

Abstract
The quest for a sound treatment on the vulnerable population suffering and dying as a result of the blood flukes, S. mansoni is on the increase because both Praziquantel and Oxamniquine widely used for the treatment of Schistosomiasis for over 51 years suffer resistance and recurrence. Here-in, chemo-informatics techniques such as QSAR modeling, pharmacokinetic, docking alongside MD simulation were harnessed in designing novel 7-keto- sempevirolsempevirol derivatives that are more competent against S. mansoni. Upon QSAR screening, compound 15, which appears to be in the model's acceptability space, emerges the best with a high predicted activity. 5 new analogues with improved activity against Schistosomiasis better than the standard drug PZQ were designed from compound 15 (template 15*) on an account of the descriptors significance from the model with robust and validated parameters. Also their pharmacokinetic profiles indicates that the designed compounds have the characteristics of a good drug. Furthermore, docking evaluation fulfilled ranges from −113.121 to −100.79 kcal/mol (moldock score), with compound U1 being the best (least moldock score of −113.121 compared to PZQ and 15* (template) having a moldock score value of (−87.21 and −83.37 kcal/mol). 100-ns MD Simulation on the U1-docked complex was run using Desmond 2019–4 package. The nature and steadiness of U1 compound within the enzyme active site was further confirmed by RMSD, RMSF, RoG and H-bond assessment. Hence, we recommend compound U1 targeting the SmCB1 enzyme (6YI7) for Schistosomiasis treatment and for further medicinal evaluation and utilization.
Keywords: Schistosomiasis, Blood flukes, QSAR, Docking-simulation, Pharmacokinetics, MD simulation, Chemo-informatics, SmCB1and diterpenoids
1. Introduction
Schistosomiasis has drawn public health attention as the second fatal parasitic disease after malaria [1,2]. The three life-threatening species to man out of the twenty-three identified species so far are Schistosoma mansoni, Schistosoma japanicum and Schistosoma haematobium [3]. It has been reported that the sub-Saharan African regions have Roughly 9/10 cases of Schistosomiasis on a global scale, with about three hundred thousand deaths yearly in most regions of the sub-Saharan Africa was reported by Marchese et al. [4]. Schistosoma mansoni disease is also widespread in tropical and subtropical regions, especially areas that are underdeveloped and without access to sanitary facilities and clean water [5,6]. The digenetic biological clock is initiated when the miracidia in the infected snails (of the genus Biomphalaria) develop into cercariae, which then creep into the human host's dermis (skin) while in contact with contaminated freshwater bodies and instantly find their way to the blood venule, heart, lungs, and full bloom in the liver [7,8], after which thousands of laid eggs progressively move to the lumen of the small intestine and are eliminated in the host's feces or urine [9]. The eggs excreted by the definite host (human) into freshwater environments or sources through feces or urine eventually hatch and let out miracidia that infect snails. This completes the infection cycle and allows the parasite to be passed from host to host. Early signs of infection are blood in the pee, feces containing blood, and stomach aches [10].
Large egg deposits by Schistosoma mansoni in the human body result in Katayama fever, glomerular disease, hepatosplenic disease, and periportal fibrosis, which progress into fatal conditions such as portal hypertension and ascites [11,12]. Praziquantel (Biltricide®) is the most recognized drug to treat Schistosomiasis at the moment [13,14], while Oxamniquine has been allowed to be used, though it is specific to some species [15]. The former is presently the most active against all species known so far but less effective against the worm's larval forms, causing recurrence and resistance [[15], [16], [17]]. Hence, the hunt to develop new drugs with the potency to overcome the re-infection and resistance menace has become necessary. A confirmed therapeutic target, cathepsin B1, obtained by isolation from the organism S. mansoni (SmCB1), is the prime enzyme that the parasite (blood fluke) uses to convert blood proteins into the nutrients that are key to its survival [18,19]. Both the larval and adult forms of Schistosoma mansoni have also been reported in the literature to be sensitive to the diterpenoids 7-keto-sempervirol [20]. In-silico techniques, which involve virtual screening of databases at low cost considering labor, time, and money for hit compound discovery, are harnessed [[21], [22], [23]]. An in-silico method is adopted to evaluate the therapeutic potency of 7-keto-sempevirol analogues on Schistosoma disease by incorporating QSAR, pharmacokinetic profiling, docking evaluation, and MD simulation. QSAR provides computational know-how for predicting compounds activities by connecting the physicochemical traits to the structural components that regulate their activity [24,25]. Computations of these compounds pharmacological and pharmacokinetic properties were carried out through the use of an online tool known as SwissADME. Molecular docking simulation accounts for all the responses obtained due to ligand-protein association so as to determine the best confirmation for a stable complex and calculate Gibb's free energy [26]. QSAR modelling, pharmacokinetic profiling, docking simulation, and further validation of the docking studies (MD simulation) were employed in designing hypothetical 7-keto-sempevirol derivatives that are more competent, effective, and safer against Schistosoma mansoni, which is the sole aim of this work. The flow chart for this findings is depicted in Fig. 1.
Fig. 1.

Flow chart for the chemo-informatics studies performed.
2. Materials and methods
2.1. Based-paper and optimization of chemical structures
Twenty (24) potent derivatives of diterpenoid-7-keto-sempivirol with inhibitory activities against S. mansoni were reported in a published literature [16] and Pubchem library in CSV file format with registration number AID 1358872 for certainty. The activities were presented in micromolar (uM) inhibitory concentrations (IC50) and converted to PIC50 harnessing equation one for discarding the disproportion that exist between the data's thereby making it suitable for this studies [27,28]. Table 1 shows the chemical structures in 2-D format, their activities, and the calculated residual values.
| (1) |
Table 1.
