [Google Scholar]
- [41].Hu Y, Ericsson I, Torseth K, et al. A combined nuclear and nucleolar localization motif in activation-induced cytidine deaminase (AID) controls immunoglobulin class switching. J Mol Biol. 2013;425(2):424–443. doi: 10.1016/j.jmb.2012.11.026 [DOI] [PubMed] [Google Scholar]
- [42].Ganesh K, Adam S, Taylor B, et al. CTNNBL1 is a novel nuclear localization sequence-binding protein that recognizes RNA-splicing factors CDC5L and Prp31. J Biol Chem. 2011;286(19):17091–17102. doi: 10.1074/jbc.M110.208769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kawaguchi Y, Nariki H, Kawamoto N, et al. SRSF1-3 contributes to diversification of the immunoglobulin variable region gene by promoting accumulation of AID in the nucleus. Biochem Biophys Res Commun. 2017;485(2):261–266. doi: 10.1016/j.bbrc.2017.02.097 [DOI] [PubMed] [Google Scholar]
- [44].Maeda K, Singh SK, Eda K, et al. GANP-mediated recruitment of activation-induced cytidine deaminase to cell nuclei and to immunoglobulin variable region DNA. J Biol Chem. 2010;285(31):23945–23953. doi: 10.1074/jbc.M110.131441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Bennett RP, Diner E, Sowden MP, et al. APOBEC-1 and AID are nucleo-cytoplasmic trafficking proteins but APOBEC3G cannot traffic. Biochem Biophys Res Commun. 2006;350(1):214–219. doi: 10.1016/j.bbrc.2006.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ito S, Nagaoka H, Shinkura R, et al. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc Natl Acad Sci U S A. 2004;101(7):1975–1980. doi: 10.1073/pnas.0307335101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Brar SS, Watson M, Diaz M. Activation-induced cytosine deaminase (AID) is actively exported out of the nucleus but retained by the induction of DNA breaks. J Biol Chem. 2004;279(25):26395–26401. doi: 10.1074/jbc.M403503200 [DOI] [PubMed] [Google Scholar]
- [48].McBride KM, Barreto V, Ramiro AR, et al. Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J Exp Med. 2004;199(9):1235–1244. doi: 10.1084/jem.20040373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Orthwein A, Zahn A, Methot SP, et al. Optimal functional levels of activation-induced deaminase specifically require the Hsp40 DnaJa1. EMBO J. 2012;31(3):679–691. doi: 10.1038/emboj.2011.417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Orthwein A, Patenaude AM, Affar el B, et al. Regulation of activation-induced deaminase stability and antibody gene diversification by Hsp90. J Exp Med. 2010;207(12):2751–2765. doi: 10.1084/jem.20101321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Aoufouchi S, Faili A, Zober C, et al. Proteasomal degradation restricts the nuclear lifespan of AID. J Exp Med. 2008;205(6):1357–1368. doi: 10.1084/jem.20070950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Uchimura Y, Barton LF, Rada C, et al. REG-gamma associates with and modulates the abundance of nuclear activation-induced deaminase. J Exp Med. 2011;208(12):2385–2391. doi: 10.1084/jem.20110856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Luo Y, Liu Y, Wu L, et al. CUL7 E3 ubiquitin ligase mediates the degradation of activation-induced cytidine deaminase and regulates the Ig class switch recombination in B lymphocytes. J Immunol. 2019;203(1):269–281. doi: 10.4049/jimmunol.1900125 [DOI] [PubMed] [Google Scholar]
