

Abstract
Introduction
Fabry disease (FD) can be undiagnosed in the context of multiple sclerosis (MS) due to similar clinical and paraclinical features. Our study aimed to determine the prevalence (and the necessity of screening) of FD among patients with possible or definite MS.
Methods
In this prospective monocentric observational study, we included consecutive patients enrolled between May 2017 and May 2019 after the first clinical event suggestive of MS. All patients underwent FD screening using dried blood spots in a stepwise manner combining genetic and enzyme testing. Patients were followed until May 2022.
Results
We included 160 patients (73.1% female, mean age 33.9 years). The 2017 revised McDonald’s criteria for definite MS were fulfilled by 74 (46.3%) patients at the time of study recruitment and 89 (55.6%) patients after 3–5 years of follow-up. None of the patients had a pathogenic GLA variant, and four (2.5%) had a variant of unknown significance (p.A143T, p.S126G, 2 × p.D313Y). In two of these patients, the intrathecal synthesis of oligoclonal bands was absent, and none had hyperproteinorachia or pleocytosis in cerebrospinal fluid. Detailed examination of FD organ manifestations revealed only discrete ocular and kidney involvement in two patients.
Conclusion
The prevalence of FD in the population of suspected or definite MS patients does not appear to be high. Our results do not support routine FD screening in all patients with a possible diagnosis of MS, but there is an urgent need to search for red flags and include FD in the differential diagnosis of MS.
Keywords: Fabry disease, A variant of unknown significance, Multiple sclerosis, Misdiagnosis, Screening, Differential diagnosis
Introduction
Multiple sclerosis (MS) is an autoimmune demyelinating disorder of the central nervous system (CNS) with a broad spectrum of clinical symptoms. Both the clinical features of the disease and laboratory investigations, such as magnetic resonance imaging (MRI) and cerebrospinal fluid (CSF) analysis, are used in the diagnosis [1]. The main aim is to establish the dissemination in space and time of the MS-typical clinical and imaging presentation caused by the lesions in the CNS and to rule out other diseases that might mimic MS. Due to the high variability of symptoms originating from many areas of the CNS and the absence of specific biomarkers confirming the diagnosis, the differential diagnosis of MS can be difficult [2]. As much as 5–10% of patients with MS are misdiagnosed due to misinterpretation and misapplication of MS clinical and radiographic diagnostic criteria with overreliance on MRI abnormalities and non-specific neurological symptoms [3, 4].
One of the recognised but sometimes underestimated mimics of MS is Fabry disease (FD). Several studies have described patients with FD who were initially misdiagnosed with MS [5–7] or later found to have both conditions [8]. FD is a progressive, X-linked inherited disorder of glycosphingolipid metabolism due to deficient or absent alpha-galactosidase-A (AGAL) activity caused by over 1000 known disease-associated variants in the GLA gene [9]. The AGAL deficiency results in the accumulation of globotriaosylceramide and other glycosphingolipids in various cell types, including renal, cardiac, nerve, and endothelial cells [10]. The estimated incidence of FD is 1 per 40,000–1 per 60,000 [11]. However, some studies indicate a possible higher prevalence [12]. The diagnosis of FD has important therapeutic implications as a specific therapy is available.
FD is a disease with a broad spectrum of heterogeneously progressive clinical phenotypes due to the different residual levels of AGAL activity, with contributions from modifying factors. The classical, and most severe, manifestation of the disease phenotype is in hemizygous males in childhood or adolescence. Symptoms include acroparesthesias due to peripheral neuropathy, angiokeratomas, hypohidrosis, gastrointestinal symptoms, corneal dystrophy (cornea verticillata), and later cardiac, renal, and CNS involvement. On the other end of the spectrum are milder late-onset or “non-classical” phenotypes. These types are mostly observed in males with a higher degree of residual GLA activity or in symptomatic heterozygous females with primarily one organ system’s impairment (e.g., the cardiac, renal, or nervous system), cerebrovascular disease, and the absence of the classical signs of the disease [10–13].
FD can be misdiagnosed as MS or remain undiagnosed in patients with MS for several reasons (Table 1). Both diseases can present clinically in a “relapsing–remitting” manner and with similar symptoms. Sensory complaints (paresthesia in MS; painful, burning acral sensations in FD) are the most common manifestations of both diseases and can easily be confused. These two diseases also coincide in the occurrence of vertigo and non-specific complaints such as fatigue, the progression of neurological disability, and accentuation of difficulties on exertion or in the heat. Additionally, some disseminated white matter MRI lesions in FD may resemble MS lesions and fulfil criteria for dissemination in space [4]. The last major factor mimicking MS is CSF analysis. The typical findings in patients with MS reflect the inflammatory nature of the disease and include mild pleocytosis, mild protein increase, and, usually, the presence of oligoclonal bands (OCBs) [14]. It was discovered that the CSF of patients with FD could also show a picture of aseptic or chronic meningitis with mild to moderate pleocytosis and mild hyperproteinorachia caused by the aseptic inflammatory process [15, 16]. Very rarely, CSF-restrictive OCBs can also be found [7, 17]. In contrast, OCBs are present in approximately 90% of MS patients [4, 18].
Table 1.