2-D compound structural drawings achieved with the aid of ChemDraw, their inhibitory activities, and residual values.
| S/No | Structure | IC50 | PIC50 | PIC50Pred | Residual |
|---|---|---|---|---|---|
| 1 |  |
21.5 | 4.6676 | 4.6863 | −0.0187 |
| 2 |  |
26.6 | 4.5751 | 4.6631 | −0.088 |
| 3 |  |
25.6 | 4.5918 | 4.6480 | −0.0562 |
| 4 |  |
28.1 | 4.5513 | 4.6247 | −0.0734 |
| 5 | 27.1 | 4.5670 | 4.5129 | 0.0541 | |
| 6 | 40.1 | 4.3969 | 4.3851 | 0.0118 | |
| 7 |  |
14.7 | 4.8327 | 4.8565 | −0.0238 |
| 8 |  |
23.5 | 4.6289 | 4.6407 | −0.0118 |
| 9 |  |
43.6 | 4.3605 | 4.4540 | −0.0935 |
| 10 |  |
13 | 4.8861 | 4.8979 | −0.0118 |
| 11 |  |
19.1 | 4.7190 | 4.5880 | 0.1310 |
| 12 |  |
18.9 | 4.7235 | 4.6471 | 0.0764 |
| 13 | 43.8 | 4.3585 | 4.2902 | 0.0683 | |
| 14 |  |
27.3 | 4.5638 | 4.7073 | −0.1435 |
| 15 |  |
6.3 | 5.2007 | 5.1127 | 0.0880 |
| 16 |  |
6.4 | 5.1938 | 5.1252 | 0.0686 |
| 17 |  |
18.2 | 4.7399 | 4.6884 | 0.0515 |
| 18 |  |
27.9 | 4.5544 | 4.6470 | −0.0926 |
| 19 |  |
17.6 | 4.7545 | 4.7645 | −0.0100 |
| 20 |  |
26.4 | 4.5784 | 4.6154 | −0.0370 |
| 21 |  |
21.9 | 4.6596 | 4.7776 | −0.1180 |
| 22 |  |
27.2 | 4.5654 | 4.4462 | 0.1192 |
| 23 | 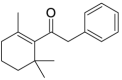 |
26.0 | 4.5850 | 4.70315 | −0.1182 |
Key: IC50: Half-Maximal Inhibitory Concentration PIC50: Negative Logarithm of IC50.
PIC50Pred: Predicted Negative Logarithm of IC50.
2-D structural drawings were achieved with ChemDraw v2.0, converted to 3D set-up in Spartan v14 software and subjected to thorough geometry optimization by exploring density functional theory (DFT) and utilizing the B3LYP/6-31G* basis set [29].
2.2. Physico-chemical properties of calculated molecular properties, pre-treatment, and data division
The descriptors are simple depictions of measurable quantities (numbers) of a compound's or atoms physical and chemical representation, which are computed with Padel [30]. The generated descriptors from the software (Padel) swiftly underwent treatment using the GUI1.2 data treatment suit so as to do away with useless parameters [31]. Also, the data is branched into two sets via data division GUI 1.2 software, namely, the confirmation set, which comprises 22 % of the data used to check the strength of the model, while 78 % of the data was used for the model construction.
2.3. Model generation and validation
In the Material Studio software interface, multi-variant equations (models) have been generated by blending both the genetic function algorithm technique (GFA) and the multi-linear regression technique (MLR) [[32], [33], [34]]. The built models using the training set were further tested by the confirmation sets to determine their predictive strength [35]. The picked model was a result of the following outstanding statistical values: Friedman's lack of fit (LOF), the squared correlation coefficient of the training set (R2), the cross-validation coefficient (Q2CV), and the adjusted squared correlation coefficient (R2adj). The statistical parameters for sound and strong predictive ability must be in line with the guidelines provided by Golbraikh and Tropsha [36]. In addition, the variance inflation factor (VIF), the confirmation set's squared correlation coefficient (Rext), the Y-randomization test (cR2p), and the mean effect (ME) are determined to check the model's reliability. These parameters were calculated using the following equations: [[37], [38], [39], [40], [41]].
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
Where Yexp, Ymintrain, and Ypred are the observed, training set average observed activity and predicted activity, while the correlation coefficient square and the correlation coefficient average are represented by R2 and, respectively. Also, the correlation coefficient for multiple regression involving I and j descriptors is represented as R2ij. Furthermore, j is the descriptor j coefficient, the count of model descriptors is indicated by m, Dj represents each training set values while the count of molecules used as the training set is represented by n.
2.4. Applicability domain (AD)
Assessing the acceptability space of a QSAR model is vital to ascertaining its robustness and consistency.
| (10) |
Where “s" is the descriptors sum and “N" is the total sum of compounds in the training set. A typical AD plot is a plot of the leverage of each compound on the Y-axis against their standardized residuals on the X-axis. Compounds that fall within the defined AD space are well predicted by the model; influential molecules exhibit higher leverage scores than the cut-off value (h*), and they affect the model's performance; outliers are molecules that fall outside the defined AD space with standardized residual values beyond the ±3 defined region; as such, they can be eliminated [42,43]. The employed leverage technique is defined by equation (10).
2.5. Ligand-based drug design (LBDD)
LBDD is an approach to developing new drugs that involves designing molecules from responses generated by the molecular descriptors of known ligands or compounds. These descriptors contains characterized info of a compound inform of calculated quantities which represents molecular properties that are useful in predicting biological activities for lead identification of compounds. This approach typically involves analyzing the structure and activity of a set of molecules, identifying common features that are important for their activity (descriptors guide), and using this information to design new molecules with high activity and specificity [44].
2.6. Pharmacokinetics prediction
Analyzing the drug-likeness and physicochemical profile of novel bioactive compounds is key to drug discovery and development [45,46]. Lipinski's rule is the gold standard accepted as a filter for assessing drug-like properties of compounds based on these four (4) parameters and their threshold values: molecular weight (MW) < 500, lipophilicity (log P) < 5, number of hydrogen bond acceptors (HBA) < 10, and hydrogen bond donors (HBD) < 5 [47]. The drug-like nature of a compound is certified only if it does not violate more than three (3) of the parameters proposed by Lipinski [48]. In addition to the four parameters proposed by Lipinski, rotatable bonds (flexibility: RB) and topological polar surface area (TPSA) proposed by Veber are as well profound and have received recognition for good drug absorption. The RB and TPSA benchmarks were suggested to be < 10 and ≤ 140 Å2 [49]. The prediction of drug likeness and physicochemical properties of compounds in this work would be carried out by utilizing the SwissADME web-based tool [50].
2.7. Docking simulation of design compounds
Docking simulation as one of the in-silico tools in drug design is utilized to forecast how small bioactive molecules (ligands) will interact with a target protein. In this work, cathepsin B1 (SmCB1), a target enzyme with a resolution of 1.29 Å retrieved from RCSB protein data bank with an identity code (6YI7) and prepared using BIOVIA Discovery Studio in order to eliminate the co-crystal ligand, solvent molecules, and cofactors. The prepared protein in 3D format is presented in Fig. 2. Docking simulations between the prepared protein and the least energy conformers of the 7-ketosempervirol ligands were performed using the v6.0 of Molegro Virtual Docker (MVD) [51]. Before the process of docking commences, the protein is imported onto the MVD work space, and all necessary preparatory processes are carried out, ranging from enzyme repair and rebuild to polar hydrogen bond addition and setting a radius of 26 Å with X, Y, and Z coordinates of 9.28, −7.20, and −16.68. The ligands were imported one after the other, and the docking process was executed using MolDock scoring functions. Analysis of the docked pulse was performed by utilizing the BIOVIA Discovery Studio software [52]. Fig. 2 represents cathepsin B1 (SmCB1) target protein in 3D format prepared.