- [54].Zaprazna K, Atchison ML. YY1 controls immunoglobulin class switch recombination and nuclear activation-induced deaminase levels. Mol Cell Biol. 2012;32(8):1542–1554. doi: 10.1128/MCB.05989-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ordinario EC, Yabuki M, Larson RP, et al. Temporal regulation of Ig gene diversification revealed by single-cell imaging. J Immunol. 2009;183(7):4545–4553. doi: 10.4049/jimmunol.0900673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang Q, Kieffer-Kwon KR, Oliveira TY, et al. The cell cycle restricts activation-induced cytidine deaminase activity to early G1. J Exp Med. 2017;214(1):49–58. doi: 10.1084/jem.20161649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Le Q, Maizels N, Aguilera A. Cell cycle regulates nuclear stability of AID and determines the cellular response to AID. PLoS Genet. 2015;11(9):e1005411. doi: 10.1371/journal.pgen.1005411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chaudhuri J, Khuong C, Alt FW. Replication protein a interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430(7003):992–998. doi: 10.1038/nature02821 [DOI] [PubMed] [Google Scholar]
- [59].Heltzel JMH, Gearhart PJ. What targets somatic hypermutation to the immunoglobulin loci? Viral Immunol. 2020;33(4):277–281. doi: 10.1089/vim.2019.0149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Storb U, Peters A, Klotz E, et al. Cis-acting sequences that affect somatic hypermutation of Ig genes. Immunol Rev. 1998;162(1):153–160. doi: 10.1111/j.1600-065X.1998.tb01438.x [DOI] [PubMed] [Google Scholar]
- [61].Pinaud E, Khamlichi AA, Le Morvan C, et al. Localization of the 3’ IgH locus elements that effect long-distance regulation of class switch recombination. Immunity. 2001;15(2):187–199. doi: 10.1016/S1074-7613(01)00181-9 [DOI] [PubMed] [Google Scholar]
- [62].Rouaud P, Vincent-Fabert C, Saintamand A, et al. The IgH 3’ regulatory region controls somatic hypermutation in germinal center B cells. J Exp Med. 2013;210(8):1501–1507. doi: 10.1084/jem.20130072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhang X, Zhang Y, Ba Z, et al. Fundamental roles of chromatin loop extrusion in antibody class switching. Nature. 2019;575(7782):385–389. doi: 10.1038/s41586-019-1723-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Le Noir S, Bonaud A, Herve B, et al. IgH 3’ regulatory region increases ectopic class switch recombination. PLoS Genet. 2021;17(2):e1009288. doi: 10.1371/journal.pgen.1009288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kenter AL. AID targeting is dependent on RNA polymerase II pausing. Semin Immunol. 2012;24(4):281–286. doi: 10.1016/j.smim.2012.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pavri R, Gazumyan A, Jankovic M, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143(1):122–133. doi: 10.1016/j.cell.2010.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wang X, Fan M, Kalis S, et al. A source of the single-stranded DNA substrate for activation-induced deaminase during somatic hypermutation. Nat Commun. 2014;5(1):4137. doi: 10.1038/ncomms5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Rajagopal D, Maul RW, Ghosh A, et al. Immunoglobulin switch mu sequence causes RNA polymerase II accumulation and reduces dA hypermutation. J Exp Med. 2009;206(6):1237–1244. doi: 10.1084/jem.20082514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Dollinger R, Gilmour DS. Regulation of promoter proximal pausing of RNA polymerase II in metazoans. J Mol Biol. 2021;433(14):166897. doi: 10.1016/j.jmb.2021.166897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Pefanis E, Wang J, Rothschild G, et al. Noncoding RNA transcription targets AID to divergently transcribed loci in B cells. Nature. 2014;514(7522):389–393. doi: 10.1038/nature13580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Rothschild G, Zhang W, Lim J, et al. Noncoding RNA transcription alters chromosomal topology to promote isotype-specific class switch recombination. Sci Immunol. 2020;5(44). doi: 10.1126/sciimmunol.aay5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Qian J, Wang Q, Dose M, et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014;159(7):1524–1537. doi: 10.1016/j.cell.2014.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Meng FL, Du Z, Federation A, et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014;159(7):1538–1548. doi: 10.1016/j.cell.2014.