The classic and overlapping findings in multiple sclerosis and Fabry disease
| Multiple sclerosis | Fabry disease | Possible overlapping findings | |
|---|---|---|---|
| Clinical findings | A clinical attack lasts at least 24 h in the absence of fever or infection; typically not sudden onset | Sudden onset of symptoms in case of cerebrovascular event | Relapsing–remitting manner of complaints, and the progression of neurological disability in time |
| Commonly relapsing–remitting manner of complaints; the progression of neurological disability in time | The progression of neurological disability in time | ||
| A common first presentation is unilateral optic neuritis | Cornea verticillata | ||
| Sensory signs (reduced fine touch, vibration sense, joint position sense, tight band-like sensation around the trunk, neuropathic pain), Lhermitte’s phenomenon | Burning acroparesthesias (peripheral small fibre neuropathy) | Sensory symptoms | |
| An upper motor neuron lesion with increased tone or spasticity, pyramidal weakness, hyperreflexia, extensor plantar responses | Cardiac, renal, gastrointestinal involvement, angiokeratomas, hypohidrosis | ||
| Brainstem syndromes (diplopia, oscillopsia, vertigo, isolated sixth nerve palsy, gaze-evoked nystagmus, internuclear ophthalmoplegia) | Vertigo, tinnitus, hearing loss | Vertigo, tinnitus, hearing loss | |
| Fatigue, difficulties on exertion or in the heat | Fatigue, difficulties on exertion or in the heat | Fatigue, difficulties on exertion or in the heat | |
| MRI | At least one T2 hypersignal lesion in two locations t of 4 typical areas of the CNS (juxtacortical/intracortical, periventricular, infratentorial, spinal cord); inflammatory aetiology | Findings consistent with affected vascular territory due to increased prevalence of cerebrovascular events; can occur cerebral micro-bleedings and basilar artery elongation and dilatation | Subcortical and periventricular white matter hyperintensities |
| Demyelinating/inflammatory lesions | Ischemic lesions | ||
|
Typical perivenular distribution, ovoid shape, perpendicular to the ventricles (Dawson fingers) Ovoid lesions in corpus callosum |
T2 hyperintense white matter lesions (subcortical, periventricular, relative sparing of midline and infratentorial structures), typically no corpus callosum | ||
| Brainstem and spinal cord lesions are typically small and usually peripherally located | Infratentorial lesions are ischemic, thus usually centrally located | ||
| Gadolinium enhancement of the new lesion for about a month | No gadolinium enhancement | ||
| Spinal cord lesions are common | No spinal cord involvement | ||
| CSF | OCBs in most of the patients, may be mild pleocytosis (typically lymphocytic), an increase in immunoglobulin concentrations | The characteristic pattern is not known | Pleocytosis, mild hyperproteinorachia, rarely OCBs |
Our study aimed to determine the prevalence of FD among patients with possible or definite MS diagnoses and to evaluate the necessity of including genetic and enzyme screening in the standard testing protocol for differential diagnosis of all suspected MS patients.
Materials and methods
Population of the study
In this prospective monocentric study, we included patients admitted between May 2017 and May 2019, after their first clinical event suggestive of MS, to the largest Czech MS centre. Patients who remained in the MS centre for follow-up until May 2022 were included in the final analysis.
Data collection
Healthcare professionals collected clinical and laboratory data (Table 2) as part of the routine examination process and patient follow-up. FD was diagnosed using dried blood spots via CentoCard (CentoGene AG) in a stepwise manner combining genetic and enzyme testing as described previously [13]. Shortly, in males: enzymatic activity, globotriaosylsphingosine (lyso-Gb3) quantification, if positive, followed by GLA gene sequencing; and in females: GLA sequencing followed by lyso-Gb3. Patients with positive screening results were referred to a specialised FD centre for further clinical and laboratory investigations and management, including family screening. All examinations in the FD centre were performed according to the internal protocol of the FD centre for initial disease evaluation as described in a previous publication [23].
Table 2.
Demographic, clinical, and laboratory characteristics
| All | Definite MS as of May 2022 | |
|---|---|---|
| Study population | 160 | 89 |
| Age in years, mean (SD) | 33.94 (8.24) | 33.94 (8.54) |
| Female (%) | 117 (73.13%) | 65 (73.03%) |
| History of sensory symptoms (%) | 88 (55.00%) | 49 (55.06%) |
| Initial CSF analysis | ||
| Two or more OCBs (%) | 111 (69.38%) | 78 (87.64%) |
| Pleocytosis (%) | 56 (35.00%) | 35 (39.33%) |
| Hyperproteinorachia (%) | 35 (21.88%) | 14 (15.73%) |
| Pathological GLA variants carriers (%) | 0 | 0 |
| GLA variants of unknown significance or probably benign carriers (%) | 4 (2.50%) | 2 (2.25%) |
MS, multiple sclerosis; CSF, cerebrospinal fluid; OCBs, oligoclonal bands
Statistical analyses
The characteristics of the population were summarised using means for continuous variables and frequencies (%) for categorical variables. Patients with genetically proven GLA variants were described separately (Table 3). Data analyses were performed by SPSS version 22.0.0.0.
Table 3.