Fig. 2.

3-D structure of the prepared cathepsin B1 (SmCB1) target protein (PDB code: 6YI7).
2.8. Molecular dynamics simulation (MDS) studies
MDS studies performed on the U1 complex of interest was to carry out further verification of docking protocol at the least atomic state and stability inspection in the binding site of the receptor [53]. Desmonds software 2019-4 was used for the 100-ns MDS study here-in while maintaining a solvent system throughout the process to examine the U1–6YI7 (ligand-receptor) complex structure stability. This complex was subjected to fitness such as root mean square deviation (RMSD), root mean square fluctuation (RMSF) and H-bond interaction counts involved considering the U1–6Y17 complex.
3. Results and discussion
3.1. QSAR model generation and internal validation
Herein, the combined use of the GFA-MLR approach, which selects VR1_Dzp, minssCH2, and gmax as the most relevant descriptors, generated four (4) sound models. Internal statistical validations of the developed models all met the agreed values aimed at selecting an acceptable model. The first model that possessed the best internal validation metrics was selected for the purpose of this research. The model expression and their respective internal validation metrics are presented in Table 2 while validation or standard threshold is shown in Table 3 (Supplementary file) [38,40,41,[54], [55], [56]] and Pearson correlation matrix for QSAR analysis which depicts the descriptors in the choosing model, class, VIF and ME values are represented in table 4 (Supplementary file). All the VIF values are less than 10, by implication, the model is reliable and non-accidental. Similarly, for multi-colinearity to exist between the descriptors, sum of values of the mean effect of the three descriptors must be equal to one (1).
Table 2.
Developed QSAR models and their respective internal validation parameters.
| S/NO | Model expression | R2 | R2adj, | Q2cv | LOF |
|---|---|---|---|---|---|
| Model 1 | pIC50 = −0.001361743* VR1_Dzp +1.309124703 * minssCH2 + 0.119928807 * gmax + 2.682043672 |
0.8650 | 0.8360 | 0.7691 | 0.0379 |
| Model 2 | pIC50 = −0.025809774 * VR2_Dzp +1.350948170 * minssCH2+ 0.119189664 * gmax + 2.644238403 |
0.8607 | 0.8309 | 0.7590 | 0.0391 |
| Model 3 | pIC50 = − 0.000751796 * VR1_Dzv +1.223274210 * minssCH2 + 0.113665119 * gmax + 2.697056488 |
0.8606 | 0.8308 | 0.7503 | 0.0391 |
| Model 4 | pIC50 = - 0.000210160 * VR1_Dzi +1.196251030 * minssCH2 + 0.110160129 * gmax + 2.665835715 |
0.8560 | 0.8252 | 0.4226 | 0.0404 |
Key: R2: Coefficient of determination R2adj: Adjusted Coefficient of determination Q2CV: Cross validation coefficient LOF: log of fit.
3.2. Model external validation
An external validation assessment was done to further ascertain the model's predictive capability. The external predicted correlation coefficient (R2ext) was 0.796 which is higher than the 0.6 bench-mark as suggested [57]. As such, the selected model is robust enough for the activity prediction of known and hypothetical 7-ketosempervirol derivatives. For replication's sake, steps used to obtain R2ext from equation (6) is presented in Table 5 (Supplementary file). In addition, the hypothetical compounds, their descriptor values, and the model used to generate their activity are presented in Table 3.
Table 3.
The hypothetical SAR related to the model and PIC50 values generated.
| Hypothetical compounds | Descriptors generated | Model 1 equation for Pred. Activity | ||
|---|---|---|---|---|
| Name | VR1_Dzp | minssCH2 | Gmax | pIC50 = −0.001361743* VR1_Dzp +1.309124703 * minssCH2 + 0.119928807 * gmax + 2.682043672 |
| u1 | 224.1311 | 1.110735 | 12.9756 | 5.387074 |
| u4 | 216.7098 | 1.134828 | 12.90768 | 5.420575 |
| u7 | 239.3999 | 1.153465 | 12.54002 | 5.369981 |
| u8 | 182.1974 | 1.151073 | 12.47048 | 5.436405 |
| 7b1 | 230.0826 | 1.166749 | 10.96824 | 5.211558 |
Key: u1: (4bR,8aR)-1-isopropyl-3,4b,8,8-tetramethyl-10-(methylamino)-6-vinyl-4b,5,6,7,8,8a,9,10-octahydrophenanthrene-4 - carbonyl chloride.
u4: (4bR,8aR)-1-isopropyl-3,4b,8,8,10-pentamethyl-6-vinyl-4b,5,6,7,8,8a,9,10-octahydrophenanthrene-4-carbonyl chloride.
u7: (4bR,8aR)-6-ethyl-3,4b,8,8-tetramethyl-4b,5,6,7,8,8a,9,10-octahydrophenanthrene-4-carbonyl chloride.
u8: (4bR,8aR)-3,4b,6,8,8-pentamethyl-4b,5,6,7,8,8a,9,10-octahydrophenanthrene-4-carbonyl chloride.
7b1:(4bR,8aR)-6-chloro-4-(dimethylamino)-1-isopropyl-4b,8,8-trimethyl-10-(methylamino)-4b,5,6,7,8,8a,9,10-octahydrophenanthren-2-ol.
While the validation parameters with minimum threshold for QSAR model are shown in Table 3 (Supplementary file). All these parameters such as R2, R2adj, Q2cv, R2-Q2cv, and cR2p passed the threshold limit, as such the choosing model (model 1) is an excellent model which was not gotten by chance.
3.3. Applicability domain
The activity predictive strength of a model is only effective on compounds within the domain's acceptability space. The warning leverage for anti-Schistosoma compounds obtained is h* = 0.67, and compound 6,10,18,19 and 23 falls beyond the threshold hat matrix h* therefore, they are considered influential molecules. Fig. 3 represents the applicability domain of anti-Schistosoma compounds.
Fig. 3.

Developed William's plot.
3.4. Significant nature of the descriptor's mean effect (ME)
The descriptors are largely influenced by the corresponding magnitude of the ME values and the sign of the relevant coefficient. The negative indicator sign and magnitude for this descriptor (VR1_Dzp) with corresponding ME value (−0.0289) signifies that the lesser this type of descriptor in the template structure, the better the anti-Schistosoma biological activity. While the two descriptors of interest (gmax and minssCH2) with their ME values (+0.533514 and + 0.495343, respectively) indicate that the more of these descriptors in the template, the better the anti-Schistosoma biological activity, these two descriptors have the same meaning and are defined as atom-type electro-topological states (E-states). Atom type E-state parameters provide information on the electron densities and distribution, spatial arrangement of electro-negative atoms in a molecule and their ability to donate or accept electron in a chemical reaction [58].