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zheng S, Vuong BQ, Vaidyanathan B, et al. Non-coding RNA generated following lariat debranching mediates targeting of AID to DNA. Cell. 2015;161(4):762–773. doi: 10.1016/j.cell.2015.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Qiao Q, Wang L, Meng FL, et al. AID recognizes structured DNA for class switch recombination. Mol Cell. 2017;67(3):361–73 e4. doi: 10.1016/j.molcel.2017.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science. 1995;267(5205):1825–1828. doi: 10.1126/science.7892607 [DOI] [PubMed] [Google Scholar]
- [77].Hein K, Lorenz MG, Siebenkotten G, et al. Processing of switch transcripts is required for targeting of antibody class switch recombination. J Exp Med. 1998;188(12):2369–2374. doi: 10.1084/jem.188.12.2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Nowak U, Matthews AJ, Zheng S, et al. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat Immunol. 2011;12(2):160–166. doi: 10.1038/ni.1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Singh SK, Maeda K, Eid MM, et al. GANP regulates recruitment of AID to immunoglobulin variable regions by modulating transcription and nucleosome occupancy. Nat Commun. 2013;4(1):1830. doi: 10.1038/ncomms2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Willmann KL, Milosevic S, Pauklin S, et al. A role for the RNA pol II-associated PAF complex in AID-induced immune diversification. J Exp Med. 2012;209(11):2099–2111. doi: 10.1084/jem.20112145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Xu Z, Fulop Z, Wu G, et al. 14-3-3 adaptor proteins recruit AID to 5’-AGCT-3’-rich switch regions for class switch recombination. Nat Struct Mol Biol. 2010;17(9):1124–1135. doi: 10.1038/nsmb.1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Li G, White CA, Lam T, et al. Combinatorial H3K9acS10ph histone modification in IgH locus S regions targets 14-3-3 adaptors and AID to specify antibody class-switch DNA recombination. Cell Rep. 2013;5(3):702–714. doi: 10.1016/j.celrep.2013.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Jeevan-Raj BP, Robert I, Heyer V, et al. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J Exp Med. 2011;208(8):1649–1660. doi: 10.1084/jem.20110118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Li G, Zan H, Xu Z, et al. Epigenetics of the antibody response. Trends Immunol. 2013;34(9):460–470. doi: 10.1016/j.it.2013.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Methot SP, Litzler LC, Subramani PG, et al. A licensing step links AID to transcription elongation for mutagenesis in B cells. Nat Commun. 2018;9(1):1248. doi: 10.1038/s41467-018-03387-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Basu U, Meng FL, Keim C, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144(3):353–363. doi: 10.1016/j.cell.2011.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wei L, Chahwan R, Wang S, et al. Overlapping hotspots in CDRs are critical sites for V region diversification. Proc Natl Acad Sci U S A. 2015;112(7):E728–37. doi: 10.1073/pnas.1500788112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Zarrin AA, Alt FW, Chaudhuri J, et al. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nat Immunol. 2004;5(12):1275–1281. doi: 10.1038/ni1137 [DOI] [PubMed] [Google Scholar]
- [89].Basu U, Chaudhuri J, Alpert C, et al. The AID antibody diversification enzyme is regulated by protein kinase a phosphorylation. Nature. 2005;438(7067):508–511. doi: 10.1038/nature04255 [DOI] [PubMed] [Google Scholar]