The genetic, biochemical, and clinical details of patients with proven GLA variants
| Patient | Sex | Age at first symptoms | EA plasma (nmol/mL/h) | EA leukocytes (nmol/mg/h) | Lyso-Gb3 DBS (ng/mL) | First neuro symptoms | OCBs |
|---|---|---|---|---|---|---|---|
| c. 427 G > A, p. Ala143Thr (p.A143T) | |||||||
| 1.0 | F | 49 | 4.59 | 33.3 | 1.0 | Combined sensory-motor | No |
| 1.1 | F | NA | 1.35 | 38.8 | NA | None | NA |
| 1.2 | M | NA | 0.80 | 25.7 | NA | None | NA |
| c. 376A > G, p. Ser126Gly (p.S126G) | |||||||
| 2.0 | F | 29 | 6.34 | 70.4 | Sensory | Yes | |
| 2.1 | F | NA | 4.63 | 46.2 | NA | None | NA |
| c. 937G > T, p. Asp313Tyr (p.D313Y) | |||||||
| 3.0 | F | 44 | 2.33 | 47.6 | 0.8 | Motor and sensory | Yes |
| 4.0 | F | 27 | NA | NA | 0.8 | Sensory | No |
EA, enzyme activity; DBS, dry blood spot; OCBs, two or more oligoclonal bands only in cerebrospinal fluid; F, female; M, male; NA, not applicable/not available; controls: alpha-galactosidase-A in leukocytes 25–103 nmol/mg/h, mean value ± SD 59.7 ± 14.6 nmol/mg/h, n = 477, in plasma 2.4–19.4 nmol/mL/h, mean value ± SD 6.1 ± 2.8 nmol/mL/h, n = 322
Results
Baseline clinical and laboratory characteristics are described in Table 2. The 2017 revised McDonald’s criteria for definite MS [1] were fulfilled by 74 out of 160 (46.3%) enrolled patients at the time of study recruitment. After 3–5 years of follow-up, this number increased to 89 (55.6%) patients. When compared to the whole study population, these 89 patients had a higher percentage with two or more OCBs (87.64%) or pleocytosis (39.33%) and a lower percentage with hyperproteinorachia (15.73%) in the initial CSF examination (Table 2). Four out of all 160 (2.50%) patients (all females) had a positive screening result. The genetic, biochemical, and clinical details of positively screened patients are provided in Table 3.
Neurological symptoms of GLA variant carriers
None of the patients had a recognised pathological variant of the GLA gene. However, four female patients had a variant of the GLA gene considered of unknown significance (p.A143T, p.S126G, 2 × p.D313Y). After a 3–5-year follow-up, only one (p.D313Y) did not fulfil the 2017 revised McDonald’s MS criteria. None of these patients had hyperproteinorachia or pleocytosis in the CSF. Three of the patients with GLA variants experienced sensory symptoms. In a patient with the p.A143T variant (patient 1.0), intrathecal OCB synthesis was absent, and in one patient with p.D313Y (patient 4.0), CSF examination showed only one non-corresponding OCB.