3.5. Modification of template via ligand-based drug design approach
Compound 15, which is the most active compound with a predicted pIC50 of 5.11, is subjected to molecular modification or design on the basis of the information unveiled by the two most influential descriptors (gmax and minssCH2) with high mean effect (ME) values from the model, coupled with its availability within the acceptable William's plot domain space and having a relatively low residual value of 0.0880, as shown in Table 1. Compound 15 and the template used for design are depicted in Fig. 4A and B. Variation of substituents such as halogen, hydroxyl, amino, formal chloride, and formaldehyde that contain electronegative atoms like oxygen, nitrogen, and chlorine pumps electrons to the ring system of the diterpenoid pharmacophore through resonance effect or electron-sharing hence, increasing the electron density. Furthermore, the ethane group observed in compounds U1 and U4 favors aromatic ring substitution, thereby combining easily with molecules in the receptor to form either cyclic or ring compounds. The iteration of the template at positions R1, R2, R3, R4, R5, and R6 generated 5 novel anti-Schistosoma derivatives with pIC50 ranging from pIC50 = 5.21 to 5.42, which is higher than that of the template (pIC50 = 5.11) and the standard drug Praziquantel (pIC50 = 4.28).
Fig. 4.

A and B shows the Template (15*) and general formula of template for design.
3.6. Pharmacokinetics results
The drug likeness profile of these SmCB1 inhibitors (novel compounds) was determined and presented in Table 4. The five novel analogues from Table 4, possessed drug-like characteristics by violating just one of Lipinski's rules (wlogp5) [59]. Additionally, they are in agreement with Veber's rule, which recommends that both RB and TPSA must be < 10 and ≤ 140 Å2 [60]. Furthermore, a bioavailability score (ABS) of 0.55 shows that all the compounds are orally bio-available and meet the drug likeness property [61], while synthetic accessibility (SA), or ease of synthesis, of all the compounds calculated falls below 5, which is within the standard range of one (1) to ten (10) [62].
Table 4.
Pharmacokinetic profile of the novel anti-Schistosoma analogues.
| S/no | HBD | HBA | RB | Wlog p | TPSA | MW | ABS | SA |
|---|---|---|---|---|---|---|---|---|
| U1 | 1 | 2 | 4 | 6.33 | 29.10 | 402.01 | ||
| 0.55 | 4.48 | |||||||
| U4 | 0 | 1 | 3 | 7.5 | 17.07 | 387 | ||
| 0.55 | 4.44 | |||||||
| U7 | 0 | 1 | 2 | 6.04 | 17.07 | 332.91 | ||
| 0.55 | 3.82 | |||||||
| U8 | 0 | 1 | 1 | 5.65 | 17.07 | 318.88 | ||
| 0.55 | 3.71 | |||||||
| 7B1 | 2 | 2 | 3 | 5.22 | 35.50 | 393.01 | 0.55 | 4.49 |
Key: HBD: Hydrogen bond donor, HBA: Hydrogen bond acceptor, RB: rotatable bond, Wlog p: weighted Partition (hydrophobicity of a compound Coefficient), TPSA: Topological Polar Surface Area, MW: molecular weight, ABS: bio-available, SA: synthetic availability.
3.7. Docking simulation results
The receptor 3D pharmacophore was presented in Fig. 2; likewise, the receptor (PDB code: 6YI7) and the SmCB1 inhibitors (novel compounds and the standard drug Praziquantel) pulse visualized by Discovery Studio was presented in Table 5. From Table 5, all novel compounds bind to the receptor in the following order: U1 (−113.121 kcal/mol) > U4 > U7 > U8 > 7B1 > PZQ >15* (−87.21 and −83.37 kcal/mol) with higher binding affinity (i.e., least moldock score) compared to the standard drug and the template (15*). In addition, there is direct proportionality between the moldock score values and the predicted activities (PIC50), as observed in Table 8 (supplementary file). This shows how well the novel compounds interact with the receptor or protein.
Table 5.
The docking portion showing 2D and 3D representations, binding interactions, and hydrogen bond lengths of the designed compounds and PZQ in the active site of the 6YI7 receptor.


Key: (4bR,8aR)-1-isopropyl-3,4b,8,8-tetramethyl-10-(methylamino)-6-vinyl-4b,5,6,7,8,8a,9,10-octahydrophenanthrene-4 - carbonyl chloride.u4: (4bR,8aR)-1-isopropyl-3,4b,8,8,10-pentamethyl-6-vinyl-4b,5,6,7,8,8a,9,10-octahydrophenanthrene-4-carbonyl chloride.u7: (4bR,8aR)-6-ethyl-3,4b,8,8-tetramethyl-4b,5,6,7,8,8a,9,10-octahydrophenanthrene-4-carbonyl chlorideu8: (4bR,8aR)-3,4b,6,8,8-pentamethyl-4b,5,6,7,8,8a,9,10-octahydrophenanthrene-4-carbonyl chloride.7b1:(4bR,8aR)-6-chloro-4-(dimethylamino)-1-isopropyl-4b,8,8-trimethyl-10-(methylamino)-4b,5,6,7,8,8a,9, 10-octahydrophenanthren-2-ol, 15*: template compound.