- [90].McBride KM, Gazumyan A, Woo EM, et al. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proc Natl Acad Sci U S A. 2006;103(23):8798–8803. doi: 10.1073/pnas.0603272103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].McBride KM, Gazumyan A, Woo EM, et al. Regulation of class switch recombination and somatic mutation by AID phosphorylation. J Exp Med. 2008;205(11):2585–2594. doi: 10.1084/jem.20081319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Pham P, Smolka MB, Calabrese P, et al. Impact of phosphorylation and phosphorylation-null mutants on the activity and deamination specificity of activation-induced cytidine deaminase. J Biol Chem. 2008;283(25):17428–17439. doi: 10.1074/jbc.M802121200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Cheng HL, Vuong BQ, Basu U, et al. Integrity of the AID serine-38 phosphorylation site is critical for class switch recombination and somatic hypermutation in mice. Proc Natl Acad Sci U S A. 2009;106(8):2717–2722. doi: 10.1073/pnas.0812304106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Chatterji M, Unniraman S, McBride KM, et al. Role of activation-induced deaminase protein kinase a phosphorylation sites in Ig gene conversion and somatic hypermutation. J Immunol. 2007;179(8):5274–5280. doi: 10.4049/jimmunol.179.8.5274 [DOI] [PubMed] [Google Scholar]
- [95].Pasqualucci L, Kitaura Y, Gu H, et al. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci U S A. 2006;103(2):395–400. doi: 10.1073/pnas.0509969103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Vuong BQ, Lee M, Kabir S, et al. Specific recruitment of protein kinase a to the immunoglobulin locus regulates class-switch recombination. Nat Immunol. 2009;10(4):420–426. doi: 10.1038/ni.1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, et al. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class-switch recombination. Nat Immunol. 2013;14(11):1183–1189. doi: 10.1038/ni.2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Tepper S, Mortusewicz O, Czlonka E, et al. Restriction of AID activity and somatic hypermutation by PARP-1. Nucleic Acids Res. 2019;47(14):7418–7429. doi: 10.1093/nar/gkz466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Tepper S, Jeschke J, Bottcher K, et al. PARP activation promotes nuclear AID accumulation in lymphoma cells. Oncotarget. 2016;7(11):13197–13208. doi: 10.18632/oncotarget.7603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kraus WL. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol. 2008;20(3):294–302. doi: 10.1016/j.ceb.2008.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ambrose HE, Papadopoulou V, Beswick RW, et al. Poly-(ADP-ribose) polymerase-1 (parp-1) binds in a sequence-specific manner at the Bcl-6 locus and contributes to the regulation of Bcl-6 transcription. Oncogene. 2007;26(42):6244–6252. doi: 10.1038/sj.onc.1210434 [DOI] [PubMed] [Google Scholar]
- [102].Pasqualucci L, Migliazza A, Fracchiolla N, et al. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci U S A. 1998;95(20):11816–11821. doi: 10.1073/pnas.95.20.11816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Casellas R, Basu U, Yewdell WT, et al. Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nat Rev Immunol. 2016;16(3):164–176. doi: 10.1038/nri.2016.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Yamane A, Resch W, Kuo N, et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol. 2011;12(1):62–69. doi: 10.1038/ni.1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Alvarez-Prado AF, Perez-Duran P, Perez-Garcia A, et al. A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. J Exp Med. 2018;215(3):761–771. doi: 10.1084/jem.20171738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Duke JL, Liu M, Yaari G, et al. Multiple transcription factor binding sites predict AID targeting in non-Ig genes. J Immunol. 