Initial neurological manifestations of patient 1.0 included sensory and motor symptoms and correlated with the focal spinal cord lesion located in the Th9/10 segment. Brain MRI revealed multiple supra- and infratentorial white matter lesions without contrast enhancement (Fig. 1). A detailed comprehensive examination revealed no other explanatory aetiology besides the GLA gene variant. After a sensitive relapse in August 2020, the clinically isolated syndrome (CIS) diagnosis was reclassified to relapsing–remitting MS.
Fig. 1.
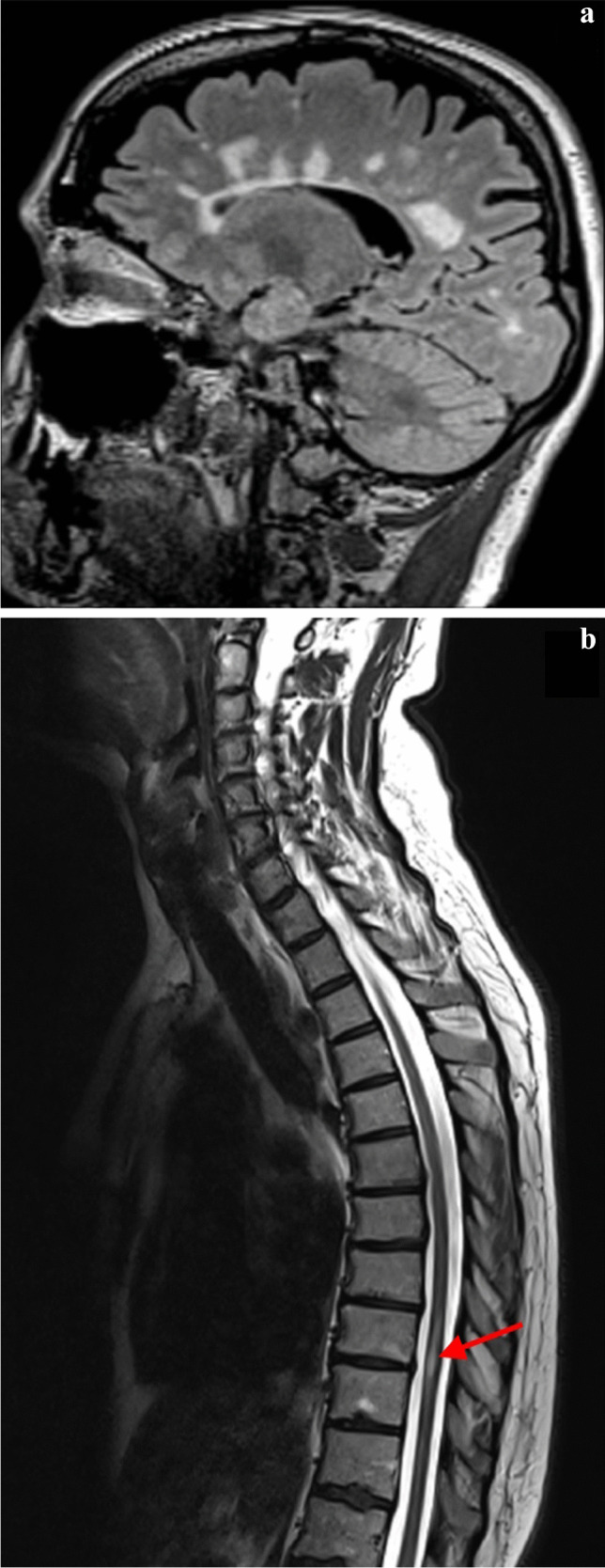
MRI of the patient 1.0. Sagittal FLAIR shows callososeptal hyperintensities radiating from the lateral ventricles with a typical perpendicular orientation (a). Sagittal T2 weighted image shows typical intramedullary lesion (b)
The second patient was diagnosed as a carrier of p.S126G (patient 2.0). Her first symptom was left upper limb hypoesthesia. Brain and spinal cord MRI showed two supratentorial and three intramedullary lesions (Fig. 2). Given the intrathecal OCB synthesis, this patient fulfilled the criteria for definitive MS. Detailed analysis of the organ manifestations of FD showed no symptoms typical of FD or Fabry organ involvement.
Fig. 2.
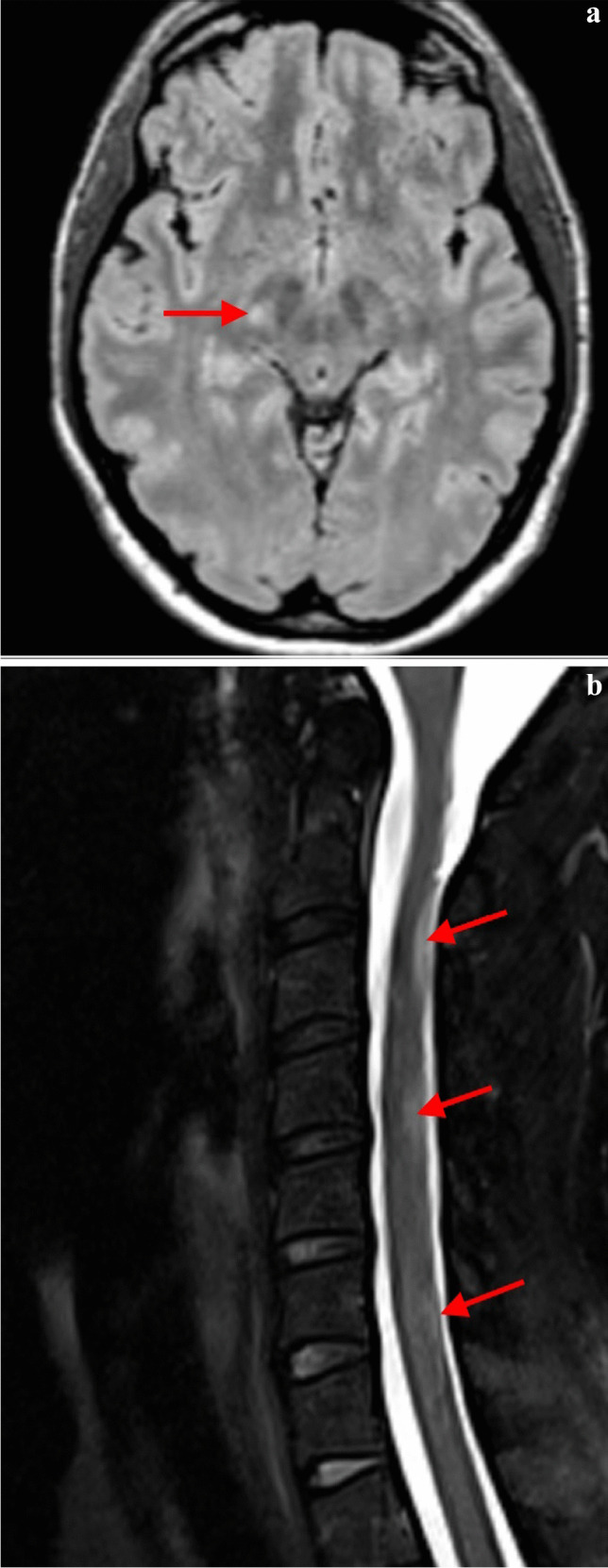
MRI of the patient 2.0. Axial FLAIR shows small lesion (a) and multiple typical laterodorsal hypersignal lesions in cervical spinal cord (b)
The two remaining patients were shown to carry the p.D313Y variant. The first one (patient 3.0) developed a sudden onset of left-sided hemiparesis and left-sided paraesthesia. Initially, stroke as an underlying course was considered. MRI of the brain and cervical spine revealed one pontine and two supratentorial lesions (Fig. 3). CSF examination showed the presence of 11 CSF-restricted OCBs, and patient 3.0 fulfilled definite MS criteria. The second patient with the p.D313Y variant (patient 4.0) contacted a physician for paraesthesia on the dorsum of the hands in 2017. These complaints resolved spontaneously within weeks and reappeared in 2019. That year, an MRI of the cervical spinal cord and brain showed two hyperintensities in the frontal lobe (Fig. 4). This patient was still diagnosed with CIS at the end of the follow-up.