3.8. Interactions between novel SmCB1 inhibitors and 6YI7 receptor
All the novel compounds display high predicted activities and more interactions compared to PZQ and 15*. The high binding affinity, stability, and specificity of the designed compounds are a function of the synergy between all the interactions. Compound U1 has one of the best predicted activity and high binding affinity (least moldock score) of 5.39 μM and −113.121 kcal/mol, which is better than PZQ, with a predicted activity of 4.28 μM and moldock score of −87.45 kcal/mol. Looking at the existing interaction between U1 analogue and 6YI7 protein, one conventional hydrogen bond and a carbon hydrogen bond involving GLN A:94 (interaction distance of 2.10 Å) and GLY A:98 (interaction distance of 1.79 Å) amino acid residue interaction were observed with the oxygen atom of the alkanoyl chloride group attached to the aromatic ring present in the diterpenoid pharmacophore. The free-moving electrons in the aromatic ring interact with two amino acid residues, TRP A:292 (5.66 Å) and CYS A:100 (3.49 Å) through the Pi-Pi T-shaped hydrophobic bond and the Pi-sulphur bond. Also, the methyl and isopropyl substituents attached to the aromatic ring interacted with four amino acid residues, namely, CYS A:100 (5.73 Å), HIS A:270 (5.13 Å), TRP A: 292 (5.23 Å) and VAL A:247 (3.78 Å) through the Pi-alkyl, Pi-alkyl, Pi-alkyl hydrophobic, and alkyl bonds, respectively. The alkene group attached to the cyclohexane ring and the cyclo-hexane ring itself interacted with five amino acid residues, where the alkene group alone is bonded to ILE A:193 (5.02 Å), ARG A:96 (3.44 Å) and CYS A:189 (4.17 Å) through an alkyl bond, while the cyclo-hexane ring interacts with HIS A:180 (4.77 Å) and CYS A:189 (5.01 Å) amino acid residues through an alkyl bond as well. Furthermore, the di-methyl substituent attached to the cyclo-hexane ring in the diterpenoid pharmacophore interacted with five amino acid residues, namely, HIS A:181 (3.50 Å and 4.35 Å) with two interactions through alkyl bonds, CYS A:189 (4.64 Å and 8.42 Å), HIS A:180 (4.18 Å) which were visible through alkyl bond formation. Finally, eleven of Vanderwaal's interactions were also observed. PZQ, an aromatic hetero-polycyclic compound, on the other hand, shows two oxygen atom interactions present in the alkanamide group of the PZQ molecular framework. One of the oxygen atoms interacts with LYS A:177 amino acid residues (bond length 2.02 Å) while the other oxygen atom interacts with ASN A:166 (bond length 2.42 Å) via a carbon hydrogen bond and a conventional hydrogen bond. Three hydrogen atoms in the PZQ pharmacophore form interactions with ILE A:91 (1.50 Å), ASP A:93 (2.60 Å) and GLU A:124 (2.73 Å) via a carbon hydrogen bond. The aromatic ring in the pharmacophore interacts with ARG A:92 (2.70 Å), ALA A:127 (4.94 Å) and SER A:162 (3.76 Å) via pi-sigma, pi-alkyl, and amide pi-stacked bonds. Furthermore, one of the cyclic rings of the pharmacophore interacts with ARG A:92 (4.33 Å) via an alkyl bond. Finally, eleven of Vanderwaal's interactions were also observed. The novel compound (U1) shows several key molecular forces and interactions compared to PZQ. This could be the reason for its high moldock score, stability, and activity. With the evidence stated earlier, the said compound (s) is brought forward for consideration and exploration as a prospective anti-Schistosoma drug candidates for the cure of Schistosomiasis caused by Schistosoma mansoni. The docking portion of the anti-Schistosoma analogues in complex with 6YI7 receptor, showing the 2D and 3D representations, binding interactions, and their respective hydrogen bond lengths, is represented in Table 5.
3.9. MD simulation results
The 100ns MDS runs presents absolute RMSD significance below 2.00 Å for both the complex and protein. The protein maintained constant RMSD value of 1.25 Å during the first 40-ns simulation from then there is sharp increase in the value to 1.75 Å as the simulation runs progress to final stage. This revealed slight conformational change in the structure of the protein. With regard to the ligand, at 10ns simulation, the value of RMSD was constant and lower than 0.75 Å and then rises to 1.00 Å which was maintained from 10ns all through the simulation period. This illustrate the ligand stability in the binding site of the protein target. RMSD plot of the protein and the ligand is presented in Fig. 5.
Fig. 5.

RMSD plot.
RMSF compute flexibility and transition of residues in the protein backbone. Protein RMSF value less than 2.4 Å during 100-ns simulation procedure is an indication of stability. Fig. 6 show the RMSF values of the protein.
Fig. 6.

Protein RMSF plot.
Formation of hydrogen bond interactions within a complex is of utmost importance in drug discovery as it play a major role in stabilizing the protein-ligand complexes. Stable nature of the complex was further examine by hydrogen bond interactions with the protein residues for 100ns runs. Fig. 7 confirms the formation of six (6) H-bonds with GLN94, CYS97, CYS100, HIS180, LEU267, and TRP292 amino acid residues, which confirms the stability of the complex as well as justifies the docking result.
Fig. 7.

H-bonds profile.
4. Conclusion
Schistosoma mansoni is on the increase because both Praziquantel (PZQ) and Oxamniquine widely used for the treatment of Schistosomiasis for over 51 years suffer resistance and recurrence. In this work, chemo-informatics techniques were employed such as QSAR modeling, evaluation of docking simulation, and pharmacokinetic profiling were harnessed in designing novel diterpenoid-7-keto-sempevirol derivatives that are more competent, effective, and safer against S. mansoni alongside MD Simulation. Here-in, upon QSAR screening, compound 15 which appears to be in the model's acceptability space emerges one of the best with high predicted activity of (PIC50pred = 5.11). Compound 15 was further used as model in designing 5 new analogues with improved activity against Schistosomiasis ranging from (5.21–5.42). Based on the generated QSAR model (model 1) that has been internally and externally validated and proven to be robust owing to certain parameters. Furthermore, these compounds having better activity compared to the standard drug PZQ were subjected to pharmacokinetic profiling before docking studies. The pharmacokinetics carried out shows that the 5 new compounds, have the characteristics of a good drug by passing the Veber's criteria and violated only one of the Lipinski's rules. While the docking evaluation further proves that all the design compound has better moldock score compared to PZQ with compound U1 being the best. Md simulation of the best compound (U1) in complex with 6YI7 has abnitio showed outstanding profile in terms of activity, binding affinity, RMSD and RMSF coupled with a well-built interaction that forms six (6) H-bonds with GLN94, CYS97, CYS100, HIS180, LEU267, and TRP292 amino acid residues at favorable bond distance, has confirmed the complex stable nature and validates the docking result. The application of chemo-informatics in the design of improved bioactive compound (s) targeting SmCB1 enzyme for Schistosomiasis treatment is open to further medicinal evaluation and utilization.
Funding statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Data availability statement
Data included in article/supp. material/referenced in article.
Additional information
No additional information is available for this paper.
CRediT authorship contribution statement
Salim Bitrus Anyubaga: Writing – original draft, Investigation, Conceptualization. Gideon Adamu Shallangwa: Supervision. Adamu Uzairu: Investigation. Stephen Eyije Abechi: Validation.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:No competing interest If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors gratefully acknowledged the technical effort of Ajala Abduljelil, Sagiru Hamza, Amina Umar Ubangari, Dr. Abdulfatai, and Dr. Zakari Yau, all of chemistry and Biology department, Ahmadu Bello University, Zaria.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2023.e23115.