2013;190(8):3878–3888. doi: 10.4049/jimmunol.1202547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Saribasak H, Maul RW, Cao Z, et al. XRCC1 suppresses somatic hypermutation and promotes alternative nonhomologous end joining in igh genes. J Exp Med. 2011;208(11):2209–2216. doi: 10.1084/jem.20111135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Wu X, Stavnezer J. DNA polymerase beta is able to repair breaks in switch regions and plays an inhibitory role during immunoglobulin class switch recombination. J Exp Med. 2007;204(7):1677–1689. doi: 10.1084/jem.20070756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Jansen JG, Langerak P, Tsaalbi-Shtylik A, et al. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J Exp Med. 2006;203(2):319–323. doi: 10.1084/jem.20052227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Rada C, Ehrenstein MR, Neuberger MS, et al. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 1998;9(1):135–141. doi: 10.1016/S1074-7613(00)80595-6 [DOI] [PubMed] [Google Scholar]
- [111].Kim N, Bozek G, Lo JC, et al. Different mismatch repair deficiencies all have the same effects on somatic hypermutation: intact primary mechanism accompanied by secondary modifications. J Exp Med. 1999;190(1):21–30. doi: 10.1084/jem.190.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Zeng X, Winter DB, Kasmer C, et al. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat Immunol. 2001;2(6):537–541. doi: 10.1038/88740 [DOI] [PubMed] [Google Scholar]
- [113].Bachl J, Ertongur I, Jungnickel B. Involvement of Rad18 in somatic hypermutation. Proc Natl Acad Sci U S A. 2006;103(32):12081–12086. doi: 10.1073/pnas.0605146103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Arakawa H, Moldovan GL, Saribasak H, et al. A role for PCNA ubiquitination in immunoglobulin hypermutation. PLoS Biol. 2006;4(11):e366. doi: 10.1371/journal.pbio.0040366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Langerak P, Nygren AO, Krijger PH, et al. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J Exp Med. 2007;204(8):1989–1998. doi: 10.1084/jem.20070902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kenter A, Wuerffel R. Immunoglobulin switch recombination may occur by a DNA end-joining mechanism a. Ann N Y Acad Sci. 1999;870(1):206–217. doi: 10.1111/j.1749-6632.1999.tb08880.x [DOI] [PubMed] [Google Scholar]
- [117].Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16(2):163–171. doi: 10.1016/j.molcel.2004.10.011 [DOI] [PubMed] [Google Scholar]
- [118].Bezzubova O, Silbergleit A, Yamaguchi-Iwai Y, et al. Reduced X-ray resistance and homologous recombination frequencies in a RAD54-/- mutant of the chicken DT40 cell line. Cell. 1997;89(2):185–193. doi: 10.1016/S0092-8674(00)80198-1 [DOI] [PubMed] [Google Scholar]
- [119].Hasham MG, Donghia NM, Coffey E, et al. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nat Immunol. 2010;11(9):820–826. doi: 10.1038/ni.1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Sale JE, Calandrini DM, Takata M, et al. Ablation of XRCC2/3 transforms immunoglobulin V gene conversion into somatic hypermutation. Nature. 2001;412(6850):921–926. doi: 10.1038/35091100 [DOI] [PubMed] [Google Scholar]
- [121].Hasham MG, Snow KJ, Donghia NM, et al. Activation-induced cytidine deaminase-initiated off-target DNA breaks are detected and resolved during S phase. J Immunol. 2012;189(5):2374–2382. doi: 10.4049/jimmunol.1200414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Hirth G, Svensson C-M, Böttcher K, et al. Regulation of the germinal center reaction and somatic hypermutation dynamics by homologous recombination. J Immunol. 2019;203(6):1493–1501. doi: 10.4049/jimmunol.1900483 [DOI] [PubMed] [Google Scholar]