Fig. 3.

MRI of the patient 3.0. Discrete pathological MRI involvements fulfil MRI criteria with typical lesions distribution for MS. Axial FLAIR show small lesions in the frontal lobe (a, b), one in the typical subcortical region (b) and in the pons (c)
Fig. 4.

MRI of the patient 4.0. Discrete pathological MRI involvements. Axial FLAIR shows two small lesion in the periventricular white matter (a, b)
FD phenotype examination, family screening
Except for patient 4.0, all patients agreed to a detailed examination by FD specialists in our referral FD centre. After mild, temporary proteinuria, patient 1.0 underwent a renal biopsy with no typical storage vacuoles being found. However, mild interstitial fibrosis and podocytopathy were shown. Moreover, increased endoplasmatic reticulum (ER) stress and signs of unfolded protein response activation were observed and were associated with alfa-GAL misprocessing in this patient. [24]. The offspring of patient 1.0 were also genetically tested. At the time of examination, the son (patient 1.1) was 27 years old, and the daughter (patient 1.2) was 29. The same GLA variant (p.A143T) was found in both offspring. Patient 1.1 also underwent the examination (except for ophthalmological, which he did not attend) at the FD centre. The tests performed did not show any FD organ manifestations. Patient 1.2 refused a detailed organ assessment. Interestingly, the proband’s father died suddenly, apparently of heart disease, at age 49.
In patient 2.0, the detailed examination did not reveal any FD organ manifestation. Family screening of patient 2.0 led to identifying the same variant in the proband’s mother. An examination in the FD centre did not show any organ involvement attributable to FD. In patient 3.0, only discrete ocular signs (tortuous conjunctival vessels and ocular changes suspicious of incipient Fabry cataract) were found. Relatives of both patients with the p.D313Y variant refused the genetic or clinical examination.
Discussion
Clinical and paraclinical findings in MS may mimic FD and vice versa. It is crucial to differentiate between the two diseases accurately or to recognise the presence of both. Early initiation of treatment before the development of irreversible tissue damage improves the quality of life and prognosis of patients with both diseases. In our 160-patient cohort, we identified four patients and three different types of GLA variants. The atypical findings not supporting MS diagnosis in these four patients included the absence of intrathecal OCB synthesis in two of these patients. Otherwise, the patients did not differ substantially from the rest of the cohort, and there is no reason to doubt the accuracy of their diagnosis of MS. All identified GLA variants are missense ones. The clinical significance of these variants has been the subject of long expert debate. It is worth mentioning that all these variants (p.A143T, p.S126G, p.D313Y) were initially considered disease-causing [25]. Later, their influence on phenotypic manifestations attributable to FD in their carriers was questioned.
The p.A143T is a relatively common variant with an incidence of 1 in every 3800 live births [26]. While some infants with a positive screening for this variant have shown a family history of organ involvement related to FD [12], its pathogenicity remains debated. The carriers of p.A143T do not develop lysosomal storage [27]. The explanation of possible organ manifestations can be the mechanism of AGALopathy, a lysosomal storage-independent pathogenetic factor in FD which was proposed recently [24]. Although the clinical course from a neurological point of view and findings on brain and spinal cord MRI support the definite diagnosis of MS in patient 1.0, the absence of OCBs in the CSF analysis, together with mild renal involvement, does not exclude the possibility that the presence of a GLA variant might potentiate neurological symptoms associated with MS. Furthermore, the enzyme analysis of the patient’s son showed an attenuated biochemical phenotype with low AGAL activity in plasma and borderline activity in leukocytes. Despite normal clinical findings, it might indicate the influence of the variant, at least at the biochemical level. Additionally, intracellular misprocessing of AGAL in p.A143T has been described [24]. However, interpretation of the effect of the variant on the clinical phenotype in the context of its interaction with other genetic, environmental, or even X chromosome inactivation factors is difficult and remains a challenge for further investigation.
Based on the enzyme activity analysis, lyso-GB3 values, and clinical symptoms in a large group of GLA gene variant carriers, the p.S126G variant (frequency 0.07% in non-Finnish Europeans [28]) was not found to be disease-causing and is currently considered a VUS [29]. Our patient 2.0 with variant p.S126G had CSF and MRI findings typical for MS, which also correlated with typical clinical sensory symptoms. Moreover, MS as the cause of the patient’s difficulties was supported by the course of the disease and the normal results of other organ examinations. Neither the patient nor her mother had any typical manifestation that could be attributed to FD.