List of Abbreviations
- Not applicable
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.De Mori R.M., Aleixo M.A., Zapata L.C., Calil F.A., Emery F.S., Nonato M.C. Structural basis for the function and inhibition of dihydroorotate dehydrogenase from Schistosoma mansoni. FEBS J. 2021;288(3):930–944. doi: 10.1111/febs.15367. [DOI] [PubMed] [Google Scholar]
- 2.Mawa P.A., Kincaid-Smith J., Tukahebwa E.M., Webster J.P., Wilson S. Schistosomiasis morbidity hotspots: roles of the human host, the parasite and their interface in the development of severe morbidity. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.635869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernández-Soto P., Avendaño C., Sala-Vizcaíno A., Crego-Vicente B., Febrer-Sendra B., García-Bernalt Diego J., Muro A. Disease markers; 2020. Molecular Markers for Detecting Schistosoma Species by Loop-Mediated Isothermal Amplification. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marchese V., Beltrame A., Angheben A., Monteiro G.B., Giorli G., Perandin F.…Bisoffi Z. Schistosomiasis in immigrants, refugees and travellers in an Italian referral centre for tropical diseases. Infectious diseases of poverty. 2018;7(3):56–65. doi: 10.1186/s40249-018-0440-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Assefa A., Erko B., Gundersen S.G., Medhin G., Berhe N. Current status of Schistosoma mansoni infection among previously treated rural communities in the Abbey and Didessa Valleys, Western Ethiopia: implications for sustainable control. PLoS One. 2021;16(2) doi: 10.1371/journal.pone.0247312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nigo M.M., Odermatt P., Salieb–Beugelaar G.B., Morozov O., Battegay M., Hunziker P.R. Epidemiology of schistosoma mansoni infection in ituri province, north-eastern democratic republic of the Congo. PLoS Neglected Trop. Dis. 2021;15(12) doi: 10.1371/journal.pntd.0009486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McManus D.P., Dunne D.W., Sacko M., Utzinger J., Vennervald B.J., Zhou X.-N. Schistosomiasis. Nature Reviews Disease Primers. 2018;4(1) doi: 10.1038/s41572-0180013-8. [DOI] [PubMed] [Google Scholar]
- 8.Zhong H., Gui X., Hou L., Lv R., Jin Y. From inflammation to fibrosis: novel insights into the roles of high mobility Group Protein Box 1 in Schistosome-induced liver damage. Pathogens. 2022;11(3):289. doi: 10.3390/pathogens11030289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McManus D.P., Bergquist R., Cai P., Ranasinghe S., Tebeje B.M., You H. Seminars in Immunopathology. vol. 42. Springer Berlin Heidelberg; 2020, June. Schistosomiasis—from immunopathology to vaccines; pp. 355–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bahri S. 2018. Risk Factors of Schistosomiasis and the Health Effect Among Patients Attending Alkalakla Health Centre-Khartoum State-Sudan. [Google Scholar]
- 11.Chen Q., Zhang J., Zheng T., Chen H., Nie H., Zheng B., Gong Q. The role of microRNAs in the pathogenesis, grading and treatment of hepatic fibrosis in Schistosomiasis. Parasites Vectors. 2019;12:1–10. doi: 10.1186/s13071-019-3866-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adu D., Ojo A.O. Chronic Renal Disease. Academic Press; 2020. Ethnicity and chronic kidney disease in Africa; pp. 149–166. [Google Scholar]
- 13.Chen S., Suzuki B.M., Dohrmann J., Singh R., Arkin M.R., Caffrey C.R. A multi-dimensional, time-lapse, high content screening platform applied to Schistosomiasis drug discovery. Commun. Biol. 2020;3(1):1–9. doi: 10.1038/s42003-020-01402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Benedetti S., Di Pisa F., Fassi E.M.A., Cretich M., Musicò A., Frigerio R.…Gourlay L.J. Structure, immunoreactivity, and in silico epitope determination of SmSPI S. Mansoni serpin for immunodiagnostic application. Vaccines. 2021;9(4):322. doi: 10.3390/vaccines9040322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rennar G.A., Gallinger T.L., Mäder P., Lange-Grünweller K., Haeberlein S., Grünweller A.…Schlitzer M. Disulfiram and dithiocarbamate analogues demonstrate promising antischistosomal effects. Eur. J. Med. Chem. 2022;242 doi: 10.1016/j.ejmech.2022.114641. [DOI] [PubMed] [Google Scholar]
- 16.Crusco A., Bordoni C., Chakroborty A., Whatley K.C., Whiteland H., Westwell A.D., Hoffmann K.F. Design, synthesis and anthelmintic activity of 7-keto-sempervirol analogues. Eur. J. Med. Chem. 2018;152:87–100. doi: 10.1016/j.ejmech.2018.04.032. [DOI] [PubMed] [Google Scholar]
- 17.Guzman M., Rugel A., Tarpley R.S., Cao X., McHardy S.F., LoVerde P.T., Taylor A.B. Molecular basis for hycanthone drug action in schistosome parasites. Mol. Biochem. Parasitol. 2020;236 doi: 10.1016/j.molbiopara.2020.111257. [DOI] [PubMed] [Google Scholar]
- 18.Caffrey C.R., Goupil L., Rebello K.M., Dalton J.P., Smith D. Cysteine proteases as digestive enzymes in parasitic helminths. PLoS Neglected Trop. Dis. 2018;12(8) doi: 10.1371/journal.pntd.0005840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jílková A., Rubesova P., Fanfrlik J., Fajtova P., Rezacova P., Brynda J.…Mares M. Druggable hot spots in the Schistosomiasis cathepsin b1 target identified by functional and binding mode analysis of potent vinyl sulfone inhibitors. ACS Infect. Dis. 2020;7(5):1077–1088. doi: 10.1021/acsinfecdis.0c00501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crusco A., Bordoni C., Chakroborty A., Whatley K.C., Whiteland H., Westwell A.D., Hoffmann K.F. Design, synthesis and anthelmintic activity of 7-keto-sempervirol analogues. Eur. J. Med. Chem. 2018;152:87–100. doi: 10.1016/j.ejmech.2018.04.032. [DOI] [PubMed] [Google Scholar]
- 21.Jabalia N., Kumar A., Kumar V., Rani R. 2021. In Silico Approach in Drug Design and Drug Discovery: an Update. Innovations And Implementations Of Computer Aided Drug Discovery Strategies In Rational Drug Design; pp. 245–271. [Google Scholar]