- [123].Sundaravinayagam D, Rahjouei A, Andreani M, et al. 53BP1 supports immunoglobulin class switch recombination independently of its DNA double-strand break end protection function. Cell Rep. 2019;28(6):1389–99 e6. doi: 10.1016/j.celrep.2019.06.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Vincendeau E, Wei W, Zhang X, et al. SHLD1 is dispensable for 53BP1-dependent V(D)J recombination but critical for productive class switch recombination. Nat Commun. 2022;13(1):3707. doi: 10.1038/s41467-022-31287-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Refaat AM, Nakata M, Husain A, et al. HNRNPU facilitates antibody class-switch recombination through C-NHEJ promotion and R-loop suppression. Cell Rep. 2023;42(3):112284. doi: 10.1016/j.celrep.2023.112284 [DOI] [PubMed] [Google Scholar]
- [126].Feng Y, Li C, Stewart JA, et al. FAM72A antagonizes UNG2 to promote mutagenic repair during antibody maturation. Nature. 2021;600(7888):324–328. doi: 10.1038/s41586-021-04144-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Rogier M, Moritz J, Robert I, et al. Fam72a enforces error-prone DNA repair during antibody diversification. Nature. 2021;600(7888):329–333. doi: 10.1038/s41586-021-04093-y [DOI] [PubMed] [Google Scholar]
- [128].Stavnezer J, Linehan EK, Thompson MR, et al. Differential expression of APE1 and APE2 in germinal centers promotes error-prone repair and A: T mutations during somatic hypermutation. Proc Natl Acad Sci U S A. 2014;111(25):9217–9222. doi: 10.1073/pnas.1405590111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Bahjat MSM, Pilzecker B, van Dam TP, et al. DNA polymerase beta prevents AID-in stigated mutagenic non-canonical mismatch DNA repair. bioRxiv 20200130926964. 2021. [Google Scholar]
- [130].Durandy A, Peron S, Taubenheim N, et al. Activation-induced cytidine deaminase: structure-function relationship as based on the study of mutants. Hum Mutat. 2006;27(12):1185–1191. doi: 10.1002/humu.20414 [DOI] [PubMed] [Google Scholar]
- [131].Ranjit S, Khair L, Linehan EK, et al. AID recruits UNG and Msh2 to Ig switch regions dependent upon the AID C terminus [corrected]. J Immunol. 2011;187(5):2464–2475. doi: 10.4049/jimmunol.1101406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Zahn A, Eranki AK, Patenaude AM, et al. Activation induced deaminase C-terminal domain links DNA breaks to end protection and repair during class switch recombination. Proc Natl Acad Sci U S A. 2014;111(11):E988–97. doi: 10.1073/pnas.1320486111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Lazzaro F, Giannattasio M, Puddu F, et al. Checkpoint mechanisms at the intersection between DNA damage and repair. DNA Repair. 2009;8(9):1055–1067. doi: 10.1016/j.dnarep.2009.04.022 [DOI] [PubMed] [Google Scholar]
- [134].Uto K, Inoue D, Shimuta K, et al. Chk1, but not Chk2, inhibits Cdc25 phosphatases by a novel common mechanism. EMBO J. 2004;23(16):3386–3396. doi: 10.1038/sj.emboj.7600328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Pan-Hammarstrom Q, Dai S, Zhao Y, et al. ATM is not required in somatic hypermutation of VH, but is involved in the introduction of mutations in the switch mu region. J Immunol. 2003;170(7):3707–3716. doi: 10.4049/jimmunol.170.7.3707 [DOI] [PubMed] [Google Scholar]
- [136].Pan-Hammarstrom Q, Lahdesmaki A, Zhao Y, et al. Disparate roles of ATR and ATM in immunoglobulin class switch recombination and somatic hypermutation. J Exp Med. 2006;203(1):99–110. doi: 10.1084/jem.20050595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Phan RT, Saito M, Kitagawa Y, et al. Genotoxic stress regulates expression of the proto-oncogene Bcl6 in germinal center B cells. Nat Immunol. 2007;8(10):1132–1139. doi: 10.1038/ni1508 [DOI] [PubMed] [Google Scholar]
- [138].Sherman MH, Kuraishy AI, Deshpande C, et al. AID-induced genotoxic stress promotes B cell differentiation in the germinal center via ATM and LKB1 signaling. Mol Cell. 2010;39(6):873–885. doi: 10.1016/j.molcel.2010.08.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Ranuncolo SM, Polo JM, Dierov J, et al. Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat Immunol. 2007;8(7):705–714. doi: 10.1038/ni1478 [DOI] [PubMed] [Google Scholar]