The p.D313Y variant is the most common of the three variants we found in the population (frequency 0.44% in non-Finnish Europeans [28]). Its contribution to phenotypic manifestations compatible with FD and the controversy regarding its pathogenicity was repeatedly discussed in the literature [30–32]. The prevalence of the p.D313Y variant among our cohort (1.25%) is higher compared with the prevalence of the variant among patients with CNS manifestations (0.59%) [13]. This variant was also the most represented in our previous FD screening among unselected patients with stroke (0.91%) [13]. Given the results of both screenings and the recent finding that D313Y can induce ER stress [24], it might be hypothesised that this variant may play a role as a non-specific low-risk or modifying factor for some CNS diseases. This fact is underscored by the fact that ER stress has already been described in several CNS diseases, including MS [33].
One of our p.D313Y patients (patient 3.0.) showed minimal signs seen in patients with FD, requiring further follow-up by an ophthalmologist. Unfortunately, neither patient 4.0 nor her relatives agreed to a detailed examination at the FD centre. However, the neurological findings, imaging results, CSF analysis, and the clinical course of the disease do not support the influence of the p.D313Y allele on our patient’s clinical symptoms to date.
Therefore, our study brings a negative answer to the question of whether we should routinely screen all patients after the first clinical event suggestive of MS for FD. We did not demonstrate pathogenic carriage of the GLA variant in any of the 160 patients. However, the prevalence of VUS in our cohort was 2.50%, which we consider at least slightly higher compared with the healthy population based on available data [34]. Although the clinical significance of the detected GLA variants is still debated, they do not represent a standard indication for initiating FD-specific treatment. It should also be considered that the stress and psychosocial impact associated with repeated examinations while monitoring FD in VUS carriers with MS can negatively influence the MS course and can be harmful from this point of view [35].
However, given the potential overlap between clinical and laboratory features, it is still necessary to identify red flags and include FD in the differential diagnosis of MS. This is particularly the case when both diagnoses can explain clinical findings and MRI fails to differentiate between white matter hyperintensities of MS or FD origin. The absence of OCBs in CSF, especially in combination with previous findings, is also cautionary (Table 1). In such patients, it is advisable to complete an essential screening of organ manifestations, including urinalysis (proteinuria), eye examination, and cardiac examinations, including ECG and echocardiography. These investigations are commonly available and performed as part of the safety monitoring of patients with MS treated with some disease-modifying drugs. In the case of non-physiological findings during these examinations or organ manifestations associated with FD in the personal or family history, we recommend genetic testing.
Some limitations of the study should be noted. First, our population was relatively small. Second, further research is required in the VUS field concerning their clinical relevance and indication for treatment. Third, our analysis does not directly compare the prevalence of GLA variants in MS and healthy populations. Fourth, one patient with VUS did not agree to a detailed examination at the FD centre. And fifth, different screening methods were used for males and females. The GLA gene sequencing was performed only in males with abnormal AGAL activity and/or elevated lyso-GB3. In contrast, all samples obtained from female patients were sequenced. This may lead to underestimated frequency of several VUS or benign variants in hemizygous males in whom a high residual enzyme activity is preserved, and lyso-Gb3 remains low. Future studies investigating the whole spectrum of genetic variants should use GLA sequencing in all male patients.
To sum up, the prevalence of FD in the population of suspected or definite MS patients seems to be very low. In general, the clinical significance of identified GLA gene variants is still debatable but is not a standard indication for FD therapy. Thus, our results do not support routine FD screening in all patients with possible MS. However, due to the frequent overlap of clinical and laboratory signs, there is still a need to look for red flags and include FD in the differential diagnosis of MS.
Acknowledgements
The authors are very grateful to all healthcare professionals from the General University Hospital and First Faculty of Medicine at Charles University in Prague for participating in the clinical and laboratory investigations and management, namely Martina Zivna and the team of Stanislav Kmoch for molecular biology investigation, Zora Dubska for ocular examination, Ondrej Kodet for skin examination, Helena Poupetova for enzyme activity assessment, and other professionals involved in the analysis of the samples. Special acknowledgements are also due Eliza Varju for language editing.
Author contribution
PR: conceptualisation, methodology, investigation, resources, data curation, writing–original draft, supervision, funding acquisition; IK: conceptualisation, investigation, resources, data curation, writing–review and editing; TU: investigation, resources, writing–review and editing; BS: investigation, resources, data curation, writing–review and editing; GD: investigation; AL: investigation, writing–review and editing; MV: resources, writing–review and editing, visualisation; DS: conceptualisation, methodology, formal analysis, investigation, writing–original draft, visualisation, supervision. All authors read and approved the final manuscript.