- 22.Ejeh S., Uzairu A., Shallangwa G.A., Abechi S.E. In silico design, drug-likeness and ADMET properties estimation of some substituted thienopyrimidines as HCV NS3/4A protease inhibitors. Chemistry Africa. 2021;4(3):563–574. [Google Scholar]
- 23.Abdullahi S.H., Uzairu A., Shallangwa G.A., Uba S., Umar A.B. In-silico activity prediction, structure-based drug design, molecular docking and pharmacokinetic studies of selected quinazoline derivatives for their antiproliferative activity against triple negative breast cancer (MDA-MB231) cell line. Bull. Natl. Res. Cent. 2022;46(1):2. [Google Scholar]
- 24.Adedirin O., Uzairu A., Shallangwa G.A., Abechi S.E. Optimization of the anticonvulsant activity of 2-acetamido-N-benzyl-2-(5-methylfuran-2-yl) acetamide using QSAR modeling and molecular docking techniques. Beni-Suef University journal of basic and applied sciences. 2018;7(4):430–440. [Google Scholar]
- 25.Ibrahim Z.Y.U., Uzairu A., Shallangwa G., Abechi S. In-silico design of aryl and aralkyl amine-based triazolopyrimidine derivatives with enhanced activity against resistant Plasmodium falciparum. Chemistry Africa. 2021;4(1):137–148. [Google Scholar]
- 26.Chen S., Suzuki B.M., Dohrmann J., Singh R., Arkin M.R., Caffrey C.R. A multi-dimensional, time-lapse, high content screening platform applied to Schistosomiasis drug discovery. Commun. Biol. 2020;3(1):1–9. doi: 10.1038/s42003-020-01402-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shrivastava B., Bhagwat M.G., Patil R., Singh M., Porwal A. Molecular modelling studies on triazole-based oxidoreductase inhibitor using 3D-QSAR. European Journal of Molecular and Clinical Medicine. 2020;7(11) [Google Scholar]
- 28.Porwal A., Shrivastava B., Bhagwat M.G., Patil R., Singh M., Porwal A. Molecular modelling studies on triazole-based oxidoreductase inhibitor using 3D-QSAR. European Journal of Molecular and Clinical Medicine. 2021;7(11):4322–4340. [Google Scholar]
- 29.Abdulrahman H.L., Uzairu A., Uba S. QSAR, ligand-based design and pharmacokinetic studies of parviflorons derivatives as anti-breast cancer drug compounds against MCF-7 cell line. Chemistry Africa. 2021;4(1):175–187. [Google Scholar]
- 30.Puratchikody A., Prabu S.L., Umamaheswari A., editors. IGI Global. 2018. Computer Applications in Drug Discovery and Development. [Google Scholar]
- 31.Adeniji S.E., Uba S., Uzairu A. Theoretical modeling for predicting the activities of some active compounds as potent inhibitors against Mycobacterium tuberculosis using GFA-MLR approach. J. King Saud Univ. Sci. 2020;32(1):575–586. [Google Scholar]
- 32.Mustapha A., Shallangwa G., Ibrahim M.T., Bello A.U., Ebuka D.A., Uzairu A., Mamza P. QSAR studies on some C14-urea tetrandrine compounds as potent anti-cancer against leukemia cell line (K562) Journal of the Turkish Chemical Society Section A: Chemistry. 2018;5(3):1387–1398. [Google Scholar]
- 33.Rasyid H., Soekamto N.H., Firdausiah S. Modelling the anticancer activity of 4-alkoxy cinnamic analogues using 3D-descriptors and genetic algorithm-multiple linear regression (GA-MLR) method. J. King Saud Univ. Sci. 2023;35(3) [Google Scholar]
- 34.Bakal R.L., Jawarkar R.D., Manwar J.V., Jaiswal M.S., Ghosh A., Gandhi A.…Lewaa I. Identification of potent aldose reductase inhibitors as antidiabetic (Anti-hyperglycemic) agents using QSAR based virtual Screening, molecular Docking, MD simulation and MMGBSA approaches. Saudi Pharmaceut. J. 2022;30(6):693–710. doi: 10.1016/j.jsps.2022.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isyaku Y., Uzairu A., Uba S. QSAR study of 2-substituted phenyl-2-oxo-, 2-hydroxyl-and 2-acylloxyethylsulfonamides as fungicides. The Journal of Engineering and Exact Sciences. 2019;5(3):283–290. [Google Scholar]
- 36.Tabti K., Elmchichi L., Sbai A., Maghat H., Bouachrine M., Lakhlifi T., Ghosh A. In silico design of novel PIN1 inhibitors by combined of 3D-QSAR, molecular docking, molecular dynamic simulation and ADMET studies. J. Mol. Struct. 2022;1253 [Google Scholar]
- 37.Umar A.B., Uzairu A., Shallangwa G.A., Uba S. Ligand-based drug design and molecular docking simulation studies of some novel anticancer compounds on MALME-3M melanoma cell line. Egyptian Journal of Medical Human Genetics. 2021;22(1):1–15. [Google Scholar]
- 38.Adeniji S.E., Shallangwa G.A., Arthur D.E., Abdullahi M., Mahmoud A.Y., Haruna A. Quantum modelling and molecular docking evaluation of some selected quinoline derivatives as anti-tubercular agents. Heliyon. 2020;6(3) doi: 10.1016/j.heliyon.2020.e03639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El Aissouq A., Toufik H., Stitou M., Ouammou A., Lamchouri F. In silico design of novel tetra-substituted pyridinylimidazoles derivatives as c-jun N-terminal kinase-3 inhibitors, using 2D/3D-QSAR studies, molecular docking and ADMET prediction. Int. J. Pept. Res. Therapeut. 2020;26:1335–1351. [Google Scholar]
- 40.Safarizadeh H., Garkani-Nejad Z. Molecular docking, molecular dynamics simulations and QSAR studies on some of 2-arylethenylquinoline derivatives for inhibition of Alzheimer's amyloid-beta aggregation: insight into mechanism of interactions and parameters for design of new inhibitors. J. Mol. Graph. Model. 2019;87:129–143. doi: 10.1016/j.jmgm.2018.11.019. [DOI] [PubMed] [Google Scholar]
- 41.Ugbe F.A., Shallangwa G.A., Uzairu A., Abdulkadir I. Activity modeling, molecular docking and pharmacokinetic studies of some boron-pleuromutilins as anti-wolbachia agents with potential for treatment of filarial diseases. Chemical Data Collections. 2021;36 [Google Scholar]
- 42.Bahmani A., Saaidpour S., Rostami A. A simple, robust and efficient computational method for n-octanol/water partition coefficients of substituted aromatic drugs. Sci. Rep. 2017;7(1):1–14. doi: 10.1038/s41598-017-05964-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adeniji S.E., Adalumo O.B. Computational modeling and ligand-based design of some novel hypothetical compound as prominent inhibitors against Mycobacterium tuberculosis. Future Journal of Pharmaceutical Sciences. 2020;6:1–11. [Google Scholar]