- [140].Ranuncolo SM, Polo JM, Melnick A. BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood Cells Mol Dis. 2008;41(1):95–99. doi: 10.1016/j.bcmd.2008.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432(7017):635–639. doi: 10.1038/nature03147 [DOI] [PubMed] [Google Scholar]
- [142].Phan RT, Saito M, Basso K, et al. BCL6 interacts with the transcription factor miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol. 2005;6(10):1054–1060. doi: 10.1038/ni1245 [DOI] [PubMed] [Google Scholar]
- [143].Ramiro AR, Jankovic M, Callen E, et al. Role of genomic instability and p53 in AID-induced c-myc-igh translocations. Nature. 2006;440(7080):105–109. doi: 10.1038/nature04495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Franco S, Gostissa M, Zha S, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21(2):201–214. doi: 10.1016/j.molcel.2006.01.005 [DOI] [PubMed] [Google Scholar]
- [145].Frankenberger S, Davari K, Fischer-Burkart S, et al. Checkpoint kinase 1 negatively regulates somatic hypermutation. Nucleic Acids Res. 2014;42(6):3666–3674. doi: 10.1093/nar/gkt1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Davari K, Frankenberger S, Schmidt A, et al. Checkpoint kinase 2 is required for efficient immunoglobulin diversification. Cell Cycle. 2014;13(23):3659–3669. doi: 10.4161/15384101.2014.964112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Bello A, Jungnickel B. Impact of Chk1 dosage on somatic hypermutation in vivo. Immunol Cell Biol. 2021;99(8):879–893. doi: 10.1111/imcb.12480 [DOI] [PubMed] [Google Scholar]
- [148].Bottcher K, Braunschmidt K, Hirth G, et al. Context-dependent regulation of immunoglobulin mutagenesis by p53. Mol Immunol. 2021;138:128–136. doi: 10.1016/j.molimm.2021.08.005 [DOI] [PubMed] [Google Scholar]
- [149].Min W, Bruhn C, Grigaravicius P, et al. Poly(adp-ribose) binding to Chk1 at stalled replication forks is required for S-phase checkpoint activation. Nat Commun. 2013;4(1):2993. doi: 10.1038/ncomms3993 [DOI] [PubMed] [Google Scholar]
- [150].Schrader CE, Guikema JE, Linehan EK, et al. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J Immunol. 2007;179(9):6064–6071. doi: 10.4049/jimmunol.179.9.6064 [DOI] [PubMed] [Google Scholar]
- [151].Bello A, Muller A, Hirth G, et al. Cell cycle–mediated regulation of secondary Ig diversification. J Immunol. 2023;210(10):1508–1518. doi: 10.4049/jimmunol.2100880 [DOI] [PubMed] [Google Scholar]
- [152].Yamane A, Robbiani DF, Resch W, et al. RPA accumulation during class switch recombination represents 5’-3’ DNA-end resection during the S-G2/M phase of the cell cycle. Cell Rep. 2013;3(1):138–147. doi: 10.1016/j.celrep.2012.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Sharbeen G, Yee CW, Smith AL, et al. Ectopic restriction of DNA repair reveals that UNG2 excises AID-induced uracils predominantly or exclusively during G1 phase. J Exp Med. 2012;209(5):965–974. doi: 10.1084/jem.20112379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Thientosapol ES, Bosnjak D, Durack T, et al. SAMHD1 enhances immunoglobulin hypermutation by promoting transversion mutation. Proc Natl Acad Sci U S A. 2018;115(19):4921–4926. doi: 10.1073/pnas.1719771115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Pilzecker B, Buoninfante OA, Pritchard C, et al. PrimPol prevents APOBEC/AID family mediated DNA mutagenesis. Nucleic Acids Res. 2016;44(10):4734–4744. doi: 10.1093/nar/gkw123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Pena-Diaz J, Bregenhorn S, Ghodgaonkar M, et al. Noncanonical mismatch repair as a source of genomic instability in human cells. Mol Cell. 2012;47(5):669–680. doi: 10.1016/j.molcel.2012.07.006 [DOI] [PubMed] [Google Scholar]