Funding
Open access publishing supported by the National Technical Library in Prague. This work was partially supported by a General University Hospital in Prague project (grant number MH CZ-DRO-VFN64165), the Charles University: Cooperatio Program in Neuroscience, a Czech Ministry of Health project (grant number NU22-04–00193), and a National Institute for Neurological Research project funded by the European Union (Next Generation EU, Programme EXCELES, ID Project No. LX22NPO5107). In addition, dry blood spot laboratory tests were financed through a grant from Takeda (formerly Shire) to the Czech Neurological Society. All other examinations of patients, as a part of standard health care, were covered by general health insurance. The authors confirm independence from the sponsors; sponsors have not influenced the article’s content.
Data Availability
Anonymised data not published within this article will be made available by request from any qualified investigator.
Declarations
Ethics approval
The study was approved by the Ethics Committee of the General University Hospital in Prague and complied with the Declaration of Helsinki and good clinical practice guidelines. All patients gave written informed consent to the study, including the genetic analyses.
Informed consent
All patients gave written informed consent to the study, including the genetic analyses.
Conflicts of interest
PR was a consultant for Takeda Pharmaceuticals. She received speaker honoraria from Takeda Pharmaceuticals and Sanofi-Genzyme, travel and accommodation support, and symposium reimbursement from Takeda Pharmaceuticals. IK received financial support for conference travel from Novartis, Teva, Merck, and Roche. TU received financial support for conference travel from Biogen, Novartis, Sanofi, Roche, and Merck and speaker honoraria from Biogen, Novartis, and Roche, as well as support for research activities from Biogen and Sanofi. BS received compensation for travelling and conference fees from Novartis, Sanofi, Biogen, Roche, and Merck, as well as support for research activities from Biogen. AL received consultation and speaker honoraria from Takeda, Amicus, and Sanofi and holds a position on the Fabry registry board and the Amicus FollowME registry board. GD has no relevant conflicts of interest to declare. MV received compensation for travelling, speaker honoraria, and consultant fees from Biogen, Novartis, Roche, Merck, and Teva. DS received financial support for conference travel and/or speaker honoraria from Novartis, Biogen, Merck, Teva, Janssen-Cilag, and Roche.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
P. Rekova and Dominika Stastna contributed equally to this work.
References
- 1.Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17:162–173. doi: 10.1016/S1474-4422(17)30470-2. [DOI] [PubMed] [Google Scholar]
- 2.Ömerhoca S, Yazici Akkaş S, Kale Içen N. Multiple sclerosis: diagnosis and differential diagnosis. Arch Neuropsychiatry. 2018;55:S1. doi: 10.29399/npa.23418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solomon AJ, Klein EP, Bourdette D. “Undiagnosing” multiple sclerosis: the challenge of misdiagnosis in MS. Neurology. 2012;78:1986–1991. doi: 10.1212/WNL.0b013e318259e1b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jurašić MJ, Bašić Kes V, Zavoreo I. Multiple sclerosis and Fabry disease - diagnostic “mixup”.". Mult Scler Relat Disord. 2019;34:112–115. doi: 10.1016/j.msard.2019.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Böttcher T, Rolfs A, Tanislav C, Bitsch A, Köhler W, Gaedeke J, Giese AK, Kolodny EH, Duning T. Fabry disease - underestimated in the differential diagnosis of multiple sclerosis? PLoS ONE. 2013 doi: 10.1371/JOURNAL.PONE.0071894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lidove O, Kaminsky P, Hachulla E, et al. Fabry disease “The New Great Imposter”: results of the French Observatoire in Internal Medicine Departments (FIMeD) Clin Genet. 2012;81:571–577. doi: 10.1111/j.1399-0004.2011.01718.x. [DOI] [PubMed] [Google Scholar]
- 7.Saip S, Uluduz D, Erkol G. Fabry disease mimicking multiple sclerosis. Clin Neurol Neurosurg. 2007;109:361–363. doi: 10.1016/j.clineuro.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 8.Invernizzi P, Bonometti MA, Turri E, Benedetti MD, Salviati A. A case of Fabry disease with central nervous system (CNS) demyelinating lesions: a double trouble? Mult Scler. 2008;14:1003–1006. doi: 10.1177/1352458508092355. [DOI] [PubMed] [Google Scholar]
- 9.Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–677. doi: 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:1–49. doi: 10.1186/1750-1172-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 12.Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ. High incidence of later-onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79:31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomek A, Petra R, Schwabová JP, et al. Nationwide screening for Fabry disease in unselected stroke patients. PLoS One. 2021;16:e0260601. doi: 10.1371/journal.pone.0260601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Domingues RB, Fernandes GBP, de Moura Leite FBV, Tilbery CP, Thomaz RB, Silva GS, Mangueira CLP, Soares CAS. The cerebrospinal fluid in multiple sclerosis: far beyond the bands. Einstein (Sao Paulo) 2017;15:100–104. doi: 10.1590/S1679-45082017RW3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tang MY, Hong YH, Zhou LX, Ni J. Fabry disease with aseptic meningitis: a case series and literature review of an underestimated clinical presentation. Curr Med Sci. 2022;42:274–279. doi: 10.1007/s11596-022-2578-4. [DOI] [PubMed] [Google Scholar]