- 44.Ajala A., Uzairu A., Shallangwa G.A., Abechi S.E. 2D QSAR, design, docking study and ADMET of some N-aryl derivatives concerning inhibitory activity against Alzheimer disease. Future Journal of Pharmaceutical Sciences. 2022;8(1):30. [Google Scholar]
- 45.Guan L., Yang H., Cai Y., Sun L., Di P., Li W.…Tang Y. ADMET-score–a comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm. 2019;10(1):148–157. doi: 10.1039/c8md00472b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gupta N., Bottino D., Simonsson U.S., Musante C.J., Bueters T., Rieger T.R.…Nayak S. Transforming translation through quantitative pharmacology for high‐impact decision making in drug discovery and development. Clinical Pharmacology and Therapeutics. 2020;107(6):1285–1289. doi: 10.1002/cpt.1667. [DOI] [PubMed] [Google Scholar]
- 47.Hammoudi N.E.H., Benguerba Y., Attoui A., Hognon C., Lemaoui T., Sobhi W.…Monari A. In silico drug discovery of IKK-β inhibitors from 2-amino-3-cyano-4-alkyl-6-(2-hydroxyphenyl) pyridine derivatives based on QSAR, docking, molecular dynamics and drug-likeness evaluation studies. J. Biomol. Struct. Dyn. 2022;40(2):886–902. doi: 10.1080/07391102.2020.1819878. [DOI] [PubMed] [Google Scholar]
- 48.Akash S., Baeza J., Mahmood S., Mukerjee N., Subramaniyan V., Islam M.R.…Abdel-Daim M.M. Development of a new drug candidate for the inhibition of Lassa virus glycoprotein and nucleoprotein by modification of evodiamine as promising therapeutic agents. Front. Microbiol. 2023;14 doi: 10.3389/fmicb.2023.1206872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sobhi W., Attoui A., Lemaoui T., Erto A., Benguerba Y. In silico drug discovery of Acetylcholinesterase and Butyrylcholinesterase enzymes inhibitors based on Quantitative Structure-Activity Relationship (QSAR) and drug-likeness evaluation. J. Mol. Struct. 2021;1229 [Google Scholar]
- 50.Dogan E.E. Computational bioactivity analysis and bioisosteric investigation of the approved breast cancer drugs proposed new design drug compounds: increased bioactivity coming with silicon and boron. Lett. Drug Des. Discov. 2021;18(6):551–561. [Google Scholar]
- 51.Ihsan A., Khera R.A., Iqbal J., Asgher M. Binding interaction of benzamide derivatives as inhibitors of DNA gyrase and Sec14p using Molegro Virtual Docker based on binding free energy. Z. Phys. Chem. 2022;236(4):561–581. [Google Scholar]
- 52.Kumar S.P., Girija A.S., Priyadharsini J.V. Targeting NM23-H1-mediated inhibition of tumour metastasis in viral hepatitis with bioactive compounds from Ganoderma lucidum: a computational study. Indian J. Pharmaceut. Sci. 2020;82(2):300–305. [Google Scholar]
- 53.Abdullahi S.H., Uzairu A., Shallangwa G.A., Uba S., Umar A.B. Molecular docking, ADMET and pharmacokinetic properties predictions of some di-aryl pyridinamine derivatives as estrogen receptor (Er+) kinase inhibitors. Egyptian Journal of Basic and Applied Sciences. 2022;9(1):180–204. [Google Scholar]
- 54.Daoui O., Elkhattabi S., Chtita S., Elkhalabi R., Zgou H., Benjelloun A.T. QSAR, molecular docking and ADMET properties in silico studies of novel 4, 5, 6, 7-tetrahydrobenzo [D]-thiazol-2-Yl derivatives derived from dimedone as potent anti-tumor agents through inhibition of C-Met receptor tyrosine kinase. Heliyon. 2021;7(7) doi: 10.1016/j.heliyon.2021.e07463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adeniji S.E., Uba S., Uzairu A. QSAR modeling and molecular docking analysis of some active compounds against mycobacterium tuberculosis receptor (Mtb CYP121) Journal of pathogens. 2018:1–24. doi: 10.1155/2018/1018694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ibrahim Z.Y., Uzairu A., Shallangwa G.A., Abechi S.E., Isyaku S. Homology modeling, docking, and ADMET studies of benzoheterocyclic 4-aminoquinolines analogs as inhibitors of Plasmodiumfalciparum. Journal of Taibah University Medical Sciences. 2023;18(6):1200. doi: 10.1016/j.jtumed.2023.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Aminu K.S., Uzairu A., Abechi S.E., Shallangwa G.A., Umar A.B. Ligand-based drug design, molecular docking and pharmacokinetic studies of some series of 1, 4-dihydropyridines derivatives as human intestinal maltase-glucoamylase inhibitor. Chemical Data Collections. 2022;41 [Google Scholar]
- 58.Grisoni F., Ballabio D., Todeschini R., Consonni V. In: Computational Toxicology (Pp. 3–53. Nicolotti O., editor. vol. 1800. Humana Press; 2018. Molecular descriptors for structure–activity applications: a hands-on approach. (Methods in Molecular Biology). [DOI] [PubMed] [Google Scholar]
- 59.Akintunde J.K., Akomolafe V.O., Taiwo O.A., Ahmad I., Patel H., Osifeso A.…Ojo O.A. Antihypertensive activity of roasted cashew nut in mixed petroleum fractions-induced hypertension: an in vivo and in silico approaches. Heliyon. 2022;8(12) doi: 10.1016/j.heliyon.2022.e12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ley-Martínez J.S., Ortega-Valencia J.E., García-Barradas O., Jiménez-Fernández M., Uribe-Lam E., Vencedor-Meraz C.I., Oliva-Ramírez J. Active compounds in Zingiber officinale as possible redox inhibitors of 5-lipoxygenase using an in silico approach. Int. J. Mol. Sci. 2022;23(11):6093. doi: 10.3390/ijms23116093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ismael M., Abdel-Mawgoud A.M.M., Rabia M.K., Abdou A. Ni (II) mixed-ligand chelates based on 2-hydroxy-1-naphthaldehyde as antimicrobial agents: synthesis, characterization, and molecular modeling. J. Mol. Liq. 2021;330 [Google Scholar]
- 62.Mishra S., Dahima R. In vitro ADME studies of TUG-891, a GPR-120 inhibitor using SWISS ADME predictor. J. Drug Deliv. Therapeut. 2019;9(2-s):366–369. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data included in article/supp. material/referenced in article.