- [157].Ulrich HD. The RAD6 pathway: control of DNA damage bypass and mutagenesis by ubiquitin and SUMO. Chembiochem. 2005;6(10):1735–1743. doi: 10.1002/cbic.200500139 [DOI] [PubMed] [Google Scholar]
- [158].Edmunds CE, Simpson LJ, Sale JE. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell. 2008;30(4):519–529. doi: 10.1016/j.molcel.2008.03.024 [DOI] [PubMed] [Google Scholar]
- [159].Kuppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001;20(40):5580–5594. doi: 10.1038/sj.onc.1204640 [DOI] [PubMed] [Google Scholar]
- [160].Johnston JM, Yu MT, Carroll WL. C-myc hypermutation is ongoing in endemic, but not all Burkitt’s lymphoma. Blood. 1991;78(9):2419–2425. doi: 10.1182/blood.V78.9.2419.2419 [DOI] [PubMed] [Google Scholar]
- [161].Pasqualucci L, Bhagat G, Jankovic M, et al. AID is required for germinal center-derived lymphomagenesis. Nat Genet. 2008;40(1):108–112. doi: 10.1038/ng.2007.35 [DOI] [PubMed] [Google Scholar]
- [162].Robbiani DF, Deroubaix S, Feldhahn N, et al. Plasmodium infection promotes genomic instability and AID-Dependent B cell lymphoma. Cell. 2015;162(4):727–737. doi: 10.1016/j.cell.2015.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Dent AL, Shaffer AL, Yu X, et al. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276(5312):589–592. doi: 10.1126/science.276.5312.589 [DOI] [PubMed] [Google Scholar]
- [164].Ye BH, Cattoretti G, Shen Q, et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat Genet. 1997;16(2):161–170. doi: 10.1038/ng0697-161 [DOI] [PubMed] [Google Scholar]
- [165].Scheller H, Tobollik S, Kutzera A, et al. C-Myc overexpression promotes a germinal center-like program in Burkitt’s lymphoma. Oncogene. 2010;29(6):888–897. doi: 10.1038/onc.2009.377 [DOI] [PubMed] [Google Scholar]
- [166].Mossadegh-Keller N, Brisou G, Beyou A, et al. Human B lymphomas reveal their secrets through genetic mouse models. Front Immunol. 2021;12:683597. doi: 10.3389/fimmu.2021.683597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Sale JE, Neuberger MS. TdT-accessible breaks are scattered over the immunoglobulin V domain in a constitutively hypermutating B cell line. Immunity. 1998;9(6):859–869. doi: 10.1016/S1074-7613(00)80651-2 [DOI] [PubMed] [Google Scholar]
- [168].Chapman CJ, Zhou JX, Gregory C, et al. VH and VL gene analysis in sporadic Burkitt’s lymphoma shows somatic hypermutation, intraclonal heterogeneity, and a role for antigen selection. Blood. 1996;88(9):3562–3568. doi: 10.1182/blood.V88.9.3562.bloodjournal8893562 [DOI] [PubMed] [Google Scholar]
- [169].Meeker T, Lowder J, Cleary ML, et al. Emergence of idiotype variants during treatment of B-cell lymphoma with anti-idiotype antibodies. N Engl J Med. 1985;312(26):1658–1665. doi: 10.1056/NEJM198506273122602 [DOI] [PubMed] [Google Scholar]
- [170].Cleary ML, Meeker TC, Levy S, et al. Clustering of extensive somatic mutations in the variable region of an immunoglobulin heavy chain gene from a human B cell lymphoma. Cell. 1986;44(1):97–106. doi: 10.1016/0092-8674(86)90488-5 [DOI] [PubMed] [Google Scholar]
- [171].Pasqualucci L, Neumeister P, Goossens T, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412(6844):341–346. doi: 10.1038/35085588 [DOI] [PubMed] [Google Scholar]
- [172].Ruckerl F, Bachl J. Activation-induced cytidine deaminase fails to induce a mutator phenotype in the human pre-B cell line nalm-6. Eur J Immunol. 2005;35(1):290–298. doi: 10.1002/eji.200425315 [DOI] [PubMed] [Google Scholar]
- [173].Yoshikawa K, Okazaki IM, Eto T, et al. AID enzyme-induced hypermutation in an actively transcribed gene in fibroblasts. Science. 2002;296(5575):2033–2036. doi: 10.1126/science.1071556 [DOI] [PubMed] [Google Scholar]
- [174].Ruckerl F, Busse B, Bachl J. Episomal vectors to monitor and induce somatic hypermutation in human Burkitt-lymphoma cell lines. Mol Immunol. 2006;43(10):1645–1652. doi: 10.1016/j.molimm.2005.09.011 [DOI] [PubMed] [Google Scholar]