- 16.Lidove O, Chauveheid MP, Caillaud C, et al. Aseptic meningitis and ischaemic stroke in Fabry disease. Int J Clin Pract. 2009;63:1663–1667. doi: 10.1111/j.1742-1241.2009.02115.x. [DOI] [PubMed] [Google Scholar]
- 17.Rost NS, Cloonan L, Kanakis AS, et al. Determinants of white matter hyperintensity burden in patients with Fabry disease. Neurology. 2016;86:1880. doi: 10.1212/WNL.0000000000002673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Link H, Huang YM. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: an update on methodology and clinical usefulness. J Neuroimmunol. 2006;180:17–28. doi: 10.1016/j.jneuroim.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Dutra-Clarke M, Tapia D, Curtin E, et al. Variable clinical features of patients with Fabry disease and outcome of enzyme replacement therapy. Mol Genet Metab Rep. 2021;26:100700. doi: 10.1016/j.ymgmr.2020.100700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ford H. Clinical presentation and diagnosis of multiple sclerosis. Clin Med. 2020;20:380. doi: 10.7861/clinmed.2020-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wattjes MP, Ciccarelli O, Reich DS, et al. 2021 MAGNIMS-CMSC-NAIMS consensus recommendations on the use of MRI in patients with multiple sclerosis. Lancet Neurol. 2021;20:653–670. doi: 10.1016/S1474-4422(21)00095-8. [DOI] [PubMed] [Google Scholar]
- 22.Cocozza S, Russo C, Pontillo G, Pisani A, Brunetti A. Neuroimaging in Fabry disease: current knowledge and future directions. Insights Imaging. 2018;9:1077. doi: 10.1007/s13244-018-0664-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reková P, Dostálová G, Kemlink D, et al. Detailed phenotype of GLA variants identified by the nationwide neurological screening of stroke patients in the Czech Republic. J Clin Med. 2021 doi: 10.3390/JCM10163543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Živná M, Dostálová G, Barešová V et al (2022) AGAL misprocessing-induced ER stress and the unfolded protein response: lysosomal storage-independent mechanism of Fabry disease pathogenesis? bioRxiv 2022.09.27.509714. 10.1101/2022.09.27.509714
- 25.Eng CM, Niehaus DJ, Enriquez AL, Burgert TS, Ludman MD, Desnick RJ. Fabry disease: twenty-three mutations including sense and antisense CPG alterations and identification of a deletional hot-spot in the α-galactosidase A gene. Hum Mol Genet. 1994;3:1795–1799. doi: 10.1093/hmg/3.10.1795. [DOI] [PubMed] [Google Scholar]
- 26.Kiesling JL (2014) Missouri’s full population pilot screening for Fabry disease and the implications for families. https://www.aphl.org/conferences/proceedings/Documents/2014/NBS/29Kiesling.pdf. Accessed 26 Dec 2022
- 27.Terryn W, Vanholder R, Hemelsoet D, et al. Questioning the pathogenic role of the GLA p.Ala143Thr “Mutation” in Fabry disease: implications for screening studies and ERT. JIMD Rep. 2013;8:101–108. doi: 10.1007/8904_2012_167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Genome Aggregation Database. https://gnomad.broadinstitute.org/. Accessed 26 Dec 2022
- 29.Duro G, Zizzo C, Cammarata G, et al. Mutations in the GLA gene and LysoGb3: is it really Anderson-Fabry disease? Int J Mol Sci. 2018 doi: 10.3390/IJMS19123726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Effraimidis G, Rasmussen ÅK, Bundgaard H, Sørensen SS, Feldt-Rasmussen U. Is the alpha-galactosidase A variant p.Asp313Tyr (p. D313Y) pathogenic for Fabry disease? A systematic review. J Inherit Metab Dis. 2020;43:922–933. doi: 10.1002/jimd.12240. [DOI] [PubMed] [Google Scholar]
- 31.Hasholt L, Ballegaard M, Bundgaard H, et al. The D313Y variant in the GLA gene - no evidence of a pathogenic role in Fabry disease. Scand J Clin Lab Invest. 2017;77:617–621. doi: 10.1080/00365513.2017.1390782. [DOI] [PubMed] [Google Scholar]
- 32.Palaiodimou L, Stefanou MI, Bakola E, et al. D313Y variant in Fabry disease: a systematic review and meta-analysis. Neurology. 2022;99:e2188–e2200. doi: 10.1212/WNL.0000000000201102. [DOI] [PubMed] [Google Scholar]
- 33.Haile Y, Deng X, Ortiz-Sandoval C, et al. Rab32 connects ER stress to mitochondrial defects in multiple sclerosis. J Neuroinflammation. 2017;14:1–13. doi: 10.1186/s12974-016-0788-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russo C, Cocozza S, Riccio E, et al. Prevalence of GLA gene mutations and polymorphisms in patients with multiple sclerosis: a cross-sectional study. J Neurol Sci. 2020 doi: 10.1016/J.JNS.2020.116782. [DOI] [PubMed] [Google Scholar]
- 35.Briones-Buixassa L, Milà R, Ma Aragonès J, Bufill E, Olaya B, Arrufat FX. Stress and multiple sclerosis: a systematic review considering potential moderating and mediating factors and methods of assessing stress. Health Psychol Open. 2015 doi: 10.1177/2055102915612271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymised data not published within this article will be made available by request from any qualified investigator.